
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Night-time oxidation at the air–water interface: co-surfactant effects in binary mixtures†
Federica
Sebastiani‡
 ab,
Richard A.
Campbell§
b and
Christian
Pfrang
*acd
ab,
Richard A.
Campbell§
b and
Christian
Pfrang
*acd
aDepartment of Chemistry, University of Reading, P. O. Box 224, RG6 6AD, Reading, UK
bInstitut Laue-Langevin, 71 avenue des Martyrs, CS20156, 38042 Grenoble Cedex 9, France
cSchool of Geography, Earth and Environmental Sciences, University of Birmingham, Edgbaston, B15 2TT, Birmingham, UK. E-mail: c.pfrang@bham.ac.uk
dDepartment of Meteorology, University of Reading, P. O. Box 243, RG6 6BB, Reading, UK
First published on 12th September 2022
Abstract
The ageing of organic-coated aqueous aerosols at night is investigated by reacting NO3 with binary surfactant mixtures floating on water. The surfactants are oleic acid (OA), methyl oleate (MO) and stearic acid (SA). Deuterated surfactants mixed with hydrogenous surfactants were studied using neutron reflectometry to determine the reaction kinetics of organic two-component monolayers with NO3 at the air–water interface for the first time. We measured the rate coefficients for OA monolayers, mixed with hydrogenous co-surfactant MO or SA to be (3 ± 1) × 10−8 cm2 per molecule per s or (3.6 ± 0.9) × 10−8 cm2 per molecule per s and MO monolayers mixed with hydrogenous co-surfactant OA or SA to be (0.7 ± 0.4) × 10−8 cm2 per molecule per s or (3 ± 1) × 10−8 cm2 per molecule per s. The initial desorption lifetimes of NO3, τd,NO3,1, were 8 ± 3 ns, 14 ± 4 ns, 12 ± 3 ns and 21 ± 10 ns. The approximately doubled desorption lifetime for MO–SA compared to the other mixtures is consistent with a more accessible double bond associated with the larger area per molecule of MO in the presence of SA facilitating NO3 attack. The significantly slower reactive loss of MO–OA compared to a MO monolayer demonstrates that multi-component surfactant mixtures need to be studied in addition to single-component monolayers. Such a retarded decay would cause the residence time to change from ca. 4 to 22 minutes associated with increased transport distances of surfactant species together with any other pollutants that may be protected underneath the surfactant film.
Environmental significanceThe most significant result was a ca. six-fold slower decay for the MO–OA binary mixture compared to a MO monolayer demonstrating that matrix effects of co-surfactants affect reaction kinetics; data from monomolecular surfactant experiments thus cannot simply be added up to understand the decay of these species in complex atmospherically relevant mixtures. Such a retardation of oxidative decay was not found for binary mixtures of surfactants with carboxylic acid headgroups. It thus seems likely that the less stable species at the air–water interface are more strongly affected by co-surfactants. This is particularly important given these species may be more likely to be encountered in the atmosphere than those with perfectly hydrophilic headgroups that are most commonly studied in the laboratory. |
1 Introduction
Atmospheric aerosols are of key importance because of their impact on Earth's radiative balance and on cloud formation,1–3 but also due to their link to air pollution.4 These aerosols originate both from natural and man-made sources. Aerosol behaviour is strongly affected by the presence of organic material both in the bulk and at the surface.5–7 Organic components of atmospheric aerosols are often surface-active including organic acids and diacids, proteins and humic-like substances (HULIS).8 Key components of surface-active organic aerosols are fatty acids;9–14 it is important to note that phase behaviour especially for fatty acids is highly temperature dependent,8 and little data are available for temperatures lower than ambient e.g. ref. 15, which are particularly relevant for surfactants in clouds.8–14 Atmospheric fatty acids include saturated (such as palmitic acid16) and unsaturated acids in particular oleic acid which is found both in marine17–19 and cooking20–22 aerosols. Cooking emissions have been proposed to contribute ca. 10% to the man-made emission of small particulate matter (PM2.5) at 320 mg per person per day based on measurements in London.23 The atmospheric lifetime of aerosol particles is largely determined by chemical ageing initiated by the oxidants nitrate radicals (NO3), hydroxyl radicals (OH) and ozone (O3). For investigation of this chemical aerosol ageing, it is crucial to study the heterogeneous reactions occurring between the aerosol and these gas-phase oxidants. Homogeneous chemistry is relatively well understood at a molecular level, while the details of heterogeneous chemistry remain largely unknown. Field measurements suggest that heterogeneous reactions substantially affect the chemical composition of particles, in particular that of their surface films.24 Such reactions may alter key particle properties such as aerosol hydrophilicity, toxicity as well as their optical properties. For example, polycyclic aromatic hydrocarbons (PAHs) as well as phthalates have been identified together with oleic acid in marine aerosols25 with several studies suggesting that surface-active coatings on organic aerosols shield these PAHs, thus increasing their ability to be transported further and cause harm.26–28 The vast majority of studies to date have investigated the heterogeneous reactions of organic aerosols with O3 and OH, which are the main oxidants during daytime. During night-time, [OH] is very low while the concentration of the photo-labile NO3 will build up, so that NO3 becomes highly significant. Therefore, while OH controls the chemistry of the daytime atmosphere, NO3 radicals have a similar role during the night.29–32 Khan and co-workers33 reported increases in NO3 of up to 15 ppt from pre-industrial times to the present day.In many cases heterogeneous reactions have been studied using organic droplets or thick films.34,35 However, it has been shown that experimental studies of organic molecules self-assembled at the surface of water rather than purely organic aerosols alone are key to understanding atmospheric ageing of aerosols covered in organic material.7,36
In the work presented here organic monolayers at the air–water interface are used as proxies for organic-coated aqueous atmospheric aerosols, and their reactions with NO3 are studied. Most of the previous work with surfactant monolayers has focussed on ozonolysis,37–40 with much less studies focussing on reaction with nitrate radicals.41–43 Furthermore, the majority of the previous investigations has described the reaction of one surfactant with the oxidant, while it is increasingly clear that it is needed to include more than one surfactant species when mimicking the organic coated atmospheric aerosols.
The surfactant species chosen are oleic acid (OA), methyl oleate (MO) and stearic acid (SA). OA,35,37,38,41,44,45 MO26,39,41,42,46–49 and SA50 are popular model systems for atmospheric surfactants. MO, the methyl ester of OA, is a main component of biodiesel (chemical name: fatty acid methyl esters or ‘FAME’),51 likely leading to an increased atmospheric abundance in the future given up to 7% of FAME is added to standard petroleum diesel in the EU to reduce greenhouse gas emissions; higher proportions of FAME in petroleum diesel (10% FAME sold as ‘B10’ and 20% FAME sold as ‘B20’) and pure FAME (‘B100’) become increasingly common fuel alternatives across a number of European countries such as Germany, France and Finland.
This range of molecules allows the investigation of the effects of head group and degree of unsaturation on the reaction kinetics and products formed. Building on our previous work on single surfactants,41 we focussed this study on the impact of co-surfactants on the NO3-initiated oxidation of these surfactants in binary mixtures.
What is lacking so far in the literature is the link between laboratory studies of single component surfactant films and real atmospheric surfactant materials. Key questions are if studies of single surfactants can be combined in modelling studies to describe more complex real surfactant compositions and also if co-surfactants will affect the loss of the reactive surfactant species. Xiao and Bertram42 investigated NO3-initiated oxidation of binary mixtures containing the unsaturated organic MO and saturated molecules (diethyl sebacate, dioctyl sebacate, and squalane) as matrix molecules. They have used a rotating-wall flow tube reactor coupled to a chemical ionization mass spectrometer (CIMS) with the aim to better understand the reactivity of unsaturated organics in multicomponent and multiphase atmospheric particles; their results suggest that for liquid binary mixtures the reactivity of methyl oleate depended on the matrix molecule. Our method of neutron reflectometry (NR) is particularly well suited for the study of these mixtures, since deuteration of one of the surfactants in the binary mixture allowed us to follow the kinetics of the deuterated component and then reverse deuteration enabled us to follow the kinetics of the other surfactant with molecular resolution (see Fig. 1). The surface excess of the organic molecule during the oxidation reaction is monitored by NR at the air–ACMW (air–contrast matched water) interface,52 and information about reaction mechanisms can even be accessed thanks to partial deuteration of the surfactant.53,54 Furthermore, the surface composition of mixed systems can be resolved in situ during dynamic processes by the selective deuteration of different components,55,56 and therefore the reaction rates of individual components in mixtures can be determined. NR is an ideal technique to resolve kinetic and dynamic processes at fluid interfaces on the second time scale:39,57 in the present work NR is used effectively to measure the surface excess of the deuterated component of binary surfactant mixtures during reactions with gas-phase NO3. NO3 is produced in situ by reacting O3 with NO2, the dependence of [NO3] on the initial [NO2] and [O3] is modelled, and to determine the concentration of NO3, the steady state concentrations of NO2 and N2O5 are measured using FTIR spectroscopy as a function of the initial [NO2].41
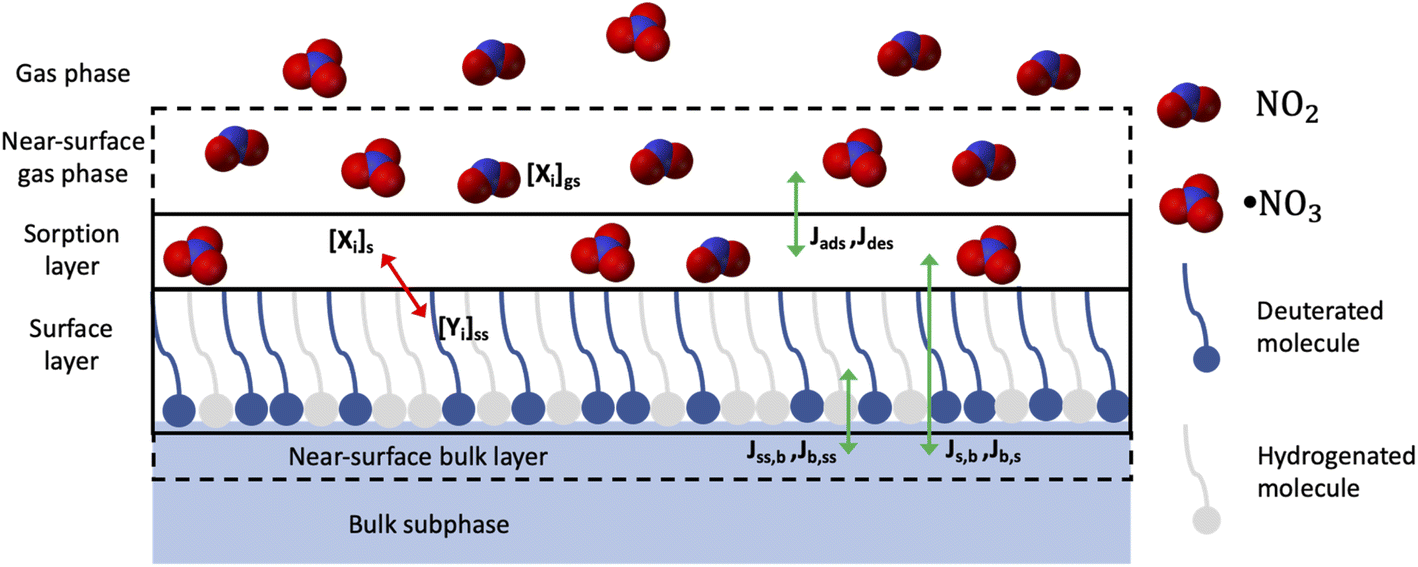 | ||
| Fig. 1 The mixture of deuterated and hydrogenous organic molecules self-assembles at the air–water interface as a monolayer and the gas species, NO3 and NO2, are expected to interact with both species. However, when monitoring the surface excess with neutron reflectometry, only the deuterated molecules are visible and hence the data collected provide information only on the reaction kinetic of the deuterated molecules. The red arrow shows chemical reaction, while the green arrows show the transport fluxes. | ||
The analysis of the kinetic experiments of the binary mixtures was based on the modelling approach developed in our previous work,41 where we took into account all the key reactions and processes. Briefly, in order to describe the NO3-initiated oxidation we used a model, which considers, in addition to reactions, other mechanisms, such as accommodation, desorption, competition for adsorption sites and transport of the gas-phase species (see Sebastiani et al.41). This model builds on the formalism and terminology of the “PRA framework” (introduced by Pöschl, Rudich and Ammann58). It is a combination of K2-SURF, kinetic double-layer surface model59 and KM-SUB, kinetic multi-layer model of aerosol surface and bulk chemistry,60 but has been adapted to a planar geometry (see Sebastiani et al.41). KM–SUB and K2–SURF have been applied to describe a range of experimental datasets and conditions.48,61,62 Both models describe the evolution of the kinetic parameters of an organic droplet exposed to oxidants. We have adapted the model to a monomolecular organic layer at the air–water interface for analysis and interpretation of the experimental data presented here. In the binary mixture, only the surface excess of the deuterated component is detected and hence the kinetic modelling describes the reaction of the deuterated surfactant. The kinetic parameters embed both the oxidant and the co-surfactant effect on the reaction with the deuterated component. The kinetic analysis of the measured surface excess decays for the four reaction systems provides information on the rate coefficients of the heterogeneous reaction as well as indirect information on the effect of the co-surfactant and the formation of surface-active products. The results obtained for the different molecules will be discussed in relation of their chemical structures. Furthermore, the comparison between NO3 and other oxidants species indicates to what extent night-time oxidation is important to atmospheric aerosol ageing. We also estimated oxidant uptake coefficients and compared those to literature data on similar organic molecules that have been studied in the condensed phase (i.e. droplets or thick films34,35).
2 Methods
2.1 Experimental
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CD(CD2)7CO2D, Sigma-Aldrich, isotopic purity ≥98%, purity 99%), oleic acid (CH3(CH2)7CHCH(CH2)7COOH, Sigma-Aldrich, purity 99%) deuterated methyl oleate (d33MO, CD3(CD2)7CDCD(CD2)7CO2CH3, custom-synthesised by the Oxford Deuteration Facility, ∼95%), methyl oleate (CH3(CH2)7CHCH(CH2)7CO2CH3, Sigma-Aldrich, purity 99%) and stearic acid (CH3(CH2)16COOH, Sigma-Aldrich, purity 98.5%); further details may be found in Section 1 of the ESI;† the chemical structures of the molecules studied are displayed in Scheme 1. The subphase was a mixture of 8.1% by volume D2O (Sigma Aldrich, 99.9%) in pure H2O (generated using a Millipore purification unit, 18.2 MΩ cm), referred to as air-contrast matched water (ACMW). Chloroform (Sigma-Aldrich, >99.8%) and O2 (Air Liquide, France, >99.9%) were used as supplied. NO2 was supplied in small gas cylinders (112 dm3) by Scientific and Technical Gases Ltd (Newcastle-under-Lyme, UK) and provided as a mixture of 1000 ppm of NO2 in synthetic air with an analytical tolerance of ±2%. The solutions of organic molecules in chloroform were prepared shortly before the experiments and then mixed in a 1
CD(CD2)7CO2D, Sigma-Aldrich, isotopic purity ≥98%, purity 99%), oleic acid (CH3(CH2)7CHCH(CH2)7COOH, Sigma-Aldrich, purity 99%) deuterated methyl oleate (d33MO, CD3(CD2)7CDCD(CD2)7CO2CH3, custom-synthesised by the Oxford Deuteration Facility, ∼95%), methyl oleate (CH3(CH2)7CHCH(CH2)7CO2CH3, Sigma-Aldrich, purity 99%) and stearic acid (CH3(CH2)16COOH, Sigma-Aldrich, purity 98.5%); further details may be found in Section 1 of the ESI;† the chemical structures of the molecules studied are displayed in Scheme 1. The subphase was a mixture of 8.1% by volume D2O (Sigma Aldrich, 99.9%) in pure H2O (generated using a Millipore purification unit, 18.2 MΩ cm), referred to as air-contrast matched water (ACMW). Chloroform (Sigma-Aldrich, >99.8%) and O2 (Air Liquide, France, >99.9%) were used as supplied. NO2 was supplied in small gas cylinders (112 dm3) by Scientific and Technical Gases Ltd (Newcastle-under-Lyme, UK) and provided as a mixture of 1000 ppm of NO2 in synthetic air with an analytical tolerance of ±2%. The solutions of organic molecules in chloroform were prepared shortly before the experiments and then mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, the concentrations are given in mM: for d34OA-hMO 4.17 mM, for d34OA-hSA 4.37 mM, for d33MO-hOA 4.05 mM and for d33MO-hSA 3.77 mM.
:1 molar ratio, the concentrations are given in mM: for d34OA-hMO 4.17 mM, for d34OA-hSA 4.37 mM, for d33MO-hOA 4.05 mM and for d33MO-hSA 3.77 mM.
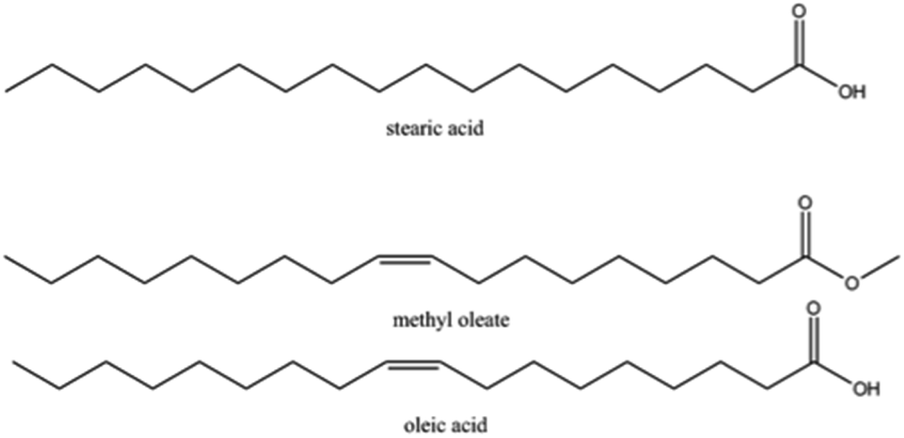 | ||
| Scheme 1 Chemical structure of the organic molecules studied. | ||
| NO3/molecule per cm3 | NO3/ppt | NO2 flow rate/dm3 min−1 |
|---|---|---|
| (3.5 ± 1.5) × 108 | 13 ± 5 | 0.360 |
| (4.2 ± 1.4) × 108 | 15 ± 5 | 0.290 |
| (6.1 ± 1.2) × 108 | 23 ± 4 | 0.200 |
| (9 ± 3) × 108 | 32 ± 10 | 0.160 |
| (10 ± 3) × 108 | 36 ± 10 | 0.130 |
| (9.3 ± 2.4) × 108 | 35 ± 9 | 0.104 |
| (2.3 ± 1.2) × 109 | 86 ± 45 | 0.08 |
| (2.6 ± 1.0) × 109 | 100 ± 40 | 0.06 |
sinϑ/λ.
Only a brief description of the physical basis of NR with reference to its application is given here while more details may be found in ref. 52, 57, 66 and 67. NR is a technique that allows measuring the surface excess of oil-like films at the air–water interface. Neutron scattering is related to the coherent cross sections of the atoms with which the neutrons interact, and these values vary non-monotonically for different isotopes of the same atom and different atoms across the periodic table. In particular, swapping hydrogen for deuterium changes significantly the scattering, and as such mixing of hydrogenous and deuterated materials enables contrast matching.
We followed the change in reflectivity of a deuterated monolayer at the air–water interface using the time-of-flight mode measuring the entire q-range stated above with respect to the time of the oxidation reaction. For a deuterated surfactant monolayer at the air–ACMW interface the reflectivity, R, can be expressed by:
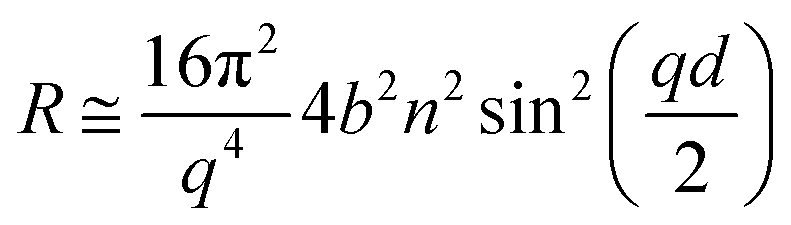 | (1) |
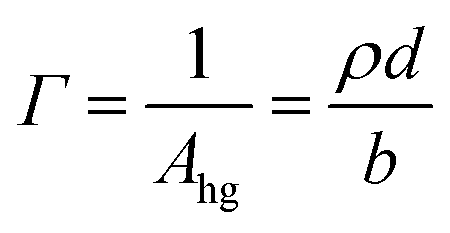 | (2) |
A stratified layer model was applied to the experimental data involving a single layer for the deuterated surfactant. It has been shown that in this low q-range (<0.07 Å−1), the value of Γ is very insensitive to specific details of the model applied.68 Fitting of the thickness with an arbitrary fixed value of the density or fitting of the density with an arbitrary fixed value of the thickness (to within reasonable bounds) gives equivalent results to within an added uncertainty of less than 2%. In our case, we chose to fit ρ while fixing d at the value obtained by fitting data recorded over a wider q-range (up to 0.25 Å−1).
Normalisation of the reflectivity data was carried out with respect to the total reflection of an air–D2O measurement. The sample stage was equipped both with passive and active anti-vibration controls. The reaction chamber was mounted on the sample stage, interfaced with the gas setup, and the trough (13 × 10 cm) was filled with 80 ml of ACMW.47 A fixed amount of solution was spread using a microlitre Hamilton syringe in order to form the monolayer following the protocol used in other NR studies of atmospheric relevance.38–41,44,47,52,53 The volume of solution spread was 26 μl for d34OA-hMO, 25 μl for d34OA-hSA, 27 μl for d33MO-hOA and 29 μl for d33MO-hSA. The solvent was allowed to evaporate before closing the chamber. Since we used a barrier-less trough the desired surface pressure (16 to 25 mN m−1 depending on the molecule) was achieved by spreading a calculated number of molecules on the surface as described earlier.41 From the surface excess obtained by NR the reproducibility is found to be within 1 to 14%, depending on the binary mixture. The choice of initial surface pressure and surface excess was based on the requirement of maximising the signal-to-noise ratio for NR measurements while having a reaction that lasts long enough to be analysed for kinetic parameters. A reduction of the initial surface pressure is not expected to affect the kinetic behaviour, i.e. the Γ(t) will start from a lower value and the curve will extend on a shorter time and less data will be available for the kinetic fitting. The monolayer was further characterised with compression–expansion isotherms with a Langmuir trough off-line, while recording Brewster-angle microscopy (BAM) images at different surface pressure values, and these results are shown in the ESI Section 1.† Data were recorded for several minutes before NO3 was admitted into the reaction chamber. The time resolution was set to 2 s. The alignment of the interface was maintained to a precision of 5 μm using an optical sensor (LK-G152, Keyence, Japan; laser class II, wavelength 650 nm, power output 0.95 mW, spot diameter 120 μm), which operated through the laser alignment window of the reaction chamber.41,47
2.2 Kinetic modelling
NO3-initiated oxidation of organic compounds proceeds generally via two reaction channels: rapid addition to the double bond of unsaturated species as well as slower abstraction of hydrogen atoms particularly relevant for saturated compounds.29 These mechanisms need to be described together with transport processes occurring in the experimental system to fit our data. Based on the PRA-framework58–60,69–72 a specific model has been developed for the heterogeneous reaction of a monomolecular organic layer at the air–water interface.41 This model was applied to the binary mixture without changes: in the binary mixture only one component is deuterated and hence visible to the NR technique. Details of the modelling approach are described in Sebastiani et al.41 and we only give a brief overview below.Two key features of our model description are: (i) the oxidant loss due to the reaction and transport to the bulk water; and (ii) the reaction products are divided into three categories: volatile, soluble and surface-active species (the branching ratios for volatile and soluble products are based on literature values, and for surface-active products an estimation was based on Γ(t) at long reaction times when the value of Γ(t) is above the detection limit). It should be noted that the technique used in this study monitors the deuterium concentration at the interface and that we could have described the reaction system by assuming only two types of products: surface active and non-surface active. However, we decided to distinguish non-surface active compounds between volatile and soluble products in order to make our model suitable for description of experimental data probing the partitioning to subphase and/or gas-phase. Due to the chosen NO3 delivery method (see Sebastiani et al.41) [NO2]/[NO3] ratios increase from 105 to 107 as [NO3] decreases from 109 to 108 molecule per cm3. NO2 can adsorb and desorb from the organic layer (compare to King et al.44), occupying reactive sites for an average time represented by the desorption lifetime, so that the loss of organic material due to reaction with NO3 could be affected: NO2 occupies a reactive site, which becomes unavailable for NO3 oxidation, and hence reduces the number of reactive sites available, in turn slowing down the apparent reaction rate. Especially for high [NO2]/[NO3] ratios the reactant loss rate will be lower than the loss rate recorded for lower [NO2]/[NO3] ratios. As a consequence, we had to include the absorption and desorption of NO2 in the model and describe this with a parameter called desorption lifetime, τd,NO2, following the approach used by Shiraiwa et al.59 The effect of N2O5 is not considered in the model, since the concentration was constant for all conditions. Experimental investigations of NO3 and N2O5 uptake73–77 have shown that NO3 uptake is much higher compared to N2O5. The reaction system is described as a gas phase (labelled g) and a near-surface gas phase (gs), above a sorption layer (s), a surface layer (ss), a near-surface bulk (nb) and the bulk (b), following the formalism of Shiraiwa et al.60 (as illustrated in Fig. 1).
Gas-phase species (NO3 and NO2) can adsorb to the sorption layer and interact with the organic molecules in the surface layer. The reaction products can stay at the surface layer, or they can be lost through solubilisation into the bulk or by evaporation into the gas phase.
The evolution of the gas species surface concentration, [Xi]s, is described by considering adsorption, desorption, transport and reaction (see ESI† for further details). The key equations that describe the reactions are discussed below (the nomenclature is based on the PRA framework58–60,69,78).
In the model, the gas-phase compound NO3 reacts with the organic layer and its loss, Lsurf,Y,NO3, is described with the second–order rate coefficient ksurf,Y,NO3:
| Lsurf,Y,NO3 = ksurf,Y,NO3[Y]S[NO3]S | (3) |
The evolution of the NO3 surface and bulk concentrations can be described as:
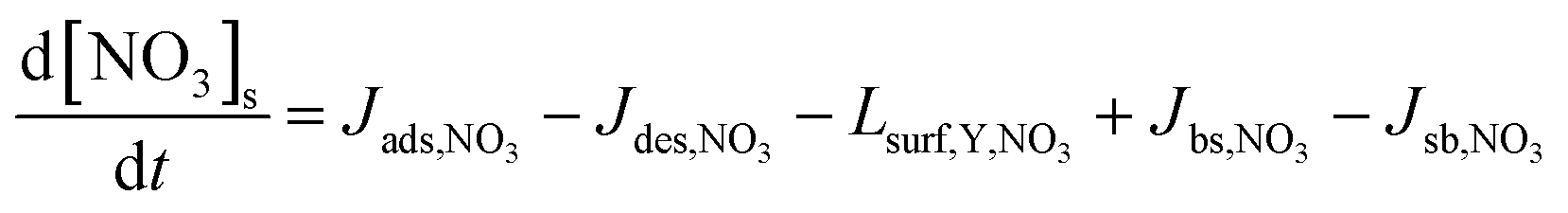 | (4) |
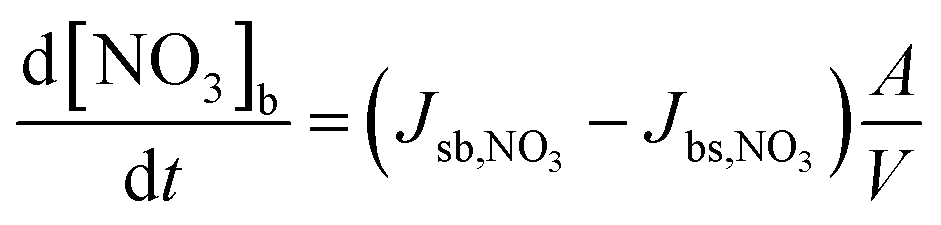 | (5) |
| Jdes,NO3 = kd,NO3[NO3]s = τd,NO3,eff−1[NO3]s | (6) |
| τd,NO3,eff−1 = θssτd,NO3,1−1 + (1 − θss)τd,NO3,2−1 | (7) |
The organic reactant, Y, (e.g. oleic acid) is lost only through reaction with NO3 at the surface, described as:
 | (8) |
The products (Z) of the heterogeneous reaction cannot be identified individually at the air–water interface in our experiments, so we described them as three key categories: surface-active (i.e. remaining at the surface and directly measurable by NR, ZS), volatile (i.e. escaping into the gas-phase, ZG) and soluble (i.e. accumulating in the water bulk, ZB) compounds. Since the surface-active products (ZS) will remain at the air–water interface, the surface–bulk transport is negligible:
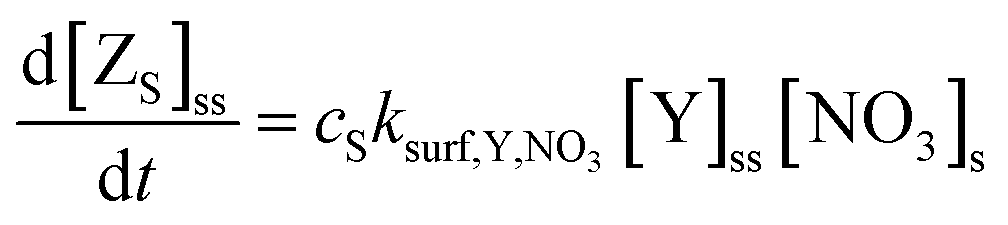 | (9) |
3 Results and discussion
Two of the organic molecules considered in this work (OA and MO) contain one unsaturated CC bond in the aliphatic tail while one molecule (SA) is fully saturated. Among the unsaturated surfactants, MO is a methyl ester in comparison with the fatty acid OA. The double bond is expected to be the key reactive site for NO3. Kinetic data on the two reactive unsaturated surfactants in the presence of a co-surfactant are presented in Sections 3.1 and 3.2. When a deuterated surfactant is mixed with a hydrogenous surfactant the reaction of the first molecule can be followed in the presence of the second. Each section will illustrate the data collected for the 1:1 mixture (by mole) of deuterated and hydrogenous same molecule, then the deuterated surfactant mixed with saturated molecule (SA) and finally the deuterated surfactant mixed with the other unsaturated molecule in its hydrogenous form.
3.1 Oleic acid (d34OA) exposed to nitrate radicals (NO3) in the presence of a co-surfactant
We investigated the time evolution of the two-component films when following the decay of deuterated oleic acid (d34OA) component mixed with hydrogenous oleic acid (dOAhOA), the saturated surfactant stearic acid (dOAhSA) and its methyl ester (dOAhMO). Fig. 2 shows the surface excess decays of d34OA monolayers at the air–ACMW interface as a function of time with respect to different [NO3] in the presence of hOA, hSA and hMO. The NO3-initiated oxidation leads to a non-zero surface excess value (4–6 × 1013 molecule per cm2) at the end of the reactive decay. This plateau value is reached following an initial decay, which lasts between 5 min and well over 1 h depending on [NO3]. [NO3] ranges from (13 ± 6) to (86 ± 45) ppt. For several gas conditions, the oxidation was carried out twice, demonstrating a good reproducibility for high [NO3] (>35 ppt), and visually higher variability for lower concentrations. However, the uncertainty in [NO3], for [NO3] < 35 ppt, is ∼30%, which means that even a small variation in concentration produces a measurable change in the rate of loss of the deuterated surfactant. The oxidant is admitted into the chamber at t = 0 s, and the decay of the surface excess starts almost immediately suggesting that the monolayer begins to be consumed as soon as the oxidant is admitted. The surface excess of d34OA was monitored also for exposure to O2 when mixed with hSA and hMO, and for exposure to NO2 only when mixed with hMO in order to assess a mechanical loss due to gas flux and isomerisation effects due to the presence of NO2 (compare to King et al.44). All blanks show a very stable surface excess for d34OA over the relevant timescale. The O2 blank for d34OA-hMO shows a surface excess fluctuation of about 5%; this may be due to some mechanical vibrations or other intermittent interference during this particular experimental run, but given the stability of this film in the presence of NO2 (and all other films in the presence of O2) and the fact that the reactive decays are all very clearly distinct from the apparent, but comparatively small fluctuations in this run, we did not explore these fluctuations further within the tight timeframe of these neutron beamtime experiments.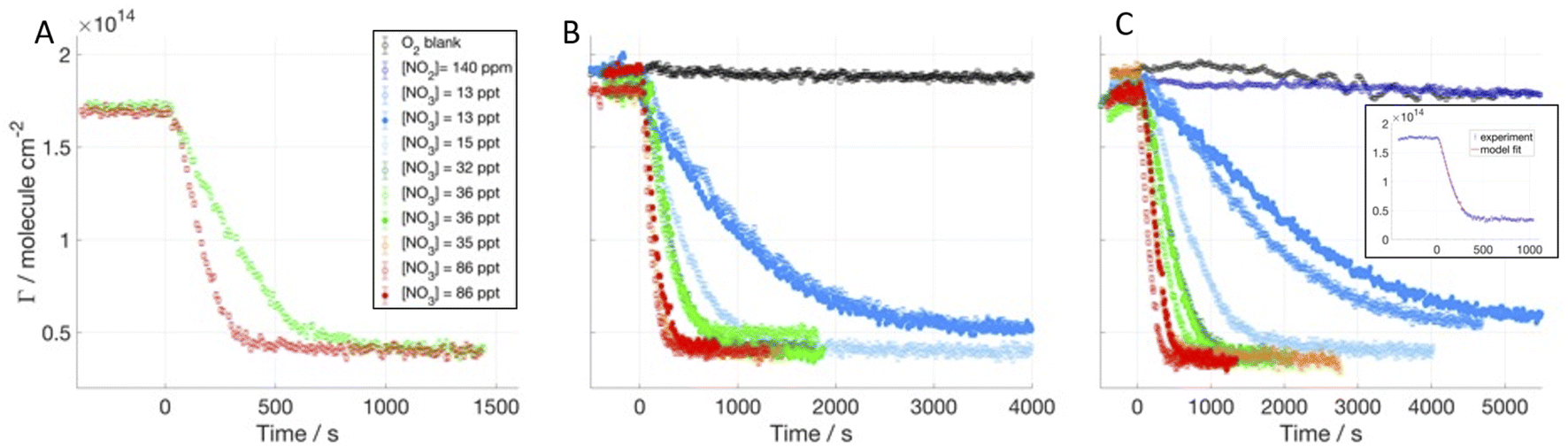 | ||
| Fig. 2 Surface excess decays of dOAhOA (A), dOAhSA (B) and dOAhMO (C) exposed to different [NO3]; mean values of NO3 mixing ratios are displayed in the legend (1 ppt = 2.7 × 107 molecule per cm3), NO3 is admitted at t = 0 s. In the insert, data and fit are shown for dOAhMO exposed to [NO3] = 86 ppt. | ||
The kinetic fitting was performed taking into account the variability of the gas concentrations (both for NO3 and NO2) and the initial surface excess was set to a value that takes into account only the d34OA, since the hydrogenous surfactant was effectively invisible to the NR technique (the scattering length of the hydrogenous material is only about 1% of the deuterated form). An example of the kinetic fit is displayed in Fig. 2C (see Section 3.2 of the ESI† for the complete data set). The modelling of the data followed the same approach as that for the single component monolayers;41 the kinetic parameters are discussed in comparison to the results for single molecule monolayers reported earlier41 with our new data allowing us now to explore the effect of the co-surfactant presence which is crucial to link the results for the frequently studied monomolecular films to complex, multi-component surfactant materials encountered in the real atmosphere.
The range of data we used for the kinetic fitting starts very close to the beginning of the reactive decay when NO3 is admitted to the chamber (t = 0 s), and ends at a surface excess value of 5 × 1013 molecule per cm2: data below this value are excluded from the fitting for two main reasons: (i) at low coverage the data become more sensitive to experimental details such as the precise background subtraction, so the parameters that affect the kinetic model are better determined without increasing sensitivity to these factors; and (ii) at low coverage some surfactants can segregate into domains which are inhomogeneous laterally (especially when mixing saturated and unsaturated surfactants), and the NR model does not have the resolution to distinguish this effect but the results are modestly affected, so again it is better to desensitize the kinetic parameters from this effect. The fitted curve, which results from the sum of the surface excesses of d34OA and the products, is shown as a solid red line in the insert of Fig. 2C. Since NR effectively measures the quantity of deuterium atoms at the air–ACMW interface, a distinction between reactant and products is not possible from NR data; hence the fitting function needs to take into account the contribution to Γ from both d34OA and its reaction products. The product yields are taken from our previous work;41 we assumed that at t = 0 s the signal is arising solely from d34OA, while the signal for long reaction time when the reactive decay has ceased (e.g. t > 1000 s for [NO3] = 86 ppt) is entirely due to the surface-active products. Also, the products43,80 are assumed to have a similar scattering length density to d34OA, since oxidation of d34OA is expected to break the molecule into two parts,43,80 which each maintains almost the same ratio between scattering length and molecular volume. The scattering length of the products is thus likely to be ca. half of the scattering length of d34OA and the product film also likely possesses ca. half of the d34OA film thickness. Given that and considering eqn (2), the resulting surface excess of the products corresponds to the value calculated with ρ, d and b of d34OA. This approximation is not valid in the extreme case of the products being only surface-active, since the packing would be two times denser than that for oleic acid, and this should be considered in the surface excess calculation and consequent modelling. In our study, the surface-active product yield is between 22% and 33%, and it has been taken into account that the total number of product molecules (surface-active, volatile and soluble) was twice the number of the reactant molecules.
Following the approach we used for the single component monolayer,41 the accommodation coefficients for the gas-phase species were fixed to one, and the desorption lifetimes were left free to vary in the range 10−9 to 10−7 s, which is in agreement with the values suggested by Shiraiwa et al.78 For the rate coefficient, ksurf, the range of variability was optimised through a sensitivity study performed by changing in the Matlab code the value of ksurf. The suitable range of values found was (0.7–8) × 10−8 cm2 per molecule per s, which is significantly higher than the best fit value provided by Shiraiwa et al.70 for abietic acid exposed to NO3 (1.5 × 10−9 cm2 per molecule per s) and wider than the range used for the single component monolayer study. The optimisation of the kinetic parameters was performed systematically by the χ2 minimisation routine FMINUIT.79 The sensitivity of the model to changes in desorption lifetimes is shown in the ESI.† Modelled evolutions of the concentrations of reactants and products are exemplified in Fig. 3 with examples taken for the mixtures dOAhMO and dMOhOA. Fig. 3A shows the model for the surface excess of dOA in the mixture dOAhMO and of dMO in the mixture dMOhOA. The steady state [NO3]s for the two mixtures reaches different values (see Fig. 3B): [NO3]s reaches a ca. 40% higher value when reacting with dOAhMO than with dMOhOA. This has two reasons: (i) the difference in the initial NO3 gas-phase concentration, in fact while both experimental runs had the same settings for the NO3 production, the uncertainty in the NO3 concentration is about 50% (see Table 1) and hence the model accounts for this variability with the best fit for the initial NO3 gas concentration being 27% higher for dOAhMO than for dMOhOA; (ii) the desorption lifetime at low coverage, τd,NO3,2, determines how long the NO3 remains at the sorption layer, and this lifetime is about 60% longer for dOAhMO compared to dMOhOA. Fig. 3C and D show the time evolution of the products; the difference in the amounts of surface-active products, ZS, and soluble products, ZB, depends on the product yields for OA and MO, while the total amount of volatile product, ZG, is comparable for the two reactants if normalized by the respective starting [Y]ss. While visually the ZG peak is significantly higher for dOAhMO compared to dMOhOA, when comparing the areas below the curves (noting the different timescales for the two reactions), the total amount of ZG for dOAhMO is about 33% higher than for dMOhOA, which is due to the initial reactant amounts differing by about 20% and the fact that reaction for dOAhMO is complete while it is not for dMOhOA (see differences in the surface excesses at t = 0 and at the last time point in Fig. 3A).
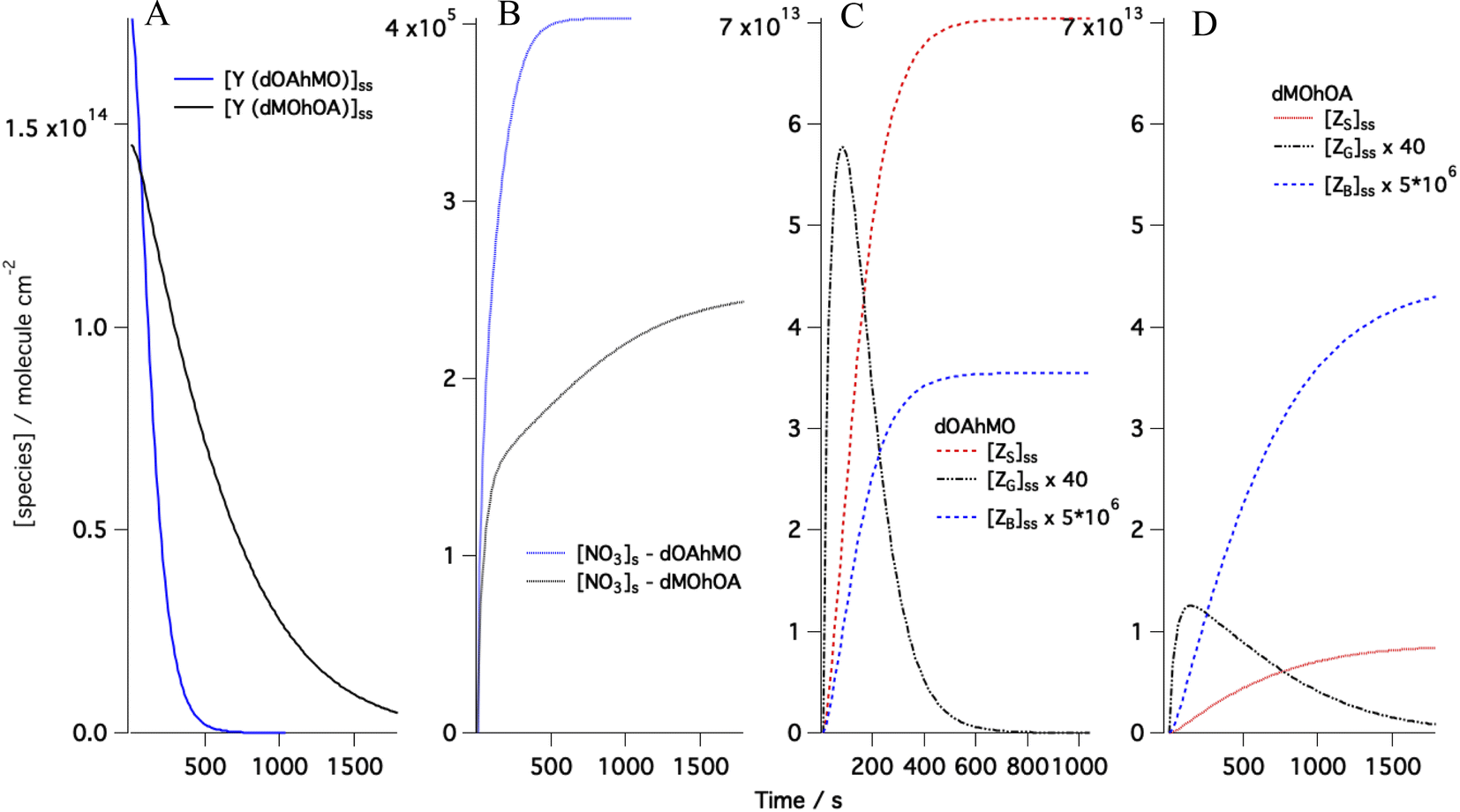 | ||
| Fig. 3 The evolution of the surface concentrations obtained from kinetic modelling using the best-fitted parameters for the data shown in Fig. 2C and 4C for (A) the organic reactant (Y) respectively dOA mixed with hMO (blue) and dMO mixed with hOA (black) exposed to [NO3] = 86 ppt; (B) the surface concentration for the gas-phase species NO3 reacting with dOAhMO (blue) and dMOhOA (black); and the surface-active (ZS), volatile (ZG) and soluble (ZB) products produced by dOAhMO (C) and dMOhOA (D). | ||
This fitting approach has been applied to all the molecules studied, while accounting for different product yields and kinetic parameter ranges (see Table 2).
| Modelled parameter | Best fit values | |||||||
|---|---|---|---|---|---|---|---|---|
| d 34OAa | d 34OA-hOAb | d 34OA-hSA | d 34OA-hMO | d 33MOa | d 33MO-hMOb | d 33MO-hSA | d 33MO-hOA | |
| a Values from Sebastiani et al.41 b Only two oxidant conditions were used to determine the kinetic parameters. | ||||||||
| k surf/108 cm2 per molecule per s (constraints) | 2.8 ± 0.7 (0.7–4) | 3.2 ± 0.2 (0.7–8) | 3.6 ± 0.9 (0.7–8) | 3 ± 1 (0.7–6) | 3.3 ± 0.6 (0.7–4) | 4.1 ± 0.8 (0.7–5) | 3 ± 1 (0.7–6) | 0.7 ± 0.4 (0.3–2) |
| τ d,NO3,1/109 s (constraints) | 8.1 ± 4.0 (5–20) | 8 ± 2 (5–40) | 14 ± 4 (5–40) | 8 ± 3 (3–20) | 8.1 ± 3.0 (5–20) | 8 ± 2 (5–20) | 21 ± 10 (5–40) | 12 ± 3 (5–20) |
| τ d,NO3,2/108 s (constraints) | 2.3 ± 0.8 (0.7–4) | 2.1 ± 0.7 (0.7–4) | 1.7 ± 0.7 (0.7–4) | 2.9 ± 1.1 (0.7–5) | 3.7 ± 1.3 (1–5) | 3.1 ± 0.7 (1–7) | 2.3 ± 0.8 (0.7–5) | 1.8 ± 0.5 (0.7–5) |
| τ d,NO2/108 s (constraints) | 2.8 ± 1.6 (0.1–6) | 3.4 ± 0.6 (0.1–6) | 4.4 ± 0.9 (0.1–6) | 4 ± 2 (0.1–6) | 2.9 ± 2.0 (0.1–6) | 4.8 ± 0.3 (0.1–6) | 3 ± 2 (0.1–6) | 3 ± 1 (0.1–6) |
The kinetic parameters related to the products, which have been used as fixed input parameters, are taken from our previous work with single component monolayers.41 The product yields were optimised to cS = 0.2 for the surface-active products, cG = 0.45 for the volatile products and cB = 0.35 for the soluble products. The best fit values for the kinetic parameters related to the heterogeneous reaction between d34OA and NO3 in the presence of hOA, hSA and hMO are summarised in Table 2. The presence of a co-surfactant can have different effects on the kinetics of the reaction occurring between d34OA and NO3: for example, the co-surfactant can have a higher reaction rate and compete for NO3 uptake. Within the monolayer, the co-surfactant will occupy a different area per molecule than d34OA; the area will be larger for hMO and smaller for hSA, thus affecting the packing and the availability of the CC double bond of d34OA for NO3 adsorption. We measured the reaction of d34OA in the presence of hOA in order to determine the effect of monitoring the surface excess of only half the molecules exposed to NO3 and we found similar results as for the fully deuterated monolayer. The rate coefficient for d34OA-hOA exposed to NO3 was found to be (3.2 ± 0.2) × 10−8 cm2 per molecule per s. The fast desorption time obtained for NO3 is (8 ± 2) × 10−9 s and the slow desorption is about 2.5 times longer, while the desorption time for NO2 (NO2 is present in our NO3 oxidation experiments together with O2) is four times longer. The introduction of two desorption times reflects the change of orientation of the organic molecules at the interface, i.e. for a highly packed monolayer the reactive site is less accessible, and the oxidant has less affinity for other parts of the molecules hence the desorption is faster. When the organic surface coverage decreases the reactive sites become more accessible and the desorption is slowed down.
The rate coefficient for d34OA-hSA exposed to NO3 was found to be (3.6 ± 0.9) × 10−8 cm2 per molecule per s. It should be noted that the loss of surfactant material due to O2 flow only (see black symbols in Fig. 2) leads to an apparent loss rate coefficient in the order of 10−11 cm2 per molecule per s, which is well within the uncertainty of the reactive rate coefficient. The fast desorption time obtained for NO3 is (14 ± 4) × 10−9 s and the slow desorption is about the same value, while the NO2 desorption time is about three times longer. The fact that τd,NO3,1 and τd,NO3,2 have similar values agrees well with the idea that the area per molecule of d34OA is larger and the CC double bond is more readily available when mixed with the same number of hSA molecules than when in a pure system, since hSA has a smaller area per molecule and packs more tightly than OA (see section 1 of ESI†).
The rate coefficient for d34OA-hMO exposed to NO3 was determined to be (3 ± 1) × 10−8 cm2 per molecule per s. The loss due to O2 flow alone leads to an apparent rate coefficient in the order of 10−11 cm2 per molecule per s, which is again well within the uncertainty of the reactive rate coefficient. The fast desorption time obtained for NO3 is (8 ± 3) × 10−9 s and the slow desorption is about three times longer, while the NO2 desorption time is about five times longer. These values are comparable with those found for d34OA-hOA which suggests that hMO has no major effect on OA oxidation kinetics, while the presence of SA slightly increases the rate coefficient and prolongs the fast desorption time both leading to a faster surface excess decay.
3.2 Methyl oleate (d33MO) exposed to nitrate radicals (NO3) in the presence of a co-surfactant
Methyl oleate (MO) differs from OA in terms of the head group: instead of a carboxylic acid it has a methyl ester (COOCH3) group. d33MO was used to study the oxidation by NO3 in the presence of a hydrogenous co-surfactant: hMO, hSA and hOA (see Table 1 in the ESI†). MO occupies a larger surface area and is less stable at the air–water interface than OA because of its less hydrophilic head group.41 However, the reactive site is in a very similar chemical environment as for OA, and any difference in reaction kinetics is expected to be related to the chain orientation and formation of different products.Fig. 4A–C display the surface excess decays of d33MO monolayers at the air–ACMW interface as a function of time with respect to [NO3] in the presence of hMO, hSA and hOA. [NO3] was varied from (13 ± 6) ppt to (100 ± 40) ppt.
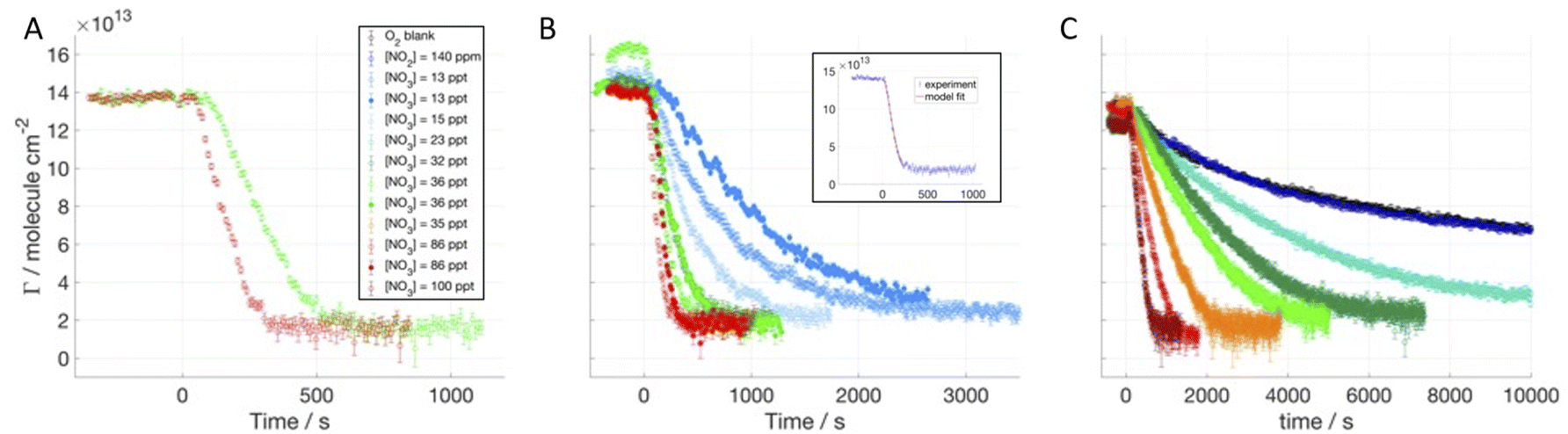 | ||
| Fig. 4 Surface excess decays of dMOhMO (A), dMOhSA (B), and dMOhOA (C) exposed to different [NO3]; mean values of NO3 mixing ratios are displayed in the legend (1 ppt = 2.7 × 107 molecule per cm3), NO3 is admitted at t = 0 s. In the insert, data and fit are shown for dMOhSA exposed to [NO3] = 86 ppt. | ||
The exposure to O2 flow leads to a non-reactive loss that is significantly larger than the one recorded for d34OA, confirming that d33MO is not as stable at the air–water interface as d34OA, even when mixed with a co-surfactant. The apparent rate coefficient obtained for the decays in the absence of NO3 is on the order of 10−10 cm2 per molecule per s. The minimum value reached by the surface excess is ≈2 × 1013 molecule per cm2, which is at the detection limit as discussed in our previous work.41 Therefore, no surface-active products are expected to remain at the interface as was also found in experiments with d33MO exposed to O3 and NO3 in the same reaction chamber.41,47 According to this finding, the product yields were chosen as for the single component film:41cS = 0.03, cG = 0.45 and cB = 0.52. The kinetic parameters were constrained to the values displayed in Table 2; for each co-surfactant a different range was optimised. An example of the fitting resulting from the kinetic modelling is displayed in the insert of Fig. 4B. The best-fit values obtained from the kinetic model are also presented in Table 2.
In the monolayer, the co-surfactant will occupy a different area per molecule than d33MO: a smaller area for both hOA and hSA, thus affecting the packing and the availability of the CC double bond of d33MO for NO3 adsorption. We measured the reaction of d33MO in the presence of hMO in order to determine the effect of monitoring the surface excess of only half the molecules exposed to NO3 and we found similar results as for the fully deuterated monolayer (also consistent with the equivalent test for d/hOA). The rate coefficient for d34MO-hMO exposed to NO3 in the presence of NO2 and O2 was found to be (4.1 ± 0.8) × 10−8 cm2 per molecule per s. The fast desorption time obtained for NO3 is (8 ± 2) × 10−9 s and the slow desorption is about four times longer, while the NO2 desorption time is six times longer. The rate coefficient for d34MO-hSA exposed to NO3 was found to be (3 ± 1) × 10−8 cm2 per molecule per s. The fast desorption time obtained for NO3 is (21 ± 10) × 10−9 s and the slow desorption is about the same value, while the NO2 desorption time is about 50% longer. As for d34OA, τd,NO3,1 and τd,NO3,2 have similar values and this agrees well with the idea that the area per molecule of d33MO is larger and CC double bond is more readily available when mixed with the same number of hSA molecules than when in a pure system, since hSA has a smaller area per molecule and packs tighter than MO (see section 1 of the ESI†).
The rate coefficient for d33MO-hOA exposed to NO3 was found to be (0.7 ± 0.4) × 10−8 cm2 per molecule per s. The loss due to O2 flow only leads to an apparent rate coefficient on the order of 10−10 cm2 per molecule per s, which is well within the uncertainty of the reactive rate coefficient. The fast desorption time obtained for NO3 is (12 ± 3) × 10−9 s and the slow desorption is about 50% longer, while the NO2 desorption time is about three times longer. Interestingly, this rate coefficient is significantly smaller than those found for d33MO-hMO and d33MO-hSA, while the desorption times are comparable for all these systems. We were thus able to quantitatively demonstrate that hOA has a significant influence on dMO oxidation kinetics. The importance in the atmospheric context of this experimental finding of a substantially altered reactive decay due to the presence of a co-surfactant will be discussed in the subsequent section on Atmospheric implications. hOA leaves surface active products upon NO3-initiated oxidation, and those products may compete with MO for the NO3 uptake or they can arrange around MO molecules making the double bonds less accessible and hence slow down the reaction. All fits are presented in the ESI.†
4 Atmospheric implications
The kinetic parameters obtained by analysing the NR data allowed investigation of the effects of co-surfactants in binary mixtures. This is a key step from the monomolecular films that are commonly studied in the laboratory environment towards an understanding of how these laboratory-based kinetic results can be linked to processes happening in the atmosphere where surfactants will exist in a complex matrix of other species including a wide range of saturated and unsaturated surfactants that are most likely to affect the structure at the air–water interface and thus potentially impact on the reactive decay of unsaturated surfactants such as oleic acid. We have carefully selected closely related surfactants that represent saturated (stearic acid) and unsaturated (oleic acid/methyl oleate) species found in the atmosphere. Thanks to our previous work in the same experimental set-up on these species as monomolecular films, we could focus this study on the quantitative differences in the presence of co-surfactant by selective deuteration of first one and then the other of the species in the binary mixtures.As outlined above, most mixtures tested do not show substantially different kinetic behaviours (ksurf is in the range of 2.8 to 4.1 × 10−8 cm2 per molecule per s) and the small differences in ksurf and the desorption times can be explained by the different structures at the air–water interface that will affect the accessibility of the CC double bond for the NO3 radicals.
The most striking result was found when following the decay of dMO in a binary mixture with hOA. We determined a ca. six times slower decay for dMOhOA compared to dMOhMO ((0.7 ± 0.4) × 10−8 cm2 per molecule per s compared to (4.1 ± 0.8) × 10−8 cm2 per molecule per s). We calculated the uptake coefficients for all the binary mixtures (see Table 3) to allow comparison with data collected in different experimental setups, where the direct result is the NO3 uptake coefficient; and again, we found a lower uptake of NO3 by dMOhOA compared to the other mixtures. This difference lies well outside the experimental uncertainty and demonstrates that matrix effects of co-surfactants can indeed affect the reaction kinetics and data from monomolecular surfactant experiments cannot simply be added up to understand the decay of these species in more complex and atmospherically relevant mixtures. Such a retardation of oxidative decay was not found for the binary mixtures of the surfactants OA and SA with a carboxylic acid headgroup. It thus seems likely that the less stable species at the air–water interface (such as MO with its methyl ester headgroup and more limited lifetime at the interface) may be more strongly affected by co-surfactant effects. These species are harder to study experimentally because of exactly this less stable nature. While there is a considerable uncertainty about the relative proportions of the head groups in surface-active material in the atmosphere, it is clear that fatty acids as well as esters are key classes (e.g. Cheng et al.83). For instance, sea spray aerosols collected over the Mediterranean Sea contained C14 to C34 fatty acid methyl esters (mainly methyl palmitate and methyl stearate).84,85 Degradation of particulate matter suspended in seawater by marine organisms has been shown to be a key source of saturated and unsaturated fatty acid methyl esters.86 Overall, it seems very likely that atmospheric surface-active materials will not be limited to perfectly hydrophilic headgroups that are most frequently studied in the laboratory. This new insight into the ageing of multi-component monolayers is thus of particular relevance for the formation of secondary organic aerosols in the atmosphere as well as clouds.
| Surfactant | k surf/cm2 per molecule per s | γ/103a | γ lit/103 | Lifetimeb/minute |
|---|---|---|---|---|
| a Calculated as ksurf × A × [Y]ss with A a gas constant in seconds and [Y]ss the surface concentration of the deuterated molecule, see ref. 41. b See ref. 41 for details on the lifetime calculation. c Values determined in our previous work.41 d Value refers to a study with a flow tube coupled to a chemical ionisation mass spectrometer.81 e Value refers to 1-octadecene uptake measured in a rotating wall flow tube.82 f Value refers to binary mixtures of MO and saturated molecules measured in a rotating wall flow tube.42 | ||||
| d 34OA | (2.8 ± 0.7) × 10−8c | 2.1 ± 0.5c (3 ± 1) × 102d [1.6 ± 0.3]e | 6.0 ± 1.5c | |
| d 34OA-hOA | (3.2 ± 0.2) × 10−8 | 2.6 ± 0.2 | 5.0 ± 0.3 | |
| d 34OA-hSA | (3.6 ± 0.9) × 10−8 | 3.4 ± 0.8 | 5 ± 1 | |
| d 34OA-hMO | (3 ± 1) × 10−8 | 2.6 ± 0.8 | 6 ± 2 | |
| d 33MO | (3.3 ± 0.6) × 10−8c | 2.1 ± 0.4c [(1.4+8.6−0.5) × 102]f | 5 ± 1c | |
| d 33MO-hMO | (4.1 ± 0.8) × 10−8 | 2.8 ± 0.5 | 4.0 ± 0.8 | |
| d 33MO-hSA | (3 ± 1) × 10−8 | 2.2 ± 0.7 | 5 ± 2 | |
| d 33MO-hOA | (0.7 ± 0.4) × 10−8 | 0.5 ± 0.3 | 22 ± 13 | |
5 Conclusion
In summary, our results provide clear evidence that multi-component mixtures need to be carefully considered when trying to establish the lifetimes of films in the atmosphere. Simply adding up kinetic data obtained for monomolecular films in the laboratory are unlikely to yield realistic residence times especially if non-perfect headgroups are present in the surface-active material of interest. We could quantify the retardation of the kinetic decay by NO3 to be ca. a factor of six for the dMO/hOA binary mixture. This would cause the residence time to change from ca. 4 to 22 minutes and could thus lead to substantially increased transport distances of surfactant species together with any other pollutants that may be protected underneath the surfactant film.Data availability
The data from the beamline experiments at FIGARO are available at Institut Laue-Langevin with DOIs: https://doi.org/10.5291/ILL-DATA.9-10-1286, https://doi.org/10.5291/ILL-DATA.9-10-1234 and https://doi.org/10.5291/ILL-DATA.9-10-1344.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank Dr Francesco Piscitelli, Dr Ernesto Scoppola and Dr Kunal Rastogi for the help during the night shifts on FIGARO. We would like to thank the Partnership for Soft Condensed Matter for access to the ellipsometer, and the ILL (Grenoble, France) for allocations of beam time on FIGARO. We are grateful for technical support at ILL from Simon Wood. FS is grateful for support from the ILL and the University of Reading in the framework of the NEATNOx studentship. CP thanks NERC (grant number NE/G000883/1) for support.References
- D. T. Shindell, G. Faluvegi, D. M. Koch, G. A. Schmidt, N. Unger and S. E. Bauer, Improved Attribution of Climate Forcing to Emissions, Science, 2009, 326, 716–718 CrossRef CAS PubMed.
- B. Stevens and G. Feingold, Untangling aerosol effects on clouds and precipitation in a buffered system, Nature, 2009, 461, 607–613 CrossRef CAS PubMed.
- T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, in Climate Change 2013: The Physical Science Basis, Cambridge University Press, 2013 Search PubMed.
- T. Petäjä, L. Järvi, V.-M. Kerminen, A. J. Ding, J. N. Sun, W. Nie, J. Kujansuu, A. Virkkula, X. Yang, C. B. Fu, S. Zilitinkevich and M. Kulmala, Enhanced air pollution via aerosol-boundary layer feedback in China, Sci. Rep., 2016, 6, 18998 CrossRef PubMed.
- S. Fuzzi, M. O. Andreae, B. J. Huebert, M. Kulmala, T. C. Bond, M. Boy, S. J. Doherty, A. Guenther, M. Kanakidou, K. Kawamura, V.-M. Kerminen, U. Lohmann, L. M. Russell and U. Pöschl, Critical assessment of the current state of scientific knowledge, terminology, and research needs concerning the role of organic aerosols in the atmosphere, climate, and global change, Atmos. Chem. Phys., 2006, 6, 2017–2038 CrossRef CAS.
- C. R. Ruehl, J. F. Davies and K. R. Wilson, An interfacial mechanism for cloud droplet formation on organic aerosols, Science, 2016, 351, 1447–1450 CrossRef CAS PubMed.
- J. Ovadnevaite, A. Zuend, A. Laaksonen, K. J. Sanchez, G. Roberts, D. Ceburnis, S. Decesari, M. Rinaldi, N. Hodas, M. C. Facchini, J. H. Seinfeld and C. O' Dowd, Surface tension prevails over solute effect in organic-influenced cloud droplet activation, Nature, 2017, 546, 637–641 CrossRef CAS PubMed.
- V. F. McNeill, N. Sareen and A. N. Schwier, in Atmospheric and Aerosol Chemistry, ed. V. F. McNeill and P. A. Ariya, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 201–259 Search PubMed.
- M. T. Latif and P. Brimblecombe, Surfactants in atmospheric aerosols, Environ. Sci. Technol., 2004, 38, 6501–6506 CrossRef CAS PubMed.
- A. Kroflič, S. Frka, M. Simmel, H. Wex and I. Grgić, Size-Resolved Surface-Active Substances of Atmospheric Aerosol: Reconsideration of the Impact on Cloud Droplet Formation, Environ. Sci. Technol., 2018, 52, 9179–9187 CrossRef.
- B. Nozière, C. Baduel and J.-L. Jaffrezo, The dynamic surface tension of atmospheric aerosol surfactants reveals new aspects of cloud activation, Nat. Commun., 2014, 5, 3335 CrossRef PubMed.
- N. Sareen, A. N. Schwier, T. L. Lathem, A. Nenes and V. F. McNeill, Surfactants from the gas phase may promote cloud droplet formation, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 2723–2728 CrossRef CAS PubMed.
- V. Gérard, B. Nozière, C. Baduel, L. Fine, A. A. Frossard and R. C. Cohen, Anionic, Cationic, and Nonionic Surfactants in Atmospheric Aerosols from the Baltic Coast at Askö, Sweden: Implications for Cloud Droplet Activation, Environ. Sci. Technol., 2016, 50, 2974–2982 CrossRef PubMed.
- V. Gérard, B. Noziere, L. Fine, C. Ferronato, D. K. Singh, A. A. Frossard, R. C. Cohen, E. Asmi, H. Lihavainen, N. Kivekäs, M. Aurela, D. Brus, S. Frka and A. Cvitešić Kušan, Concentrations and Adsorption Isotherms for Amphiphilic Surfactants in PM(1) Aerosols from Different Regions of Europe, Environ. Sci. Technol., 2019, 53, 12379–12388 CrossRef PubMed.
- B. Woden, M. W. A. Skoda, A. Milsom, C. Gubb, A. Maestro, J. Tellam and C. Pfrang, Ozonolysis of fatty acid monolayers at the air–water interface: organic films may persist at the surface of atmospheric aerosols, Atmos. Chem. Phys., 2021, 21, 1325–1340 CrossRef CAS.
- E. Adams and H. Allen, Palmitic Acid on Salt Subphases and in Mixed Monolayers of Cerebrosides: Application to Atmospheric Aerosol Chemistry, Atmosphere, 2013, 4, 315–336 CrossRef.
- H. Tervahattu, J. Juhanoja and K. Kupiainen, Identification of an organic coating on marine aerosol particles by TOF-SIMS, J. Geophys. Res. Atmos., 2002, 107, ACH-18 Search PubMed.
- H. Tervahattu, K. Hartonen, V.-M. Kerminen, K. Kupiainen, P. Aarnio, T. Koskentalo, A. F. Tuck and V. Vaida, New evidence of an organic layer on marine aerosols, J. Geophys. Res. Atmos., 2002, 107, AAC-1 Search PubMed.
- P. Q. Fu, K. Kawamura, J. Chen, B. Charrière and R. Sempéré, Organic molecular composition of marine aerosols over the Arctic Ocean in summer: contributions of primary emission and secondary aerosol formation, Biogeosciences, 2013, 10, 653–667 CrossRef.
- J. D. Allan, P. I. Williams, W. T. Morgan, C. L. Martin, M. J. Flynn, J. Lee, E. Nemitz, G. J. Phillips, M. W. Gallagher and H. Coe, Contributions from transport, solid fuel burning and cooking to primary organic aerosols in two UK cities, Atmos. Chem. Phys., 2010, 10, 647–668 CrossRef CAS.
- Q. Wang, X. He, M. Zhou, D. D. Huang, L. Qiao, S. Zhu, Y. Ma, H. Wang, L. Li, C. Huang, X. H. H. Huang, W. Xu, D. Worsnop, A. H. Goldstein, H. Guo and J. Z. Yu, Hourly Measurements of Organic Molecular Markers in Urban Shanghai, China: Primary Organic Aerosol Source Identification and Observation of Cooking Aerosol Aging, ACS Earth Space Chem., 2020, 4, 1670–1685 CrossRef CAS.
- A. M. P. Vicente, S. Rocha, M. Duarte, R. Moreira, T. Nunes and C. A. Alves, Fingerprinting and emission rates of particulate organic compounds from typical restaurants in Portugal, Sci. Total Environ., 2021, 778, 146090 CrossRef CAS PubMed.
- R. Ots, M. Vieno, J. D. Allan, S. Reis, E. Nemitz, D. E. Young, H. Coe, C. Di Marco, A. Detournay, I. A. Mackenzie, D. C. Green and M. R. Heal, Model simulations of cooking organic aerosol (COA) over the UK using estimates of emissions based on measurements at two sites in London, Atmos. Chem. Phys., 2016, 16, 13773–13789 CrossRef CAS.
- A. L. Robinson, N. M. Donahue and W. F. Rogge, Photochemical oxidation and changes in molecular composition of organic aerosol in the regional context, J. Geophys. Res., 2006, 111, D03302 Search PubMed.
- M. Kang, F. Yang, H. Ren, W. Zhao, Y. Zhao, L. Li, Y. Yan, Y. Zhang, S. Lai, Y. Zhang, Y. Yang, Z. Wang, Y. Sun and P. Fu, Influence of continental organic aerosols to the marine atmosphere over the East China Sea: Insights from lipids, PAHs and phthalates, Sci. Total Environ., 2017, 607–608, 339–350 CrossRef CAS.
- A. Milsom, A. M. Squires, J. A. Boswell, N. J. Terrill, A. D. Ward and C. Pfrang, An organic crystalline state in ageing atmospheric aerosol proxies: spatially resolved structural changes in levitated fatty acid particles, Atmos. Chem. Phys., 2021, 21, 15003–15021 CrossRef CAS.
- Q. Mu, M. Shiraiwa, M. Octaviani, N. Ma, A. Ding, H. Su, G. Lammel, U. Pöschl and Y. Cheng, Temperature effect on phase state and reactivity controls atmospheric multiphase chemistry and transport of PAHs, Sci. Adv., 2022, 4, eaap7314 CrossRef PubMed.
- M. Shrivastava, S. Lou, A. Zelenyuk, R. C. Easter, R. A. Corley, B. D. Thrall, P. J. Rasch, J. D. Fast, S. L. Massey Simonich, H. Shen and S. Tao, Global long-range transport and lung cancer risk from polycyclic aromatic hydrocarbons shielded by coatings of organic aerosol, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 1246–1251 CrossRef CAS.
- R. P. Wayne, I. Barnes, P. Biggs, J. P. Burrows, C. E. Canosa-Mas, J. Hjorth, G. Le Bras, G. K. Moortgat, D. Perner, G. Poulet, G. Restelli and H. Sidebottom, The nitrate radical: physics, chemistry, and the atmosphere, Atmos. Environ. Part A Gen. Top., 1991, 25, 1–203 CrossRef.
- N. Mora-Diez and R. J. Boyd, A Computational Study of the Kinetics of the NO3 Hydrogen-Abstraction Reaction from a Series of Aldehydes (XCHO:X = F, Cl, H, CH3), J. Phys. Chem. A, 2002, 106, 384–394 CrossRef CAS.
- N. L. Ng, S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams and R. A. Zaveri, Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 2017, 17, 2103–2162 CrossRef CAS PubMed.
- M. Takeuchi and N. L. Ng, Chemical composition and hydrolysis of organic nitrate aerosol formed from hydroxyl and nitrate radical oxidation of α-pinene and β-pinene, Atmos. Chem. Phys., 2019, 19, 12749–12766 CrossRef CAS.
- M. A. H. Khan, M. C. Cooke, S. R. Utembe, A. T. Archibald, R. G. Derwent, P. Xiao, C. J. Percival, M. E. Jenkin, W. C. Morris and D. E. Shallcross, Global modeling of the nitrate radical (NO3) for present and pre-industrial scenarios, Atmos. Res., 2015, 164–165, 347–357 CrossRef CAS.
- S. Gross, R. Iannone, S. Xiao and A. K. Bertram, Reactive uptake studies of NO3 and N2O5 on alkenoic acid, alkanoate, and polyalcohol substrates to probe nighttime aerosol chemistry, Phys. Chem. Chem. Phys., 2009, 11, 7792–7803 RSC.
- M. D. King, K. C. Thompson and A. D. Ward, Laser Tweezers Raman Study of Optically Trapped Aerosol Droplets of Seawater and Oleic Acid Reacting with Ozone: Implications for Cloud-Droplet Properties, J. Am. Chem. Soc., 2004, 126, 16710–16711 CrossRef CAS.
- O. Vesna, S. Sjogren, E. Weingartner, V. Samburova, M. Kalberer, H. W. Gäggeler and M. Ammann, Changes of fatty acid aerosol hygroscopicity induced by ozonolysis under humid conditions, Atmos. Chem. Phys., 2008, 8, 4683–4690 CrossRef CAS.
- J. Zahardis and G. A. Petrucci, The oleic acid-ozone heterogeneous reaction system: products, kinetics, secondary chemistry, and atmospheric implications of a model system; a review, Atmos. Chem. Phys., 2007, 7, 1237–1274 CrossRef CAS.
- M. D. King, A. R. Rennie, K. C. Thompson, F. N. Fisher, C. C. Dong, R. K. Thomas, C. Pfrang and A. V Hughes, Oxidation of oleic acid at the air–water interface and its potential effects on cloud critical supersaturations, Phys. Chem. Chem. Phys., 2009, 11, 7699–7707 RSC.
- C. Pfrang, F. Sebastiani, C. O. M. Lucas, M. D. King, I. D. Hoare, D. Chang and R. A. Campbell, Ozonolysis of methyl oleate monolayers at the air-water interface: oxidation kinetics, reaction products and atmospheric implications, Phys. Chem. Chem. Phys., 2014, 16, 13220–13228 RSC.
- M. W. A. Skoda, B. Thomas, M. Hagreen, F. Sebastiani and C. Pfrang, Simultaneous neutron reflectometry and infrared reflection absorption spectroscopy (IRRAS) study of mixed monolayer reactions at the air-water interface, RSC Adv., 2017, 7, 34208–34214 RSC.
- F. Sebastiani, R. A. Campbell, K. Rastogi and C. Pfrang, Night-time oxidation of surfactants at the air–water interface: effects of chain length, head group and saturation, Atmos. Chem. Phys., 2018, 18, 3249–3268 CrossRef CAS.
- S. Xiao and A. K. Bertram, Reactive uptake kinetics of NO3 on multicomponent and multiphase organic mixtures containing unsaturated and saturated organics, Phys. Chem. Chem. Phys., 2011, 13, 6628–6636 RSC.
- H. M. Hung, Y. Katrib and S. T. Martin, Products and mechanisms of the reaction of oleic acid with ozone and nitrate radical, J. Phys. Chem. A, 2005, 109, 4517–4530 CrossRef CAS PubMed.
- M. D. King, A. R. Rennie, C. Pfrang, A. V Hughes and K. C. Thompson, Interaction of nitrogen dioxide (NO2) with a monolayer of oleic acid at the air–water interface – A simple proxy for atmospheric aerosol, Atmos. Environ., 2010, 44, 1822–1825 CrossRef CAS.
- C. Pfrang, K. Rastogi, E. R. Cabrera-Martinez, A. M. Seddon, C. Dicko, A. Labrador, T. S. Plivelic, N. Cowieson and A. M. Squires, Complex three-dimensional self-assembly in proxies for atmospheric aerosols, Nat. Commun., 2017, 8, 1724 CrossRef CAS PubMed.
- J. D. Hearn, A. J. Lovett and G. D. Smith, Ozonolysis of oleic acid particles: evidence for a surface reaction and secondary reactions involving Criegee intermediates, Phys. Chem. Chem. Phys., 2005, 7, 501–511 RSC.
- F. Sebastiani, R. A. Campbell and C. Pfrang, Complementarity of neutron reflectometry and ellipsometry for the study of atmospheric reactions at the air–water interface, RSC Adv., 2015, 5, 107105–107111 RSC.
- T. Berkemeier, A. Mishra, C. Mattei, A. J. Huisman, U. K. Krieger and U. Pöschl, Ozonolysis of Oleic Acid Aerosol Revisited: Multiphase Chemical Kinetics and Reaction Mechanisms, ACS Earth Space Chem., 2021, 5, 3313–3323 CrossRef CAS.
- A. Milsom, A. M. Squires, B. Woden, N. J. Terrill, A. D. Ward and C. Pfrang, The persistence of a proxy for cooking emissions in megacities: a kinetic study of the ozonolysis of self-assembled films by simultaneous small and wide angle X-ray scattering (SAXS/WAXS) and Raman microscopy, Faraday Discuss., 2021, 226, 364–381 RSC.
- S. Sobanska, J. Barbillat, M. Moreau, N. Nuns, I. De Waele, D. Petitprez, Y. Tobon and C. Bremard, Influence of stearic acid coating of the NaCl surface on the reactivity with NO2 under humidity, Phys. Chem. Chem. Phys., 2015, 17, 10963–10977 RSC.
- Y. Wang, F. S. Cannon, M. Salama, D. A. Fonseca and S. Giese, Characterization of Pyrolysis Products from a Biodiesel Phenolic Urethane Binder, Environ. Sci. Technol., 2009, 43, 1559–1564 CrossRef CAS PubMed.
- J. R. Lu, R. K. Thomas and J. Penfold, Surfactant layers at the air/water interface: structure and composition, Adv. Colloid Interface Sci., 2000, 84, 143–304 CrossRef CAS PubMed.
- K. C. Thompson, A. R. Rennie, M. D. King, S. J. O. Hardman, C. O. M. Lucas, C. Pfrang, B. R. Hughes and A. V. Hughes, Reaction of a Phospholipid Monolayer with Gas-Phase Ozone at the Air–Water Interface: Measurement of Surface Excess and Surface Pressure in Real Time, Langmuir, 2010, 26, 17295–17303 CrossRef CAS PubMed.
- K. C. Thompson, S. H. Jones, A. R. Rennie, M. D. King, A. D. Ward, B. R. Hughes, C. O. M. Lucas, R. A. Campbell and A. V. Hughes, Degradation and Rearrangement of a Lung Surfactant Lipid at the Air–Water Interface during Exposure to the Pollutant Gas Ozone, Langmuir, 2013, 29, 4594–4602 CrossRef CAS PubMed.
- R. A. Campbell, A. Tummino, B. A. Noskov and I. Varga, Polyelectrolyte/surfactant films spread from neutral aggregates, Soft Matter, 2016, 12, 5304–5312 RSC.
- D. Ciumac, R. A. Campbell, H. Xu, L. A. Clifton, A. V. Hughes, J. R. P. Webster and J. R. Lu, Implications of lipid monolayer charge characteristics on their selective interactions with a short antimicrobial peptide, Colloids Surf. B Biointerfaces, 2017, 150, 308–316 CrossRef CAS PubMed.
- R. A. Campbell, Recent advances in resolving kinetic and dynamic processes at the air/water interface using specular neutron reflectometry, Curr. Opin. Colloid Interface Sci., 2018, 37, 49–60 CrossRef CAS.
- U. Pöschl, Y. Rudich and M. Ammann, Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions & ndash; Part 1: general equations, parameters, and terminology, Atmos. Chem. Phys., 2007, 7, 5989–6023 CrossRef.
- M. Shiraiwa, R. M. Garland and U. Pöschl, Kinetic double-layer model of aerosol surface chemistry and gas-particle interactions (K2-SURF): degradation of polycyclic aromatic hydrocarbons exposed to O3, NO2, H2O, OH and NO3, Atmos. Chem. Phys., 2009, 9, 9571–9586 CrossRef CAS.
- M. Shiraiwa, C. Pfrang and U. Pöschl, Kinetic multi-layer model of aerosol surface and bulk chemistry (KM-SUB): the influence of interfacial transport and bulk diffusion on the oxidation of oleic acid by ozone, Atmos. Chem. Phys., 2010, 10, 3673–3691 CrossRef CAS.
- P. A. Alpert, P. Corral Arroyo, J. Dou, U. K. Krieger, S. S. Steimer, J.-D. Förster, F. Ditas, C. Pöhlker, S. Rossignol, M. Passananti, S. Perrier, C. George, M. Shiraiwa, T. Berkemeier, B. Watts and M. Ammann, Visualizing reaction and diffusion in xanthan gum aerosol particles exposed to ozone, Phys. Chem. Chem. Phys., 2019, 21, 20613–20627 RSC.
- C. Pfrang, M. Shiraiwa and U. Pöschl, Chemical ageing and transformation of diffusivity in semi-solid multi-component organic aerosol particles, Atmos. Chem. Phys., 2011, 11, 7343–7354 CrossRef CAS.
- B. Woden, M. W. A. Skoda, M. Hagreen and C. Pfrang, Night-Time Oxidation of a Monolayer Model for the Air–Water Interface of Marine Aerosols—A Study by Simultaneous Neutron Reflectometry and in Situ Infra-Red Reflection Absorption Spectroscopy (IRRAS), Atmosphere, 2018, 9(12), 471 CrossRef CAS.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, Inc., 2nd edn, 2006 Search PubMed.
- R. Campbell, H. Wacklin, I. Sutton, R. Cubitt and G. Fragneto, FIGARO: The new horizontal neutron reflectometer at the ILL, Eur. Phys. J. Plus, 2011, 126, 107 CrossRef.
- L. Braun, M. Uhlig, R. von Klitzing and R. A. Campbell, Polymers and surfactants at fluid interfaces studied with specular neutron reflectometry, Adv. Colloid Interface Sci., 2017, 247, 130–148 CrossRef CAS.
- T. Narayanan, H. Wacklin, O. Konovalov and R. Lund, Recent applications of synchrotron radiation and neutrons in the study of soft matter, Crystallogr. Rev., 2017, 23, 160–226 CrossRef CAS.
- X. Liu, C. Counil, D. Shi, E. E. Mendoza-Ortega, A. V Vela-Gonzalez, A. Maestro, R. A. Campbell and M. P. Krafft, First quantitative assessment of the adsorption of a fluorocarbon gas on phospholipid monolayers at the air/water interface, J. Colloid Interface Sci., 2021, 593, 1–10 CrossRef CAS PubMed.
- C. Pfrang, M. Shiraiwa and U. Pöschl, Coupling aerosol surface and bulk chemistry with a kinetic double layer model (K2-SUB): oxidation of oleic acid by ozone, Atmos. Chem. Phys., 2010, 10, 4537–4557 CrossRef CAS.
- M. Shiraiwa, U. Pöschl and D. A. Knopf, Multiphase Chemical Kinetics of NO3 Radicals Reacting with Organic Aerosol Components from Biomass Burning, Environ. Sci. Technol., 2012, 46, 6630–6636 CrossRef CAS.
- A. Milsom, A. M. Squires, A. D. Ward and C. Pfrang, The impact of molecular self-organisation on the atmospheric fate of a cooking aerosol proxy, Atmos. Chem. Phys., 2022, 22, 4895–4907 CrossRef CAS.
- A. Milsom, A. Lees, A. M. Squires and C. Pfrang, MultilayerPy (v1.0): A Python-based Framework for Building, Running and Optimising Kinetic Multi-Layer Models of Aerosols and Films, EGUsphere, 2022, 1–20 Search PubMed.
- S. Gross and A. K. Bertram, Reactive Uptake of NO3, N2O5, NO2, HNO3, and O3 on Three Types of Polycyclic Aromatic Hydrocarbon Surfaces, J. Phys. Chem. A, 2008, 112, 3104–3113 CrossRef CAS PubMed.
- S. Gross and A. K. Bertram, Products and kinetics of the reactions of an alkane monolayer and a terminal alkene monolayer with NO3 radicals, J. Geophys. Res., 2009, 114, D02307 CrossRef.
- P. Zhang, W. Sun, N. Li, Y. Wang, J. Shu, B. Yang and L. Dong, Effects of Humidity and [NO3]/[N2O5] Ratio on the Heterogeneous Reaction of Fluoranthene and Pyrene with N2O5/NO3/NO2, Environ. Sci. Technol., 2014, 48, 13130–13137 CrossRef CAS PubMed.
- G. Gržinić, T. Bartels-Rausch, T. Berkemeier, A. Türler and M. Ammann, Viscosity controls humidity dependence of N2O5 uptake to citric acid aerosol, Atmos. Chem. Phys., 2015, 15, 13615–13625 CrossRef.
- C. Pfrang, R. S. Martin, C. E. Canosa-Mas and R. P. Wayne, Gas-phase reactions of NO3 and N2O5 with (Z)-hex-4-en-1-ol, (Z)-hex-3-en-1-ol ('leaf alcohol’), (E)-hex-3-en-1-ol, (Z)-hex-2-en-1-ol and (E)-hex-2-en-1-ol, Phys. Chem. Chem. Phys., 2006, 8, 354–363 RSC.
- M. Shiraiwa, C. Pfrang, T. Koop and U. Pöschl, Kinetic multi-layer model of gas-particle interactions in aerosols and clouds (KM-GAP): linking condensation, evaporation and chemical reactions of organics, oxidants and water, Atmos. Chem. Phys., 2012, 12, 2777–2794 CrossRef CAS.
- G. Allodi, FMINUIT - A binding to Minuit for Matlab, Octave & Scilab, 2011, http://www.fis.unipr.it/~giuseppe.allodi/Fminuit/Fminuit_intro.html Search PubMed.
- K. S. Docherty and P. J. Ziemann, Reaction of Oleic Acid Particles with NO3 Radicals: Products, Mechanism, and Implications for Radical-Initiated Organic Aerosol Oxidation, J. Phys. Chem. A, 2006, 110, 3567–3577 CrossRef CAS PubMed.
- Z. Zhao, S. Husainy, C. T. Stoudemayer and G. D. Smith, Reactive uptake of NO3 radicals by unsaturated fatty acid particles, Phys. Chem. Chem. Phys., 2011, 13, 17809–17817 RSC.
- T. Moise, R. K. Talukdar, G. J. Frost, R. W. Fox and Y. Rudich, Reactive uptake of NO3 by liquid and frozen organics, J. Geophys. Res. Atmos., 2002, 107, AAC-6 CrossRef.
- S. Cheng, S. Li, N. T. Tsona, C. George and L. Du, Insights into the Headgroup and Chain Length Dependence of Surface Characteristics of Organic-Coated Sea Spray Aerosols, ACS Earth Space Chem., 2019, 3, 571–580 CrossRef CAS.
- M.-A. Sicre, J.-C. Marty and A. Saliot, n-Alkanes, fatty acid esters, and fatty acid salts in size fractionated aerosols collected over the Mediterranean Sea, J. Geophys. Res. Atmos., 1990, 95, 3649–3657 CrossRef CAS.
- A. Cincinelli, A. M. Stortini, M. Perugini, L. Checchini and L. Lepri, Organic pollutants in sea-surface microlayer and aerosol in the coastal environment of Leghorn—(Tyrrhenian Sea), Mar. Chem., 2001, 76, 77–98 CrossRef CAS.
- M. Ehrhardt, Ch. Osterroht and G. Petrick, Fatty-acid methyl esters dissolved in seawater and associated with suspended particulate material, Mar. Chem., 1980, 10, 67–76 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ea00056c |
| ‡ Current address: Biofilms – Research Center for Biointerfaces, Department of Biomedical Science, Faculty of Health and Society, Malmö University. |
| § Current address: Division of Pharmacy & Optometry, Faculty of Medicine, Biology & Health, University of Manchester, Oxford Road, Manchester M13 9PT, UK. |
| This journal is © The Royal Society of Chemistry 2022 |