
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in applying C–H functionalization and naturally sourced building blocks in organic semiconductor synthesis
Liwen
Xing
a and
Christine K.
Luscombe†
 *b
*b
aMolecular Engineering & Sciences Institute, University of Washington, Seattle, WA 98195, USA
bDepartment of Material Science & Engineering, University of Washington, Seattle, WA 98195, USA
First published on 15th November 2021
Abstract
Organic electronics is a rising field, with novel applications including but not limited to stretchable solar cells, flexible display screens, and biosensors. The high performance of these organic electronics is enabled by the outstanding optoelectronic and thermomechanical features of organic semiconducting materials. However, the production of the promising organic semiconducting materials at industrial scales has not yet become feasible, due to huge energy and capital costs in the large-scale synthesis as well as the potential damage to the environment and human health caused by vast hazardous chemical waste released. This review summarizes recent research advances in improving the environmental friendliness of the organic semiconducting material synthesis by appying atom economical C–H functionalization-based synthetic routes, minimizing hazardous chemical waste, lowering the energy consumption, and employing safe and abundant chemicals including naturally sourced semiconducting building blocks. This review showcases the remarkable progress that has been made towards the environmentally friendly organic semiconductor synthesis and provides insight for researchers developing green synthetic strategies and organic semiconductor building blocks in the future.
1. Introduction
Organic semiconducting materials (OSMs) are promising materials for organic electronics in many applications, such as photovoltaics (OPVs),1,2 light-emitting diodes (LEDs),3–5 organic (OFETs),6–9 and biomedical electronic devices.10,11 They are intrinsically more flexible and stretchable compared to inorganic semiconducting materials such as silicon. In addition, their solution processibility makes them accessible to inexpensive and efficient manufacturing processes such as roll-to-roll printing, while their biocompatibility allows them to be applied as implantable medical devices in living organisms. The various chemical structures of the OSMs enable their wide range of electronic and optical tunability.Despite these advantages of OSMs, there are some obstacles that hinder the large-scale manufacture and commercial viability of the OSMs. Besides the high material costs, one severe obstacle is the amplified negative environmental impacts associated with the conventional synthetic methods of OSMs in industrial scales, for example, large amounts of chemicals, energy, money, and time wasted by lengthy synthetic routes, energy-intensive conditions employed, toxic and hazardous reagents used. Scheme 1 shows a few examples of OSMs and their conventional synthetic routes (Scheme 1).12–15
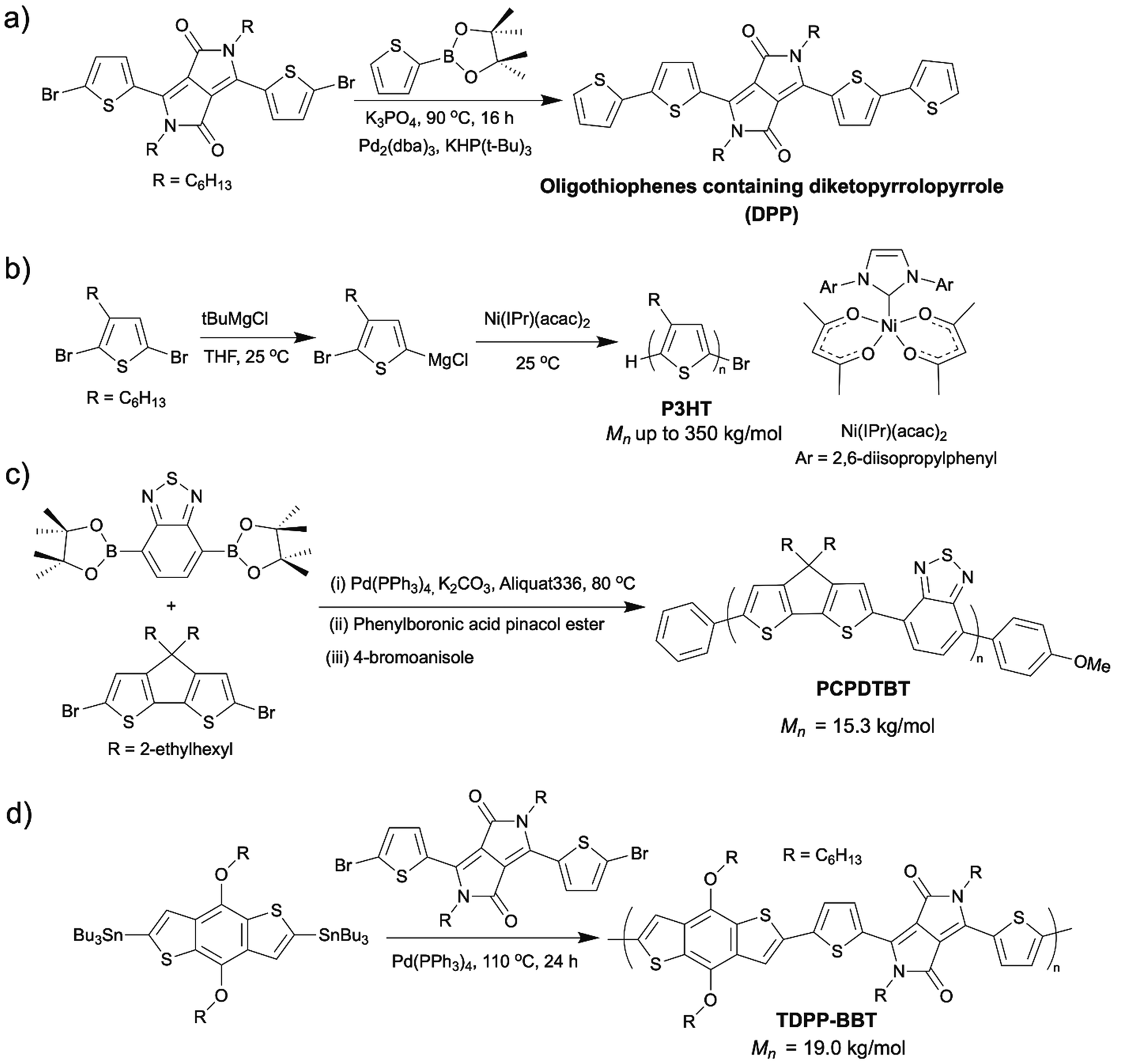 | ||
| Scheme 1 (a) Synthesis of diketopyrrolopyrrole (DPP) containing oligothiophenes via Suzuki coupling.12 (b) Synthesis of P3HT via Kumada catalyst transfer polycondensation (KCTP).13 (c) Synthesis of poly[4,4-bis(2-ethylhexyl)-4Hcyclopenta[2,1-b;3,4-b′]dithiophene-2,6-diyl-alt-2,1,3-benzothiadiazole-4,7-diyl] (PCPDTBT) via Suzuki coupling.14 (d) Synthesis of DPP containing copolymers (TDPP–BBT) via Stille coupling.15 | ||
Thiophene and its derivatives are some of the most common building blocks in OSMs due to their excellent optical and electronic properties as well as their high thermal stability.16,17 Poly(3-hexylthiophene) (P3HT) is a ubiquitous OSM that has been widely applied as an electron donor in OPV devices.18,19 However, the current production process of thiophene is not environmentally friendly. It is synthesized via a vaper phase reaction that occurs at an exceedingly high temperature (∼500 °C), which is highly energy-intensive and imposes safety issues.20 Moreover, thiophene itself is toxic. Its release to the environment can cause long lasting harmful effects on to aquatic life.21–23 Similarly, other common building blocks in OSMs, including furan, azoles, and other cyclic aromatic structures, also have certain levels of toxicity to living organisms and are prepared by harmful industrial processes,22–25 which negatively impacts the environment.
The conventional synthetic methods for OSMs, which include Suzuki, Kumada, Negishi, and Stille couplings, are often multi-step processes which involve pre-functionalization of the starting materials (Scheme 1). The pre-functionalization steps include halogenation, boronation, stannylation, and other metalation reactions depending on the type of coupling. The long synthetic process of OSMs results in waste of time and money and generation of a large amount of hazardous chemical waste and toxic byproducts, particularly the organotin byproducts generated from Stille coupling are virulent to humans.26–28 Additionally, the use of scarce and toxic metal catalysts is environmentally unsustainable.
Many scientific endeavors have been made to improve the environmental impact of OSMs, including shortening the synthetic routes by adopting C–H functionalization, applying green solvents, replacing the toxic and scarce metal catalysts with earth abundant (first row) transition metal catalysts, reducing the energy consumption by performing the synthesis under ambient conditions, as well as utilizing naturally sourced building blocks.29–43 This review highlights the progress on enhancing the environmental friendliness of synthetic protocols, consisting of synthetic routes, reagents, reaction conditions, and building blocks of OSMs, made in the past four years to facilitate the large-scale production and commercialization of OSMs. Specifically, the purification and processing of OSMs are outside the scope of this review.
2. Recent advances in developing alternative synthetic methods
2.1 Atom-economical syntheses – C–H functionalization
The concept of atom economy was first proposed by Trost in Science in 1991.44 The atom economy of a chemical process is measured by the ratio of mass of the desired product to the total mass of all the products generated, including byproducts. The lower the weight of byproduct(s) a chemical process generates, the more atom-economical the process is. The optimal atom economy is 100%. Reducing the number of steps and avoiding using large functional groups in a synthetic process are effective ways to decrease the generation of wasted byproducts. C–H activation on sp2 carbons of aromatic rings has become a promising tool to synthesize OSMs, in which the C–H bonds are activated, and subsequently C–C bonds are formed in situ. The development of aryl–aryl coupling via C–H activation was first driven by pharmaceutical industry, because biaryl structures are very common in many drug compounds. Direct arylation and oxidative C–H/C–H coupling are the two C–H activation involved synthetic routes for OSMs as green alternatives to the conventional synthetic methods, such as Suzuki and Stille couplings, due to their high atom economy (Scheme 2).29–43 For instance, the atom economy of a Suzuki coupling shown in Scheme 1a is calculated to be 60%. If direct arylation and oxidative C–H/C–H coupling were used instead to synthesize the same organic semiconducting product in Scheme 1a, respectively, then the values of atom economy would be 80% for direct arylation and 99% for oxidative C–H/C–H coupling.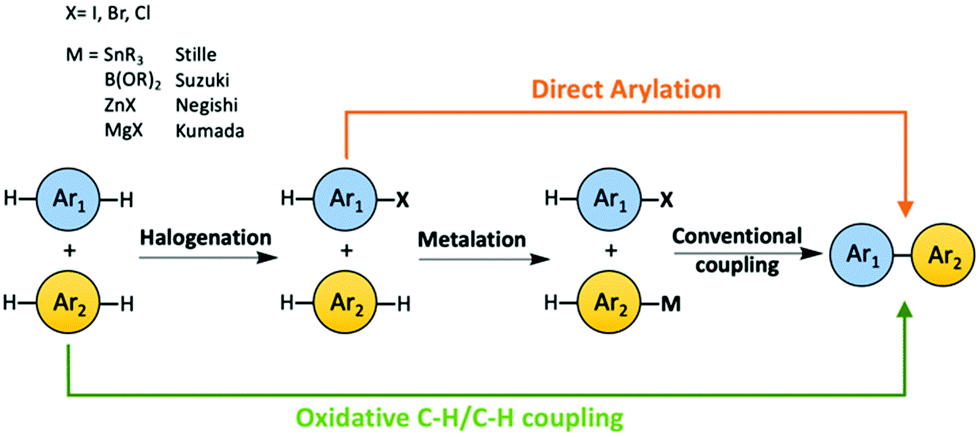 | ||
| Scheme 2 Comparison among the conventional methods, direct arylation and oxidative C–H/C–H coupling to synthesize OSMs. | ||
In the direct arylation reaction, only one of the two coupling partners needs to be pre-halogenated, and the necessity of pre-metalation step of the other coupling partner is avoided. Direct arylation has been widely studied as a new method to synthesize OSMs, especially semiconducting polymers.35,36 Oxidative C–H/C–H coupling, also referred as C–H oxidative direct arylation, is a more ideal synthetic route for OSMs than direct arylation in terms of atom economy, because neither of the coupling partners needs to be pre-functionalized. The unfunctionalized C–H bonds on both coupling partners will be activated and form a C–C bond subsequently during the material synthesis. However, due to the inert nature of C–H bonds, it is more challenging for direct arylation and oxidative C–H/C–H coupling to obtain high-yielding coupling products with desired chemo- and regioselectivity compared to the conventional coupling methods.
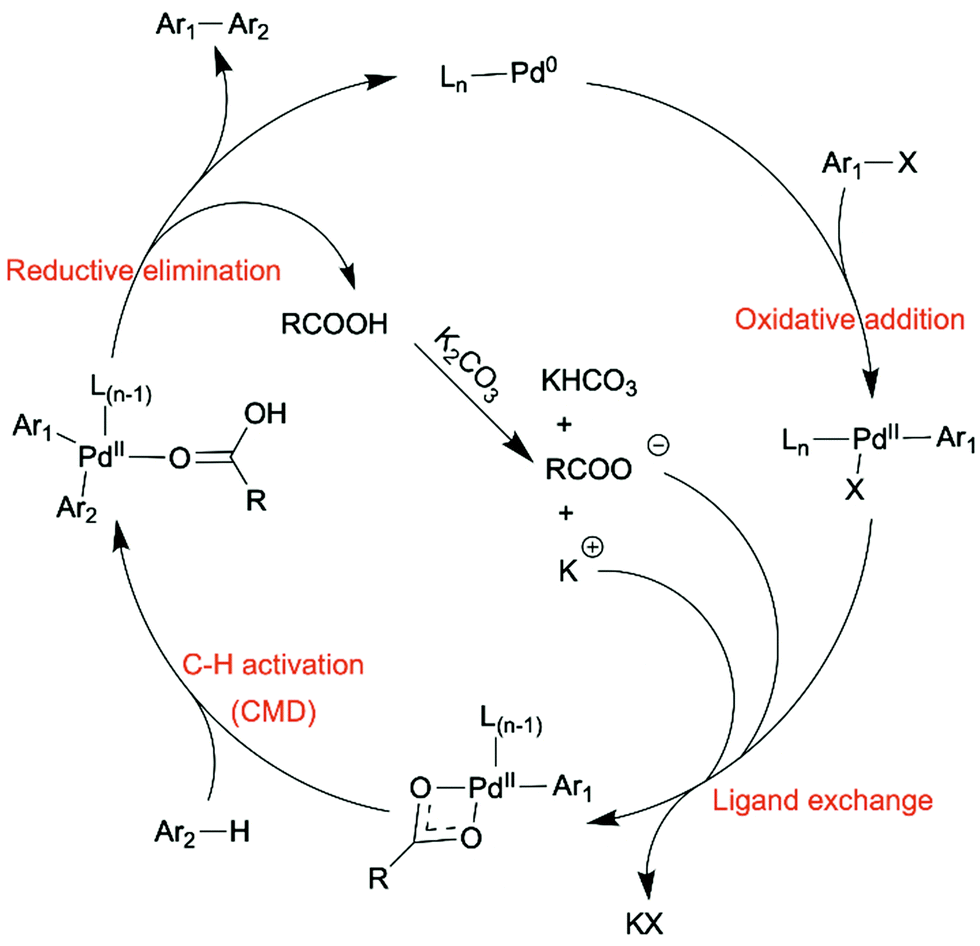 | ||
| Scheme 3 A plausible catalytic cycle of direct arylation. | ||
In the past few years, most studies using direct arylation to synthesize OSMs focused on synthesizing semiconducting polymers using direct arylation polymerization (DArP). The reports on the synthesis of small molecule organic semiconducting materials via direct arylation are less common. Therefore, we will devote a large segment of this section to illustrating some recent advances in semiconducting polymer synthesis by DArP, and briefly discuss about the syntheses of some small molecule OSMs using direct arylation at the end of this section.
DArP, as an emerging subject in the field of OSM synthesis, has been reviewed many times in the past few years.29–43 The semiconducting polymers synthesized via DArP are mostly linear homopolymers and alternating copolymers. When homopolymers are prepared using DArP, the monomer has a C–X (X = Cl, Br, I) bond and a C–H bond to proceed a head-to-tail polycondensation (Scheme 4, top). While, in DArP for alternating copolymers, one of the comonomers is required to possess two reactive C–X bonds and the other comonomer has two C–H bonds (Scheme 4, bottom).
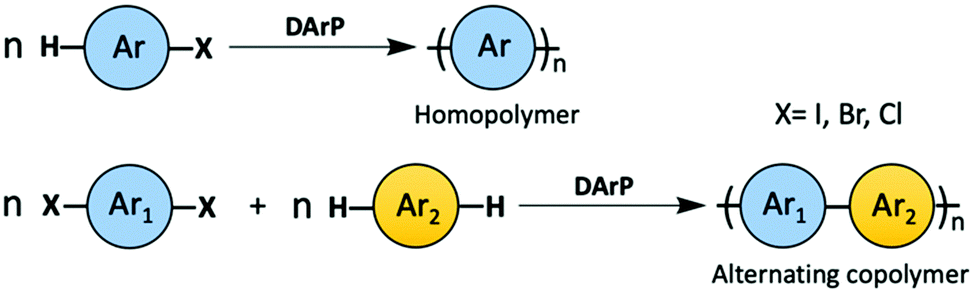 | ||
| Scheme 4 A general scheme of DArP to synthesize semiconducting homopolymer and alternating copolymer. | ||
Donor–acceptor (D–A) semiconducting copolymers are very attractive semiconducting materials as their backgrounds demonstrate a “push–pull” mechanism of electron transport. Moreover, these materials display lowered bandgaps, which enables high charge mobility as well as broader ranges for light absorption – useful properties in electronic devices.1,2 In order to acquire the D–A copolymers with high electronic device performance, the synthesized D–A copolymers need to have perfectly alternating donor and acceptor chemical structures and high molecular weights. Any homo-coupling defect on the polymer backbone limits the material's electronic performance.46,47 DArP has emerged as a promising and greener synthetic strategy for defect-free D–A copolymers, in contrast to the conventional coupling methods.48–50
Synthesizing D–A copolymers via conventional coupling methods can be difficult because the pre-functionalization to prepare the required electron-poor comonomers is challenging due to either their unresponsiveness to electrophilic substitution or their decomposition during the functionalization reactions.51–53 In this case, DArP, where C–H activation of one of the comonomers is a crucial part, appears to be a favorable synthetic method for D–A copolymers. A collaborative work performed by Scott, Luscombe, Marder, and Blakey groups on synthesizing 5,6-dicyano[2,1,3]benzothiadiazole (DCBT)-containing D–A copolymers via DArP was reported in 2018.48 In this work, DCBT was incorporated in D–A copolymers as an electron-poor unit by C–H arylation on DCBT for the first time, and three high molecular weight and perfectly alternating D–A polymers were successfully prepared after condition optimization (Scheme 5a). All three resultant polymers were characterized by broad light absorption and low LUMO levels, and they were proven to be n-type materials for OFET applications with an average mobility of 1.2 × 10−3 cm2 V−1 s−1. Recently, Ozawa et al. also reported some syntheses of D–A copolymers with well-defined structures using DArP.49,50 They prepared a D–A copolymer using 1,2-dithienylethene (DTE) (donor) and dibromoisoindigo (acceptor) as comonomers and a catalytic system consisting of Pd2(dba)3·CHCl3, P(2-MeOC6H4)3, pivalic acid, and Cs2CO3.49 The resulted D–A copolymer exhibited a high molecular weight (Mn was up to 44.9 kg mol−1) and less than 0.1% homocoupling defect. Moreover, no branching defect was observed, which is important for conjugated polymers containing non-alkylated thiophene as branching and crosslinking, which are results of C–H activation at β-position of thiophene moieties, can lead to the formation of insoluble materials. In addition, the charge transfer properties in OFETs of the D–A copolymer synthesized from DArP was demonstrated to be superior to those of the same D–A copolymer synthesized from Stille coupling. This is likely because the DArP product possessed less structural defect than Stille coupling product and was free of residual tin-containing impurities which may deteriorate the device performance.54 In another report published by Ozawa et al., they synthesized D–A copolymers via DArP using unsubstituted 2,2′-bithiophene as the donor comonomer without generating insoluble materials (Scheme 5b).50 In this study, they applied a mixed ligand catalyst (P(2-MeOC6H4)3 and tetramethylethylenediamine (TMEDA) as ligands and Pd2(dba)3·CHCl3 as palladium source) to DArP and successfully suppressed the formation of insoluble materials resulting from branching side reactions. The resulted D–A copolymers displayed high molecular weight (up to 88.1 kg mol−1), low content of homocoupling defects (up to 1.1%), as well as high device performance (power conversion efficiency up to 9.0%).
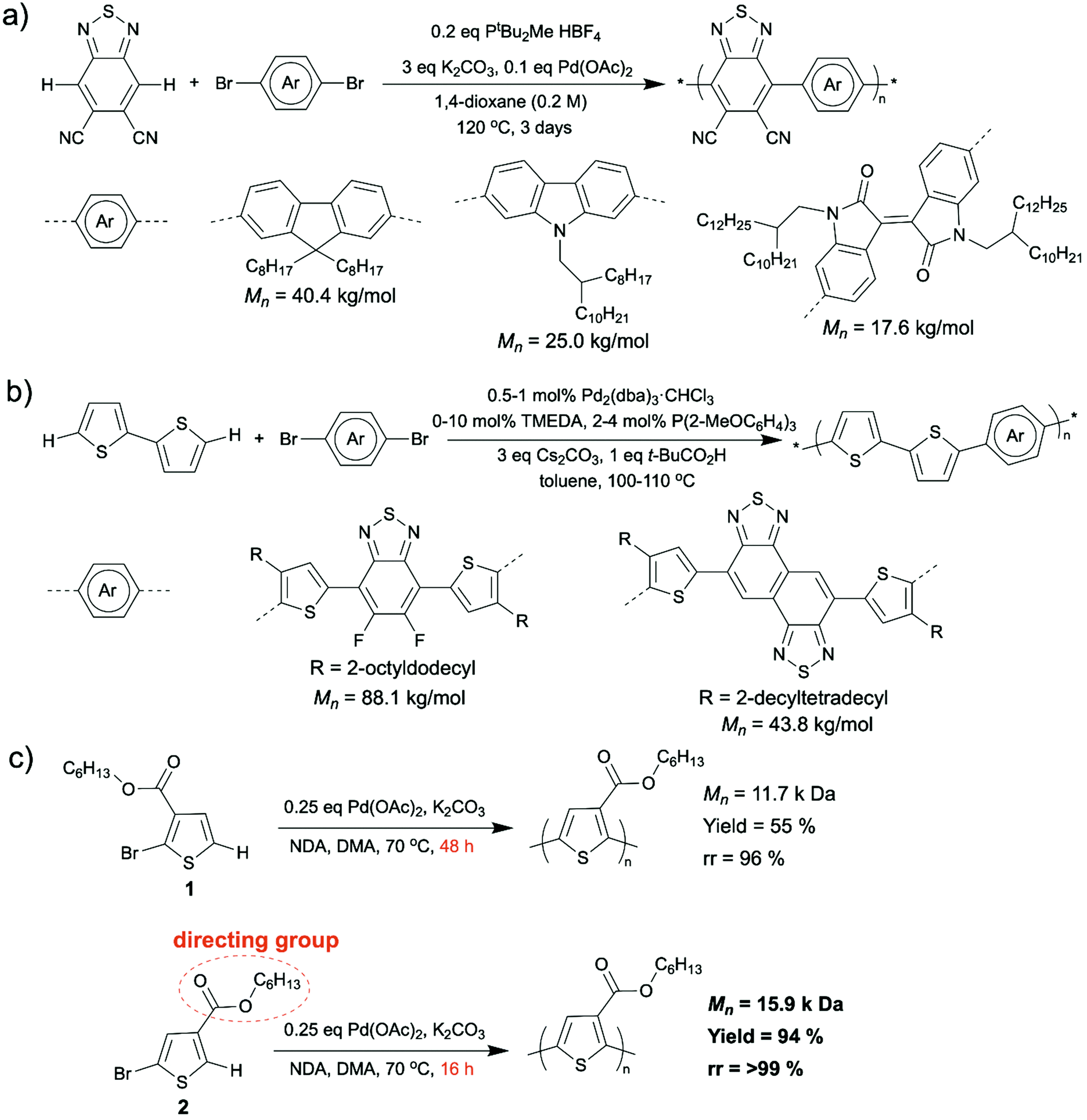 | ||
| Scheme 5 (a) Synthesis of DCBT containing conjugated D–A copolymers via DArP. (b) Synthesis of unsubstituted bithiophene containing D–A copolymers via DArP using a mixed ligand catalyst to suppress branching side reactions. (c) Synthesis of highly regioregular P3HET via DArP with the ester functionality as the directing group to promote regioregularity. | ||
The control of regioselectivity (head-to-tail) in homopolymer synthesis via DArP has always been a big challenge. Activating undesired C–H bonds in DArP can cause structural defects (head-to-head, tail-to-tail, and branching defects) in resultant homopolymers, which diminishes their electronic performance. Utilization of directing groups was demonstrated to be an effectual way to avoid the formation of structural defects during certain DArPs. Thompson et al. have recently uncovered that using an ester as a directing group installed directly on the thiophene monomer improves the regioregularity (head-to-tail coupling) of the resulted polymer.55 They installed a hexyl ester directing group adjacent to the C–H bond that was designated for activation on the 2-bromothiophene monomer 2 (Scheme 5c, bottom). Compared to the similar monomer 1 (Scheme 5c, top), where the ester group was installed adjacent to the bromine, the regioregularity of produced poly(3-hexyl ester thiophene-2,5-diyl) (P3HET) improved from 96% to >99%.56 In addition, the reaction time was reduced from 48 h to 16 h for a similar sized polymer.
DArP normally occurs using a polycondensation mechanism, which is not a controlled polymerization. Conjugated polymers prepared by a polycondensation mechanism will have a broad molecular weight distribution, and the molecular weight can be hard to reproduce. Our group recently achieved chain DArP to synthesize P3HT with a low dispersity.57 We applied a silver-carboxylate co-catalyst to promote the C–H activation step and PEPPSI–iPr as the palladium source. The association of the polymer chain with the large π-conjugated N-heterocyclic carbene (NHC) ligand on PEPPSI–iPr allowed chain walking of the palladium catalyst along the polymer backbone while adding monomers to the chain ends one by one (Scheme 6). This was also the first report where a dual-catalytic system was implemented in DArP.
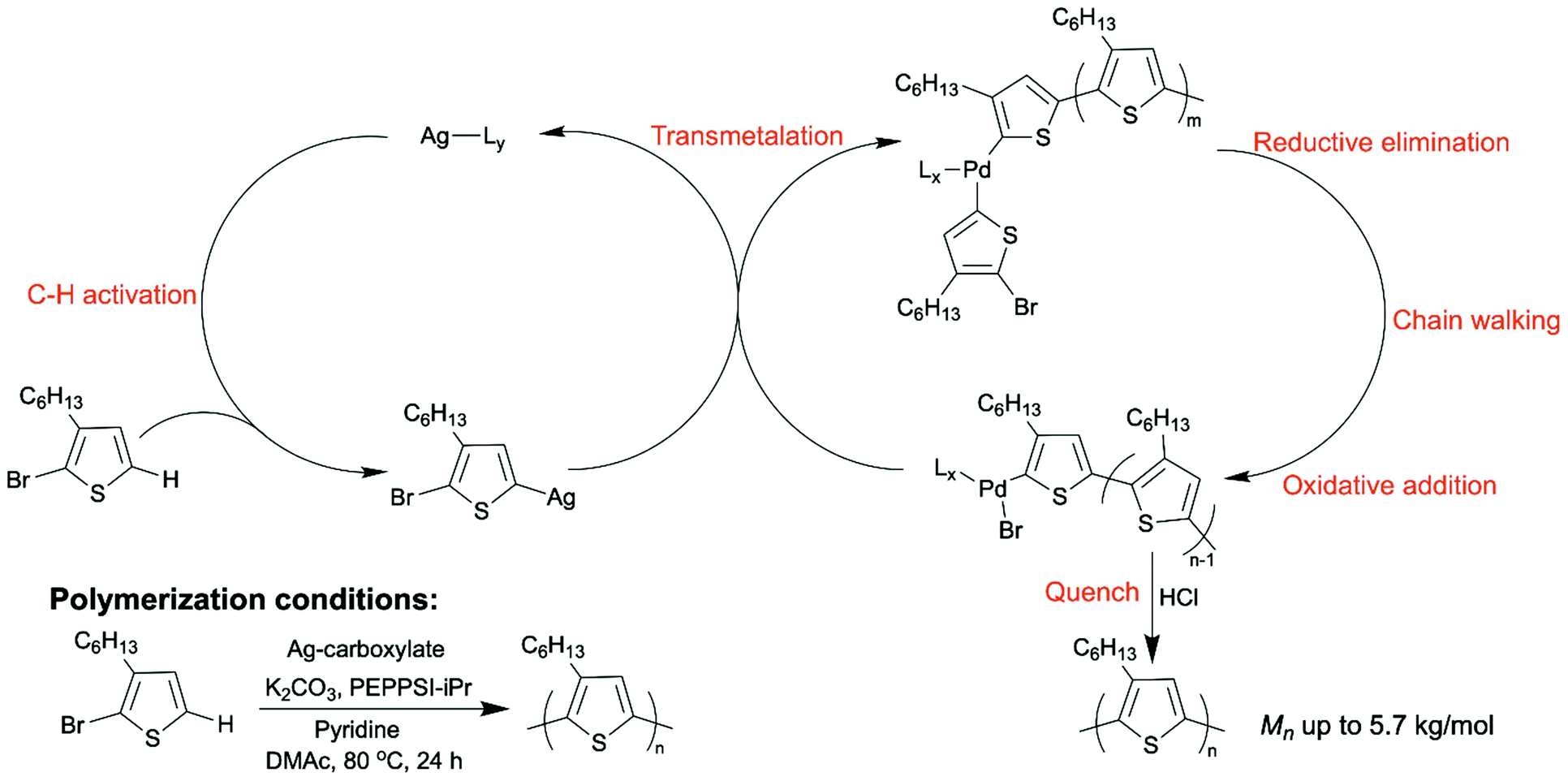 | ||
| Scheme 6 Dual catalytic DArP of P3HT to achieve chain-growth mechanism. | ||
In recent years, there have been reports on synthesis of small molecule OSMs using direct arylation. For example, Ching et al. successfully applied a two-step synthetic route, direct arylation followed by Knoevenagel condensation, to prepare a new A–D–A–D–A semiconducting small molecule based on benzothiadiazole and thiophene (Scheme 7a).58 Welch et al. published a personal account which summarized their achievements over the last few years in molecule design of organic dye-based non-fullerene acceptors for OPV applications using direct arylation.59 Direct arylation benefited their research significantly by enabling a facile synthetic access to various molecular structures and easy structural modifications. With this powerful tool, they synthesized isoindigo (ISI)- and diketopyrrolopyrrole (DPP)-based non-fullerene acceptors (Scheme 7b). Perylene diimide–diketopyrrolopyrrole–perylene diimide (PDI–DPP–PDI) was identified as the most promising dye-based framework, the OPV device made from which exhibited a PCE as high as ∼6%.60,61 Our group also reported small molecule organic luminophores with high photoluminescence quantum yields prepared by direct arylation, which will be detailed in the later section.62
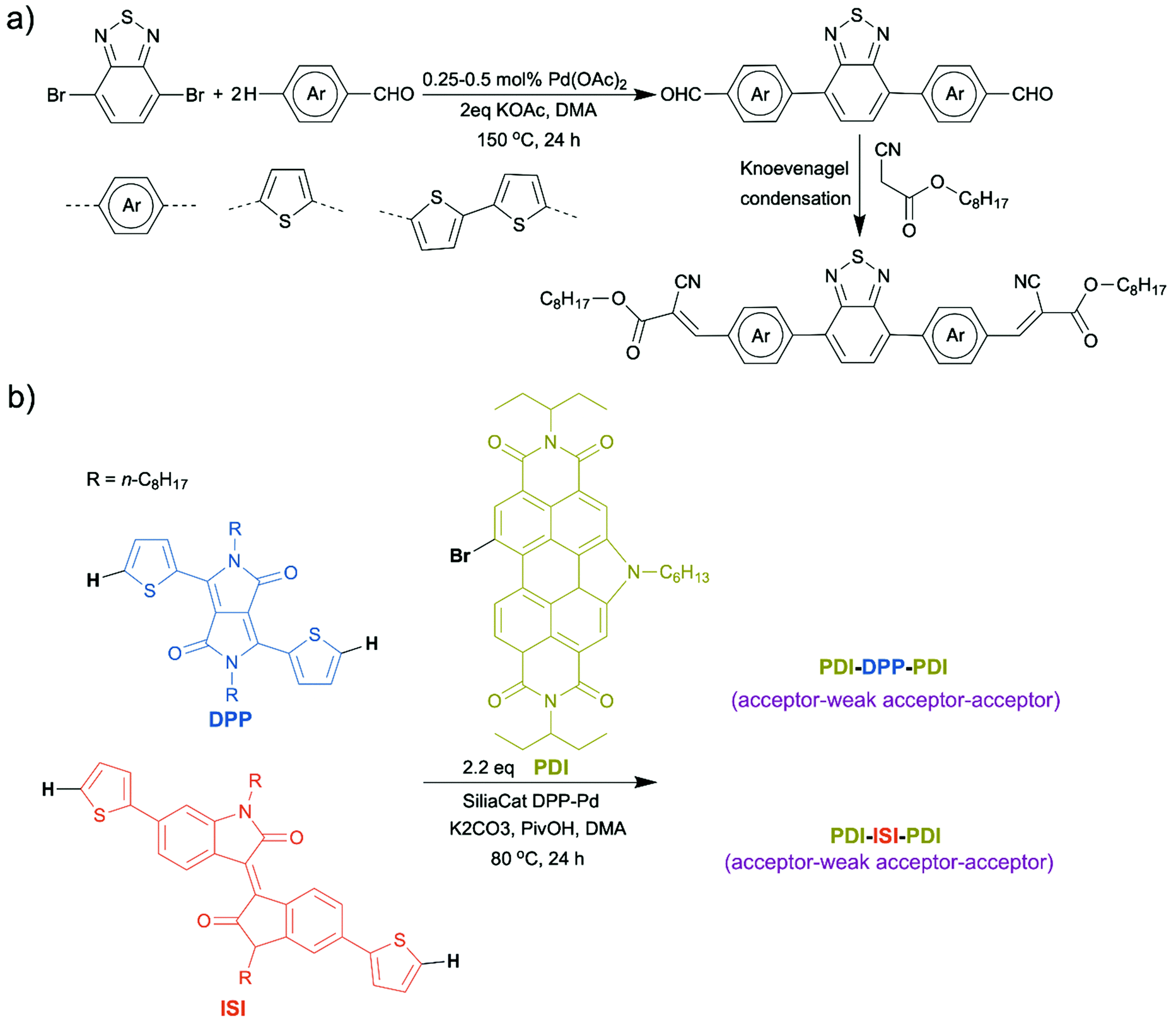 | ||
| Scheme 7 (a) Synthesis of small molecule OSMs via direct arylation. (b) Synthesis of isoindigo (ISI)- and diketopyrrolopyrrole (DPP)-based non-fullerene acceptors via direct arylation. | ||
 | ||
| Scheme 8 (a) A general synthetic scheme of D–A semiconducting copolymers via CDC. (b) Palladium/silver catalyzed CDC polymerization. (c) Synthesis of fluorinated benzotriazole containing D–A conjugated copolymers VIA CDC. (d) The CDC polymerizations of various D–A conjugated copolymers. | ||
Kanbara et al. conducted a palladium/silver dual catalyzed CDC polymerization (Scheme 8b).66,68 The D–A copolymer they synthesized, a copolymer of 2,2′,3,3′,5,5′,6,6′-octafluorobiphenyl and 3,3′-dihexyl-2,2′-bithiophene, was applied as a light emitting material in OLEDs. Three years later, Lu et al. expanded the monomer scope of Kanbara's palladium/silver catalytic CDC polymerization by preparing a series of fluorinated benzotriazole-based D–A conjugated copolymers (Scheme 8c).69 Chen and co-workers accomplished CDC polymerization of various D–A conjugated copolymers with a similar palladium catalytic system to Kanbara's (Scheme 8d).67 The highly alternating and regioregular structures of the copolymers were demonstrated by comparing the 1H NMR spectra of the same copolymers synthesized by Stille coupling, showing the robustness of the CDC method compared to the conventional method.
Our group also published a study on D–A copolymer synthesis via a gold/silver catalytic CDC in early 2019.65 In this study, we attempted to adapt a highly reactive and highly chemo-selective gold/silver catalyzed small molecule CDC reaction reported by Larrosa into a CDC polymerization.70 We used the same comonomers as Kanbara did, 2,2′,3,3′,5,5′,6,6′-octafluorobiphenyl and 3,3′-dihexyl-2,2′-bithiophene. The resulted copolymers showed low molecular weights (7.5 kg mol−1) and ∼70% degree of alternation, but we gained a great insight into the factors that determine the chemo-selectivity and readiness of chain propagation during the CDC polymerization, which were mentioned in the early discussion about the challenges in D–A copolymer synthesis via CDC.
Apart from poly(arylene)s, another type of promising semiconducting polymers which was successfully synthesized via oxidative C–H/C–H coupling are luminescent poly(arylene–vinylene)s (Scheme 9a).71–73 In 2016, the Kanbara group depicted the first oxidative C–H/C–H polymerization of a diethenyl aromatic monomer and an arene monomer, namely dehydrogenative direct alkenylation polycondensation, to synthesize several 1-(2-pyrimidinyl)pyrrole-based poly(arylene–vinylene)s with a rhodium catalyst (Scheme 9b, top).71 After that, they reported a palladium catalyzed dehydrogenative direct alkenylation polymerization of polyfluoroarylene-based poly(arylene–vinylene)s (Scheme 9b, bottom).72 In this report, they eliminated the necessity of installing the directing 2-pyrimidinyl group. This year, they published a synthesis of 1-(2-pyrimidinyl)pyrrole-based poly(arylene–vinylene)s from aromatic diynes by polyaddition via a cobalt-catalyzed hydroarylation of alkynes (Scheme 9c).73 Technically this does not fall into the oxidative C–H/C–H coupling category since it is an addition reaction instead of condensation, discussed here for its extraordinary atom-economy (no byproducts), enriching the toolbox of green synthesis for OSMs. Although the polymers that were synthesized via the polyaddition contained a small number of 1,1-vinylidene units (7%), which are problematic for using these materials in electronic devices, this cobalt-catalyzed hydrogenative polyaddition is still very appealing because it proceeded at near room temperature (30 °C), avoided using stoichiometric amounts of oxidants, and applied a first-row transition metal (cobalt) catalyst.
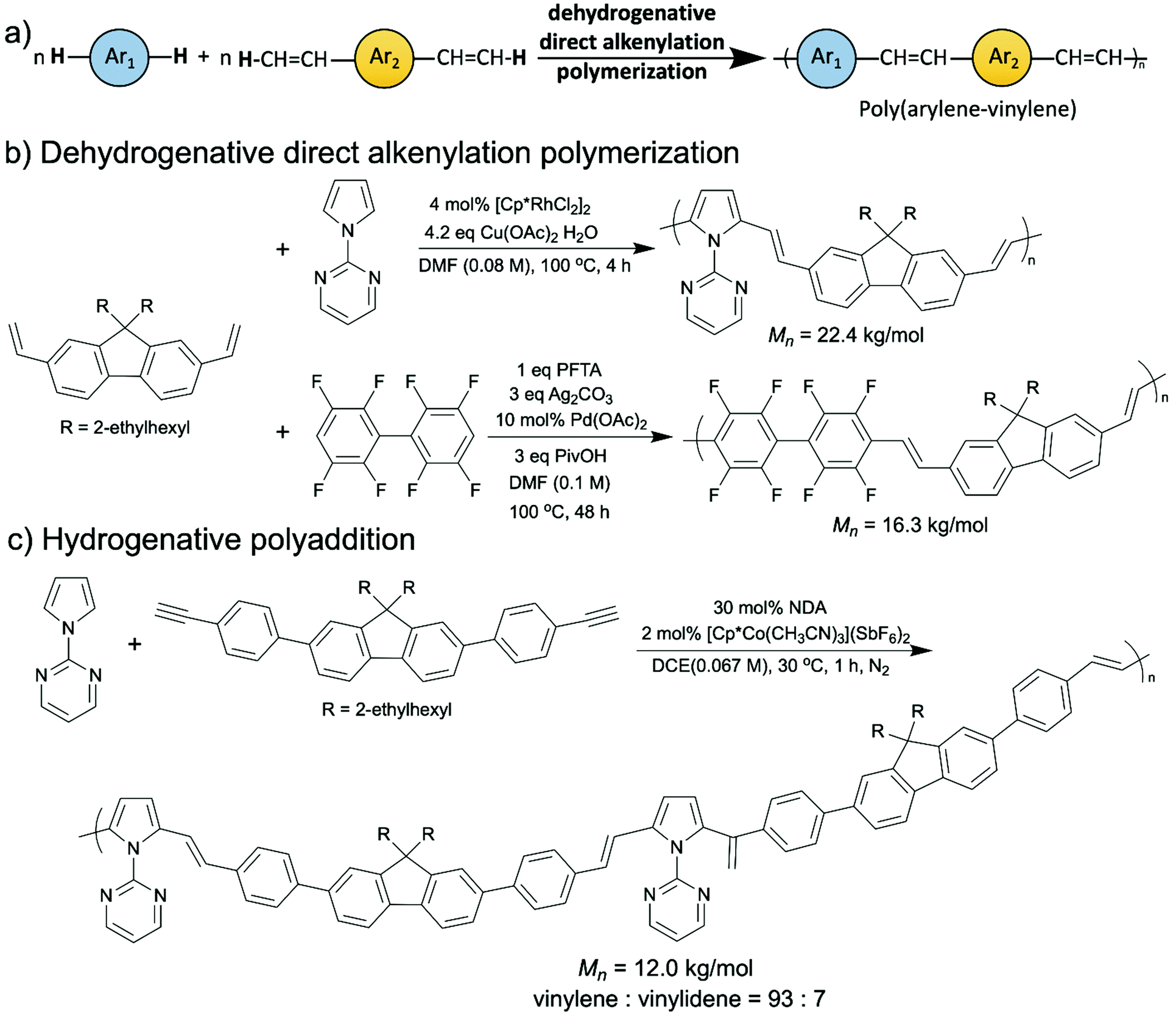 | ||
| Scheme 9 (a) A general scheme of dehydrogenative direct alkenylation polymerization to synthesize poly(arylene–vinylene)s. (b) Rhodium- and palladium catalyzed dehydrogenative direct alkenylation polymerizations to synthesize poly(arylene–vinylene)s. (c) Cobalt-catalyzed hydrogenative polyaddition to synthesize poly(arylene–vinylene)s. | ||
2.2 Energy-efficient conditions in atom-economical syntheses
Minimizing energy input in the synthetic process of OSMs is an important part of improving the environmental friendliness and scalability of OSM production. As we discussed before, most atom-economical synthetic methods (direct arylation and oxidative C–H/C–H coupling) for OSMs are carried out under inert atmosphere and at relatively elevated temperatures (∼60 °C to ∼120 °C). Conducting the synthetic reactions at room temperature can lessen the thermal energy that were put into the OSM production significantly. In addition, a mild reaction temperature can also lower the risk of dangerous accidents during manufacturing. The synthetic reactions that can tolerate the aerobic conditions can avoid the long and energy-intensive degassing processes to remove moisture and oxygen. Moreover, commercially available and cheap reagent-grade chemicals can be directly used for the moisture- and oxygen-tolerant reactions.The reports on room-temperature synthesis of OSMs are not very common compared to those on optimizing other aspects of OSM synthesis, such as additives, catalysts, and solvents. This is likely because when the C–H bond, which is less reactive than other organometallic functionalities, needs to be activated, the introduction of an external energy source, such as heat, or employment of highly reactive additives is usually required.74 The first attempt to synthesize OSMs by room-temperature DArP was reported by Thompson et al.75 They prepared P3HT using DArP at 20 °C and obtained a polymer with a molecular weight of 14 kg mol−1 but a low yield of only 9%. To our knowledge, the second attempt to achieve room-temperature DArP was made recently by our group. In this study, we synthesized a low molecular weight and branched semiconducting homopolymer from 5-iodo-1-octylindole monomer using room-temperature DArP as shown in Scheme 10.76 Notably, the mechanistic study indicated that this polymerization was governed by a light-mediated radical process, which was a significant discovery that can inspire future developments of mild reaction conditions for DArP. Also, the aforementioned polyaddition via a cobalt-catalyzed hydroarylation of alkynes reported by Kanbara group is another example of near room temperature synthesis of OSMs.73
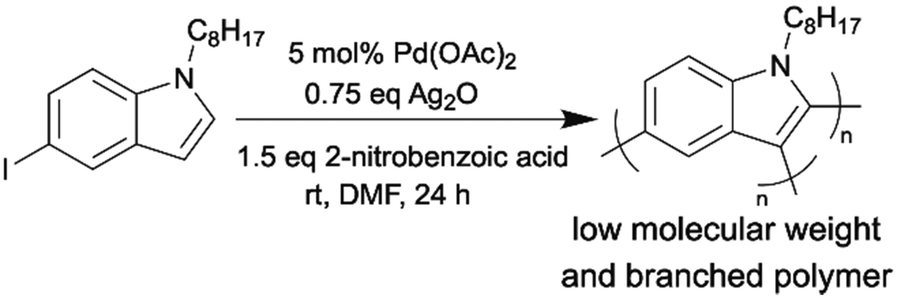 | ||
| Scheme 10 Room-temperature DArP of 5-iodo-1-octylindole monomer. | ||
Direct arylation and oxidative C–H/C–H coupling are usually performed under inert gas because most of their catalysts and generated intermediates are air and moisture sensitive.77,78 In recent years, Kanbara et al. discovered that refluxing the solvent (toluene, o-xylene, or DMF) during the polymerization allows certain DArP reactions to proceed under aerobic conditions.79,80 The aerobic DArP of 5-(2-ethylhexyl)thieno-[3,4-c]-pyrrole-4,6-dione (TPD) with 2,7-dibromo-9,9-dioctylfluorene was accomplished in the refluxed toluene with a molecular weight up to 176.8 kg mol−1.79 In their other report, a copolymer of 3,4-ethylenedioxythiophene (EDOT) and 2,7-dibromo-9,9-dioctylfluorene with a molecular weight of 54.2 kg mol−1 was successfully synthesized via DArP under aerobic conditions by refluxing the solvent DMF.80 The authors attributed this to efficient degassing of dissolved oxygen from the reaction mixture and prevention of resolubilizing oxygen.79,80 Aside from DArP, the Kanbara group also reported a few oxidative C–H/C–H polymerizations carried out under aerobic conditions. In the same paper where they achieved the initial oxidative C–H/C–H polymerization to synthesize D–A copolymers, they were able to conducted the same polymerization under aerobic atmosphere as well, obtaining a D–A copolymer with high molecular weight (23.2 kg mol−1) and only 2% homo-coupling defects.66 In the same year, their study of copper-catalyzed aerobic oxidative C–H/C–H homo-polymerization of dithiazole-based monomer was published, where dioxygen acted as the oxidant.81 Notably, a copper catalyst was applied in this polymerization, which made this polymerization protocol better than other palladium catalyzed ones in terms of environmental friendliness. This will be detailed in the Section 2.3.2.
2.3 Safe and abundant chemicals for atom-economical syntheses
Thompson and colleagues conducted a solvent optimization for the DArP of poly[2,5-bis(2hexyldecyloxy)phenylene-alt-(4,7-dithiophen-2-yl)benzo[c][1,2,5]thiazole] (PPDTBT) synthesis using the four green organic solvents aforementioned.90 Among these solvents, CPME gave preeminent results. The molecular weight of PPDTBT synthesized in CPME was measured to be 41 kg mol−1 by size exclusion chromatography (SEC), which surpassed the previously reported PPDTBT prepared under THF conditions (15 kg mol−1). In addition, they also performed DArP to synthesize P3HT in CPME, which provided a P3HT with the molecular weight of 12 kg mol−1 and regioregularity of 93%. Grisorio, et al. also investigated the possibility of using another sustainable solvent, anisole in DArP to synthesize P3HT.91 Anisole is a low-cost, non-toxic, biodegradable, and highly recommended organic solvent based on recent solvent selection guides.92 The best P3HT they obtained was endowed with the molecular weight of 17.2 kg mol−1 and regioregularity of 93%. Thompson and colleagues reported more syntheses of other semiconducting polymers via DArP performed in green solvents.93,94 They used anisole and CPME in the synthesis of diester functionalized bithiophene-containing polymers, poly[5,5′-bis(2-butyloctyl)-(2,2′-bithiophene)-4,4′dicarboxylate-alt-5,5′-2,2′-bithiophene] (PDCBT) (13.8 kg mol−1), poly[5,5′-bis(2-butyloctyl)-(2,2′-bithiophene)-4,4′-dicarboxylate-alt-2,5-[3,2-b]thienothiophene] (PDCTT) (26.4 kg mol−1), and poly[5,5′-bis(2-butyloctyl)-(2,2′-bithiophene)-4,4′-dicarboxylate-alt-5,5′-2,2′-bithiazole] (PDCBTz) (4.9 kg mol−1).93 They also synthesized a series of amide-functionalized semiconducting polymers using CPME.94
2-MeTHF is a popular green alternative to THF. Pappenfus et al. successfully scaled up P3HT synthesis to 10 g via DArP solvated by 2-MeTHF.95 The P3HT prepared in 2-MeTHF exhibited a molecular weight of 22.8 kg mol−1, high regioregularity, and zero β-branching defects. Recently, Thompson introduced a new, naturally sourced aromatic solvent, p-cymene, for DArP as a green alternative to toluene and xylenes.96p-Cymene is a side product generated in large quantities in citrus fruit processing.97 They demonstrated that p-cymene outperformed toluene, DMA, and THF in the synthesis of some D–A copolymers using DArP (Scheme 11).
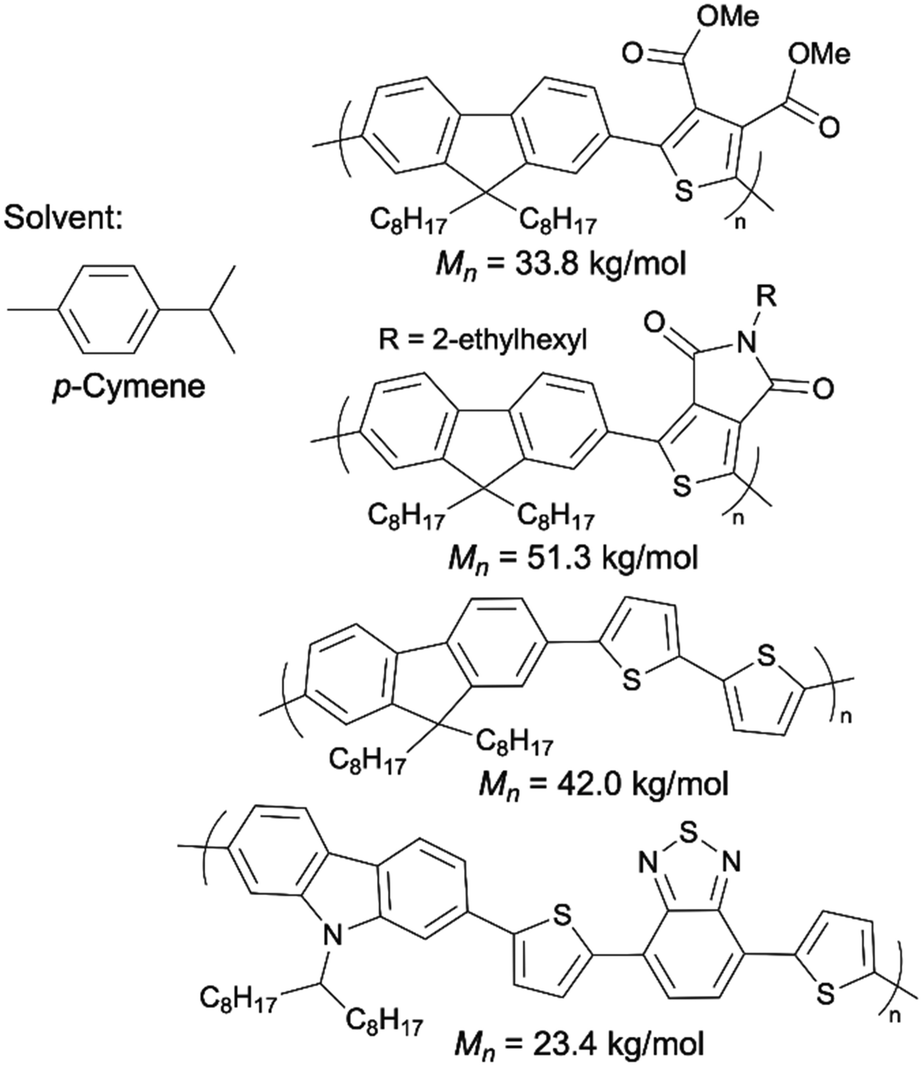 | ||
| Scheme 11 Semiconducting polymers synthesized by DArP with p-cymene as the solvent. | ||
Lately, there are also some exciting breakthroughs in OSM preparation in water. In 2017, Leclerc et al. developed water/toluene (1/1) biphasic DArP conditions for the synthesis of a wide range of semiconducting polymers (thienyl and phenyl-based) without reducing the polymer molecular weight and other important properties, which is the first example where water was used as a co-solvent for DArP.98 In their research, tetrabutylammonium chloride (TBAC) was added as phase transfer agent. Delightfully, their water/toluene biphasic DArP was also demonstrated to be air tolerant. In 2018, Yu et al. reported DArPs to synthesize water-soluble alkoxysulfonate- and carboxylic acid-functionalized poly(3,4-ethylenedioxythiophene)s (PEDOTs) in DMF and water, respectively.99 Tetrabutylammonium bromide (TBAB) was used a phase transfer agent in the water solvated DArP. The DArP conducted in water provided a PEDOTS polymer with molecular weight of 6.4 kg mol−1 and a yield of 80%. In 2020, Liu group reported an aqueous DArP to prepare water-soluble semiconducting polymers from ammonium bromide- and sulfonate-functionalized thiophene monomers, 6-(2-(2-bromothiophen-3-yl)ethoxy)hexyl trimethylammonium bromide and 4-(2-(2-bromothien-3-yl)ethoxy)butylsulfonate.100 The molecular weights were up to 10.0 kg mol−1.
Although the investigations into replacing noble transition-metals with environmentally benign first-row transition-metals in OSM synthesis emerged decades ago, challenges remain in first-row transition-metal catalyzed direct arylation and oxidative C–H/C–H coupling. Thus far, among the first-row transition-metals, only copper was successfully applied as a catalyst in DArP and oxidative C–H/C–H polymerization. However, the monomers in the copper-catalyzed C–H polymerizations are often limited to nitrogen-containing heterocycles, such as thiazole, oxazole, and imidazole. This is because the nitrogen atoms are good coordinating sites to the copper-center, and by coordinating to the copper-center, the energy barrier to activate the adjacent C–H bonds can be decreased.105 In this section, we will summarize all the copper catalyzed DArPs and oxidative C–H/C–H coupling polymerizations that have been published so far.
The first OSM synthesis via copper catalyzed oxidative C–H/C–H polymerization was reported by the You group in 2014. Various regioregular polybenzodiimidazoles (PBDIs) were prepared with the 20 mol% copper catalyst (Scheme 12a, top).106 The PBDIs they obtained exhibited high regioregularity and were blue emitters. Afterwards, at the beginning of 2018 Kanbara et al. published the second example of copper catalyzed oxidative C–H/C–H coupling polymerization using a dithiazole-based monomer with between 10–20 mol% catalyst loadings, which was mentioned in section 2.2 (Scheme 12a, bottom).81
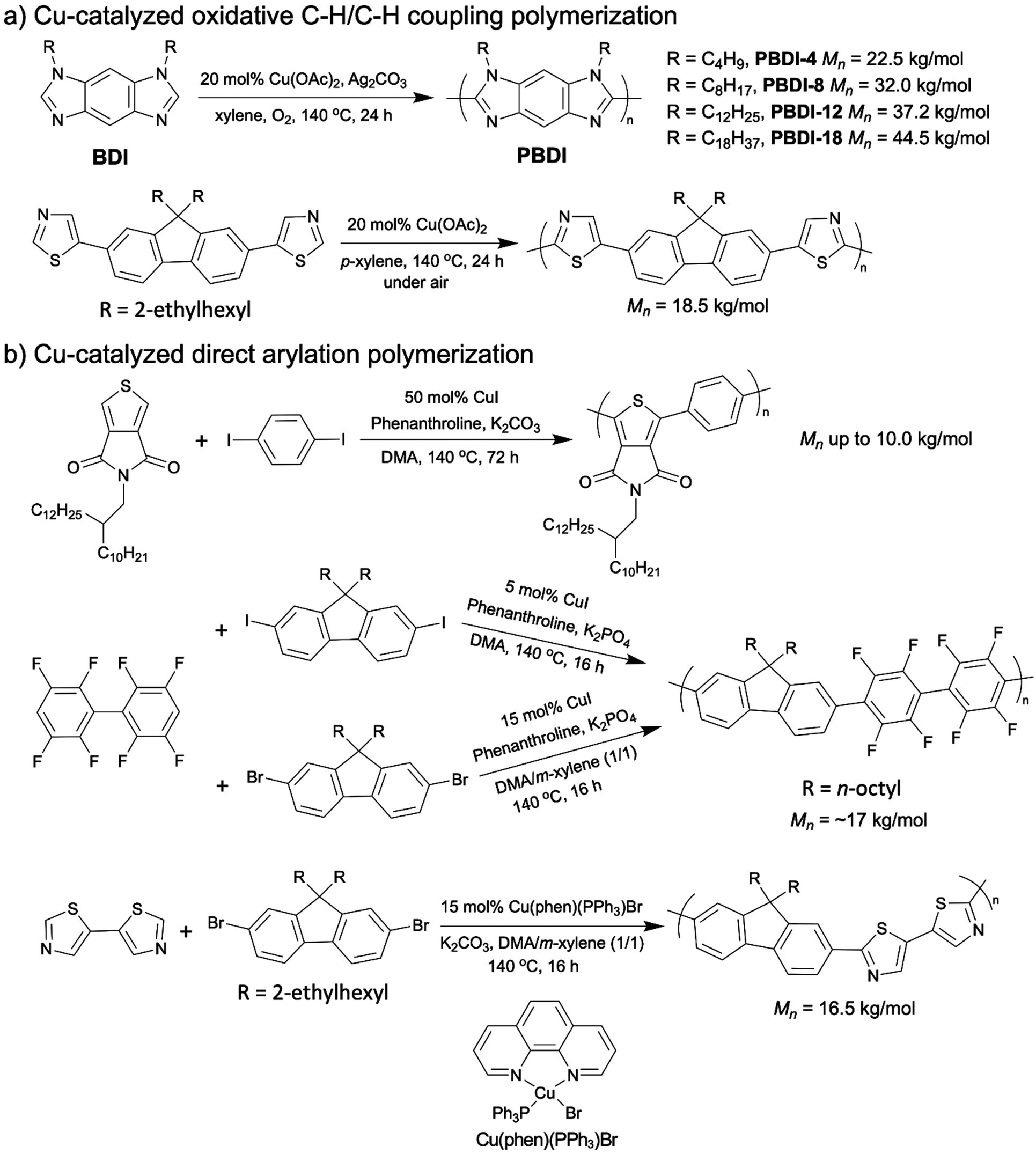 | ||
| Scheme 12 (a) Cu-catalyzed oxidative C–H/C–H coupling polymerizations. (b) Cu-catalyzed direct arylation polymerizations. | ||
The above two examples are restricted to homo-polymerization of nitrogen-containing heterocyclic monomers. Thompson and colleagues published a series of studies on copper catalyzed DArP to synthesize D–A copolymers.107–110 The first copper catalyzed DArP for D–A copolymer synthesis was reported in 2018 by Thompson. Thieno[3,4-c]pyrrole-4,6-dione (TPD)-containing D–A copolymers were prepared with the catalyst loading of 50 mol% and the molecular weights of ∼ 4k to ∼10 kg mol−1 (Scheme 12b, top).107 In the same year, they achieved a synthesis of a D–A copolymer of fluorene and fluoroarenes (Mn = 16.4 kg mol−1) via DArP by employing only 5 mol% copper catalyst (Scheme 12b, middle).108 The above two examples of copper catalyzed DArP both required the use of aryl iodides as the comonomers whose preparation processes are normally more challenging and less sustainable than those of aryl bromides.111 Thus, in their next report on copper catalyzed DArP in 2019, they developed a new catalytic system for aryl-bromides, which led to the same fluorene–fluoroarene copolymer with a good molecular weight of 17.3 kg mol−1 (Scheme 12b, middle).109 It is worth noting that the only modification they made to accomplish the copper catalyzed DArP for aryl bromides was the solvent. In the new catalytic system for aryl-bromides, a DMA/m-xylene (1/1) co-solvent system was adopted, while in the DArP for aryl iodides the solvent was DMA solely. In 2020, Thompson and colleagues further upgraded their copper catalyzed DArP for aryl bromides by substituting the original CuI catalyst with a soluble and stable Cu(I) pre-catalyst, Cu(phen)(PPh3)Br.110 With the new copper catalyst, they synthesized a fluorene and dithiazole-based D–A copolymer with molecular weight of 16.5 kg mol−1 within 16 h (Scheme 12b, bottom).
3. Naturally sourced building blocks for OSM syntheses
One of the attempts to reduce the harm of a synthetic process to the environment is to use naturally sourced products as the building blocks and develop synthetic protocols that mimic the naturally occurring processes.112 Naturally occurring products and processes are innocuous and compatible to the environment. In addition, most natural organic chemicals are intrinsically biocompatible, bio-renewable and biodegradable.113,114 Whereas, as discussed in the introduction, the current building blocks of OSM, such as thiophene, are mostly toxic and often produced by environmentally harmful and energy intensive synthetic procedures. Shifting the building blocks of OSMs to these environmentally compatible and benign natural products is conducive to enhancing the environmental friendliness of OSMs. Herein, we advocate three promising naturally sourced organic materials that have displayed potential to be applied as OSMs and OSM building blocks, while being non-toxic to humans. They are indigo, melanin, and caffeine analogues.3.1 Indigo
Indigo (2,2′-bis(2,3-dihydro-3-oxoindolyliden)) is a cheap natural blue dye (a few USDs per kg) with a rich history of at least 6000 years. It was initially obtained from indigo plants (Indigofera tinctoria, Indigofera suffruticosa, Isatis tinctoria, etc.). Its major application is dyeing denim cloth and blue jeans. Although indigo is now primarily produced synthetically with a one-pot Heumann–Pfleger reaction on large industrial scales,115 its environmental compatibility (non-toxicity, biocompatibility, and biodegradability) still allows it to be considered a relatively green building block for OSMs. It was in 2006 that the use of indigo and its derivatives as OSMs was initially proposed in a Japanese patent.116 Since then significant efforts towards incorporating indigo in the organic electronics have emerged.115,117–130Indigo has exhibited very promising semiconducting behaviors. It has balanced ambipolar charge transport, stemming from its reversible redox states and displays good charge mobility (1 × 10−2 cm2 V−1 s−1).115,117 The intra- and intermolecular hydrogen bonding between N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups contributes to the high charge mobility of indigo. Intramolecular hydrogen bonding results in a highly trans-planar structure, which allows extensive delocalization of electrons (Scheme 13a). The intermolecular hydrogen bonding leads to the indigo molecules tightly stacking together, which explains high crystallinity of indigo thin films formed on aliphatic nonpolar substrate upon vacuum evaporation.115,117,118 Indigo thin films have been extensively used in OFET devices, organic inverters, and photodiode as active layers.117–121 Indigo is electronic deficient and has been used as an non-fullerene acceptor in blends with the donor P3HT for use in OPVs.122 Besides acting as a small molecule semiconductor itself, indigo is also an electron accepting building block in semiconducting D–A copolymers.123–126 Indigo is a striking OSM or a OSM building block not only because of its excellent optical and electronic properties, but also because it is non-toxic, air stable, biocompatible, and biodegradable, enabling its applications in the biomedical field.131,132
O groups contributes to the high charge mobility of indigo. Intramolecular hydrogen bonding results in a highly trans-planar structure, which allows extensive delocalization of electrons (Scheme 13a). The intermolecular hydrogen bonding leads to the indigo molecules tightly stacking together, which explains high crystallinity of indigo thin films formed on aliphatic nonpolar substrate upon vacuum evaporation.115,117,118 Indigo thin films have been extensively used in OFET devices, organic inverters, and photodiode as active layers.117–121 Indigo is electronic deficient and has been used as an non-fullerene acceptor in blends with the donor P3HT for use in OPVs.122 Besides acting as a small molecule semiconductor itself, indigo is also an electron accepting building block in semiconducting D–A copolymers.123–126 Indigo is a striking OSM or a OSM building block not only because of its excellent optical and electronic properties, but also because it is non-toxic, air stable, biocompatible, and biodegradable, enabling its applications in the biomedical field.131,132
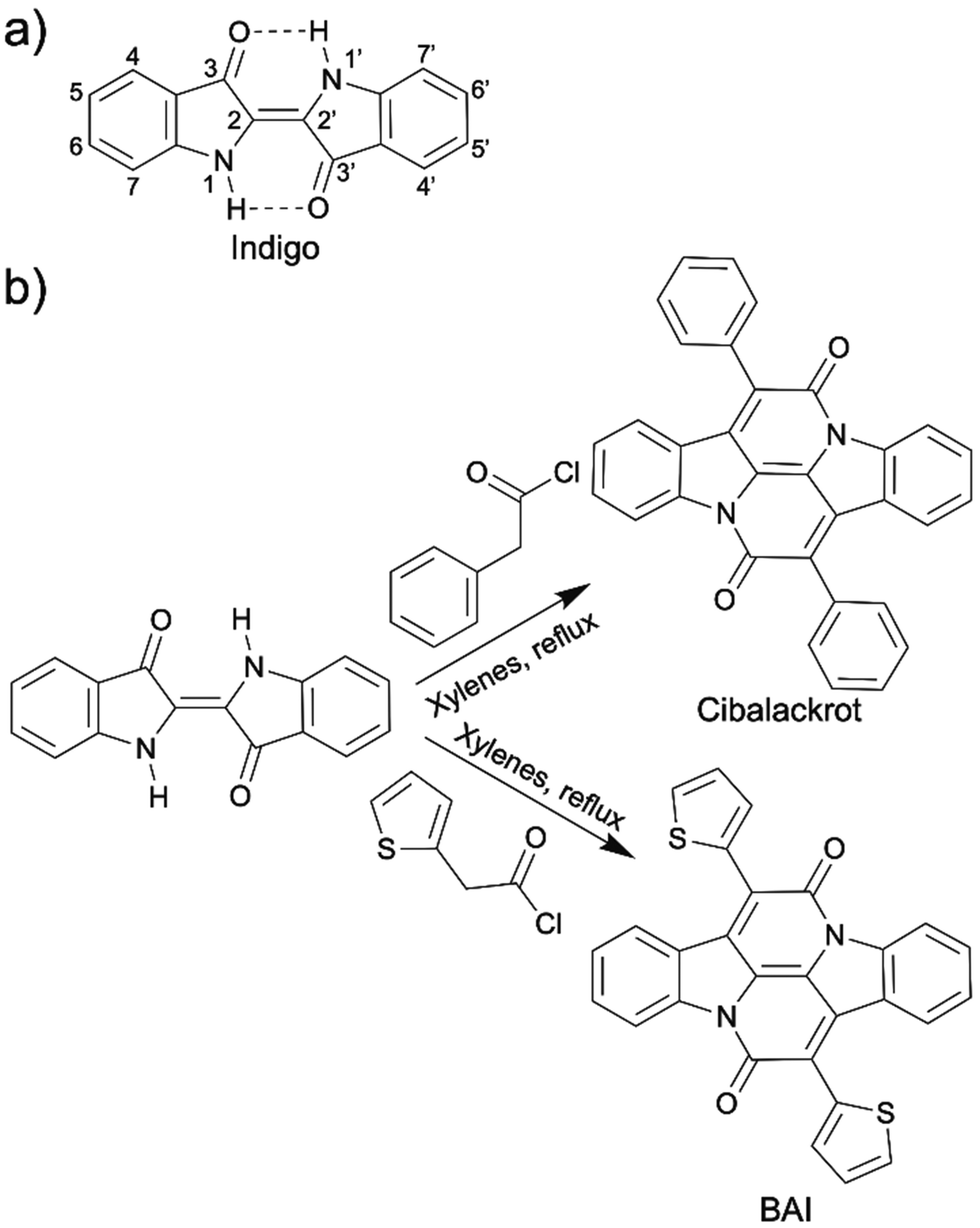 | ||
| Scheme 13 (a) Chemical structure of indigo. (b) Annulation reaction of indigo to synthesize cibalackrot and BAI. | ||
Indigo, however, has very poor solubility in organic solvents owing to its hydrogen bonds, which prevents it from being solution processible like some of other OSMs. The poor solubility of indigo in organic solvents also limits the synthetic chemistry using indigo as a starting material. Functionalizing the indigo molecule with solubilizing groups is an applicable approach to improving its solubility. However, carelessly attaching any solubilizing functional groups to random positions on indigo molecule may cause distortion of the planarity of the molecule, which will hamper its charge mobility. For example, in the work on synthesis of indigo-based D–A copolymers by Li et al., their indigo monomer had an acyl side chain as the solubilizing group, which led to twisted polymer backbones in the produced D–A copolymers and reduced effective main chain conjugation length.123
One of the most effective ways to improve solubility of indigo without harming its electronic properties is to disrupt the hydrogen bonding reversibly, which is so called protection–deprotection. Before processing or reactions, a photo-, acid-, and/or thermo-labile protection group, for example tert-butoxy carbonyl (tBOC) group, has been attached to the nitrogen atom on the indigo to break the inter-molecular hydrogen bonding temporarily, which makes the protected indigo soluble. After the solution processing or chemical reactions, light, heat and/or acid treatment can be carried out to deprotect and regenerate the indigo or indigo moieties.122,124,125 However, this method is not flawless. The solution processed indigo thin films after deprotection have contained large hydrogen-bonded crystallites, causing morphological defects (rough and discontinuous films).122 In addition, the protection and deprotection are extra steps in the synthetic/fabrication processes, which does not help with atom-economy. There is also work that has been done to improve the solubility of indigo without interrupting the N–H and CO hydrogen bonding. Recently, the Li group reported a synthesis of a D–A copolymer using an indigo-based monomer with a long alkoxyl solubilizing group (2-octyldodecyloxy) attached to the 7,7′-positions.126 Although the synthesized polymer displayed the highest hole mobility (up to 0.016 and 0.028 cm2 V−1 s−1) for indigo-based polymers and was demonstrated to be an excellent material for OFET based fluoride sensors, the synthetic route for this polymer contained five steps and four of them are for synthesizing the functionalized indigo monomer alone.
Another practical solution to the insolubility of indigo is to replace the intra-molecular N–H and CO hydrogen bonding with annulated rings, which not only reinforce the coplanarity of the chemical structure but also opens up another electron conducting direction that is nearly orthogonal to the original conducting direction of indigo (Scheme 13b). This annulated indigo was first synthesized in 1914 from indigo and phenylacetyl chloride. The product was called cibalackrot.133 In 2014, instead of reacting indigo with phenylacetyl chloride, Liu et al. synthesized an annulated indigo from 2-thienylacetyl chloride, which is named bay-annulated indigo (BAI) because the annulation occurs at the bay positions on the indigo.134 Delightedly, these annulated indigos only take one step to synthesize from indigo and thiopheneacetyl or phenylacetyl chlorides. The solubility of them was slightly improved compared to indigo because hydrogen bonding was removed, but it is still relatively low due to strong π–π interaction. Nevertheless, annulated indigos have more suitable functionalization sites for installation of a solubilizing group without sacrificing their electronic properties compared to indigo. Another strategy is to pre-install the solubilizing group (alkyl chain) on the thiopheneacetyl or phenylacetyl chloride before the annulation reaction.135,136 The applications of cibalackrot and BAI as organic OSMs or OSM building blocks have been shown in many organic electronic fields, such as OPVs, OFETs, organic lasers, and OLEDs.135–138
3.2 Melanin
The term “melanin” represents a class of polymeric natural pigments, which are ubiquitously present in animals such as bird feathers, cuttlefish ink, insect exoskeletons, human skin, and even in human brains. They are highly biocompatible and biodegradable. Some melanin macromolecules can also be found in wounded plant tissues and microorganisms. Naturally occurring melanin consists of five basic types: eumelanin, pheomelanin, allomelanin, pyomelanin, and neuromelanin.139,140 At present, when most researchers talk about melanin, they are referring to the black-brown eumelanin, which is the most common and easy to access type of natural melanin.141 Although the existence of melanin was discovered in 19th century, the exact chemical structure has always been debated due to its complexity. Generally, melanin macromolecules are extensively conjugated two-dimensional polymeric sheets that are primarily composed of 5,6-dihydroxyindole (DHI), 5,6-dihydroxyindole-2-carboxylic acid (DHICA), and their redox derivatives.142 The biosynthesis of melanin in vivo happens in melanocyte cells, starting with oxidation of tyrosine which gives DHI and DHICA. Then the polymerization occurs to form melanin. The synthesis of melanin in vitro is mostly done by oxidating tyrosine with hydrogen peroxide, followed by polymerization.143 The melanin synthesized in vitro is called synthetic melanin. Melanins generated from different sources or environments have different contents of each subunit, resulting in melanin's irregular chemical structure. A generic chemical structure of part of melanin is shown in Scheme 14.139,142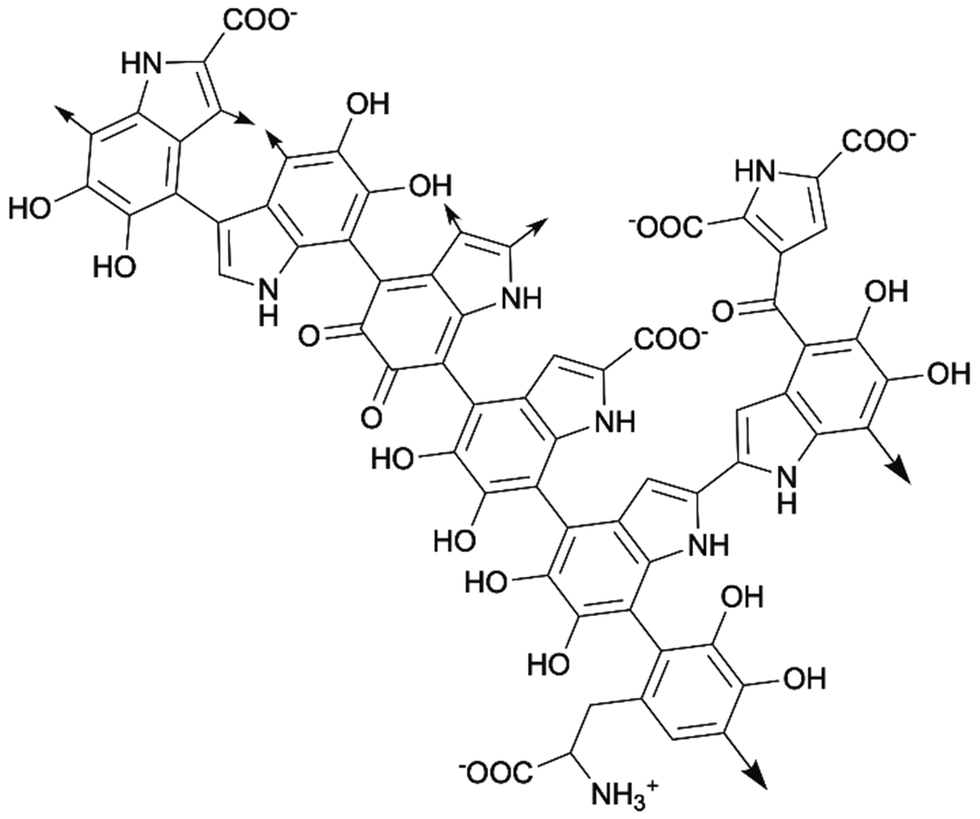 | ||
| Scheme 14 Part of chemical structure of two-dimensional melanin sheet (arrows means the positions where the sheet could extend from). | ||
Melanin, as a natural biomaterial, has a broadband light absorption range, spanning across the UV, visible, and near-infrared (NIR) regions. In addition, melanin has multiple bandgaps ranging from 2.0 to 3.7 eV because of the complex nature of its chemical structure.144,145 The outstanding optical property of melanin is associated with its photoprotective function in human skin. In 1970s, McGinness et al. as well as Powell and Rosenberg reported the electrical conductivity of melanin, which unlocked the applications of melanin in organic electronic field.146,147 It was found that melanin is amorphous in the solid state due to the irregularity of its chemical structures. McGinness et al. proposed that the origin of charge transport in melanin was explained by the amorphous semiconductor model.146 However, around 40 years later Mostert et al. showed evidence pointing that the melanin was a hybrid ionic–electronic semiconductor instead of a typical amorphous semiconductor, and the charge carriers in melanin were electrons and protons.148,149 The optical and electronic properties have made melanin a great OSM for OPV,150,151 OLED,152 and other optoelectronic applications.153 The hybrid ionic–electronic conductivity endowed melanin with a prominent position in organic electrochemical transistors (OECT).154,155 It is worth mentioning that the electrical conductivity of melanin strongly and positively correlates to its hydration state,146,147 enabling melanin's application in humidity sensors.156,157 It can also be used as a pH sensor, ascribed to the H+ ion binding ability of carboxylate groups in melanin.158 The viability of melanin as implanted bioelectronics has also been examined.159,160 Moreover, there are reports showing the potential of melanin to be used in batteries.161–163 The applications of melanin as an OSM was also carefully reviewed by Shankar et al.142
Compared to indigo, developments of organic electronics using melanin are still in its early stages. There are two major barriers remaining. One is the industrial scalability limited by the scarcity of melanin. The most common natural source of melanin is from animals, specifically cuttlefish ink, which is not as accessible as plant-derived chemicals. This leads to the high price of commercially available melanin which according to Sigma-Aldrich is 464.00 USD per gram in 2021. It is imperative for the ongoing and future research to develop low-cost and effective methods to produce or extract melanin. A plausible solution is biosynthesis of melanin in bacteria.164 The other barrier is the limited electrical conductivity of melanin. In recent years, some progresses have been made to address this issue. Shim et al. achieved electrical conductivities as high as 1.17 ± 0.13 S cm−1 by preparing a composite with melanin nanoparticles in a poly(vinyl alcohol) matrix.165 This was a huge improvement in contrast to the original conductivity of significantly hydrated melanin which was reported to be 10−3 S cm−1.166 Meredith et al. reported a novel metal (copper(II) ions) doping strategy to enhance and control the proton conductivity of melanin for OECT application.155 However, there is still much work to do to improve the conductivity of melanin so that it becomes a more competitive OSM for high-performance electronic devices.
3.3 Caffeine analogues
Last but not least, caffeine and some of its analogues, such as theobromine and theophylline, are classified as xanthine alkaloids that were originally discovered in plants, such as cacao plants, tea plants, and coffee beans. They exist in a number of common foods and beverages, including coffee, tea, and chocolate. Currently, these chemicals are industrially produced by decaffeination or biosynthesis from a nucleoside, xanthosine.167 They are also inexpensive, less than one USD per gram. The xanthine core of caffeine and its analogues contains a five-membered ring, imidazole, fused with a six-membered ring, pyrimidinedione. As the amide functionalities are typically in their zwitterionic resonance forms, the xanthine core is a conjugated planar structure where electrons are delocalized. (Scheme 15).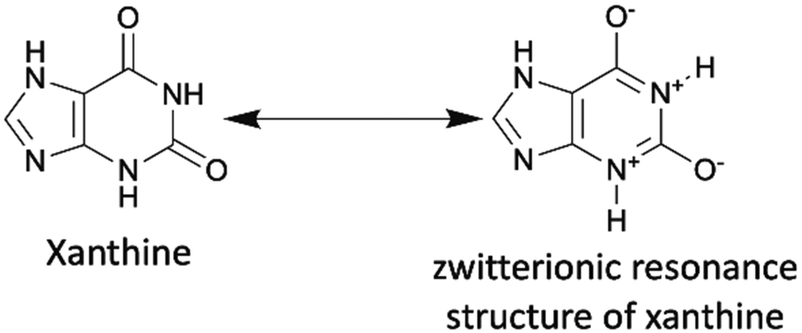 | ||
| Scheme 15 Xanthine and its zwitterionic resonance structure. | ||
In sharp contrast to indigo and melanin, the application of caffeine and its naturally occurring analogues in organic electronics has a very short history. In fact, the first OSM synthesized from theobromine was reported by our group in 2019. In that report, we attached alkylated theobromine onto a pyrene luminophore by direct arylation. This worked to suppress aggregation caused quenching (ACQ) in pyrene's solid state (Scheme 16a).62 The resulted pyrene–theobromine thin films exhibited a photoluminescence quantum yield (PLQY) of almost 100%. After that, we synthesized theobromine-based organic dyes (Theo-green and Theo-red) (Scheme 16b) using alkylated theobromine via direct arylation.168 We demonstrated the stability of Theo-green and Theo-red against photoreduction due to their “acceptor–acceptor” skeleton resulting from electron-withdrawing theobromine and thiadiazole. The organic LED light converter made from theobromine-based organic dyes (1 wt%) and a commercial polymer, poly(styrene–butadiene–styrene) (SBS) (99 wt%), were transparent because of the high solubility of theobromine-based dyes in SBS. The high transparency endowed the organic light converter with high efficiency due to minimal light scattering loss. Additionally, the PLQY of the organic light converter was as high as 90% under ambient conditions. In our other study, we incorporated three organic dyes (Theo-blue, Theo-green, and Theo-ruby) synthesized via direct arylation (Scheme 16b) into a light converter in a hybrid LED.169 Compared to the incumbent inorganic phosphor light converters made from rare-earth elements, the new organic theobromine-based light converter offered improved performance as measured by the color rendering index and color fidelity index, resulting in more continuous and uniform emitted light across visible wavelengths. In addition, owing to the affordability and abundance of the starting material and the excellent atom-economy of the synthetic route, the price of the light converters could be reduced to 0.013 USD per 1 W LED from approximately 0.192 USD of the commercial products. Overall, our development of novel theobromine-based organic dyes provided a new strategy to produce organic dyes with good structural and spectral tunability using environmentally compatible and abundant natural products.
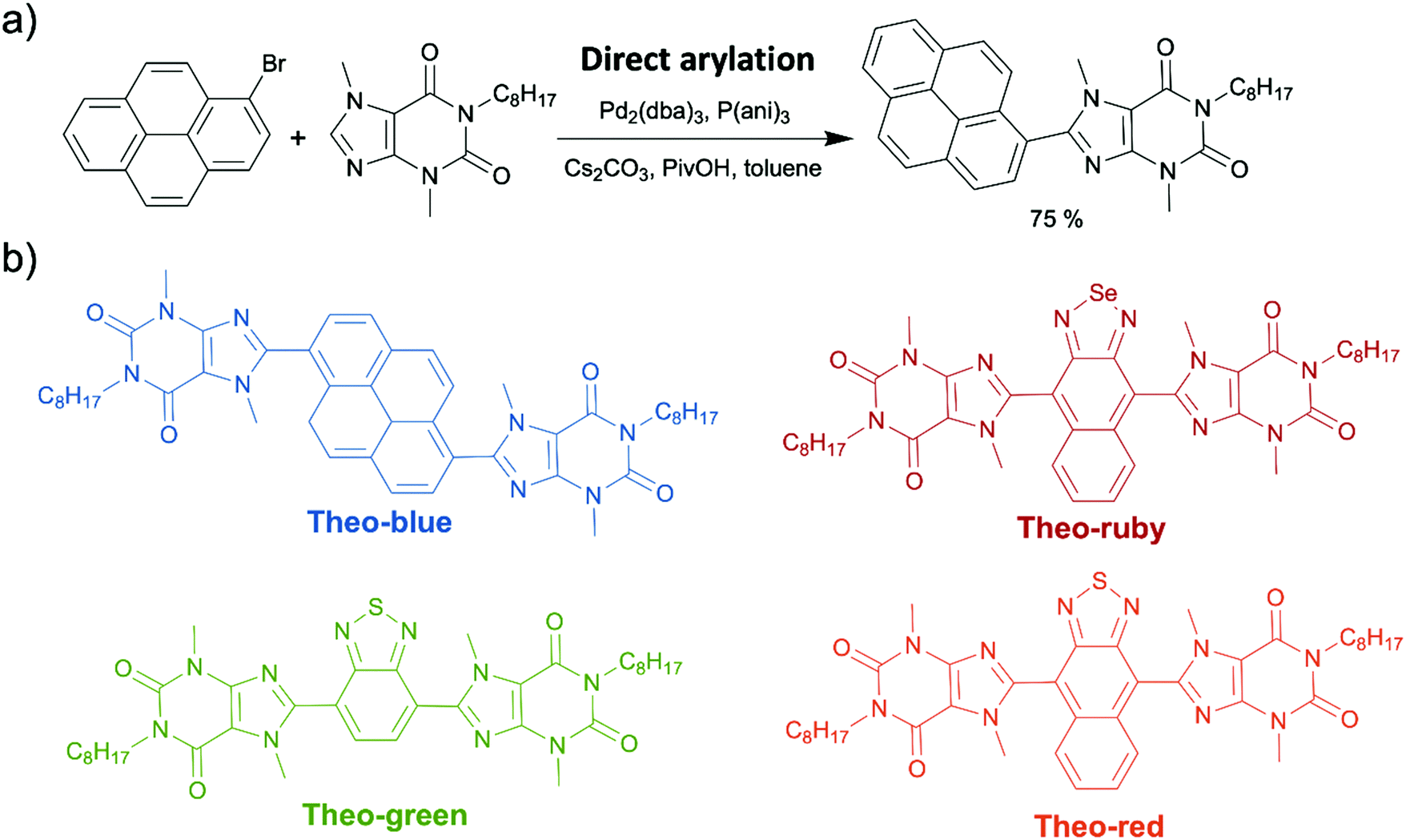 | ||
| Scheme 16 (a) Direct arylation to attach alkylated theobromine onto pyrene. (b) The structures of the four synthesized theobromine-based organic dyes (Theo-blue, Theo-green, Theo-ruby, and Theo-red). | ||
4. Summary and perspective
OSMs have received much attention in the electronics industry due to the advantages they have over the inorganic semiconducting materials. They not only allow for more flexible, stretchable, and lightweight materials, but also are solution processible. More importantly, their properties are tunable by simple modifications to their chemical structures, which enables tailoring properties for a specific application. However, the conventional syntheses of OSMs are not environmentally friendly, which poses severe problems with industrial scale production, where the negative environmental impacts are amplified. In order to improve the environmental friendliness of OSMs, many efforts have been made to minimize chemical waste generation, toxicity of the chemicals used, and energy consumption of the overall synthetic procedures. Despite all the advances made during the past few years as summarized in this review, there is still a long way to go in the pursuit of high-performance and truly environmentally friendly OSMs.First, oxidative CH/CH coupling has not reached widespread use yet compared to direct arylation and other conventional coupling methods. Future studies should be focused on overcoming the challenges in reactivity and selectivity control, while there is a simultaneous need to expand the substrate scope of the oxidative CH/CH coupling. Controlled DArPs and oxidative CH/CH polymerizations are desired to achieve consistent semiconducting polymer products every batch with narrow molecular weight distributions. Secondly, all the chemicals used in the synthesis of OSMs, including solvents, catalysts, and starting materials, need to be non-toxic and innocuous to human health and the environment. Future research should focus on applying earth abundant, bio-based, biocompatible and/or biodegradable substances in OSM syntheses. In addition, the energy consumption of OSM syntheses could be lessened further if the syntheses were able to be conducted under ambient conditions (at room temperature, under atmospheric pressure, and in the air). Finally, the work presented here can provide the foundation and guidance for the future development of eco-friendly, cost-effective, and well-functioning organic electronics.
Author contributions
All authors contributed to the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF under the CCI Center for Selective CH Functionalization, CHE-1700982. Yunping Huang and Amy L. Mayhugh are gratefully acknowledged for proofreading the manuscript and for helpful discussions.References
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Chem. Soc. Rev., 2016, 45, 4825 RSC.
- S. Holliday, Y. Li and C. K. Luscombe, Prog. Polym. Sci., 2017, 70, 34 CrossRef CAS.
- M. U. Chaudhry, K. Muhieddine, R. Wawrzinek, J. Li, S.-C. Lo and E. B. Namdas, ACS Photonics, 2018, 5, 2137 CrossRef.
- V. Ahmad, A. Shukla, J. Sobus, A. Sharma, D. Gedefaw, G. G. Andersson, M. R. Andersson, S.-C. Lo and E. B. Namdas, Adv. Opt. Mater., 2018, 6, 1800768 CrossRef.
- B. G. A. L. Borges, M. Gioti, R. S. Correa, A. K. Andreopoulou, A. G. Veiga, A. Laskarakis, J. K. Kallitsis, S. Logothetidis and M. L. M. Rocco, Opt. Mater., 2021, 117, 111145 CrossRef CAS.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319 CrossRef CAS PubMed.
- S. Riera-Galindo, F. Leonardi, R. Pfattner and M. Mas-Torrent, Adv. Mater. Technol., 2019, 4, 1900104 CrossRef CAS.
- H. Chen, M. Hurhangee, M. Nikolka, W. Zhang, M. Kirkus, M. Neophytou, S. J. Cryer, D. Harkin, P. Hayoz, M. Abdi-Jalebi, C. R. McNeill, H. Sirringhaus and I. McCulloch, Adv. Mater., 2017, 29, 1702523 CrossRef PubMed.
- J. Yang, Z. Zhao, S. Wang, Y. Guo and Y. Liu, Chem, 2018, 4, 2748 CAS.
- S. Nambiar and J. T. W. Yeow, Biosens. Bioelectron., 2011, 26, 1825 CrossRef CAS PubMed.
- D. C. Martin, MRS Commun., 2015, 5, 131 CrossRef CAS.
- A. B. Tamayo, M. Tantiwiwat, B. Walker and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 15543 CrossRef CAS.
- X. Shi, A. Sui, Y. Wang, Y. Li, Y. Geng and F. Wang, Chem. Commun., 2015, 51, 2138 RSC.
- J. Kettle, M. Horie, L. A. Majewski, B. R. Saunders, S. Tuladhar, J. Nelson and M. L. Turner, Sol. Energy Mater. Sol. Cells, 2011, 95, 2186 CrossRef CAS.
- C. Kanimozhi, P. Balraju, G. D. Sharma and S. Patil, J. Phys. Chem. B, 2010, 114, 3095 CrossRef CAS PubMed.
- B. S. Ong, Y. Wu, Y. Li, P. Liu and H. Pan, Chem. – Eur. J., 2008, 14, 4766 CrossRef CAS PubMed.
- V. Malytskyi, J.-J. Simon, L. Patrone and J.-M. Raimundo, RSC Adv., 2015, 5, 354 RSC.
- L. Hrostea, M. Girtan, R. Mallet and L. Leontie, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 374, 012015 CrossRef.
- T. Fukuda, A. Toda, K. Takahira, K. Suzuki, Y. Liao, M. Hirahara, M. Saito and I. Osaka, Thin Solid Films, 2016, 612, 373 CrossRef CAS.
- J. Swanston, Ullmann's Encycl. Ind. Chem., 2006, 36, 657 Search PubMed.
- Thiophene. Safety Data Sheet number T31801 [online]. Sigma-Aldrich: St. Louis, MO, April 13, 2020. https://www.sigmaaldrich.com/US/en/sds/aldrich/t31801 (accessed August 14, 2021).
- C. K. Jaladanki, N. Taxak, R. A. Varikoti and P. V. Bharatam, Chem. Res. Toxicol., 2015, 28, 2364 Search PubMed.
- N. Le Dang, T. B. Hughes, G. P. Miller and S. J. Swamidass, Chem. Res. Toxicol., 2017, 30, 1046 Search PubMed.
- A. L. Harreus, Ullmann's Encycl. Ind. Chem., 2000, 30, 615 Search PubMed.
- H. E. Hoydonckx, W. M. Van Rhijn, W. Van Rhijn, D. E. De Vos and P. A. Jacobs, Ullmann's Encycl. Ind. Chem., 2007, 16, 285 Search PubMed.
- T. P. Osedach, T. L. Andrewb and V. Bulović, Energy Environ. Sci., 2013, 6, 711 RSC.
- R. Po, A. Bernardi, A. Calabrese, C. Carbonera, G. Corso and A. Pellegrino, Energy Environ. Sci., 2014, 7, 925 RSC.
- A. Marrocchi, A. Facchetti, D. Lanari, C. Petruccia and L. Vaccaro, Energy Environ. Sci., 2016, 9, 763 RSC.
- I. A. Stepek and K. Itami, ACS Mater. Lett., 2020, 2, 951 CrossRef CAS.
- J. Kuwabara and T. Kanbara, Macromol. Rapid Commun., 2021, 42, 2000493 CrossRef CAS PubMed.
- Y. Huang and C. K. Luscombe, Chem. Rec., 2019, 19, 1039 CrossRef CAS PubMed.
- J. Kuwabara and T. Kanbara, Bull. Chem. Soc. Jpn., 2019, 92, 152 CrossRef CAS.
- M. Mooney, A. Nyayachavadi and S. Rondeau-Gagné, J. Mater. Chem. C, 2020, 8, 14645 RSC.
- R. M. Pankow and B. C. Thompson, Polym. Chem., 2020, 11, 630 RSC.
- K. Nakabayashi, Polym. J., 2018, 50, 475 CrossRef CAS.
- J. Kuwabara, Polym. J., 2018, 50, 1099 CrossRef CAS.
- M. Leclerc, S. Brassard and S. Beaupré, Polym. J., 2020, 52, 13 CrossRef CAS.
- M. Wakioka and F. Ozawa, Asian J. Org. Chem., 2018, 7, 1206 CrossRef CAS.
- M. Mainville and M. Leclerc, ACS Appl. Polym. Mater., 2021, 3, 2 CrossRef CAS.
- N. S. Gobalasingham and B. C. Thompson, Prog. Polym. Sci., 2018, 83, 135 CrossRef CAS.
- J. Zhang, L. J. Kang, T. C. Parker, S. B. Blakey, C. K. Luscombe and S. R. Marder, Molecules, 2018, 23, 922 CrossRef PubMed.
- S. Phan and C. K. Luscombe, Trends Chem., 2019, 1, 670 CrossRef CAS.
- Y. Huang, D. L. Elder, A. L. Kwiram, S. A. Jenekhe, A. K. Y. Jen, L. R. Dalton and C. K. Luscombe, Adv. Mater., 2021, 33, 1904239 CrossRef CAS PubMed.
- B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS PubMed.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef.
- K. H. Hendriks, W. Li, G. H. L. Heintges, G. W. P. van Pruissen, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 11128 CrossRef CAS PubMed.
- T. Vangerven, P. Verstappen, J. Drijkoningen, W. Dierckx, S. Himmelberger, A. Salleo, D. Vanderzande, W. Maes and J. V. Manca, Chem. Mater., 2015, 27, 3726 CrossRef CAS.
- Q. Shi, W. Tatum, J. Zhang, C. Scott, C. K. Luscombe, S. R. Marder and S. B. Blakey, Asian J. Org. Chem., 2018, 7, 1419 CrossRef CAS.
- M. Wakioka, N. Yamashita, H. Mori, Y. Nishihara and F. Ozawa, Molecules, 2018, 23, 981 CrossRef PubMed.
- M. Wakioka, H. Morita, N. Ichihara, M. Saito, I. Osaka and F. Ozawa, Macromolecules, 2020, 53, 158 CrossRef CAS.
- J. Zhang, W. Chen, A. J. Rojas, E. V. Jucov, T. V. Timofeeva, T. C. Parker, S. Barlow and S. R. Marder, J. Am. Chem. Soc., 2013, 135, 16376 CrossRef CAS PubMed.
- J. L. Bon, D. Feng, S. R. Marder and S. B. Blakey, J. Org. Chem., 2014, 79, 7766 CrossRef CAS PubMed.
- Q. Shi, S. Zhang, J. Zhang, V. F. Oswald, A. Amassian, S. R. Marder and S. B. Blakey, J. Am. Chem. Soc., 2016, 138, 3946 CrossRef CAS PubMed.
- Ö. Usluer, M. Abbas, G. Wantz, L. Vignau, L. Hirsch, E. Grana, C. Brochon, E. Cloutet and G. Hadziioannou, ACS Macro Lett., 2014, 3, 1134 CrossRef.
- R. M. Pankow, L. Ye and B. C. Thompson, Macromolecules, 2020, 53, 3315 CrossRef CAS.
- N. S. Gobalasingham, S. Noh and B. C. Thompson, Polym. Chem., 2016, 7, 1623 RSC.
- J. A. Lee and C. K. Luscombe, ACS Macro Lett., 2018, 7, 767 CrossRef CAS.
- C. Chen, D. H. Maldonado, D. Le Borgne, F. Alary, B. Lonetti, B. Heinrich, B. Donnioe and K. I. M. Ching, New J. Chem., 2016, 40, 7326 RSC.
- S. M. McAfee and G. C. Welch, Chem. Rec., 2019, 19, 989 CrossRef CAS PubMed.
- S. M. McAfee, A.-J. Payne, S. V. Dayneko, G. P. Kini, C. E. Song, J.-C. Lee and G. C. Welch, J. Mater. Chem. A, 2017, 5, 16907 RSC.
- S. M. McAfee, S. V. Dayneko, P. Josse, P. Blanchard, C. Cabanetos and G. C. Welch, Chem. Mater., 2017, 29, 1309 CrossRef CAS.
- Y. Huang, Y. Liu, P. J. W. Sommerville, W. Kaminsky, D. S. Ginger and C. K. Luscombe, Green Chem., 2019, 21, 6600 RSC.
- D. R. Stuart and K. Fagnou, Science, 2007, 316, 1172 CrossRef CAS PubMed.
- J.-R. Liu, Y.-Q. Duan, S.-Q. Zhang, L.-J. Zhu, Y.-Y. Jiang, S. Bi and X. Hong, Org. Lett., 2019, 21, 2360 CrossRef CAS PubMed.
- L. J. Kang, L. Xing and C. K. Luscombe, Polym. Chem., 2019, 10, 486 RSC.
- H. Aoki, H. Saito, Y. Shimoyama, J. Kuwabara, T. Yasuda and T. Kanbara, ACS Macro Lett., 2018, 7, 90 CrossRef CAS.
- Q. Zhang, M. Chang, Y. Lu, Y. Sun, C. Li, X. Yang, M. Zhang and Y. Chen, Macromolecules, 2018, 51, 379 CrossRef CAS.
- Y. Shimoyama, J. Kuwabara and T. Kanbara, ACS Catal., 2020, 10, 3390 CrossRef CAS.
- J. Li, D. Han, Q. Zhang, Z. He and Y. Lu, J. Polym. Sci., 2021, 59, 240 CrossRef CAS.
- X. C. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636 CrossRef CAS PubMed.
- H. Saito, J. Kuwabara, T. Yasuda and T. Kanbara, Polym. Chem., 2016, 7, 2775 RSC.
- H. Saito, J. Kuwabara, T. Yasuda and T. Kanbara, Macromol. Rapid Commun., 2018, 39, 1800414 CrossRef PubMed.
- R. Iwamori, R. Sato, J. Kuwabara, T. Yasuda and T. Kanbara, Macromol. Rapid Commun., 2021, 2100283 CrossRef CAS PubMed.
- S. Murai, Activation of Unreactive Bonds and Organic Synthesis, Springer, New York, 1999 Search PubMed.
- P. P. Khlyabich, B. Burkhart, A. E. Rudenko and B. C. Thompson, Polymer, 2013, 54, 5267 CrossRef CAS.
- A. L. Mayhugh and C. K. Luscombe, Beilstein J. Org. Chem., 2020, 16, 384 CrossRef CAS PubMed.
- L. Boisvert and K. I. Goldberg, Acc. Chem. Res., 2012, 45, 899 CrossRef CAS PubMed.
- M. L. Scheuermann and K. I. Goldberg, Chem. – Eur. J., 2014, 20, 14556 CrossRef CAS PubMed.
- A. Ichige, H. Saito, J. Kuwabara, T. Yasuda, J.-C. Choi and T. Kanbara, Macromolecules, 2018, 51, 6782 CrossRef CAS.
- X. Chen, A. Ichige, J. Chen, I. Fukushima, J. Kuwabara and T. Kanbara, Polymer, 2020, 207, 122927 CrossRef CAS.
- A. Faradhiyani, Q. Zhang, K. Maruyama, J. Kuwabara, T. Yasudab and T. Kanbara, Mater. Chem. Front., 2018, 2, 1306 RSC.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaksh and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686 CrossRef CAS PubMed.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546 RSC.
- D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156 CrossRef CAS.
- J. J. Dong, J. Roger, C. Verrier, T. Martin, R. Le Goff, C. Hoarau and H. Doucet, Green Chem., 2010, 12, 2053 RSC.
- K. Beydoun and H. Doucet, ChemSusChem, 2011, 4, 526 CrossRef CAS PubMed.
- G. Strappaveccia, E. Ismalaj, C. Petrucci, D. Lanari, A. Marrocchi, M. Drees, A. Facchetti and L. Vaccaro, Green Chem., 2015, 17, 365 RSC.
- J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164 RSC.
- R. M. Pankow, L. Ye, N. S. Gobalasingham, N. Salami, S. Samal and B. C. Thompson, Polym. Chem., 2018, 9, 3885 RSC.
- D. Conelli, N. Margiotta, R. Grisorio and G. P. Suranna, Macromol. Chem. Phys., 2021, 222, 2000382 CrossRef CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879 RSC.
- R. M. Pankow, L. Ye and B. C. Thompson, Polym. Chem., 2019, 10, 4561 RSC.
- L. Ye, R. M. Pankow, M. Horikawa, E. L. Melenbrink, K. Liu and B. C. Thompson, Macromolecules, 2019, 52, 9383 CrossRef CAS.
- T. M. Pappenfus, F. Almyahi, N. A. Cooling, E. W. Culver, S. C. Rasmussen and P. C. Dastoor, Macromol. Chem. Phys., 2018, 219, 1800272 CrossRef.
- L. Ye and B. C. Thompson, ACS Macro Lett., 2021, 10, 714 CrossRef CAS.
- J. H. Clark, D. J. Macquarrie and J. Sherwood, Green Chem., 2012, 14, 90 RSC.
- F. Grenier, K. Goudreau and M. Leclerc, J. Am. Chem. Soc., 2017, 139, 2816 CrossRef CAS PubMed.
- H. Ayalew, T.-L. Wang, T.-H. Wang, H.-F. Hsu and H.-H. Yu, Synlett, 2018, 2660 CAS.
- Y.-J. Lin, H.-S. Sun, H.-R. Yang, Y.-Y. Lai, K.-Y. Hou and Y.-H. Liu, Macromol. Rapid Commun., 2020, 41, 2000021 CrossRef CAS PubMed.
- R. J. Lundgren and M. Stradiotto, Chem. – Eur. J., 2012, 18, 9758 CrossRef CAS PubMed.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147 CrossRef CAS.
- A. Kumbhar, J. Organomet. Chem., 2017, 848, 22 CrossRef CAS.
- L. Giraud, S. Grelier, E. Grau, G. Hadziioannou, C. Brochon, H. Cramail and E. Cloutet, J. Mater. Chem. C, 2020, 8, 9792 RSC.
- J. Huang, J. Chan, Y. Chen, C. J. Borths, K. D. Baucom, R. D. Larsen and M. M. Faul, J. Am. Chem. Soc., 2010, 132, 3674 CrossRef CAS PubMed.
- Q. Huang, X. Qin, B. Li, J. Lan, Q. Guo and J. You, Chem. Commun., 2014, 50, 13739 RSC.
- R. M. Pankow, L. Ye and B. C. Thompson, Polym. Chem., 2018, 9, 4120 RSC.
- R. M. Pankow, L. Ye and B. C. Thompson, ACS Macro Lett., 2018, 7, 1232 CrossRef CAS.
- L. Ye, R. M. Pankow, A. Schmitt and B. C. Thompson, Polym. Chem., 2019, 10, 6545 RSC.
- L. Ye, A. Schmitt, R. M. Pankow and B. C. Thompson, ACS Macro Lett., 2020, 9, 1446 CrossRef CAS.
- E. B. Merkushev, Synthesis, 1988, 923 CrossRef CAS.
- H. I. Patil, M. C. Singh, P. Gaikwad, K. S. Lade, N. A. Gadhave and S. D. Sawant, J. Pharm. Res., 2011, 4, 4798 Search PubMed.
- V. R. Feig, H. Tran and Z. Bao, ACS Cent. Sci., 2018, 4, 337 CrossRef CAS PubMed.
- Y. Cao and K. E. Uhrich, J. Bioact. Compat. Polym., 2019, 34, 3 CrossRef CAS.
- E. D. Głowacki, G. Voss, L. Leonat, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, Isr. J. Chem., 2012, 52, 540 CrossRef.
- Y. Hiroyuki, Organic transistor, JP 098454A, 2008.
- M. Irimia-Vladu, E. D. Głowacki, P. A. Troshin, G. Schwabegger, L. Leonat, D. K. Susarova, O. Krystal, M. Ullah, Y. Kanbur, M. A. Bodea, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Adv. Mater., 2012, 24, 375 CrossRef CAS PubMed.
- M. Irimia-Vladu, Y. Kanbur, F. Camaioni, M. E. Coppola, C. Yumusak, C. V. Irimia, A. Vlad, A. Operamolla, G. M. Farinola, G. P. Suranna, N. Gonzalez-Benitez, M. C. Molina, L. F. Bautista, H. Langhals, B. Stadlober, E. D. Głowacki and N. S. Sariciftci, Chem. Mater., 2019, 31, 6315 CrossRef CAS PubMed.
- D. V. Anokhin, L. I. Leshanskaya, A. A. Piryazev, D. K. Susarova, N. N. Dremova, E. V. Shcheglov, D. A. Ivanov, V. F. Razumov and P. A. Troshin, Chem. Commun., 2014, 50, 7639 RSC.
- M. A. Manthrammel, I. S. Yahia, M. Shkir, S. AlFaify, H. Y. Zahran, V. Ganesh and F. Yakuphanoglu, Solid State Sci., 2019, 93, 7 CrossRef CAS.
- A. Rivalta, C. Albonetti, D. Biancone, M. D. Ciana, S. d’Agostino, L. Biniek, M. Brinkmann, A. Giunchi, T. Salzillo, A. Brillante, R. G. D. Valle and E. Venuti, Surf. Interfaces, 2021, 24, 101058 CrossRef CAS.
- E. D. Głowacki, G. Voss, K. Demirak, M. Havlicek, N. Sünger, A. C. Okur, U. Monkowius, J. Gąsiorowski, L. Leonat and N. S. Sariciftci, Chem. Commun., 2013, 49, 6063 RSC.
- C. Guo, B. Sun, J. Quinn, Z. Yan and Y. Li, J. Mater. Chem. C, 2014, 2, 4289 RSC.
- C. Guo, J. Quinn, B. Sun and Y. Li, J. Mater. Chem. C, 2015, 3, 5226 RSC.
- C. Liu, S. Dong, P. Cai, P. Liu, S. Liu, J. Chen, F. Liu, L. Ying, T. P. Russell, F. Huang and Y. Cao, ACS Appl. Mater. Interfaces, 2015, 7, 9038 CrossRef CAS PubMed.
- J. H. L. Ngai, G. Y. Chang, X. Gao, X. Zhou, A. D. Hendsbee and Y. Li, RSC Adv., 2019, 9, 26230 RSC.
- E. D. Głowacki, G. Voss and N. S. Sariciftci, Adv. Mater., 2013, 25, 6783 CrossRef PubMed.
- I. V. Klimovich, L. I. Leshanskaya, S. I. Troyanov, D. V. Anokhin, D. V. Novikov, A. A. Piryazev, D. A. Ivanov, N. N. Dremova and P. A. Troshin, J. Mater. Chem. C, 2014, 2, 7621 RSC.
- S.-F. Zhang, X.-K. Chen, J.-X. Fan and A.-M. Ren, Org. Electron., 2015, 24, 12 CrossRef CAS.
- O. Pitayatanakul, T. Higashino, T. Kadoya, M. Tanaka, H. Kojima, M. Ashizawa, T. Kawamoto, H. Matsumoto, K. Ishikawa and T. Mori, J. Mater. Chem. C, 2014, 2, 9311 RSC.
- M. Irimia-Vladu, E. D. Głowacki, G. Voss, S. Bauer and N. S. Sariciftci, Mater. Today, 2012, 15, 340 CrossRef CAS.
- K.-Y. Choi, Dyes Pigm., 2020, 181, 108570 CrossRef CAS.
- G. Engi, Angew. Chem., 1914, 27, 144 CrossRef CAS.
- B. He, A. B. Pun, D. Zherebetskyy, Y. Liu, F. Liu, L. M. Klivansky, A. M. McGough, B. A. Zhang, K. Lo, T. P. Russell, L. Wang and Y. Liu, J. Am. Chem. Soc., 2014, 136, 15093 CrossRef CAS PubMed.
- M. A. Kolaczkowski and Y. Liu, Chem. Rec., 2019, 19, 1062 CrossRef CAS PubMed.
- K. J. Fallon and H. Bronstein, Acc. Chem. Res., 2021, 54, 182 CrossRef CAS PubMed.
- G. Summers and E. Gibson, ChemPhotoChem, 2018, 2, 498 CrossRef CAS.
- A. Shukla, N. R. Wallwork, X. Li, J. Sobus, V. T. N. Mai, S. K. M. McGregor, K. Chen, R. J. Lepage, E. H. Krenske, E. G. Moore, E. B. Namdas and S.-C. Lo, Adv. Opt. Mater., 2020, 8, 1901350 CrossRef CAS.
- F. Solano, New J. Sci., 2014, 2014, 1 CrossRef.
- V. P. Grishchuk, S. A. Davidenko, I. D. Zholner, A. B. Verbitskii, M. V. Kurik and Y. P. Piryatinskii, Tech. Phys. Lett., 2002, 28, 896 CrossRef CAS.
- P. Meredith and T. Sarna, Pigm. Cell Res., 2006, 19, 572 CrossRef CAS PubMed.
- E. Vahidzadeh, A. P. Kalra and K. Shankar, Biosens. Bioelectron., 2018, 122, 127 CrossRef CAS PubMed.
- T. Ligonzo, M. Ambrico, V. Augelli, G. Perna, L. Schiavulli, M. A. Tamma, P. F. Biagi, A. Minafra and V. Capozzi, J. Non-Cryst. Solids, 2009, 355, 1221 CrossRef CAS.
- P. R. Crippa, V. Cristofoletti and N. Romeo, Biochim. Biophys. Acta, Gen. Subj., 1978, 538, 164 CrossRef CAS.
- R. Micillo, L. Panzella, M. Iacomino, G. Prampolini, I. Cacelli, A. Ferretti, O. Crescenzi, K. Koike, A. Napolitano and M. d’Ischia, Sci. Rep., 2017, 7, 41532 CrossRef CAS PubMed.
- J. McGinness, P. Corry and P. Proctor, Science, 1974, 183, 853 CrossRef CAS PubMed.
- M. R. Powell and B. Rosenberg, J. Bioenerg., 1970, 1, 493 CrossRef CAS.
- A. B. Mostert, B. J. Powell, I. R. Gentle and P. Meredith, Appl. Phys. Lett., 2012, 100, 093701 CrossRef.
- A. B. Mostert, B. J. Powell, F. L. Pratt, G. R. Hanson, T. Sarna, I. R. Gentle and P. Meredith, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8943 CrossRef CAS PubMed.
- G. Mula, L. Manca, S. Setzu and A. Pezzella, Nanoscale Res. Lett., 2012, 7, 377 CrossRef PubMed.
- A. Antidormi, C. Melis, E. Canadell and L. Colombo, J. Phys. Chem. C, 2017, 121, 11576 CrossRef CAS.
- L. Migliaccio, S. Aprano, L. Iannuzzi, M. G. Maglione, P. Tassini, C. Minarini, P. Manini and A. Pezzella, Adv. Electron. Mater., 2017, 3, 1600342 CrossRef.
- N. Gogurla, B. Roy, K. Min, J.-Y. Park and S. Kim, Adv. Mater. Technol., 2020, 5, 1900936 CrossRef CAS.
- M. Sheliakina, A. B. Mostert and P. Meredith, Mater. Horiz., 2018, 5, 256 RSC.
- A. B. Mostert, S. B. Rienecker, M. Sheliakina, P. Zierep, G. R. Hanson, J. R. Harmer, G. Schenk and P. Meredith, J. Mater. Chem. B, 2020, 8, 8050 RSC.
- T.-F. Wu and J.-D. Hong, Sens. Actuators, B, 2016, 224, 178 CrossRef CAS.
- T.-F. Wu, B.-H. Wee and J.-D. Hong, Adv. Mater. Interfaces, 2015, 2, 1500203 CrossRef.
- M. P. da Silva, J. C. Fernandes, N. B. de Figueiredo, M. Congiu, M. Mulato and C. F. de Oliveira Graeff, AIP Adv., 2014, 4, 037120 CrossRef.
- C. J. Bettinger, J. P. Bruggeman, A. Misra, J. T. Borenstein and R. Langer, Biomaterials, 2009, 30, 3050 CrossRef CAS PubMed.
- M. Piacenti-Silva, A. A. Matos, J. V. Paulin, R. A. d. S. Alavarce, R. C. de Oliveira and C. F. Graeff, Polym. Int., 2016, 65, 1347 CrossRef CAS.
- Y. J. Kim, W. Wu, S.-E. Chun, J. F. Whitacre and C. J. Bettinger, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20912 CrossRef CAS PubMed.
- Y. J. Kim, W. Wu, S.-E. Chun, J. F. Whitacre and C. J. Bettinger, Adv. Mater., 2014, 26, 6572 CrossRef CAS PubMed.
- H. Coskun, A. Aljabour, T. Greunz, M. Kehrer, D. Stifter and P. Stadler, Adv. Sustainable Syst., 2021, 5, 2000176 CrossRef CAS.
- M. E. Pavan, N. I. López and M. J. Pettinari, Appl. Microbiol. Biotechnol., 2020, 104, 1357 CrossRef CAS PubMed.
- T. Eom, J. Jeon, S. Lee, K. Woo, J. E. Heo, D. C. Martin, J. J. Wie and B. S. Shim, Part. Part. Syst. Charact., 2019, 36, 1900166 CrossRef CAS.
- J. Wünsche, Y. X. Deng, P. Kumar, E. Di Mauro, E. Josberger, J. Sayago, A. Pezzella, F. Soavi, F. Cicoira, M. Rolandi and C. Santato, Chem. Mater., 2015, 27, 436 CrossRef.
- H. Ashihara, T. Yokota and A. Crozier, Adv. Bot. Res., 2013, 68, 111 CAS.
- Y. Huang, T. A. Cohen, P. J. W. Sommerville and C. K. Luscombe, J. Mater. Chem. C, 2021, 9, 7274 RSC.
- Y. Huang, T. A. Cohen and C. K. Luscombe, Adv. Sustainable Syst., 2021, 2000300 CrossRef.
Footnote |
| † Current address: pi-Conjugated Polymers Unit, Okinawa Institute of Science and Technology Graduate University, Okinawa, 904-0495, Japan. E-mail: christine.luscombe@oist.jp |
| This journal is © The Royal Society of Chemistry 2021 |