
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A family of mono-, di-, and tetranuclear DyIII complexes bearing the ligand 2,6-diacetylpyridine bis(picolinoylhydrazone) and exhibiting slow relaxation of magnetization†
Alexandros S.
Armenis‡
 a,
Georgia P.
Bakali‡
a,
ChristiAnna L.
Brantley
b,
Catherine P.
Raptopoulou
c,
Vassilis
Psycharis
c,
Luís
Cunha-Silva
d,
George
Christou
b and
Theocharis C.
Stamatatos
*ae
a,
Georgia P.
Bakali‡
a,
ChristiAnna L.
Brantley
b,
Catherine P.
Raptopoulou
c,
Vassilis
Psycharis
c,
Luís
Cunha-Silva
d,
George
Christou
b and
Theocharis C.
Stamatatos
*ae
aDepartment of Chemistry, University of Patras, 26504 Patras, Greece. E-mail: thstama@upatras.gr; Tel: +30 2610 997732
bDepartment of Chemistry, University of Florida, Gainesville, Florida 32611-7200, USA
cInstitute of Nanoscience and Nanotechnology, NCSR “Demokritos”, 15310 Aghia Paraskevi Attikis, Greece
dLAQV/REQUIMTE & Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, 4169-007 Porto, Portugal
eInstitute of Chemical Engineering Sciences, Foundation for Research and Technology – Hellas (FORTH/ICE – HT), Platani, P.O. Box 1414, 26504, Patras, Greece
First published on 2nd November 2022
Abstract
The systematic investigation of the general reaction scheme DyIII/X−/LH2, where X− = Cl−, CF3SO3−, ClO4−, MeCO2−, and LH2 is the pocket-type ligand 2,6-diacetylpyridine bis(picolinoylhydrazone), resulting from the condensation of 2,6-diacetylpyridine with picolinic acid hydrazide, has led to a new family of mono-, di-, and tetranuclear metal complexes of the formulae [DyCl2(LH2)(MeOH)]Cl (1), [Dy2(O3SCF3)2(LH)2(MeOH)1.42(H2O)0.58](O3SCF3)2 (2), [Dy2(LH)2(MeOH)2(H2O)2](ClO4)4 (3), and [Dy4(OH)2(O2CMe)6(L)2] (4), respectively. The organic chelate undergoes metal-assisted amide–iminol tautomerism and adopts the neutral zwitterionic, and single- and double-deprotonated forms, respectively, upon coordination with the metal center(s). Interestingly, the different forms of the ligand LH2/LH−/L2− act independently as penta-, hexa-, and heptadentate, either as single-chelating or chelating and bridging, thus yielding new DyIII compounds of various nuclearities and different magnetic properties. All complexes 1–4 exhibit frequency-dependent, out-of-phase (χ′′M) tails of signals in zero external dc field, characteristic of the onset of quantum tunnelling of magnetization. Attempts to suppress the tunnelling through the application of an external dc field were mostly successful in the case of complex 1, where entirely visible peaks of χ′′M have been observed and rendered possible the fit of the data to the Arrhenius equation, thus yielding the parameters: Ueff = 10.9(1) K and τ0 = 1.9(1) × 10−6 s, where Ueff is the effective energy barrier for the magnetization reversal and τ0 is the pre-exponential factor. The combined results demonstrate the ability of pyridyl-bis(acylhydrazone) ligands to yield chemically, structurally, and magnetically interesting compounds through their rich interconversion between various amide–iminol resonance forms.
Introduction
Single-molecule magnets (SMMs) are coordination compounds which retain their magnetization in the absence of an external magnetic field, and they exhibit frequency- and temperature-dependent out-of-phase (χ′′M) signals and hysteresis loops below a certain temperature, the so-called blocking temperature (TB).1 SMMs have been proposed as excellent candidates for potential applications in ultra-high density data storage, quantum computing and spintronics.2 There are four major classes of SMMs extensively developed to date: polynuclear metal complexes,3 dinuclear4 and mononuclear5 compounds, and organometallic systems.6Paramagnetic transition metals in moderate-to-high oxidation states have attracted most of the interest over the past three decades or so, mostly due to their ability to form aesthetically pleasing structures with new motifs and topologies,7 and fascinating magnetic properties, such as high-spin molecules,8 SMMs with large energy barriers for magnetization reversal,9 SMMs with step-like features at periodic fields of the hysteresis loop due to quantum tunnelling of the magnetization (QTM),10 and molecular magnetic refrigerants.11 However, the longstanding quest for 3d-metal based SMMs with improved intrinsic properties, such as higher ground-state spin values (S) and larger magnetoanistropies (D), has lagged a bit behind in meeting the needs for some practical applications, which require higher blocking temperatures12 and more flexible in engineering molecular systems, such as monomers and oligonuclear compounds.13
The new era of the field of SMMs has leaned on 4f-metal ions acting as ‘protagonists’14 in the quest for new molecules with enhanced magnetic dynamics and higher blocking temperatures, and hybrid multifunctional materials15 (i.e., SMMs@graphenes and SMMs@CNTs) with potential applications in the areas of information technology16 and molecular electronics.17 In particular, the DyIII ion has had a dominant position over the other lanthanide ions in the pursuit of efficient SMMs.18 This is mainly due to: (i) the odd number of electrons (4f9), ensuring a bistable ground state (Kramers doublet) irrespective of the symmetry of the ligand field,19 and (ii) the large spin–orbit coupling and subsequently large magnetic anisotropy, which gives rise to 6H15/2 (2S+1LJ) multiplets with 16-fold (2J + 1) degeneracy; these are further split into ±mj sublevels due to the removal of the degeneracy from the surrounding crystal field.20 The main obstacle towards the preparation of high-performance DyIII SMMs still retains the effect of QTM, which allows the electronic spins to tunnel through the energy barrier following a non-thermally activated pathway.21 Suppression of the QTM is mainly achieved by either chemical/structural means, such as the synthesis of highly symmetric structures and ideal lanthanide coordination geometries (i.e. D5h and D6h geometries),22 or the application of external direct current (dc) fields in alternating current (ac) magnetic dynamics.
Every ‘protagonist’ – in any demanding field of art and science – to unveil the anticipated skills and performance in action must be surrounded by the appropriate group of “side characters”. Organic chelates are key “side characters” and very important players in the field of lanthanide SMMs;23 they provide thermodynamic stability and crystallinity to the molecular systems, the ability to fine-tune the chemical, structural and electronic properties of the coordination compounds, while the ligand and crystal field effects appear to inherently affect the SMM efficacy, and the quantum tunnelling rates.24 Hydrazones of the general formula R1R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-NH offer a stable scaffold to chemically manipulate and transform them into pocket-type ligands with high affinity for binding to 4f-metal ions.25 Along these lines, bis(acylhydrazones) originating from the condensation of 2,6-pyridinedicarbonyl groups and acyl hydrazides appear to be a promising class of polydentate chelates which – upon deliberate R-substitution at the acyl groups – could also act as bridging ligands, facilitating both the formation of mononuclear and polynuclear metal complexes. This is corroborated by the amide–imino tautomerism that these ligands can undergo, thus enhancing their coordinating capabilities and structural versatilities towards various metal ions and topologies, respectively.
N-NH offer a stable scaffold to chemically manipulate and transform them into pocket-type ligands with high affinity for binding to 4f-metal ions.25 Along these lines, bis(acylhydrazones) originating from the condensation of 2,6-pyridinedicarbonyl groups and acyl hydrazides appear to be a promising class of polydentate chelates which – upon deliberate R-substitution at the acyl groups – could also act as bridging ligands, facilitating both the formation of mononuclear and polynuclear metal complexes. This is corroborated by the amide–imino tautomerism that these ligands can undergo, thus enhancing their coordinating capabilities and structural versatilities towards various metal ions and topologies, respectively.
In this work, we have employed the ligand 2,6-diacetylpyridine bis(picolinoylhydrazone) (LH2, Scheme 1) in DyIII chemistry, in conjunction with various supporting anions and in the absence or presence of an external base, as a means of preparing new molecular magnetic compounds bearing different resonance forms of the ligand LH2. Indeed, the systematic investigation of the tertiary system DyIII/X−/LH2, where X− = Cl−, CF3SO3−, ClO4−, and MeCO2−, has led us to the synthesis, structural, spectroscopic, and magnetic characterization of a family of mono-, di-, and tetranuclear complexes, all exhibiting slow relaxation of the magnetization and containing the neutral zwitterionic, and single- and double-deprotonated forms of the ligand LH2, respectively.
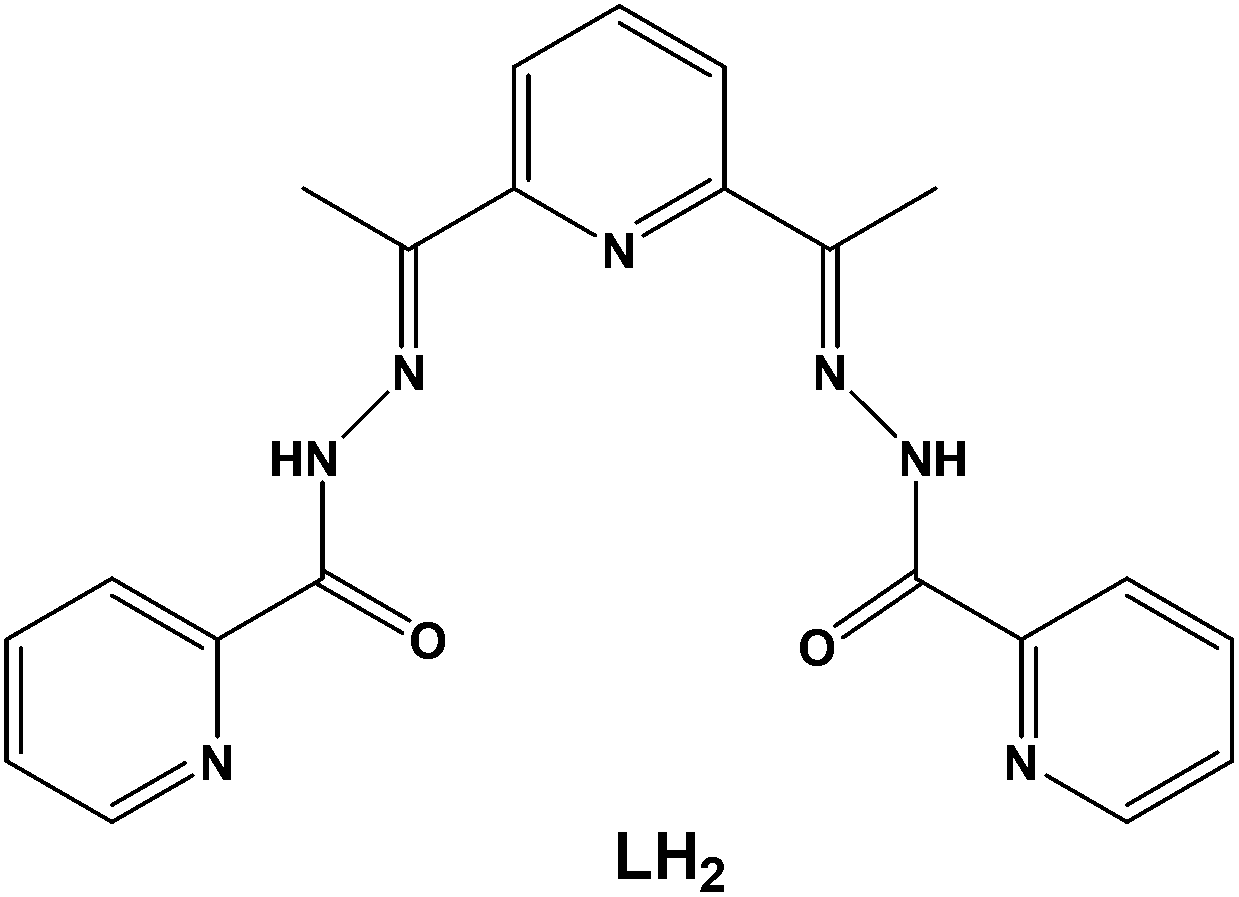 | ||
| Scheme 1 The structure of the pocket-type ligand 2,6-diacetylpyridine bis(picolinoylhydrazone) (LH2) used in the present work. | ||
Results and discussion
Synthetic comments and IR characterization
The structural diversity of the reported coordination compounds (1–4) is mainly dependent upon the nature of the employed ancillary anions (i.e., Cl−, CF3SO3−, ClO4− and MeCO2−) and the absence or presence of an external organic base (i.e., NEt3). In all cases, the versatility of the pentadentate, pocket-type ligand LH2 towards metal ion-assisted amide–iminol tautomerism has rendered possible the stabilization and crystallization of a family of mono-, di- and tetranuclear DyIII complexes bearing different resonance forms of the neutral zwitterionic LH2 (in complex 1) and the single- (in complexes 2 and 3) and double-deprotonated (in complex 4) forms of LH− and L2−, respectively (Scheme 2). The crystal structures of 1–4 have allowed us to determine the actual positions of the ligand's H-atoms with respect to the amide or pyridine N-atoms, while the deviations in the carbon–nitrogen and carbon–oxygen bond distances of the amide groups, in conjunction with charge-balance considerations for each of the reported coordination compounds, helped us to further support the assigned resonance forms.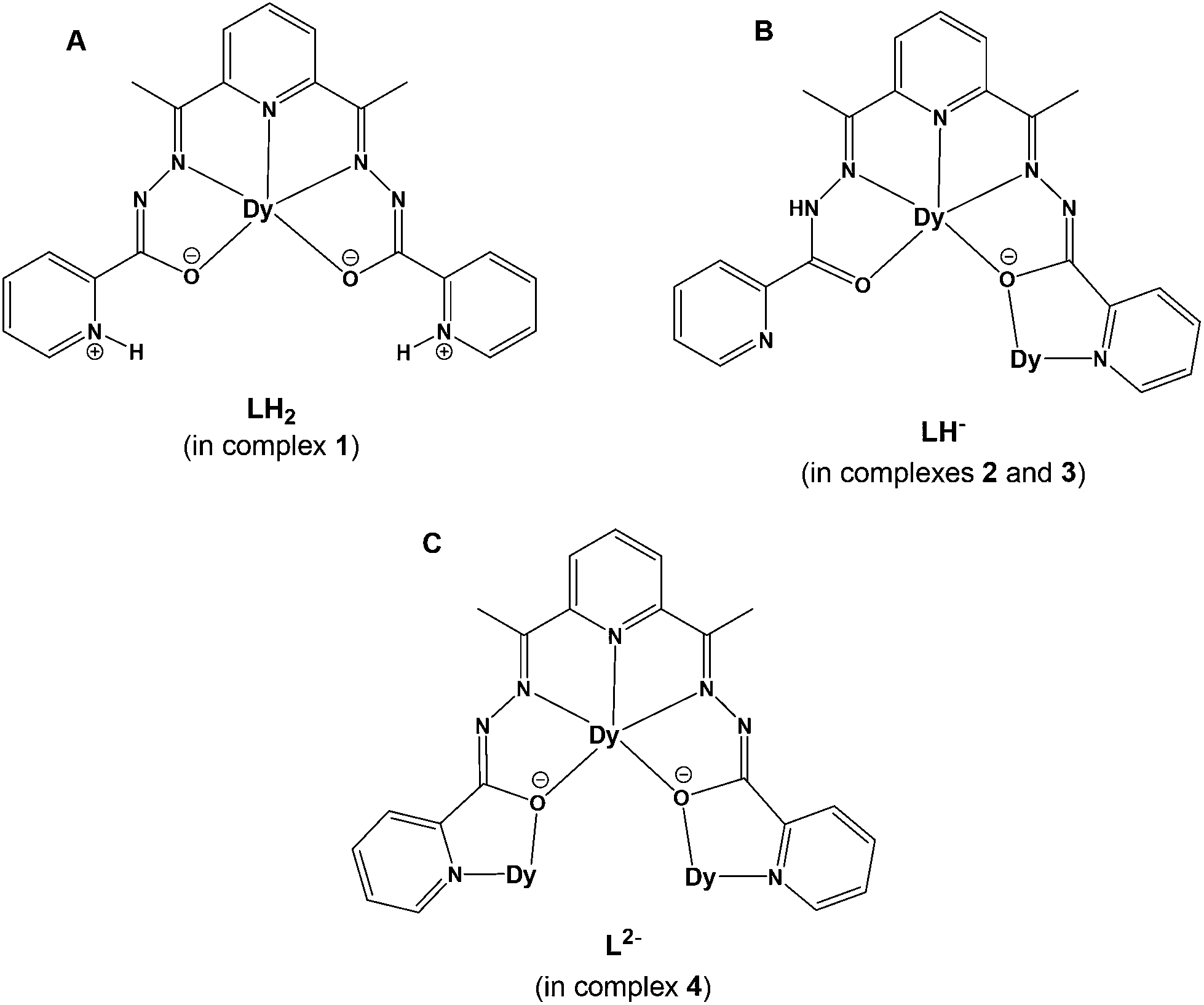 | ||
| Scheme 2 The crystallographically established coordination modes of the neutral (A), single- (B) and double-deprotonated (C) forms of the ligand LH2 in complexes 1–4. The depicted charges on specific N- and O-donor atoms in A, B, and C serve to comprehend the different resonance forms of the ligand LH2 in its zwitterionic (A) or deprotonated (B and C) forms. | ||
The systematic exploration of the ability of the ligand LH2 to foster the formation of structurally and magnetically interesting DyIII complexes included as a starting point the DyCl3 precursor. The Cl− ions are well-known for their ability to coordinate to 4f-metal ions, either as terminal monodentate groups or bridging ligands.26 Therefore, the reaction between DyCl3·6H2O and LH2 in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, in a solvent mixture comprising MeOH and CHCl3, afforded yellow-orange prismatic crystals of the mononuclear complex [DyCl2(LH2)(MeOH)]Cl (1), upon layering with Et2O, in 65% yield. As noticed from the chemical formula of 1, the experimental molar ratio of 1:2 between DyCl3·6H2O and LH2 has turned out to be dissimilar to the stoichiometric 1:1. When the same reaction was performed in a 1:1 molar ratio between the two reagents, microcrystalline solid (and not single-crystals) of 1 was obtained in yields as high as 85%; the chemical identity of this microcrystalline solid was confirmed by means of IR spectral comparison with the IR spectrum of the original 1. Furthermore, the reaction solvent mixture of MeOH and CHCl3 was essential for the formation and crystallization of 1. In the absence of MeOH or its replacement by other alcohols, such as EtOH, n-PrOH or n-BuOH, only amorphous yellow solids were obtained, which were probably mixtures of unreacted LH2 and unspecified inorganic-based materials (based on the recorded IR spectra). Although CHCl3 does not appear in the crystal structure of 1 as a lattice solvent (vide infra), its use as a co-solvent in this reaction scheme was found to yield the best quality single-crystals of 1 compared to similar solvents, such as CH2Cl2 and CHBr3.
:2, in a solvent mixture comprising MeOH and CHCl3, afforded yellow-orange prismatic crystals of the mononuclear complex [DyCl2(LH2)(MeOH)]Cl (1), upon layering with Et2O, in 65% yield. As noticed from the chemical formula of 1, the experimental molar ratio of 1:2 between DyCl3·6H2O and LH2 has turned out to be dissimilar to the stoichiometric 1:1. When the same reaction was performed in a 1:1 molar ratio between the two reagents, microcrystalline solid (and not single-crystals) of 1 was obtained in yields as high as 85%; the chemical identity of this microcrystalline solid was confirmed by means of IR spectral comparison with the IR spectrum of the original 1. Furthermore, the reaction solvent mixture of MeOH and CHCl3 was essential for the formation and crystallization of 1. In the absence of MeOH or its replacement by other alcohols, such as EtOH, n-PrOH or n-BuOH, only amorphous yellow solids were obtained, which were probably mixtures of unreacted LH2 and unspecified inorganic-based materials (based on the recorded IR spectra). Although CHCl3 does not appear in the crystal structure of 1 as a lattice solvent (vide infra), its use as a co-solvent in this reaction scheme was found to yield the best quality single-crystals of 1 compared to similar solvents, such as CH2Cl2 and CHBr3.
As a part of the reactivity studies on 1, we wondered whether it was possible to obtain new DyIII/LH2 complexes with different resonance forms of LH2 by omitting the ancillary Cl− ions and replacing them with anions that have negligible coordination affinity with DyIII centers, such as CF3SO3− and ClO4−. Indeed, the 1:2 reaction between Dy(O3SCF3)3 and the ligand LH2 in refluxing MeOH (to enhance the solubility of the starting materials) led to yellow prismatic crystals of the dinuclear complex [Dy2(O3SCF3)2(LH)2(MeOH)1.42(H2O)0.58](O3SCF3)2 (2), upon layering with Et2O, in 60% yield. Interestingly, the CF3SO3− ions were found to be a part of both the first- and second-coordination sphere of 2, acting as terminal monodentate ligands and counterions, respectively. Terminally-bound CF3SO3− ions are rare, albeit of precedence, in DyIII coordination chemistry.27 In contrast to 2, the use of Dy(ClO4)3·xH2O in place of Dy(O3SCF3)3, under the same conditions (i.e., DyIII/LH2 molar ratio, reaction solvent and crystallization process), led to yellow plate-like crystals of the dinuclear complex [Dy2(LH)2(MeOH)2(H2O)2](ClO4)4·2MeOH (3·2MeOH) in similar yields (∼55%). In spite of the structural similarities between 2 and 3 in most of their chemical features, the ClO4− ions in 3 were found to solely act as counterions, thus allowing H2O molecules to occupy the remaining coordination sites. It is likely that steric reasons merely facilitate the coordination of CF3SO3− groups (over ClO4−) with the DyIII centers. As with many reactions in trivalent lanthanide chemistry, the solution likely contained a mixture of species in equilibrium, and what crystallizes out is determined by the relative solubilities, the nature of counterions, lattice energies, and related factors.28 In both 2 and 3, the coordinated organic ligand appears to be in its single-deprotonated form (B form in Scheme 2) because of partial amide-to-iminol tautomerism.
Within this context, our last synthetic attempts were focused on studying the effect of an external organic base (i.e., NEt3) on the organic ligand's resonance forms and subsequently on the stabilization and crystallization of DyIII complexes with new structural motifs. Various reactions within the tertiary DyX3/LH2/NEt3 scheme were performed for X− = halides, NO3−, ClO4− and CF3SO3−, but all failed to produce any crystalline material after many modifications in molar ratios, reaction solvents and crystallization processes. However, when MeCO2− ions were employed in the form of Dy(O2CMe)3·4H2O, and this reacted with LH2 and NEt3 in a 2:1:5 molar ratio and a solvent mixture of MeOH and CH2Cl2, a new tetranuclear complex [Dy4(OH)2(O2CMe)6(L)2]·2CH2Cl2 (4·2CH2Cl2) was obtained as yellow plate-like crystals (upon layering with Et2O) in very good yields (∼70%). Complex 4 is very stable under the prevailing basic conditions, and its identity was neither affected by the nature of the external base nor by the Dy(O2CMe)3:LH2:NEt3 molar ratio. The same product, but in much lower yields and poorer crystallinities, was obtained when using NMe3, NPr3, Me4NOH, or other similar organic bases. A very large excess of NEt3 yielded white amorphous solids which were DyIII/oxido/hydroxido mixed species. The reaction solvent MeOH was necessary for the enhancement of the solubility of all starting reagents; reactions in other alcoholic media failed to yield any crystalline material. On the other hand, the use of CH2Cl2 as a co-solvent was found to be decisive for the crystallization of 4; this is also supported by its presence as the crystal lattice solvent in the crystal structure of 4·2CH2Cl2. Finally, the basic conditions applied within the aforementioned synthetic route have fostered the complete deprotonation of the ligand LH2 (C form in Scheme 2) and contributed towards the stabilization of its iminol form, thus rendering L2− as an unusual heptadentate chelating and bridging ligand within complex 4.
Tang and coworkers have employed the chemically similar ligand 2,6-(picolinoylhydrazone)pyridine (H2php), which contains H-atoms in place of the Me-groups in LH2, for the synthesis of a family of tetranuclear [Ln4(μ3-OH)2(php)2(O2CMe)6(H2O)2] (LnIII = GdIII, TbIII, DyIII and HoIII) complexes with the same structural topology as that of 4 but with dissimilar metric parameters (bond distances and angles) and magnetic properties (vide infra).29 The ligand LH2 has been previously used by Pelizzi and coworkers for the synthesis and structural characterization of mononuclear MnII,30 CoII,31 and SnIV complexes,32 and a tetranuclear {CuII4} cluster with a ring-like topology.33
The IR spectra of all complexes 1–4 exhibit medium- or strong-intensity broad bands in the region ∼3300–3400 cm−1, attributed to the ν(OH) vibration of the coordinated solvent molecules (MeOH, H2O, or both) or the OH− bridging groups (in 4), and to the ν(NH+)/ν(NH) vibrations of the zwitterionic LH2 or single-deprotonated LH− ligands (in 1 or 2/3).34 In addition, the medium-to-strong bands in the region ∼1600–1650 cm−1, common for all 1–4, are assigned to the ν(CN) and pyridyl stretching vibrations.35 In the IR spectrum of complex 2, the distinguishment of the bands denoted to the terminally coordinated and uncoordinated triflate ions is clearly unfeasible. Regardless of this drawback, the presence of the triflate ions in the structure of 2 is confirmed by the following bands: (i) a medium broad band at 1288 cm−1 attributed to the asymmetric stretching frequency of the SO3 group, (ii) a medium broad band at 1228 cm−1 corresponding to the symmetric stretching frequency of the CF3 moiety, (iii) a medium band at 1162 cm−1 attributed to νas(CF3), (iv) a very strong band at 1031 cm−1 corresponding to νs(SO3), (v) a strong band at 640 cm−1 attributed to the bending vibration of the SO3 group, and (vi) a weak and a medium-strong band at 574 and 516 cm−1 corresponding to the δas(CF3) and δas(SO3) vibrations, respectively.36 The IR spectrum of 3 exhibits a strong band at 1116–1084 cm−1 and a medium-intensity band at 630 cm−1, attributable to the IR-active ν3(F2)[νd(Cl–O)] and ν4(F2)[δd(OClO)] vibrations of the uncoordinated (Td geometry) ClO4− counterions, respectively.37 Several bands appear in the 1600–1370 cm−1 range in the IR spectrum of complex 4. These are due to contributions from the stretching vibrations of the imino groups, the pyridyl rings and the MeCO2− ligands. Since overlap cannot be ruled out, the bands most probably do not represent pure vibrations, and this renders exact assignments and studies of the coordination shifts tentative. The bands at 1568 and 1420 cm−1 in the spectrum of 4 are assigned to the νas(CO2) and νs(CO2) vibrations, respectively, of the acetato ligands, coupled with ring stretching vibrations.38 The difference Δ, where Δ = νas(CO2) − νs(CO2), is 148 cm−1, less than that for NaO2CMe (164 cm−1), as expected for the bidentate bridging mode of the carboxylate ligation. The asymmetric and the symmetric carboxylate stretches of the monodentate MeCO2− in 4 can be observed at 1545 and 1378 cm−1, respectively, giving a Δ value of 167 cm−1, slightly higher than that of NaO2CMe; usually higher Δ values as expected for monodentate acetato ligands.39 The two strong intramolecular hydrogen bonds (vide infra) between the uncoordinated carboxylate oxygen and the H-atom of the bridging OH− group withdraws the electron cloud of the C–O bond closer to the oxygen atom. The C–O bond order slightly decreases which shifts the asymmetric vibration to lower wavenumbers.
The thermal decomposition of complexes 1–4 was studied using thermogravimetric analysis (TGA) techniques under nitrogen. TG data and plots for 1–4 are shown in Fig. S1.† Complex 1 decomposes fast without a clear plateau to progressively lose the coordinated MeOH, the bound and lattice Cl− ions (in the form of HCl), and the organic chelate LH2 (from 30 °C to 410 °C). In complex 2, there is an initial weight loss of ∼7% from 30 °C to 130 °C, which perfectly agrees with the loss of four MeOH solvate molecules (calcd: 6.9%). Above 130 °C, the continuous loss of the organic chelates and the triflate ions (in the form of CF3SO3H) is observed in the temperature range of 130–400 °C and >400 °C, respectively.40 The decomposition of complex 3 begins with the loss of lattice MeOH and coordinated MeOH and H2O molecules to yield a weight loss of ∼9.8% up to a temperature of 280 °C (calcd: 9.7%). In temperatures above 280 °C, the gradual loss of the organic chelates and the ClO4− ions (in the form of HClO4) is detected. For cluster compound 4, the loss of the lattice CH2Cl2 solvate molecules and the bridging OH− ions (in the form of H2O) is obvious in the temperature region 30 °C to 290 °C (experimental weight loss: 9.9%; calcd: 10.1%), while further decomposition is associated with the loss of the coordinated MeCO2− groups (in the form of MeCO2H) and the organic chelates.
Description of structures
Selected interatomic distances and angles for complexes 1–4 are listed in Tables S1–S4,† respectively. For the sake of brevity, only important metric parameters will be discussed in the main text. The structure of 1 contains [DyCl2(LH2)(MeOH)]+ complex cations and Cl− counterions in a 1:1 ratio. The structure of the [DyCl2(LH2)(MeOH)]+ cation of 1 is shown in Fig. 1. It contains a DyIII atom surrounded by an overall neutral, pentadentate chelate LH2, which is coordinated as its zwitterionic form (A in Scheme 2). The pocket-type ligand LH2 makes use of the two enolate O-atoms (O1 and O2), the two imino N-atoms (N3 and N5) and a pyridyl N-atom (N4) for binding to the DyIII center. The C16–O2 and C6–O1 bond lengths within LH2 are 1.280(4) and 1.276(4) Å, respectively, further supporting the enolate resonance form of the zwitterionic chelate. Peripheral ligation about the DyIII center is provided by two and one terminally bound Cl− ions (Cl1 and Cl2) and MeOH (O1M) molecules, respectively. Thus, the metal ion is 8-coordinate, possessing a distorted hexagonal bipyramidal geometry.
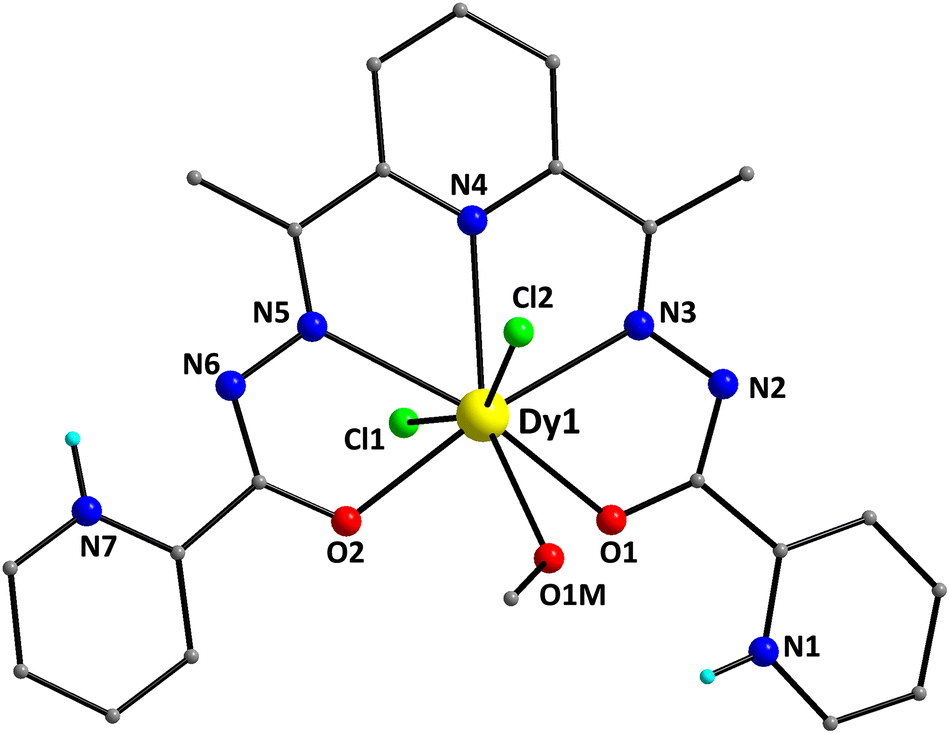 | ||
| Fig. 1 The structure of the [DyCl2(LH2)(MeOH)]+ cation that is present in the crystal of 1. The H-atoms, except those bonded to the pyridyl N-atoms, are omitted for clarity. Color scheme: DyIII, yellow; O, red; N, blue; Cl, green; C, grey; and H, cyan. | ||
This was confirmed by the so-called continuous shape measures (CShM) approach of the SHAPE program,41 which allows one to numerically evaluate by how much a particular polyhedron deviates from the ideal shape. The best fit was obtained for the hexagonal bipyramid (CShM value = 5.29; Fig. S2 and Table S5†) with the Cl-atoms occupying the apical positions. Values of CShM larger than 3 correspond to a significant distortion from the ideal geometry. This is also evidenced by the large deviation of the Cl1–Dy1–Cl2 angle (155.6°) from the ideal value of 180.0°. The Dy–O, Dy–Cl and Dy–N bond lengths fall in the range of 2.342(2)–2.486(3), 2.668(8)/2.704(9) and 2.531(3)–2.581(3) Å, and are typical of DyIII complexes containing this set of donor atoms.42 The bond lengths of DyIII to the deprotonated alkoxido oxygen atoms [2.342(2) and 2.382(2) Å] are noticeably shorter than the distance to the methanol oxygen atom [2.486(3) Å].
The coordinated ligand LH2 is not strictly planar; this deviation is imposed by the opposite twist of the two pyridyl groups of the picolynoylhydrazone moieties. The two opposite pyridine groups form an angle of 8.9°. As a result, the pyridyl N-atom (N7) gets closer to the uncoordinated imino N-atom (N6), while the other pyridyl N-atom (N1) appears to be closer to the coordinated alkoxido O-atom (O1). The DyIII atom is displaced by 0.25 Å out of the best-mean-equatorial plane that is formed by the coordinated atoms of the ligand LH2. The intermolecular H-bonding interactions that appeared in the crystal structure of 1 have a pronounced effect on the spatial arrangements of the ligands’ atoms (Fig. 2). These interactions involve the coordinated Cl− ion (Cl2) and the uncoordinated pyridinium N–H+ moiety (N7),43 as well as the Cl− counterion (Cl3) and the set of coordinated MeOH solvent and the other uncoordinated pyridinium N–H+ moiety (N1). The dimensions of these hydrogen bonds are: N7⋯Cl2# = 3.130(3) Å, O1M⋯Cl3 = 3.194(3) Å, and N1⋯Cl3 = 3.045(3) Å. Interestingly, the hydrogen-bonded monomers of 1 are intermolecularly linked to each other forming {Dy2} dimers rather than pseudo-polymeric chains of repeating monomers. In addition, the H-bonded dimers are only very weakly interacting with their neighboring units via π–π stacking interactions between the pyridyl groups of picolynoylhydrazone units of adjacent compounds (centroid-to-centroid distances are 4.694 and 4.979 Å). The shortest intermolecular Dy⋯Dy separation in the crystal of 1 is 8.230 Å (between the DyIII atoms of the H-bonded dimers).
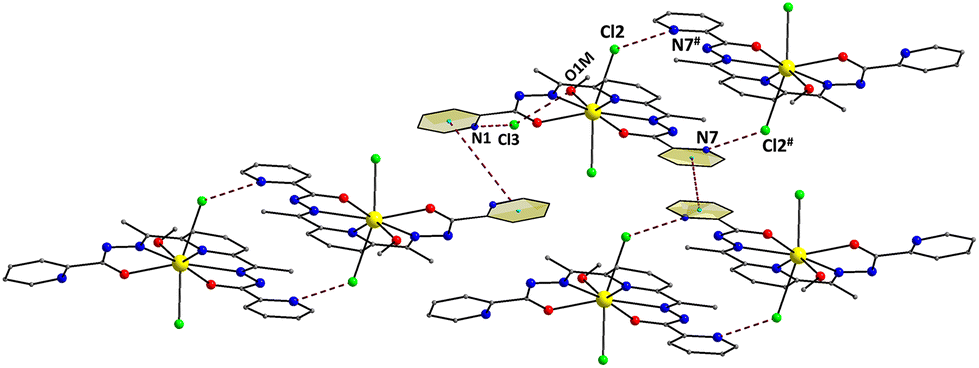 | ||
| Fig. 2 A portion of the repeating H-bonded dimers in the crystal of 1 and their interactions with their neighboring units through weak π–π stacking interactions. Both H-bonds and π–π stacking interactions are indicated by dashed lines. Symmetry code: # = 2 − x, 2 − y, 1 − z. Color scheme as in Fig. 1. | ||
The structures of the dinuclear complexes [Dy2(O3SCF3)2(LH)2(MeOH)1.42(H2O)0.58](O3SCF3)2 (2) and [Dy2(LH)2(MeOH)2(H2O)2](ClO4)4·2MeOH (3·2MeOH) are very similar to each other, and therefore only that of 3·2MeOH will be described in detail as a representative example. The major difference between 2 (Fig. S3†) and 3 is the coordination of two terminal triflate ions, one to each DyIII center, in the dicationic 2 over the coordination of two water molecules (in place of the two triflates) in the tetracationic 3. The most noticeable similarities between the two compounds are (i) the same binding mode of the single-deprotonated ligand LH− (B in Scheme 2), (ii) the same {Dy2(μ-OR)2}4+ planar core, and (iii) the same coordination number nine, donor N4O5 set and coordination polyhedra of all DyIII atoms.
The structure of 3·2MeOH contains [Dy2(LH)2(MeOH)2(H2O)2]4+ complex cations, ClO4− counterions and lattice MeOH solvent molecules in a 1:4:2 ratio. The structure of the centrosymmetric [Dy2(LH)2(MeOH)2(H2O)2]4+ cation of 3 is shown in Fig. 3. The two DyIII atoms (Dy1 and Dy1′) are linked to each other through the bridging alkoxido O-atoms (O1 and O1′) of two single deprotonated LH− ligands; the latter adopt the η1:η1:η1:η1:η2:μ binding mode acting as pentadentate chelating/bridging groups. One of the pyridyl groups (N3 and N3′) of each LH− remains unbound and interacts weakly with the lattice MeOH molecules. The C8–O8 and C22–O1 bond lengths within each coordinated LH− are 1.243 and 1.307 Å, respectively, as expected for a keto–enol resonance form of the chelate. Peripheral ligation about the planar {Dy2(μ-OR)2}4+ core (highlighted in Fig. 3) is provided by the chelating part of the LH− ligands, and two terminally bound H2O (O4 and O4′) and MeOH (O6 and O6′) molecules, one on each DyIII center. The Dy1⋯Dy1′ separation in 3 is 3.958(4) Å and the Dy1–O1/O1′–Dy1′ angle within its core is 114.6(1)°.
 | ||
| Fig. 3 Partially labelled representation of the [Dy2(LH)2(MeOH)2(H2O)2]4+ cation that is present in the crystal of 3·2MeOH, highlighting the {Dy2(μ-OR)2}4+ core with yellow thick bonds. H atoms are omitted for clarity. Color scheme as in Fig. 1. Symmetry code: (′) 1 − x, 1 − y, 1 − z. | ||
The DyIII atoms are 9-coordinate possessing a distorted muffin-like geometry (CShM value = 2.44; Fig. S4 and Table S6†), with the basal trigonal plane formed by N4, O6, and O1′ atoms, the equatorial pentagonal plane made of N1, N7, O8, N2, and O1, and the O4 atom occupies the vertex of the muffin.44 The bond lengths of DyIII atoms to the deprotonated alkoxido oxygen atoms [2.315(2) and 2.389(2) Å] are much shorter than the distances to the ketone [2.399(2) Å], methanol [2.406(2) Å], and aqua [2.409(2) Å] oxygen donor atoms. The corresponding CShM value for the 9-coordinate DyIII atoms in 2 with a muffin-like geometry is 2.50 (Table S6†).
Finally, there is an extended network of intermolecular H-bonding interactions in the crystal of 3·2MeOH, which include the coordinated aqua and methanol groups as donors and the ClO4− counterions as acceptors (Fig. S5†). These interactions serve to link the dinuclear complexes into a pseudo-1-D chain along the a-axis. Their dimensions are O4⋯O7 = 2.804 (7) Å, O4⋯O13 = 2.780(6) Å, and O6⋯O11 = 2.761(5) Å. Furthermore, the lattice MeOH solvate molecules are weakly H-bonded to the ClO4− ions and the uncoordinated pyridyl and imino N-atoms of LH−, resulting in an overall 2-D hydrogen-bonded network of repeating {Dy2} complexes (Fig. S5†). The shortest intermolecular Dy⋯Dy separation in the crystal of 2 is 9.744 Å.
The structure of 4·2CH2Cl2 consists of a tetranuclear [Dy4(OH)2(O2CMe)6(L)2] cluster (Fig. 4) and two lattice CH2Cl2 molecules; the latter will not be further discussed. Complex 4·2CH2Cl2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with the {Dy4} molecule lying on an inversion center. The four DyIII atoms (Dy1, Dy1′, Dy2, and Dy2′) are located at the four vertices of a defective dicubane, i.e. two cubanes sharing a face [Dy(1)O(1)Dy(1′)O(1′)] and each missing one metal vertex. The DyIII centers are primarily bridged by two μ3-OH− (O1 and O1′) and four μ-OR− (O2, O2′, O3, and O3′) groups from two η1:η2:η1:η1:η1:η2:η1:μ3 L2− ligands (C in Scheme 2), which are double deprotonated and act as heptadentate chelating and bridging groups utilizing all available for binding donor atoms. Thus, the core of 4 is {Dy4(μ3-OH)2(μ-OR)4}6+ (highlighted in Fig. 4), where RO− belongs to L2−. Peripheral bridging ligation about the {Dy4} core is provided by four syn,syn-η1:η1:μ MeCO2− groups, while terminal ligation is granted by two η1 MeCO2− and the chelating part of L2−. The dangling acetate O atoms (O7 and O7′) are intramolecularly H-bonded to the μ3-OH− groups [O1⋯O7 = 2.728(6) Å]. The bridging OH− groups are displaced by 0.839 Å from the corresponding Dy3 best-mean-planes towards the monodentate acetate groups.
with the {Dy4} molecule lying on an inversion center. The four DyIII atoms (Dy1, Dy1′, Dy2, and Dy2′) are located at the four vertices of a defective dicubane, i.e. two cubanes sharing a face [Dy(1)O(1)Dy(1′)O(1′)] and each missing one metal vertex. The DyIII centers are primarily bridged by two μ3-OH− (O1 and O1′) and four μ-OR− (O2, O2′, O3, and O3′) groups from two η1:η2:η1:η1:η1:η2:η1:μ3 L2− ligands (C in Scheme 2), which are double deprotonated and act as heptadentate chelating and bridging groups utilizing all available for binding donor atoms. Thus, the core of 4 is {Dy4(μ3-OH)2(μ-OR)4}6+ (highlighted in Fig. 4), where RO− belongs to L2−. Peripheral bridging ligation about the {Dy4} core is provided by four syn,syn-η1:η1:μ MeCO2− groups, while terminal ligation is granted by two η1 MeCO2− and the chelating part of L2−. The dangling acetate O atoms (O7 and O7′) are intramolecularly H-bonded to the μ3-OH− groups [O1⋯O7 = 2.728(6) Å]. The bridging OH− groups are displaced by 0.839 Å from the corresponding Dy3 best-mean-planes towards the monodentate acetate groups.
 | ||
| Fig. 4 Partially labelled representation of the [Dy4(OH)2(O2CMe)6(L)2] cluster that is present in the crystal of 4·2CH2Cl2, highlighting the {Dy4(μ3-OH)2(μ-OR)4}6+ core with yellow thick bonds. The dashed lines indicate the intramolecular H-bonds (see the text for the corresponding discussion). H atoms are omitted for clarity. Color scheme as in Fig. 1. Symmetry code: (′) 1 − x, 1 − y, 1 − z. | ||
The two edge-sharing {Dy3} triangles in 4 form a dihedral angle of 142.9° between them. The triangular {Dy3} units are near isosceles within the 3σ-criterion; the Dy1⋯Dy1′ and Dy1⋯Dy2 distances are 3.720(5) and 3.793(4) Å, respectively, while the Dy1⋯Dy2′ separation is 3.946(5) Å. Complex 4 can alternatively be described as containing a ‘butterfly’ of DyIII sites with the ‘body’ of the ‘butterfly’ (Dy1 and Dy1′) bridged by the O-atoms of two μ3-OH− groups; these O atoms also bridge two DyIII ‘wing-tip’ sites (Dy2 and Dy2′). The long ‘wing-tip’ Dy2⋯Dy2′ separation is 6.788(5) Å. Each of the four edges of the closed-type ‘butterfly’ is bridged by a μ-OR group from the chelate ligands L2− and a η1:η1:μ MeCO2− group.
The Dy1/Dy1′ and Dy2/Dy2′ atoms in 4 are 8- and 9-coordinate, possessing distorted biaugmented trigonal prismatic (CShM value = 2.22) and muffin-like geometries (CShM value = 3.16), respectively (Fig. 5 and Table S7†). The biaugmented trigonal prismatic geometry (Dy1 and Dy1′) is a trigonal prism capped on two of the three rectangular faces;45 the latter faces consist of the atoms O5/O8/O3/O1, O8/O3/N6/N7, and O5/O1/N6/N7, while the capping atoms are the O2 and O1′. In the muffin-like geometry of Dy2 (and Dy2′), the basal trigonal plane is formed by O2, O4, and N2 atoms, the equatorial pentagonal plane is made of O6, O1, O3, N4, and N3, and the O9′ atom occupies the vertex of the muffin; the Dy2–O9′ distance of 2.370(5) Å is the shortest one within the muffin polyhedron.
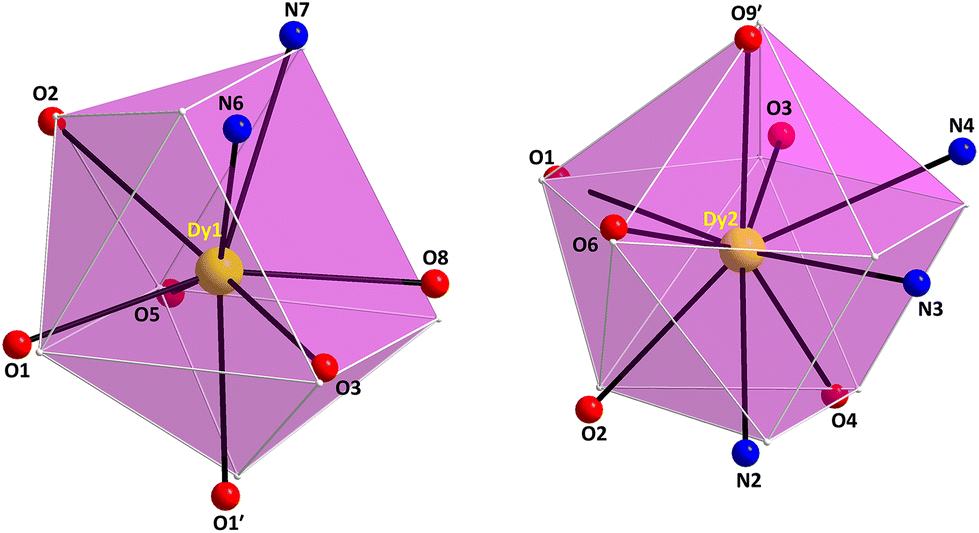 | ||
| Fig. 5 The biaugmented trigonal prism (left) and muffin (right) coordination polyhedra of the DyIII centers in complex 4·2CH2Cl2 with the corresponding atom labelling scheme as discussed in the text. The smaller white spheres define the vertices of the corresponding ideal polyhedra. Color scheme as in Fig. 1. Symmetry code: (′) 1 − x, 1 − y, 1 − z. | ||
From a supramolecular point of view, the {Dy4} clusters in the crystal of 4·2CH2Cl2 are nicely packed with each other through an extended network of intermolecular π–π stacking interactions between the pyridyl groups of the L2− ligands, thus forming 2-D sheets with cavities, in which the lattice CH2Cl2 solvate molecules are resided (Fig. S6†). The centroid-to-centroid distances of the π–π stacking interactions are 3.409 and 3.497 Å along the a- and b-axis, respectively. The shortest intermolecular Dy⋯Dy separation in the crystal of 4·2CH2Cl2 is 9.296 Å.
Although there are many tetranuclear {Dy4} clusters reported in the literature, possessing the {Dy4(μ3-OH)2(μ-OR)4}6+ ‘butterfly’-like core,46 complex 4 is structurally more reminiscent to the complex [Dy4(μ3-OH)2(php)2(O2CMe)6(H2O)2], featuring the dianion of 2,6-(picolinoylhydrazone)pyridine (H2php).29 However, a closer inspection of the two structural motifs reveals some noticeable differences, which comprise no coordinated aqua ligands in 4, the different binding modes of the acetate groups, and the dissimilar coordination numbers and geometries of the DyIII centers (9- and 10-coordinate in the {Dy4}/php2− cluster). This illustrates further the rich coordination and structural chemistry of bis(acylhydrazone) ligands, even in cases of ligands with many chemical similarities between them.
Magnetic susceptibility studies
Direct current (dc) magnetic susceptibility measurements were performed on microcrystalline samples of 1–4 in the 1.8–300 K range under an applied magnetic field of 0.1 T (Fig. 6). Note that the samples used for the magnetic studies have been readily analyzed by elemental analyses, which confirmed their chemical purity and allowed for the accurate determination of their exact molecular weights. The room-temperature χMT values of 13.98 (1), 28.40 (2), 28.50 (3), and 56.45 (4) cm3 K mol−1 are very close to the theoretical values of 14.17 (1), 28.34 (2 and 3), and 56.68 (4) cm3 K mol−1 for one, two and four non-interacting DyIII ions (6H15/2, S = 5/2, L = 5 and g = 4/3), respectively.47 For all complexes, χMT steadily decreases on cooling until ∼5 K, and then more sharply, reaching values of 9.40 (1), 15.70 (2), 10.54 (3), and 14.73 (4) cm3 K mol−1 at 1.8 K. The abrupt decline of χMT for all complexes as the temperature is lowered is primarily due to the depopulation of the mj sublevels of the ground J state,48 and possibly the presence of some weak intramolecular antiferromagnetic interactions between the DyIII ions (in 2, 3, and 4).14,49 The most noticeable, low temperature decrease of χMT is that observed for the tetranuclear compound 4; this could be attributed to a larger separation between the ground and excited mj states and a greater magnetic anisotropy (vide infra).50 Reduced magnetization (M/NμBvs. H/T) studies were also performed (Fig. S7†) for all four complexes in various dc fields (0.1–7.0 T) and low temperatures (1.8–10.0 K). In all cases, the isofield lines do not superimpose into a single master curve, suggesting the presence of unquenched magnetic anisotropy. This is further supported by the fact that the magnetization values at the maximum applied fields (∼5NμB in 1, ∼11NμB in 2, ∼10NμB in 3, and ∼20NμB in 4) are significantly lower than the expected values for one (10NμB), two (20NμB), and four (40NμB) isolated DyIII centers.51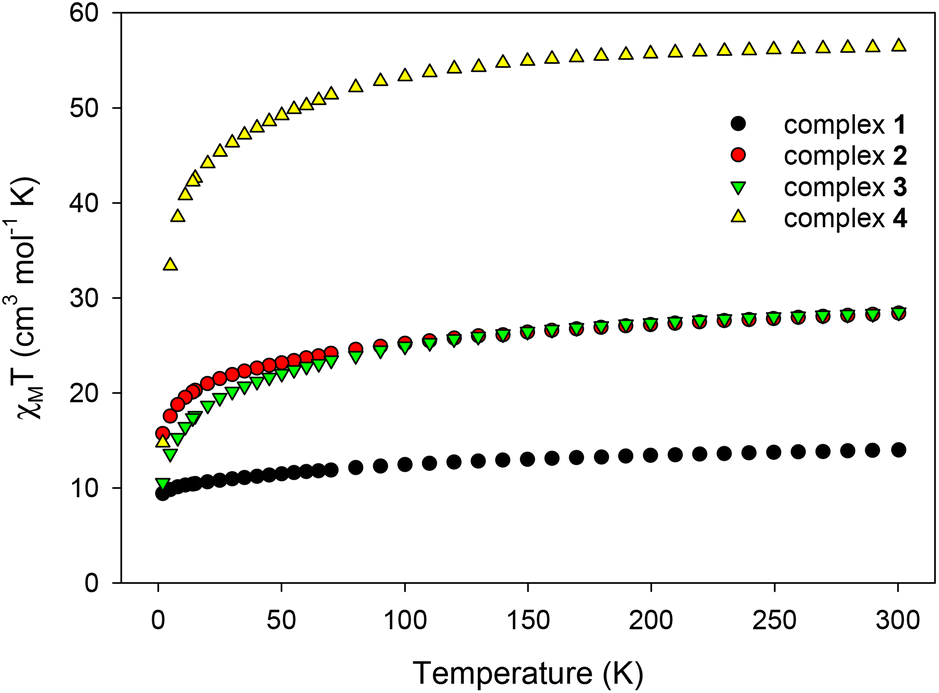 | ||
| Fig. 6 Temperature dependence of the χMT products for complexes 1–4 at 0.1 T. | ||
The evaluation of the magnetic dynamics of complexes 1–4 was probed by alternating current (ac) magnetic susceptibility measurements at zero applied dc field, under a weak ac field of 3.5 G at oscillation frequencies of 50–1500 Hz. The common feature of all compounds is the appearance of frequency-dependent tails of peaks in the out-of-phase (χ′′M) susceptibilities vs. T plots at temperatures below ∼10 (for 1–3) and ∼6 K (for 4), suggesting the onset of slow magnetization relaxation and SMM behavior (Fig. 7). These broad tails of signals are clear indications of fast quantum tunneling of magnetization (QTM), and consequently small energy barriers for magnetization reversal. This is a common phenomenon in mono-, di- and polynuclear 4f-SMMs with low-symmetry structures, in which the metal centers adopt various distorted coordination geometries with a random orientation of their single-ion anisotropies, with respect to the molecular easy-axis.52
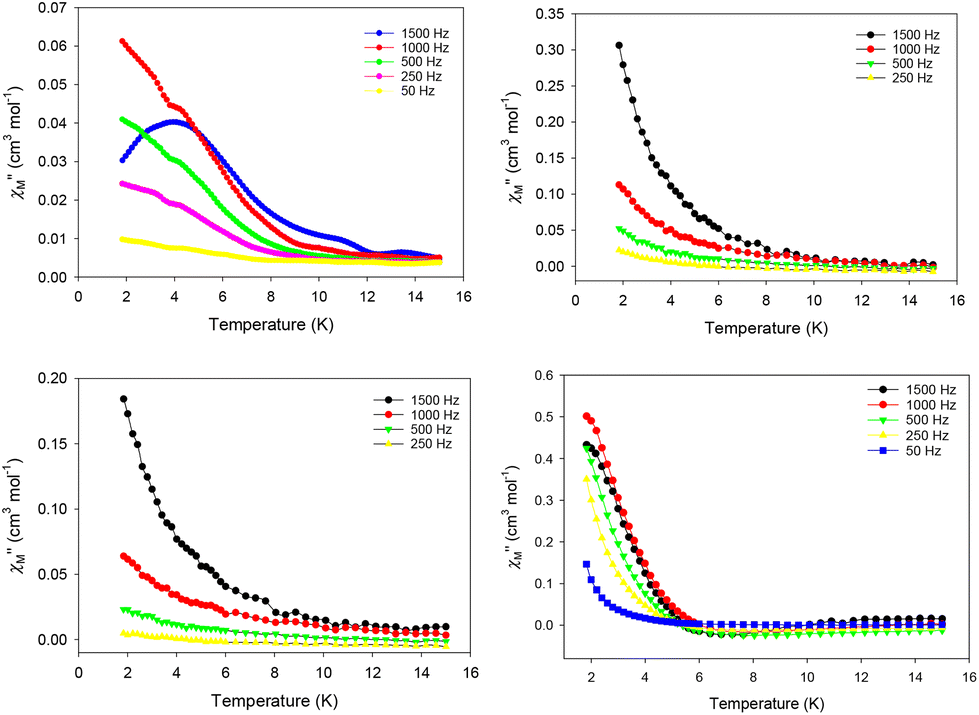 | ||
| Fig. 7 Temperature dependence of the out-of-phase (χ′′M) ac magnetic susceptibilities in zero dc field for 1 (top left), 2 (top right), 3 (bottom left), and 4 (bottom right) measured in a 3.5 G ac field oscillating at frequencies of 50–1500 Hz. The solid lines are guides only. | ||
In particular, for Kramers ions such as DyIII in the majority of coordination environments, the presence of easy-axis anisotropy, and dipole–dipole and hyperfine interactions allows the mixing of the individual DyIII ground states in zero dc field, thus amplifying the QTM mechanism over the thermally-assisted relaxation processes (Orbach and Raman).53 Although the hexagonal bipyramidal geometry of the DyIII center is an appealing coordination environment for the preparation of air-stable mononuclear SMMs with large energy barriers,54 this turned out not to be the case for 1. This is readily explained on the basis of the large deviation of DyIII in 1 from the ideal D6h geometry, the orientation of the hard- and soft-donor ligands around the equatorial plane and the axial positions, and subsequently the operating crystal field effects which do not allow for specific terms of the crystal field Hamiltonian (i.e., the Bkq term), responsible for the mitigation of QTM, to vanish and ideally quench the fast relaxation of the magnetization.55 A search in the Cambridge Structural Database (CSD) revealed the absence of mononuclear DyIII complexes bearing a N3O3Cl2 set of donor atoms. The closest environment to that of 1 can be resembled by the 8-coordinate compounds [DyCl(bpy)2(H2O)3]Cl2 (DyN4O3Cl chromophore) and [DyCl(terpy)(H2O)4]Cl2 (DyN3O4Cl chromophore),56 in which no magnetic data were reported (bpy and terpy are the ligands 2,2′-bipyridine and 2,2′:6′,2′′-terpyridine, respectively), while no results were derived from the search of the DyN4O2Cl2 and DyN2O4Cl2 chromophores.
To tackle the dominant QTM pathway, a common strategy is the application of an external dc field in ac magnetometry, targeting the shift of the χ′′M signals to higher temperatures and the observation of entirely visible peaks. Indeed, the application of an external dc field of 0.1 T in the ac measurements of 1 revealed clear, frequency-dependent out-of-phase, χ′′M, signals in the temperature range of 1.8–4 K (Fig. 8, top), which is indicative of a thermally-assisted relaxation process. The χ′′Mvs. T data were used to construct an Arrhenius-type plot of ln(τ) vs. 1/T, where τ is the relaxation time as deduced from the equation: τ = (2πf)−1 for every ac frequency f, and T is the temperature value (peak maximum) derived from the fit of χ′′M signals. The linearity of the ln(τ) vs. 1/T plot (Fig. 8, bottom) allowed us to fit the data according to the Arrhenius law [ln(τ) = ln(τ0) + Ueff/kBT]; the fit yielded a small effective energy barrier, Ueff, of 10.9(1) K and a pre-exponential factor, τ0, of 1.9(1) × 10−6 s.
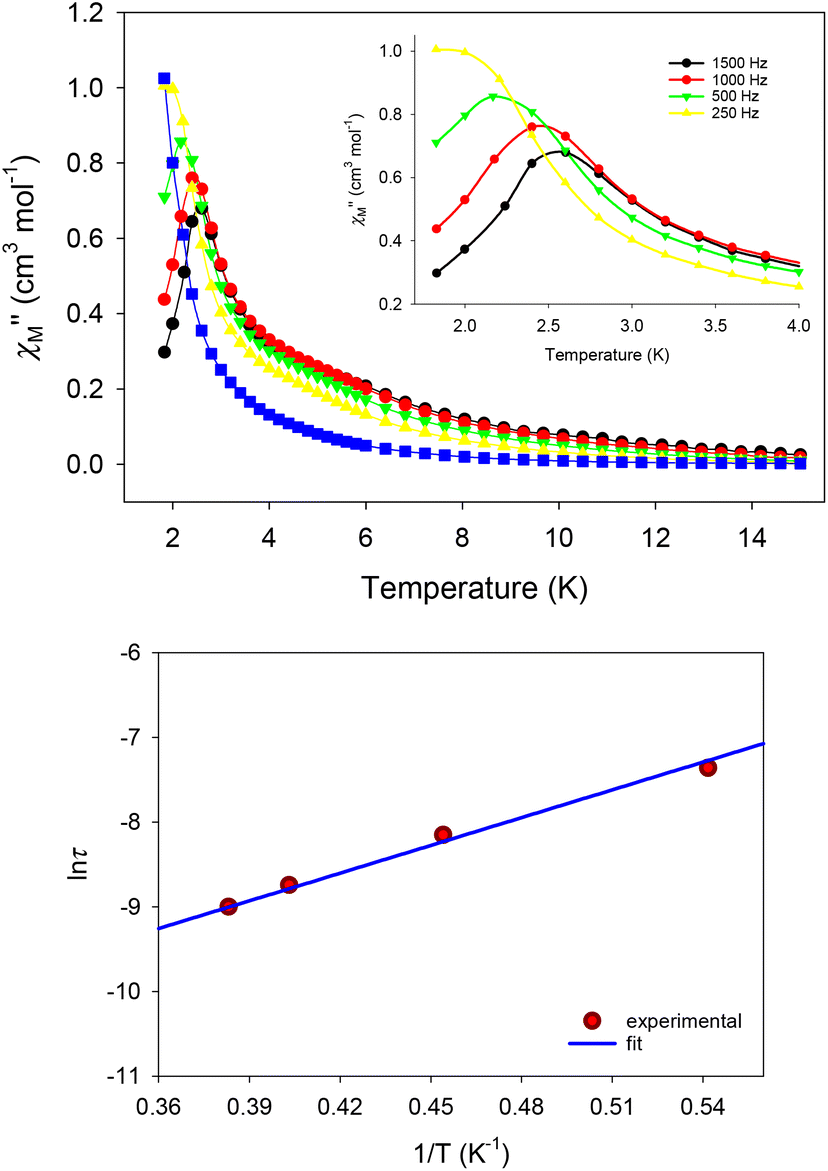 | ||
| Fig. 8 (Top) χ′′Mvs. T plot for 1 at 0.1 T dc field, measured under a 3.5 G ac field oscillating at the indicated frequencies; the inset highlights the peak maxima obtained at the indicated temperature region. (Bottom) Arrhenius-type plot of lnτ vs. 1/T for 1, constructed from the χ′′Mvs. T data under the applied dc field of 0.1 T; the solid blue line represents the fit that gave the Ueff and τ0 values mentioned in the text. | ||
Unfortunately, the application of an external dc field did not alter the ac profiles of complexes 2, 3, and 4, and no peaks of signals were obtained, suggesting that QTM is still the most dominating mechanism for the magnetization relaxation of these compounds. We have thus focused on quantifying the expectedly small energy barriers of 2–4 using the ac data at zero dc field. Assuming that the magnetization relaxation has only one characteristic time that corresponds to a Debye relaxation process, the SMM parameters can be deduced by applying the Kramers–Kronig equations,57 which result in the combined eqn (1):
| ln(χ′′/χ′) = ln(ωτ0) + Ueff/kBT, | (1) |
The corresponding best-fit parameters for the tetranuclear complex 4 (Fig. 9) were Ueff = 7.8(2) K and τ0 = 6.1(2) × 10−7 s. The structurally similar complex [Dy4(μ3-OH)2(php)2(O2CMe)6(H2O)2] does not exhibit any out-of-phase ac signals in the absence of an external dc field, but it is a field (dc = 0.12 T)-induced SMM with two relaxation phases corresponding to the high- and low-frequency regions. No Ueff and τ0 parameters were reported for this compound. The differences in the magnetic dynamics of 4 over the {Dy4}/php2− cluster29 can be tentatively assigned to the dissimilar coordination numbers and geometries of the individual DyIII ions, as well as the intra- and intermolecular structural and supramolecular variations, respectively.
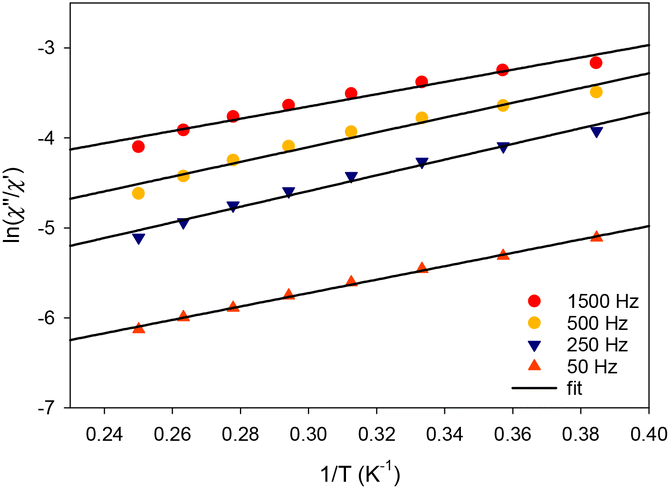 | ||
| Fig. 9 Debye plots of complex 4 in the temperature range 1.8–5.2 K for the indicated ac frequencies. The solid lines correspond to the fit of the data by applying eqn (1); see the text for the fit parameters. | ||
Concluding comments and perspectives
In conclusion, we have herein reported a family of mono-, di-, and tetranuclear DyIII complexes resulting from the structural flexibility of the pocket-type ligand 2,6-diacetylpyridine bis(picolinoylhydrazone) (LH2) under different synthetic conditions, including the use of various ancillary anions with coordinating or non-coordinating ability, in the absence or presence of an external base. As a result of this systematic study, the organic versatile ligand LH2 was proved to act as (i) pentadentate chelate under its neutral, zwitterionic form in the mononuclear complex 1, (ii) hexadentate chelating and bridging ligand under its single-deprotonated, amide–iminol tautomerism form in the dinuclear compounds 2 and 3, and (iii) heptadentate chelating and bis-bridging ligand under its double-deprotonated, iminol form in the tetranuclear cluster 4. From a magnetism viewpoint, all the reported compounds exhibit slow magnetization relaxation under zero external dc field albeit with small energy barriers, because of the efficient quantum tunneling of the magnetization between the lowest mj sublevels of the ground J state. Attempts to switch-off the tunnelling through the application of an external dc field, all failed to significantly improve the SMM properties. Only the mononuclear complex 1, which possesses a very distorted D6h geometry, showed entirely visible out-of-phase ac signals under a dc field of 0.1 T, but still the energy barrier was significantly small when compared to the huge energy barriers reported for hexagonal bipyramidal DyIII monomers with ideal D6h geometry.Work in progress includes the chemical modulation of the coordination geometry of 1 by either removing the coordinating MeOH solvate molecule and replacing the axial Cl− ions with strongly binding anions, such as Ph3SiO− or derivatives of phenolate groups, or replacing the MeOH molecule of the equatorial plane with bulkier coordinating solvents, as a means of exerting a “chemical pressure” to the system towards adopting a closer to the ideal D6h geometry. These short-term objectives aim at the synthesis of air-stable, mononuclear single-molecule magnets with large energy barriers for magnetization reversal.
Experimental section
General, physical measurements and spectroscopic studies
All manipulations were performed under aerobic conditions using materials (reagent grade) and solvents as received. The organic ligand LH2 was prepared and characterized according to the literature methods described elsewhere.60 The reaction is a simple hydrazone condensation of one equivalent of 2,6-diacetylpyridine with two equivalents of picolinic acid hydrazide in refluxing MeOH (yellowish solid; 90% isolated yield). Elemental analyses were performed by the microanalytical service of the University of Patras. FT-IR spectra (4000–400 cm−1) were recorded using a Thermo Scientific Nicolet iS20 spectrometer with samples prepared as KBr pellets. Thermogravimetric analysis (TGA) was conducted on a Labys TG (Setaram Instrumentation) under N2 flow from 25 °C to 600–800 °C with a heating rate of 10 °C min−1. Direct current (dc) and alternating current (ac) magnetic susceptibility studies were performed at the University of Florida Chemistry Department on a Quantum Design MPMS-XL SQUID susceptometer equipped with a 7 T magnet and operating in the 1.8–400 K range. Samples were embedded in solid eicosane to prevent torquing. Alternating current magnetic susceptibility measurements were performed in an oscillating ac field of 3.5 G and either a zero or an external dc field when required. Oscillation frequencies were in the 50–1000 Hz range. Pascal's constants were used to estimate the diamagnetic correction, which was subtracted from the experimental susceptibility to give molar paramagnetic susceptibility (χM).61Synthetic details
Single-crystal X-ray crystallography
Yellow crystals (0.32 × 0.18 × 0.15 mm) of 1 and 2 (0.20 × 0.17 × 0.14 mm) were taken directly from the mother liquor and immediately cooled to 180 and 190 K, respectively. X-ray diffraction data were collected on a Rigaku R-AXIS SPIDER Image Plate diffractometer using graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation. Data collection (ω-scans) and processing (cell refinement, data reduction and empirical absorption correction) were performed using the CrystalClear program package.62 The structures of 1 and 2 were solved by direct methods using SHELXS ver. 2013/163 and refined by full-matrix least-squares techniques on F2 with SHELXL ver. 2014/6.64 H atoms were either located by difference maps and refined isotropically or were introduced at calculated positions as riding on their respective bonded atoms. All non-H atoms were refined anisotropically. The SQUEEZE procedure65 was used for the analysis of the structures of 1 and 2; in both 1 and 2 the unit cells contained two methanol solvent molecules, which have been treated as a diffuse contribution to the overall scattering without specific atom positions. With respect to compound 2, the site labeled as O1M is occupied by two ligands; the estimated value for the occupancy of this site by a methanol molecule is 0.71(1) and by a water one 0.29(1). Sites O1M are shared by oxygen atoms of both the hydroxyl group of methanol and water solvate ligands. Also, the sites of H1MB hydrogen atoms are occupied by one hydrogen atom of the water ligand and the hydrogen atom from the hydroxyl group of methanol ligands. The sites H1MA are occupied only by the second hydrogen atom of water ligands with an estimated occupancy of 0.29(1). C24 atom sites and the respective hydrogen atoms of the methyl group have 0.71(1) occupancy. The description of disorder of this model at these sites is completed by considering the presence of methanol lattice molecules at the sites O25 and C26 with an occupancy of 0.29(1). Also, the coordinated triflate anions occupy two sites with 0.902(3) and 0.098(3) occupancies.
Single-crystal X-ray diffraction data of complex 3·2MeOH were collected on a yellow plate-like crystal (0.06 × 0.05 × 0.04 mm) using a Rigaku Oxford Diffraction XtaLAB Synergy diffractometer equipped with a HyPix-6000HE area detector at 173 K and utilizing Cu Kα (λ = 1.54184 Å) radiation from a PhotonJet micro-focus X-ray source. The structure was solved using SHELXT ver. 2018/2 and refined by full-matrix least-squares techniques against Fo2 using the SHELXL ver. 2018/3 program63 through the OLEX2 interface.66 The non-hydrogen atoms were successfully refined using anisotropic displacement parameters, and hydrogen atoms bonded to the carbon, nitrogen and oxygen atoms of the ligands were placed at their idealized positions using appropriate HFIX instructions in SHELXL. All these atoms were included in subsequent refinement cycles in riding-motion approximation with isotropic thermal displacement parameters (Uiso) fixed at 1.2 or 1.5 × Ueq of the relative atom.
A yellow crystal (0.20 × 0.07 × 0.07 mm) of 4·2CH2Cl2 was mounted on a CryoLoop with the assistance of a stereomicroscope.67 Diffraction data were collected on a Bruker D8 diffractometer (Mo Kα graphite-monochromated radiation, λ = 0.71073 Å) with the acquisition controlled by the APEX2 software package.68 The temperature of data acquisition was 150(2) K, and it was set up with a cryosystem by the Oxford Cryosystems Series 700. Images were processed with the software SAINT+,69 and absorption effects were corrected by the multi-scan method implemented in SADABS.70 The structure was solved using SHELXTL incorporated in the Bruker APEX-III software package and refined through SHELXLE and PLATON programs.71,72 All the non-hydrogen atoms of the structures were successfully refined using anisotropic displacement parameters. H-atoms bound to carbon atoms were placed at geometrical positions using suitable HFIX instructions in SHELXL and included in subsequent refinement cycles in riding-motion approximation with isotropic thermal displacement parameters (Uiso) fixed at the carbon atom to which they are attached. Some electron density was found in the data of the crystal structure, probably due to additional disordered solvent molecules occupying the spaces created by the packing arrangement of the cluster compound. Efforts to accurately locate, model and refine these residues were ineffective, and the investigation of the total potential solvent area using the software package PLATON73,74 confirmed the existence of cavities with potential solvent accessible void volume. Thus, the original data sets were treated with the program SQUEEZE,75 which is used to calculate the contribution of the smeared electron density in the lattice voids and adds this to the calculated structure factors from the structural model when refining against the hkl file.
Various figures of the structures were created, using Diamond 3 and Mercury software packages.76,77 Additional structural drawings of the four complexes with thermal ellipsoids at 50% probability are shown in Fig. S9.† The unit cell parameters, structure solution, and refinement details of 1, 2, 3·2MeOH, and 4·2CH2Cl2 are summarized in Table S8.† Further crystallographic details can be found in the corresponding CIF files provided in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Andreas Mentzelopoulos Foundation in the form of a scholarship to G. P. B. G. C. was supported by the Center for Molecular Magnetic Quantum Materials (M2QM), an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Basic Energy Sciences, under award DE-SC0019330. This research work also received financial support from Portuguese national funds (FCT/MCTES, Fundação para a Ciência e a Tecnologia and Ministério da Ciência, Tecnologia e Ensino Superior) through the strategic projects UIDB/50006/2020 (for LAQV/REQUIMTE). L. C.-S. also thanks FCT/MCTES for funding through the Individual Call to Scientific Employment Stimulus (ref. CEECIND/00793/2018).References
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, MS, USA, 2006 Search PubMed.
- M. Atzori and R. Sessoli, J. Am. Chem. Soc., 2019, 141, 11339 CrossRef CAS PubMed.
- For representative reviews, see: (a) R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011 RSC; (b) C. J. Milios and R. E. P. Winpenny, Struct. Bonding, 2015, 164, 1 CAS.
- F. Habib and M. Murugesu, Chem. Soc. Rev., 2013, 42, 3278 RSC.
- (a) J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470–2491 RSC; (b) M. Feng and M.-L. Tong, Chem. – Eur. J., 2018, 24, 7574 CrossRef CAS PubMed.
- M. J. Heras Ojea, L. C. H. Maddock and R. A. Layfield, Lanthanide Organometallics as Single-Molecule Magnets, in Organometallic Magnets. Topics in Organometallic Chemistry, ed. V. Chandrasekhar and F. Pointillart, Springer, Cham, 2019, vol. 64 Search PubMed.
- C. Papatriantafyllopoulou, E. E. Moushi, G. Christou and A. J. Tasiopoulos, Chem. Soc. Rev., 2016, 45, 1597 RSC.
- A. M. Ako, I. J. Hewitt, V. Mereacre, R. Clerac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 4926 CrossRef CAS PubMed.
- C. J. Milios, A. Vinslava, W. Wernsdorder, S. Moggach, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2006, 129, 2754 CrossRef PubMed.
- Th. C. Stamatatos, D. Foguet-Albiol, S.-C. Lee, C. C. Stoumpos, C. P. Raptopoulou, A. Terzis, W. Wernsdorfer, S. O. Hill, S. P. Perlepes and G. Christou, J. Am. Chem. Soc., 2007, 129, 9484 CrossRef CAS PubMed.
- For a very recent review, see: P. Konieczny, W. Sas, D. Czernia, A. Pacanowska, M. Fitta and R. Pełka, Dalton Trans., 2022, 51, 12762 RSC.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400 CrossRef CAS PubMed.
- N. F. Chilton, Inorg. Chem., 2015, 54, 2097 CrossRef CAS PubMed.
- J. Tang and P. Zhang, Lanthanide Single Molecule Magnets, Springer-Verlag, Heidelberg, Germany, 2015 Search PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed.
- D. Aguilà, L. A. Barrios, V. Velasco, O. Roubeau, A. Repollés, P. J. Alonso, J. Sesé, S. J. Teat, F. Luis and G. Aromí, J. Am. Chem. Soc., 2014, 136, 14215 CrossRef PubMed.
- M. Ganzhorn, S. Klyatskaya, M. Ruben and W. Wernsdorfer, ACS Nano, 2013, 7, 6225 CrossRef CAS PubMed.
- P. Zhang, Y.-N. Guo and J. Tang, Coord. Chem. Rev., 2013, 257, 1728 CrossRef CAS.
- Z. Zhu, M. Guo, X.-L. Li and J. Tang, Coord. Chem. Rev., 2019, 378, 350 CrossRef CAS.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078 RSC.
- E. Moreno-Pineda, G. Taran, W. Wernsdorfer and M. Ruben, Chem. Sci., 2019, 10, 5138 RSC.
- J.-L. Liu, Y.-C. Chen and M.-L. Tong, Chem. Soc. Rev., 2018, 47, 2431 RSC.
- K. S. Pedersen, D. N. Woodruff, J. Bendix and R. Clérac, in Lanthanides and Actinides in Molecular Magnetism, ed. R. A. Layfield and M. Murugesu, Wiley-VCH, Berlin, Germany, 1st edn, 2015, pp. 125–152 Search PubMed.
- Y. Gil, A. Castro-Alvarez, P. Fuentealba, E. Spodine and D. Aravena, Chem. – Eur. J., 2022, 28, e202200336 CAS.
- P. Zhang, L. Zhang and J. Tang, Curr. Inorg. Chem., 2013, 3, 101 CrossRef CAS.
- For example, see: (a) J. Zhang, Z. Zhang, Z. Chen and X. Zhou, Dalton Trans., 2012, 41, 357 RSC; (b) H. Tian, B.-L. Wang, J. Lu, H.-T. Liu, J. Su, D. Li and J. Dou, Chem. Commun., 2018, 54, 12105 RSC; (c) T. Bereta, A. Mondal, K. Ślepokura, Y. Peng, A. K. Powell and J. Lisowski, Inorg. Chem., 2019, 58, 4201 CrossRef CAS PubMed.
- For example, see: (a) M. Guo, Y. Xu, J. Wu, L. Zhao and J. Tang, Dalton Trans., 2017, 46, 8252 RSC; (b) C. P. Burns, B. O. Wilkins, C. M. Dickie, T. P. Latendresse, L. Vernier, K. R. Vignesh, N. S. Bhuvanesh and M. Nippe, Chem. Commun., 2017, 53, 8419 RSC; (c) M. Guo, Y.-Q. Zhang, Z. Zhu and J. Tang, Inorg. Chem., 2018, 57, 12213 CrossRef CAS PubMed; (d) F. Pointillart, B. Le Guennic, S. Golhen, O. Cador and L. Ouahab, Chem. Commun., 2013, 49, 11632 RSC.
- V. S. Sastri, J. R. Perumareddi, V. Ramachandra Rao, G. V. S. Rayudu and J.-C. Bunzli, in Modern Aspects of Rare Earths and their Complexes, 1st edn, 2003 Search PubMed.
- S. Xue, L. Zhao, Y.-N. Guo, R. Deng, Y. Guo and J. Tang, Dalton Trans., 2011, 40, 8347 RSC.
- C. Pelizzi, G. Pelizzi, G. Predieri and S. Resola, J. Chem. Soc., Dalton Trans., 1982, 1349 RSC.
- C. Pelizzi, G. Pelizzi and F. Vitali, Transition Met. Chem., 1986, 11, 401 CrossRef CAS.
- D. Delledonne, G. Pelizzi and C. Pelizzi, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 1502 CrossRef.
- A. Bonardi, S. Ianelli, C. Pelizzi and G. Pelizzi, Inorg. Chim. Acta, 1991, 187, 167 CrossRef CAS.
- I. Mylonas-Margaritis, D. Maniaki, J. Mayans, L. Ciammaruchi, V. Bekiari, C. P. Raptopoulou, V. Psycharis, S. Christodoulou, A. Escuer and S. P. Perlepes, Magnetochemistry, 2018, 4, 45 CrossRef CAS.
- E. Pilichos, I. Mylonas-Margaritis, A. P. Kontos, V. Psycharis, N. Klouras, C. P. Raptopoulou and S. P. Perlepes, Inorg. Chem. Commun., 2018, 94, 48 CrossRef CAS.
- W. Harhouri, C. Mchiri, S. Najmudin, C. Bonifácio and H. Nasri, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2016, 72, 720 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, NY, USA, 4th edn, 1986 Search PubMed.
- (a) G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS; (b) D. Martinez, M. Motevalli and M. Watkinson, Dalton Trans., 2010, 446 RSC.
- V. Zeleňák, Z. Vargová and K. Györyová, Spectrochim. Acta, Part A, 2007, 66, 262 CrossRef PubMed.
- X. Sun, Y.-K. Yang and F. Lu, Macromolecules, 1998, 31, 4291 CrossRef CAS.
- M. Llunell, D. Casanova, J. Girera, P. Alemany and S. Alvarez, SHAPE, Continuous Shape Measures Calculation; Version 2.0, Universitat de Barcelona, Barcelona, Spain, 2010 Search PubMed.
- (a) A. K. Bar, P. Kalita, J.-P. Sutter and V. Chandrasekhar, Inorg. Chem., 2018, 57, 2398 CrossRef CAS PubMed; (b) P. Rey, A. I. Smolentsev and K. E. Vostrikova, Molecules, 2022, 27, 1626 CrossRef CAS PubMed; (c) B.-Y. Chen, M.-Y. Tsai, Y.-C. Su, P.-H. Lin and J. Long, CrystEngComm, 2021, 23, 8343 RSC.
- For an excellent paper discussing N–H⋯Cl hydrogen bonds, see: J. V. Gavette, C. M. Klug, L. N. Zakharov, M. P. Shores, M. M. Haley and D. W. Johnson, Chem. Commun., 2014, 50, 7173 RSC.
- (a) P. Antal, B. Drahoš, R. Herchel and Z. Trávníček, Dalton Trans., 2016, 45, 15114 RSC; (b) L. Mandal, S. Biswas and M. Yamashita, Magnetochemistry, 2019, 5, 56 CrossRef CAS.
- E. C. Manickas, M. Zeller and C. M. Zaleski, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2020, 76, 1213 CrossRef CAS PubMed.
- For example, see: (a) G. Abbas, Y. Lan, G. E. Kostakis, W. Wernsdorfer, C. E. Anson and A. K. Powell, Inorg. Chem., 2010, 49, 8067 CrossRef CAS PubMed; (b) Y.-X. Zhang, Y.-H. Zhang, B.-Y. Liu, W.-M. Wang, G.-P. Tang, H.-Y. Wei and Z.-L. Wu, New J. Chem., 2018, 42, 18305 RSC; (c) K. Zhang, G.-P. Li, V. Montigaud, O. Cador, B. Le Guennic, J. Tang and Y.-Y. Wang, Dalton Trans., 2019, 48, 2135 RSC; (d) H. Ke, P. Gamez, L. Zhao, G.-F. Xu, S. Xue and J. Tang, Inorg. Chem., 2010, 49, 7549 CrossRef CAS PubMed.
- G. G. Morgan and I. A. Kühne, in Practical Approaches to Biological Inorganic Chemistry, ed. R. R. Crichton and R. O. Louro, 2nd edn, 2020, pp. 69–119 Search PubMed.
- For an excellent comprehensive review, see: D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS PubMed.
- T. G. Ashebr, H. Li, X. Ying, X.-L. Li, C. Zhao, S. Liu and J. Tang, ACS Mater. Lett., 2022, 4, 307 CrossRef CAS.
- Z. Zhu, C. Zhao, T. Feng, X. Liu, X. Ying, X.-L. Li, Y.-Q. Zhang and J. Tang, J. Am. Chem. Soc., 2021, 143, 10077 CrossRef CAS PubMed.
- (a) D. I. Alexandropoulos, A. A. Alaimo, D. Sun and Th. C. Stamatatos, Magnetochemistry, 2018, 4, 48 CrossRef CAS; (b) E. C. Mazarakioti, J. Regier, L. Cunha-Silva, W. Wernsdorfer, M. Pilkington, J. Tang and Th. C. Stamatatos, Inorg. Chem., 2017, 56, 3568 CrossRef CAS PubMed; (c) K. N. Pantelis, P. S. Perlepe, S. Grammatikopoulos, C. Lampropoulos, J. Tang and Th. C. Stamatatos, Molecules, 2020, 25, 2191 CrossRef CAS PubMed.
- Y.-N. Guo, G.-F. Xu, W. Wernsdorfer, L. Ungur, Y. Guo, J. Tang, H.-J. Zhang, L. F. Chibotaru and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 11948 CrossRef CAS PubMed.
- S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655 RSC.
- For example, see: (a) Z. Zhu, C. Zhao, T. Feng, X. Liu, X. Ying, X.-L. Li, Y.-Q. Zhang and J. Tang, J. Am. Chem. Soc., 2021, 143, 10077 CrossRef CAS PubMed; (b) Z.-H. Li, Y.-Q. Zhai, W.-P. Chen, Y.-S. Ding and Y.-Z. Zheng, Chem. – Eur. J., 2019, 25, 16219 CrossRef CAS PubMed; (c) A. B. Canaj, S. Dey, E. R. Martí, C. Wilson, G. Rajaraman and M. Murrie, Angew. Chem., Int. Ed., 2019, 58, 14146 CrossRef CAS PubMed.
- (a) J. Li, S. Gómez-Coca, B. S. Dolinar, L. Yang, F. Yu, M. Kong, Y.-Q. Zhang, Y. Song and K. R. Dunbar, Inorg. Chem., 2019, 58, 2610 CrossRef CAS PubMed; (b) B. Ali, X.-L. Li, F. Gendron, B. Le Guennic and J. Tang, Dalton Trans., 2021, 50, 5146 RSC.
- L. I. Semenova and A. H. White, Aust. J. Chem., 1999, 52, 571 CrossRef CAS.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341 CrossRef CAS.
- Y.-N. Guo, G.-F. Xu, Y. Guo and J. Tang, Dalton Trans., 2011, 40, 9953 RSC.
- For example, see: (a) N. Qiao, X.-X. Li, Y. Chen, X.-Y. Xin, C. Yang, S.-S. Dong, Y.-Z. Wang, X.-J. Li, Y.-P. Hua and W.-M. Wang, Polyhedron, 2022, 215, 115675 CrossRef CAS; (b) Y. Peng, V. Mereacre, C. E. Anson and A. K. Powell, Inorganics, 2016, 4, 2 CrossRef; (c) J. Wang, Z.-L. Wu, L.-R. Yang, M.-M. Xue, Z.-X. Fang, S.-C. Luo and W.-M. Wang, Inorg. Chim. Acta, 2021, 514, 120015 CrossRef CAS; (d) J. Wu, X.-L. Li, L. La Droitte, O. Cador, B. Le Guennic and J. Tang, Dalton Trans., 2021, 50, 15027 RSC; (e) M. U. Anwar, A. Al-Harrasi, J. M. Rawson, E. L. Gavey, J. Regier, D. Alexandropoulos, M. Pilkington and L. K. Thompson, Dalton Trans., 2019, 48, 14269 RSC.
- C. Pelizzi and G. Pelizzi, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 126 CrossRef.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- CrystalClear, Rigaku/MSC Inc., The Woodlands, TX, USA, 2005 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615 CrossRef.
- APEX2. Data Collection Software Version 2012.4, Bruker AXS, Delft, The Netherlands, 2012 Search PubMed.
- SAINT+, Data Integration Engine v. 7.23a ©, Bruker AXS, Madison, Wisconsin, USA, 1997–2005 Search PubMed.
- G. M. Sheldrick, SADABS v.2.01, Bruker/Siemens Area Detector Absorption Correction Program, Bruker AXS, Madison, Wisconsin, USA, 1998 Search PubMed.
- APEX-III, Bruker AXS Inc., Madison, Wisconsin, USA, 2016 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, C34 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- P. Van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- K. Bradenburg, DIAMOND, Release 3.1f, Crystal Impact GbR, Bonn, Germany, 2008 Search PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Various physicochemical and structural figures (Fig. S1–S6, and S9), magnetism plots (Fig. S7 and S8), and crystallographic data and metrical parameters (Tables S1–S8). CCDC 2205399, 2205400, 2205298 and 2205303 for complexes 1, 2, 3·2MeOH, and 4·2CH2Cl2. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02921a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |