
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Real-time regeneration of a working zeolite monitored via operando X-ray diffraction and crystallographic imaging: how coke flees the MFI framework†
Georgios N.
Kalantzopoulos
a,
Daniel
Rojo Gama
a,
Dimitrios K.
Pappas
 a,
Iurii
Dovgaliuk
cd,
Unni
Olsbye
a,
Pablo
Beato
b,
Lars F.
Lundegaard
b,
David S.
Wragg
a and
Stian
Svelle
*a
a,
Iurii
Dovgaliuk
cd,
Unni
Olsbye
a,
Pablo
Beato
b,
Lars F.
Lundegaard
b,
David S.
Wragg
a and
Stian
Svelle
*a
aCenter for Materials Science and Nanotechnology (SMN), Department of Chemistry, University of Oslo, P.O. Box 1033, Blindern, N-0315 Oslo, Norway. E-mail: stian.svelle@kjemi.uio.no
bHaldor Topsøe A/S, Haldor Topsøes Allé 1, 2800 Kgs, Lyngby, Denmark
cSwiss-Norwegian Beamline at the European Synchrotron Facility, 71 avenue des Martyrs, F-38000, Grenoble, France
dInstitut des Matériaux Poreux de Paris, Département de Chimie, ENS - UMR 8004 CNRS-ENS-ESPCI, 24 rue Lhomond, 75005 Paris, France
First published on 19th October 2022
Abstract
We have monitored the regeneration of H-ZSM-5 via operando time-resolved powder X-Ray diffraction (PXRD) coupled with mass spectroscopy (MS). Parametric Rietveld refinements and calculation of the extra-framework electronic density by differential Fourier maps analysis provide details on the mode of coke removal combined with the corresponding sub-unit cell changes of the zeolite structure. It is clear that the coke removal is a complex process that occurs in at least two steps; a thermal decomposition followed by oxidation. In a coked zeolite, the straight 10-ring channel circumference is warped to an oval shape due to structural distortion induced by rigid aromatic coke species. The data presented explain why the difference in length between the a-vector and the b-vector of the MFI unit cell is a robust descriptor for bulky coke, as opposed to the unit cell volume, which is affected also by adsorbed species and thermal effects. Our approach holds the promise to quantify and identify coke removal (and formation) in structurally distinct locations within the zeolite framework.
Introduction
More than four decades have passed since the conversion of Methanol to Gasoline (MTG) over acidic zeolites was first reported by researchers from Mobil.1 During these 45 years, the scientific and industrial community has gained a comprehensive understanding of several aspects of the MTG process.2,3 Some of the most recent achievements have been the disentanglement of the reaction mechanism4–6 and the study of the influence of reaction parameters and zeolite structure in the product distribution.7–11 Intrinsic structural characteristics such as the role of the porosity12–14 and acidity15,16 on the catalytic performance have also been investigated. Moreover, the deactivation of zeolites has been analysed with both experimental and theoretical approaches.17–19One of the main weaknesses of zeolite frameworks as MTG catalysts is the loss of catalytic performance over time. When exposed to a methanol feed, the zeolite deactivates as the reaction proceeds; carbon-containing species accumulate in the zeolite pores and on the external surface preventing the reaction from taking place.17,20 The nature of deposited coke species has been reported to correlate with the reaction conditions as well as the catalyst used.21 Once the coke coverage reaches a critical level where the diffusion through the pores is restricted and the reactants no longer have access to the acidic centres, the zeolite shows total performance loss.18,22,23 To reverse this process, the catalyst material is regenerated by exposure to oxidizing gas flow at a high temperature (500–600 °C).18,24–26 More specifically, due to the large amount of catalyst loaded in an industrial reactor, the amount of oxidizing gas has to be increased carefully until the removal of bulk coke is achieved, in order to avoid thermal runaway.26 Still, each reaction-regeneration cycle leads to slight dealumination of the zeolite.27 This dealumination results in the loss of Brønsted acidity, which has been reported to contribute in the coke oxidation.28When repeated many times, the regeneration procedure leads to reduced overall catalyst performance, making this step an extremely important operation in the industrial process. In laboratory studies, the regeneration of coked zeolites is usually performed as a Temperature Programmed Oxidation (TPO) to gain insights into coke characteristics and location.7,17,29 The coke on the external surface of the zeolite crystals is more resistant to oxidation than the intracrystalline coke, which is preferentially removed during oxidation.28
H-ZSM-5 (Fig. 1) is the archetype zeolite for MTG due to its three dimensional structure consisting of straight and sinusoidal channels lying perpendicular to each other that enhance the diffusion of products and improve the resistance to deactivation. The material crystallizes in the orthorhombic space group Pnma (MFI topology) with typical unit cell dimensions: a = 20.078 Å, b = 19.894 Å and c = 13.372 Å.30 The loss of activity of H-ZSM-5 during the MTG reaction and its spatial distribution along the catalyst bed is reported to follow the “burning cigar” model proposed by Haw.7,11,31 The H-ZSM-5 deactivation as well as its effect on zeolite properties, such as acid site density, surface area and coke content, has been directly correlated to the local transformations occurring in the crystalline structure of the catalyst.32 Previously, we have followed with operando X-Ray Diffraction (XRD) the MTG conversion in the time- and space-domain capturing the build-up of coke in the intracrystalline structure and thereby extracting the kinetics of coke-based deactivation for H-ZSM-5.33
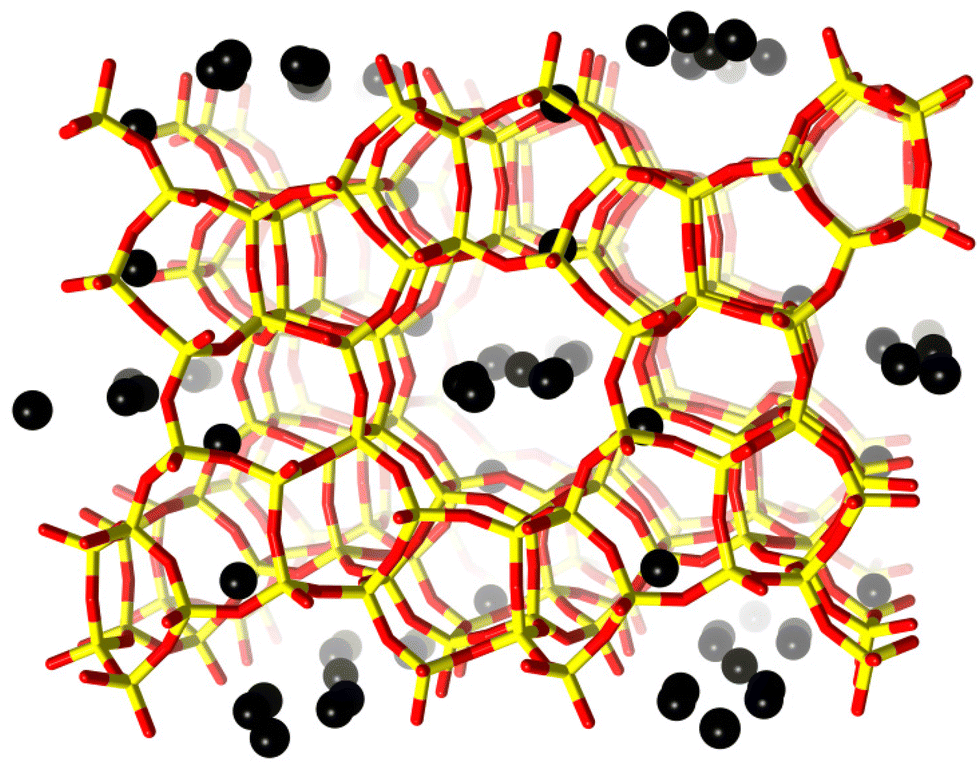 | ||
| Fig. 1 H-ZSM-5 unit cell viewed along the b-axis. Framework oxygen-atoms and T-atoms are indicated in red and yellow, respectively. Black spheres indicate preferential sites of coke residing in the framework. The coke sites have been identified by difference Fourier map analysis. A detailed analysis of the precise location of these dummy atoms is presented in the ESI.† | ||
In this contribution, we have studied the local structural transformations of the H-ZSM-5 catalyst during the removal of coke by TPO with 20% of O2 utilizing operando PXRD. The recent developments in synchrotron instrumentation and detector technology have previously allowed us to study the effect of isolated thermal effects within a zeolite framework, such as the template removal,34 the local response of the acidic centre to hydration,35 or the dynamics of nonhomogeneous water sorption36 with unprecedented detail. Here, the geometrical response of the material has been investigated by operando time-resolved synchrotron PXRD allowing us to follow the structural changes on a timescale of seconds.
Experimental
Materials
An in house prepared H-ZSM-5 catalyst sample was used. The material was synthesized according to procedure found in the literature37 resulting in a catalyst with micron sized particles and an Si/Al ratio of 37. Further basic characterization of the catalysts is provided in the ESI (Tables S1 and S2, Fig. S1†).Operando MTG and TPO monitored by SR-XRD
Operando MTG and TPO experiments were performed at the Swiss–Norwegian beamline (station BM01A/BM01) of The European Synchrotron (ESRF) with a wavelength of 0.7743 Å, using a Dectris Pilatus 2M photon counting pixel area detector38 and then integrated with the SNBL Bubble software.38,39 A Huber goniometer head was used for alignment and phi rotations. When collecting data during the MTG reaction, the reactor was moved across the beam (z-scans), and diffractograms were acquired at different positions of the bed at fixed time and distance intervals.33,40 After completing a full scan of the capillary, the reactor was moved back with no data collection.TPO data were collected at a single point at the center of the capillary reactor.
The patterns were analyzed by parametric Rietveld refinement41using TOPAS42 to extract the unit cell parameters, framework atom positions and the coke occupancy for each individual coke position in the MFI framework as a function of time-on-stream. The precise locations of these coke positions are described in the ESI.† A single B-iso parameter was refined for all oxygen atoms. Vesta43 was used for illustrating the crystal structures and the extra-framework electronic density calculated with the difference Fourier maps.
For the catalytic reaction and following catalyst regeneration, the H-ZSM-5 catalyst powder was pressed into pellets, crushed and sieved to a size between 212–250 μm. The catalyst was placed in a 0.5 mm quartz capillary and mounted on a SNBL flow cell bracket that provides controlled atmosphere. Reaction gases (argon, 20% oxygen in helium, or argon bubbled through methanol held at controlled room temperature) were fed with a flow rate of 2.2 mL min−1 (Weight Hourly Space Velocity, WHSV = 20 gMeOH gcatalyst−1 h−1). The reactor was heated with an Osram Sylvania heat blower. The temperature profile across an empty capillary reactor was measured inserting a thermocouple, and the temperature variation was ±5 °C for a 10 mm catalyst bed. This has a minor influence on the catalytic reaction, where spatially resolved data were recorded, but it can safely be assumed that the thermal gradient was negligible in the TPO experiment, which was carried out for a single point of the catalyst bed. Prior to reaction, the catalyst was activated at 500 °C for 1 h using a stream of 20% oxygen in argon. Then, the temperature was lowered to 400 °C, the reaction temperature. For the regeneration, the reactor was first cooled down to RT under flow of He and was then heated up to 700 °C at 5 °C min−1 under 2.5 ml min−1 flow of 20% O2 in He.
A Pfeiffer Omnistar GSD 301 T3 Quadrupole mass spectrometer was coupled at the exhaust line of the flow cell (sample to instrument distance <30 cm) for mass spectrometry data acquisition during the activation and MTG reaction and the TPO experiment. The total acquisition and read out time of each of the acquired mass spectra was <0.8 s.
Results and discussion
Structural transformations of the MFI framework during the MTG reaction
Previously, Alvarez et al.44 suggested that H-ZSM-5 can undergo a structural transformation upon deactivation. The XRD patterns of their deactivated H-ZSM-5 samples showed a structural change from orthorhombic space group Pnma (No. 62) to tetragonal P41212 (No. 94). The deactivated catalysts exhibited a shrinkage in the a- axis and a simultaneous extension in the b- axis, which thereby eventually become identical. This observation was fully reversible after catalyst regeneration and it was attributed to structural changes due to accumulation of coke in the framework pores.44 Recently Rojo-Gama et al.32 demonstrated that this apparent symmetry is a metric coincidence and appears in some H-ZSM-5 samples. However, the difference in length of the a- and b- axes can be used as a global descriptor for the deactivation of the H-ZSM-5 zeolite catalyst, referred to as the a minus b parameter, or simply (a − b).32,33,45Thus, we have previously shown that during the MTG reaction, (a − b) is proportional to the amount of coke and consecutively to the degree of the H-ZSM-5 catalyst deactivation.32,33 As the coke level increases, it starts occupying preferential sites within the MFI framework, equalizing the a and b-axes length from 20.17 Å and 20.01 Å, respectively, to 20.04 Å. This procedure changes the apparent H-ZSM-5 unit cell symmetry from orthorhombic towards tetragonal, as shown for several H-ZSM-5 catalysts in our previous work.32 However, this is a metric coincidence and does not in reality correspond to an actual symmetry change.
The H-ZSM-5 catalyst employed here behaves very similarly to that previously reported for the commercial H-ZSM-5, and the data are available in the ESI (Fig. S2†). Briefly, as the TOS increases, the pore network becomes gradually filled up with coke, as illustrated by the coke occupancy data (Fig. S2, bottom†). The further away from the reactor inlet, the later the coke accumulation begins, corresponding to the burning cigar mentioned above. The amount of coke is strongly connected with the change in the a minus b parameter (Fig. S2, top†). Exceptions to this gradual deactivation behavior are the first two slices at the reactor inlet. Unlike the rest of the slices, the a- and b-axis of the catalyst found in the first part of the reactor do not change much. This is because only the initiation of the MTG reaction occurs in this part of the catalyst bed, and no actual MTG conversion and related coke formation. Instead, a minor expansion is seen. This is most likely a physical reaction to the presence of the influent gases and/or reaction products/intermediates. Overall, the behavior is in very high agreement with what we have observed and quantified already for the H-ZSM-5 system in the past.32,33
Regeneration of the catalyst – temperature programmed oxidation (TPO)
When exposing a coked catalyst to oxidative conditions, the coke will be oxidized and gradually abandon the zeolite framework until it is completely removed. Fig. 2 shows the evolution of CO2, the oxidation product, as monitored with MS, the change in the unit cell volume, as well as the a minus b parameter during temperature ramp from 380 to 700 °C. Data for the full temperature range from RT are presented in the ESI (Fig. S3†).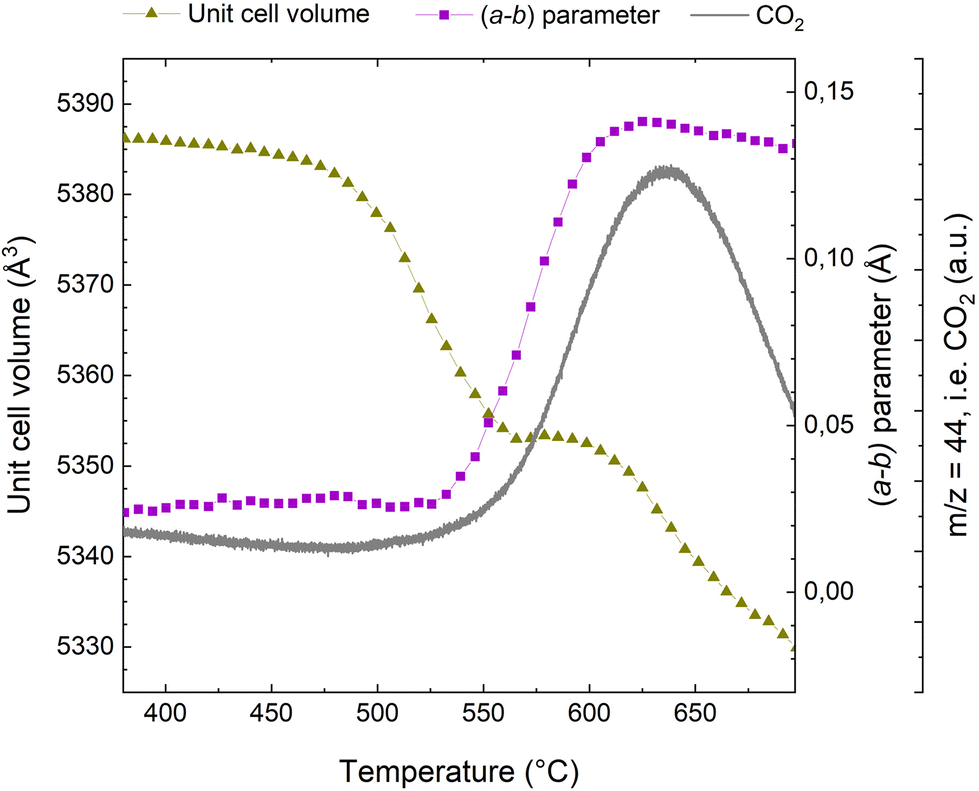 | ||
| Fig. 2 MS signal for CO2 (grey line), evolution of the (a − b) parameter (connected purple squares), and unit cell volume evolution (connected dark yellow triangles) during the course of regeneration of the H-ZSM-5 catalyst. | ||
Turning first to the unit cell volume, it becomes evident that the catalyst regeneration appears to be a multi-step process. We observe an initial slight reduction of the unit cell volume followed by a more rapid decrease starting at ∼475 °C. At ∼560 °C, the unit cell volume levels off and remains quite stable until ∼600 °C. Beyond ∼600 °C, we again observe a rapid reduction in unit cell volume. The a minus b parameter, which is linked to bulky coke species that distort the framework (see ref. 10, 11, 32 and 33 and below), is fairly stable until 530 °C, at which a very rapid increase is seen. Interestingly, appreciable CO2 evolution appears at approximately the same temperature. Thus, the unit cell volume changes earlier than the CO2 evolution and the change in the a minus b parameter, which occur at a quite similar temperature. The plateau in the unit cell volume occurs distinctly later. The major change in the a minus b parameter is concluded at ∼620 °C, where it has returned to a value similar to that seen for the fresh catalyst.32 This occurs fairly close to temperature of the maximum of the CO2 evolution, seen at ∼635 °C.
Superimposed to the oxidation processes thermal effects have to be considered. Up to 450 °C (see Fig. S3 of the ESI†) the unit cell volume exhibits an incremental increase of ∼3 Å3. This should be attributed to the thermal expansion of the rigid coke within the pores and not to intrinsic properties of the zeolite material, which is known to have a negative thermal expansion coefficient above 300 °C.46 This is similar to the incremental unit cell expansion that has been observed in SAPO-37 during the template removal process before the combustion of the organic compounds.34 Above 600 °C, we observe a reduction in the unit cell volume, which we attribute to negative thermal expansion, which has been reported to be quite profound for MFI at elevated temperatures.46,47 Also, removal of silanol nests and thermal dealumination cannot be ruled out.
A more qualitative representation of the structural changes occurring in H-ZSM-5 during coke removal is shown in Fig. 3 below. The removal of coke leads to a clear change in the shape of the straight channel, converting it from elliptical to almost circular. Further details on this subtle change is provided in Fig. S8 of the ESI.† We note that a very similar ellipsoidal distortion has been directly observed upon both benzene48 and para-xylene49 adsorption in H-ZSM-5 using (scanning) transmission electron microscopy ((S)TEM). It is therefore proposed that aromatic molecules, be it ordered adsorbed species48,49 or coke as in the present study, induce this structural change of the MFI framework through steric strain.
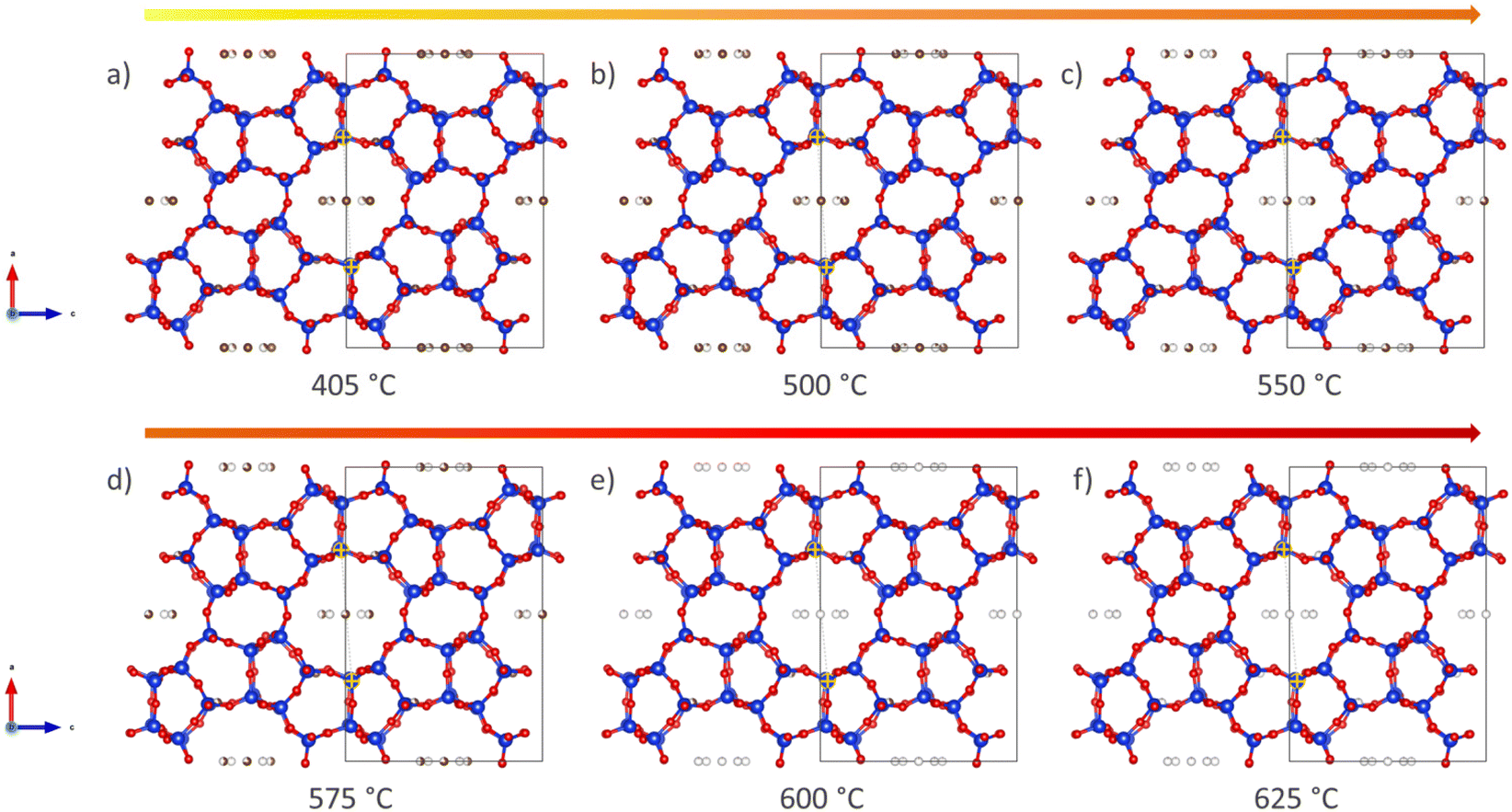 | ||
| Fig. 3 Graphical illustration of the structural re-arrangement taking place during the TPO of the H-ZSM-5 framework. The time frames have been extracted from the calculated cif files and represent the framework view along the straight channel at 405, 500, 550, 575, 600 and 625 °C (a–f). | ||
This transition from elliptical to circular can be quantified in terms of a 10 ring aspect ratio, as defined in,48,49 and this key structural parameter is shown in Fig. 4. A large numerical value of the aspect ratio corresponds to an oval 10-ring, whereas a lower value describes a more circular ring shape (Fig. 3). Also presented in Fig. 4 is the total coke content, as inferred from the refined occupancy of dummy carbon atoms during the Rietveld refinement analysis in TOPAS (see also Fig. 1). Finally, the a minus b parameter is superimposed on these two characteristics.
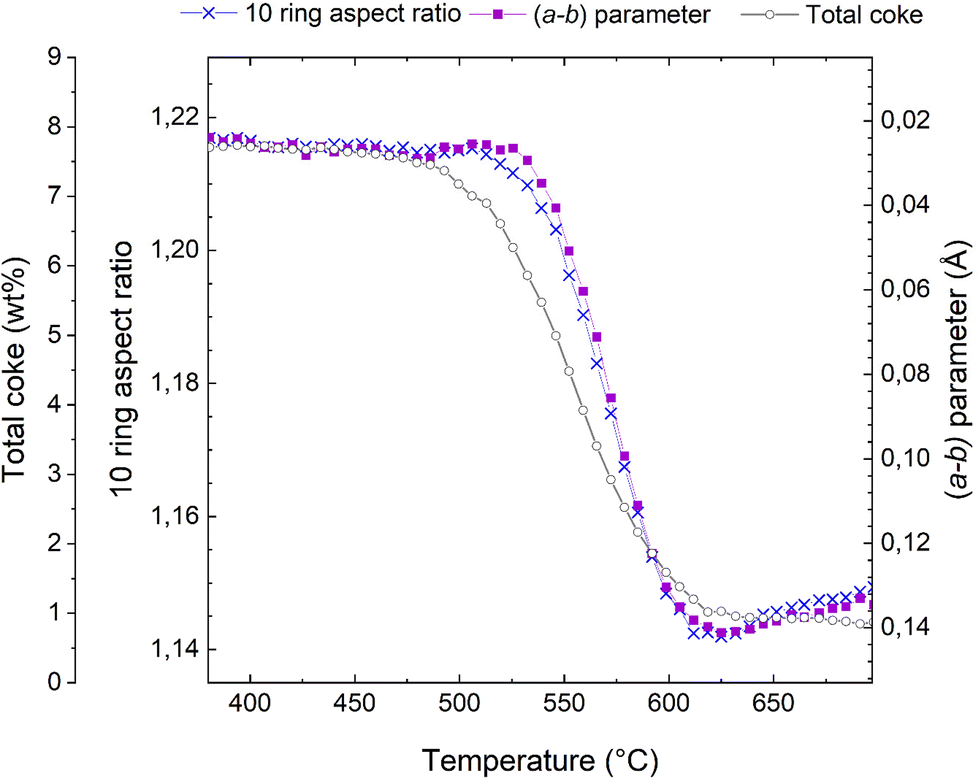 | ||
| Fig. 4 Total coke content from dummy atom occupancies (connected grey open circles), evolution of the (a − b) parameter (connected purple squares, note that the scale is inverted relative to Fig. 2), and 10 ring aspect ratio (connected blue crosses) during the course of regeneration of the H-ZSM-5 catalyst. | ||
It is evident from Fig. 4 that these three characteristics follow each other closely, but not exactly. The reduction of the total coke content occurs earliest, at ∼475 °C; exactly the same temperature at which the reduction in unit cell volume starts (Fig. 2). The changes in the a minus b parameter and the 10 ring aspect ratio occur at slight higher temperatures and are very nearly perfectly superimposed.
Most likely, the first rapid reduction of the coke content (Fig. 4) and concurrent reduction of the unit cell volume (Fig. 2) cannot be due to coke oxidation, as very little CO2 is observed (Fig. 2). The a minus b parameter and the 10 ring aspect ratio are stable during most of these initial processes, meaning that the species leaving the catalyst pores cannot be framework distorting rigid coke molecules. Rather, is seems very likely that the initial species lost from the framework are adsorbed compounds or eliminated substituents on aromatic coke species.
This analysis also sheds considerable light on why the a minus b parameter is such a powerful descriptor for coke content and catalyst deactivation. The near identical behaviour of (a − b) and the 10 ring aspect ratio known to be sensitive to rigid aromatic molecules,48,49 means that (a − b) is insensitive to species that have little to do with catalyst deactivation, such as adsorbed volatile products, substituents on aromatic rings, and the presence of water. Water is not discussed here, but the insensitivity of (a − b) and the 10 ring aspect ratio can easily be demonstrated in hydration-dehydration experiments (not shown).
More detail can be gleaned from inspecting the occupancy of coke as shown in Fig. 5. Five extra framework dummy carbon atom sites (see Fig. S5–S7 of the ESI†) were used to fit the extra-framework electron density distribution in the framework. We initially point out that through this approach, it is possible to quantify the overall amount of coke as well as the amount of coke in specific locations. Typically, the total coke from the electron density agrees very well with coke from thermogravimetric analysis (not carried out here due to the operando nature of the experiment and the very small amount of catalyst in the capillary reactor). When first reduction of total coke is clearly related to reduced coke occupancies in four out of five locations in the framework (C1, C2, C4, and C5) (Fig. 5). At ∼575 °C, the occupancies of C2, C4, and C5 have been reduced to zero. At the same temperature, the occupancy of C3 starts to be reduced, clearly delayed relative to the other dummy positions. The occupancy at C1 continues to be monotonously decreased. At approximately 620 °C, the occupancies are negligible at all dummy atom positions (the non-zero final value for the occupancy at C1 and total coke is an artefact).
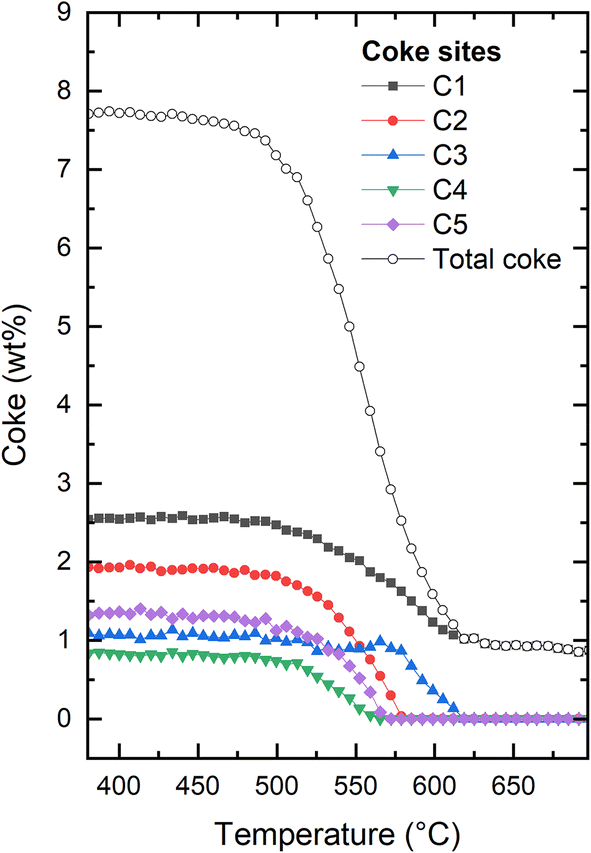 | ||
| Fig. 5 Evolution of the total coke and the individual coke occupancies within the H-ZSM-5 framework at increasing temperature for H-ZSM-5. The figure illustrating the full evolution of the experiment can be found in the ESI (Fig. S3†). Details regarding the exact placements of the coke dummy atoms can be found in Fig. S5–S7 of the ESI.† | ||
It is tempting to try to relate the different behaviour seen for the dummy atoms to their positions within the framework. Clearly, the onset of reduction in occupancy at C3 occurs at higher temperature than the other four positions, and the occupancy of C1 reaches its final stable value at the same temperature as C3, which is higher than that seen for C2, C4, and C5. However, it should be kept in mind that the positions of the dummy atoms were chosen to best match the electron density obtained from Fourier difference analysis of a fully coked catalyst, which does not necessarily lead to structurally distinct sitings. Also, this means that there is a risk that the locations chosen become increasingly less relevant as the coke is gradually removed. Finally, although individually refined, the occupancies are to an unknown extent coupled, as reduction in the occupancy of one site will lead to an increase in the others, for a given coke content. Thus, there is a clear risk of data over-interpretation in the following. Nevertheless, it would be an omission not to comment on this.
We note that C1 is placed in the middle of straight channel that runs along b-direction. C2 is placed close to the center of the channel intersection, in the most spacious region of the pore structure. C3 is also placed in the intersection, but very close to the wall of the straight channel, away from where the zig zag channels connect to the intersection. The distance between C3 and the nearest framework oxygen is ∼3.0 Å. In contrast, it ranges from 3.6–3.9 Å for the other dummy sites. C4 and C5 definitely describe the coke located in the zig zag channels, but are placed approximately in the 10-rings connecting the straight and zig zag channels. See Fig. 3 and further detail in Fig. S5–S7.† We speculate that the high temperature of onset of reduction seen for C3 is due to a tight interaction with the zeolite. Following this, the late depletion of C1 and C3 might then indicate a slower coke removal from the straight channel relative to the zig zag channel and intersections. Indeed, Weckhuysen and co-workers have shown that the straight channels favour the formation of internal coke, whereas the zig zag channels favour production of aromatics that grow and migrate to form external coke.50 Obviously, external coke is not detectable by our XRD methods employed here.
Finally, we point out that we have conducted regeneration under identical conditions on other H-ZSM-5 catalysts with different Si/Al ratio and different textural characteristics. Despite these differences in the characteristics, the overall coke removal profiles were very similar (not shown here). Based on our results, we posit that the framework topology dictates the mode of coke oxidation. With this work, we have described, in unprecedented detail, how a typical H-ZSM-5 network will be regenerated under TPO conditions. Further similar studies in other frameworks are expected to shed light on the structural characteristics that can affect the framework regeneration.
Conclusions
We have closely monitored the structural rearrangements taking place during the regeneration of an H-ZSM-5 catalyst, which has been coked during the conversion of methanol to gasoline/hydrocarbons. By combining parametric Rietveld refinements and calculation of the extra-framework electronic density by difference Fourier maps analysis, we could reveal the evolution of the coke species, monitor the regeneration of the catalyst, and study the geometrical response of different framework domains as the solid coke species combust and abandon their preferential residing sites. A small amount of adsorbed molecular species and alkyl side chains are leaving the zeolite pores first, followed by the actual oxidation products of the majority of the coke. During the oxidation process, the straight 10-ring channels revert from a highly distorted oval shape, induced by aromatic coke, to a more circular shape. Likewise, the a minus b parameter is sensitive specifically to the presence of rigid aromatic species, and thus functions as a deactivation descriptor that is largely insensitive to thermal effects and the presence of smaller molecules. In contrast, the unit cell volume displays more complex changes during temperature programmed oxidation. This work reveals the powerful nature of crystallographic analysis, that combined with the advances on data collection and processing can allow identification of specific coke locations within a three dimensional zeolite framework and provide new insight into the industrially highly important regeneration step in the MTH process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge ESRF for granting beamtime at SNBL (BM01). This work is partially funded by the European Industrial Doctorate Project “ZeoMorph” (Grant Agreement No. 606965) and the Research Council of Norway via the TomoCAT researcher project (project no. 301619).References
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249–259 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS.
- M. Stöcker, Microporous Mesoporous Mater., 1999, 29, 3–48 CrossRef.
- S. Svelle, U. Olsbye, F. Joensen and M. Bjørgen, J. Phys. Chem. C, 2007, 111(49), 17981–17984 CrossRef CAS.
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K. P. Lillerud, S. Kolboe and M. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS.
- H. Schulz, Catal. Today, 2010, 154, 183–194 CrossRef CAS.
- E. Borodina, H. S. Harun Kamaluddin, F. Meirer, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, J. Ruiz-Martinez and B. M. Weckhuysen, ACS Catal., 2017, 7(8), 5268–5281 CrossRef CAS.
- E. Borodina, F. Meirer, I. Lezcano-González, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, J. Ruiz-Martinez and B. M. Weckhuysen, ACS Catal., 2015, 5(2), 992–1003 CrossRef CAS.
- D. Rojo-Gama, M. Signorile, F. Bonino, S. Bordiga, U. Olsbye, K. P. Lillerud, P. Beato and S. Svelle, J. Catal., 2017, 351, 33–48 CrossRef CAS.
- D. Rojo-Gama, S. Etemadi, E. Kirby, K. P. Lillerud, P. Beato, S. Svelle and U. Olsbye, Faraday Discuss., 2017, 197, 421–446 RSC.
- D. Verboekend and J. Perez-Ramirez, Catal. Sci. Technol., 2011, 1(6), 879–890 RSC.
- J. Pérez-Ramírez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37(11), 2530–2542 RSC.
- F. L. Bleken, K. Barbera, F. Bonino, U. Olsbye, K. P. Lillerud, S. Bordiga, P. Beato, T. V. W. Janssens and S. Svelle, J. Catal., 2013, 307, 62–73 CrossRef CAS.
- M. Westgård Erichsen, S. Svelle and U. Olsbye, Catal. Today, 2013, 215, 216–223 CrossRef.
- M. Mortén, Ł. Mentel, A. Lazzarini, I. A. Pankin, C. Lamberti, S. Bordiga, V. Crocellà, S. Svelle, K. P. Lillerud and U. Olsbye, ChemPhysChem, 2018, 19(4), 484–495 CrossRef PubMed.
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155–7176 RSC.
- T. V. W. Janssens, J. Catal., 2009, 264, 130–137 CrossRef CAS.
- T. V. W. Janssens, S. Svelle and U. Olsbye, J. Catal., 2013, 308, 122–130 CrossRef CAS.
- A. Devaraj, M. Vijayakumar, J. Bao, M. F. Guo, M. A. Derewinski, Z. Xu, M. J. Gray, S. Prodinger and K. K. Ramasamy, Sci. Rep., 2016, 6, 37586 CrossRef CAS.
- A. T. Aguayo, A. G. Gayubo, A. Atutxa, M. Olazar and J. Bilbao, J. Chem. Technol. Biotechnol., 1999, 74(11), 1082–1088 CrossRef CAS.
- A. de Lucas, P. Cañizares and A. Durán, Appl. Catal., A, 2001, 206(1), 87–93 CrossRef CAS.
- D. M. Bibby, N. B. Milestone, J. E. Patterson and L. P. Aldridge, J. Catal., 1986, 97(2), 493–502 CrossRef CAS.
- M. Castilla, A. G. Gayubo, A. T. Aguayo, J. M. Arandes and J. Bilbao, Ind. Eng. Chem. Res., 1998, 37(6), 2383–2390 CrossRef CAS.
- P. L. Benito, A. T. Aguayo, A. G. Gayubo and J. Bilbao, Ind. Eng. Chem. Res., 1996, 35(7), 2177–2182 CrossRef CAS.
- D. M. Bibby, R. F. Howe and G. D. McLellan, Appl. Catal., A, 1992, 93(1), 1–34 CrossRef CAS.
- Z. Wan, G. K. Li, C. Wang, H. Yang and D. Zhang, Appl. Catal., A, 2018, 549, 141–151 CrossRef CAS.
- S.-J. Jong, A. R. Pradhan, J.-F. Wu, T.-C. Tsai and S.-B. Liu, J. Catal., 1998, 174(2), 210–218 CrossRef CAS.
- D. Chen, K. Moljord and A. Holmen, Microporous Mesoporous Mater., 2012, 164, 239–250 CrossRef CAS.
- H. Van Koningsveld, Acta Crystallogr., Sect. B: Struct. Sci., 1990, B46, 731–735 CrossRef CAS.
- J. F. Haw and D. M. Marcus, Top. Catal., 2005, 34, 41–48 CrossRef CAS.
- D. Rojo-Gama, M. Nielsen, D. S. Wragg, M. Dyballa, J. Holzinger, H. Falsig, L. F. Lundegaard, P. Beato, R. Y. Brogaard, K. P. Lillerud, U. Olsbye and S. Svelle, ACS Catal., 2017, 7, 8235–8246 CrossRef CAS.
- D. Rojo-Gama, L. Mentel, G. N. Kalantzopoulos, D. K. Pappas, I. Dovgaliuk, U. Olsbye, K. P. Lillerud, P. Beato, L. F. Lundegaard, D. S. Wragg and S. Svelle, J. Phys. Chem. Lett., 2018, 9, 1324–1328 CrossRef CAS PubMed.
- G. N. Kalantzopoulos, F. Lundvall, A. Lind, B. Arstad, D. Chernyshov, H. Fjellvåg and D. S. Wragg, Catal., Struct. React., 2017, 3, 79–88 CAS.
- G. N. Kalantzopoulos, F. Lundvall, S. Checchia, A. Lind, D. S. Wragg, H. Fjellvåg and B. Arstad, ChemPhysChem, 2018, 19(4), 519–528 CrossRef CAS.
- G. N. Kalantzopoulos, F. Lundvall, K. Thorshaug, A. Lind, P. Vajeeston, I. Dovgaliuk, B. Arstad, D. S. Wragg and H. Fjellvåg, Chem. Mater., 2020, 32(4), 1495–1505 CrossRef CAS.
- M. Bjørgen, F. Joensen, M. Spangsberg Holm, U. Olsbye, K.-P. Lillerud and S. Svelle, Appl. Catal., A, 2008, 345, 43–50 CrossRef.
- V. Dyadkin, P. Pattison, V. Dmitriev and D. Chernyshov, J. Synchrotron Radiat., 2016, 23, 825–829 CrossRef CAS.
- J. Kieffer and D. Karkoulis, J. Phys.: Conf. Ser., 2013, 425, 202012 CrossRef.
- D. S. Wragg, F. L. Bleken, M. G. O'Brien, M. Di Michiel, H. Fjellvag and U. Olsbye, Phys. Chem. Chem. Phys., 2013, 15, 8662–8671 RSC.
- G. W. Stinton and J. S. Evans, J. Appl. Crystallogr., 2007, 40, 87 CrossRef CAS.
- A. Coelho, J. Appl. Crystallogr., 2018, 51(1), 210–218 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- A. G. Alvarez, H. Viturro and R. D. Bonetto, Mater. Chem. Phys., 1992, 32, 135–140 CrossRef CAS.
- D. S. Wragg, G. N. Kalantzopoulos, D. K. Pappas, I. Pinilla-Herrero, D. Rojo-Gama, E. Redekop, M. Di Michiel, P. Beato, L. F. Lundegaard and S. Svelle, J. Catal., 2021, 401, 1–6 CrossRef CAS.
- S. Sen, R. R. Wusirika and R. E. Youngman, Microporous Mesoporous Mater., 2006, 87(3), 217–223 CrossRef CAS.
- B. A. Marinkovic, P. M. Jardim, A. Saavedra, L. Y. Lau, C. Baehtz, R. R. de Avillez and F. Rizzo, Microporous Mesoporous Mater., 2004, 71, 117–124 CrossRef CAS.
- H. Xiong, Z. Liu, X. Chen, H. Wang, W. Qian, C. Zhang, A. Zheng and F. Wei, Science, 2022, 376, 491–496 CrossRef CAS PubMed.
- B. Shen, X. Chen, D. Cai, H. Xiong, X. Liu, C. Meng, Y. Han and F. Wei, Adv. Mater., 2020, 32, 1906103 CrossRef CAS.
- D. Fu, O. van der Heijden, K. Stanciakova, J. E. Schmidt and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15502–15506 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt02845j |
| This journal is © The Royal Society of Chemistry 2022 |