
Coacervation-driven instant paintable underwater adhesives with tunable optical and electrochromic properties†
Qiongyao
Peng
a,
Jingsi
Chen
 a,
Tao
Wang
a,
Lu
Gong
a,
Xuwen
Peng
a,
Meng
Wu
a,
Yuhao
Ma
b,
Feiyi
Wu
a,
Diling
Yang
a,
Hao
Zhang
a and
Hongbo
Zeng
*a
a,
Tao
Wang
a,
Lu
Gong
a,
Xuwen
Peng
a,
Meng
Wu
a,
Yuhao
Ma
b,
Feiyi
Wu
a,
Diling
Yang
a,
Hao
Zhang
a and
Hongbo
Zeng
*a
aDepartment of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta T6G 1H9, Canada. E-mail: hongbo.zeng@ualberta.ca; Fax: +1-780-492-2881; Tel: +1-780-492-1044
bDepartment of Biomedical Engineering, University of Alberta, Edmonton, Alberta T6G 2V2, Canada
First published on 5th April 2021
Abstract
Coacervation generally refers to liquid–liquid phase separation when mixing oppositely charged polyelectrolytes in an aqueous solution, which produces a polyelectrolyte-dense coacervate phase in a dilute solution phase. Coacervation plays a crucial role in many biological processes such as the versatile underwater adhesion of sessile organisms, cellular compartmentalization, and cell replication, which inspires a surge of biomimetic designs of underwater adhesives. However, conventional coacervate-based underwater adhesives are usually weak and lack functionalities, limiting their practical applications in biomedical and engineering fields. Herein, a novel instant underwater adhesive is presented based on coacervation via one-step mixing of aqueous solutions of silicotungstic acid (SiW) and poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (P123) micelles, driven by hydrogen-bonding and hydrophobic interactions. The as-prepared adhesive possesses instant and excellent underwater adhesion (up to 479.6 kPa on poly(methyl methacrylate)) and can be facilely painted underwater on diverse substrates, showing resistance to water flush as well as repeatable stretching and bending of the substrates, superior to many previously reported coacervates. The adhesive also exhibits outstanding stability in high-salt aqueous solutions up to 3 M for at least 1200 h. The introduction of P123 endows the adhesives with thermo-responsive optical properties, while the innate reduction-related color switch of SiW offers electrochromic properties. The functional adhesives hold great promise in bioengineering applications and for the fabrication of novel flexible electronics such as smart aqueous batteries and low-power electrochromic windows.
1. Introduction
Underwater adhesion is a great challenge for the development of universal adhesives, as water can hinder the intimate contact between the adhesives and the substrates by forming hydration layers on surfaces and weakening various adhesive bonds.1,2 Sessile organisms, such as sandcastle worms and mussels, can survive in the turbulent seawater environment and have attracted extensive and intensive research interest for their innate and excellent underwater adhesion on various substrates.3–7 Coacervation is considered to play a significant role in the secretion process of the underwater adhesives from sessile organisms,8–10 and it also contributes to the formation of biological tissues such as extracellular matrices11 and gradient materials like squid beak.12,13 Coacervation usually occurs when aqueous solutions of oppositely charged proteins, polymers and colloids are mixed, yielding a polyelectrolyte-rich liquid phase named a coacervate which coexists with an immiscible polyelectrolyte-sparse supernatant.14–16 Water-borne coacervates generally possess low interfacial energy to easily spread on different substrates, but further environment-triggered curing is required to fulfill the underwater adhesion. Moreover, their fabrication usually suffers from complex synthesis, specific stoichiometry of the components, and the adjustment of ionic strength and pH. Therefore, instant underwater adhesives that can be facilely prepared and delivered are highly desired for practical applications in complex environments (e.g., physiological environment), especially where ideal curing conditions can hardly be achieved.Various types of intermolecular interactions can induce coacervation, including electrostatic interaction, cation–π interaction, hydrogen-bonding interaction, and hydrophobic interaction. Sandcastle worm secretes two oppositely charged proteins from different glands interacting with each other via electrostatic interaction to generate a coacervate, which is crosslinked by Ca2+ and Mg2+ ions in the surrounding seawater and oxidized by the pH jump from the acidic glands to the basic seawater to achieve underwater adhesion.17 Adhesives mimicking the underwater adhesion of sandcastle worm have been developed, where complicated synthesis and specific environmental triggers such as temperature or pH jump are usually indispensable.5 Short-range cation–π interaction was found to drive the complex coacervation between like-charged mussel-inspired polyelectrolytes9 and in a single cationic recombinant mussel foot protein.10 A high salt concentration (at a seawater level of about 0.7 M) was needed for the coacervation of the cationic recombinant mussel foot protein10 and no macroscopic adhesive property was reported. Hydrogen-bonding interaction has been proved to contribute to the formation of adhesives based on the coacervation between tannic acid/silicotungstic acid and poly(ethylene glycol), which could serve as effective hemostatics.18,19 The preparation of such adhesives was very facile, but they were still unable to accomplish robust underwater adhesion. It is expected that the synergy of different intermolecular interactions could endow coacervation systems with strong underwater adhesion. A previous study showed that introducing thermo-responsive poly(N-isopropylacrylamide) (PNIPAM) into oppositely charged polyelectrolytes could endow the electrostatic interaction-driven coacervate with thermo-responsiveness, which turned the coacervate into an underwater adhesive hydrogel upon the increase of temperature, owing to the hydrophobic interaction among methyl groups of PNIPAM.20 In another study, a hydrophobic interaction-actuated tropoelastin-mimetic coacervate was equipped with mussel-inspired 3,4-dihydroxyphenylalanine (DOPA) groups to acquire adhesive property, and UV crosslinking was employed to achieve suitable cohesion.21 This charge-free coacervate could rapidly achieve underwater adhesion in less than 300 s despite a relatively low adhesion strength of less than 100 kPa.21 The synergetic effect of electrostatic and hydrogen-bonding interaction and other interactions between polyamidoamine-epichlorohydrin and tannic acid produced an instant underwater adhesive with antibacterial properties.22
Smart and functional underwater adhesives that can simultaneously achieve underwater adhesion and stimuli-responsive functionalities are intriguing and offer great promise for the development of next-generation electronic or biomedical devices. For example, adhesives with electrochemical properties can act as paintable/printable electrodes toward safe, portable and environment-friendly energy storage devices such as aqueous batteries.23 Electrochromic devices have been widely studied for their tunable optical properties which have great potential in applications including energy-saving smart windows, visual-comfortable displays, intelligent wearable electronics, eye-friendly rear mirrors and sunglasses, and so on.24–26 Electrochromic aqueous batteries attracted much research attention due to their charge/discharge-accompanied reversible color switch, showing great potential when used as user/device interfaces.23,27,28 However, achieving suitable flexibility still remains a challenge in the development of electrochromic aqueous batteries, as conventional electrochromic electrodes are usually fabricated by depositing nanostructured electrochromic materials on rigid transparent conductors via high-temperature treatment.24,27,29–32 3D printing has been demonstrated to be an efficient and low-cost solution, but until now, extra elastic polymer binders are required to attach the electrochromic nanomaterials on the conductive substrates, during which toxic organic solvents are usually used.33 Aqueous-based inks are desirable for 3D printing thanks to their inexpensiveness and environment-friendliness, however, organic solvents are still widely exploited to acquire good wettability.34 Therefore, water-borne adhesive coacervates with low interfacial tension, liquid-like properties and instant adhesiveness are ideal candidates for 3D printing to fabricate flexible devices, such as electrochromic electrodes used in the aqueous environment.23,35
In this work, we report the fabrication of a series of novel instant underwater electrochromic adhesives via simple mixing of silicotungstic acid (SiW, H4[Si(W3O10)4]) and poly(ethylene glycol)19-b-poly(propylene glycol)69-b-poly(ethylene glycol)19 (PEG-PPG-PEG, P123) aqueous solutions. The coacervation was mainly driven by the hydrogen bonding between SiW clusters and the PEG shells of P123 micelles, where the hydrophobic cores of P123 micelles provided a second crosslinking to reinforce the network. The synergy of the hydrogen bonding and hydrophobic interactions effectively enhanced the mechanical properties and the adhesiveness of the coacervates. The as-prepared coacervates could be painted on and bind various substrates underwater with instant and excellent adhesion strength up to 479.6 kPa on poly(methyl methacrylate) (PMMA). Additionally, the thermo-responsive property of P123 micelles endowed the coacervates with tunable optical properties in response to temperature. Owing to the well-known reduction reaction-related color change of SiW36 and the excellent stability of the coacervates in high-salinity aqueous solution, the coacervates exhibited quick and reversible electrochromic responsiveness to both self-powered electrochemical reaction and external voltages, making them promising electrode materials for flexible electronics.
2. Experimental section
2.1 Materials
Silicotungstic acid (SiW), Pluronic® P123 (poly(ethylene glycol)19-b-poly(propylene glycol)69-b-poly(ethylene glycol)19, average Mn ∼ 5800), Pluronic® F68 (poly(ethylene glycol)77-b-poly(propylene glycol)29-b-poly(ethylene glycol)77, average Mn ∼ 8400), indium tin oxide (ITO) coated glass slide (rectangular, surface resistivity 70–100 Ω sq−1, slide), poly(ethylene glycol) (BioUltra, for molecular biology, 8000), and LiOH were purchased from Sigma-Aldrich and used as received. Lithium chloride (LiCl, 98.5%), sodium chloride, potassium chloride, and calcium chloride were purchased from Fisher Scientific and used as received. Poly(methyl methacrylate) (PMMA) round disks with a diameter of 18.5 mm, wood and glass round disks with a diameter of 25 mm were purchased from Amazon Canada.2.2 Preparation of coacervates
Silicotungstic acid (SiW) aqueous solutions with weight percentages of 0.1%, 1%, 10%, 20% and 40% were prepared using deionized water. P123/F68 aqueous solutions with weight percentages of 0.1%, 1%, 10%, 15% and 20% were prepared using deionized water. In a typical preparation process of the coacervate, SiW aqueous solution and P123/F68 aqueous solution were directly mixed at the volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, followed by vigorously shaking and centrifugation at the speed of 5000 rpm for 10 min. The coacervate was acquired as the dense bottom phase. Meanwhile, a corresponding phase diagram of coacervation was obtained using SiW and P123 aqueous solutions of various concentrations. The coalescence process of coacervate droplets was observed under a confocal microscope, and the sample was prepared by dropping 5 μL of 40 wt% SiW aqueous solution on a glass slide, followed by the addition of 5 μL of 10 wt% P123 aqueous solution on the previous drop, which was then covered by a coverslip.
:1, followed by vigorously shaking and centrifugation at the speed of 5000 rpm for 10 min. The coacervate was acquired as the dense bottom phase. Meanwhile, a corresponding phase diagram of coacervation was obtained using SiW and P123 aqueous solutions of various concentrations. The coalescence process of coacervate droplets was observed under a confocal microscope, and the sample was prepared by dropping 5 μL of 40 wt% SiW aqueous solution on a glass slide, followed by the addition of 5 μL of 10 wt% P123 aqueous solution on the previous drop, which was then covered by a coverslip.
2.3 Rheological properties of coacervates
The rheological properties of the coacervates were studied via a TA Instruments AR-G2 rheometer using a 20 mm (diameter) 2° cone geometry with a gap of 53 μm. Oscillatory angular frequency sweep ranging from 0.1 rad s−1 to 100 rad s−1 at a fixed strain of 2% was carried out at 20 °C to characterize the shear moduli of the coacervates. The viscosity of the coacervates was characterized with shear rate ranging from 0.1 s−1 to 100 s−1. For SiW and P123 aqueous solutions, the rheological properties were measured in a DIN concentric cylinder with an operating gap of 5917.1 μm, and the test parameters were consistent with those of the coacervates. To investigate the effect of salts on the viscosity of the coacervates, selected salts (i.e., LiCl, NaCl, KCl, and CaCl2) were first added into SiW and P123 aqueous solutions to a desired concentration before mixing the solutions. All the rheological properties were studied with three independently prepared samples.2.4 Underwater adhesion properties of coacervates
Underwater adhesion properties of the coacervates were characterized on an AGS-X universal tensile testing machine (Shimadzu, Japan) at room temperature. A water container was attached to the compression cylinders to provide an underwater environment. PMMA, wood and glass round disks were glued on the center of the two compression cylinders with super glue, respectively, until they totally dried in air. Then the two compression cylinders were mounted onto the tensile testing machine with a 5000 N load cell and immersed underwater. Subsequently, 100 μL coacervate was injected on the lower cylinder and compressed by the upper cylinder with a force of 30 N or 300 N for 10 s. The adhesion strength was then obtained by lifting the upper cylinder at a constant speed of 100 mm min−1. The underwater adhesion tests in different salt solutions (Milli-Q water, 0.1 M LiCl, NaCl, KCl, CaCl2 and 1.0 M LiCl) were carried out between a stainless steel substrate (upper surface) with diameter of 12 mm and a transparent PMMA disk (lower surface) with a diameter of 18.5 mm. 50 μL of the coacervate was injected onto the PMMA surface and compressed by the upper surface with a force of 30 N for 10 s. The adhesion strength was then obtained by lifting the upper cylinder at a constant speed of 5 mm min−1. All the instant underwater adhesion strengths were determined based on at least 8 samples.2.5 Thermo-responsive optical properties of coacervates
The thermo-responsive optical properties of the coacervates were evaluated by alternately immersing the coacervate-painted glass slide in a 60 °C water bath and an ice-water bath for several cycles. The temperature-induced variation of transparency was quantified using a microplate reader (Cytation 5, Biotek, USA) at different temperatures in the visible light range (400–800 nm), with samples of 6.5 mm diameter and 1 mm thickness. The thermo-induced optical properties of the coacervates were quantified on a UV-vis spectrometer (Evolution 300, Thermo Scientific, CA) with a temperature controller (Isotemp, Fisher Scientific, CA) and a sample with a thickness of 10 mm. The repeatable heat–cool cycles were conducted between 40 and 55 °C. To unveil the mechanism of the transparency change, the size of 0.1 wt% P123 micelles at various temperatures ranging from 10 to 60 °C was characterized by dynamic light scattering (DLS) with a Malvern Zetasizer Nano ZSP. The morphology of P123 micelles at 20 °C and 60 °C was examined using a Dimension Icon atomic force microscope (AFM) system (Bruker, Santa Barbara, CA, USA) operating in the tapping mode in air. The samples were prepared by dropping 10 μL of 0.1 wt% P123 aqueous solution (stored at the desired temperature) on a clean silicon wafer substrate till fully dried in air at the corresponding temperature.2.6 Electrochromic properties of coacervates
The electrochromic properties were characterized by sandwiching the coacervate between two ITO-coated glass slides, where the external voltages applied on the outer surfaces of the bonded glass slides were provided by an electrochemical workstation (CHI920, CH Instruments, USA) with a Multi-Potential Steps mode. Coacervates before and after electrochromism studies were freeze-dried and characterized by X-ray photoelectron spectroscopy (XPS) to reveal the binding energies of the oxidized and reduced states of tungsten.3. Results and discussion
3.1 Fabrication and intermolecular interactions of the SiW-P123 coacervate
The fabrication process of the SiW-P123 coacervate adhesives was very facile. For example, 10 wt% P123 (ref. 37) aqueous solution was directly mixed with 40 wt% SiW aqueous solution at a volume ratio of 1:1, leading to a turbid solution after vigorous shaking (Fig. 1a). Coacervate droplets with sizes ranging from several to tens of micrometers were dispersed in the aqueous environment (Fig. 1b), where small coacervate droplets could coalesce into bigger ones as shown in Movie S1.† The cryogenic transmission electron microscopy (cryo-TEM) image of the coacervate droplet exhibited a densely packed continuous sponge-like network structure (Fig. 1c), suggesting stable and strong physical interactions between SiW clusters and P123 micelles (Fig. 1a). After centrifugation at 5000 rpm for 10 minutes, the solution could completely separate into a white coacervate phase and a corresponding transparent supernatant, from which mass production of the adhesive coacervate could be readily achieved.
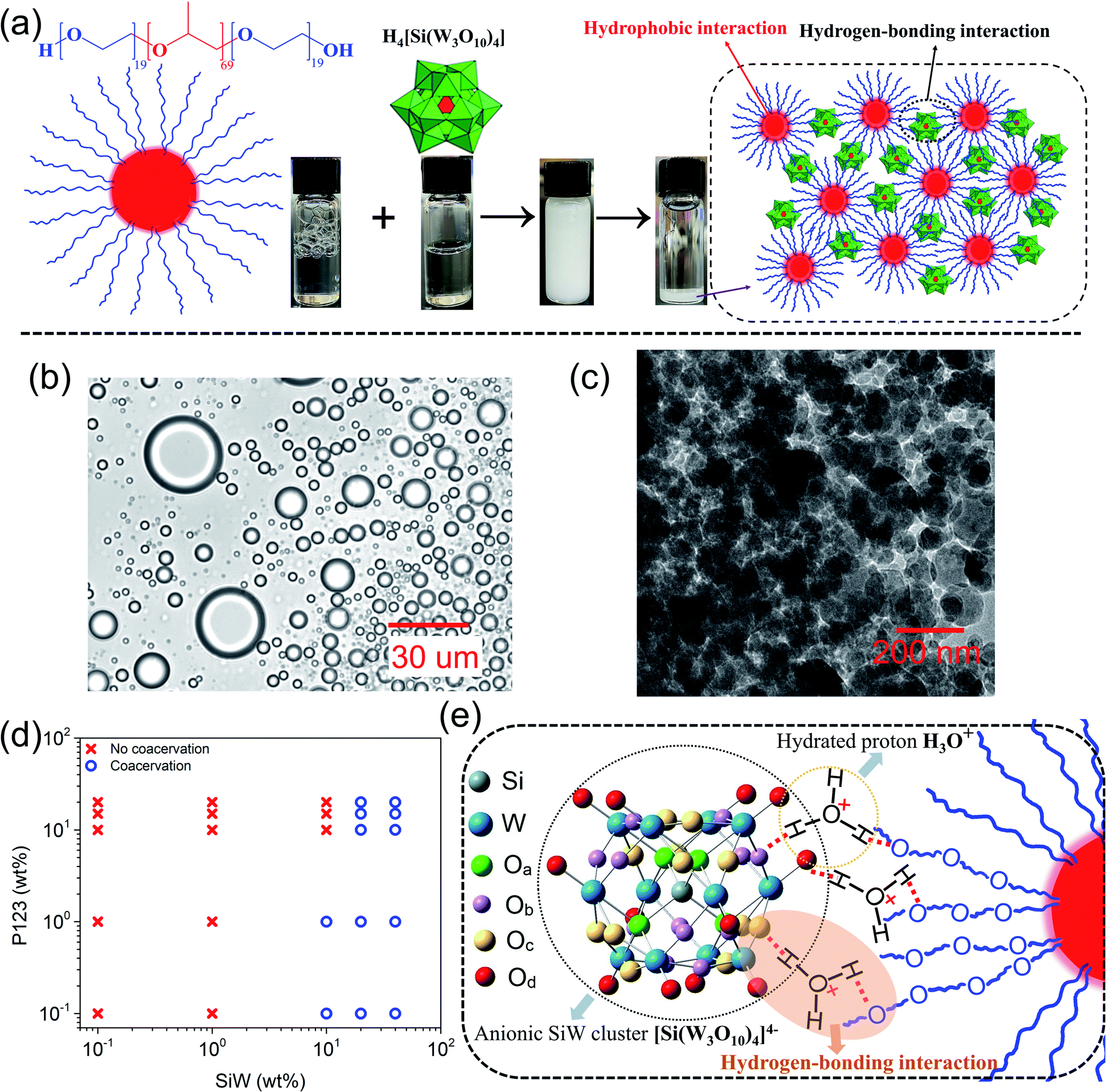 | ||
| Fig. 1 Fabrication and intermolecular interaction of the SiW-P123 coacervate. (a) Schematic illustration of the fabrication process of the SiW40-P123-10 coacervate. (b) Distribution of newly formed SiW40-P123-10 coacervate droplets observed under an optical microscope. (c) Cryo-TEM image of the SiW40-P123-10 coacervate. (d) Phase diagram of coacervation between SiW and P123 aqueous solutions. (e) Schematic illustration of the proposed interaction mechanism between SiW and P123 for the coacervate formation. | ||
Different concentrations of SiW and P123 aqueous solutions with a fixed volume ratio of 1:1 were employed to yield a series of coacervates denoted as SiWx-P123-y (x and y represent the weight percentage of SiW aqueous solution and P123 aqueous solution, respectively), and their phase behaviors are shown in Fig. 1d. When the concentration of SiW was 0.1 wt% and 1 wt%, no coacervation occurred with P123 solution ranging from 0.1 wt% to 20 wt%, which might be because of the sparse distribution of the small SiW clusters. When the concentration of SiW was increased to 10 wt%, a coacervate could be detected only with 0.1 wt% and 1 wt% P123, suggesting that the quantity of SiW clusters was still not sufficient to crosslink concentrated P123 micelles. On further increasing the concentration of SiW to 20 wt% and above, coacervates could be formed with P123 at all the concentrations used (0.1 to 20 wt%). The simple binary phase diagram of the SiW-P123 coacervation system holds great potential to understand and develop platforms with liquid–liquid phase separation. As shown in Fig. S1,† the yields of SiW40-P123-10/15/20 coacervates were relatively high after mixing, among which SiW40-P123-10 exhibited the highest viscosity (Fig. S2†) to resist the flow of the material underwater. Therefore, SiW40-P123-10 was employed for further characterization in this work if not specified.
Our previous study demonstrated that the coacervation between SiW and PEG most likely originated from the hydrogen bonding between the oxygens of SiW and etheric oxygens of PEG bridged by hydrated protons.19 As the P123 micelle contains a hydrophobic PPG core and hydrophilic PEG shell, a similar coacervation mechanism is expected to occur between SiW and PEG shells. According to the FTIR spectra of SiW powder and freeze-dried SiW40-P123-10 coacervate (Fig. S3†), the Keggin structure of SiW was well maintained before and after coacervation, with typical stretching vibration bands of W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Od, Si–Oa, W–Ob–W (edge shared), and W–Oc–W (corner shared) shifted from 1015, 977, 910, and 743 cm−1 to 1008, 967, 911, and 771 cm−1, originating from intermolecular interactions with P123 (Fig. 1e and S3†).38 As for P123, C–O–C shifted from 1103 to 1070 cm−1 after coacervation, indicating that etheric oxygens were occupied due to intermolecular interactions. Besides, antisymmetric and symmetric stretching of C–H as well as symmetric deformation of –CH3 were observed at 2970, 2870 and 1372 cm−1, respectively, in the coacervate, suggesting that the hydrophobic interaction among methyl groups of P123 micelles was maintained after coacervation.39 SiW is usually regarded as a strong acid with fully ionized protons.40 According to our previous study, the primary driving force for coacervation was most likely the hydrogen-bonding interactions (WO⋯H3O+⋯O) between oxygens (both terminal Od atoms (predominant) and bridging oxygens Ob and Oc) of SiW and etheric oxygens of PEG bridged by hydrated protons, which was demonstrated not only by FTIR spectra but also by changing the end groups of PEG, adjusting the pH of the solutions and modulating the solvents.19 As both hydrogen-bonding interaction and hydrophobic interaction tend to be affected by temperature,41,42 the viscosities of SiW40-P123-10 (driven by hydrogen-bonding interaction and hydrophobic interaction) and SiW40-PEG8000-10 (actuated by hydrogen-bonding interaction, where PEG with a molecular weight of 8000 was used instead of P123) coacervates as well as 10 wt% P123 aqueous solution (assembled by hydrophobic interaction) were characterized with temperature ranging from 20 °C to 45 °C. The results (Fig. S4†) revealed that the viscosities of all the samples decreased with increasing the temperature, while the highest viscosity and the most noticeable decrease were detected for the SiW40-P123-10 coacervate, which was attributed to the weakened hydrogen-bonding interaction between SiW clusters and P123 micelles as well as between the PEG shells of P123.43 The role of hydrated protons in the coacervation was also investigated by neutralizing SiW aqueous solution with saturated LiOH aqueous solution. The pH of 40 wt% SiW aqueous solution was measured to be 1.14; however, when it was adjusted to 1.75, the SiW aqueous solution could not form a coacervate with 10 wt% P123 (Fig. S5†). The above result demonstrated that the coacervation of the SiW-P123 system was highly dependent on the presence of hydrated protons, and the pH required for the formation of the SiW-P123 coacervate was much lower than that of the SiW-PEG coacervate system (pH < 4.3) we reported previously.19 A possible reason was that the chain length of PEG on the shells of P123 was shorter than that of the previously used linear PEG, therefore more proton crosslinkers would be required to bind the P123 micelles and SiW clusters. PEG is a well-known nonionic polymer and a hydrogen bonding accepter, while P123 was reported to carry slight negative charges in water (zeta potential of −6.8 mV at a pH of 7.4).44 During the formation of the coacervate, concentrated SiW aqueous solution was employed (e.g., 40 wt%), and P123 micelles were dispersed in water with a high concentration of hydrated protons, where the Debye length of the electric double-layer was calculated to be
Od, Si–Oa, W–Ob–W (edge shared), and W–Oc–W (corner shared) shifted from 1015, 977, 910, and 743 cm−1 to 1008, 967, 911, and 771 cm−1, originating from intermolecular interactions with P123 (Fig. 1e and S3†).38 As for P123, C–O–C shifted from 1103 to 1070 cm−1 after coacervation, indicating that etheric oxygens were occupied due to intermolecular interactions. Besides, antisymmetric and symmetric stretching of C–H as well as symmetric deformation of –CH3 were observed at 2970, 2870 and 1372 cm−1, respectively, in the coacervate, suggesting that the hydrophobic interaction among methyl groups of P123 micelles was maintained after coacervation.39 SiW is usually regarded as a strong acid with fully ionized protons.40 According to our previous study, the primary driving force for coacervation was most likely the hydrogen-bonding interactions (WO⋯H3O+⋯O) between oxygens (both terminal Od atoms (predominant) and bridging oxygens Ob and Oc) of SiW and etheric oxygens of PEG bridged by hydrated protons, which was demonstrated not only by FTIR spectra but also by changing the end groups of PEG, adjusting the pH of the solutions and modulating the solvents.19 As both hydrogen-bonding interaction and hydrophobic interaction tend to be affected by temperature,41,42 the viscosities of SiW40-P123-10 (driven by hydrogen-bonding interaction and hydrophobic interaction) and SiW40-PEG8000-10 (actuated by hydrogen-bonding interaction, where PEG with a molecular weight of 8000 was used instead of P123) coacervates as well as 10 wt% P123 aqueous solution (assembled by hydrophobic interaction) were characterized with temperature ranging from 20 °C to 45 °C. The results (Fig. S4†) revealed that the viscosities of all the samples decreased with increasing the temperature, while the highest viscosity and the most noticeable decrease were detected for the SiW40-P123-10 coacervate, which was attributed to the weakened hydrogen-bonding interaction between SiW clusters and P123 micelles as well as between the PEG shells of P123.43 The role of hydrated protons in the coacervation was also investigated by neutralizing SiW aqueous solution with saturated LiOH aqueous solution. The pH of 40 wt% SiW aqueous solution was measured to be 1.14; however, when it was adjusted to 1.75, the SiW aqueous solution could not form a coacervate with 10 wt% P123 (Fig. S5†). The above result demonstrated that the coacervation of the SiW-P123 system was highly dependent on the presence of hydrated protons, and the pH required for the formation of the SiW-P123 coacervate was much lower than that of the SiW-PEG coacervate system (pH < 4.3) we reported previously.19 A possible reason was that the chain length of PEG on the shells of P123 was shorter than that of the previously used linear PEG, therefore more proton crosslinkers would be required to bind the P123 micelles and SiW clusters. PEG is a well-known nonionic polymer and a hydrogen bonding accepter, while P123 was reported to carry slight negative charges in water (zeta potential of −6.8 mV at a pH of 7.4).44 During the formation of the coacervate, concentrated SiW aqueous solution was employed (e.g., 40 wt%), and P123 micelles were dispersed in water with a high concentration of hydrated protons, where the Debye length of the electric double-layer was calculated to be  (c is the concentration of electrolytes with the unit of mol L−1).45 Due to the short Debye length, the long-range electrostatic interaction between the negatively charged SiW clusters and P123 molecules tends to be significantly suppressed, while electrostatic interaction between the molecules might also play a role in the short range in addition to the strong hydrogen bonding and hydrophobic interaction within the SiW40-P123-10 coacervate. The coacervation of the SiW-P123 system was proposed to be mainly driven by hydrogen-bonding interaction between oxygens of SiW and etheric oxygens of P123 shells bridged by hydrated protons, where the hydrophobic interaction, which drove the self-assembly of P123 micellar structures, played a synergetic role in strengthening the coacervate as illustrated in Fig. 1a and e.
(c is the concentration of electrolytes with the unit of mol L−1).45 Due to the short Debye length, the long-range electrostatic interaction between the negatively charged SiW clusters and P123 molecules tends to be significantly suppressed, while electrostatic interaction between the molecules might also play a role in the short range in addition to the strong hydrogen bonding and hydrophobic interaction within the SiW40-P123-10 coacervate. The coacervation of the SiW-P123 system was proposed to be mainly driven by hydrogen-bonding interaction between oxygens of SiW and etheric oxygens of P123 shells bridged by hydrated protons, where the hydrophobic interaction, which drove the self-assembly of P123 micellar structures, played a synergetic role in strengthening the coacervate as illustrated in Fig. 1a and e.
3.2 Rheological properties
Rheological properties including viscosity η, storage modulus G′ and loss modulus G′′ of the SiW-P123 coacervates were characterized with a rheometer. Compared to the low viscosities of 40 wt% SiW (∼0.00129 Pa s) and 10 wt% P123 (∼0.0057 Pa s) aqueous solutions, the viscosity of the SiW40-P123-10 coacervate drastically increased to ∼193.77 Pa s, indicating the strong intermolecular interactions between SiW clusters and P123 micelles (Fig. 2a). It is noted that when the concentration of P123 varied from 10 wt% to 20 wt%, the viscosities of the formed coacervates slightly decreased (Fig. 2b) while the volume of the coacervate phase increased by 86.4% (Fig. S6†), which might be due to the slightly weakened intermolecular interaction among P123 micelles and SiW clusters. To illustrate the effect of the hydrophobic cores of polymeric micelles on the coacervates, F68 was also used to form the coacervates, as it contains more hydrophilic PEG and less hydrophobic PPG compared to P123. As displayed in Fig. 2b, the viscosities of SiW40-P123-10/15/20 coacervates were much larger than those of SiW40-F68-10/15/20 coacervates, which was mainly attributed to the stronger hydrophobic interactions among the PPG cores of P123 micelles. Moreover, the viscosity of the SiW40-PEG8000-10 coacervate was only about 10% that of the SiW40-P123-10 coacervate (Fig. S7†), indicating that stable hydrophobic micelle cores could act as crosslinking points to enhance the mechanical properties of the coacervates. | ||
| Fig. 2 Rheological properties of the SiW-P123 coacervates. (a) Viscosities of 40 wt% SiW aqueous solution, 10 wt% P123 aqueous solution, and the corresponding coacervate. (b) Viscosities of SiW40-P123-10/15/20 coacervates and SiW40-F68-10/15/20 coacervates at a shear rate of 10 s−1. (c) and (d) Shear moduli and viscosity (at a shear rate of 10 s−1) of the SiW40-P123-20 coacervate with 0, 0.1, 0.5 and 1.0 M LiCl. (e) Shear moduli and (f) viscosity (at a shear rate of 0.1 s−1) of the SiW40-P123-20 coacervate with 0.1 M LiCl, NaCl, KCl and CaCl2. | ||
Traditional coacervates are driven by electrostatic interactions between oppositely charged molecules, where salt plays a crucial role in balancing the electrostatic interaction and tuning the rheological properties.14 Generally, above a critical salt concentration, polyelectrolyte systems are always miscible and coacervation cannot occur due to the screening of electrostatic interaction.14,46,47 In addition, the formation of coacervates based on cation–π interaction requires high salt concentration to shield the repulsive electrostatic interaction between like-charged molecules.9,10 In our work, the SiW-P123 coacervation system was triggered by the synergy of hydrogen-bonding interaction and hydrophobic interaction, and the coacervates could form in a salt-free environment. However, their properties can be affected by the addition of salts. As shown in Fig. 2b, c and S8,† without the addition of salt, the SiW40-P123-20 coacervate exhibited low shear modulus and viscosity. With the addition of LiCl ranging from 0.1 to 1.0 M, the coacervate still maintained its liquid-like property, but the rheological properties were strengthened with increasing the salt concentration (Fig. 2c and d). With the introduction of 0.1 M LiCl, the viscosity of the coacervate increased from ∼166.8 Pa s to ∼209.3 Pa s (at a shear rate of 10 s−1), although their shear moduli were comparable. On further raising the LiCl concentration to 0.5 M and 1.0 M, both shear moduli and viscosity of the coacervate were substantially enhanced. The storage modulus, loss modulus (at an angular frequency of 10 rad s−1) and viscosity (at a shear rate of 10 s−1) of the coacervate were significantly enhanced by 5205%, 1201% and 1145%, respectively, with the addition of 1.0 M LiCl as compared to the case without salt. Therefore, the presence of LiCl up to 1.0 M was still safe for the formation of the SiW-P123 coacervation system. Meanwhile, the effect of different salts including LiCl, NaCl, KCl and CaCl2 on the rheological properties of the coacervate was also studied (Fig. 2e and f). By varying the different types of salts at a fixed concentration 0.1 M, the salt-induced enhancement of shear moduli and viscosity of the coacervate followed the trend: LiCl < NaCl < KCl < CaCl2, where LiCl, NaCl and KCl could salt out PEG from aqueous solution following the Hofmeister series while CaCl2 was capable of interacting with the electronegative oxygens of SiW clusters.48,49 Therefore, varying the concentration and type of salts provides a feasible way to tune the mechanical properties of the SiW-P123 coacervates over several orders of magnitude for diverse practical applications.
3.3 Underwater adhesive properties
The as-prepared SiW40-P123-10 coacervate could be facilely painted on a glass slide underwater with designed shapes and it immediately resisted the water flush, suggesting its excellent underwater adhesive property (Fig. 3a and Movie S2†). Meanwhile, the coacervate could facilely bind different materials such as wood, coin, stone, and glass slide as well as a plastic substrate underwater (Fig. 3b and Movie S3†). As shown in Fig. 3c, the coacervate instantly and firmly bound two weights together underwater and easily supported a mass of 100 g (Movie S4a†). Besides, the coacervate could also instantly bind two porcine skins underwater which could resist repeatable movement and swing (Fig. 3d and Movie S4b†), holding great potential in the field of biomedical tissue glue. The underwater adhesion of the coacervate on different substrates was quantitatively examined and the results are presented in Fig. 3e, showing the highest adhesion strength of 479.6 kPa to poly(methyl methacrylate) (PMMA, contact angle of 67.3°). It was most likely attributed to the synergetic effect of hydrogen bonding and hydrophobic interactions, where the electronegative oxygen atoms of –(CO)–OCH3 groups served as proton acceptors to form hydrogen bonds and the extensive methyl groups participated in hydrophobic interaction with the coacervate. The instant adhesion strength of the coacervate on the glass substrate (contact angle of 15.1°) was determined to be 297.1 kPa because only hydrogen bonding contributed to the adhesion. The coacervate on the wood surface exhibited a relatively low adhesion of 128.5 kPa, which was possibly due to the existence of micropores and microtubes on the wood surface that absorbed the coacervate (Fig. S9†), decreasing the effective contact area between the wood surfaces. Additionally, the underwater adhesion of the coacervate was pressure-sensitive as illustrated in Fig. 3f. When two glass substrates bonded by the coacervate were compressed with a force of 300 N for 10 s, the adhesion strength of the coacervate was measured to be 442.4 kPa, which was increased by 48.9% compared to that of the sample under 30 N compression for 10 s. The pressure-sensitivity of the underwater adhesion might originate from two factors: first, the rearrangement of P123 polymer chains within the coacervate phase under pressure could affect the molecular interactions within the coacervate and at the coacervate/substrate interface;50 second, the water pockets or air bubbles trapped within the coacervate or at the coacervate/substrate interface tended to be removed by compression or through the reconfiguration of PEG polymer chains, creating a larger contact area and leading to a stronger adhesion.51,52 Moreover, as salt is ubiquitous in the physiological environment of humans,53 seawater,5,10 and aqueous batteries,23 it is important to study the effect of salts on the underwater adhesion strength of the coacervate. As shown in Fig. 3g, when the salt concentration was fixed at 0.1 M, the adhesion strength of the coacervate increased following the trend LiCl < NaCl < KCl < CaCl2, which was consistent with the variation of rheological properties (Fig. 2f). The variation of instant underwater adhesion strength of the coacervate in LiCl (0.1 and 1.0 M) aqueous solutions also agreed with the change of rheological properties (Fig. 2d). In Milli-Q water, the coacervate exhibited the highest instant adhesion strength, which might suggest that the addition of saline ions could impair the electrostatic interactions at the coacervate/substrate interfaces.54 Herein, the instant underwater adhesion strength of the as-prepared coacervate is superior to that of previously reported coacervates (<200 kPa for all cases18 and ∼420 kPa for PMMA substrate22) and even some adhesive hydrogels (≤180 kPa55,56). A detailed summary of the reported wet/underwater adhesives is presented in Table S1,† demonstrating the facile fabrication as well as instant and strong underwater adhesion of the SiW40-P123-10 coacervate, which shows greater advantages than all the listed adhesives. The instant and robust underwater adhesion of the coacervate makes it a promising adhesive for various applications, especially those in aqueous or wet environments.
 | ||
| Fig. 3 Underwater adhesive properties of the SiW40-P123-10 coacervate. (a) The SiW40-P123-10 coacervate was injected and painted on a glass slide underwater (left) and was then immediately flushed with running water which could not noticeably affect the painted patterns (right). (b) The SiW40-P123-10 coacervate can firmly bind various substrates such as wood, coin, stone, and glass slide to a plastic substrate underwater. (c) The SiW40-P123-10 coacervate could instantly and firmly bind two weights together underwater and easily support a mass of 100 g. (d) The SiW40-P123-10 coacervate could instantly and firmly bind two porcine skins together underwater. (e) Instant underwater adhesion strength of the SiW40-P123-10 coacervate on different substrates including PMMA, wood and glass. The inset images show the contact angles of water on PMMA, wood and glass substrates. (f) Instant underwater adhesion strength of the SiW40-P123-10 coacervate on a glass substrate under different compression forces of 30 N and 300 N. (g) Effect of salts on the underwater adhesion strength of the SiW40-P123-10 coacervate. The inset image shows the set-up of the test with a stainless steel upper surface and a PMMA lower surface. | ||
3.4 Thermo-responsive optical properties
The optical properties of the SiW40-P123-10 coacervate could be readily adjusted with temperature, whereby it was white and opaque at 0 °C and turned transparent when heated to 60 °C (Fig. 4a). The transition process was highly reversible and showed a rapid response to temperature change as displayed in Movie S5,† where the coacervate painted on a glass slide was successively immersed in a 60 °C water bath and 0 °C ice-water bath for several cycles. The variation of transparency was quantitatively determined using an optical microplate reader in the visible light range (400–800 nm). As demonstrated in Fig. 4b and S10,† the transmittance of the coacervate increased with the increase of temperature, showing an extremely low value at 0 °C and reached about 70% at 60 °C. Meanwhile, transmittance variation of the coacervate under heat–cool cycles between 40 and 55 °C is presented in Fig. 4c, showing excellent repeatability of the thermo-tunable optical property of the coacervate. It is noted that as the thickness of the coacervate sample used for the characterization was 10 mm, the transmittance measured at 55 °C was lower than that measured with a microplate reader (Fig. 4b, sample thickness of 1 mm). The adjustable optical property of the coacervate originated from the size change of P123 micelles with temperature, which was characterized by DLS (Fig. 4d). With temperature ranging from 0 °C to 60 °C, P123 micelles presented thermo-sensitive size distribution. At low temperatures (<30 °C), the micelle size was polydisperse and large aggregates could be detected, while increasing the temperature gradually led to monodisperse micelles. The change of size distribution of the micelles was most likely due to the enhanced hydrophobic interaction within PPG micelle cores with increase of temperature, which tended to induce the dissociation of random aggregates and the formation of densely packed micelles.39 The thermo-responsive size evolution of P123 micelles was also in accordance with AFM topography images as shown in Fig. 4e and f. At 20 °C, the existence of large aggregates could prevent the transmittance of light, while relatively small and homogeneously distributed micelles were observed at 60 °C, yielding a clear solution. Besides, structural transition of P123 with increase of temperature could also weaken the intermolecular interaction among the micelles, which was in agreement with the lower shear modulus and viscosity of P123 at the elevated temperature (Fig. S11†). | ||
| Fig. 4 Thermo-responsive properties of the SiW40-P123-10 coacervate. (a) Repeatable transparency transition of the SiW40-P123-10 coacervate painted on a glass slide which was alternately immersed in a 60 °C water bath and ice-water bath. (b) Transmittance variation of the SiW40-P123-10 coacervate at temperatures of 0, 37 and 60 °C. (c) Transmittance variation of the SiW40-P123-10 coacervate under heat–cool cycles between 40 and 55 °C. (d) Size distributions of P123 micelles at various temperatures ranging from 0 to 60 °C. (e) and (f) AFM topography images of P123 micelles deposited on silica surfaces at 20 and 60 °C. | ||
3.5 Electrochromic properties
The SiW40-P123-10 coacervate adhesive could be directly painted on different substrates and displayed excellent resistance to mechanical deformation such as stretching and bending (Movie S6,† the coacervate was dyed with carbon nanotubes (CNTs)). Despite repeatable stretching, bending and vigorously shaking underwater, the coacervate coating can be kept intact, indicating its excellent tolerance to turbulent underwater environment. Besides, the hydrophobic cores of P123 micelles endowed the SiW-P123 coacervation system with the capability to load hydrophobic species like CNTs (Fig. S12†), providing great versatility to the platform. The coacervate coating also exhibited exemplary stability in high-salt aqueous solutions (e.g., 3 M LiCl, NaCl or KCl) for at least 1200 h (Fig. 5a). It was observed that the adhesive coacervate changed from opaque to transparent within 5 h, which could have originated from the enhanced hydrophobic interaction within PPG cores and the shrinkage of PEG shells due to the decrease of free water molecules in the coacervate in the presence of salt.43,57 | ||
| Fig. 5 Electrochromic properties of the SiW40-P123-10 coacervate. (a) Pictures showing the excellent stability of the SiW40-P123-10 coacervate in 3 M LiCl, NaCl and KCl solutions. (b) Schematic illustration of the working mechanism for the self-powered electrochromic device with the SiW40-P123-10 coacervate painted on an ITO-coated glass slide. (c) The SiW40-P123-10 coacervate painted on the ITO-coated glass slide as a smiling face turned deep blue by forming a circuit with Al foil, which was then bleached to light blue by disconnecting the circuit as well as adding H2O2. (d)–(g) The SiW40-P123-10 coacervate sandwiched between two ITO-coated glass slides changed to deep blue by applying a reduction voltage of −0.25 V, followed by bleaching triggered by an oxidation voltage of +0.8 V, which was further bleached to translucent in air. (h) and (i) XPS spectra of the as-prepared coacervate before and after reduction. | ||
Combining the facile paintability and excellent stability of the coacervate underwater with the innate redox-driven color-switching behavior of SiW, a self-powered electrochromic device can be built up by connecting a coacervate-painted indium tin oxide (ITO) glass slide with Al foil through a salt bridge and metal wire (Fig. 5b). The associated reduction–oxidation reactions are also illustrated in Fig. 5b. Once the circuit was completed, the coacervate coating instantaneously changed from white to deep blue, and then the blue color could be bleached with the disconnection of the circuit and addition of H2O2 (30 wt%) (Fig. 5c and Movie S7†). The color-switching property of the coacervate can also be triggered by external voltages. Two ITO-coated glass slides were bonded together with the coacervate and connected to an external voltage provided by an electrochemical workstation (Fig. 5d). When a reduction potential of −0.25 V was applied, the coacervate immediately turned deep blue (Fig. 5e and Movie S8†), while a subsequent oxidation potential of +0.8 V could bleach the color to light blue (Fig. 5f). The coacervate would turn translucent by further oxidation of air (Fig. 5g). The electrochromic behavior of the coacervate was ascribed to the reduction of tungsten, which was confirmed by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 5h, the coacervate exhibited two peaks at 38.1 eV and 35.9 eV before electrochemical/electrical reduction, corresponding to the binding energies of W6+ 4f5/2 and W6+ 4f7/2. After reduction (Fig. 5i), the peak at 38.1 eV split into two peaks at 38.2 eV and 36.9 eV, which were assigned to the binding energies of W6+ 4f5/2 and W5+ 4f5/2, respectively. Meanwhile, the peak at 35.9 eV also split into two peaks at 36.1 eV and 34.8 eV, attributed to the binding energies of W6+ 4f7/2 and W5+ 4f7/2. The results are consistent with the reported reducible polyoxometalates, which are named “heteropoly blues” for their innate color-switching property enabled by specific triggers.23,38,58
4. Conclusions
In this work, we have developed a novel instant underwater adhesive with tunable optical and electrochromic properties via a facile one-step mixing of SiW and P123 aqueous solutions, where the formation of the adhesive coacervate was driven by the synergy of hydrogen-bonding interaction and hydrophobic interaction. The coacervation process is mainly actuated by the intermolecular hydrogen bonding between the oxygens of SiW and etheric oxygens of P123 micelles bridged by hydrated protons, and the hydrophobic cores of P123 micelles offer an additional crosslinking, significantly enhancing the mechanical properties (e.g., shear moduli, viscosity) of the coacervates. The rheological properties of as-prepared coacervates can be modulated over several orders of magnitude for practical applications by introducing different types and concentrations of salts (e.g., LiCl, NaCl, KCl and CaCl2), and they are able to maintain the integrity in saline aqueous solutions with a concentration up to 3 M for at least 1200 h. Meanwhile, the coacervates show robust and instant underwater adhesion on various substrates with an adhesion strength up to 479.6 kPa on PMMA, which can also resist water flush and repeatable stretching and bending of the substrate. Such a wet adhesion performance of the developed SiW-P123 coacervate is superior to that of a lot of previously reported coacervate materials, even some hydrogels and functionalized surfaces. Moreover, the SiW-P123 coacervate exhibits thermo-responsive optical properties, which are attributed to the configuration change of P123 micelles. The innate reduction-related color switch of SiW endows the coacervate with electrochromic properties in response to both chemical oxidation–reduction reactions and external voltages, suggesting the great potential of the coacervate in electrochromic devices such as smart batteries. The SiW-P123 coacervation system provides an ideal platform for the development of color-switching underwater adhesives, holding great promise in wearable electronics and energy-saving buildings. It is noted that the SiW can be facilely modified or replaced by other kinds of polyoxometalates,59 and other polymers in the Pluronic family37 can be used to substitute P123, further expanding the versatility and functionality of the SiW-P123 coacervation system.Author contributions
The manuscript was written through contributions of all the authors. All the authors have given approval to the final version of the manuscript. The more specific contributions of the authors are as follows. Conceptualization: Q. Peng, H. Zeng; investigation: Q. Peng; writing – original draft: Q. Peng; methodology, validation and writing – review & editing: Q. Peng, J. Chen, T. Wang, L. Gong, X. Peng, M. Wu, Y. Ma, F. Wu, D. Yang, H. Zhang, H. Zeng; resources, funding acquisition and supervision: H. Zeng.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the financial support from the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Foundation for Innovation (CFI) and the Canada Research Chairs Program (H. Zeng).References
- G. P. Maier, M. V. Rapp, J. H. Waite, J. N. Israelachvili and A. Butler, Science, 2015, 349, 628–632 CrossRef CAS PubMed.
- A. M. Smith, Biological Adhesives, Springer, Berlin, Germany, 2016 Search PubMed.
- J. H. Waite, Nat. Mater., 2008, 7, 8–9 CrossRef CAS PubMed.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed.
- H. Shao and R. J. Stewart, Adv. Mater., 2010, 22, 729–733 CrossRef CAS PubMed.
- Q. Ye, F. Zhou and W. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC.
- C. Zhang, B. Wu, Y. Zhou, F. Zhou, W. Liu and Z. Wang, Chem. Soc. Rev., 2020, 49, 3605–3637 RSC.
- R. J. Stewart, J. C. Weaver, D. E. Morse and J. H. Waite, J. Exp. Biol., 2004, 207, 4727–4734 CrossRef CAS PubMed.
- S. Kim, J. Huang, Y. Lee, S. Dutta, H. Y. Yoo, Y. M. Jung, Y. Jho, H. Zeng and D. S. Hwang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E847–E853 CrossRef CAS PubMed.
- S. Kim, H. Y. Yoo, J. Huang, Y. Lee, S. Park, Y. Park, S. Jin, Y. M. Jung, H. Zeng, D. S. Hwang and Y. Jho, ACS Nano, 2017, 11, 6764–6772 CrossRef CAS PubMed.
- G. C. Yeo, F. W. Keeley and A. S. Weiss, Adv. Colloid Interface Sci., 2011, 167, 94–103 CrossRef CAS PubMed.
- A. Miserez, T. Schneberk, C. Sun, F. W. Zok and J. H. Waite, Science, 2008, 319, 1816–1819 CrossRef CAS PubMed.
- A. Miserez, Y. Li, J. H. Waite and F. Zok, Acta Biomater., 2007, 3, 139–149 CrossRef CAS PubMed.
- C. E. Sing and S. L. Perry, Soft Matter, 2020, 16, 2885–2914 RSC.
- D. Priftis and M. Tirrell, Soft Matter, 2012, 8, 9396–9405 RSC.
- R. Chollakup, W. Smitthipong, C. D. Eisenbach and M. Tirrell, Macromol, 2010, 43, 2518–2528 CrossRef CAS.
- H. Zhao, C. Sun, R. J. Stewart and J. H. Waite, J. Biol. Chem., 2005, 280, 42938–42944 CrossRef CAS PubMed.
- K. Kim, M. Shin, M. Koh, J. H. Ryu, M. S. Lee, S. Hong and H. Lee, Adv. Funct. Mater., 2015, 25, 2402–2410 CrossRef CAS.
- Q. Peng, J. Chen, Z. Zeng, T. Wang, L. Xiang, X. Peng, J. Liu and H. Zeng, Small, 2020, 16, 2004132 CrossRef CAS PubMed.
- M. Dompe, F. J. Cedano-Serrano, O. Heckert, N. van den Heuvel, J. van der Gucht, Y. Tran, D. Hourdet, C. Creton and M. Kamperman, Adv. Mater., 2019, 31, 1808179 CrossRef PubMed.
- A. Narayanan, J. R. Menefee, Q. Liu, A. Dhinojwala and A. Joy, ACS Nano, 2020, 14, 8359–8367 CrossRef CAS PubMed.
- Z. Wang, S. Zhang, S. Zhao, H. Kang, Z. Wang, C. Xia, Y. Yu and J. Li, Chem. Eng. J., 2021, 404, 127069 CrossRef CAS.
- X. Li, Z. Du, Z. Song, B. Li, L. Wu, Q. Liu, H. Zhang and W. Li, Adv. Funct. Mater., 2018, 28, 1800599 CrossRef.
- G. Yang, Y. M. Zhang, Y. Cai, B. Yang, C. Gu and S. X. Zhang, Chem. Soc. Rev., 2020, 49, 8687–8720 RSC.
- A. Cannavale, U. Ayr, F. Fiorito and F. Martellotta, Energies, 2020, 13, 1449 CrossRef CAS.
- W. Wu, M. Wang, J. Ma, Y. Cao and Y. Deng, Adv. Electron. Mater., 2018, 4, 1800185 CrossRef.
- J. Zhao, Y. Tian, Z. Wang, S. Cong, D. Zhou, Q. Zhang, M. Yang, W. Zhang, F. Geng and Z. Zhao, Angew. Chem., 2016, 128, 7277–7281 CrossRef.
- J. Wang, L. Zhang, L. Yu, Z. Jiao, H. Xie, X. W. Lou and X. W. Sun, Nat. Commun., 2014, 5, 4921 CrossRef CAS PubMed.
- Y. Tian, W. Zhang, S. Cong, Y. Zheng, F. Geng and Z. Zhao, Adv. Funct. Mater., 2015, 25, 5833–5839 CrossRef CAS.
- Y. Tian, S. Cong, W. Su, H. Chen, Q. Li, F. Geng and Z. Zhao, Nano Lett., 2014, 14, 2150–2156 CrossRef CAS PubMed.
- Y. Zhang, S. W. Ng, X. Lu and Z. Zheng, Chem. Rev., 2020, 120, 2049–2122 CrossRef CAS PubMed.
- J. Zhang, W. Zhang, Z. Yang, Z. Yu, X. Zhang, T. C. Chang and A. Javey, Sens. Actuators, B, 2014, 202, 708–713 CrossRef CAS.
- W. Wu, Nanoscale, 2017, 9, 7342–7372 RSC.
- Y. Aleeva and B. Pignataro, J. Mater. Chem. C, 2014, 2, 6436–6453 RSC.
- D. S. Hwang, H. Zeng, A. Srivastava, D. V. Krogstad, M. Tirrell, J. N. Israelachvili and J. H. Waite, Soft Matter, 2010, 6, 3232–3236 RSC.
- S. M. Wang, J. Hwang and E. Kim, J. Mater. Chem. C, 2019, 7, 7828–7850 RSC.
- A. Pitto-Barry and N. P. E. Barry, Polym. Chem., 2014, 5, 3291–3297 RSC.
- J. Xu, X. Li, J. Li, X. Li, B. Li, Y. Wang, L. Wu and W. Li, Angew. Chem., Int. Ed., 2017, 56, 8731–8735 CrossRef CAS PubMed.
- Y. Su, J. Wang and H. Liu, Langmuir, 2002, 18, 5370–5374 CrossRef CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed.
- J. Chen, Q. Peng, T. Thundat and H. Zeng, Chem. Mater., 2019, 31, 4553–4563 CrossRef CAS.
- J. Chen, B. Yan, X. Wang, Q. Huang, T. Thundat and H. Zeng, Polym. Chem., 2017, 8, 3066–3073 RSC.
- R. Ganguly, A. Kunwar, B. Dutta, S. Kumar, K. C. Barick, A. Ballal, V. K. Aswal and P. A. Hassan, Colloids Surf., B, 2017, 152, 176–182 CrossRef CAS PubMed.
- N. Suthiwangcharoen and R. Nagarajan, RSC Adv., 2014, 4, 10076–10089 RSC.
- J. N. Israelachvili, Intermolecular and Surface Forces, 2nd edn, Academic Press, New York, 1992 Search PubMed.
- Q. Wang and J. B. Schlenoff, Macromolecules, 2014, 47, 3108–3116 CrossRef CAS.
- J. T. G. Overbeek and M. J. Voorn, J. Cell. Comp. Physiol., 1957, 49, 7–26 CrossRef CAS.
- K. P. Ananthapadmanabhan and E. D. Goddard, Langmuir, 1987, 3, 25–31 CrossRef CAS.
- P. Zhang, O. Gourgas, A. Lainé, M. Murshed, D. Mantovani and M. Cerruti, Cryst. Growth Des., 2020, 20, 7170–7179 CrossRef CAS.
- D. S. Hwang, H. Zeng, Q. Lu, J. N. Israelachvili and J. H. Waite, Soft Matter, 2012, 8, 5640–5648 RSC.
- P. Karnal, P. Roberts, S. Gryska, C. King, C. Barrios and J. Frechette, ACS Appl. Mater. Interfaces, 2017, 9, 42344–42353 CrossRef CAS PubMed.
- S. Baik, D. W. Kim, Y. Park, T.-J. Lee, S. H. Bhang and C. Pang, Nature, 2017, 546, 396–400 CrossRef CAS PubMed.
- J. P. Jones, M. Sima, R. G. O'Hara and R. J. Stewart, Adv. Healthcare Mater., 2016, 5, 795–801 CrossRef CAS PubMed.
- L. Han, B. Yan, L. Zhang, M. Wu, J. Wang, J. Huang, Y. Deng and H. Zeng, Colloids Surf., A, 2018, 539, 37–45 CrossRef CAS.
- H. Fan, J. Wang and J. P. Gong, Adv. Funct. Mater., 2020, 2009334 Search PubMed.
- H. Fan, J. Wang, Z. Tao, J. Huang, P. Rao, T. Kurokawa and J. P. Gong, Nat. Commun., 2019, 10, 5127 CrossRef PubMed.
- K. Yamazoe, Y. Higaki, Y. Inutsuka, J. Miyawaki, Y. T. Cui, A. Takahara and Y. Harada, Langmuir, 2017, 33, 3954–3959 CrossRef CAS PubMed.
- T. Yamase, Chem. Rev., 1998, 98, 307–325 CrossRef CAS PubMed.
- D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta01658j |
| This journal is © The Royal Society of Chemistry 2021 |