DOI:
10.1039/D1TA06759A
(Paper)
J. Mater. Chem. A, 2021,
9, 25391-25398
Nitride MXenes as sulfur hosts for thermodynamic and kinetic suppression of polysulfide shuttling: a computational study†
Received
9th August 2021
, Accepted 6th October 2021
First published on 7th October 2021
Abstract
The practical applications of lithium–sulfur (Li–S) batteries are greatly hindered by the poor conductivity of sulfur, the shuttling of lithium polysulfides (LiPSs), and the sluggish kinetics in the charge–discharge process. In order to solve these problems, here we propose the surface-functionalized V2N MXenes as the host materials to improve the electrochemical performance of Li–S batteries. Based on the density functional theory (DFT) calculations, we found that both the bare and functionalized V2NT2 (T = O, F, OH, and S) exhibit metallicity, and three of them (V2NO2, V2NF2, and V2NS2) possess moderate LiPS adsorption strength, which thermodynamically benefits the suppression of the dissolution and shuttling of LiPSs. Besides, V2NS2 shows the lowest Gibbs free energy barrier for the sulfur reduction reaction (0.49 eV) during discharge, which kinetically suppresses the dissolution and shuttling of LiPSs by expediting the decomposition process from soluble LiPSs to insoluble ones. Moreover, surface functionalized V2NT2 also exhibits outstanding catalytic ability for Li2S decomposition during charge, which decreases the energy barrier from 3.64 eV (bare V2N) to 1.55 (V2NO2) and 1.19 eV (V2NS2), and increases the charging kinetics. Based on these results, V2NS2 monolayers are suggested as promising host materials for S cathodes due to the fast charge/discharge kinetics and effective suppression of LiPS shuttling. This theoretical study provides further insight into the application of nitride MXenes and other two-dimensional materials as conductive anchoring materials for Li–S batteries.
1. Introduction
In recent years, battery technology has made considerable progress which greatly promotes the application of large-scale electrical energy storage in our daily life.1–6 Lithium–sulfur (Li–S) batteries, because of their high theoretical specific capacity of 1675 mA h g−1 and high theoretical specific energy of 2600 W h kg−1, have received a great deal of attention.7–11 Their practical energy density is expected to be 500–600 W h kg−1, which is around twice that of traditional lithium-ion batteries. Moreover, Li–S batteries show an inherently low competitive cost because of the abundance of sulfur.
Despite these promising features, practical applications of Li–S batteries are hindered by several obstacles.12–15 One of the major issues is the short cycle life stemming from the dissolution of lithium polysulfides (LiPSs) into liquid electrolytes and the migration of LiPSs between the anode and the cathode, which causes the so-called “shuttle effect” and would induce low discharge energy capacity and low coulombic efficiency. Besides, the relatively poor electrical conductivity of sulfur and the ∼80% volume expansion during the cycling of sulfur will lead to low utilization of active materials and rapid aging of electrodes. Last but not least, the large energy barriers of the sulfur reduction reaction (SRR) during discharge and Li2S decomposition during charge lead to slow kinetics in Li–S batteries, which greatly affects their performance.16,17 Therefore, the rational design of Li–S host materials should take into consideration not only the suppression of LiPS shuttling, but also the catalytic activity of the SRR and Li2S decomposition, the latter being long overlooked.
Two-dimensional (2D) materials have been demonstrated to be effective sulfur hosts with improved electrochemical performance for Li–S batteries.18–20 Among them, 2D transition metal carbides and nitrides, named MXenes, have attracted much attention due to their excellent electronic conductivity, high surface area, controllable hierarchical structures, and physical confinement for electrode volume changes.21–26 For instance, Liang et al. demonstrated that 70 wt% S/Ti2C composites exhibited long-term stability as cathodes and showed excellent cycling performance with a specific capacity close to 1200 mA h g−1 at C/5 current rate;27 Zhang et al. used a phase engineering strategy to fabricate Ti3C2 MXene/1T-2H MoS2–C nanohybrids which greatly improved the capacity (1194.7 mA h g−1 at 0.1C), rate ability (677.2 mA h g−1 at 2C), and cycling stability (0.07% per cycle) in Li–S batteries.28 Nevertheless, their bifunctional catalytic activities toward the SRR and Li2S decomposition have not been investigated yet, and until now, the majority of studies about the application of MXenes in Li–S batteries are focused on the carbides; more than 20 types of carbide MXenes have been synthesized while only a few nitride MXenes with higher conductivity than corresponding carbides are reported.21,29,30 Recently, the successful synthesis of 2D V2N and Mo2N further expanded the family of experimentally achievable 2D transition-metal nitrides.29 Particularly, V2N MXenes are worthy of attention because the VN/graphene composite has been experimentally proven to have a number of desirable properties as a Li–S host material in a pioneering report.31 For the vanadium element, its multiple-valence-state feature (from V2+ to V5+), stable crystalline framework when forming compounds, and rich distribution in the Earth's crust32 endow V2N MXenes with great potential as sulfur hosts.
Except for the surface terminations (–O, –F, and –OH) which are commonly introduced during the MXene synthesis process,33–35 non-native functional groups, such as –S, also inevitably exist on the surface of MXenes when they are used in Li–S batteries. It is also reported that S-functionalized MXenes may exhibit lower Li2S decomposition barrier because of the trapping effect between Li atoms and the S-containing surface.16 Thus, the understanding of these surface groups, including the distribution and arrangement of surface terminations, is important in both theoretical studies and the interpretation of the resultant properties. In this work, we select the V2N monolayer as a typical nitride MXene to systematically investigate its potential as a host material by means of DFT calculations. The electronic properties, anchoring strength, catalytic ability, and vacancy effect are comprehensively studied. Our calculations reveal that the anchoring strength and bifunctional catalytic ability of V2N can be modulated by the surface terminating groups (T = O, F, and S). The computational scheme in this work can open up a new avenue for the rational design of other 2D materials as hosts for S cathodes.
2. Computational methods
First-principles calculations were performed in the framework of DFT using the Vienna ab initio simulation package (VASP).36 The exchange-correlation energy was described by the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional.37 For geometry optimization, the plane-wave cut-off energy was set as 500 eV. The Brillouin-zone integration was performed using a Monkhorst–Pack grid of k-point sampling, and the Γ-centered meshes of 15 × 15 × 1 and 3 × 3 × 1 were used for the unit cell and the 4 × 4 × 1 supercell, respectively. During geometric relaxation, the convergence criteria for energy and force were set at 10−5 eV and 0.01 eV Å−1, respectively, while for self-consistent field calculations, an energy convergence criterion of 10−5 eV was also used.
The van der Waals (vdW) interaction between V2N/V2NT2 and Li2Sn (S8) (n = 1, 2, 4, 6, and 8) was considered by using the semiempirical DFT-D2 (ref. 38) approach. A vacuum layer of 20 Å along the c-axis was added to the slabs to avoid the interlayer interaction. Furthermore, charge differential analysis was performed to estimate the charge redistribution and transfer. The climbing image nudged elastic band (CI-NEB) method39 was applied to calculate the minimum diffusion energy barrier and decomposition barriers. To investigate the dynamical stability, the phonopy code was used to calculate the phonon dispersion spectra.40 During all the calculations, all the atoms were fully relaxed.
In the discharge process of Li–S batteries, the overall reaction of the reduction of one S8 molecule (SRR) to form eight Li2S molecules is a 16-electron process:17
| S8 + 16(Li+ + e−) → 8Li2S |
and the elementary steps in the reaction pathway for generating one Li
2S molecule is:
41,42| *S8 + 2(Li+ + e−) → *Li2S8 |
where * denotes active sites on the catalyst surface. The Gibbs free energy change Δ
G of each step was calculated using:
where Δ
E, ΔZPE, and
TΔ
S represent the changes of the DFT-calculated total energy, zero-point energy (ZPE), and entropic contribution, respectively. The ZPE and entropic contribution were calculated from the vibrational frequencies, and a temperature of 298 K was used.
3. Results and discussion
3.1 Structural and electronic properties of V2N and V2NT2 monolayers
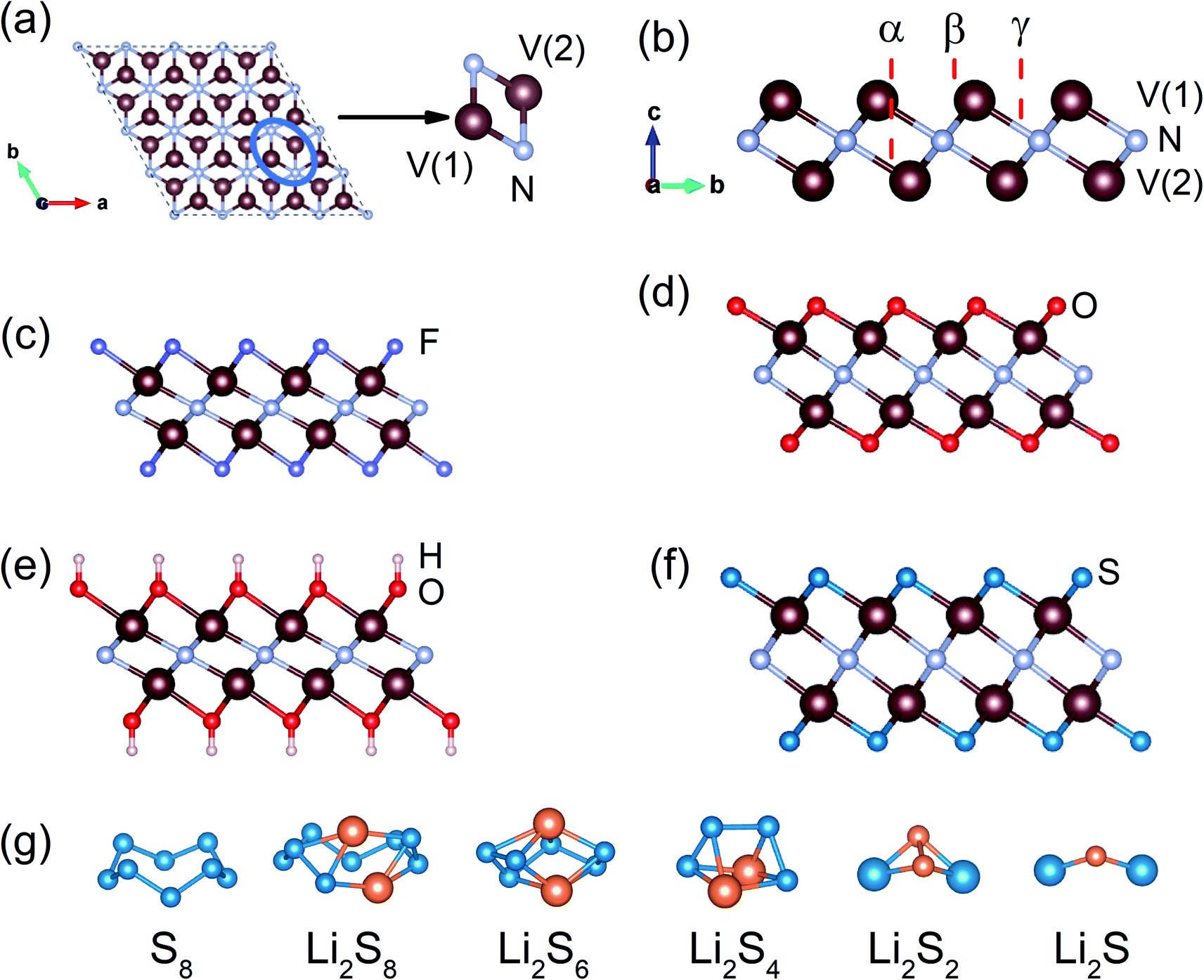
The V2N monolayer is built up of a sandwich structure containing one N and two V layers in the sequence of V(1)–N–V(2) (Fig. 1a and b). The optimized V2N monolayer has the lattice parameters of a = b = 2.89 Å and a thickness of c = 2.08 Å. In the phonon dispersion spectra, the absence of imaginary phonon modes in the first Brillouin zone indicates that the V2N monolayer is dynamically stable (Fig. S1†).40,43 For the V2NT2 monolayer, the most favorable sites for terminating groups T are determined by performing the total energy calculations. Three possible high-symmetry sites α, β, and γ are considered (Fig. 1b), and the total energies of all possible V2NT2 phases are listed in Table S1.† Based on the analysis, the α site is the most favorable adsorption site (Fig. 1c–f). With the terminal groups, both V(1)–N and V(2)-N bonds are elongated (Table S2†), implying that the terminal atoms strongly interact with the original V2N monolayer. The thermal stability of V2NT2 was verified by ab initio molecular dynamics (AIMD) simulations (Fig. S2†). Moreover, we also obtain the lowest energy configuration of the isolated S8 and Li2Sn molecules by DFT calculations, as shown in Fig. 1g and S3.† The structural parameters and symmetries of our configurations are very close to those obtained by B3LYP/6-311G(3df)-level quantum chemistry calculations44 and structure prediction using the particle-swarm optimization algorithm45 with negligible differences.
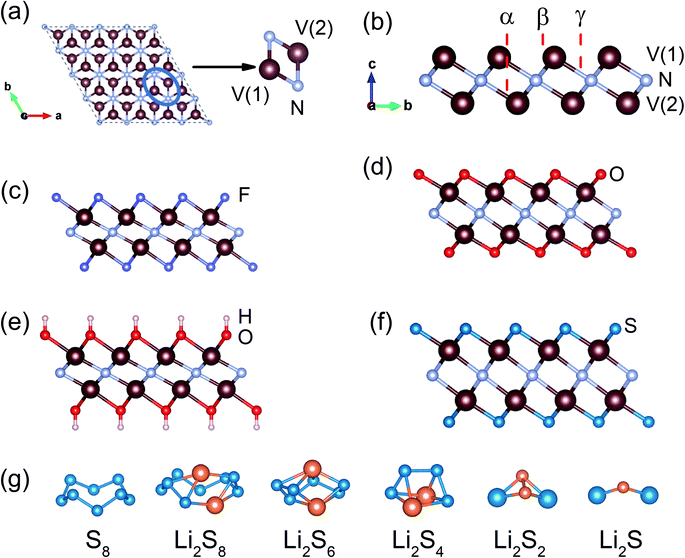 |
| | Fig. 1 DFT-optimized structures. (a) Top view of V2N; (b) side view of V2N, (c) V2NF2, (d) V2NO2, (e) V2N(OH)2; (f) V2NS2; (g) S8 and Li2Sn (n = 1, 2, 4, 6 and 8) structures. V, N, O, F, H, S and Li atoms are represented in brown, grey, red, purple, pink, blue and orange colours respectively. | |
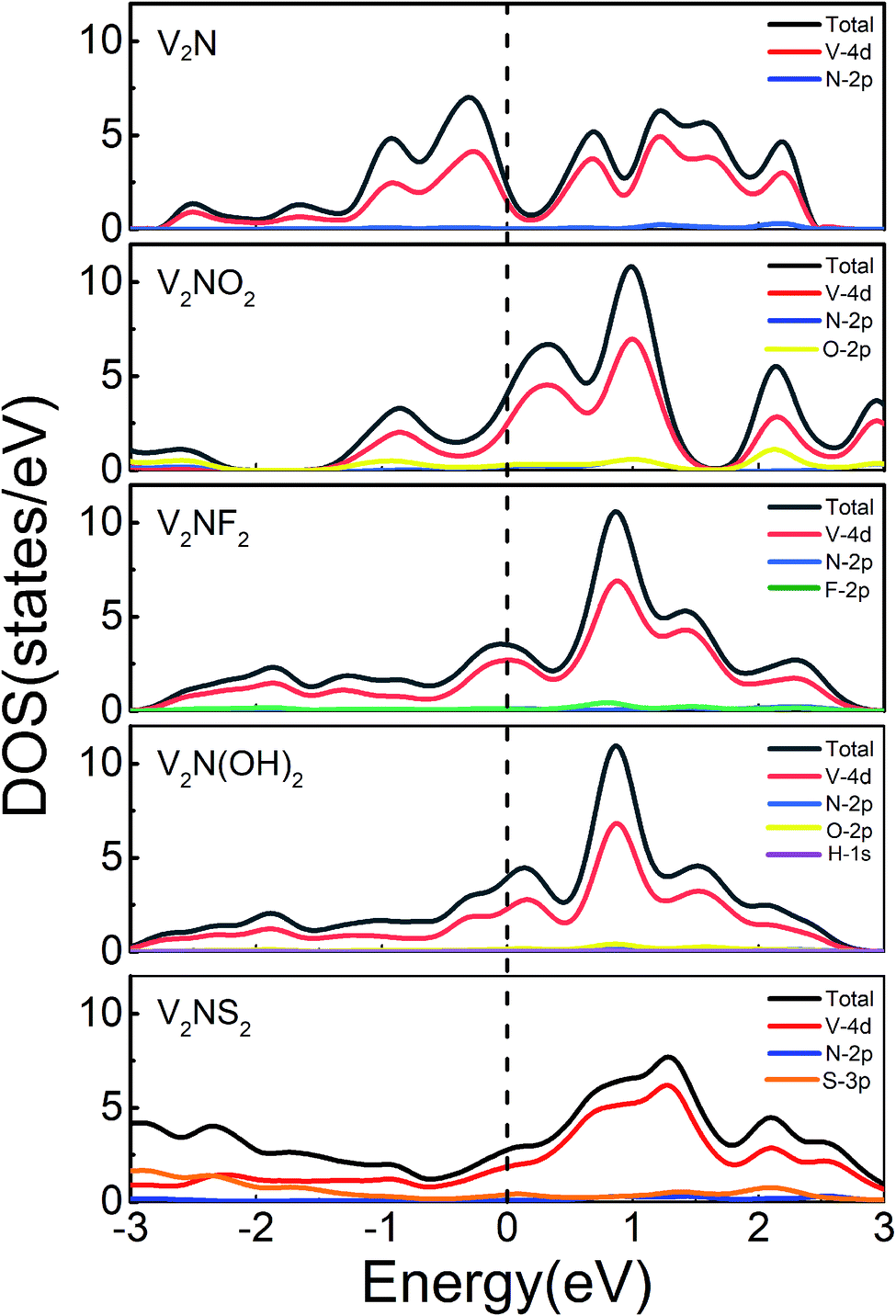
The metallicity of the sulfur host material is an important factor to consider for Li–S battery applications. Based on the calculated electronic density of states (DOS) of V2N and V2NT2, both the bare and terminated (V2N and V2NT2) phases exhibit excellent metallicity and the majority of electron states at the Fermi level originate from the V 4d orbitals (Fig. 2), with very little contribution from the non-metal atoms. As a result, the excellent metallic characters of V2N and V2NT2 are expected to improve the electrochemical performance of Li–S cells. Since sulfur and short-chain LiPSs (Li2S2 and Li2S) exhibit insulating behavior,42 the metallicity of V2N and V2NT2 is particularly significant for the electron transfer in the bifunctional catalytic reaction (SRR and Li2S decomposition).
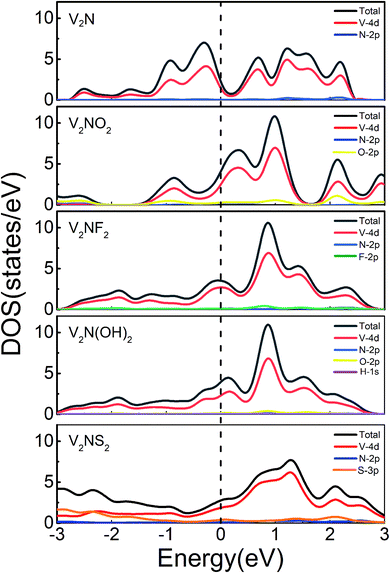 |
| | Fig. 2 Total and partial density of states of 2D V2N and V2NT2 (T = O, F, OH, and S). Fermi levels are set to be zero (denoted as black dotted lines). | |
3.2 Anchoring ability for S8 and Li2Sn
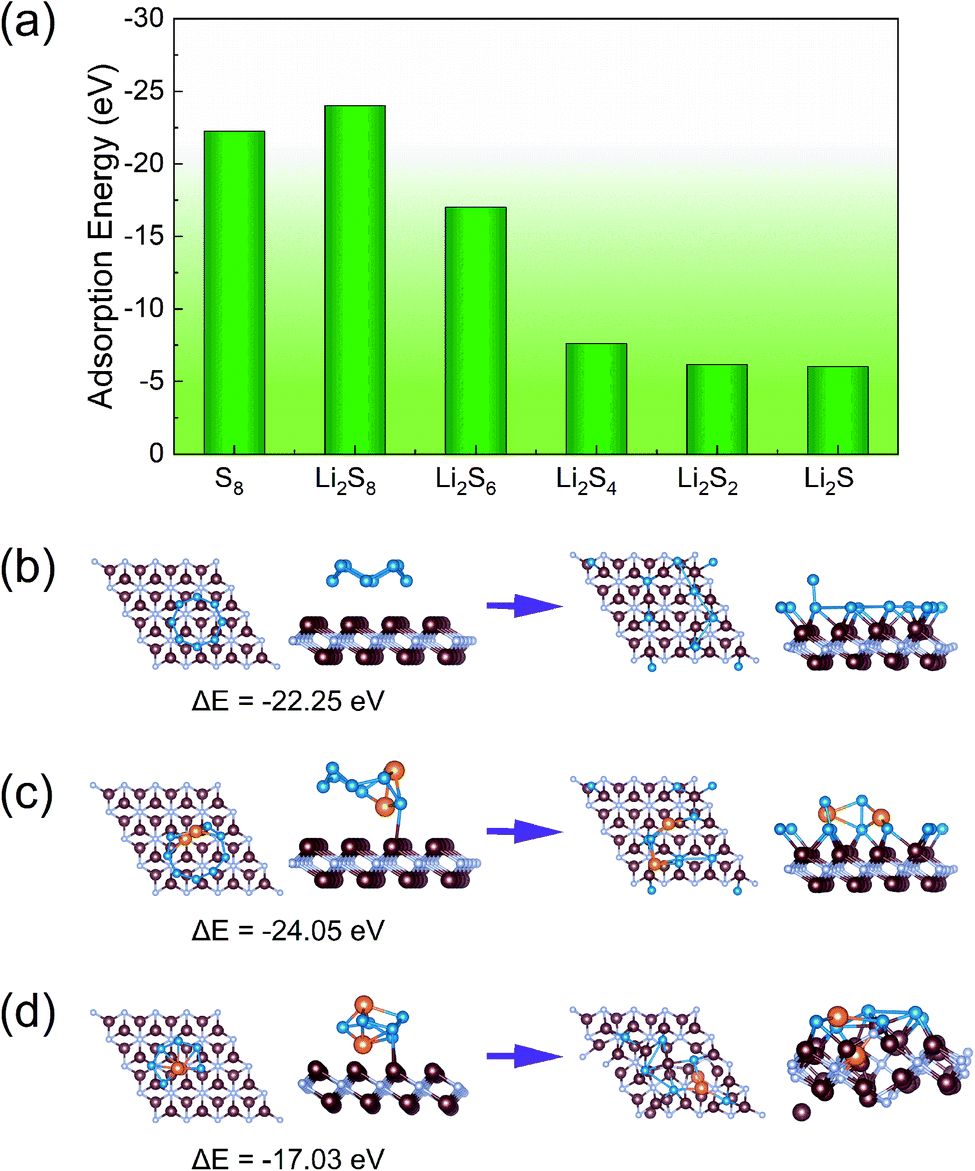
The anchoring effect of V2N and V2NT2 monolayers is evaluated by calculating the adsorption energies. We have conducted a comprehensive search for the most energy favorable adsorption sites for each S8 and Li2Sn species. To be specific, we have tested three symmetry unique sites (α, β, and γ) and two different rotational orientations on the host surface (Fig. S4,† with a total number of 180 different structures) for each compound to obtain the most stable adsorption site and configuration. The results showed that the adsorption strength of S8 and Li2Sn on bare V2N is extremely strong, as indicated by the large adsorption energy values in the range from −5 to −25 eV (Fig. 3a). However, with such a large adsorption strength, S8 and Li2Sn are easily decomposed into single S (and Li) atoms, which are then tightly captured by the outer V layer of V2N monolayers (Fig. 3b–d). Thus, bare V2N is not suitable for direct application in Li–S batteries, and it will automatically transform into S-functionalized V2N upon S8 adsorption, so it is not discussed in the following sections.
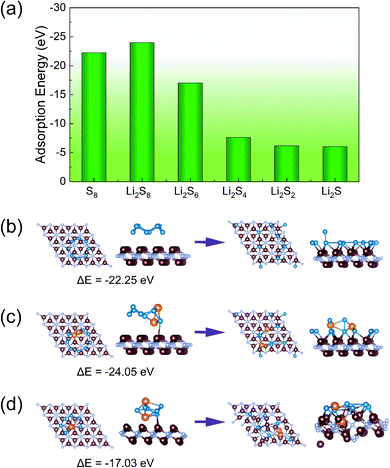 |
| | Fig. 3 (a) Adsorption energies of S8 and Li2Sn on V2N; adsorption structures of (b) S8, (c) Li2S8, and (d) Li2S6 on V2N before (left) and after (right) optimization. The V, N, Li, and S atoms are distinguished by brown, gray, orange, and blue colors, respectively. | |
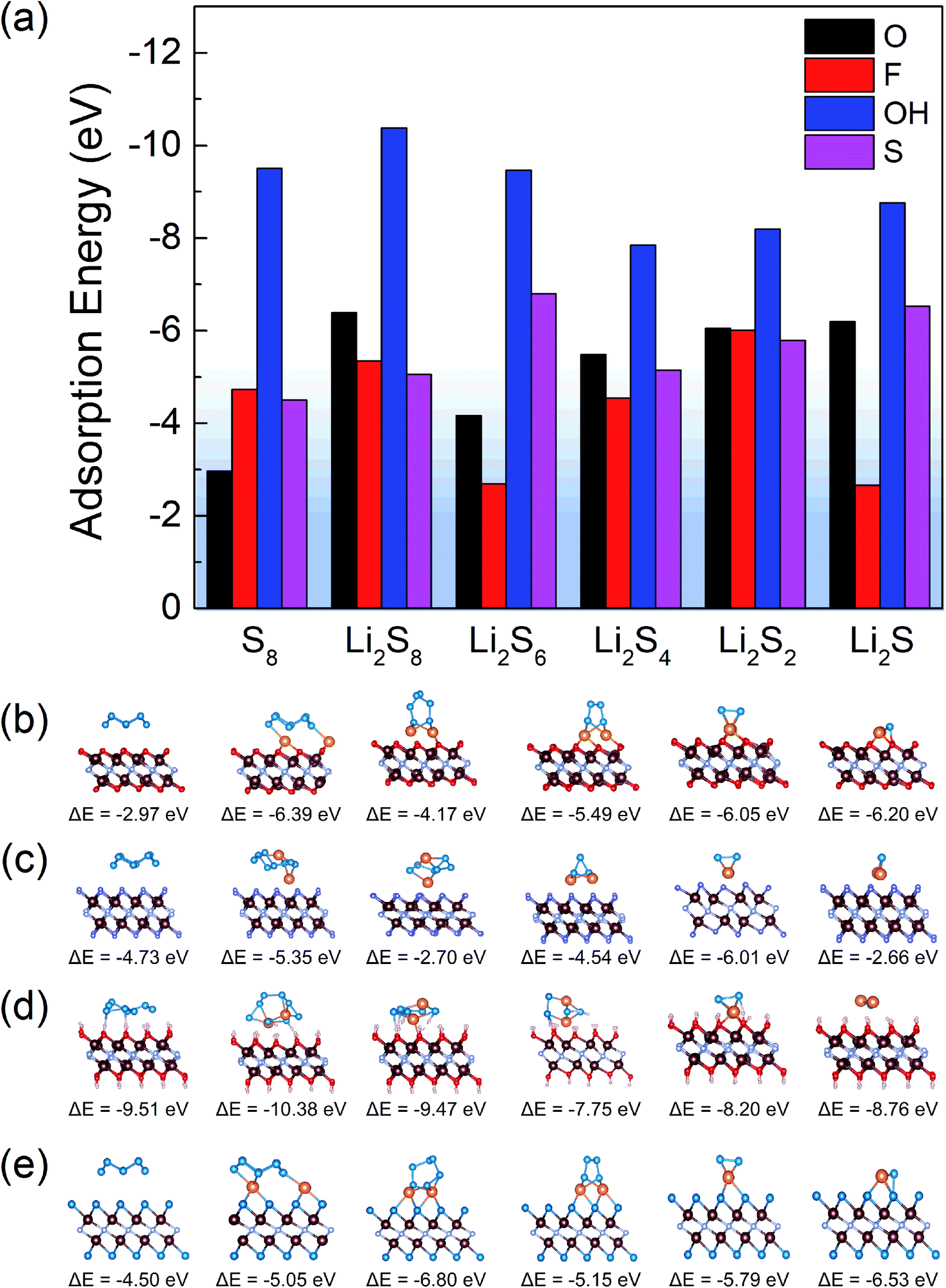
For all the functionalized V2NT2 structures, the adsorption strengths of S8 and Li2Sn on V2N(OH)2 are relatively stronger than those on V2NO2, V2NF2, and V2NS2 during the whole lithiation process (Fig. 4a). However, V2N(OH)2 becomes unstable after the adsorption of Li2Sn species, where H atoms spontaneously migrate toward S atoms in Li2Sn while the remaining O atoms interact with Li atoms, leading to the breakage of the Li2Sn species eventually (Fig. 4d). For V2NO2, V2NF2, and V2NS2 monolayers, in contrast, the structures remain stable, which indicates the relatively mild adsorption strength (Fig. 4b, c, and e). We also identify the interactions between V2NT2 and S8/Li2Sn by comparing the adsorption energies with and without vdW corrections. From the results of the ratio of vdW 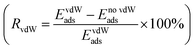 (Fig. S5†), we can conclude that physical interactions (vdW) play a major role in the adsorption of S8/Li2Sn on V2NS2, exhibiting RvdW values higher than 90%. V2NF2 and V2NO2, on the other hand, with relatively lower RvdW values especially for S8 and long-chain LiPSs, imply that chemical interactions also contribute a lot to the S8/Li2Sn adsorption. In short, the vdW interaction should be considered when studying the adsorption of LiPSs on V2NT2 monolayers.
(Fig. S5†), we can conclude that physical interactions (vdW) play a major role in the adsorption of S8/Li2Sn on V2NS2, exhibiting RvdW values higher than 90%. V2NF2 and V2NO2, on the other hand, with relatively lower RvdW values especially for S8 and long-chain LiPSs, imply that chemical interactions also contribute a lot to the S8/Li2Sn adsorption. In short, the vdW interaction should be considered when studying the adsorption of LiPSs on V2NT2 monolayers.
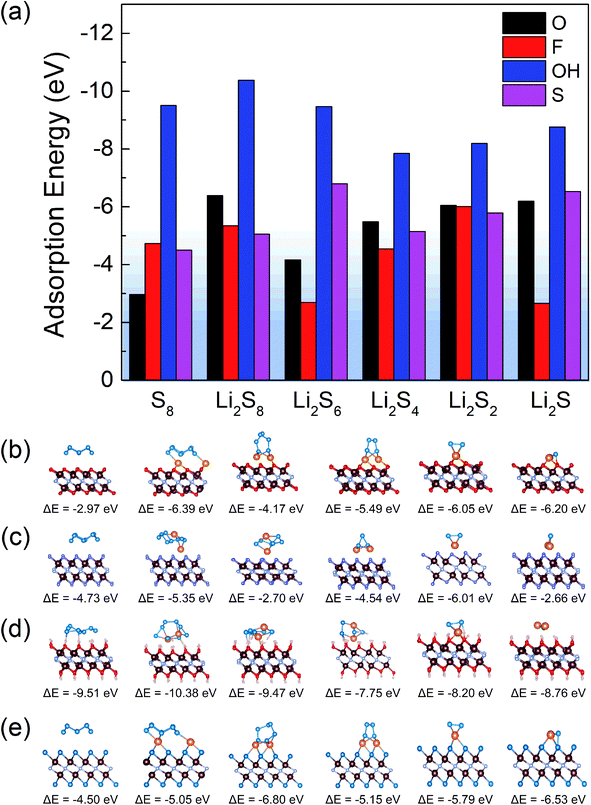 |
| | Fig. 4 (a) Adsorption energies of S8 and Li2Sn on V2NT2 (T = O, F, OH, and S), and the corresponding optimized structures of S8 and Li2Sn on the surface of (b) V2NO2, (c) V2NF2, (d) V2N(OH)2, and (e) V2NS2. The adsorption energy values are marked under each structure. The V, N, O, F, H, Li, and S atoms are distinguished by brown, gray, red, purple, pink, orange, and blue colors, respectively. | |
In addition, since there are usually small amounts of vacancies on the surface of MXenes, we also investigate the vacancy effect based on the structure of V2NO2−x/V2NF2−x/V2NS2−x (x = 1/16). Similar to intact V2NO2/V2NF2/V2NS2, none of the adsorbed Li2Sn structures exhibit a tendency of decomposition, and the adsorption of Li2Sn (S8) on V2NO2−x/V2NF2−x/V2NS2−x (x = 1/16) is energetically favorable (Fig. S6†). This can be attributed to the electron-rich regions with lone-pair electrons induced by the O/F/S vacancy, as indicated by the electron localization function (ELF) results (marked by red arrows in Fig. S7†),46 and after Li2Sn (S8) species are adsorbed on the surface of V2NT2−x, they can form chemical bonds with the vacancy sites through those electrons, which leads to the easy bonding with S of Li2Sn (S8). Consequently, for nitride-based MXenes, the shuttle suppression ability would not be hampered by the small amounts of vacancies in Li–S batteries.
To verify the ability of surface terminations in suppressing the LiPS shuttling, it is necessary to evaluate the binding effects between polysulfides and 1,3-dioxolane (DOL)/1,2-dimethoxyethane (DME) electrolyte.47,48 Through comparing the two different types of adsorption energies (EV2NT2+S8/Li2Sn and EDOL/DME+S8/Li2Sn), we found that S8 and Li2Sn prefer to be adsorbed on V2NT2 rather than DOL/DME molecules since the adsorption energies between LiPSs and DOL/DME molecules were between −1 and 0 eV (Fig. S8†), larger than those between LiPSs and V2NT2 monolayers (Fig. 4a). Based on the above discussion, V2NO2, V2NF2, and V2NS2 can not only fix S8 and Li2Sn on their surfaces but also maintain their own structures stably. Thus, they exhibit great potential in preventing polysulfide dissolution and shuttling in the electrolyte.
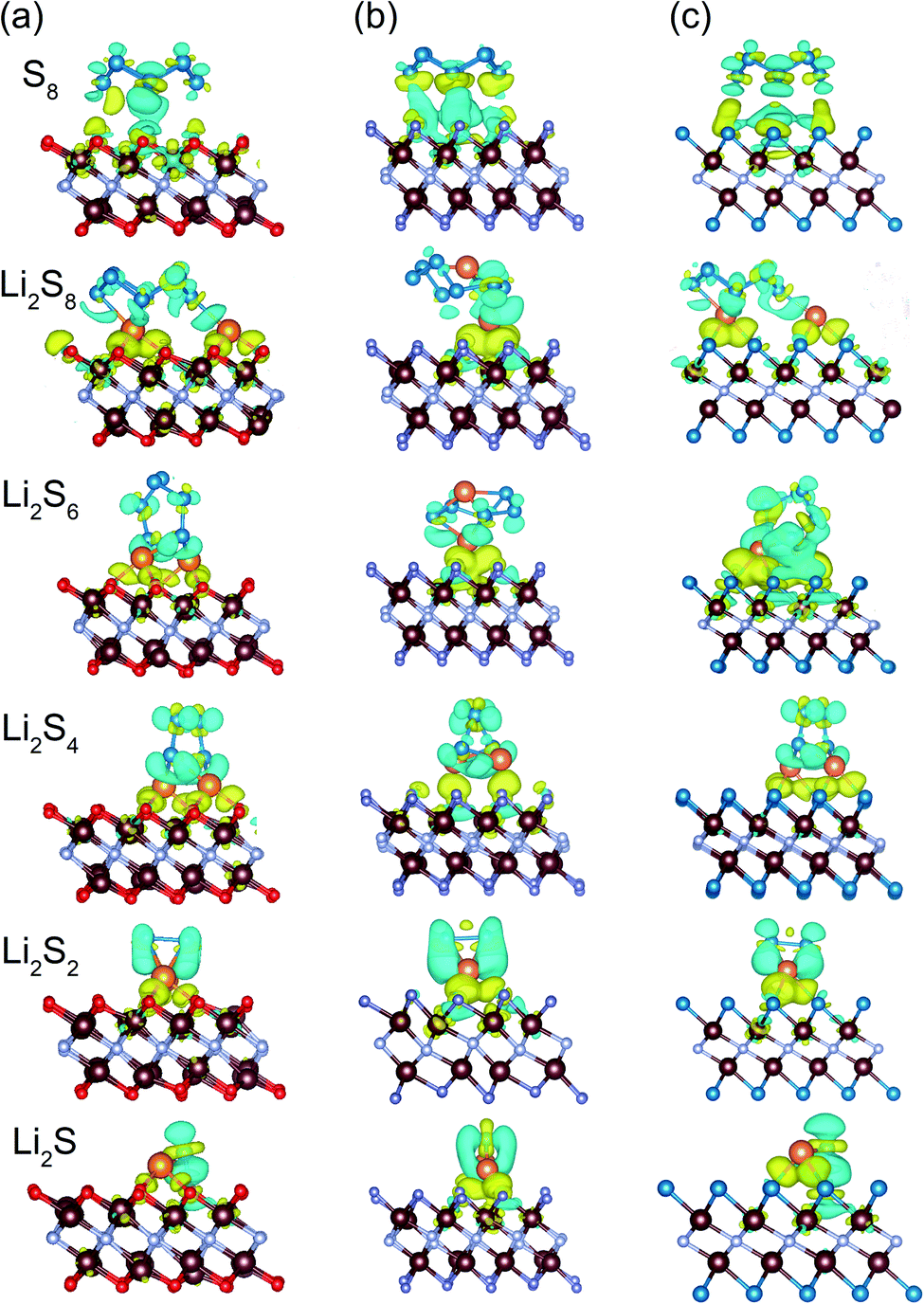
To assess the nature of the interaction between the host material and polysulfides, the charge density difference is also analyzed. Taking V2NO2 as an example (Fig. 5a), for S8, obvious charge transfer occurs between S and V, indicating the formation of polar covalent bonds between V and S. For other Li2Sn structures, electron accumulation regions (yellow) appear between the Li atoms and the outer O layer, while electron depletion (blue) happens around the Li–S and S–S bonds, explaining the binding behavior of Li2Sn on V2NO2 with the formation of Li–O bonds. A similar phenomenon can be observed in V2NF2 and V2NS2 (Fig. 5b and c).
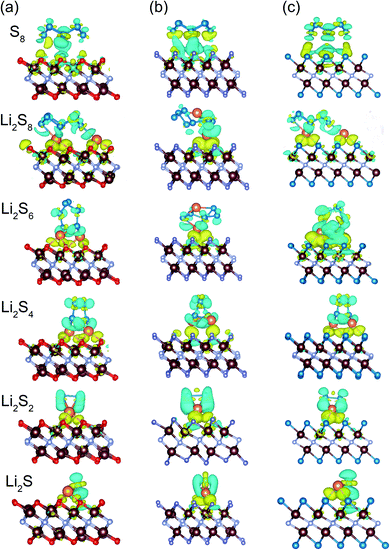 |
| | Fig. 5 Charge density difference of S8 and Li2Sn adsorbed on (a) V2NO2, (b) V2NF2, and (c) V2NS2. Yellow and blue regions indicate electron accumulation and depletion, respectively. The V, N, O, F, Li and S atoms are distinguished by brown, gray, red, purple, orange, and blue colors, respectively. | |
3.3 Bifunctional catalytic performance and Li+ diffusion kinetics for Li–S batteries
Apart from the anchoring ability, the diffusion properties and catalytic effect are also key indicators for Li–S batteries. In the discharge process, the fast kinetics of S8 reduction to long-chain soluble LiPSs (Li2S8, Li2S6, and Li2S4), and finally short-chain insoluble LiPSs (Li2S2 and Li2S) could shorten the LiPS accumulation time in the cathode and thus reduce the LiPS dissolution. The final discharge product, Li2S, which suffers from low Li+ diffusivity and high decomposition energy barrier greatly affects the conversion between the polysulfides and will cause high overpotential and low-rate capability. Thus, the decomposition kinetics of LiPSs during discharge and that of Li2S during charge processes, as well as Li+ diffusion on hosts, are all vital for the kinetic performance of Li–S batteries.
To analyze the decomposition kinetics of Li2S on V2NT2, the CI-NEB method is used. The decomposition process can be described by the following equation: Li2S → LiS + Li+ + e−. That is, a Li2S molecule can be decomposed into a LiS cluster by releasing a single Li atom, which corresponds to the charging process. The natural decomposition barrier of Li2S is calculated to be 3.64 eV, while the decomposition barriers on V2NO2, V2NF2, and V2NS2 are remarkably reduced to 1.55, 2.31, and 1.19 eV (Fig. S9†), respectively. Thus, surface functionalization can efficiently catalyze the Li2S decomposition and reduce the overpotential of Li–S batteries, especially for V2NO2 and V2NS2 (Table 1). Besides, the diffusion barriers were studied to discuss the Li+ diffusion on host materials. The energy profiles along the Li+ diffusion coordinates show much smaller barriers than decomposition, where the minimum Li+ diffusion barriers on V2NO2, V2NF2, and V2NS2 were 0.21, 0.17, and 0.16 eV, respectively (Fig. S10†). These values are comparable to or smaller than those for other MXenes such as V2CO2 (0.26 eV), Ti3CNO2 (0.26 eV), and Ti2NS2 (0.17 eV) (Table S3†).45,49–51 The low Li2S decomposition and Li+ diffusion barriers imply the improved kinetics during Li2S decomposition and Li+ diffusion, which can accelerate the whole charging process in the Li–S batteries.
Table 1 Calculated Li2S decomposition barriers, Li+ diffusion barriers, and ΔG of the rate-limiting step during the SRR on V2NO2, V2NF2, and V2NS2
| Substrate |
Li2S decomposition barrier (eV) |
Li+ diffusion barrier (eV) |
ΔG of the rate-limiting step during the SRR (eV) |
| V2NO2 |
1.55 |
0.21 |
1.88 |
| V2NF2 |
2.31 |
0.17 |
2.14 |
| V2NS2 |
1.19 |
0.16 |
0.49 |
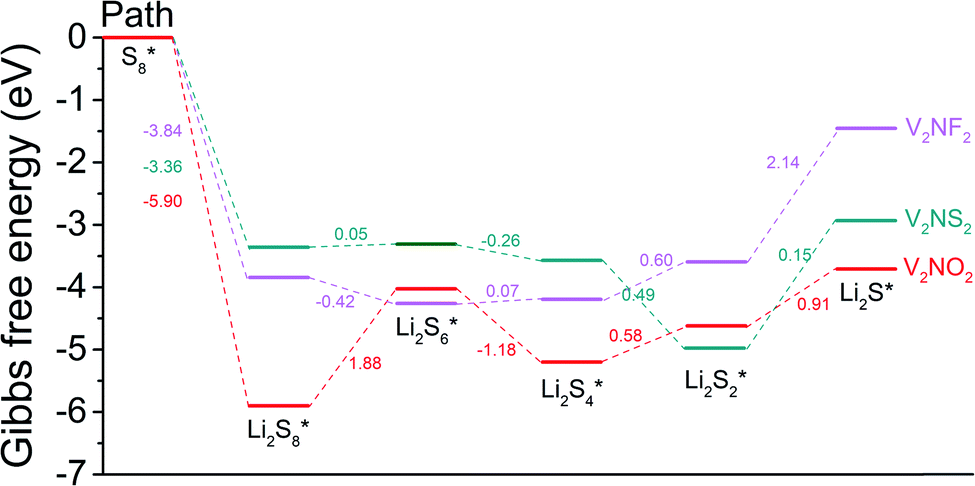
To gain a deeper understanding of the discharge reaction kinetics of polysulfides on the hosts (i.e. SRR), the overall reactions based on the reversible formation of Li2S from S8 and Li bulk were considered (Fig. 6). The Gibbs free energies for all the reaction steps in the SRR are calculated and the evolution profiles (from S8 to Li2S species) for V2NO2, V2NF2, and V2NS2 are exhibited.
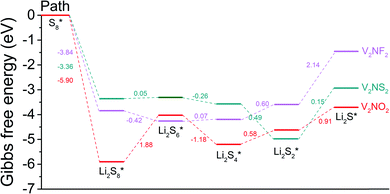 |
| | Fig. 6 Gibbs free energy profiles for the sulfur reduction reaction on V2NO2, V2NF2, and V2NS2. | |
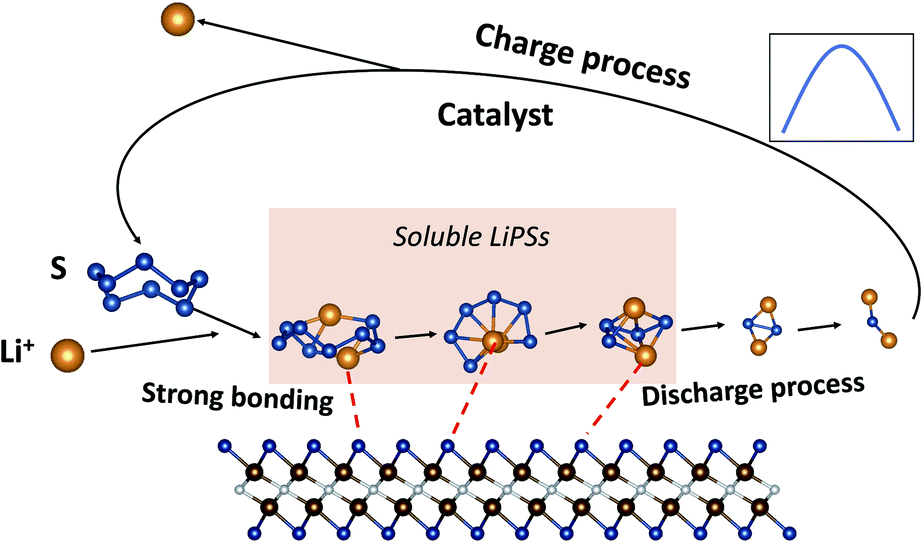
For V2NO2, it can be seen that the reduction steps from S8 to the Li2S8 and from Li2S6 to the Li2S4 are exothermic, with the Gibbs free energy change (ΔG) values of −5.90 and −1.18 eV, respectively. The other three reduction steps, from Li2S8 to Li2S6, Li2S4 to Li2S2, and Li2S2 to Li2S, are all endothermic, and among them, the step from Li2S8 to Li2S6 exhibits the largest ΔG value of 1.88 eV, which is determined as the rate-limiting step (RLS). For V2NF2, two exothermic reaction steps include those from S8 to Li2S8 and from Li2S8 to Li2S6, while the other three steps are endothermic steps, and RLS is the last step (Li2S2 to Li2S) with a ΔG of 2.14 eV. The significant differences in ΔG of step Li2S8 → Li2S6 and step Li2S2 → Li2S for the three systems are related to the differences in their anchoring abilities of LiPSs: for V2NO2, the Li2S8 adsorption is the strongest among the three systems, and its Li2S6 anchoring ability is relatively weak (Fig. 4), leading to its high Li2S8 → Li2S6 barrier; for V2NF2, the adsorption strength of Li2S is significantly decreased compared with that of Li2S2, while for V2NO2 and V2NS2, the Li2S2 and Li2S adsorption energies are relatively close to each other (Fig. 4). Among all three structures, V2NS2 exhibits the smallest ΔG value for RLS in the SRR (0.49 eV, Li2S4 to Li2S2, see Table 1) due to moderate LiPS adsorption. Thus, its outstanding bifunctional catalytic activity during charge (Li2S decomposition) and discharge (SRR) can be achieved simultaneously. In combination with its metallic behavior and moderate LiPS anchoring ability, V2NS2 can be an outstanding host material for Li–S batteries (Fig. 7).
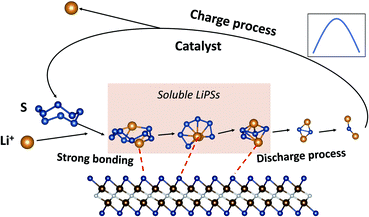 |
| | Fig. 7 Schematics of 2D V2NS2 MXenes as promising sulfur hosts for Li–S battery applications. | |
4. Conclusions
In summary, we have explored the potential applications of 2D V2N/V2NT2 monolayers as the S cathode host for Li–S batteries by means of DFT calculations. It was shown that both bare and functionalized V2NT2 monolayers exhibited excellent metallic character. However, bare V2N cannot be directly used in Li–S batteries because of the extremely strong V–S or V–Li bond. Among the four functional groups (O/F/OH/S) studied, the V2NS2 monolayer is identified as the most promising candidate through a comprehensive examination of its metallicity, LiPS anchoring ability, Li2S decomposition barrier, Li+ diffusion barrier, and SRR thermodynamics. This study gives insightful guidance for further experimental work on nitride MXenes and other novel 2D materials as promising host materials for Li–S batteries.
Author contributions
Ke Fan: conceptualization, data curation, investigation, software, methodology, writing – original draft; Yiran Ying: data curation, writing – review & editing; Xin Luo: supervision, writing – review & editing; Haitao Huang: funding acquisition, supervision, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. PolyU152178/20E), the Hong Kong Polytechnic University (Project No. RHA3), and Science and Technology Program of Guangdong Province of China (Project No. 2019A050510012 and 2020A0505090001). X. L. acknowledges support from NSFC (No. 11804286, No. 12172386), the National Natural Science Foundation of Guangdong Province, China (Grant No. 2021B1515020021), and the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (Grant No. 2021qntd27).
References
- Y. Yang, S. Bremner, C. Menictas and M. Kay, Renew. Sustain. Energy Rev., 2018, 91, 109–125 CrossRef
 .
.
- H. Chen, T. N. Cong, W. Yang, C. Tan, Y. Li and Y. Ding, Prog. Nat. Sci., 2009, 19, 291–312 CrossRef CAS .
- D. G. Ladha, Mater. Today Chem., 2019, 11, 94–111 CrossRef CAS .
- Z. Rao and S. Wang, Renew. Sustain. Energy Rev., 2011, 15, 4554–4571 CrossRef CAS .
- Q. He, B. Yu, Z. Li and Y. Zhao, Energy Environ. Mater., 2019, 2, 264–279 CrossRef CAS .
- X. Li, K. Li, S. Zhu, K. Fan, L. Lyu, H. Yao, Y. Li, J. Hu, H. Huang and Y. W. Mai, Angew. Chem., Int. Ed., 2019, 58, 6239–6243 CrossRef CAS PubMed .
- M. Barghamadi, A. Kapoor and C. Wen, J. Electrochem. Soc., 2013, 160, A1256–A1263 CrossRef CAS .
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC .
- A. Fotouhi, D. J. Auger, L. O'Neill, T. Cleaver and S. Walus, Energies, 2017, 10, 1937 CrossRef .
-
S. Walus, Université Grenoble Alpes, 2015 Search PubMed .
- X.-B. Cheng, J.-Q. Huang and Q. Zhang, J. Electrochem. Soc., 2017, 165, A6058 CrossRef .
- M. R. Busche, P. Adelhelm, H. Sommer, H. Schneider, K. Leitner and J. Janek, J. Power Sources, 2014, 259, 289–299 CrossRef CAS .
- D. Liu, C. Zhang, G. Zhou, W. Lv, G. Ling, L. Zhi and Q. H. Yang, Adv. Sci., 2018, 5, 1700270 CrossRef PubMed .
- G. Gao, F. Zheng, F. Pan and L.-W. Wang, Adv. Energy Mater., 2018, 8, 1801823 CrossRef .
- L. Fan, M. Li, X. Li, W. Xiao, Z. Chen and J. Lu, Joule, 2019, 3, 361–386 CrossRef CAS .
- G. Zhou, H. Tian, Y. Jin, X. Tao, B. Liu, R. Zhang, Z. W. Seh, D. Zhuo, Y. Liu and J. Sun, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 840–845 CrossRef CAS PubMed .
- L. Peng, Z. Wei, C. Wan, J. Li, Z. Chen, D. Zhu, D. Baumann, H. Liu, C. S. Allen and X. Xu, Nat. Catal., 2020, 3, 762–770 CrossRef CAS .
- M. Jana, R. Xu, X.-B. Cheng, J. S. Yeon, J. M. Park, J.-Q. Huang, Q. Zhang and H. S. Park, Energy Environ. Sci., 2020, 13, 1049–1075 RSC .
- L. Zhou, D. L. Danilov, R. A. Eichel and P. H. L. Notten, Adv. Energy Mater., 2020, 11, 2001304 CrossRef .
- X. Chen, H.-J. Peng, R. Zhang, T.-Z. Hou, J.-Q. Huang, B. Li and Q. Zhang, ACS Energy Lett., 2017, 2, 795–801 CrossRef CAS .
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed .
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS .
- Z. Xiao, Z. Li, X. Meng and R. Wang, J. Mater. Chem. A, 2019, 7, 22730–22743 RSC .
- C. Zhang, L. Cui, S. Abdolhosseinzadeh and J. Heier, InfoMat, 2020, 2, 613–638 CrossRef CAS .
- K. Fan, Y. Ying, X. Li, X. Luo and H. Huang, J. Phys. Chem. C, 2019, 123, 18207–18214 CrossRef CAS .
- X. Xu, Y. Zhang, H. Sun, J. Zhou, F. Yang, H. Li, H. Chen, Y. Chen, Z. Liu and Z. Qiu, Adv. Electron. Mater., 2021, 7, 2000967 CrossRef CAS .
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed. Engl., 2015, 54, 3907–3911 CrossRef CAS PubMed .
- Y. Zhang, Z. Mu, C. Yang, Z. Xu, S. Zhang, X. Zhang, Y. Li, J. Lai, Z. Sun and Y. Yang, Adv. Funct. Mater., 2018, 28, 1707578 CrossRef .
- P. Urbankowski, B. Anasori, K. Hantanasirisakul, L. Yang, L. Zhang, B. Haines, S. J. May, S. J. L. Billinge and Y. Gogotsi, Nanoscale, 2017, 9, 17722–17730 RSC .
- X. Xiao, H. Yu, H. Jin, M. Wu, Y. Fang, J. Sun, Z. Hu, T. Li, J. Wu and L. Huang, ACS Nano, 2017, 11, 2180–2186 CrossRef CAS PubMed .
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H. M. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef PubMed .
- M. Chen, Q. Liu, Z. Hu, Y. Zhang, G. Xing, Y. Tang and S. L. Chou, Adv. Energy Mater., 2020, 10, 2002244 CrossRef CAS .
- S. Lai, J. Jeon, S. K. Jang, J. Xu, Y. J. Choi, J.-H. Park, E. Hwang and S. Lee, Nanoscale, 2015, 7, 19390–19396 RSC .
- M. Naguib, R. R. Unocic, B. L. Armstrong and J. Nanda, Dalton Trans., 2015, 44, 9353–9358 RSC .
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed .
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed .
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed .
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed .
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS .
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS .
- R. S. Assary, L. A. Curtiss and J. S. Moore, J. Phys. Chem. C, 2014, 118, 11545–11558 CrossRef CAS .
- X. Song, Y. Qu, L. Zhao and M. Zhao, ACS Appl. Mater. Interfaces, 2021, 13, 11845–11851 CrossRef CAS PubMed .
- K. Fan, Y. Ying, X. Luo and H. Huang, Phys. Chem. Chem. Phys., 2020, 22, 16665–16671 RSC .
- L. Wang, T. Zhang, S. Yang, F. Cheng, J. Liang and J. Chen, J. Energy Chem., 2013, 22, 72–77 CrossRef CAS .
- D. Wang, F. Li, R. Lian, J. Xu, D. Kan, Y. Liu, G. Chen, Y. Gogotsi and Y. Wei, ACS Nano, 2019, 13, 11078–11086 CrossRef CAS PubMed .
- S. Zhang, Q. Wang, Y. Kawazoe and P. Jena, J. Am. Chem. Soc., 2013, 135, 18216–18221 CrossRef CAS PubMed .
- S. S. Zhang, J. Power Sources, 2013, 231, 153–162 CrossRef CAS .
- J. Scheers, S. Fantini and P. Johansson, J. Power Sources, 2014, 255, 204–218 CrossRef CAS .
- Y. Wang, J. Shen, L.-C. Xu, Z. Yang, R. Li, R. Liu and X. Li, Phys. Chem. Chem. Phys., 2019, 21, 18559–18568 RSC .
- V. Shukla, N. K. Jena, S. R. Naqvi, W. Luo and R. Ahuja, Nano Energy, 2019, 58, 877–885 CrossRef CAS .
- X. Chen, Z. Kong, N. Li, X. Zhao and C. Sun, Phys. Chem. Chem. Phys., 2016, 18, 32937–32943 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta06759a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Yiran
Ying
a,
Yiran
Ying
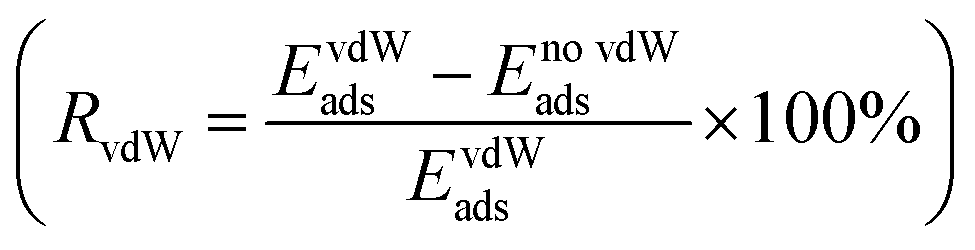 (Fig. S5†), we can conclude that physical interactions (vdW) play a major role in the adsorption of S8/Li2Sn on V2NS2, exhibiting RvdW values higher than 90%. V2NF2 and V2NO2, on the other hand, with relatively lower RvdW values especially for S8 and long-chain LiPSs, imply that chemical interactions also contribute a lot to the S8/Li2Sn adsorption. In short, the vdW interaction should be considered when studying the adsorption of LiPSs on V2NT2 monolayers.
(Fig. S5†), we can conclude that physical interactions (vdW) play a major role in the adsorption of S8/Li2Sn on V2NS2, exhibiting RvdW values higher than 90%. V2NF2 and V2NO2, on the other hand, with relatively lower RvdW values especially for S8 and long-chain LiPSs, imply that chemical interactions also contribute a lot to the S8/Li2Sn adsorption. In short, the vdW interaction should be considered when studying the adsorption of LiPSs on V2NT2 monolayers.