
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The kinetics and mechanism of H2O2 decomposition at the U3O8 surface in bicarbonate solution
John McGrady *a,
Yuta Kumagai*a,
Masayuki Watanabea,
Akira Kirishimab,
Daisuke Akiyamab,
Akira Kitamurac and
Shingo Kimuroc
*a,
Yuta Kumagai*a,
Masayuki Watanabea,
Akira Kirishimab,
Daisuke Akiyamab,
Akira Kitamurac and
Shingo Kimuroc
aNuclear Science and Engineering Center, Japan Atomic Energy Agency (JAEA), Tokai, Ibaraki, 319-1195, Japan. E-mail: mcgrady.john@jaea.go.jp; kumagai.yuta@jaea.go.jp
bInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 1-1 Katahira, 2-chome, Aoba-ku, Sendai 980-8577, Japan
cRadionuclide Migration Research Group, Japanese Atomic Energy Agency (JAEA), Tokai, Ibaraki, 319-1195, Japan
First published on 31st August 2021
Abstract
In the event of nuclear waste canister failure in a deep geological repository, groundwater interaction with spent fuel will lead to dissolution of uranium (U) into the environment. The rate of U dissolution is affected by bicarbonate (HCO3−) concentrations in the groundwater, as well as H2O2 produced by water radiolysis. To understand the dissolution of U3O8 by H2O2 in bicarbonate solution (0.1–50 mM), dissolved U concentrations were measured upon H2O2 addition (300 μM) to U3O8/bicarbonate mixtures. As the H2O2 decomposition mechanism is integral to the dissolution of U3O8, the kinetics and mechanism of H2O2 decomposition at the U3O8 surface was investigated. The dissolution of U3O8 increased with bicarbonate concentration which was attributed to a change in the H2O2 decomposition mechanism from catalytic at low bicarbonate (≤5 mM HCO3−) to oxidative at high bicarbonate (≥10 mM HCO3−). Catalytic decomposition of H2O2 at low bicarbonate was attributed to the formation of an oxidised surface layer. Second-order rate constants for the catalytic and oxidative decomposition of H2O2 at the U3O8 surface were 4.24 × 10−8 m s−1 and 7.66 × 10−9 m s−1 respectively. A pathway to explain both the observed U3O8 dissolution behaviour and H2O2 decomposition as a function of bicarbonate concentration was proposed.
Introduction
The current strategy for the disposal of spent nuclear fuel is in a deep geological repository according to the majority of the international community. The repositories provide a long-term storage solution, yet the release of radioactive species from spent nuclear fuel into the environment from the repository is projected to occur in the future upon failure of the repository barriers. Therefore, it is necessary to develop safety models for the repositories to predict their performance when failure occurs and nuclear material is exposed to the local environment. The main pathway for radionuclide release is predicted to be caused by the ingress of groundwater into the repository and interaction of the groundwater with the surface of the spent fuel. Understanding the reaction mechanisms between groundwater and spent fuel is integral to the development of safety models. Such interactions between the groundwater and spent fuel will lead to dissolution of the UO2 matrix which constitutes the majority of the spent fuel.1 The solubility of U in groundwater is governed by the form of U (U(IV), U(V) and U(VI)), with the hexavalent U(VI) form being more soluble than U(IV) and U(V).2–4 Therefore, the presence of U(VI) facilitates U dissolution into the groundwater upon canister failure.Under the reducing, anoxic conditions typically found in groundwater at repository depths, the solubility of U(IV) is very low,5–7 and so significant dissolution of the UO2 spent fuel may not be expected. However, radiation from the spent fuel will cause radiolysis of fuel adjacent water leading to the formation of a complex water chemistry involving radical, ionic and molecular species in the form of both reductants (eaq−, H·, H2) and oxidants (OH·, H2O2).8 This will significantly affect the local redox chemistry of the water and the oxidation state of U.
Of the oxidants generated by radiolysis, it has been shown that H2O2 is the dominant species in regards to U dissolution under deep geological repository conditions.9,10 The interaction of H2O2 with the UO2 surface has been thoroughly studied due to its importance for U dissolution, and it has been proposed that there are two competing pathways, both of which involve the decomposition of H2O2 at the UO2 surface.11,12 The first involves catalytic H2O2 decomposition forming H2O2 and O2 where the UO2 surface acts as a catalyst (ads = adsorbed):
| (H2O2)ads → 2(OH˙)ads | (1) |
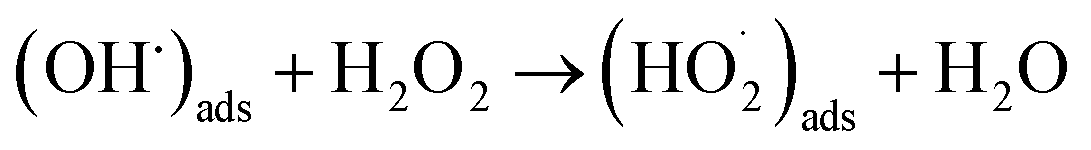 | (2) |
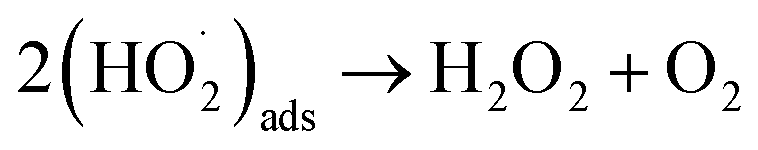 | (3) |
In this case, the H2O2 decomposition does not directly cause U dissolution. The second is an oxidative decomposition reaction where H2O2 oxidises U(IV) to U(V) (eqn (4)) and U(V) to U(VI) (eqn (5)) while itself being reduced to OH− (eqn (6)) in a redox couple.
| U(IV)O2 → U(V)O2+ + e− | (4) |
| U(V)O2 → U(VI)O22+ + e− | (5) |
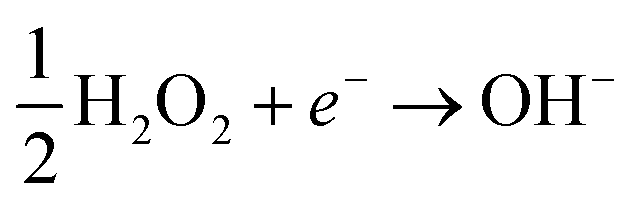 | (6) |
Typically, groundwater also contains bicarbonate (HCO3−) which has been shown to enhance the dissolution of U due to favourable complexation with U(VI) and stabilisation of the dissolution products:13–16
| UIV + HCO3− → UV(HCO3)ads + e− | (7) |
| UV(HCO3)ads + OH− → UVI(CO3)ads + e− + H2O | (8) |
| UVI(CO3)ads + HCO−3 → (UVIO2(CO3)2)2− + H+ | (9) |
Therefore, the concentration of bicarbonate is believed to have a significant effect on U dissolution and the rate of H2O2 decomposition at the UO2 surface.
Due to the importance of developing models to predict U dissolution into groundwater, various studies have been undertaken with UO2 in simulated groundwater. A recent study by Kumagai et al.17 has shown that increasing the oxygen content from UO2 to UO2.3 increased U dissolution and reduced the rate of H2O2 decomposition at the oxide surface. As the dissolution of U is governed by the redox behaviour of the U atoms, it follows that the ratio of U(IV), U(V) and U(VI) will have a significant impact on both U dissolution as well as the H2O2 decomposition pathway. Therefore, the form of uranium oxide that exists on the spent fuel oxide will have a large effect on the dissolution of U into the environment. Due to the radiolysis of spent fuel surface adjacent groundwater and the elevated temperatures from spent fuel decay, the formation of highly oxidised forms of U is expected i.e., where x > 0.3 for UO2+x. However, there is still a lack of knowledge regarding the impact of higher oxidised forms of U on the mechanism of U dissolution and H2O2 decomposition.
To investigate this, we adopted U3O8 as an extreme case, which corresponds to UO2.66 containing two U(V) atoms and one U(VI) atom.18–20 U3O8 has been observed on used nuclear fuel both in wet21 and air22,23 environments, and can be used as a highly oxidised form of uranium oxide for an examination of the effects of U valence on U dissolution. As the complexation of bicarbonate with U(VI) is thought to drive U dissolution by favourable complexation, the effect of U oxidation state on the dissolution of U in bicarbonate solution can be investigated by using U3O8. The H2O2 decomposition mechanism is dependent on U oxidation and so can also be investigated using U3O8 for comparison with UO2. The concentration of bicarbonate in groundwater is dependent on the location of the deep geological repository, and can range from ∼10−4 M (Tono, Japan),24 to ∼10−3 M (Daejeon, South Korea),25,26 to ∼10−2 M (Forsmark, Sweden)27 and so it is necessary to understand U dissolution and H2O2 decomposition at uranium oxide surfaces over a range of bicarbonate concentrations.
Therefore, in this work, U dissolution from U3O8 suspensions with H2O2 as a function of sodium bicarbonate (NaHCO3) has been investigated, and the mechanism of H2O2 decomposition at the U3O8 surface has been elucidated.
Experimental
Materials
Two samples of U3O8 powder were used in this study to investigate the reproducibility of the U dissolution tests. The first U3O8 powder (sample 1) was prepared by heating UO2 powder to 750 °C for 3 hours under a continuous flow of air. The second (sample 2) was prepared by dissolving U metal in 13 M HNO3 (Fujifilm Wako Pure Chemical, 60%) to form UO2(NO3)2·(H2O)n, which was then heated under identical conditions to give a 96% yield of U3O8. The formation of U3O8 was confirmed by XRD and the data was refined using the Rietveld method.28 The average crystallite size was measured using the Scherrer equation:29
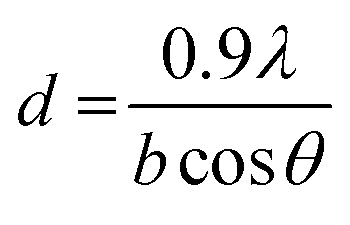 | (10) |
Dissolution experiments
The effect of NaHCO3 (Alfa Aesar) on the dissolution of U3O8 powder by reaction with H2O2 (Fujifilm Wako Pure Chemical, 30%) was investigated by monitoring the U and H2O2 concentration as a function of reaction time. A suspension of U3O8 was prepared at concentrations of NaHCO3 between 0.1–50 mM (pH 8.2–9.7), and the suspensions were purged with Ar for approximately 18 hours to ensure removal of O2 to imitate the anoxic conditions of groundwater. Into the suspension, H2O2 was added to initiate the reaction. The concentration of H2O2 added was 300 μM which has been shown to be optimal to study oxidative dissolution on UO2.17 Ar purging was continued throughout the experiment. Experiments were conducted under atmospheric pressure at a temperature of 25 °C which was maintained with a coolant system. Samples of the suspension were taken at intervals over the course of the reaction. The samples were immediately filtered through a 0.45 μm filter to stop the reaction, and then analysed for H2O2 and U. For determination of the pseudo-first order rate constants, 50 mg of U3O8 (sample 1) was added to 50 ml bicarbonate solution, whilst for the second-order rate constant measurements, 50, 100, 150 and 200 mg of U3O8 (sample 2) were added to 70 ml bicarbonate solution. Error in the experimental methodology was estimated as <5% by conducting a set of dissolution experiments in triplicate and taking the standard deviation of the H2O2 pseudo-first order decay constants.Analytical techniques
The concentration of U was measured by ICP-OES using a PerkinElmer Avio-200 spectrometer. Calibration was conducted using U standards and measurements were done in triplicate. The standard deviations of the measurements were typically <1% of the measured values. The concentration of H2O2 was measured by the Ghormley triiodide method where the iodide ion (I−) reacts with H2O2 and is converted to triiodide (I3−) using ammonium heptamolybdate ((NH4)6Mo7O24) and an acidic buffer (KHC8H4O4).33,34 The concentration of H2O2 was then determined from the absorbance spectra of I3− at 350 nm using a Shimadzu UV-3600 Plus UV-Vis-NIR spectrophotometer. Raman analysis of the oxide surface was conducted with a JASCO NRS-4500 Raman spectrometer. A 532 nm laser was introduced through a 20× objective lens, and 3 spectra of 10 seconds each were recorded and averaged for each sample.Results and discussion
U dissolution
The dissolution of U upon addition of H2O2 to U3O8 suspensions as a function of NaHCO3 was investigated by measuring dissolved U concentrations over the reaction time. Fig. 1 shows the dissolved uranium (U) minus the dissolved U concentration prior to H2O2 addition (U0). The dissolution of U changed over the experimental time and showed a clear effect of bicarbonate on the dissolution of U3O8. At 0.1 < [NaHCO3] < 5 mM, the dissolution was low. At > 5 mM the extent of dissolution significantly increased with bicarbonate. This is due to a change in the H2O2 decomposition mechanism as discussed later. The magnitude of U dissolution decreased with increasing bicarbonate concentration (i.e., from 5 to 10 mM bicarbonate the increase in U dissolution was ∼0.3 mM, and from 20 to 50 mM was ∼0.1 mM) indicating a complex relationship.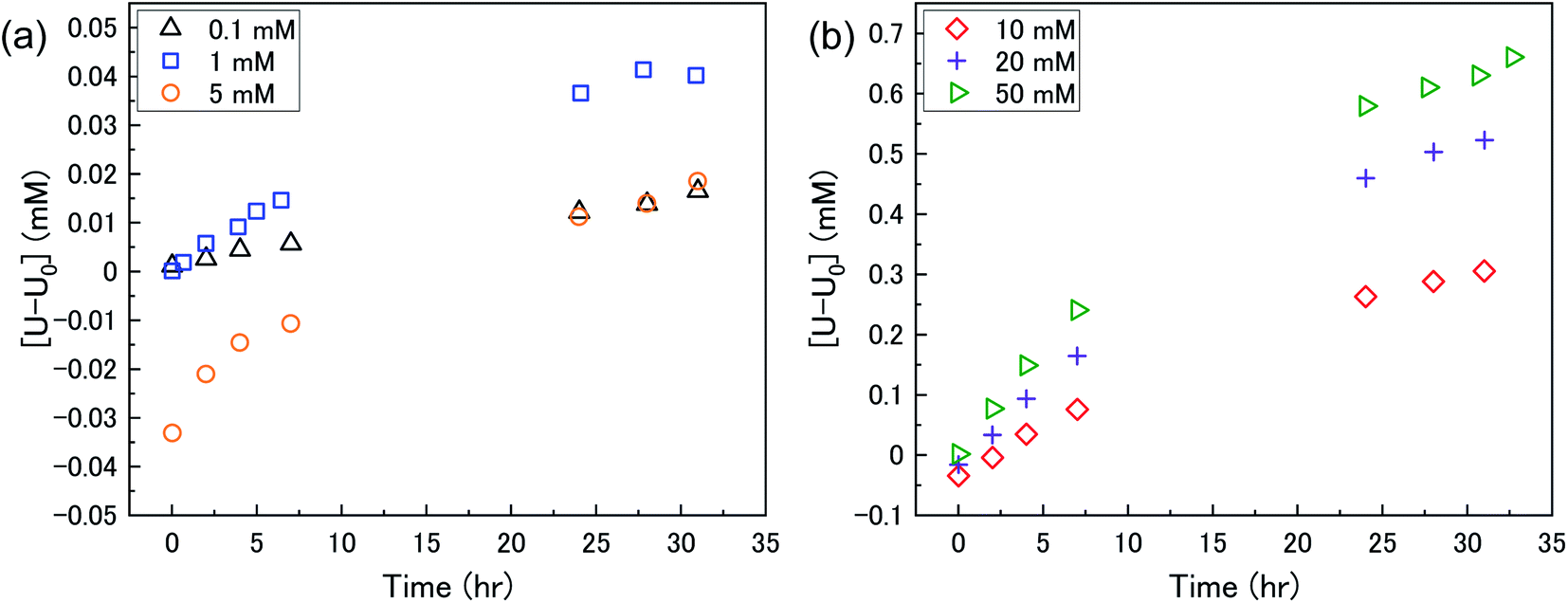 | ||
| Fig. 1 The dissolution of U as a function of time in a 50 mg suspension of U3O8 (sample 1) in (a) 0.1, 1 and 5 mM bicarbonate and (b) 10, 20 and 50 mM bicarbonate solution after addition of 300 μM H2O2. | ||
At t = 1 min, the value of [U–U0] became negative at certain bicarbonate concentrations, and so the measured values of [U–U0]t=1min were plotted as a function of bicarbonate concentration (Fig. 2). A decrease in the concentration of dissolved U can be seen in 5, 10 and 20 mM NaHCO3 solution, suggesting deposition from solution of U onto the U3O8 surface after the initial addition of H2O2 as highlighted by the second y-axis. As the extent of deposition increased with bicarbonate, it can be predicted that the deposits are uranium carbonates. Under the experimental conditions, the stable form of U in solution is UO2(CO3)34− and so the deposits may be UO2(CO3)3. Another possibility is the formation of uranyl peroxide (UO2(O2)), where the increase in deposition with bicarbonate is due to an increase in dissolved U with bicarbonate and, therefore, an increase in uranyl peroxide.
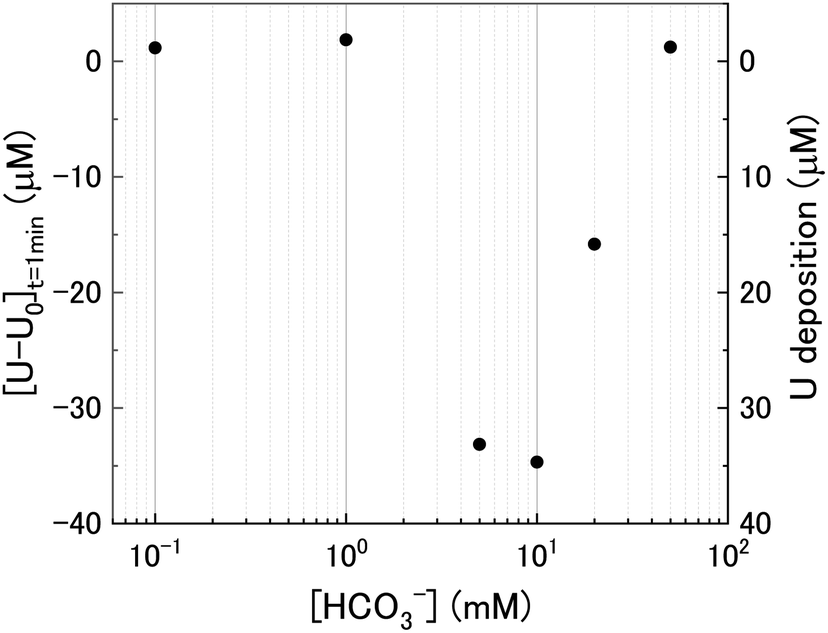 | ||
| Fig. 2 The initial change in U concentration one minute after H2O2 addition to the U3O8 suspension as a function of bicarbonate concentration. | ||
Kinetics of H2O2 decomposition
The dissolution of U from U3O8 was induced by the addition of H2O2 to the bicarbonate solution. Therefore, to understand the observed U dissolution behaviour from U3O8, the kinetics and mechanism of H2O2 decomposition at the U3O8 surface was studied. The kinetics of the reaction between H2O2 and U3O8 as a function of NaHCO3 were investigated by measuring the concentration of H2O2 over the reaction time. Fig. 3 shows the concentration of H2O2 after adding 300 μM to a 50 mg suspension of U3O8 at different bicarbonate concentrations as a function of time. The H2O2 concentration decreased quickly at low bicarbonate concentration (0.1 mM), but the decomposition slowed down as the bicarbonate concentration increased to 5 mM. Further increases in the bicarbonate concentration up to 50 mM caused the rate of H2O2 decomposition to gradually increase again, until the H2O2 concentration profile in 50 mM bicarbonate was similar to that in 0.1 mM bicarbonate. The results in Fig. 3 clearly show an effect of bicarbonate on H2O2 decomposition on U3O8.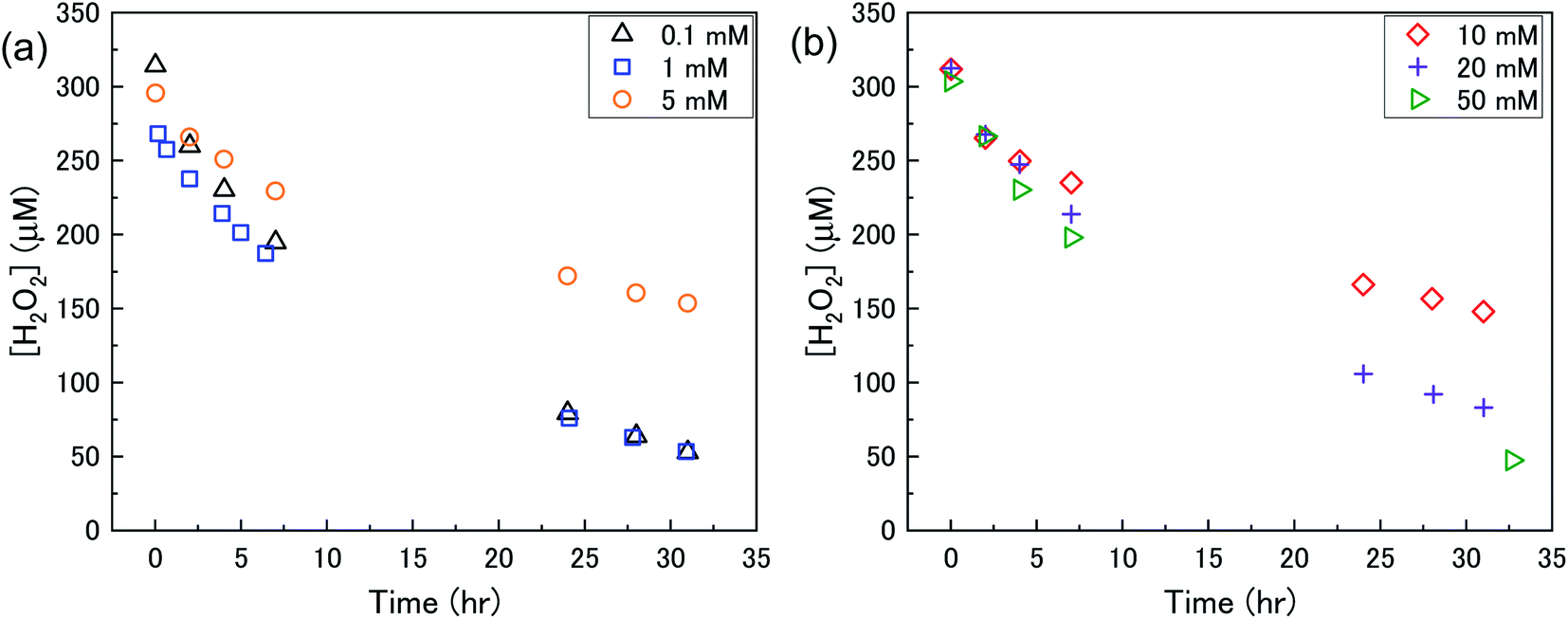 | ||
| Fig. 3 The concentration of H2O2 as a function of time in a 50 mg suspension of U3O8 (sample 1) in (a) 0.1, 1 and 5 mM bicarbonate and (b) 10, 20 and 50 mM bicarbonate solution after addition of 300 μM H2O2. | ||
To investigate the mechanism of H2O2 decomposition at the U3O8 surface, the kinetics of decomposition was investigated. Previous studies on H2O2 decomposition at the surface of uranium oxides have shown that the reaction follows first order kinetics with respect to H2O2. As the U3O8 surface is in excess relative to H2O2, the reaction can be modelled as a pseudo-first order reaction. Therefore, the rate of H2O2 decomposition can be explained by,
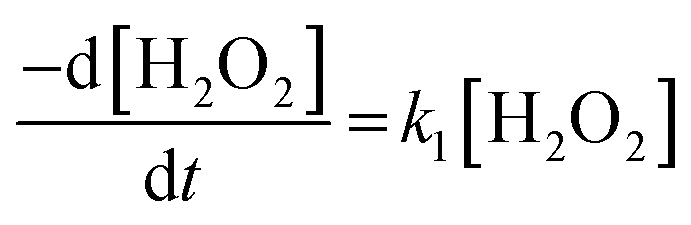 | (11) |
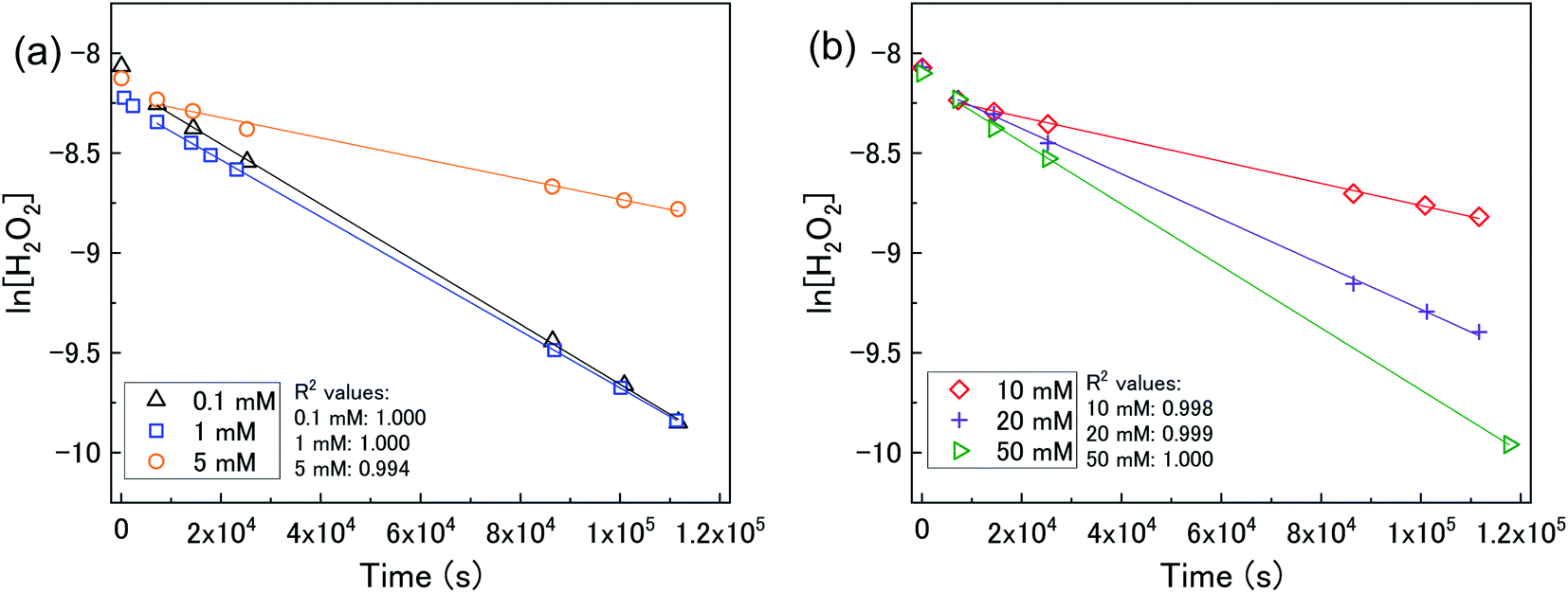 | ||
| Fig. 4 (a) A plot of ln[H2O2] vs. time as a function of bicarbonate concentration with 50 mg U3O8 (sample 1) in (a) 0.1, 1 and 5 mM bicarbonate and (b) 10, 20 and 50 mM bicarbonate solution showing pseudo-first order behaviour. | ||
The calculated values of k from the linear region (from t = 2 hours to the experiment end) are plotted against bicarbonate concentration in Fig. 5 and the value of k was found to be in the range between 0.4 to 1.6 × 10−5 s−1. The decrease in the pseudo-first order rate constant coincided with U deposition from solution indicating that the secondary phases that deposit on the surface of the U3O8 may block the approach of H2O2 to the surface. As the plots in Fig. 4 show linear behaviour, this suggests that these deposits are stable over the experimental timescale.
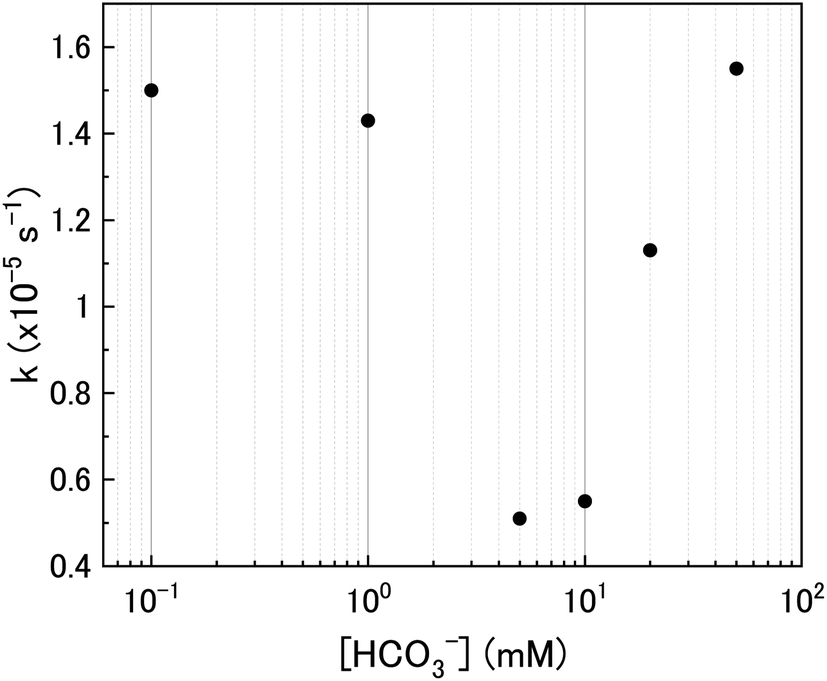 | ||
| Fig. 5 The pseudo-first order rate constants for H2O2 decomposition. | ||
U dissolution with H2O2 decomposition
The mechanism of U dissolution via H2O2 decomposition can be investigated by analysing the extent of U dissolution as a function of H2O2 decomposition. To illustrate this, Fig. 6 shows a plot of the amount of dissolved U against the amount of consumed H2O2 for each bicarbonate concentration. The U dissolution per H2O2 decomposition shows linear behaviour. If we consider the oxidative pathway for H2O2 decomposition at the U3O8 surface, U(V) is oxidised to U(VI) leading to decomposition of the U3O8 unit since the net charge in the lattice is no longer neutral. Therefore, each H2O2 decomposition event via oxidative decomposition will lead to a U3O8 dissolution event (U3O8 + H2O2 → 3UO22+(aq)), and a gradient of 3 may be expected from the plots in Fig. 6. If we consider only catalytic decomposition of H2O2, no U dissolution would occur giving an ideal gradient of 0. Therefore, the measured gradients provide a ratio of oxidative to catalytic H2O2 decomposition at each bicarbonate concentration, assuming these pathways are the only pathways for H2O2 decomposition (i.e. for 50 mM bicarbonate the ratio of oxidative dissolution is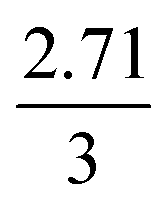 and catalytic decomposition is
and catalytic decomposition is 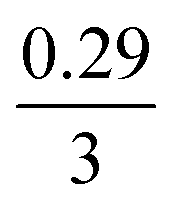 ). The dissolution of U from U3O8 in 10 mM bicarbonate gave a gradient of 2.68. By comparison with the gradients measured from the dissolution of U from UO2 (∼0.4) and UO2.3 (∼1) with H2O2 addition in 10 mM bicarbonate,17 this shows that the oxidative dissolution of U increases with increased oxidation state of the uranium oxide. As the complexation of U(VI) with bicarbonate drives the dissolution of U from the surface, it follows that the two-step oxidation of U(IV) → U(V) → U(VI) for UO2 would result in less dissolution of U than the one-step oxidation for U(V) → U(VI) in U3O8, and for intermediate UO2+x stoichiometries the dissolution rate would increase with increasing values of x. Another point of consideration regarding U dissolution is the crystal structure of UO2 (cubic fluorite) and U3O8 (orthorhombic). As the crystal structures are different, the number of surface sites for H2O2 decomposition will impact U dissolution. The surface site densities for UO2 and U3O8 have been reported as between 126 (ref. 35) to 165 (ref. 36) sites per nm2 for UO2 and 48 (ref. 36) sites per nm2 for U3O8. As the surface sites are ∼3 times lower for U3O8, the observed increase in U dissolution from U3O8 relative to UO2 is more pronounced than the measured dissolved U concentrations suggest.
). The dissolution of U from U3O8 in 10 mM bicarbonate gave a gradient of 2.68. By comparison with the gradients measured from the dissolution of U from UO2 (∼0.4) and UO2.3 (∼1) with H2O2 addition in 10 mM bicarbonate,17 this shows that the oxidative dissolution of U increases with increased oxidation state of the uranium oxide. As the complexation of U(VI) with bicarbonate drives the dissolution of U from the surface, it follows that the two-step oxidation of U(IV) → U(V) → U(VI) for UO2 would result in less dissolution of U than the one-step oxidation for U(V) → U(VI) in U3O8, and for intermediate UO2+x stoichiometries the dissolution rate would increase with increasing values of x. Another point of consideration regarding U dissolution is the crystal structure of UO2 (cubic fluorite) and U3O8 (orthorhombic). As the crystal structures are different, the number of surface sites for H2O2 decomposition will impact U dissolution. The surface site densities for UO2 and U3O8 have been reported as between 126 (ref. 35) to 165 (ref. 36) sites per nm2 for UO2 and 48 (ref. 36) sites per nm2 for U3O8. As the surface sites are ∼3 times lower for U3O8, the observed increase in U dissolution from U3O8 relative to UO2 is more pronounced than the measured dissolved U concentrations suggest.
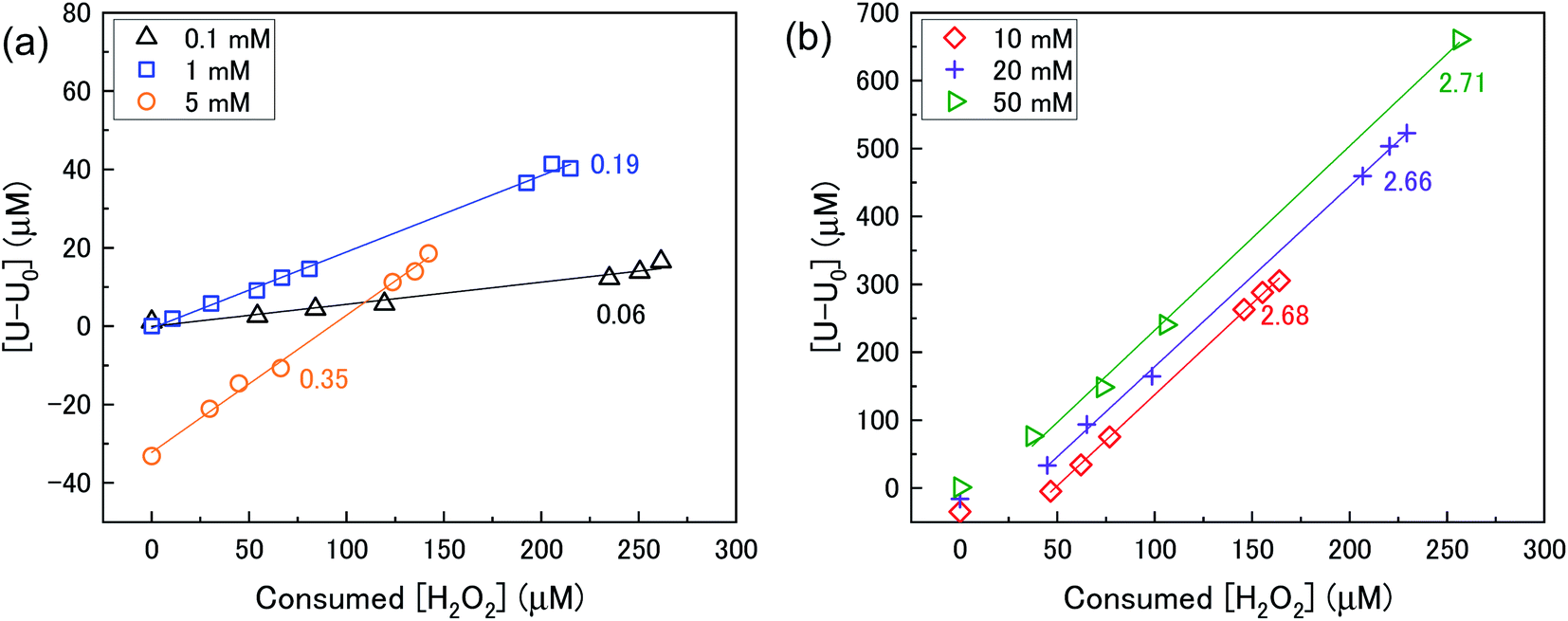 | ||
| Fig. 6 U dissolution from a 50 mg U3O8 (sample 1) suspension as a function of consumed H2O2 in (a) 0.1, 1 and 5 mM bicarbonate and (b) 10, 20 and 50 mM bicarbonate solution. | ||
Using the ratios taken from the gradients, the contributions of catalytic (kcat) and oxidative (kox) decomposition to the measured pseudo-first order rate constant can be found and are plotted in Fig. 7 as a function of bicarbonate.
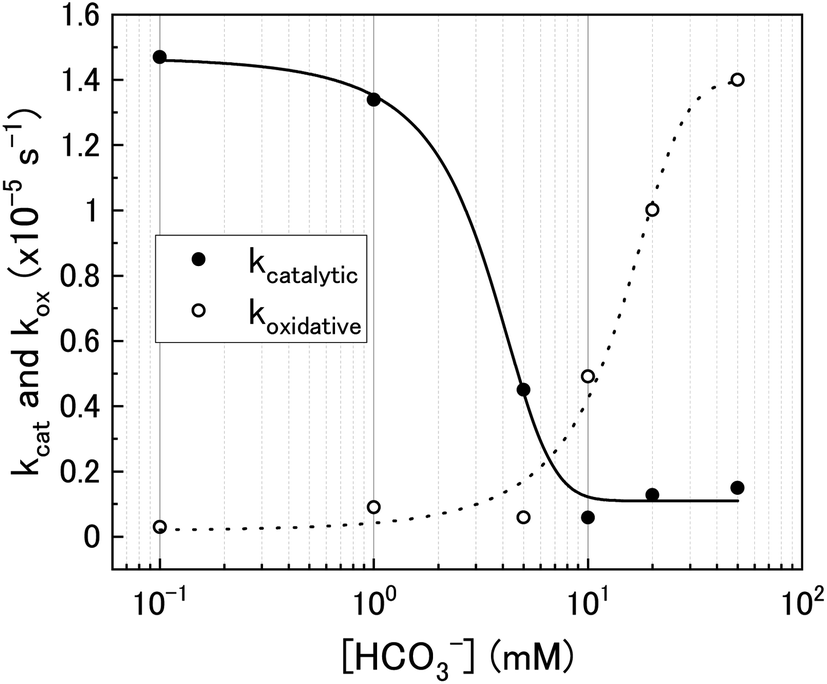 | ||
| Fig. 7 The catalytic (kcat) and oxidative (kox) pseudo-first order rate constants for H2O2 decomposition on U3O8 as a function of bicarbonate concentration. | ||
At low bicarbonate concentrations, the main pathway for H2O2 decomposition is the catalytic decomposition mechanism as there is little U dissolution associated with H2O2 decomposition. As kox is low, this indicates that the U3O8 is protected from H2O2 by a surface layer. It is postulated that upon addition of H2O2 to the bicarbonate solution, oxidative dissolution proceeds on the bare U3O8 surface (Fig. 8). As this involves the oxidation of U(V) to U(VI), it is likely that U(VI) forms a surface layer on the U3O8 which protects against further oxidative dissolution as U(VI) is already fully oxidised, and due to the low concentration of bicarbonate the surface layer is stable.
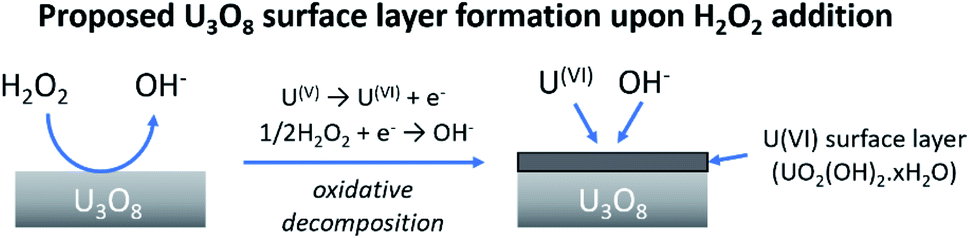 | ||
| Fig. 8 The formation of a protective surface layer on the surface of U3O8 due to oxidative decomposition of H2O2. | ||
The composition of the surface layer is thought to be in the hydroxide form (UO2(OH)2·xH2O) due to the formation of hydroxide from the oxidative decomposition of H2O2. Raman analysis of the U3O8 surface after removal from solution and vacuum drying showed spectra representative of U3O8 only (Fig. 9). Peaks relating to U3O8 were observed including the U–O A1g stretching modes at 335 and 410 cm−1, and the U–O Eg stretching mode at 475 cm−1.37 As U3O8 was the only phase observed, any surface layer that formed had been removed prior to Raman analysis. If the surface layer is in the hydroxide form, it is expected to decompose upon drying which would explain the observed results. Further studies are required to elucidate the composition of the surface.
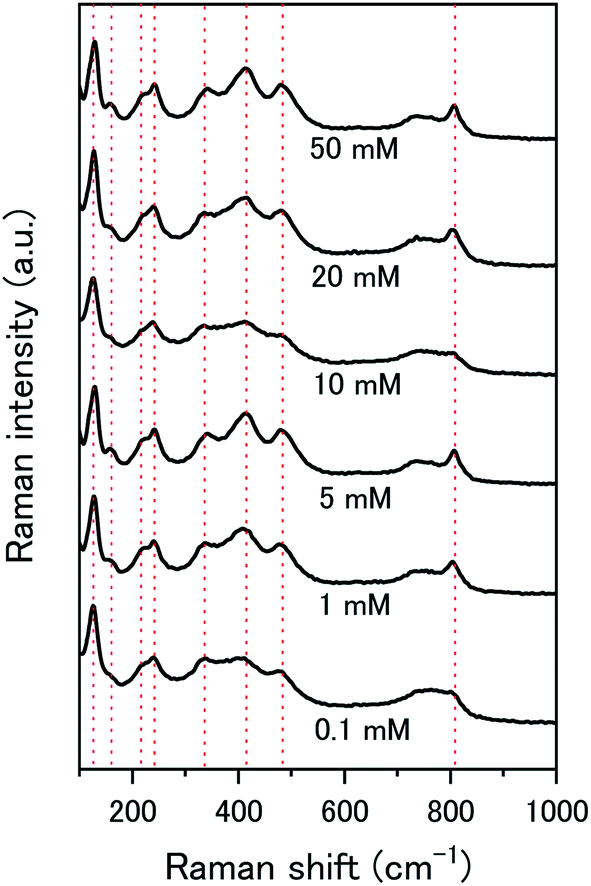 | ||
| Fig. 9 Raman spectra of U3O8 (sample 1) after the dissolution tests for different concentrations of NaHCO3. | ||
As the bicarbonate concentration increases from 0.1 to 5 mM, the rate of catalytic H2O2 decomposition decreases. This is caused by an increase in deposition from solution as seen in Fig. 2. As the pseudo-first order rate constant decreases up to 5 mM, it can be said that the deposits do not catalyse H2O2 decomposition to the extent that U3O8 does.
Increasing the bicarbonate concentration > 5 mM changes the main H2O2 decomposition mechanism pathway from catalytic to oxidative. At NaHCO3 concentrations of 10 mM and above, at least 90% of the H2O2 decomposed via oxidation of U(V) to U(VI). This is due to increased dissolution of the U(VI) surface layer and exposure of the U3O8 surface beneath leading an increase in kox. As the value of k increases with bicarbonate, this suggests that the dissolution step is rate determining rather than the redox reaction. Therefore, dissolution experiments in solutions of higher bicarbonate concentrations are required to elucidate the true value of k for oxidative dissolution in this system. Interestingly, a study by Nilsson et al. on UO2 dissolution in 10 mM NaHCO3 with H2O2 addition using pellets showed that ∼14% of H2O2 decomposition events occurred via oxidative dissolution while the value was even lower (∼2%) on SIMFUEL.38 This suggests that kcat is high in the case of the pellets indicating that the surface oxide that forms on the pellets is more protective than on the powders. The large discrepancy between the H2O2 decomposition behaviour between UO2 pellets and U3O8 powder (and UO2 powder) is a point that requires investigation.
To clarify the dependence of the catalytic and oxidative mechanisms on U3O8, the second order rate constants for H2O2 decomposition were obtained for 0.1 mM and 50 mM solutions with U3O8 (sample 2). The second order rate equation,
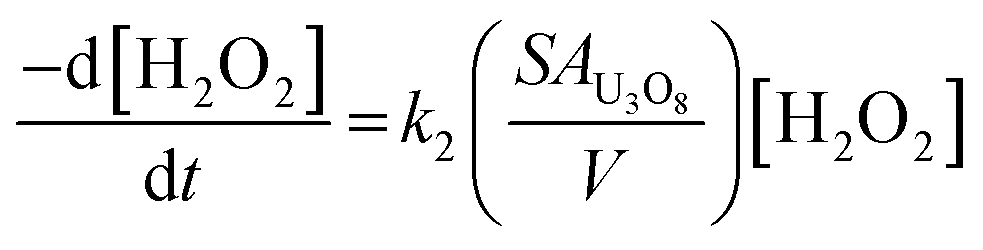 | (12) |
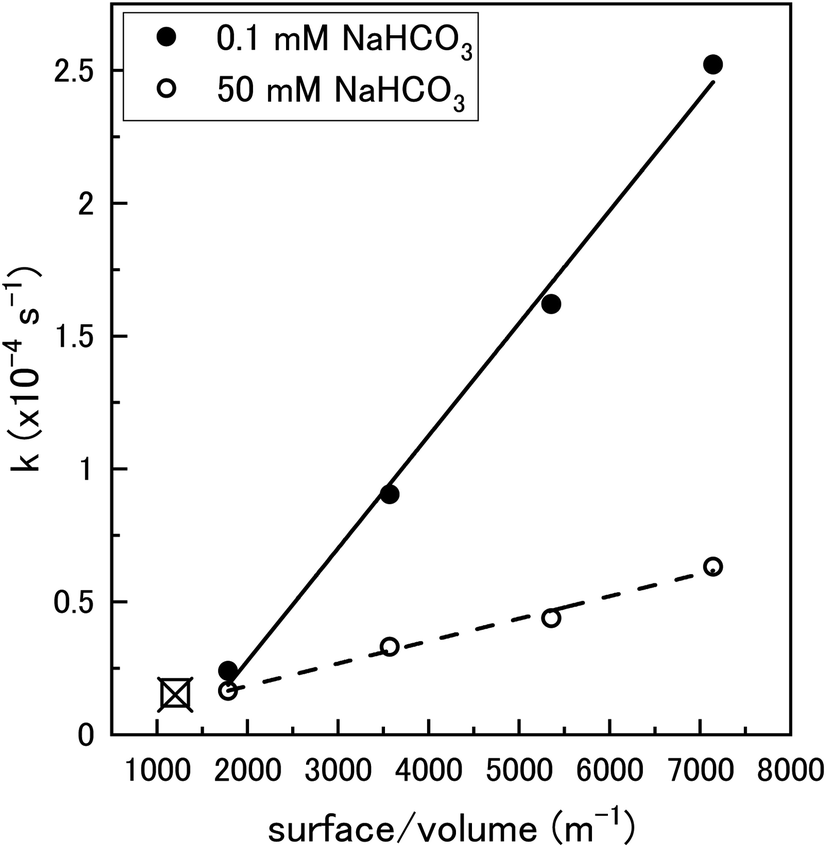 | ||
| Fig. 10 The pseudo-first order rate constant, k, plotted against the U3O8 (sample 2) surface area to solution volume ratio (m−1) for 0.1 mM and 50 mM HCO3− solutions (70 ml solution). The data for 0.1 mM (□) and 50 mM (x) calculated using U3O8 sample 1 shown in Fig. 5 is included to show reproducibility (50 mg U3O8 in 50 ml solution). | ||
Proposed pathway for U3O8 dissolution by H2O2 in NaHCO3 solution
From the experimental results, a proposed pathway to explain the observed behaviour of U3O8 in bicarbonate solution with H2O2 is summarized, and a schematic is provided in Fig. 11. At low bicarbonate concentrations upon H2O2 addition, oxidative decomposition of H2O2 occurs at the exposed U3O8 surface forming a surface layer comprised of U(VI) that provides protection against further oxidative dissolution. The decomposition of H2O2 proceeds via catalytic decomposition, and so the rate of U dissolution is low. The surface layer protects the U3O8 in bicarbonate concentrations up to 5 mM, and the H2O2 decomposition mechanism remains catalytic and U dissolution remains low. At 10 mM bicarbonate, the concentration of bicarbonate is sufficient to induce dissolution of the surface layer, and the surface layer does not fully protect the U3O8 which is exposed leading to oxidative decomposition of H2O2 and an increase in U dissolution. At higher bicarbonate concentrations, the surface layer is further dissolved, and oxidative decomposition of H2O2 and dissolution of U proceeds at higher rates.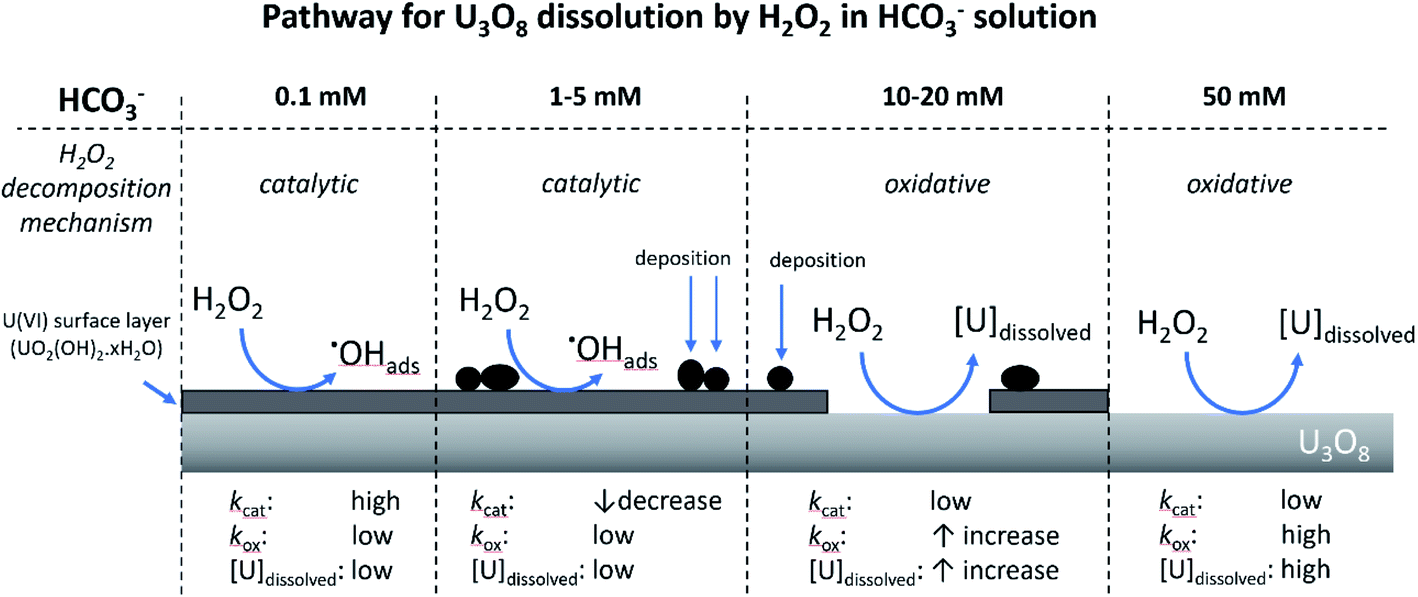 | ||
| Fig. 11 The proposed pathway for U3O8 dissolution upon H2O2 addition as a function of bicarbonate concentration. | ||
Conclusions
Based on the presented results, the effect of bicarbonate on U dissolution from U3O8 with H2O2 addition can be split into 3 sections:(1) [NaHCO3] < 5 mM: H2O2 decomposition occurs via catalytic decomposition at the U3O8 surface, and the dissolution of U into solution is low.
(2) 5 mM < [NaHCO3] < 20 mM: secondary phases deposit onto the surface of the U3O8 upon H2O2 addition. The mechanism of decomposition changes from catalytic to oxidative, causing dissolution of U.
(3) [NaHCO3] > 20 mM: the decomposition mechanism of H2O2 is >90% oxidative, leading to significant dissolution of U.
The concentration of bicarbonate and form of uranium oxide has a large influence on U dissolution and H2O2 decomposition. Significant dissolution of U from U3O8 was observed at bicarbonate concentrations > 5 mM, and the extent of U dissolution was found to be larger on U3O8 than for UO2 which was attributed to the one-step oxidation of U(V) to U(VI) for U3O8 compared to the two-step oxidation for UO2 from U(IV) to U(V) to U(VI). The rate of H2O2 decomposition on U3O8 was comparable to literature data for UO2. However, the mechanism of H2O2 decomposition on U3O8 showed a strong dependence on the concentration of bicarbonate in solution with catalytic preferred at low bicarbonate and oxidative at high bicarbonate. The increase in catalytic activity at low bicarbonate was attributed to oxidation of the U3O8 surface and formation of a surface oxide.
Predicting the dissolution behaviour of spent fuel in the far future upon deep geological repository failure is a challenging task that requires significant experimental data for the development of accurate predictive models. In this work, elucidation of the mechanism of H2O2 decomposition on U3O8 and its effect on U dissolution was achieved, along with H2O2 decay constants as a function of simulated groundwater bicarbonate concentration. In groundwater containing high bicarbonate concentrations, significant dissolution of U from U3O8 is expected. This provides contributions to the development of such models for safety assessment of deep geological repositories. By demonstrating that the form of U will play a major role in the rate of U dissolution into the environment, the need for further studies regarding the effect of spent fuel composition on radionuclide dissolution into groundwater has been highlighted.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was performed as part of “Project on Research and Development of Spent Fuel Direct Disposal as an Alternative Disposal Option (2020FY)” funded by the Ministry of Economy, Trade and Industry of Japan.References
- I. Casas, J. de Pablo, J. Giménez, M. E. Torrero, J. Bruno, E. Cera, R. J. Finch and R. C. Ewing, Radiochim. Acta, 2009, 97, 485 CAS.
- S. J. Romaniello, A. D. Herrmann and A. D. Anbar, Chem. Geol., 2013, 362, 305 CrossRef CAS.
- Y. Kolodny, A. Torfstein, K. Weiss-Sarusi, Y. Zakon and L. Halicz, Chem. Geol., 2017, 451, 1 CrossRef CAS.
- N. E. Jemison, A. E. Shiel, T. M. Johnson, C. C. Lundstrom, P. E. Long and K. H. Williams, Environ. Sci. Technol., 2018, 52, 3422 CrossRef CAS.
- E. Ekeroth, O. Roth and M. Jonsson, J. Nucl. Mater., 2006, 355, 38 CrossRef CAS.
- A. Traboulsi, J. Vandenborre, G. Blain, B. Humbert, J. Barbet and M. Fattahi, J. Phys. Chem. C, 2014, 118, 1071 CrossRef CAS.
- R. Springell, S. Rennie, L. Costelle, J. Darnbrough, C. Stitt, E. Cocklin, C. Lucas, R. Burrows, H. Sims, D. Wermeille, J. Rawle, C. Nicklin, W. Nuttall, T. Scott and G. Lander, Faraday Discuss., 2015, 180, 301 RSC.
- S. Le Caër, Water, 2011, 3, 235 CrossRef.
- S. Sunder, N. H. Miller and D. W. Shoesmith, Corros. Sci., 2004, 46, 1095 CrossRef CAS.
- J. S. Goldik, J. J. Noël and D. W. Shoesmith, J. Electroanal. Chem., 2005, 582, 241 CrossRef CAS.
- L. Wu and D. W. Shoesmith, Electrochim. Acta, 2014, 137, 83 CrossRef CAS.
- L. Bauhn, N. Hansson, C. Ekberg, P. Fors and K. Spahiu, J. Nucl. Mater., 2018, 507, 38 CrossRef CAS.
- D. W. Shoesmith, J. Nucl. Mater., 2000, 282, 1 CrossRef CAS.
- M. M. Hossain, E. Ekeroth and M. Jonsson, J. Nucl. Mater., 2006, 358, 202 CrossRef CAS.
- J. De Pablo, I. Casas, J. Giménez, V. Martí and M. E. Torrero, J. Nucl. Mater., 1996, 232, 138 CrossRef CAS.
- S. Röllin, K. Spahiu and U. B. Eklund, J. Nucl. Mater., 2001, 297, 231 CrossRef.
- Y. Kumagai, A. Barreiro Fidalgo and M. Jonsson, J. Phys. Chem. C, 2019, 123, 9919 CrossRef CAS.
- G. Leinders, R. Bes, J. Pakarinen, K. Kvashnina and M. Verwerft, Inorg. Chem., 2017, 56, 6784 CrossRef CAS.
- K. O. Kvashnina, S. M Butorin, P. Martin and P. Glatzel, Phys. Rev. Lett., 2013, 111, 1 CrossRef.
- K. Sanyal, A. Khooha, G. Das, M. K. Tiwari and N. L. Misra, Anal. Chem., 2017, 89, 871 CrossRef CAS.
- P. Taylor, D. D. Wood, D. G. Owen and G. Park, J. Nucl. Mater., 1991, 183, 105 CrossRef CAS.
- A. O. Allen, C. J. Hochanadel, J. A. Ghormley and T. W. Davis, J. Phys. Chem., 1952, 56, 575 CrossRef.
- T. K. Campbell, E. R. Gilbert, C. K. Thornhill and B. J. Wrona, Nucl. Technol., 1989, 84, 182 CrossRef CAS.
- K. Fukuda, Y. Watanabe, H. Murakami, Y. Amano, D. Aosai, Y. Kumamoto and T. Iwatsuki, Hydrochemical Investigation at the Mizunami Underground Research Laboratory – Compilation of Groundwater Chemistry Data in the Mizunami Group and the Toki Granite, Japanese Atomic Energy Agency, 2020 Search PubMed.
- B. Y. Kim, J. Y. Oh, M. H. Baik and J. I. Yun, Nucl. Eng. Technol., 2010, 42, 552 CrossRef CAS.
- S. S. Kim, M. H. Baik, J. W. Choi, H. S. Shin and J. I. Yun, J. Radioanal. Nucl. Chem., 2010, 286, 91 CrossRef CAS.
- L. F. Auque, M. J. Gimeno and J. B. Gomez, Groundwater chemistry around a repository for spent nuclear fuel over a glacial cycle, Swedish Nuclear Fuel and Waste Managemnt Co, 2006 Search PubMed.
- H. M. Rietveld, J. Appl. Crystalogr., 1969, 2, 65 CrossRef CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- P. W. H. Bragg and W. L. Bragg, Proc. R. Soc. London. Ser. A, Contain. Pap. a Math. Phys. Character, 1913, 17, 428 Search PubMed.
- B. O. Loopstra, J. Inorg. Nucl. Chem., 1977, 39, 1713 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309 CrossRef CAS.
- A. O. Allen, C. J. Hochanadel, J. A. Ghormley and T. W. Davis, J. Phys. Chem., 1952, 56, 575 CrossRef.
- T. C. J. Overton and W. T. Rees, Analyst, 1950, 75, 204 RSC.
- M. M. Hossain and M. Jonsson, J. Nucl. Mater., 2008, 373, 186 CrossRef CAS.
- F. Clarens, J. de Pablo, I. Casas, J. Giménez and M. Rovira, MRS Online Proc. Libr., 2003, 1, 730 Search PubMed.
- D. Manara and B. Renker, J. Nucl. Mater., 2003, 321, 233 CrossRef CAS.
- S. Nilsson and M. Jonsson, J. Nucl. Mater., 2011, 410, 89 CrossRef CAS.
- T. E. Eriksen, D. W. Shoesmith and M. Jonsson, J. Nucl. Mater., 2012, 420, 409 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |