
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmine-functionalized porous organic polymers for carbon dioxide capture
Ali K.
Sekizkardes
 *ab,
Ping
Wang
a,
James
Hoffman
a,
Samir
Budhathoki
a and
David
Hopkinson
a
*ab,
Ping
Wang
a,
James
Hoffman
a,
Samir
Budhathoki
a and
David
Hopkinson
a
aU.S. Department of Energy National Energy Technology Laboratory, Pittsburgh, PA 15236, USA. E-mail: ali.sekizkardes@netl.doe.gov
bNETL Support Contractor, Pittsburgh, PA 15236, USA
First published on 15th June 2022
Abstract
Recent developments in CO2 capture using porous organic polymers (POPs) have received accrescent attention due to their sorbent properties such as high CO2 uptake capacity and selectivity, tunable chemical structure and permanent porosity. POPs are constructed using two and/or three-dimensional organic monomers (building blocks) linked to each other through covalent bonding, creating high porosity. The pore structure in POPs is exceptionally stable, which leads to their cyclable CO2 adsorption performance. However, POPs generally suffer from low CO2 uptake and selectivity due to their interaction with CO2 in physisorption limits (20–40 kJ mol−1). Similar to that in other physisorbents, the CO2 uptake capacity of POPs further decreases under humid conditions. Pursuant to these limitations, amine functionalization in POPs has resulted in enhanced CO2 uptake performance with improved CO2 selectivity over non-polar gases such as N2. More importantly, several types of amine-functionalized POPs showed that the CO2 uptake could remain intact under humid conditions such as in post-combustion flue gas. This review article covers recent developments in amine-functionalized porous organic polymers. Three main categories of amine functionalization, such as direct amine synthesis, amine impregnation and amine grafting, were investigated in detail by considering the effect of amines on the sorbent properties and CO2 capture performance of POPs. The recent findings in amine-functionalized POPs were investigated including porous polymeric networks (PPNs), covalent organic frameworks (COFs), amine linked POPs, hyper-crosslinked polymers (HCPs), conjugated microporous polymers (CMPs), benzimidazole linked polymers (BILPs), porous aromatic frameworks (PAFs) and polymers of intrinsic microporosity (PIMs).
 Ali K. Sekizkardes | Dr. Ali K. Sekizkardes is a materials scientist employed by Batelle at the U.S. Department of Energy National Energy and Technology Laboratory (NETL). His current research entails the development of polymeric sorbent and membrane technology for CO2 capture and separation applications. He received his PhD in Materials Chemistry at Virginia Commonwealth University in 2014 under the supervision of Prof. Hani El-Kaderi. He synthesized and functionalized porous organic polymers and covalent organic frameworks for CO2 capture and hydrogen storage. He later joined NETL as an ORISE postdoctoral fellow in 2015 and worked on polymeric and mixed matrix membrane development for gas separation. |
 Ping Wang | Dr. Ping Wang is a Principal Investigator at National Energy Technology Laboratory (NETL) in Pittsburgh, PA. She has been working on advanced thermochemical processes (pyrolysis, gasification and combustion) of converting carbonaceous materials (coal, biomass and waste plastic) to energy, in the form of solid, liquid and gaseous fuels. Her research focuses on the development of technologies and materials for CO2 emission control. Dr Wang obtained her BS and MS in chemical engineering from Zhejiang University, China. She earned her PhD in Agricultural and Biological Engineering from the University of Illinois at Urbana-Champaign (UIUC). |
 James Hoffman | James Hoffman is a research engineer with 31 years of experience within the Research & Innovation Center of the U.S. Department of Energy, National Energy Technology Laboratory. His primary areas of research have centered on environmental controls for the use of fossil fuels in electric generation. He currently serves as Task Technical Coordinator for post combustion CO2 capture using solid sorbents. He has a BS in Chemical Engineering, MS & PhD in Mechanical Engineering, all from Penn State University. |
 Samir Budhathoki | Dr. Samir Budhathoki received his PhD in chemical engineering from the University of Notre Dame. His PhD research focused on investigating the thermodynamic and transport properties of ionic liquids and solute gases: carbon dioxide, methane, and hydrogen in bulk and under confinement in nanoporous materials using molecular dynamics and Monte Carlo simulation methods. Then, he worked as a research fellow at the National Technology Lab (NETL), Pittsburgh, and as a research scientist at AECOM, investigating metal–organic frameworks and mixed matrix membranes for carbon dioxide separation from flue gas. Currently, he is a research scientist at Battelle Memorial Institute, Pittsburgh, where he uses machine learning and molecular simulation to evaluate various materials for direct air capture (DAC) and water purification applications. |
David Hopkinson is the Technical Portfolio Lead for Point Source Carbon Capture at the U.S. Department of Energy, National Energy Technology Laboratory. Prior to joining NETL, Dr. Hopkinson was a technology analyst for the Innovations for Existing Plants program at the U.S. Department of Energy, Office of Fossil Energy in Washington, D.C. Dr. Hopkinson completed his BS and MS degrees in mechanical engineering from Georgia Tech in 2002 and 2003, and earned his PhD in mechanical engineering from Virginia Tech in 2008. |
1. Introduction
Carbon capture and storage (CCS) is a viable alternative to reduce the emissions of the greenhouse gas carbon dioxide (CO2) from large point sources.1 It holds the potential to provide significant reductions in greenhouse gas emissions.2 CCS is nominally a two-step process where the separation of carbon dioxide from a mixed gas stream is followed by permanent storage. Of particular interest are power generation and other industrial point sources that use fossil fuels. Since nearly one-third of the anthropogenic CO2 emissions are produced by these facilities, conventional coal and natural gas-burning power plants, as well as industrial sources, present opportunities where carbon can be removed and then permanently stored.3 Coal-fired steam cycles have been the predominant electric power generation technology in the United States, but have been supplanted by natural gas-fired plants.4 Post-combustion technologies for capturing CO2 will need to be applied for both power generation and industrial sources such as cement and steel production that will continue to be a notable source of CO2 emissions. The capture step for carbon dioxide represents a major cost in the overall process.5The removal of carbon dioxide (CO2) from a gas stream is the most energy intensive step in the overall carbon sequestration process.6 Capture techniques could be retrofitted onto existing conventional fossil fuel power plants or integrated into new power generation facilities.5 Although the separation of CO2 from other gas constituents can be approached using different methods, the ultimate goal is for CO2 to be concentrated into a process stream that is more amenable for further carbon sequestration treatment, including compression, transportation, and storage.7 The selection of technology for a given capture application depends on many factors, including the partial pressure of CO2 in the gas stream, temperature, total system pressure, the process gas composition, the extent of CO2 recovery, sensitivity to impurities, such as acid gases and particles, the purity of the recovered CO2 product, capital and operating costs of the process, and environmental impacts.8
In addition to the point source capture of CO2, there is more recent interest in capturing CO2 that is already present in the atmosphere. This strategy of negative emission technology (NET) has led to the introduction of direct air capture (DAC).9–11 For DAC applications, CO2 is present at much lower concentration levels (∼400 ppm). It is anticipated that a stronger binding affinity will be required for trace capture, and hence chemisorption rather than physisorption processes will likely dominate as DAC solutions are pursued.12
The conventional CO2 removal technology that was the comparative baseline for all other CO2 capture technologies utilizes monoethanolamine (MEA) as a liquid solvent.13 This wet scrubbing process removes the CO2 in an absorption column and then regenerates the CO2 rich solution in a vessel by heating with plant steam.14 Although there have been large-scale commercial demonstrations of this technology, the process has several disadvantages, such as high heat of the reaction, low working capacity, corrosivity of the solution, the susceptibility of being poisoned by minor contaminants, and, most notably, its need to be in aqueous solution. This latter results in increased energy to regenerate the spent solution, especially the sensible heating of water which is about 70 wt% of the solution. Another energy loss while regenerating the spent MEA solution includes the evaporative heat loss from vaporizing liquid water.15
Alternatively, the adsorption process is an attractive CO2 capture technology because of its low energy requirement.16–18 The adsorption is physical or chemical interaction between the adsorbate, i.e., CO2, and the surface of the solid adsorbent. Desorption of CO2 can be achieved by decreasing the pressure or increasing the temperature.19 Solid porous adsorbents have great potential due to their attractive properties such as high CO2 uptake capacity, selectivity, tunable chemical structure, and adjustable porosity.20 In general, the porous adsorbent portfolio consists of zeolites,21 silica,22 porous carbon,23 metal–organic frameworks (MOFs),24 and, more recently, porous organic polymers (POPs).25,26
As a relatively new CO2 adsorbent, porous organic polymers (POPs) have recently attracted a considerable amount of research attention because of their very high porosity coupled with functionality and scalability.27–29 Although not as high as in MOFs, the surface area of POPs can also be extremely high.30 The high porosity in POPs is stabilized by strong covalent bonding between the building blocks (monomers) of the polymer.31 By contrast, in MOFs, the building blocks are connected to each other through relatively weaker coordination bonds.32 Given the strong covalent bonding between the building blocks, the pore structure of POPs is exceptionally stable, resulting in stable CO2 capture performance with absorption and desorption cycling.
The CO2 uptake capacity of POPs is much higher than that of conventional polymers.33 On the other hand, POPs generally suffer from low CO2 uptake and selectivity compared to chemical sorbents and solvents due to their relatively weak interaction with CO2 (within physisorption limits, 20–40 kJ mol−1).34 The CO2 uptake capacity of POPs further decreases under humid conditions. The recent developments have shown that amine functionalization in POPs drastically increases the CO2 capture performance with improved CO2 selectivity over non-polar gases such as N2.35,36 Furthermore, some amine-functionalized POPs demonstrated that the CO2 capture capacity could remain high under humid conditions such as in post-combustion flue gas and direct air capture.37–39
In this review article, we present a concise evaluation of recent developments in amine-functionalized porous organic polymers. Three types of amine functionalization are reviewed: direct amine synthesis, amine grafting, and amine impregnation (Table 1). In the literature, direct amine synthesis, also classified as pre-synthetic functionalization, is amine functionalization during the polymer synthesis. Thus, either the monomer and/or the linker contains amine functionality. Amine grafting, on the other hand, entails the post-synthetic functionalization of sorbents through covalent bonding. Amine impregnation is the physical blending of the sorbent with an amine to boost the CO2 uptake capacity and lacks any chemical bonds.
| Amine functionalization methods in porous organic polymers | |||
|---|---|---|---|
| Direct synthesis | Amine grafting | Amine impregnation | |
| Amine type | Aromatic amines | Aromatic amines | Alkylamines |
| Alkylamines | |||
| Surface area | On monomers, linkers | On pores | In pores and surface |
| CO2 capture | High | Moderate to high | High |
| CO2 selectivity | Moderate | High | High |
| CO2 cyclability | High | High | Moderate |
| CO2 desorption energy | Low | Low to medium | High |
The recent findings in amine-functionalized POPs are examined including porous aromatic frameworks (PAFs),40 covalent organic frameworks (COFs),41 amine linked POPs,42 hyper-crosslinked polymers (HCPs),43 conjugated microporous polymers (CMPs),44 benzimidazole linked polymers (BILPs),45 and polymers of intrinsic microporosity (PIMs).46 The polymer structure and property relationship was investigated for CO2 capture applications via experimental and computational studies, with properties summarized for a large variety of POPs (Table 2).
| Porous organic polymer | Amine functionalization method | Amine type/concentration | SABET (m2 g−1) | Pore size (nm) | CO2 uptake (mmol g−1) | CO2/N2 selectivity | ΔHCO2 (kJ mol−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Porous aromatic frameworks (PAFs) | ||||||||
| PAF-33-NH2 | Direct synthesis | Aromatic amine | 370 | — | 0.33 | 79.8 | 32.9 | 89 |
| 0.15 bar, 298 K | ||||||||
| PEI (40 wt%) ⊂ PAF-5 | Amine impregnation | Polyethylenimine/40 wt% | 40.3 | — | 2.6 | 1200 | 68.7 | 58 |
| 0.15 bar, 298 K | ||||||||
| PPN-6-SO3NH4 | Amine grafting | SO3NH4 | 593 | 1.78 | 196 | 40 | 90 | |
| 0.15 bar, 298 K | ||||||||
| PPN-6-DETA | Amine grafting | Alkylamine (DETA) | 555 | 3.08 | 442 | 55 | 52 | |
| 0.15 bar, 298 K | ||||||||
| PPN-125-DETA | Amine grafting | Alkylamine (DETA) | 229 | 1.43 | — | 62 | 55 | |
| 0.15 bar, 298 K | ||||||||
| Covalent organic frameworks (COFs) | ||||||||
| ILCOF-1 | Direct synthesis | Imine | 2723 | 2.3 | 29.3 | — | 18.3 | 68 |
| 40 bar, 298 K | ||||||||
| TpPa-1 | Direct synthesis | Keto−enamine | 535 | 1.25 | 3.48 | 69 | ||
| 1 bar, 273 K | ||||||||
| COF-JLU2 | Direct synthesis | Azine | 410 | 0.96 | 4.93 | 77 | 31 | 91 |
| 1 bar, 273 K | ||||||||
| [EtNH2]75-H2P-COF | Amine grafting | Ethylamine | 568 | 1.6 | 3.6 | — | 20.8 | 72 |
| 1 bar, 273 K | ||||||||
| FCTF-1-600 | Direct synthesis | Triazine | — | — | 1.76 | 77 | 31 | 92 |
| 0.1 bar, 273 K | ||||||||
| Amine linked porous organic polymers | ||||||||
| BILP-10 | Direct synthesis | Imidazole | 787 | 0.73 | 0.73 | 57 | 38.2 | 79 |
| 0.15 bar, 273 K | ||||||||
| TBILP-1 | Direct synthesis | Imidazole, triazine | 330 | 0.55 | 0.55 | 63 | 35 | 80 |
| 0.15 bar, 273 K | ||||||||
| SNW-1 | Direct synthesis | Melamine | 821 | 0.67 | 0.67 | 50 | 35 | 77 |
| 0.15 bar, 273 K | ||||||||
| TNP-4 | Direct synthesis | Triazole | 1348 | 0.66 | 2.87 | 27 | 36.5 | 84 |
| 1 bar, 298 K | ||||||||
| COP-97A | Amine impregnation | Polyethyleneimine | 118 | — | 1.74 | 965 | — | 87 |
| 0.15 bar, 298 K | ||||||||
| Hyper crosslinked polymers (HCPs) | ||||||||
| xPEI@XAD-4-pc | Amine impregnation | Polyethyleneimine/30 wt% | 334 | 9.89 | 3.24 | 1103 | 53.01 | 93 |
| 0.1 bar, 298 K | ||||||||
| HCP-MAAMs | Direct synthesis | NH2 | 298 | 2 | 1.56 | 104 | 35 | 94 |
| 0.15 bar, 273 K | ||||||||
| xTEPA@PDVBpc | Amine impregnation | Tetraethylenepentamine/30 wt% | 330 | 6.44 | 3.11 | — | — | 95 |
| 0.1 bar, 298 K | ||||||||
| PBTP-(x)-DETA | Amine grafting | Diethylenetriamine (DETA) | 600 | 4 | 2.27 | 1064 | 35 | 37 |
| 0.15 bar, 298 K | ||||||||
| Conjugated microporous polymers (CMPs) | ||||||||
| CMP-1 NH2 | Direct synthesis | NH2 | 710 | — | 0.96 | 14.6 | 29.5 | 96 and 97 |
| 1 bar, 298 K | ||||||||
| NaNO3@CMP-PTPA | Direct synthesis | NH | 1028 | — | 3.6 | — | — | 98 |
| 1 bar, 273 K | ||||||||
| NCMP-I | Direct synthesis | NH | 945 | 2 | 1.43 | — | 38 | 99 |
| 1 bar, 298 K | ||||||||
| NCMPIII | Direct synthesis | NH | 593 | 2 | 0.4 | — | 28 | 99 |
| 1 bar, 298 K | ||||||||
| KCMP-M3 | Amine grafting | NH2 | 1321 | — | 0.52 | — | 28 | 100 |
| 0.15 bar, 298 K | ||||||||
| POP (2) | Amine grafting | NH2 and NH | 485 | — | 0.27 | 155 | 50 | 101 |
| 0.15 bar, 298 K | ||||||||
| Polymers of intrinsic micro porosity (PIMs) | ||||||||
| 21 wt% PEI-PIM-1 | Amine impregnation | Polyethyleneimine/21 wt% | — | — | 1.24 | — | — | 102 |
| 0.15 bar, 298 K | ||||||||
| PIM-1-C3-TA | Amine impregnation | TAEA/15 wt% | — | — | 1.62 | — | — | 103 |
| 0.15 bar, 298 K | ||||||||
| PIM-1-AO-deta | Amine impregnation | DETA/12 wt% | — | — | 1.75 | — | — | 104 |
| 0.15 bar, 298 K | ||||||||
| Computationally designed porous organic polymers | ||||||||
| TrzPOP-1 | Direct synthesis | Triazine, secondary amine | 995 | — | 3.53 | 27 | 29 | 105 |
| 1 bar, 298 K | ||||||||
| TrzPOP-2 | Direct synthesis | Triazine, secondary amine | 868 | — | 4.52 | 72 | 34 | 105 |
| 1 bar, 298 K | ||||||||
2. Amine functionalization in porous aromatic frameworks
Porous aromatic frameworks (PAFs) are one of the most studied classes of porous polymers.47,48 The polymer structure of PAFs is constructed from rigid aromatic monomers, which are linked to each other by a cross coupling reaction. The high surface area and porosity can be created within PAFs by the targeted topology design of the monomers (building blocks).49 For example, PAF-1 was synthesized by homocoupling of 3-dimensional tetrahedral monomers with a very high surface area of 5640 m2 g−1. The high porosity of PAFs has been utilized in many different applications as well as in CO2 capture applications.50 For CO2 capture, the porosity in PAFs presents a prodigious opportunity to reach high CO2 uptake capacity in a given volume. In addition to the porosity and surface area, the pore functionality also plays an important role to increase the CO2 affinity of the sorbent.51 Accordingly, PAFs have been functionalized using various amine groups for advanced CO2 capture.47Zhou et al. have developed several amine-functionalized PAFs, which are also referred to as porous polymeric networks (PPNs).35 Specifically, PPN-6 has been studied intensively due to its very high surface area of 4023 m2 g−1. PPN-6 was post-synthetically functionalized (grafted) via alkyl amines such as diethylenetriamine (DETA) (Fig. 1A).52 The amine groups were yielded from a reaction of amines with the intermediate methyl halide groups in PPN-6. The amine functionalization in PPN-6 resulted in porosity loss in the polymer. However, the CO2 uptake performance of PPN-6-CH2-DETA was reported to be at least 15 fold larger (3.08 mmol g−1 at 298 K 0.15 bar) compared to the unfunctionalized polymer (PPN-6). Intriguingly, the CO2 capture capacity was reported to be stable over many adsorption–desorption cycles. This stability can be attributed to the covalently bonded DETA in PPN-6, which impedes the amines leaching from the polymer.
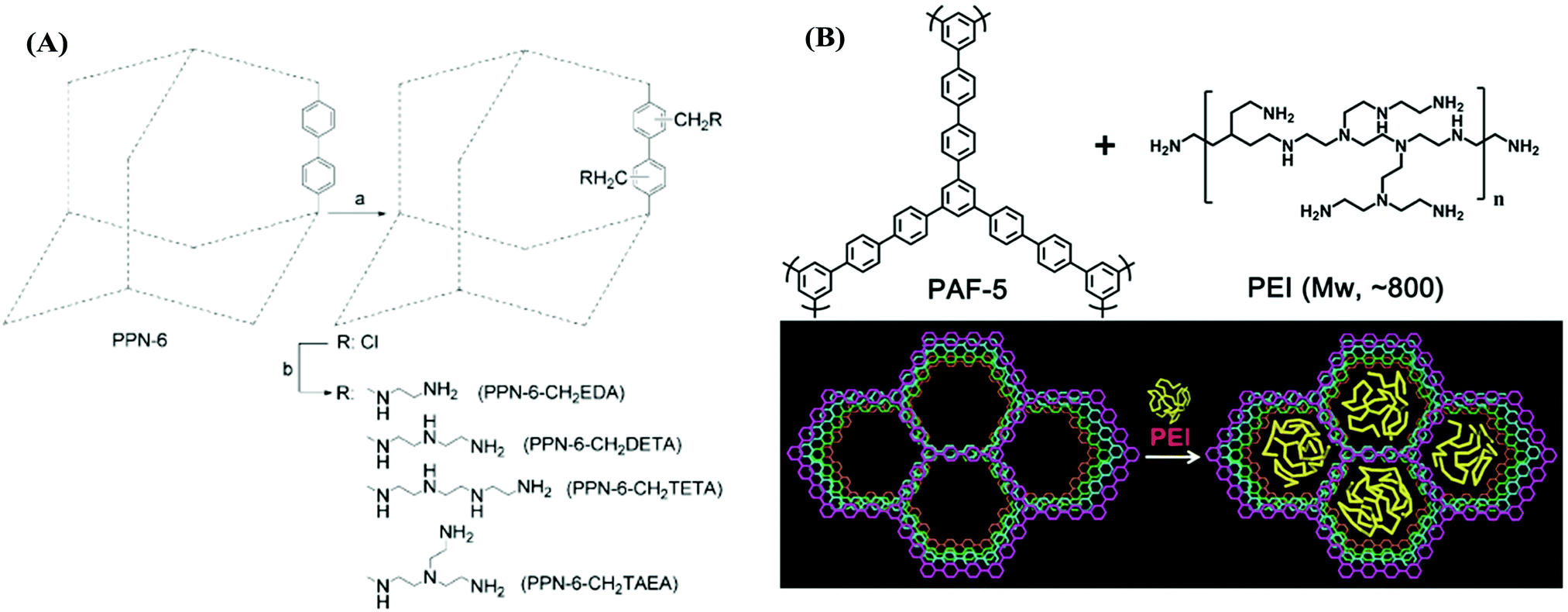 | ||
| Fig. 1 (A) Post-synthetic amine functionalization of PPN-6. (B) Amine (PEI) impregnation in PAF-1. Reproduced with permission from ref. 52 and 58, respectively. | ||
Other alkylamines including ethylenediamine (EDA) and triethylenetetramine (TETA) have also been incorporated in PPNs (Fig. 1A) and a similar trend in CO2 uptake enhancement was also observed.52 Studies have pointed out that the selection of the alkylamine plays a significant role in tuning the CO2 uptake in PPNs as well as in other sorbents such as MOFs.53 For example, triamines such as tris(2-aminoethyl)amine (TAEA) showed a higher CO2 adsorption capacity compared to diamines (EDA) at a low partial pressure of CO2, which may be advantageous in low concentration CO2 capture applications such as DAC.
One of the major drawbacks of PAFs is the cost of polymerization and scalability. The reaction protocol is heavily dependent on the use of expensive heterogeneous catalysts.54 PPN-125 has been alternatively offered with a relatively cost-efficient preparation method, circumventing the use of expensive catalysts.55 Post-synthetic functionalization of PPN-125 with DETA yielded a good CO2 adsorption capacity for PPN-125-DETA, despite a lowered surface area of 229 m2 g−1. The CO2 absorption/desorption cycling of PPN-6-CH2-DETA did not result in any notable reduction in the CO2 uptake capacity.
In another study, PPN-6 has been functionalized with amine groups using acid–base interaction between the polymer and amines.56 First, the polymer was functionalized using a sulfonic acid (PPN-6-SO3H). In the second step, PPN-6-SO3H was treated with amines to afford PPN-6-NH4. The acid–base interaction concept between the polymer and amines could be applied to many other future PAFs. PAF-1 was also functionalized using alkylamines and the dramatic CO2 uptake improvement was recorded over the non-functionalized PAF-1 at a low partial pressure of CO2. In PAF-1, the amine groups were created by deprotection of phthalimidomethyl groups which were intermediately placed in PAF-1.57
The amine impregnation method has also been adopted in PAFs. Polyethylene amine (PEI) was impregnated in PAF-5, resulting in a depletion of the porosity but with a dramatic increase in the CO2 adsorption capacity and CO2/N2 selectivity (Fig. 1B and Table 2).58
Besides PAFs and PPNs, several other types of other POPs have been also synthesized by cross-coupling reactions and then post-synthetically functionalized using amines.59 For example, Islamoglu et al. reported a nanoporous organic framework (NPOF-4) by the coupling reaction of the adamantane based monomer (TAPA) (Fig. 2).60 NPOF-4 shows lower surface area compared to PAF-1 and PPN-4. However, polymerization does not entail the use of an expensive catalyst. In addition, the amine functionalization method of NPOFs showed that benzene groups in PAFs can be easily nitrated and subsequently aminated.
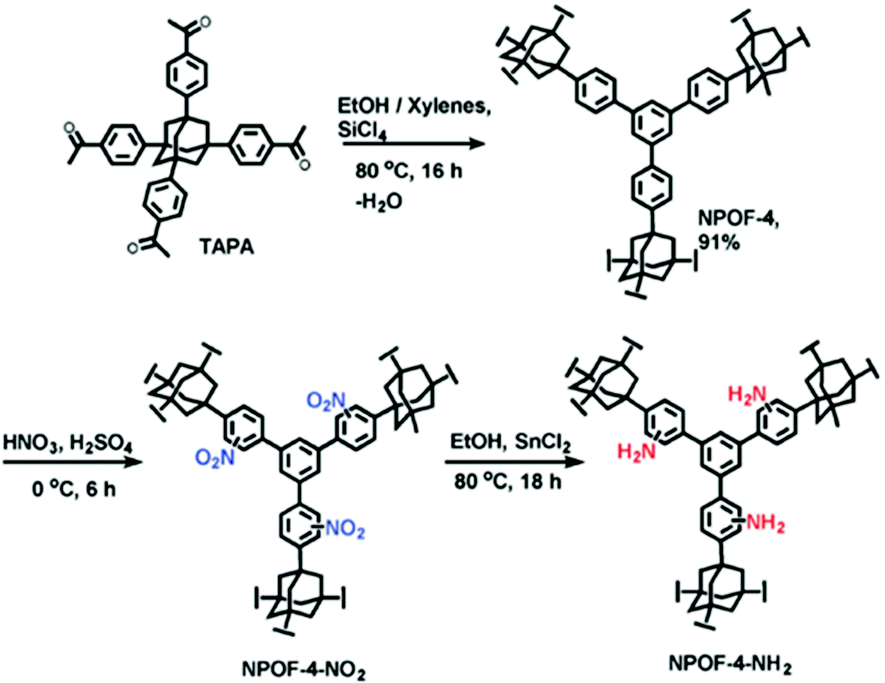 | ||
| Fig. 2 Post-synthetic functionalization of NPOF-4 with amines. Reproduced with permission from ref. 60. | ||
3. Amine functionalization in covalent organic frameworks and amine linked porous organic polymers
Covalent organic frameworks (COFs) are a highly studied class of POPs for CO2 capture and other applications.61 In general, COFs are synthesized by a dynamic equilibrium reaction from aromatic monomers with 2D and/or 3D topology.62 The rigid and multidimensional monomers and reversibility of the reaction deliver high porosity and the crystalline polymer structure of COFs, respectively. Given their crystalline structures, control over the functional groups can be very precise in COFs.63,64Amine functionalization in COFs has been achieved through the direct amine synthesis method (Table 1). Pioneered by Yaghi, imine-linked COFs have been the frontrunner COF in CO2 capture.65 In imine-linked COFs, the amine functionality (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– bonds) is yielded by the Schiff base polycondensation reaction between aryl aldehyde and arylamine monomers.66 The high surface area coupled with the imine functionality results in high CO2 capture capacity at high CO2 pressure.67 For example, Rabbani et al. reported an imine-linked COF, ILCOF-1 (Fig. 3), exhibiting a very high surface area of 2723 m2 g−1 and a remarkable CO2 uptake capacity of 29.3 mmol g−1 at 298 K and 40 bar (Table 2).68
N– bonds) is yielded by the Schiff base polycondensation reaction between aryl aldehyde and arylamine monomers.66 The high surface area coupled with the imine functionality results in high CO2 capture capacity at high CO2 pressure.67 For example, Rabbani et al. reported an imine-linked COF, ILCOF-1 (Fig. 3), exhibiting a very high surface area of 2723 m2 g−1 and a remarkable CO2 uptake capacity of 29.3 mmol g−1 at 298 K and 40 bar (Table 2).68
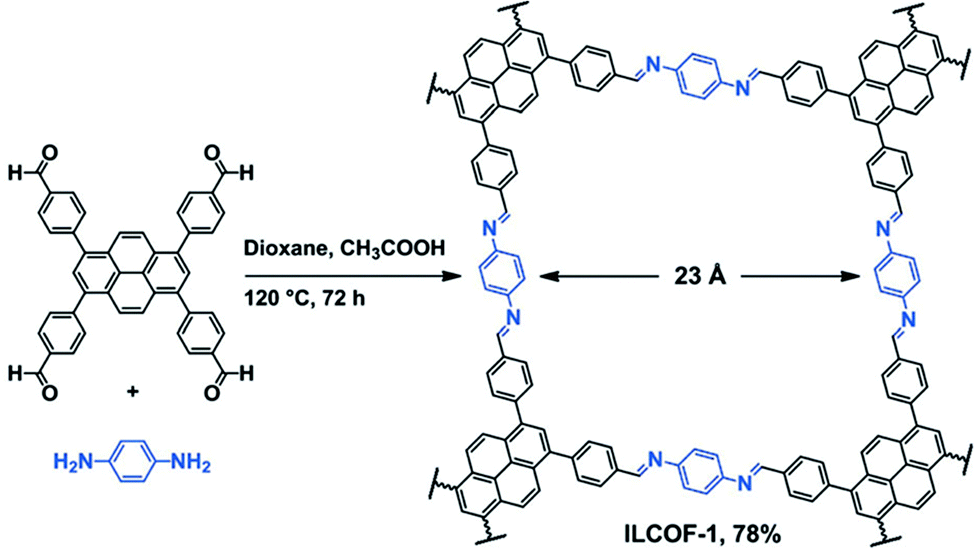 | ||
| Fig. 3 Synthetic scheme of the imine linked COF (ILCOF-1). Reproduced with permission from ref. 68. | ||
The reduction of imine linked COFs has also been demonstrated as an alternative route to increase the low concentration CO2 uptake in COFs. Accordingly, TpPa-1 and TpPA-2 were synthesized by the polycondensation reaction of 1,3,5-triformylphloroglucinol (Tp) with p-phenylenediamine (Pa-1) and 2,5-dimethyl-p-phenylenediamine (Pa-2) (Fig. 4).69 In this reaction, irreversible proton tautomerism converted imine groups into stronger Lewis basic secondary amine groups.
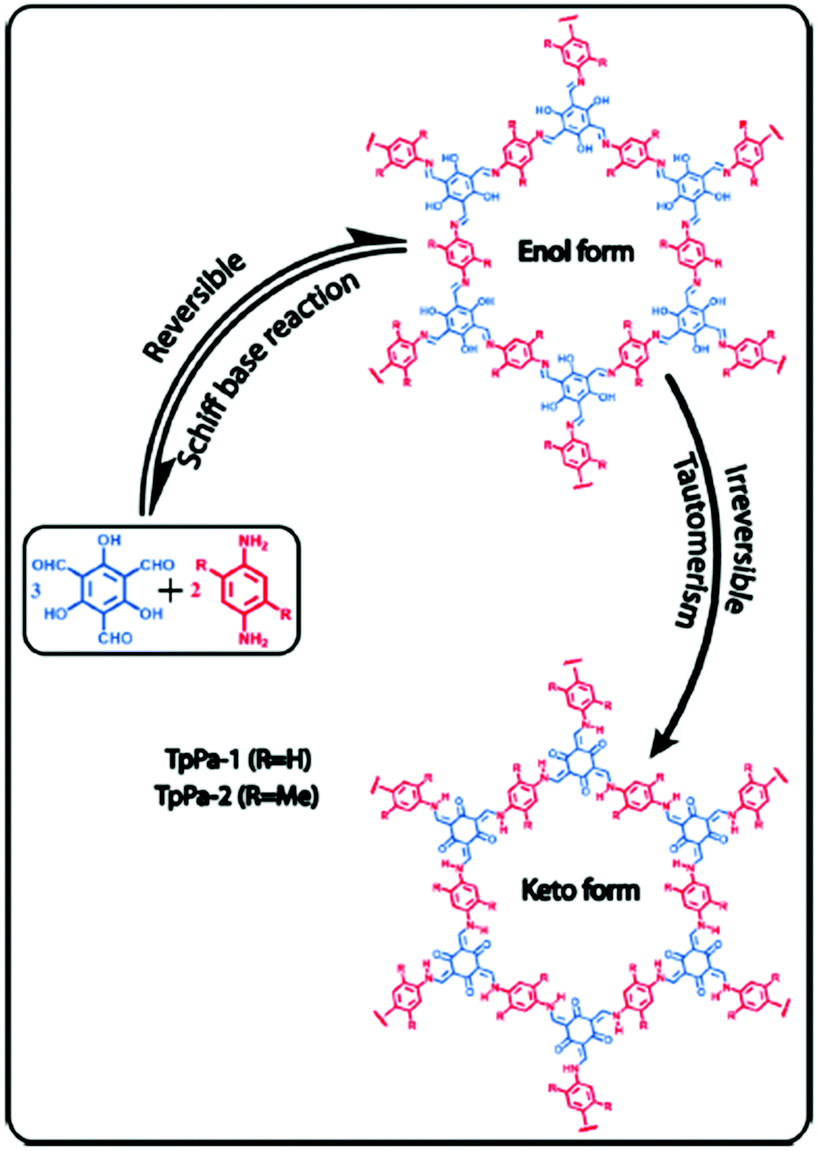 | ||
| Fig. 4 Synthesis of TpPA-1 and TpPA-2 and the reduction of imine groups to amines by enol keto tautomerism. Reproduced with permission from ref. 69. | ||
Another series of amine functional COFs is covalent triazine frameworks (CTFs). The cyclotrimerization of various benzonitrile monomers has rendered many of the CTFs reported to date.70 CTFs are mostly microporous polymers. The small pore size favors the interaction of CO2 with triazine sites in CTFs. Therefore, CO2 selectivity over other inert gases such as N2 is usually high in CTFs.
Another example of direct amine functionalization in COFs was introduced using a mixed monomer approach including the amine-functionalized monomer (DtATH).71 The amine functionalized COF, named amine–coCOF–OH, showed an improved CO2 capture capacity compared to the parent COF, coCOF–OH. COFs have also been post-synthetically functionalized, so-called “grafted”, with amine groups. Click chemistry was implemented between ethynyl units and azide compounds yielding the ethylamine functionalized COF, [EtNH2]100-H2P-COF], with a high CO2 capacity (80 cc g−1 at 273 K and 1 bar) (Fig. 5).72 Consequently, the amine functionalization resulted in surface area and pore size reductions in COFs. Conversely, the CO2/N2 selectivity was reported to increase. The chemical and pore stabilities of COFs are essential factors for practical carbon capture applications.73 As a result, research has been concentrated on triazine and imine-based COFs where the pore stability is stronger compared to other COFs such as boron-linked COFs.74 On the other hand, the CO2 uptake under humid conditions is also an important parameter which has not been explored in most COF studies to date.71
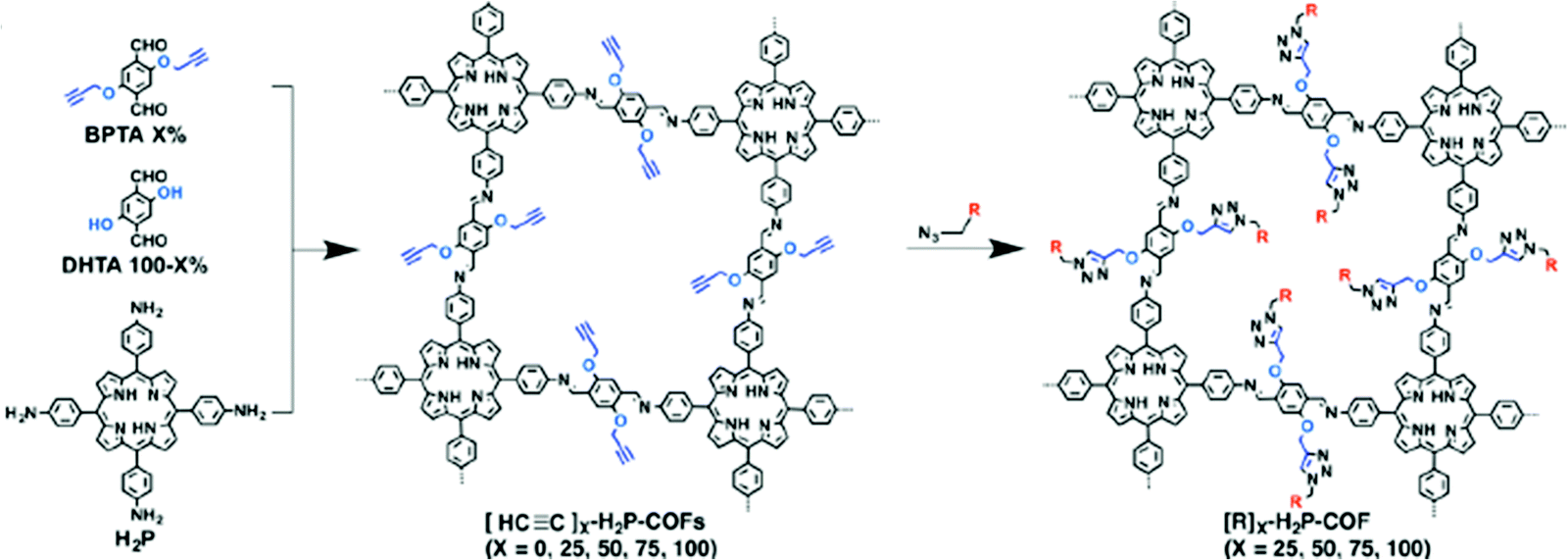 | ||
| Fig. 5 Post-synthetic amine functionalization of COFs using click chemistry between ethynyl units and azide compounds yielding the ethylamine functionalized COF, [EtNH2]100-H2P-COF. R = EtNH2. Reproduced with permission from ref. 72. | ||
Apart from imine linked COFs, numerous amorphous imine-linked POPs were introduced with imine functionality as well.75 Compared to mesoporous imine-linked COFs (such as ILCOF-1), the surface area and pore size in imine-linked POPs were calculated to be smaller.76 Therefore, low concentration CO2 uptake applications such as in post-combustion flue gas was considered more suitable for these POPs rather than high-pressure CO2 capture. For example, the porous organic polymer SNW-1 was synthesized by the polycondensation reaction of the relatively simple monomers melamine and terepthalaldehyde.77 In addition to the simple preparation method, SNW-1 possesses a high surface area and 40 wt% nitrogen content. Dual amine functionality (triazine and amine) in SNW-1 leads to high CO2 uptake capacity at relatively low pressure (Table 2).
Benzimidazole-linked polymers (BILPs) are another type of amine (imidazole) linked POPs.78 BILPs such as polybenzimidazoles (PBIs) already found in industrial applications. However, compared to conventional PBIs, the surface area in BILPs is very high and the polymer structure is highly microporous.79 The amine functionality in BILPs consists of secondary and tertiary amines in the imidazole linker. The high CO2 uptake selectivity over nitrogen was reported in BILPs such as BILP-10 and TBILPs for post-combustion CO2 capture.80 Similar to SNW-1, triazine functionalities were incorporated in BILPs (TBILPs) through the monomer. On the other hand, triptycene based BILP-12 showed potential for high pressure CO2 capture applications such as natural gas purification and landfill gas capture, with high CO2 uptake.80
Similar to TBILPs, carbazole and triazine based porous organic polymers were also studied as bifunctional POPs.81 Porous polycarbazoles are high surface area (as high as 2220 m2 g−1) adsorbents structured from an amine containing (carbazole) polymer backbone.82 With the combination of triazine, the CO2 affinity in the sorbent (TSP-2) showed a CO2 affinity of 30.2 kJ mol−1 with a CO2 uptake capacity of 2.6 mmol g−1 and a CO2/N2 selectivity of 24.81
Triazole-linked POPs are also amine-functionalized by direct synthesis.83 Click chemistry between azide and alkynyl functionalized monomers shows a facile reaction route to create a library of different triazole-based POPs. Given the high basicity of amines (in triazole), the high binding affinity for CO2 (34.8–38.5 kJ mol−1) was reported for polymers such as TNP-4.84
New azo-linked covalent organic polymers (azo-COPs) have been reported by Yavuz and Coskun.85 The synthesis of Azo COPs is based on the facile and catalyst-free polycondensation reaction between arylamines and aryl nitro functionalized monomers. The nitrogen-rich linker of azo-COPs coupled with a high surface area (729 m2 g−1) resulted in a CO2 uptake capacity of 1.52 mmol g−1 at 1 bar and 298 K for azo-COP-2. More intriguingly, the CO2/N2 selectivity of azo-COP-2 was reported to be 109.6 which was attributed to the “nitrogen phobic” nature of the azo groups linking the monomers of the POP.
Another type of COP, COP-122, was also reported by the same group.86 This time, the industrial monomers acrylonitrile (AN) and divinylbenzene (DVB) were employed in suspension polymerization with a radical initiator. COP-122 offers a cost-efficient preparation method with great potential for scalability. Equally important, it also contains alkyl nitrile functional groups. Accordingly, the nitrile groups were post-synthetically reduced into alkylamines in the presence of boranes. Compared to neat COP-122, the reduced polymer, COP-122-G1, showed over five-fold higher CO2 adsorption capacity. Also, the heat of adsorption of CO2 was doubled to 98 kJ mol−1 after the reduction.
The amine impregnation method was used in COPs as well.87 COP-19 was an amine impregnated sorbent sharing the same polymer structure with the aforementioned polymer, SNW-1 (Fig. 6). Several different polyethyleneimine loadings were studied in COP-19. Higher amine loading in COP-19 drastically improved the CO2 uptake and CO2/N2 selectivity (Table 2). However, an increased level of amine impregnation was found to be blocking the pores of the polymer, resulting in a lower surface area. Similar to the reported amine impregnated sorbents,88 higher amine loading in COP-19 resulted in lower CO2 adsorption kinetics. Therefore, amine loading was optimized at 24.1 wt% polyethyleneimine in COP-19 (COP-97b) to achieve high CO2 uptake with good kinetics. COP-97b also showed higher CO2 uptake performance under humidity conditions which is applicable to post-combustion flue gas.
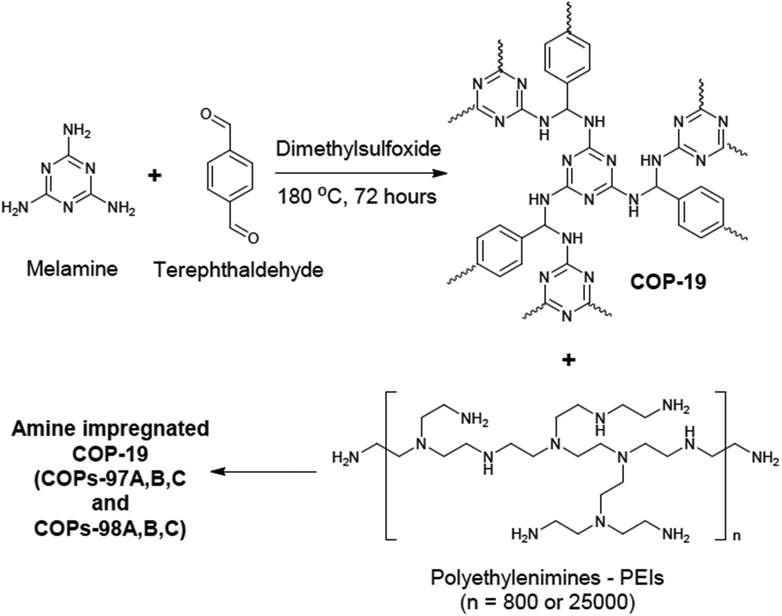 | ||
| Fig. 6 COP-19 synthesis and polyethylenimine impregnation in COP-19. Reproduced with permission from ref. 87. | ||
4. Amine functionalization hyper-crosslinked polymers (HCPs)
Hyper-crosslinked polymers (HCPs) have been developed rapidly over the past two decades and have been extensively studied for CO2 capture.106,107 HCPs consist of polymer chains thar are crosslinked and are made from light elements (C, H, N, and O). Their advantages are moderate synthetic conditions, inexpensive monomers and reagents, easy functionalization, microporosity and high surface area, robust structures, and good thermal and chemical stabilities.108,109 HCPs can be prepared via the Friedel–Crafts reaction from (i) the crosslinking of polystyrene-type precursors in their swollen state,110,111 (ii) one-step self-polycondensation,112,113 and (iii) external crosslinking strategies.109,114–116 Using these methodologies, HCPs can be tailored into efficient porous adsorbents with customized porosity and functionalities for CO2 capture applications. Recently, the HCPs prepared by the solvent knitting method showed a Brunauer–Emmett–Teller (BET) surface area of 3002 m2 g−1. A pore volume of 1.53 cm3 g−1 and porous structures ranging from ultramicropores to mesopores (0.5 to 3.8 nm) were reported.117 HCPs generally have a hydrophobic surface that is not favored for polar adsorbates such as CO2.118 This limitation can be overcome by introducing polar nucleophilic amine groups.119 Similar to other sorbents, the amine groups have been incorporated in HCPs by direct amine synthesis,120–123 amine grafting106,124,125 and amine impregnation.126,127Fayemiwo et al. investigated a series of amine-functionalized HCPs (HCP-MAAMs) for low-pressure CO2 capture such as from coal-fired power plant flue gas.94 The synthesis of HCPs is based on the co-polymerization of methacrylamide (MAAM) and ethylene glycol dimethacrylate (EGDMA) using azobisisobutyronitrile (AIBN) as an initiator (Fig. 7). The HCPs with a MAAM-to-EGDMA molar ratio (S) from 0.3 to 0.9 were (directly) amine-functionalized and exhibited a high affinity towards CO2 at low pressures. At a CO2 partial pressure of 0.02–0.15 bar, the CO2/N2 selectivity increased (Table 2). The isosteric heats of the adsorption (Qst) properties of HCPs were calculated to be as high as 35 kJ mol−1, resulting in an electrostatic interaction of CO2 molecules with the amine moieties. The CO2 adsorption capacity was reduced by increasing the ratio of MAAM-to-EGDMA due to lower surface areas. The HCP with S = 0.3 had a maximum CO2 adsorption of 1.56 mmol g−1 at 273 K that may be further improved using amine-based cross-linkers of N,N-methylenebis(acrylamide). The cycle stability of the CO2 capacity for the HCP with S = 0.6 was tested under adsorption conditions of 298 K and 0.15 bar CO2 partial pressure and desorption conditions of 393 K and N2. The CO2 adsorption capacity was reduced by 1.9% over five cycles. Further studies are needed to gain a full understanding of the cycle stability.
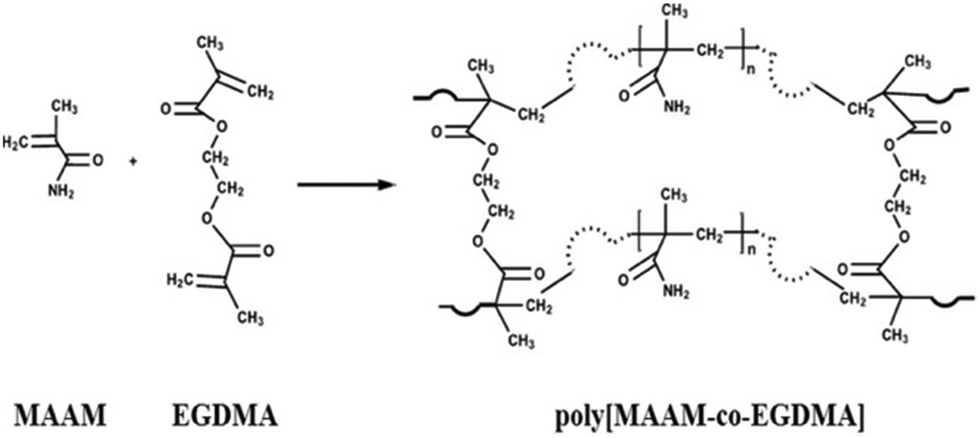 | ||
| Fig. 7 Synthesis of amine-functionalized HCP-MAAMs by co-polymerization of MAAM and EGDMA. Reproduced with permission from ref. 94. | ||
Nitrogen-rich HCPs, triptycene based nitrogen-rich HCPs (TNHCPs),118 polytriphenylamine HCPs (PTPAs)128 and octaphenylcyclotetrasiloxane (OPCTS) based nitrogen-rich HCPs (ONHCPs)129 were also investigated. The TNHCPs were synthesized by cross-linking commercially available nitrogen-rich heterocycles such as isomeric pyrazine, pyridazine and pyrimidine with triptycene molecules. On the other hand, PTPAs were prepared using tris-functionalized crosslinkers (trimethyl orthoformate, trimethyl orthoacetate, triethyl orthoacetate, and triisopropyl orthoformate) via a FeCl3-promoted one-step oxidative coupling reaction and Friedel–Crafts alkylation. In a different route, the synthesis of ONHCPs involved two steps: the polymer was prepared using OPCTS as the organic–inorganic hybrid monomer, an amine containing monomer like cyanuric chloride (CC) as the cross-linker, and AlCl3 as the catalyst through the Friedel–Crafts reaction. By changing the CC content, the porosity of the polymer, and the N content, the porosity of the ONHCPs was significantly enhanced, which is preferred for the physical adsorption of CO2. All three HCPs exhibited large surface areas, high porosity and high CO2 adsorption capacity. Compared to the HCP-MAAM, the HCPs with N in the backbone of the polymer showed the lower CO2 adsorption capacity and selectivity for CO2 at low CO2 pressures.
Apart from the aforementioned rigid monomers, low-cost coal tar with nitrogen groups was utilized to generate HCPs through a one-step Friedel–Crafts reaction.130 The maximum BET surface area of the prepared HCPs could reach up to 929 m2 g−1. Owing to the high affinity between the heteroatoms on the coal-tar building blocks and the CO2 molecules, the adsorption capacity of CTHPs towards CO2 reached up to 14.2 wt% with a CO2N2 selectivity of 32 (at 1 bar, 273 K). Later, another carbon source, waste polystyrene, was transformed into HCPs using 1,2-dichloroethane.131 Organosolv lignin separated from wood (renewable resource) was developed for HCPs by the reaction with formaldehyde dimethyl acetal (FDA).132 These low-cost HCPs may have potential in mass production and practical use. Liu et al. developed amine-functionalized HCPs (PEI@XAD-4-pc) by synthesizing the HCP (XRD-4-pc) through a Friedel–Crafts reaction of a commercial polystyrene resin (XAD-4) and subsequently functionalizing it with polyethyleneimine (PEI Mn = 600) (Fig. 8).93 PEI@XAD-4-pc featured enhanced CO2 adsorption compared to other carbon based HCPs because of the strong interaction between CO2 and PEI. The CO2 adsorption capacity increased from 1.14 to 3.24 mmol g−1 with a higher ratio of PEI (increased from 10 to 30 wt%) in the HCP. The higher PEI loading (over 30 wt%) in the sorbent significantly decreased the CO2 uptake capacity. The CO2 diffusion into the inner pores was reduced due to the excess amine functionalities. The CO2/N2 selectivity was 1103 for 30 wt% PEI in XAD-4-pc at 10 °C and 0.02 bar CO2 partial pressure. The heat of adsorption of 30 wt% PEI@XAD-4-pc was calculated to be 53 kJ mol−1.
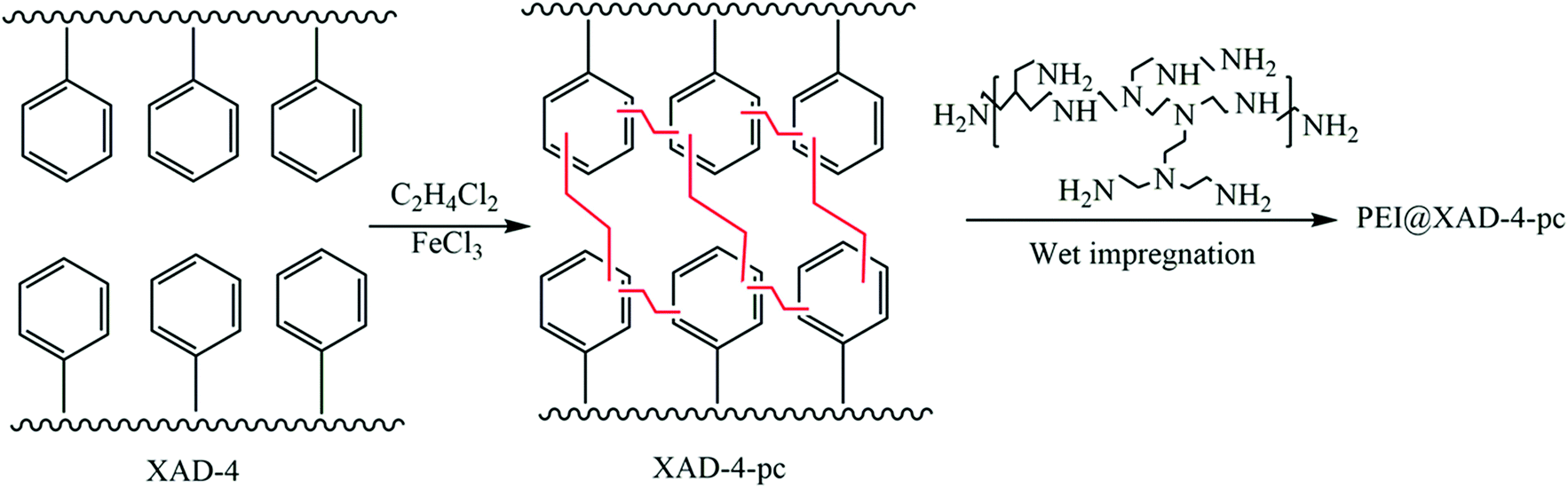 | ||
| Fig. 8 Synthesis of PEI-functionalized hyper-crosslinked polymers by co-polymerization of methacrylamide (MAAM) and ethylene glycol dimethacrylate (EGDMA). Reproduced with permission from ref. 93. | ||
The CO2 capacity of 30 wt% PEI in XAD-4-pc (3.24 mmol g−1) is 1.7 times higher than that of 30 wt% PEI in XAD-4 (1.87 mmol g−1). This may be attributed to the large surface area and higher pore volume of XAD-4-pc (1239 m2 g−1 and 1.87 cm3 g−1) compared to the porosity of XRD-4 (852 m2 g−1 and 1.18 cm3 g−1).
Several other types of molecular amines such as triethylenetetramime (TEPA) were also impregnated in HCPs. For example; TEPA functionalized HCP (TEPA@PDVBpc) was prepared by wet impregnation of TEPA on PDVBpc that was synthesised by a Friedel–Crafts alkylation reaction of polydivinylbenzene (PDVB).95 PDVBpc showed a higher surface area (1369 m2 g−1) and pore volume (1.27 cm3 g−1) compared to the PDVB (672 m2 g−1 and 0.58 cm3 g−1). In line with the other amine functionalized sorbents, the large surface area and pore volume properties were found to be beneficial for the loading of amines and adsorption of CO2.133 The CO2 adsorption capacity of 30TEPA@PDVBpc (30 wt% TEPA loading) was 3.11 mmol g−1 at 298 K with 10 vol% CO2 at 1 bar and 2.6 times higher than that of 30TEPA@PDVB. The stability of 30TEPA@PDVBpc was tested in five cycles of CO2 adsorption at 298 K and 0.1 bar CO2 with desorption at 75 °C. After the five cycles, the CO2 adsorption capacity decreased by 7.4% compared to the first cycle. This may be because a proportion of TEPA was vaporized or decomposed during the desorption process. DETA and TETA were also loaded on PDVBpc to investigate the impact of the different amine species on the adsorption of CO2. The CO2 adsorption capacities (at 0.1 bar and 298 K) of three different amines loaded on the PDVBpc of 30TEPA@PDVBpc, 30TETA@PDVBpc and 30DETA@PDVBpc increased with the length of the amine chain (30TEPA@PDVBpc > 30TETA@PDVBpc > 30DETA@PDVBpc).
The impact of the TEPA@PDVBpc loading (10 to 50 wt%) on the CO2 adsorption capacity was studied because the chemical adsorption is mainly determined by the number of accessible amine groups in the adsorbent. To simply quantify the density of amine groups in the adsorbent, an elemental analysis of nitrogen (N) (wt% or mmol g−1) was conducted. The amine efficiency was calculated using the molar ratio of the adsorbed CO2 to the amine groups in the adsorbent. The CO2 adsorption capacity and amine efficiency significantly increased from 1.0 mmol g−1 and 0.35 (mmol CO2/mmol N) to 3.11 mmol g−1 and 0.46 (mmol CO2/mmol N) as the TEPA content increased from 10 wt% to 30 wt%, respectively. The TEPA content further increased to 50 wt% and the CO2 adsorption capacity and amine efficiency significantly decreased to 0.61 mmol g−1 and 0.06 (mmol CO2/mmol N), respectively. This was attributed to the excess TEPA blocking the pore channels and preventing CO2 molecules from reaching internal amine groups.
Yang et al. reported a series of novel amine functionalized HCPs prepared by the amine grafting method.37 First, porous organic copolymers (PBTP-(x)) were synthesised via Friedel–Crafts alkylation using FeCl3 as a catalyst, which is the common catalyst choice in HCPs. The copolymers were subsequently functionalized using alkylamines DETA and TEPA, labelled as PBTP-(x)-DETA and PBTP-(x)-TEPA, respectively (Fig. 9). The molar ratio (x) of BCMBP-to-TCB significantly impacted both the porosity and Cl content of the copolymers. As the ratios varied from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, 1:2, to 2:1, the BET surface areas (970, 1160, and 1600 m2 g−1) and total pore volumes of the copolymers increased, and Cl contents decreased due to the lower Cl content of BCMBP. In the copolymers with a higher Cl content, the amine functionality level increased, improving the CO2 uptake and CO2/N2 selectivity. The PBTP-(x)-DETA at x = 1:2 and 1:4 exhibited a higher CO2 adsorption capacity than the PBTP-(x)-TEPA at the same x despite the higher amine content of the latter. This might be attributed to the higher surface area of PBTP-(x)-DETA. Hence, adsorbents with DETA amine groups displayed higher amine efficiencies than their counterparts with TEPA groups.38 PBTP-(x)-TEPA had higher CO2/N2 selectivity than PBTP-(x)-DETA because a significant reduction of N2 accessible pores in PBTP-(x)-TEPA probably offsets the effect of their lower CO2 adsorption capacity.
:4, 1:2, to 2:1, the BET surface areas (970, 1160, and 1600 m2 g−1) and total pore volumes of the copolymers increased, and Cl contents decreased due to the lower Cl content of BCMBP. In the copolymers with a higher Cl content, the amine functionality level increased, improving the CO2 uptake and CO2/N2 selectivity. The PBTP-(x)-DETA at x = 1:2 and 1:4 exhibited a higher CO2 adsorption capacity than the PBTP-(x)-TEPA at the same x despite the higher amine content of the latter. This might be attributed to the higher surface area of PBTP-(x)-DETA. Hence, adsorbents with DETA amine groups displayed higher amine efficiencies than their counterparts with TEPA groups.38 PBTP-(x)-TEPA had higher CO2/N2 selectivity than PBTP-(x)-DETA because a significant reduction of N2 accessible pores in PBTP-(x)-TEPA probably offsets the effect of their lower CO2 adsorption capacity.
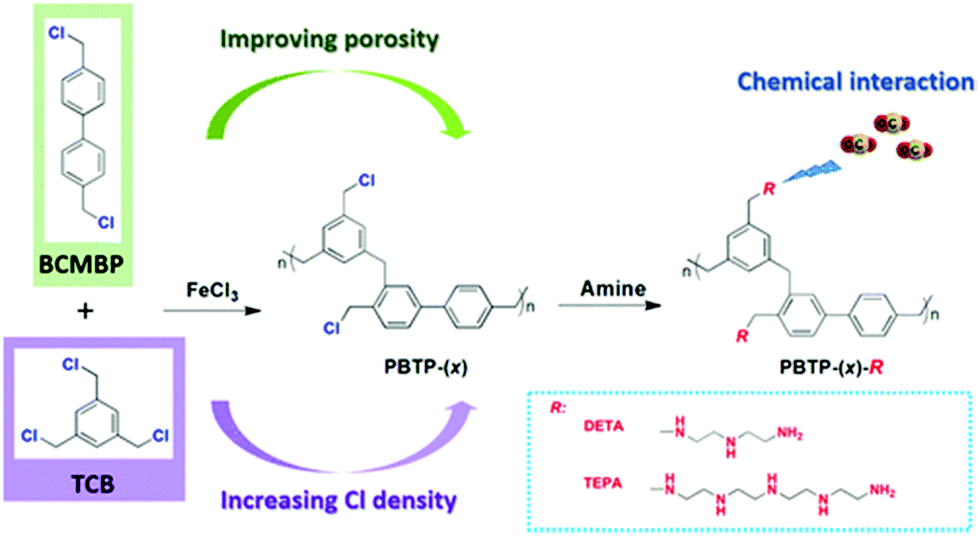 | ||
| Fig. 9 Synthesis of porous organic copolymers (PBTP-(x)) and alkylamine-functionalised hyper-crosslinked polymers (PBTP-(x)-R) by postsynthetic grafting. Reproduced with permission from ref. 37. | ||
Overall, PBTP-(1:4)-DETA is a promising sorbent candidate considering its high CO2 adsorption capacity (2.27 mmol g−1), large CO2/N2 selectivity (1064) and fast adsorption kinetics (5.8 min to reach 90% of saturation capacity) (at 298 K and 0.15 bar CO2). Ten adsorption (298 K)–desorption (393 K) cycles were conducted with and without water vapor at 0.2 bar CO2/N2 under dynamic flow conditions were performed on the sorbents. The CO2 adsorption performance remained intact during the dry and humid cycles, showing good stability.134 In addition, the accumulation of water in the sorbent was insignificant over the cycles due to the hydrophobic nature of the PBTP-(x) support. In this study, two representative MOFs of HKUST-1 and Ni2 dobdc (Ni-MOF-74)135 were synthesized and tested by repetitive cycling under humid conditions. PBTP-(1:4)-DETA outperformed HKUST-1 Ni2 dobdc (Ni-MOF-74). HKUST-1 had a lower CO2 working capacity. Ni2 dobdc (Ni-MOF-74) decreased in CO2 adsorption over 10 cycles.
The Jiang group designed and synthesized a nitrogen-rich HCP (FCDTPA) based on the knitting N,N,N′,N′-tetraphenylbiphenyl-4,4′-diamine (DTPA) monomer.136 Carbonized materials were obtained by the pyrolysis of FCDTPA with/without potassium hydroxide at high temperatures. The carbon materials with potassium hydroxide activation (FCDPA-k-x) exhibited higher surface areas and enhanced CO2 adsorption capacity compared to the carbon materials without potassium hydroxide activation (FCDPA-x). The activated carbon material of FCDPA-k-700 showed a high surface area of 2065 m2 g−1 and a CO2 uptake of 6.51 mmol g−1 at 1.13 bar and 273 K compared to the precursor of FCDTPA (871 m2 g−1 and 2.82 mmol g−1).
5. Conjugated microporous polymers (CMPs)
Conjugated microporous polymers (CMPs) are unique porous materials that combine extended π-conjugation with a permanently microporous skeleton.137 The number of studies has been growing since its discovery in 2007 because of attractive properties such as the conjugation, high porosity, tunable chemistry, and good chemical and thermal stabilities.138 CO2 adsorption by CMPs has been of interest because the adsorption capacity and selectivity of CMPs can be improved by synthetic control over structure properties such as surface area, pore size, pore volume and functional groups.96 The functionalization of CMPs using amine functional groups have enhanced the CO2 adsorption capacities.139 In general, the amine groups have been incorporated in CMPs by direct amine synthesis,140–142 and amine grafting.101 CMPs are typically synthesized via the palladium-catalyzed Sonogashira–Hagihara polymerization of terminal alkynes and/or aryl halides.143 For example, Dawson et al. have reported amine-functionalized CMPs (CMP-1-NH2) by the Sonogashira–Hagihara palladium cross-coupling reaction of 1,3,5-triethynylbenzene with 2,5-dibromoaniline (Fig. 10).96 The amine groups in the CMP-1-NH2 increased the isosteric heats of the adsorption of CO2 from 26 kJ mol−1 to 29.5 kJ mol−1 compared to the unfunctionalized CMP-1 (Table 2). Accordingly, CMP-NH2 showed higher CO2/N2 selectivity than CMP-1. For enhancing the CO2 sorption at lower pressures, increasing the surface area may not be the correct strategy but factors like the isosteric heats of adsorption are more likely to dominate both the sorption and the energy penalty for sorbent regeneration.144,145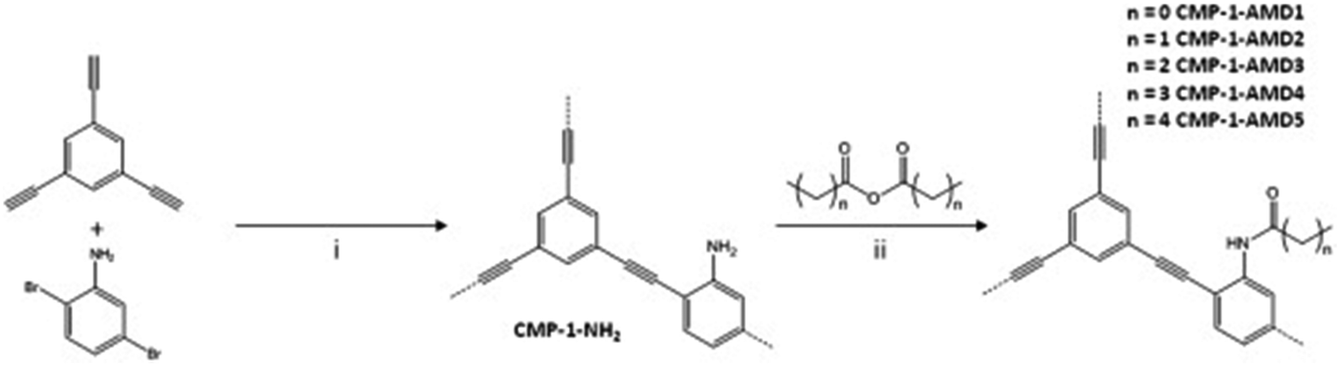 | ||
| Fig. 10 Step (i): Synthesis of CMP-1-NH2: Pd(PPh3)4, CuI, DMF, NEt3, 100 °C, 24 h under a N2 atmosphere. Step (ii): Post-synthetic modification using anhydrides, n = 0–4 and 8 (i.e., C1–C5 and C9): 24 h, 30 °C. Reproduced with permission from ref. 96. | ||
Following the amine functionalized CMP (CMP-1-NH2), Ratvijitvech et al. reported the post-synthetic modification of amines into amides with different alkyl chains (CMP-1-AMDn, n = 0–4) (Fig. 10).146 As the alkyl chain length in the amide was increased, the BET surface areas and pore volumes of the CMP-1-AMDn were decreased, which resulted in a reduction of the CO2 uptake and selectivity.
Chen et al. used the addition of simple inorganic salts with different sized cations or anions (NaNO3 and NaF) to improve the BET surface area and porosity of the polytriphenylamine functionalized polymer, CMP-PTPA, that was synthesized by the Buchwald–Hartwigh (BH) cross-coupling reaction (Fig. 11).98 The addition of salts may act similarly to the addition of porogen, helping in adjusting and optimizing the Hansen solubility parameters (HSPs) of solvents for the growing polymer. The solvents that are compatible with the polymer networks could contribute to phase separation at a much later stage during polymerization, thus producing a polymer with lower average pore diameters and a larger surface area. The surface areas of the N-rich CMP were significantly gradually decreased by increasing the ionic radius of salts. Ions with smaller radii have a greater influence on the permanent dipole interaction and the hydrogen-bonding interactions of the solvent by adjusting the polarity and hydrogen bonding of the solvents, thus resulting in a more effective adjustment of the compatibility of the HSPs with the polymer. The addition of NaNO3 increased the surface area of CMP-PTPA eighteen times from 58 to 1028 m2 g−1 which led to improved CO2 uptake by five times from 0.7 to 3.48 mmol g−1 at 273 K at 1 bar. With the addition of salts, the pore size distributions were narrowed to the micropore range from the broader distribution of micropores, mesopores, and macropores in the neat polymers. The addition of NaF increased the total pore volume, the micropore volume and ultramicropore volume in the polymers. The salts affected the porosity of CMP-PTPA by changing the Hansen solubility parameters (HSPs) of solvents used in the polymerization of CMPs without changing the chemical structure of the polymer.
 | ||
| Fig. 11 Synthesis of salt-tunable CMP-PTPA by Buchwald–Hartwogj (BH) cross-coupling. Reproduced with permission from ref. 146. | ||
Mu et al. synthesized N-rich CMPs (NCMPs) by palladium-catalyzed Sonogashira–Hagihara cross-coupling condensation of 1,3,5-triethynylbenzene and bis(4-bromophenyl)amine.99 The results showed that the choice of reaction solvents had a major influence on the porosity of NCMPs. NCMP-I (0.61% N) using toluene as a solvent resulted in a specific surface area of 945 m2 g−1 and a pore volume of 0.55 cm3 g−1 (45% micropores). NCMP-III (1.63% N) using 1,4-ioxane as a solvent had a surface area of 593 m2 g−1 and a pore volume of 0.96 cm3 g−1 (6% micropores). The CO2 uptake at 289 K at 1 bar for NCMP-I (1.42 mmol g−1) was higher than that of the NCMP-III (0.4 mmol g−1). The isosteric heats of adsorption of NCMP-I and NCMP-III were 23–38 kJ mol−1 and 21–28 kJ mol−1, respectively.
Bao et al. synthesized amine functional CMPs (NCMPs) containing pyridine units and amine groups by the Sonogashira–Hagihara cross-coupling reaction using 1,3,5-triethynylbenzene and 2-amino-3,5-di-bromopyridine as monomers.143 The molar ratio of the ethynyl monomer during the polymerization significantly affected the morphology, BET surface areas, and pore volumes of the resulting NCMP networks. When the molar ratio of ethynyl-to-halogen functionalities was 5:1, tubular-shaped NCMPs (NMCP-3) with a diameter of 100–200 nm were obtained. The BET surface areas and total pore volumes of the resulting polymers were increased with the increase of the molar ratios (1, 3, 5 and 9) of ethynyl in the networks. For NCMP-3, the CO2 uptake was calculated as 1.04 mmol g−1 at 298 K and 1 bar.
Wang et al. synthesized N-rich CMPs with azo-fused structures (azo-CMPs) via palladium-catalyzed Suzuki or Sonogashira cross-coupling reaction from diamino-grafted or azo-fused monomers.147 The azo-fused polymers with a rigid conformation showed an enhanced surface area up to 1146 m2 g−1 compared with the diamino-grafted polymers. Azo-CMP1 had the highest surface area among the reported polymers and exhibited a CO2 uptake of 3.72 mmol g−1 (1.13 bar and 273 K) with a CO2/N2 selectivity of 42.1 at 273 K. The highly N2-phobic nature of –NN– functional groups can be attributed to the high CO2 selectivity over N2.148 The results showed that the polymer with higher micropore surface area has higher CO2 adsorption.148 All of the polymers show high isosteric heats of CO2 adsorption (∼30 kJ mol−1) because the incorporation of nitrogen atoms into the skeleton of the conjugated microporous polymers led to an enhanced interaction between the pore wall and CO2 molecules. Due to the high surface area, good physicochemical stability, and high CO2 sorption performance, these nitrogen-rich polymer networks are promising candidates for applications in CO2 capture technologies.149
Liu et al. developed a post knitting method to increase the surface area and CO2 uptake of CMPs.100 CMPs were knitted using two different cross-linkers via a Friedel–Crafts reaction to prepare CMP-based HCPs with reconstructed nanopores (KCMPs). The KCMPs showed higher surface areas and increased CO2 uptake compared to the corresponding CMPs. In the dichloromethane (DCM) knitted KCMP-Ms, KCMP-M3 with NH2 groups had a surface area of 1321 m2 g−1 and a CO2 uptake of 2.54 mmol g−1 at 298 K and 1 bar, which was 16 times and 4.5 times higher than the corresponding CMPs. KCMP-M3 exhibited the highest CO2 uptake of 0.52 mmol g−1 at 198 K and 0.15 bar even with the lowest surface area of 1321 m2 g−1 compared to other KCMP-Ms without NH2 groups.
Guillerm et al. reported deliberate amine grafting (aldehyde conversion to imine) via a one-step post-synthetic modification process.101 First, the POP (1) was synthesized by the Sonogashira–Hagihara coupling of tri-connected 1,3,5-triethynylbenzene (TEB) and 2.6-dibromo-4-trimehynylbenzaldehyde (Br2TMSBA). The amine functionalized POP (2) was prepared by grafting ethylenediamine to the POP (1). The POP (2) had a surface area of 485 m2 g−1, which was 25% lower compared to the POP (1) as expected. Its CO2 uptake was 0.27 mmol g−1 at 0.15 bar and 0.95 mmol g−1 at 1 bar CO2 at 298 K, which was higher than that POP (1) (0.15 mmol g−1 at 0.15 bar and 0.78 mmol g−1 at 1 bar). The isosteric heat of POP (2) increased to 50 kJ mol−1 from the 30 kJ mol−1 of POP (1). As expected, the CO2 selectivity of POP (2) was 155 and higher than the 14 of POP (1). Therefore, the amine grafting enhanced the CO2 adsorption properties.
6. Amine functionalization in polymers of intrinsic microporosity (PIMs)
Polymers of intrinsic microporosity (PIMs) are a unique class of porous polymers. Virtually, all other POPs covered here are crosslinked polymers in which two or three-dimensional monomers create the porosity and high surface area.150 Whereas, in PIMs, the monomers are bulky, contorted, and one-dimensional.151 The inefficient packing of these monomer chains creates the porosity in PIMs rather than the usual case of monomer crosslinking.152 PIMs have the advantage of being processable into films, fibers, or other morphologies while maintaining the porosity.153 Accordingly, the focus in the literature has been on the membrane applications of PIMs including gas and liquid separation.154 Recent studies, however, have shown that processible polymers can be advantageous as solid sorbents, for example to reduce the reactor pressure drop.155 PIMs are less likely to be considered for use as a CO2 capture sorbent, on the other hand, because the polymer weakly binds to CO2 in the physisorption range.156 There have been several amine functionalization studies for PIMs, mostly to functionalize PIM-1 with primary and secondary amine moieties. However, the amine functionalities were utilized in membrane formation rather than as solid sorbents.157 Recently, PIM-1 was impregnated with polyethyleneimine in the solid sorbent form.102 Various concentrations of amines were loaded into PIM-1 solution and the subsequent solvent removal afforded PIM-1-PEI sorbents. Compared to neat PIM-1, a very high CO2 capture capacity (1.24 mmol g−1 at 0.15 bar and 298 K) was observed with the polyethyleneimine impregnated PIMs.102 Several sorbent geometries such as fibers and monoliths have been demonstrated. In one study, PIM-1 was processed into hollow fibers from various PIM-1/solvent dope solutions (Fig. 12).158 Then, polyethyleneimine was impregnated into the PIM-1 fiber post-fabrication. The results showed that the increased PEI content resulted in a higher CO2 uptake in the PIM-1 sorbents. However, increased PEI loading in the sorbents can also worsen the kinetics of CO2 adsorption.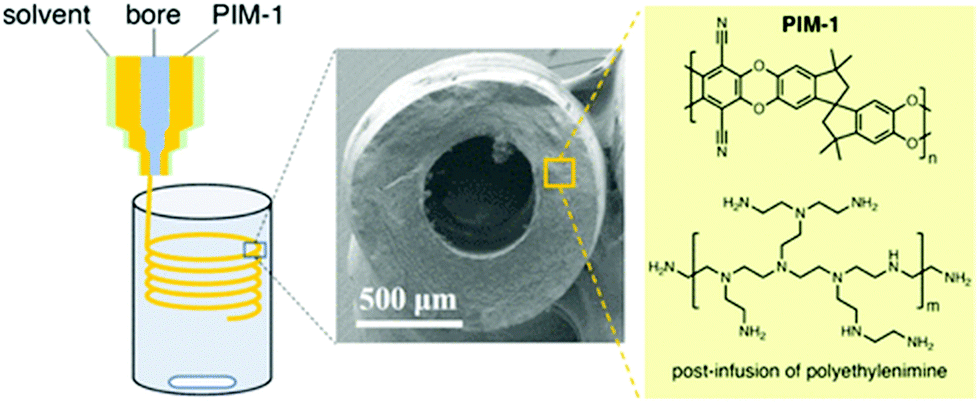 | ||
| Fig. 12 Shaping PIM-1 into hollow fiber sorbents and polyethylene impregnation in PIM-1. Reproduced with permission from ref. 158. | ||
In another study, PIM-1 was first functionalized with carboxylic acid and amide to increase the interaction of PIM-1 with the guest amine groups.103,159 In the following step, the acid functionalized PIM-1 (PIM-1-C) was impregnated with molecular amines such as TAEA, yielding an acid–base and hydrogen bonding interaction between the amine and the functionalized PIM-1. A control study in which the acid functionalization step was omitted showed that neat PIM-1 was not able to bind strongly to amines by the same impregnation method, resulting in amine leaching. PIM-1-C-TA had a high CO2 uptake (1.62 mmol g−1 at 0.15 bar and 298 K) which was retained for several cycles under humid conditions. More recently, PIM-1 with amidoxime functionality (PIM-1-AO) was utilized to bind molecular amines such as diethylenetriamine by strong hydrogen bonding and weak acid–base reaction (Fig. 13).104 The DETA impregnation in PIM-1-AO resulted in a high CO2 uptake (1.75 mmol g−1 at 0.15 bar and 298 K). PIM-1-AO-DETA loaded with CO2 was regenerated after heating to 80 °C. The low regeneration temperature for PIM-1-AO-DETA can be attributed to relatively low amine loading (<15%) in the polymer.
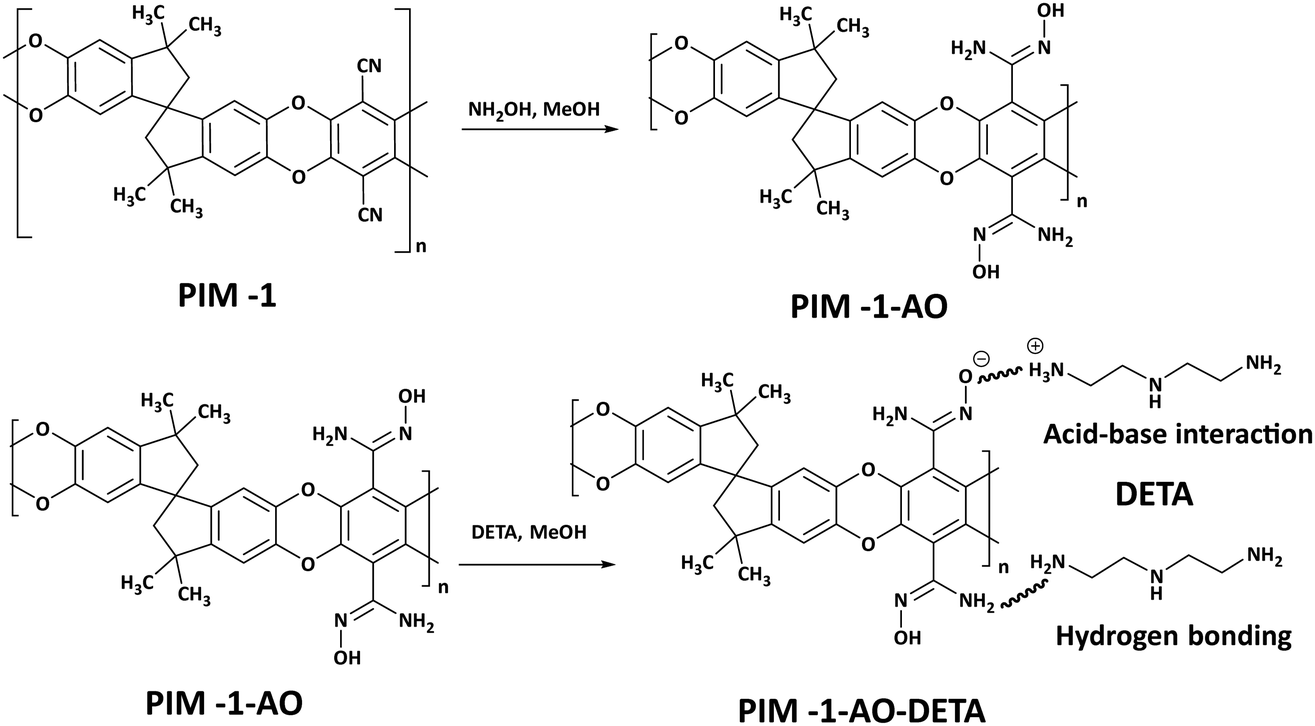 | ||
| Fig. 13 Post-synthetic functionalization of PIM-1 with amidoxime and alklyamines (DETA). Reproduced with permission from ref. 104. | ||
Besides PIMs, there have been reports on other types of processible polymer sorbents such as cellulose acetate160 and matrimid.161 However, these polymers are not categorized as a porous organic polymer because the porosity in these polymers is predominately macroporous (>50 nm). Also, the porosity in the polymers is usually created by a template method during fabrication, whereas, in PIMs, the porosity is a material property of the rigid and contorted monomers. Consequently, the surface areas of these conventional polymers are not comparable with those of PIMs. Nevertheless, these polymers can serve as a “molecular basket” for immobilizing amines and perform as CO2 adsorbents, which is the scope of another review article.162
7. Computationally designed amine functionalized porous organic polymers
There are a limited number of computational studies on amine functionalized porous organic frameworks for CO2 capture. Jiang et al.163 designed new functional porous aromatic frameworks (PAFs) by incorporating functional groups such as –NH2, –OCH3, and –CH2OCH2– in PAF-1 and investigated their separation efficiencies of CO2/N2, CO2/CH4 and CO2/H2 mixtures using the grand-canonical Monte Carlo (GCMC) simulation. Pure gas isotherms showed that CO2 sorption and selectivity over N2, CH4, and H2 decreased in the order –CH2OCH2– PAF-1 > –OCH3-PAF-1 > –NH2-PAF-1 > PAF-1 at 298 K and pressure up to 1 bar. The same trend was observed for mixed gas isotherms for 15:85 mixtures of CO2/N2, CO2/CH4 and CO2/H2 at 298 K and pressures up to 1 bar. Similarly, Lu et al.164 investigated the CO2 adsorption and separation properties of four alkyl amine functionalized triphenylamine-based covalent organic frameworks (TPAC-xC-NH2, x = 1–4) using the GCMC simulation. The computed CO2 isotherms at 273 K and 298 K up to 1 bar showed that increasing the alkylamine chain length (x = 2–4) in TPA-COFs led to an increase in CO2 adsorption, reduction in N2 adsorption, and consequently enhanced CO2/N2 selectivity. Furthermore, isosteric heat analysis showed that narrow pores in TPA-COFs due to long alkylamines led to a higher CO2–adsorbent interaction. And coulomb/non-coulomb interaction analysis showed the CO2–TPAC–xC–NH2 interaction to be dominant over CO2–CO2 interaction.
Ab initio quantum chemical calculations and atoms in molecule (AIM) analysis165 methods were used to explore the CO2 adsorption mechanism in a series of triazine-based porous organic polymers (TrzPoP): TrzPOP-1, TrzPoP-2 and TrzPop-3. TrzPOP-1 contains only a triazine moiety and secondary amine linkages while TrzPoP-2 and TrzPOP-3 contain additional phenolic –OH groups. The investigation confirmed the existence of strong N–H–O and O–H–O hydrogen-bonding interactions between the TrzPOPs and CO2 molecules along with weak N–C, O–C and C–H–O interactions. The presence of a large number of weak interactions was attributed to the higher CO2 uptake in the series of TrzPOPs. However, a larger number of phenolic–OH and N-rich surfaces in TrzPOP-2 and TrzPOP-3 were found to be directly responsible for the higher CO2 uptake and CO2/N2 selectivity than TrzPOP-1. Vogiatzis et al.166 screened functionalized calix[4]arenes for their CO2-philicity. The structures were constructed by automated, high-throughput structure generation through directed modifications to a molecular scaffold. In total, 13 different functional groups were chosen for the study, of which 10 were placed at 4 different sites (para, axial, equatorial and bottom) of a calix[4]arene structure, and the remaining larger 3 functional groups were placed at the para position to generate 40 different structures (Fig. 14). Overall, 41 different structures were considered including non-functionalized calix[4]arene. Density functional theory (DFT) and symmetry-adapted perturbation theory (SAPT) were applied to compute the CO2 interaction energy for the set of 41 calix[4]arenes. It was found that the top performing structures, the ones with the largest CO2 interaction energy, had functional groups at the para-site, and the top four functional groups were found to be methoxypyridine, methoxyphenyl, methoxyamine, and ethoxyamine, respectively. Methoxyamine functionalized calix[4]arene was found to be more promising because the terminal amino group led to the formation of a molecular cage via hydrogen bonds, which led to the increased CO2-interaction compared to the thoroughly studied tert-butyl functional group.
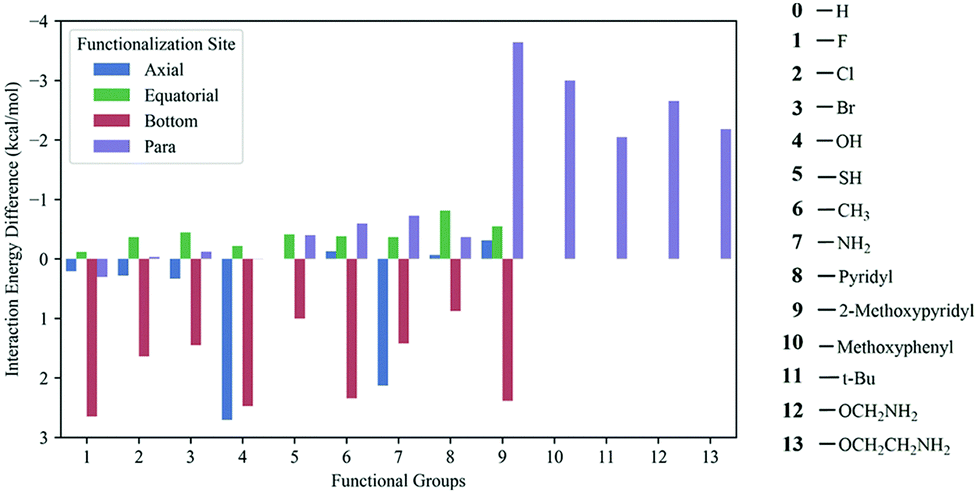 | ||
| Fig. 14 Calculated CO2 interaction energies of functionalized calixarenes. All energies are given as a difference from the interaction energy of the unfunctionalized calixarenes (−6.82 kcal mol−1). Reproduced with permission from ref. 166. | ||
The CO2 sorption mechanism in the amine functionalized polymer of intrinsic microporosity (PIM-1) was investigated using molecular dynamics simulation and quantum mechanics (QM).157 Molecular dynamics simulations were performed to obtain the radial distribution function, g(r), the local probability density of finding one atom at distance r from another atom. The radial distribution function between PIM-1 and CO2 molecules showed a strong peak at about 3 Å from the amino groups, a sign that there is a strong interaction between the amine groups and polarized CO bonds of the CO2 molecules. In contrast, the radial distribution function between the dioxane–O functional groups of PIM-1 and CO2 was smoother, indicating a weak interaction. The interaction energy between the amine-PIM-1 monomer and CO2, N2 and CH4 molecules was computed by quantum mechanics methods, which showed that the CO2-amine had an interaction energy of about −2 kcal mol−1, which was in the range of weak hydrogen bonding. N2 and CH4 did not interact significantly with the monomer, which is proof of the selective interaction between the amine group in PIM-1 and CO2. Hamouz et al.167 recently synthesized a novel porous organic polymer, KFUPM-5, via the acid catalyzed polycondensation of polar aromatic monomers, melamine and pyrrole with p-formaldehyde as the cross-linking agent, and investigated its efficacy for the selective adsorption of CO2 and H2S for natural gas sweetening. They used quantum chemical reactivity descriptors, AIM topology analysis, and reduced density gradient (RDG) isosurface analysis to elucidate the adsorption mechanism of H2S in a vacuum and CO2 in a vacuum and aqueous environments (Fig. 15). The quantum chemical reactivity descriptors, energy of the highest occupied molecular orbital (EHOMO), energy of the lowest unoccupied molecular orbital (ELUMO), band gap (ΔE), electronegativity (χ), global hardness (η) and dipole moment (μ) were estimated for the polymer under vacuum and aqueous conditions. Under aqueous conditions, there was no notable difference in the chemical reactivity descriptors except for the dipole moment, suggesting that the van der Waals attracting power power of the polymer is enhanced in the presence of water molecules. Furthermore, the condensed Fukui analysis identified the adsorption sites of CO2 and H2S to be centered in melamine carbon atoms and pyrrole carbon atoms of KFUMP-5. The electron density and Laplacian electron density were computed at several bond critical points (BCPs) between KFUPM-5 and gas molecules using AIM topology analysis. Higher electron density and Laplacian density, which correspond to a greater interaction strength, were obtained for C–N and O–C interactions between CO2 and KFUMP-5, and S–H, H–N and H–C interactions between H2S and KFUMP-5.
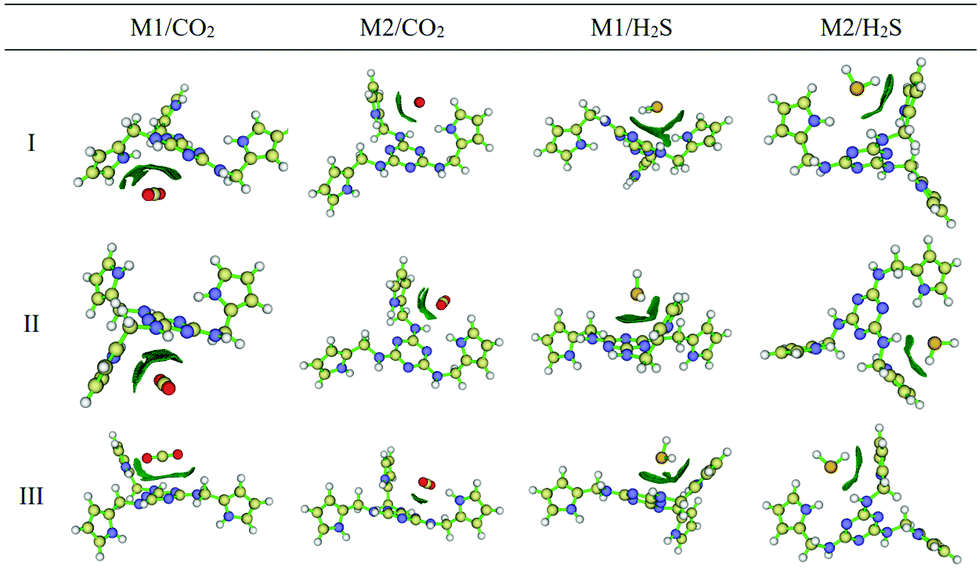 | ||
| Fig. 15 The reduced density gradient (RDG) isosurface analyses of KFUPM-5 core unit (P1) interactions with gas molecules. The blue regions correspond to a strong hydrogen bond; the red regions indicate strong steric effects, while the green regions describe strong van der Waals interactions. M1 and M2 represent the most electrophilic and nucleophilic atoms in the polymer unit. Reproduced with permission from ref. 167. | ||
An examination of CO2 adsorption in an aqueous environment showed an increase in the electron density and Laplacian electron density values suggesting the enhancement of the interaction between CO2 and the polymer and a consequent increase in the adsorption capacity. RDG analysis was performed to visualize the interaction regions between the polymer and gas molecules. It showed that the gas–polymer interaction is predominantly van der Waals, which is in agreement with the chemical reactivity descriptor and the AIMS topology analyses.
Simulation studies have mostly focused on quantifying the interaction strength between the CO2 molecules and amine functionalized POPs, and in a few cases computing CO2 adsorption isotherms. The majority of the functionalized POPs reviewed above have aromatic amines, which interact with CO2 through hydrogen bonding and van der Waals interaction. However, it is well known that alkyl amines have a superior CO2 sorption capacity compared with aromatic amines because CO2 sorption in alkyl amines occurs through a chemical reaction in which CO2 is adsorbed as a carbamate (primary and secondary amines) or bicarbonate (tertiary amines). Since alkyl amine functionalized POPs are garnering a lot of interest, it is imperative that computational research on these types of materials should also focus on the reaction mechanism between CO2 and the adsorbent. Additionally, no computational studies have been performed to investigate the gas diffusion in these classes of PoPs. Diffusion is important for understanding the kinetics of adsorption and desorption of gases in such materials, which is another avenue where the computational modelling could play a major role in advancing the research.
8. Conclusion
Porous organic polymers have shown great potential in carbon capture applications and are the subject of increasing research activities, although they are yet to find their way into commercial use for this purpose. Versatile synthetic methods and functional groups studied to date have provided a large opportunity to create different types of POPs with various sorbent properties. Here, we have covered the recent literature on POPs for carbon capture. We have observed that microporosity and high surface area make POPs a strong candidate. However, as physical sorbents, amine functionalization is critical for this application of POPs. Multiple methods were reviewed for this, including direct amine synthesis, amine grafting, and amine impregnation.Each classification of POP has unique features, with a few noted here in brief summary. COFs have been studied extensively and are known for high porosity and crystalline polymer structures which are amenable to imine functionalization. Triazine and imine functionalized COFs have excellent pore stability, making them suitable for adsorption/desorption cycling in practical applications. Several COFs have been reported with a very high CO2 capacity, such as TpPa-1 and [EtNH2]75-H2P-COF. However, the chemical stability of COFs should be further studied under industrial conditions, including humidity. For POPs that were prepared by direct synthesis, including COFs, the focus to date has been on aromatic amines which have a lower CO2 affinity at low concentrations of CO2 compared to alkyl amines. Therefore, future studies may consider the incorporation of amines with a higher Lewis basicity.
The PPN type POPs were grafted with alkylamines such as DETA and TAEA using the post-synthetic amine functionalization method. This has the advantage of strong covalent bonding with amines, which is a potential solution to the amine leaching problem often associated with amine-based sorbents. The major drawback of PPNs and PAFs could be their high-cost synthesis, which should be altered with more cost-efficient reaction methods to be considered for widespread commercial use. The HCP class of POPs, on the other hand, could offer scalable and less expensive sorbent preparation. Compared to PAFs and PPNs, the surface area range in HCPs is relatively smaller. However, some of the HCPs such as PBTP-(x)-DETA can reach high CO2 uptake capacity and high CO2/N2 selectivity at low CO2 concentrations.
PIMs are also noteworthy because of their unique morphology, consisting of bulky, contorted, one-dimensional monomers which lead to inefficient packing and high porosity. Interestingly, they can be cast into films, fibers, pellets, or other shapes without the use of binder materials. This feature can be exploited to reduce the pressure drop in reactor systems as compared with powder sorbents, which is a major factor in the cost of capture. By contrast, non-processible sorbents are usually loaded into unreactive polymeric or ceramic substrate materials, which can drastically reduce the CO2 adsorption capacity. While other solution processible polymers have been studied as sorbents, they lack the inherent porosity of PIMs. Recent studies have shown that the acid and amidoxime functionalization of PIMs combined with amine functionalization leads to a strong interaction with CO2 and cycle stability. This set of properties is very promising for CO2 capture applications.
Amine-functionalization strategies of POPs covered in this review are important to boost the CO2 capture using POPs. In addition to the high CO2 uptake, future studies should also focus on faster CO2 adsorption and desorption cycles without high energy. High amine loadings in POPs could increase the CO2 uptake performance. However, it can also cause longer adsorption and desorption cycles because of smaller mass and heat transfer in the sorbent media. At this point, the amine type, size and concentration in POPs should be optimized accordingly for the future POP candidates.
Another important consideration in the amine functionalization is the selection of an amine type. Primary amines are mostly incorporated in the sorbents due to their higher CO2 affinity compared to the secondary and tertiary amines. On the other hand, secondary and tertiary amines can be advantageous to be used as their stabilities over oxidation are higher compared to primary amines. For all degrees of amines, alkylamines showed drastically higher CO2 uptake capacity and selectivity than aromatic amines in POP functionalization studies.
Lastly, other sorbent properties such as amine stability, scalability and processibility must be considered for future studies to compete with the best performing sorbents. Processible and scalable POPs such as PIMs are a promising class of POPs as they can be shaped into sorbent geometries such as fibers and pellets. In terms of high porosity and amine stability, amine grafted PPNs also show great potential moving forward to the widespread use of POPs in carbon capture. In the past decade, many new amine functionalized POPs have been studied and reported in the ever-growing literature on CO2 capture. There is significant potential to commercialize POPs for CO2 capture applications using a comprehensive approach considering all necessary sorbent properties discussed in this review.
Disclaimer
This project was funded by the United States Department of Energy, National Energy Technology Laboratory, in part, through a site support contract. Neither the United States Government nor any agency thereof, nor any of their employees, nor the support contractor, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.Conflicts of interest
There are no conflicts to declare.References
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- C. M. White, B. R. Strazisar, E. J. Granite, J. S. Hoffman and H. W. Pennline, J. Air Waste Manage. Assoc., 2003, 53, 645–715 CrossRef CAS PubMed.
- P. Psarras, J. He, H. Pilorgé, N. McQueen, A. Jensen-Fellows, K. Kian and J. Wilcox, Environ. Sci. Technol., 2020, 54, 6272–6280 CrossRef CAS PubMed.
- J. G. Vitillo, B. Smit and L. Gagliardi, Chem. Rev., 2017, 117, 9521–9523 CrossRef CAS PubMed.
- S. Roussanaly, N. Berghout, T. Fout, M. Garcia, S. Gardarsdottir, S. M. Nazir, A. Ramirez and E. S. Rubin, Int. J. Greenhouse Gas Control, 2021, 106, 103263 CrossRef CAS.
- J. Y. Lai, L. H. Ngu and S. S. Hashim, Greenhouse Gases: Sci. Technol., 2021, 11, 1076–1117 CrossRef CAS.
- L. Joos, J. M. Huck, V. Van Speybroeck and B. Smit, Faraday Discuss., 2016, 192, 391–414 RSC.
- Y. Deng, J. Li, Y. Miao and D. Izikowitz, Energy Rep., 2021, 7, 3506–3516 CrossRef.
- H. Azarabadi and K. S. Lackner, Appl. Energy, 2019, 250, 959–975 CrossRef.
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- D. Aaron and C. Tsouris, Sep. Sci. Technol., 2005, 40, 321–348 CrossRef CAS.
- X. Wu, Y. Yu, Z. Qin and Z. Zhang, Energy Procedia, 2014, 63, 1339–1346 CrossRef CAS.
- A. Sayari, Y. Belmabkhout and R. Serna-Guerrero, Chem. Eng. J., 2011, 171, 760–774 CrossRef CAS.
- C.-H. Yu, C.-H. Huang and C.-S. Tan, Aerosol Air Qual. Res., 2012, 12, 745–769 CrossRef CAS.
- S. Sjostrom and H. Krutka, Fuel, 2010, 89, 1298–1306 CrossRef CAS.
- A. Sattari, A. Ramazani, H. Aghahosseini and M. K. Aroua, J. CO2 Util., 2021, 48, 101526 CrossRef CAS.
- H. A. Patel, J. Byun and C. T. Yavuz, ChemSusChem, 2017, 10, 1303–1317 CrossRef CAS PubMed.
- B. Li, Y. Duan, D. Luebke and B. Morreale, Appl. Energy, 2013, 102, 1439–1447 CrossRef CAS.
- S. Choi, M. L. Gray and C. W. Jones, ChemSusChem, 2011, 4, 628–635 CrossRef CAS PubMed.
- Z. Zhang, Z. P. Cano, D. Luo, H. Dou, A. Yu and Z. Chen, J. Mater. Chem. A, 2019, 7, 20985–21003 RSC.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- R. L. Siegelman, E. J. Kim and J. R. Long, Nat. Mater., 2021, 20, 1060–1072 CrossRef CAS PubMed.
- J. S. Hoffman, S. Hammache, M. L. Gray, D. J. Fauth and H. W. Pennline, Fuel Process. Technol., 2014, 126, 173–187 CrossRef CAS.
- W. Wang, M. Zhou and D. Yuan, J. Mater. Chem. A, 2017, 5, 1334–1347 RSC.
- M. G. Mohamed, A. F. M. EL-Mahdy, M. G. Kotp and S.-W. Kuo, Mater. Adv., 2022, 3, 707–733 RSC.
- X. Kong, S. Li, M. Strømme and C. Xu, Nanomaterials, 2019, 9, 1020 CrossRef CAS PubMed.
- P. Bhanja, A. Modak and A. Bhaumik, ChemCatChem, 2019, 11, 244–257 CrossRef CAS.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- C. Xu and N. Hedin, Mater. Today, 2014, 17, 397–403 CrossRef CAS.
- J. Wang, L. Wang, Y. Wang, D. Zhang, Q. Xiao, J. Huang and Y.-N. Liu, Chin. J. Chem. Eng., 2022, 42, 91–103 CrossRef.
- L. Zou, Y. Sun, S. Che, X. Yang, X. Wang, M. Bosch, Q. Wang, H. Li, M. Smith, S. Yuan, Z. Perry and H. Zhou, Adv. Mater., 2017, 29, 1700229 CrossRef PubMed.
- S. Rochat, M. Tian, R. Atri, T. J. Mays and A. D. Burrows, Multifunct. Mater., 2021, 4, 025002 CrossRef CAS.
- Y. Yang, C. Y. Chuah and T.-H. Bae, ACS Sustainable Chem. Eng., 2021, 9, 2017–2026 CrossRef CAS.
- Y. Yang, C. Y. Chuah and T.-H. Bae, Chem. Eng. J., 2019, 358, 1227–1234 CrossRef CAS.
- N. McQueen, K. V. Gomes, C. McCormick, K. Blumanthal, M. Pisciotta and J. Wilcox, Prog. Energy, 2021, 3, 032001 CrossRef.
- T. Ben and S. Qiu, CrystEngComm, 2013, 15, 17–26 RSC.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- Y. Zhu, H. Long and W. Zhang, Chem. Mater., 2013, 25, 1630–1635 CrossRef CAS.
- M. Khakbaz, A. Ghaemi and G. Mir Mohamad Sadeghi, Polym. Chem., 2021, 12, 6962–6997 RSC.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- A. K. Sekizkardes, T. İslamoğlu, Z. Kahveci and H. M. El-Kaderi, J. Mater. Chem. A, 2014, 2, 12492–12500 RSC.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 CrossRef CAS.
- M. Li, H. Ren, F. Sun, Y. Tian, Y. Zhu, J. Li, X. Mu, J. Xu, F. Deng and G. Zhu, Adv. Mater., 2018, 30, 1804169 CrossRef PubMed.
- Y. Yuan and G. Zhu, ACS Cent. Sci., 2019, 5, 409–418 CrossRef CAS PubMed.
- Y. Yuan, Y. Yang and G. Zhu, EnergyChem, 2020, 2, 100037 CrossRef.
- J.-S. Sun, L.-P. Jing, Y. Tian, F. Sun, P. Chen and G. Zhu, Chem. Commun., 2018, 54, 1603–1606 RSC.
- W. Zhang, Y. Cheng, C. Guo, C. Xie and Z. Xiang, Ind. Eng. Chem. Res., 2018, 57, 10985–10991 CrossRef CAS.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H.-C. Zhou, Angew. Chem., 2012, 124, 7598–7602 CrossRef.
- Y. Hu, W. M. Verdegaal, S.-H. Yu and H.-L. Jiang, ChemSusChem, 2014, 7, 734–737 CrossRef CAS PubMed.
- L.-B. Sun, A.-G. Li, X.-D. Liu, X.-Q. Liu, D. Feng, W. Lu, D. Yuan and H.-C. Zhou, J. Mater. Chem. A, 2015, 3, 3252–3256 RSC.
- W. Lu, M. Bosch, D. Yuan and H.-C. Zhou, ChemSusChem, 2015, 8, 433–438 CrossRef CAS PubMed.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H.-C. Zhou, J. Am. Chem. Soc., 2011, 133, 18126–18129 CrossRef CAS PubMed.
- S. J. Garibay, M. H. Weston, J. E. Mondloch, Y. J. Colón, O. K. Farha, J. T. Hupp and S. T. Nguyen, CrystEngComm, 2013, 15, 1515–1519 RSC.
- S. Sung and M. P. Suh, J. Mater. Chem. A, 2014, 2, 13245–13249 RSC.
- T. Islamoglu, T. Kim, Z. Kahveci, O. M. El-Kadri and H. M. El-Kaderi, J. Phys. Chem. C, 2016, 120, 2592–2599 CrossRef CAS.
- T. İslamoğlu, M. Gulam Rabbani and H. M. El-Kaderi, J. Mater. Chem. A, 2013, 1, 10259 RSC.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- Q. Liao, C. Ke, X. Huang, D. Wang, Q. Han, Y. Zhang, Y. Zhang and K. Xi, Angew. Chem., Int. Ed., 2021, 60, 1411–1416 CrossRef CAS PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- Y. Jin, Y. Zhu and W. Zhang, CrystEngComm, 2013, 15, 1484–1499 RSC.
- E. Vitaku and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 12911–12914 CrossRef CAS PubMed.
- M. G. Rabbani, A. K. Sekizkardes, Z. Kahveci, T. E. Reich, R. Ding and H. M. El-Kaderi, Chem. – Eur. J., 2013, 19, 3324–3328 CrossRef CAS PubMed.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed.
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS.
- K. Gottschling, L. Stegbauer, G. Savasci, N. A. Prisco, Z. J. Berkson, C. Ochsenfeld, B. F. Chmelka and B. V. Lotsch, Chem. Mater., 2019, 31, 1946–1955 CrossRef CAS PubMed.
- N. Huang, R. Krishna and D. Jiang, J. Am. Chem. Soc., 2015, 137, 7079–7082 CrossRef CAS PubMed.
- F. Haase and B. V. Lotsch, Chem. Soc. Rev., 2020, 49, 8469–8500 RSC.
- X. Li, C. Yang, B. Sun, S. Cai, Z. Chen, Y. Lv, J. Zhang and Y. Liu, J. Mater. Chem. A, 2020, 8, 16045–16060 RSC.
- P. Pandey, A. P. Katsoulidis, I. Eryazici, Y. Wu, M. G. Kanatzidis and S. T. Nguyen, Chem. Mater., 2010, 22, 4974–4979 CrossRef CAS.
- P. Arab, A. Verlander and H. M. El-Kaderi, J. Phys. Chem. C, 2015, 119, 8174–8182 CrossRef CAS.
- X. Gao, X. Zou, H. Ma, S. Meng and G. Zhu, Adv. Mater., 2014, 26, 3644–3648 CrossRef CAS PubMed.
- M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2012, 24, 1511–1517 CrossRef CAS.
- M. G. Rabbani, A. K. Sekizkardes, O. M. El-Kadri, B. R. Kaafarani and H. M. El-Kaderi, J. Mater. Chem., 2012, 22, 25409 RSC.
- A. K. Sekizkardes, S. Altarawneh, Z. Kahveci, T. İslamoğlu and H. M. El-Kaderi, Macromolecules, 2014, 47, 8328–8334 CrossRef CAS.
- X. Zhu, S. M. Mahurin, S.-H. An, C.-L. Do-Thanh, C. Tian, Y. Li, L. W. Gill, E. W. Hagaman, Z. Bian, J.-H. Zhou, J. Hu, H. Liu and S. Dai, Chem. Commun., 2014, 50, 7933 RSC.
- Q. Chen, M. Luo, P. Hammershøj, D. Zhou, Y. Han, B. W. Laursen, C.-G. Yan and B.-H. Han, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef CAS PubMed.
- T. Muller and S. Bräse, Angew. Chem., Int. Ed., 2011, 50, 11844–11845 CrossRef CAS PubMed.
- S. Mondal and N. Das, J. Mater. Chem. A, 2015, 3, 23577–23586 RSC.
- H. A. Patel, S. Hyun Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Nat. Commun., 2013, 4, 1357 CrossRef PubMed.
- N. A. Dogan, Y. Hong, E. Ozdemir and C. T. Yavuz, ACS Sustainable Chem. Eng., 2019, 7, 123–128 CrossRef CAS.
- H. A. Patel and C. T. Yavuz, Faraday Discuss., 2015, 183, 401–412 RSC.
- T. Gelles and F. Rezaei, AIChE J., 2020, 66, 16785–16789 CrossRef.
- R. Yuan, H. Ren, Z. Yan, A. Wang and G. Zhu, Polym. Chem., 2014, 5, 2266 RSC.
- W. Lu, W. M. Verdegaal, J. Yu, P. B. Balbuena, H.-K. Jeong and H.-C. Zhou, Energy Environ. Sci., 2013, 6, 3559 RSC.
- Z. Li, Y. Zhi, X. Feng, X. Ding, Y. Zou, X. Liu and Y. Mu, Chem. – Eur. J., 2015, 21, 12079–12084 CrossRef CAS PubMed.
- Y. Zhao, K. X. Yao, B. Teng, T. Zhang and Y. Han, Energy Environ. Sci., 2013, 6, 3684 RSC.
- F. Liu, W. Fu and S. Chen, J. Appl. Polym. Sci., 2020, 137, 48479 CrossRef CAS.
- K. A. Fayemiwo, G. T. Vladisavljević, S. A. Nabavi, B. Benyahia, D. P. Hanak, K. N. Loponov and V. Manović, Chem. Eng. J., 2018, 334, 2004–2013 CrossRef CAS.
- F. Liu, S. Wang, G. Lin and S. Chen, New J. Chem., 2018, 42, 420–428 RSC.
- T. Ratvijitvech, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Polymer, 2014, 55, 321–325 CrossRef CAS.
- R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173 RSC.
- J. Chen, W. Yan, E. J. Townsend, J. Feng, L. Pan, V. Del Angel Hernandez and C. F. J. Faul, Angew. Chem., Int. Ed., 2019, 58, 11715–11719 CrossRef CAS PubMed.
- P. Mu, H. Sun, Z. Zhu, W. Liang, J. Liu and A. Li, Macromol. Mater. Eng., 2016, 301, 451–456 CrossRef CAS.
- Y. Liu, S. Wang, X. Meng, Y. Ye, X. Song and Z. Liang, Mater. Chem. Front., 2021, 5, 5319–5327 RSC.
- V. Guillerm, J. Weseliński, M. Alkordi, M. I. H. Mohideen, Y. Belmabkhout, A. J. Cairns and M. Eddaoudi, Chem. Commun., 2014, 50, 1937 RSC.
- S. H. Pang, M. L. Jue, J. Leisen, C. W. Jones and R. P. Lively, ACS Macro Lett., 2015, 4, 1415–1419 CrossRef CAS PubMed.
- A. K. Sekizkardes, S. Hammache, J. S. Hoffman and D. Hopkinson, ACS Appl. Mater. Interfaces, 2019, 11, 30987–30991 CrossRef CAS PubMed.
- A. Miles, W. C. Wilfong, D. Hopkinson and A. K. Sekizkardes, Energy Technol., 2020, 8, 2000419 CrossRef CAS.
- S. K. Das, P. Bhanja, S. K. Kundu, S. Mondal and A. Bhaumik, ACS Appl. Mater. Interfaces, 2018, 10, 23813–23824 CrossRef CAS PubMed.
- J. Huang, J. Zhu, S. A. Snyder, A. J. Morris and S. R. Turner, Polymer, 2018, 154, 55–61 CrossRef CAS.
- A. A. Olajire, Greenhouse Gases: Sci. Technol., 2017, 7, 399–459 CrossRef CAS.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- B. Li, R. Gong, Y. Luo and B. Tan, Soft Matter, 2011, 7, 10910 RSC.
- H. Ramezanipour Penchah, A. Ghaemi and H. Ghanadzadeh Gilani, Polym. Bull., 2022, 79, 3681–3702 CrossRef CAS.
- C. F. Martín, E. Stöckel, R. Clowes, D. J. Adams, A. I. Cooper, J. J. Pis, F. Rubiera and C. Pevida, J. Mater. Chem., 2011, 21, 5475 RSC.
- R. Dawson, E. Stöckel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239 RSC.
- R. Vinodh, P. Hemalatha, M. Ganesh, M. M. Peng, A. Abidov, M. Palanichamy, W. S. Cha and H.-T. Jang, RSC Adv., 2014, 4, 3678–3684 RSC.
- D. Zhang, L. Tao, J. Ju, Y. Wang, Q. Wang and T. Wang, Polymer, 2015, 60, 234–240 CrossRef CAS.
- M. Ansari, R. Bera and N. Das, J. Appl. Polym. Sci., 2022, 139, 51449 CrossRef CAS.
- D. Chen, S. Gu, Y. Fu, X. Fu, Y. Zhang, G. Yu and C. Pan, New J. Chem., 2017, 41, 6834–6839 RSC.
- A. Hassan, S. Goswami, A. Alam, R. Bera and N. Das, Sep. Purif. Technol., 2021, 257, 117923 CrossRef CAS.
- H. Ouyang, K. Song, J. Du, Z. Zhan and B. Tan, Chem. Eng. J., 2022, 431, 134326 CrossRef CAS.
- Y. Qiao, Z. Zhan, Y. Yang, M. Liu, Q. Huang, B. Tan, X. Ke and C. Wu, Mater. Today Commun., 2021, 27, 102338 CrossRef CAS.
- P. Su, X. Zhang, Z. Xu, G. Zhang, C. Shen and Q. Meng, New J. Chem., 2019, 43, 17267–17274 RSC.
- C. Xu, Z. Bacsik and N. Hedin, J. Mater. Chem. A, 2015, 3, 16229–16234 RSC.
- H. R. Penchah, P. Najafi, A. Ghaemi and H. Ghanadzadeh Gilani, Environ. Prog. Sustainable Energy, 2020, 40, 13586–13590 Search PubMed.
- S. Krishnan and C. V. Suneesh, J. Solid State Chem., 2021, 299, 122152 CrossRef CAS.
- P. Najafi, H. Ramezanipour Penchah and A. Ghaemi, Environ. Technol. Innovation, 2021, 23, 101746 CrossRef CAS.
- H. Zhou, C. Rayer, A. R. Antonangelo, N. Hawkins and M. Carta, ACS Appl. Mater. Interfaces, 2022, 14, 20997–21006 CrossRef CAS PubMed.
- L. B. Hamdy, A. Gougsa, W. Y. Chow, J. E. Russell, E. García-Díez, V. Kulakova, S. Garcia, A. R. Barron, M. Taddei and E. Andreoli, Mater. Adv., 2022, 3, 3174–3191 RSC.
- D. Zhang, L. Tao, Q. Wang and T. Wang, Polymer, 2016, 82, 114–120 CrossRef CAS.
- Z. Tian, J. Huang, Z. Zhang, G. Shao, A. Liu and S. Yuan, Microporous Mesoporous Mater., 2016, 234, 130–136 CrossRef CAS.
- H. Gao, L. Ding, H. Bai and L. Li, ChemSusChem, 2017, 10, 618–623 CrossRef CAS PubMed.
- Z. Fu, J. Jia, J. Li and C. Liu, Chem. Eng. J., 2017, 323, 557–564 CrossRef CAS.
- Q. B. Meng and J. Weber, ChemSusChem, 2014, 7, 3312–3318 CrossRef CAS PubMed.
- X. Yan, L. Zhang, Y. Zhang, G. Yang and Z. Yan, Ind. Eng. Chem. Res., 2011, 50, 3220–3226 CrossRef CAS.
- F. Raganati, F. Miccio and P. Ammendola, Energy Fuels, 2021, 35, 12845–12868 CrossRef CAS.
- C. Y. Chuah, W. Li, Y. Yang and T.-H. Bae, Chem. Eng. J. Adv., 2020, 3, 100021 CrossRef CAS.
- X. Yang, M. Yu, Y. Zhao, C. Zhang, X. Wang and J.-X. Jiang, J. Mater. Chem. A, 2014, 2, 15139–15145 RSC.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- R. Dawson, A. Laybourn, R. Clowes, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2009, 42, 8809–8816 CrossRef CAS.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. AlBahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- A. F. Saber, K.-Y. Chen, A. F. M. EL-Mahdy and S.-W. Kuo, J. Polym. Res., 2021, 28, 430 CrossRef CAS.
- L. Wang, C. Yao, W. Xie, G. Xu, S. Zhang and Y. Xu, New J. Chem., 2021, 45, 19636–19640 RSC.
- Y. Xu, Z. Li, F. Zhang, X. Zhuang, Z. Zeng and J. Wei, RSC Adv., 2016, 6, 30048–30055 RSC.
- L. Bao, H. Sun, Z. Zhu, W. Liang, P. Mu, J. Zang and A. Li, Mater. Lett., 2016, 178, 5–9 CrossRef CAS.
- A. K. Sekizkardes, J. T. Culp, T. Islamoglu, A. Marti, D. Hopkinson, C. Myers, H. M. El-Kaderi and H. B. Nulwala, Chem. Commun., 2015, 51, 13393–13396 RSC.
- Z. Kahveci, A. K. Sekizkardes, R. K. Arvapally, L. Wilder and H. M. El-Kaderi, Polym. Chem., 2017, 8, 2509–2515 RSC.
- T. Ratvijitvech, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Polymer, 2014, 55, 321–325 CrossRef CAS.
- X. Wang, Y. Zhao, L. Wei, C. Zhang and J.-X. Jiang, J. Mater. Chem. A, 2015, 3, 21185–21193 RSC.
- A. Mukhtar, S. Saqib, N. B. Mellon, M. Babar, S. Rafiq, S. Ullah, M. A. Bustam, A. G. Al-Sehemi, N. Muhammad and M. Chawla, J. Nat. Gas Sci. Eng., 2020, 77, 103203 CrossRef CAS.
- H. Gao, Q. Li and S. Ren, Curr. Opin. Green Sustainable Chem., 2019, 16, 33–38 CrossRef.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- P. M. Budd and A. B. Foster, Curr. Opin. Chem. Eng., 2022, 36, 100792 CrossRef.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675 RSC.
- B. Satilmis, Curr. Opin. Chem. Eng., 2022, 36, 100793 CrossRef.
- Y. Wang, B. S. Ghanem, Y. Han and I. Pinnau, Curr. Opin. Chem. Eng., 2022, 35, 100755 CrossRef.
- Y. H. Lee, J. Jeong, K. Kim, T. Hyun, A. Jamal and D.-Y. Koh, Chem. Mater., 2020, 32, 7081–7104 CrossRef CAS.
- H. A. Patel and C. T. Yavuz, Chem. Commun., 2012, 48, 9989 RSC.
- B. Satilmis, M. Lanč, A. Fuoco, C. Rizzuto, E. Tocci, P. Bernardo, G. Clarizia, E. Esposito, M. Monteleone, M. Dendisová, K. Friess, P. M. Budd and J. C. Jansen, J. Membr. Sci., 2018, 555, 483–496 CrossRef CAS.
- W. Quan, F. Zhang, B. L. Hamlett, M. G. Finn, C. W. Abney, S. C. Weston, R. P. Lively and W. J. Koros, Ind. Eng. Chem. Res., 2021, 60, 12709–12718 CrossRef CAS.
- D. Hopkinson and A. K. Sekizkardes, US Pat., 20220032268A1, 2022, 7/20/2021 Search PubMed.
- N. A. D. Ho and C. P. Leo, Environ. Res., 2021, 197, 111100 CrossRef CAS PubMed.
- A. R. Sujan, D.-Y. Koh, G. Zhu, V. P. Babu, N. Stephenson, A. Rosinski, H. Du, Y. Luo, W. J. Koros and R. P. Lively, Ind. Eng. Chem. Res., 2018, 57, 11757–11766 CrossRef CAS.
- F. Rezaei, R. P. Lively, Y. Labreche, G. Chen, Y. Fan, W. J. Koros and C. W. Jones, ACS Appl. Mater. Interfaces, 2013, 5, 3921–3931 CrossRef CAS PubMed.
- R. Babarao, S. Dai and D. Jiang, Langmuir, 2011, 27, 3451–3460 CrossRef CAS PubMed.
- M. Wang, S. Wei, Z. Wu, S. Zhou, Z. Wang, J. Wang and X. Lu, Mater. Lett., 2018, 230, 28–31 CrossRef CAS.
- V. Tognetti and L. Joubert, Phys. Chem. Chem. Phys., 2014, 16, 14539 RSC.
- J. H. Hymel, J. Townsend and K. D. Vogiatzis, J. Phys. Chem. A, 2019, 123, 10116–10122 CrossRef CAS PubMed.
- M. M. Abdelnaby, K. E. Cordova, I. Abdulazeez, A. M. Alloush, B. A. Al-Maythalony, Y. Mankour, K. Alhooshani, T. A. Saleh and O. C. S. Al Hamouz, ACS Appl. Mater. Interfaces, 2020, 12, 47984–47992 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |