
Kinetic control of chirality and circularly polarized luminescence in G-quartet materials†
Jingqi
Chen
,
Chenqi
Gao
,
Zhiwei
Zhang
,
Xiaowei
Liu
,
Yingying
Chen
 and
Lingyan
Feng
*
and
Lingyan
Feng
*
Materials Genome Institute, and Department of Chemistry, College of Sciences, Shanghai University, Shanghai 200444, China. E-mail: lingyanfeng@t.shu.edu.cn
First published on 12th May 2021
Abstract
The formation of chirality of G-quartet materials has been of concern for a long time, however, the helix-handedness of G-quartet materials is still ambiguous, as well as the novel circularly polarized luminescence (CPL) properties. Here, we demonstrated that the handedness of G-quartet materials highly depends on their formation kinetics. By controlling the temperature or the initial concentration of reactants, we found that right-handed helical G-quartet nanostructures were synthesized in the slow process, while left-handed structures were synthesized in the fast process via orderly stacking. The phenomenon can be explained by the theory of kinetic trapping, in which a slow process leads to the thermodynamic equilibrium, while a fast process results in the kinetic trap state. Furthermore, the first kinetic trapping-controlled reversal CPL system was designed in G-quartet materials via chirality transfer, which has potential applications in CPL materials design and application.
Molecular self-assembly is the orderly association of molecules into larger structures through noncovalent interactions and it has become one of the most important ways to design novel structures.1 Recently, self-assembly of biological related molecules has attracted a lot of attention due to their structural diversity and biocompatibility.2–4 Due to their non-covalent interactions, nucleobases have been considered as promising building blocks for generating functional nanomaterials.5,6 The G-quartet, a cyclic planar tetramer which is typically formed by the self-assembly of metal ions and four guanosine residues, plays an important role in biological processes.7 The orderly G-quartet unit has also been proposed as a powerful scaffold to construct functional nanostructures such as hydrogels,8 nanofibers9 and nanodots.10 It is worth mentioning that since 1962, researchers have found that G-quartet nanostructures can form interesting helical structures,11 and these helical structures were also applied in the field of chirality.12 However, to date, the helix-handedness of G-quartet materials is still ambiguous, and in 2017 researchers asked, “Is the helix structure left- or right-handed?”.13 Our group recently reported the right- and left-helical G-quartet nanostructures through the self-assembly of guanosine monophosphate (GMP), and found that their helix-handedness is regulated by metal ions.14 However, the reason for the reversal of helix-handedness is still a mystery, and more research is needed.
Very recently, a kinetically controlled chiral supramolecular polymerization with helical inversion has been reported.15–17 Since non-covalent interactions are relatively weak, the formation and destruction of supramolecular systems have no significant activation barriers. Therefore, the thermodynamic equilibrium of these self-assembled architectures will be influenced by kinetic processes and finally lead to a kinetically trapped state.18 Under kinetics control, researchers have designed many self-assembled systems with distinct structures and functions.19,20 Usually, fast processes lead to kinetic traps while slow processes result in thermal equilibrium, and the kinetically trapped states possess higher potential energy. According to these principles, many achievements have been made in the study of kinetic traps.21 However, there are only a few reports about self-assembled helix inversion induced by kinetic traps to avoid complex enantiomeric synthesis. This method is an inspiration for us to reverse the helix of G-quartet nanostructures and further explain the reason of helix chirality inversion.
Recently, the research of circularly polarized luminescence (CPL) has become an important direction in the field of chirality due to its numerous potential applications in 3D displays,22 optical security systems,23 and so on. CPL refers to the different intensities between the left- and right-handed circularly polarized light emitted by the luminophore. Various self-assembled materials have been endowed with CPL properties.24 Our group has demonstrated that G-quadruplex DNA and G-quartet nanostructures can be used as excellent CPL templates with a large luminescence dissymmetry factor (glum).14,25 In addition to glum, another characteristic that CPL-active materials should have is switchability, because the switch property is an indispensable part of applications.26 The switch between thermodynamic equilibrium state and kinetically trapped state may be a good way to design switchable CPL-active materials. However, to date, there are no reports yet.
Herein we design reversible helix-handedness of G-quartet nanostructures controlled by kinetic factors, such as metal ions, temperature, concentration and cooling rate. To be specific, we found that the fast formation process results in a left-handed G-quartet structure corresponding to the kinetic trap state, while the slow formation process leads to a right-handed G-quartet structure corresponding to the thermodynamic equilibrium. Furthermore, the first kinetic trapping-controlled reversal CPL system was designed in G-quartet materials via chirality transfer (Scheme 1).
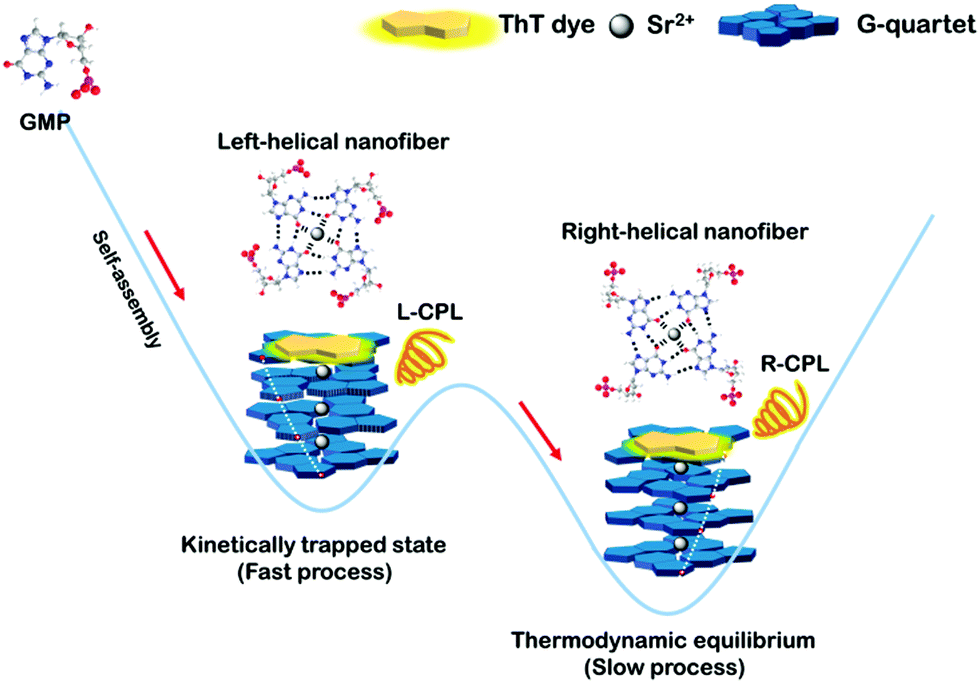 | ||
| Scheme 1 Illustration of the structures of g-nanofibers in the kinetically trapped state and thermodynamic equilibrium. | ||
It is well known that metal ions such as Sr2+, K+, and Na+ are effective inducers and stabilizers of G-quartet structures. In a typical process of Sr2+-induced G-quartet self-assembly reported by Hu et al., the assembly of 20 mM GMP can be induced by 8 mM Sr2+ at low temperature.9 Our group previously reported the right-handed helix G-quartet structure assembled by 50 mM GMP and 8 mM Sr2+ in a 4 °C refrigerator, and found that low temperature is the necessary condition for assembly at this concentration.14 Also, temperature is an important factor in self-assembly, which not only determines the degree of self-assembly, but also determines the self-assembly rate. Therefore, in this study, the self-assembly temperature was taken as the research object, and the effect of self-assembly temperature on helix-handedness was discussed.
Firstly, the self-assembly systems were synthesized at different temperatures by using the same concentration of GMP and Sr2+. In the process of synthesis, we found that the lower the temperature, the faster the assembly formation (1–10 °C). After 2 hour's incubation, the chirality of the assembly was measured by circular dichroism (CD) spectroscopy, which characterizes the chirality of the ground state electronic conformation. As shown in Fig. 1A, we were surprised to see that the chirality of the assembly decreased with the decrease of synthesis temperature, and even a small chiral inversion occurred at lower temperature, which indicates that the synthesis temperature is an important factor affecting the stacking chirality of the G-quartet. In order to verify the authenticity of the CD signal real reaction chirality, scanning electron microscopy (SEM) was used to observe the morphology of the assembly. As shown in Fig. 1C and D, the assembly synthesized at 10 °C showed obvious right-handed helical g-quartet nanofibers, while the assembly synthesized at 3 °C had no obvious helical architecture, which was consistent with the results of CD spectra, indicating that CD spectra can correctly reflect the helical chirality of g-nanofibers. K+ is another effective inducer and stabilizer of G-quartet structures. In our previous studies, we found that the appropriate amount of K+ can reverse the helical chirality of Sr2+-based G-quartet nanofibers. As shown in Fig. 1B, the Sr2+/K+-based g-nanofiber exhibits the same rule as the Sr2+-based nanofiber, that is, with the decrease of synthesis temperature, the chirality of g-fibers changes from right-handed helix to left-handed helix (10–15 °C). The difference is that the existence of K+ increases the critical temperature of chiral inversion. We also observed an interesting phenomenon that K+ accelerates the formation of g-nanofibers at the same synthesis temperature. Therefore, we wonder whether the formation rate of g-fibers determines the stacking chirality of g-fibers.
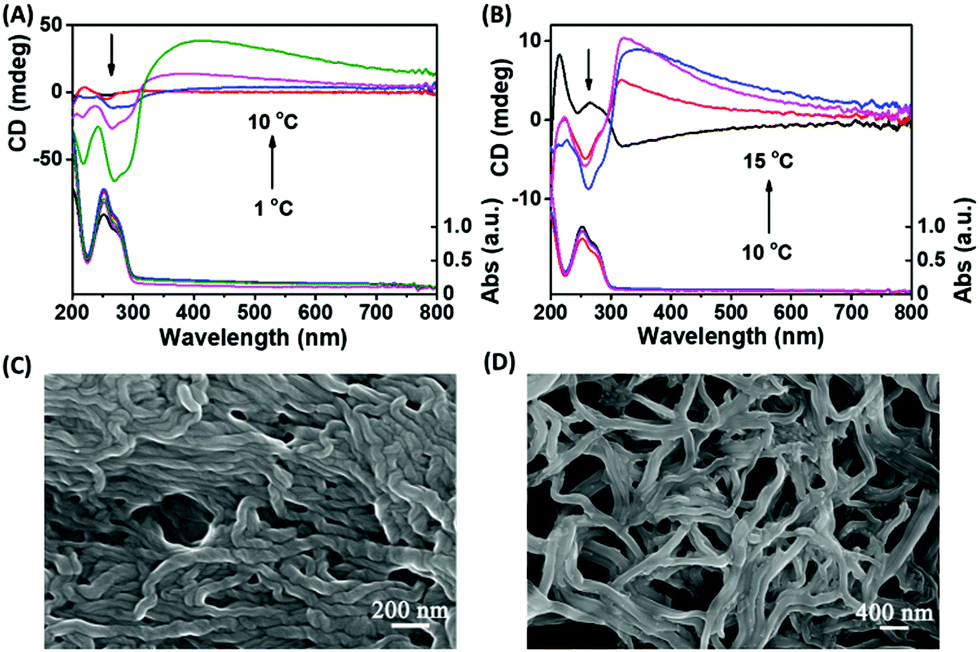 | ||
| Fig. 1 CD spectra of (A) Sr2+-stabilized G-quartet nanofibers and (B) Sr2+/K+-stabilized G-quartet nanofibers synthesized at different temperatures; SEM images of Sr2+-stabilized G-quartet nanofibers synthesized at (C) 10 °C and (D) 3 °C. | ||
In order to determine the basic unit of the two chiral stacked assemblies, X-ray diffraction (XRD) and Fourier-transform infrared (FTIR) spectra were measured. As shown in Fig. 2A, the XRD spectra of left- and right-handed assembly were almost overlapped and displayed an obvious peak centre at 27.4°, which is related to the G-quartet stack between adjacent vertical G–G stacks and demonstrate the similar basic unit of the two chiral assemblies.10Fig. 2B shows the similar FTIR spectra between the two different chiral assemblies. Also, the fact that the band at 1675 cm−1 moved to 1695 cm−1 should be assigned to the C-6![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, which occurred in the formation of the g-quartet. The fact that the band at 1493 cm−1 exhibited a shift to low wavenumbers should be assigned to the imidazole ring N-7–C-8 stretching and C-8–H bending when the formation of the g-quartet occurred.
O stretching vibration, which occurred in the formation of the g-quartet. The fact that the band at 1493 cm−1 exhibited a shift to low wavenumbers should be assigned to the imidazole ring N-7–C-8 stretching and C-8–H bending when the formation of the g-quartet occurred.
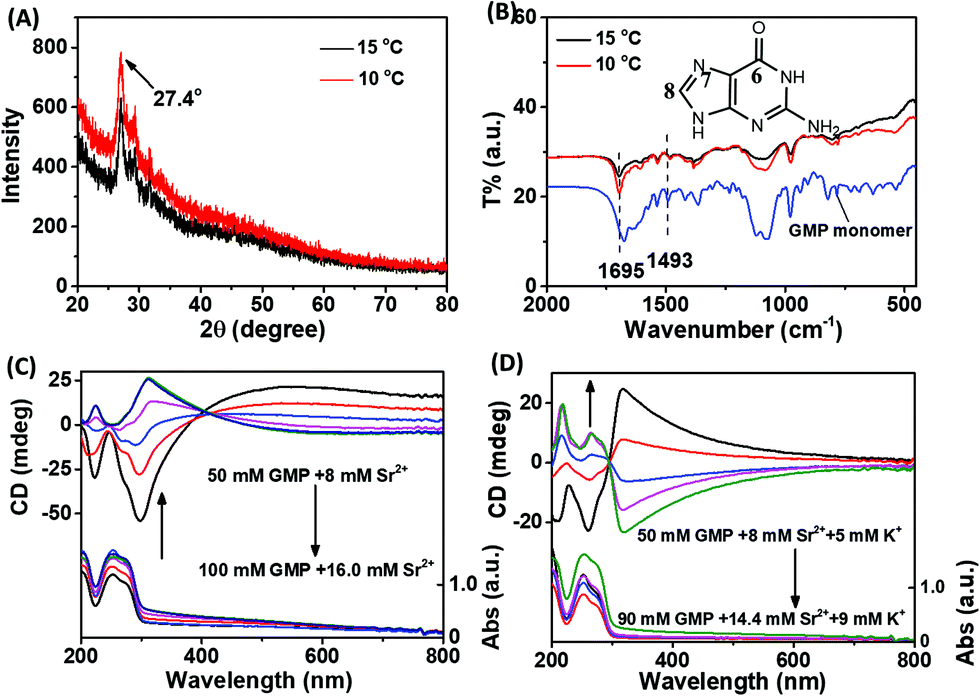 | ||
| Fig. 2 (A) XRD patterns and (B) FTIR spectra of Sr2+/K+-stabilized G-quartet nanofibers synthesized at 10 °C and 15 °C; CD spectra of (C) Sr2+-stabilized G-quartet nanofibers and (D) Sr2+/K+-stabilized G-quartet nanofibers synthesized at different concentrations. (15 °C) | ||
In addition to temperature, the concentration of reactants also plays an important role in the reaction rate. In order to explore whether the fast assembly process leads to the left-handed G-quartet chiral stacking, and the slow assembly process results in the right-handed G-quartet chiral stacking, the relationship between the initial concentration of reactants and the chirality of the assembly was studied. To avoid the structural influence caused by the different ratios of GMP to metal ions, the ratio of GMP to Sr2+ was fixed. As shown in Fig. 2C, we were glad to see that with the increase of initial reactant concentration, the chirality of the assembly was reversed from right-handed to left-handed, indicating that the fast assembly process corresponds to the left-handed G-quartet chiral stacking, and the slow assembly process corresponds to the right-handed G-quartet chiral stacking. A similar phenomenon was also observed in the Sr2+/K+ stabilized G-quartet system. As shown in Fig. 2D, with the increase of initial reactant concentration, the helix chirality of the assembly was also reversed from right-handed to left-handed. It is worth noting that the CD signal of the G-quartet structure with different initial reactant concentrations shows mirror spectra, demonstrating that there are opposite chiral stacking between the two nanostructures. Compared with Fig. 2C and D, we can find that the addition of K+ made the chiral inversion concentration change from 80 mM GMP to 70 mM GMP. Combined with the phenomenon that K+ accelerates the formation of g-fibers, we can conclude from another point of view that the fast process is conducive to the formation of left-handed G-quartet stacking. XRD and FTIR spectra also demonstrated the formation of G-quartet nanostructures under multiple concentrations (Fig. S1A and B, ESI†).
In the field of supramolecular self-assembly, kinetical trapping was proposed to explain the kinetics effect on molecular packing. In the kinetical trapping, the self-assembled architecture is not in the thermodynamic equilibrium state and has higher potential energy, thus forming self-assembly systems with distinct structures and functions. Usually, fast processes lead to kinetic traps while slow processes result in thermal equilibrium. In the current G-quartet self-assembly system, fast processes lead to the left-handed helix chirality, while slow processes result in the right-handed helix chirality. Therefore, left-handed self-assembly corresponded to the kinetically trapped state and right-handed self-assembly corresponded to the thermodynamic equilibrium state. In a previous study on G-quartets, researchers found that the quartets are stacked in a right-handed way when all quartets were in the same orientation, such as a head-to-tail stacking (homopolar). Meanwhile, when quartets stack through the same face (heteropolar, head-to-head or tail-to-tail), the CD signals are opposite to that displayed in homopolar stacking in the specific wavelength range.14,27 Therefore, we believe that the ordered head-to-tail stacking occurred in the slow formation process, while heteropolar stacking occurred in the fast formation process.
Fluorescent dye thioflavin T (ThT) is an efficient inducer for G-quadruplex DNA and a molecular chaperone for G-quartet nanostructures. In order to endow G-quartets with fluorescence properties, ThT and G-quartet fibers were co-assembled. As shown in Fig. 3A, the helix chirality of G-quartet nanostructures was changed by relatively less ThT. Compared with Fig. 1A, we can see that ThT significantly enhanced the left-handed helix chirality of g-fibers formed at low temperature (3 °C) and decreased the right-handed helix chirality at high temperature (10 °C), demonstrating that the involvement of ThT is beneficial for the formation of left-handed G-quartet stacking. More importantly, a new 450 nm positive peak appeared when ThT and g-fibers were co-assembled at 3 °C (left-handed g-fbers), while a new 450 nm negative peak arose when ThT and g-fibers were co-assembled at 15 °C (right-handed g-fibers), demonstrating that the chirality twist of ThT was induced after intercalation into g-fibers and its chirality is completely dependent on the helix chirality of g-fibers.
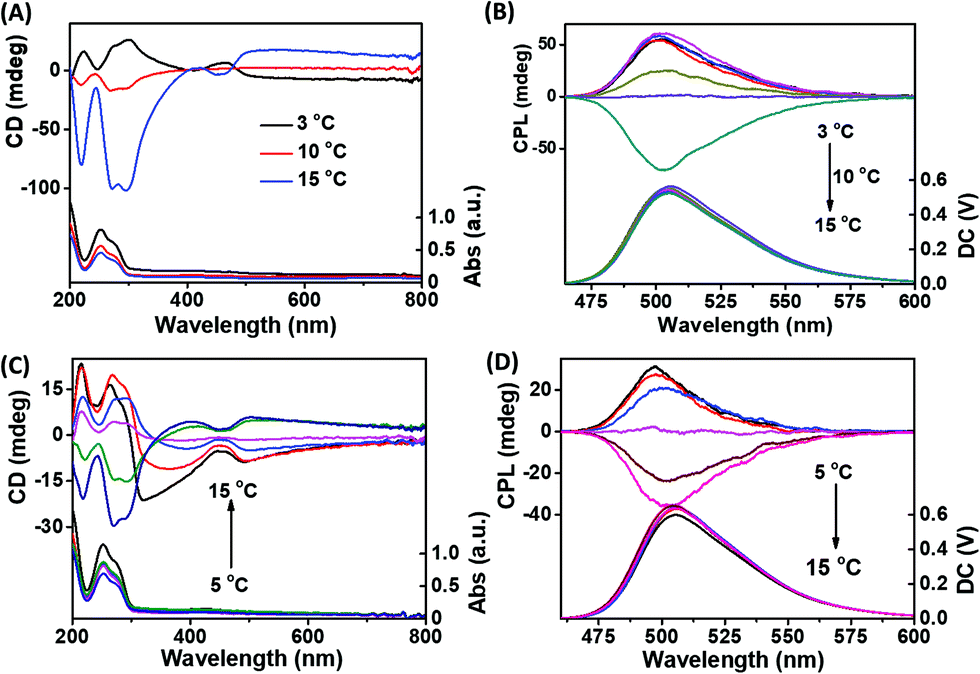 | ||
| Fig. 3 (A) CD and (B) CPL spectra of Sr2+-stabilized ThT-doped G-quartet nanofibers synthesized at different temperatures; (C) CD and (D) CPL spectra of Sr2+/K+-stabilized ThT-doped G-quartet nanofibers synthesized at different temperatures. (CGMP = 50 mM, CSr2+ = 8 mM, Ck+ = 5 mM, CThT = 100 μM) | ||
For a long time, how to simply construct reversible CPL-active materials has been a difficult problem. Due to the reversible helix-handedness controlled by a kinetic process, G-quartet nanostructures may be an ideal template to solve this problem. From Fig. 3B, we can clearly see that the CPL signal decreased gradually and finally realized the inversion when the synthesis temperature decreased from 15 °C to 3 °C. Compared with Fig. 3A and B, we can see that the chirality of CPL signals is consistent with the helical chirality of G-quartet structures, showing that both left-handed and right-handed CPL can be obtained from this system by changing the synthesis temperature. A similar result was also obtained in the Sr2+/K+ stabilized G-quartet system (Fig. 3C and D). The relationship between the initial concentration of reactants and the chiral properties of CPL emitted by fluorescent g-fibers was also studied. As shown in Fig. S2 (ESI†), with the increase of initial reactant concentration, the chiral properties of CPL were reversed from right-handed to left-handed, showing that both left-handed and right-handed CPL can be obtained from this system by controlling the initial reactant concentration.
In order to prove the effect of the kinetic process on CPL more strictly, the fluorescent g-fibers synthesized at different cooling rates were studied. As shown in Fig. 4, with the cooling rate changing from 30 °C min−1 (fast process) to 0.25 °C min−1 (slow process), the chiral properties of CPL were reversed from right-handed to left-handed, which was consistent with the conclusion that left-handed self-assembly corresponds to the kinetically trapped state and right-handed self-assembly corresponds to the thermodynamic equilibrium state.
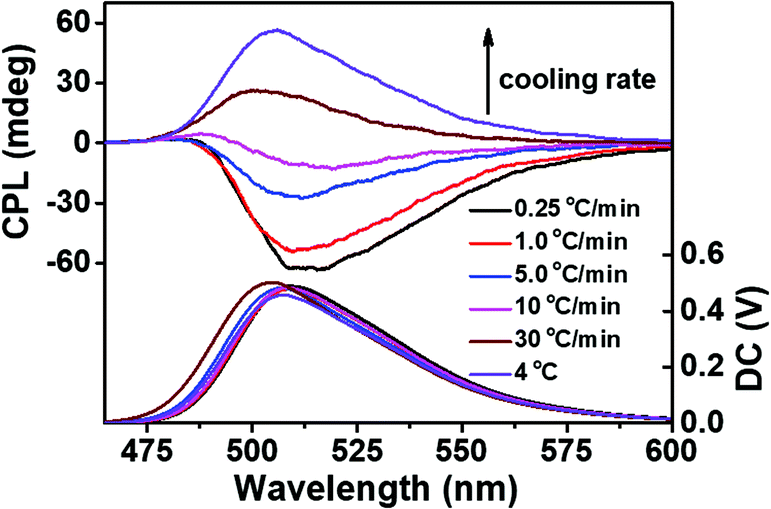 | ||
| Fig. 4 CPL spectra of Sr2+-stabilized ThT doped G-quartet nanofibers synthesized at different cooling rates. | ||
In summary, we report here that the handedness and CPL property of G-quartet materials highly depend on its formation kinetics. In GMP/Sr2+ and GMP/Sr2+/K+ self-assembly systems, by controlling the temperature or the initial concentration of reactants, right-handed helical G-quartet nanostructures were synthesized in slow processes, while left-handed structures were synthesized in fast processes. The reason is that the slow process leads to thermodynamic equilibrium, and the fast process results in the kinetic trap state. Furthermore, the corresponding opposite CPL phenomenon in G-quartet materials via chirality transfer was also controlled by kinetic factors such as temperature, concentration and cooling rate, which has potential design and application prospects. Especially in the field of drug loading and treatment, the most popular research field,28–30 chiral G-quartet nanostructures may be a good drug carrier.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China [No. 21705106]; the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning [No. TP2016023]; the Shanghai Natural Science Foundation [No. 18ZR1415400]; the Shanghai Rising-Star Program [No. 20QA1403400]; and the Shanghai Sailing Program [No. 20YF1413000].Notes and references
- J.-M. Lehn, C. R. Chim., 2011, 14, 348–361 Search PubMed.
- J. Li, R. Xing, S. Bai and X. Yan, Soft Matter, 2019, 15, 1704–1715 Search PubMed.
- H. Acar, S. Srivastava, E. J. Chung, M. R. Schnorenberg, J. C. Barrett, J. L. LaBelle and M. Tirrell, Adv. Drug Delivery Rev., 2017, 110–111, 65–79 Search PubMed.
- G.-B. Qi, Y.-J. Gao, L. Wang and H. Wang, Adv. Mater., 2018, 30, 1703444 Search PubMed.
- G. M. Peters and J. T. Davis, Chem. Soc. Rev., 2016, 45, 3188–3206 Search PubMed.
- F. Pu, J. Ren and X. Qu, Chem. Soc. Rev., 2018, 47, 1285–1306 Search PubMed.
- G. W. Collie and G. N. Parkinson, Chem. Soc. Rev., 2011, 40, 5867–5892 Search PubMed.
- R. Zhong, Q. Tang, S. Wang, H. Zhang, F. Zhang, M. Xiao, T. Man, X. Qu, L. Li, W. Zhang and H. Pei, Adv. Mater., 2018, 30, 1706887 Search PubMed.
- D. Hu, J. Ren and X. Qu, Chem. Sci., 2011, 2, 1356 Search PubMed.
- A. Ghosh, B. Parasar, T. Bhattacharyya and J. Dash, Chem. Commun., 2016, 52, 11159–11162 Search PubMed.
- M. Gellert, M. N. Lipsett and D. R. Davies, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 2013–2018 Search PubMed.
- Y. Huang, X. Vidal and A. E. Garcia-Bennett, Angew. Chem., Int. Ed., 2019, 58, 10859–10862 Search PubMed.
- G. Wu, I. C. M. Kwan, Z. Yan, Y. Huang and E. Ye, J. Nucleic Acids, 2017, 2017, 1–7 Search PubMed.
- J. Chen, X. Liu, Z. Suo, C. Gao, F. Xing, L. Feng, C. Zhao, L. Hu, J. Ren and X. Qu, Chem. Commun., 2020, 56, 7706–7709 Search PubMed.
- X. Wang, M. Li, P. Song, X. Lv, Z. Liu, J. Huang and Y. Yan, Chem. – Eur. J., 2018, 24, 13734–13739 Search PubMed.
- H. Choi, S. Heo, S. Lee, K. Y. Kim, J. H. Lim, S. H. Jung, S. S. Lee, H. Miyake, J. Y. Lee and J. H. Jung, Chem. Sci., 2020, 11, 721–730 Search PubMed.
- Q. Li, G. Zhang, Y. Wu, Y. Wang, Y. Liang, X. Yang, W. Qi, R. Su and Z. He, J. Colloid Interface Sci., 2021, 583, 234–242 Search PubMed.
- U. Mazur and K. W. Hipps, Chem. Commun., 2015, 51, 4737–4749 Search PubMed.
- A. Langenstroer, K. K. Kartha, Y. Dorca, J. Droste, V. Stepanenko, R. Q. Albuquerque, M. R. Hansen, L. Sánchez and G. Fernández, J. Am. Chem. Soc., 2019, 141, 5192–5200 Search PubMed.
- L. Herkert, J. Droste, K. K. Kartha, P. A. Korevaar, T. F. A. de Greef, M. R. Hansen and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 11344–11349 Search PubMed.
- Y. Yan, J. Huang and B. Z. Tang, Chem. Commun., 2016, 52, 11870–11884 Search PubMed.
- M. Schadt, Annu. Rev. Mater. Sci., 1997, 27, 305–379 Search PubMed.
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 Search PubMed.
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1900110 Search PubMed.
- J. Chen, Y. Chen, L. Zhao, L. Feng, F. Xing, C. Zhao, L. Hu, J. Ren and X. Qu, J. Mater. Chem. C, 2019, 7, 13947–13952 Search PubMed.
- Y. Gao, C. Ren, X. Lin and T. He, Front. Chem., 2020, 8, 458 Search PubMed.
- S. Masiero, R. Trotta, S. Pieraccini, S. De Tito, R. Perone, A. Randazzo and G. P. Spada, Org. Biomol. Chem., 2010, 8, 2683 Search PubMed.
- Y. Kuang, T. Li, T. Jia, A. Gulzar, C. Zhong, S. Gai, F. He, P. Yang and J. Lin, Small, 2020, 16, 2003799 Search PubMed.
- J. Xu, J. Zhou, Y. Chen, P. Yang and J. Lin, Coord. Chem. Rev., 2020, 415, 213328 Search PubMed.
- Z. Wang, T. Jia, Q. Sun, Y. Kuang, B. Liu, M. Xu, H. Zhu, F. He, S. Gai and P. Yang, Biomaterials, 2020, 228, 119569 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb00683e |
| This journal is © The Royal Society of Chemistry 2021 |