
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the PET radioisotope universe utilizing solid targets on small medical cyclotrons
K. J. H. George
ab,
S. Borjian
e,
M. C. Cross
e,
J. W. Hicks
ab,
P. Schaffer
defg and
M. S. Kovacs
 *abc
*abc
aLawson Health Research Institute, 268 Grosvenor Street, London, ON N6A 4V2, Canada. E-mail: mkovacs@lawsonimaging.ca
bMedical Biophysics, Western University, 1151 Richmond Street N., London, ON N6A 5C1, Canada
cMedical Imaging, Western University, 1151 Richmond Street N., London, ON N6A 5C1, Canada
dLife Sciences, TRIUMF, 4004 Wesbrook Mall, Vancouver, BC V6T 2A3, Canada
eARTMS, 301-4475 Wayburn Drive, Burnaby, BC V5G 4X4, Canada
fRadiology, University of British Columbia, 2775 Laurel St, Vancouver, BC V5Z 1M9, Canada
gChemistry, Simon Fraser University, 8888 University Dr, Burnaby, BC V5A 1S6, Canada
First published on 21st September 2021
Abstract
Molecular imaging with medical radioisotopes enables the minimally-invasive monitoring of aberrant biochemical, cellular and tissue-level processes in living subjects. The approach requires the administration of radiotracers composed of radioisotopes attached to bioactive molecules, the pairing of which considers several aspects of the radioisotope in addition to the biological behavior of the targeting molecule to which it is attached. With the advent of modern cellular and biochemical techniques, there has been a virtual explosion in potential disease recognition antigens as well as targeting moieties, which has subsequently opened new applications for a host of emerging radioisotopes with well-matched properties. Additionally, the global radioisotope production landscape has changed rapidly, with reactor-based production and its long-defined, large-scale centralized manufacturing and distribution paradigm shifting to include the manufacture and distribution of many radioisotopes via a worldwide fleet of cyclotrons now in operation. Cyclotron-based radioisotope production has become more prevalent given the commercial availability of instruments, coupled with the introduction of new target hardware, process automation and target manufacturing methods. These advances enable sustained, higher-power irradiation of solid targets that allow hospital-based radiopharmacies to produce a suite of radioisotopes that drive research, clinical trials, and ultimately clinical care. Over the years, several different radioisotopes have been investigated and/or selected for radiolabeling due to favorable decay characteristics (i.e. a suitable half-life, high probability of positron decay, etc.), well-elucidated chemistry, and a feasible production framework. However, longer-lived radioisotopes have surged in popularity given recent regulatory approvals and incorporation of radiopharmaceuticals into patient management within the medical community. This review focuses on the applications, nuclear properties, and production and purification methods for some of the most frequently used/emerging positron-emitting, solid-target-produced radioisotopes that can be manufactured using small-to-medium size cyclotrons (≤24 MeV).
 K. J. H. George | Keller Joshell Hadassah George obtained her BSc in chemical engineering at the Illinois Institute of Technology in 2013. She then obtained her MESc and PhD at Western University in 2015 and 2019 respectively. She currently holds a Mitacs Accelerate Postdoctoral Fellowship at Western University/ARTMS, and her research focuses on the optimization of separation methods for cyclotron-produced 68Ga on solid targets. |
 S. Borjian | Sogol Borjian received a BSc (2005) and MSc (2007) in Polymer Engineering from Amirkabir University of Technology in Tehran, Iran. She then received a PhD (2014) in Organometallic Chemistry from Queen's University in Kingston, ON, where her research focused on developing new palladium catalysts for pharmaceutical applications. She followed with an Industrial Postdoctoral Fellowship in collaboration with ABB (Zurich) where her research focused on developing novel thin films for selective chemical sensing using micro-optical devices. Sogol joined ARTMS Inc. in 2018 and is currently responsible for managing the Chemistry and Target Production teams, overseeing the development and commercialization of medical radioisotopes used in cancer diagnosis and treatment. |
 M. C. Cross | Michael Cross is a co-founder and COO of ARTMS Inc. where he is responsible for overseeing operational activities, establishing regulatory and clinical strategies, and innovator and research collaborations. Previously Michael was General Manager at the Centre for Probe Development and Commercialization (McMaster University). Michael has over 20 years of healthcare and life science industry experience. Michael co-founded and was COO/CBO of OncoSec Medical and was also the SVP and co-lead of a $330 M life sciences venture fund. Michael received his PhD in physiology and MBA from the University of Toronto and was a post-doctoral fellow with the Department of National Defence. |
 J. W. Hicks | Justin Hicks is a research scientist focusing on radiochemistry for molecular imaging applications to investigate pressing biomedical questions. He received a MSc in 99mTc chemistry under Dr John Valliant at McMaster University (2010). He then completed a PhD on PET tracer development with Drs Alan Wilson and Neil Vasdev at the University of Toronto (2015). Dr Hicks then joined the Lawson Health Research Institute cyclotron as a radiochemist to translate clinical PERS, followed in 2016 by appointments as a Lawson Scientist and Assistant Professor in Medical Biophysics at Western University. |
 P. Schaffer | Paul Schaffer obtained his PhD (Chemistry) in 2003 from McMaster University, after which he transitioned to become a Research Associate at McMaster Nuclear Reactor. This was followed by a role of Lead Scientist at GE Global Research. Since 2009, he has served as Associate Laboratory Director, Life Sciences at TRIUMF since 2012. Dr Schaffer is an Associate Professor in Radiology at the University of British Columbia, an adjunct professor in Chemistry at Simon Fraser University and is affiliated with the Research Center for Nuclear Physics at Osaka University. Dr Schaffer now serves as Chief Technology of ARTMS, Inc.; a spin-off company resulting from his research in large-scale cyclotron-based production of 99mTc in response to the 2007–2009 supply crises. |
 M. S. Kovacs | Michael Kovacs received his PhD (Medicinal Inorganic Chemistry) from the University of British Columbia in 2001. Under an NSERC Industrial Fellowship, he worked at Advanced Cyclotron Systems Inc (ACSI) where he lead research and development work on automated radiochemistry synthesis and cyclotron targetry. He was recruited as a Scientist by the Lawson Health Research Institute in 2003 to create a PET cyclotron and radiochemistry research program (founded 2010). Dr Kovacs is currently the Director of the Lawson Cyclotron and is Assistant Professor in Medical Imaging and Medical Biophysics at Western University. His research interests include the production and application of metal ions to PET imaging. |
Introduction
Positron Emission Tomography (PET) is one of several diagnostic imaging tools used to non-invasively assess metabolic activity in body tissue. PET scanning is sensitive enough to quantify changes in biological processes, making it an ideal candidate for assessing the efficacy of an early treatment plan or newly developed drugs.1 PET makes use of radiotracers which permit the visual isolation of metabolic processes within specific tissues and cells. These radiotracers are composed of a radioisotope attached to a bioactive molecule (e.g. a small molecule, peptide, antibody, etc.), as demonstrated in Fig. 1. The choice of bioactive targeting molecule mainly depends on the process to be visualized but also on the molecule's ability to incorporate the radioisotope into the chemical structure of the pharmaceutical compound. However, choosing the radioisotope requires consideration of biocompatibility in addition to the assessment of physical and biological half-life suitability for the metabolic process under investigation.2–4 Over the years, the suitability of several PET radioisotopes for use as tracers has been advanced, along with their unique methods of production.2,5 There are three major methods that can be used to produce radiometals – parent/progeny generators, nuclear reactors and cyclotrons.2 For radioisotope production utilizing cyclotrons, radioisotopes can be produced using gas, liquid, or solid phase targets.2,6 This review will focus on recent advancements in the use of solid targets.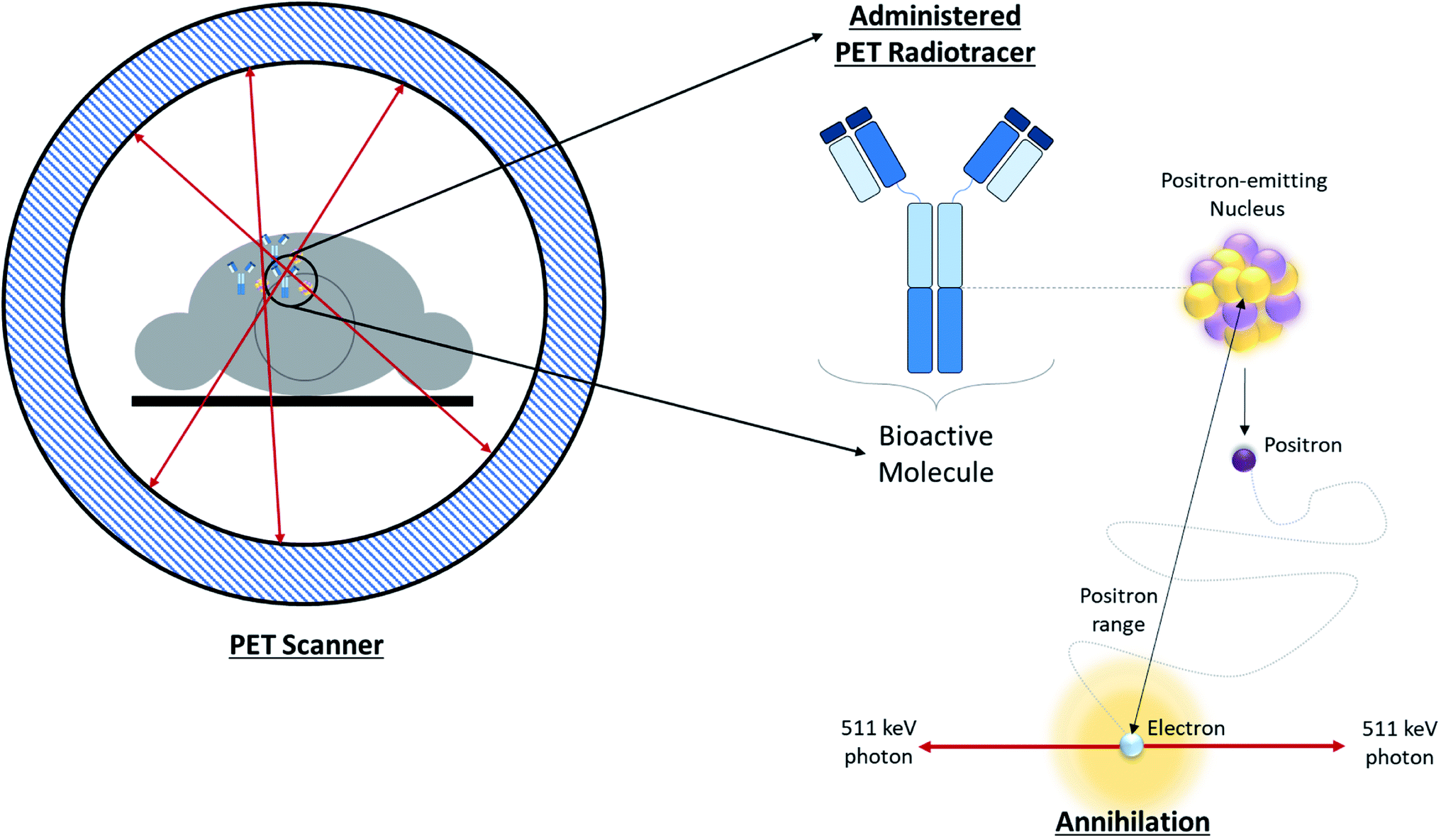 | ||
| Fig. 1 Overview of PET. Schematic depicting positron annihilation in a patient undergoing a PET scan. | ||
The design of a solid target requires several considerations, and these factors influence its configuration and performance. Targets may be composed entirely of enriched and natural elements and alloys, and may be prepared as foils, disks or composites (through the electroplating, sputtering, sedimenting, or pressing of alloys and elements onto supporting metal substrates (target backing) such as gold, silver, tantalum, titanium, rhodium or niobium).7–10 The specific design selected depends on factors such as substrate bond, heat transfer efficiency, ease of product isolation from the target material after irradiation, ease of target material recovery for enriched-material targets, product sensitivity to impurities and nuclear cross-section data.8 An ideal target should be stable, allow a high rate of heat transfer, and possess favorable melting properties6 since proton-induced irradiation generates significant amounts of heat in a small target area. When heat transfer and melting point are not suitable, innovations such as an oblique target arrangement as well as target alloying practices have been implemented to pacify these limitations.6 The separation chemistry employed should be fast (i.e. less than one half-life of the product radionuclide) and efficient at separating the product from both the target material and co-produced impurities.11 Commonly used separation techniques include are ion exchange chromatography, solid phase extraction (SPE) and thermochromatographic dry distillation, the schematics for which can be seen in Fig. 2, Fig. 3 and Fig. 4 respectively.
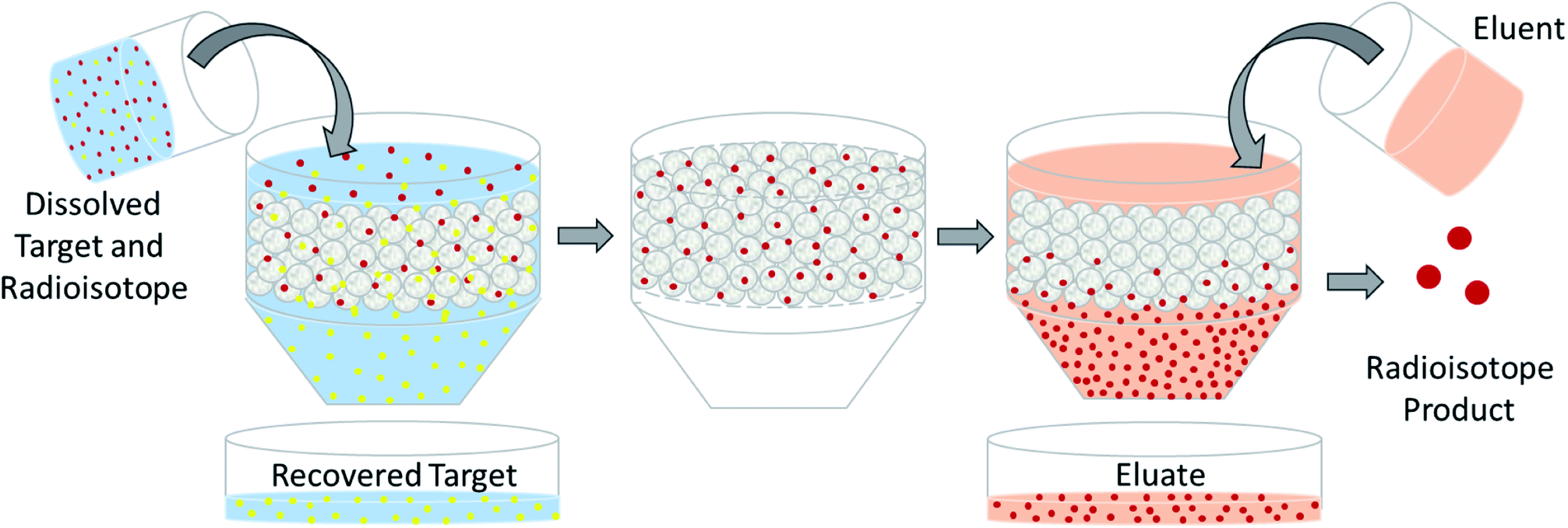 | ||
| Fig. 2 Ion exchange chromatography. Schematic depicting the separation of dissolved PET radioisotopes from dissolved, unreacted target material after passage through an ion exchange column. The resin retains the PET radioisotope, and an eluent is used for its elution. | ||
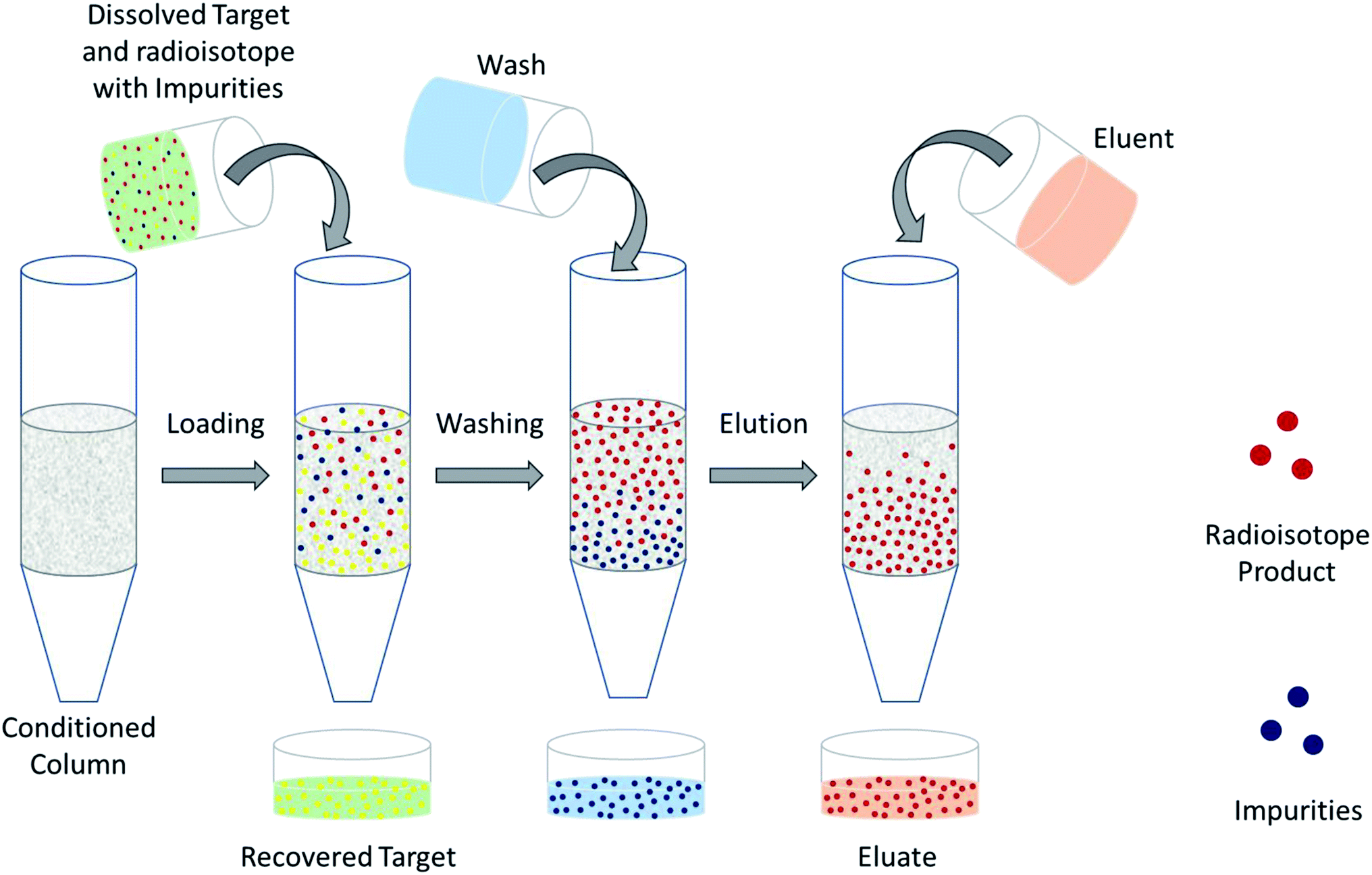 | ||
| Fig. 3 Solid phase extraction. Schematic depicting the separation of dissolved PET radioisotopes from dissolved, unreacted target material after passage through a solid phase extraction column. The column retains the PET radioisotope and any impurities. Impurities are washed-off before elution is performed to obtain the PET radioisotope. | ||
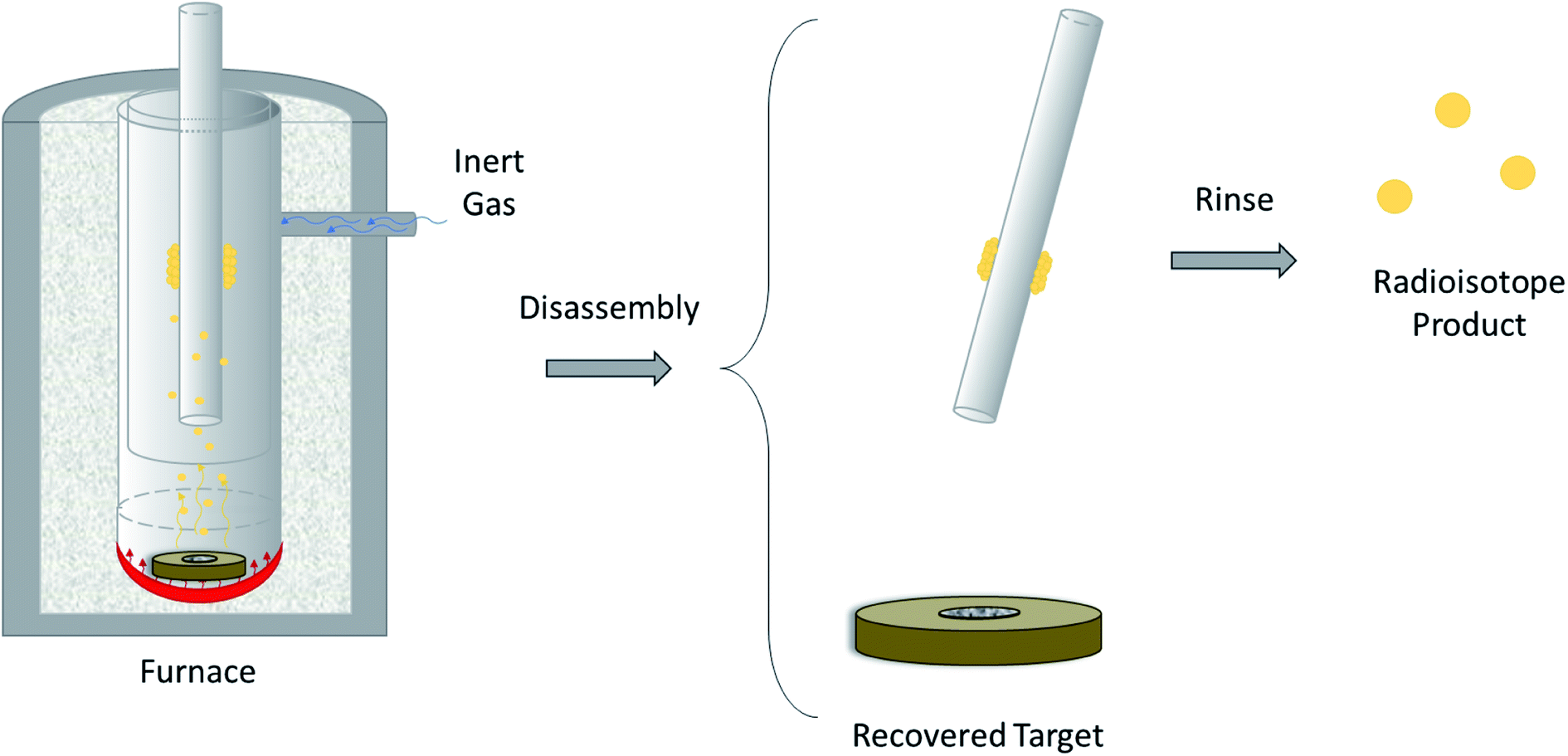 | ||
| Fig. 4 Thermochromatographic dry distillation. Schematic depicting the separation of volatile PET radioisotopes from non-volatile target material after heating is performed in a furnace. The PET radioisotope condenses on a removable, cooler surface within the furnace. The radioisotope is removed from the surface using a rinse. | ||
The radioisotopes covered in this review (see Table 1) can be produced on solid targets using small-to-medium sized, proton-accelerating cyclotrons with a maximum particle acceleration energy of 24 MeV. Production using this class of cyclotron was selected since these machines are the most prevalent in medical and research institutions. While the list of radioisotopes reviewed is a comprehensive one, the authors would like to highlight other PET radioisotopes such as 110mIn12,13, 152Tb,13,14 51Mn,15,16 62Cu16–18 and 132La19,20 which are also available for production on the aforementioned cyclotrons, but were excluded from this review due to limitations. This review briefly discusses the biomedical application of each radionuclide while focusing mainly on the specifics of its production and purification procedures. Several recent reviews can provide more details on the applications of established and emerging PET radionuclides discussed herein ref. 21–23. A summary of the major nuclear and production properties of each radioisotope is provided in Table 1 for convenience. The nuclear information presented in this review was retrieved from the National Nuclear Data Center website by Brookhaven National Laboratory (https://www.bnl.gov).
| Element | Isotope | Nuclear properties | Production conditions | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Half-life | Mode of decay (%) | Avg. β+ energy, keV | β+ endpoint energy, keV (%) | Principal γ energies, keV (Abs. %) | Major nuclear reactions | Beam energy, MeV | Target | Max. reported yield, MBq (μA h)−1 | ||
| Arsenic (As) | 72 | 26.0 h | β+ (87.8) | 1117 | 2500 (64.2) 3334 (16.3) | 511 (176.4), 630 (8.1), 834 (81.0), 1464 (1.13) | 72Ge(p,n)72As | 18–8 | Natural or enriched GeO2 on Cu or Nb, Ge foil | 90 |
| EC (12.2) | 1528 | |||||||||
| Bromine (Br) | 75 | 96.7 min | β+ (75.0) | 773, 708, 904 | 1753 (53.0), 1612 (4.9), 2040 (4.0) | 511 (149.1),141 (6.6), 287 (88.0), 428 (4.4) | 76Se(p,2n)75Br | 24–21.5 | Enriched Se, enriched selenides (i.e. Ag2Se, CuAgSe, Cu2Se, PbSe) | 1480 |
| EC (25.0) | ||||||||||
| 76 | 16.2 h | β+ (55.0) | 1532, 375, 1800 | 3382 (25.8), 871 (6.3), 3941 (6.0) | 511 (109), 559 (74.0), 657 (15.9), 1854 (147) | 76Se(p,n)76Br | 16–8 | Enriched Se, enriched selenides (i.e. Cu2Se) | 70 | |
| EC (45.0) | ||||||||||
| Copper (Cu) | 60 | 23.7 min | β+ (93.0) | 872, 1325, 840 | 1982 (49.0), 2947 (15.0), 1912 (11.6) | 511 (185.0), 826 (21.7), 1333 (88.0), 1792 (45.4) | 60Ni(p,n)60Cu | 14.7 | Natural or enriched Ni on Au | 2146 |
| EC (7.0) | ||||||||||
| 61 | 3.3 h | β+ (61.0) | 524, 399 | 1216 (51.0), 933 (5.8) | 511 (123.0), 67 (4.2), 282 (12.2), 656 (10.8) | 61Ni(p,n)61Cu | 14.7–9 | Enriched Ni on Au | 281 | |
| EC (39.0) | 64Zn(p,α)61Cu | 17.6–11.7 | Natural or enriched Zn | 13 | ||||||
| 64 | 12.7 h | β+ (17.6) | 278 | 653 (17.6) | 511 (35.2), 1346 (0.5) | 64Ni(p,n)64Cu | 15–10 | Enriched Ni on Au or Rh | 5883 | |
| β− (38.5) | ||||||||||
| EC (43.9) | ||||||||||
| Cobalt (Co) | 55 | 17.5 h | β+ (76.0) | 649, 436 | 1499 (46.0), 1021 (25.6) | 511 (152.0), 477 (20.2), 931 (75.0), 1409 (16.9) | 56Fe(p,2n)55Co | 23.5–16 | Enriched Fe | 236 |
| EC (24.0) | 58Ni(p,α)55Co | 16–15 | Enriched Ni, enriched Ni on Ag or Au | |||||||
| Gallium (Ga) | 66 | 9.5 h | β+ (57.0) | 1904, 397 | 4153 (51.0), 924 (3.7) | 511 (114.0), 834 (5.9), 1039 (37.0), 2752 (22.7) | 66Zn(p,n)66Ga | 15–6 | Natural or enriched Zn on Au or Ag | 700 |
| EC (43.0) | ||||||||||
| 68 | 67.7 min | β+ (88.9) | 836, 353 | 1899 (87.7), 822 (1.2) | 511 (177.8), 1077 (3.2), 1261 (0.1), 1883 (0.1) | 68Zn(p,n)68Ga | 14–11 | Enriched Zn, enriched Zn on Al, Au or Ag | 1380 | |
| EC (11.1) | ||||||||||
| Iodine(I) | 124 | 4.2 d | β+ (22.7) | 687, 975 | 1535 (11.7), 2138 (10.7) | 511 (45.0), 603 (62.9), 723 (10.4), 1691 (11.2) | 124,125Te(p,xn)124I | 22–5 | Natural or enriched Te, natural or enriched TeO2 | 111 |
| EC (77.3) | ||||||||||
| Manganese (Mn) | 52g | 5.6 d | β+ (29.4) | 242 | 575 (29.4) | 511 (58.8), 744 (90.0), 936 (94.5), 1434 (100.0) | 52Cr(p,n)52gMn | 20–10 | Natural Cr, natural Cr on Cu or Ag | 9.6 |
| EC (70.6) | ||||||||||
| Niobium (Nb) | 90 | 14.6 h | β+ (53.0) | 662, 726 | 1500 (51.0), 1641 (2.0) | 511 (102.0), 142 (66.8), 1129 (92.7), 2319 (82.0) | 90,91Zr(p,xn)90Nb | 19–7 | Natural Zr, enriched ZrO2 on Cu | 596 |
| EC (47.0) | ||||||||||
| Scandium (Sc) | 44g | 4.0 h | β+ (94.3) | 632 | 1474 (94.3) | 511 (188.5), 1157 (99.9), 1499 (0.9) | 44Ca(p,n)44g,mSc | 18–6 | Natural or enriched Ca, enriched CaO3 | 50 |
| EC (5.7) | ||||||||||
| Technetium (Tc) | 94m | 52 min | β+ (70.2) | 1094, 639, 405 | 2439 (67.6), 1446 (0.9), 917 (0.9) | 511 (140.3), 871 (94.2) | 94Mo(p,n)94mTc | 13–6 | Natural Mo, enriched MoO3 | 111 |
| EC (29.8) | ||||||||||
| Titanium (Ti) | 45 | 184.8 min | β+ (84.8) | 439 | 1040 (84.8) | 511 (169.6), 720 (0.2), 1408 (0.1) | 45Sc(p,n)45Ti | 16–8 | Natural Sc | 422 |
| EC (15.2) | ||||||||||
| Yttrium (Y) | 86g | 14.7 h | β+ (31.9) | 535, 681, 883 | 1221 (11.9), 1545 (5.6), 1988 (3.6) | 511 (64.0), 628 (32.6), 1077 (82.5), 1153 (30.5) | 86Sr(p,n)86gY | 15.1–11 | Enriched SrCO3 or SrO | 166 |
| EC (68.2) | ||||||||||
| Zinc (Zn) | 63 | 38.5 min | β+ (92.7) | 1042, 733, 600 | 2345 (80.3), 1675 (7.0), 1382 (4.9) | 511 (185.5), 670 (8.2), 962 (6.5), 1412 (0.8) | 63Cu(p,n)63Zn | 16–6 | Natural or enriched Cu | 2470 |
| EC (7.3) | ||||||||||
| Zirconium (Zr) | 89 | 78.4 h | β+ (22.7) | 396 | 902 (22.7) | 511 (45.5), 909 (99.0), 1713 (0.7), 1745 (0.1) | 89Y(p,n)89Zr | 14–9 | Natural Y, natural Y2O3 on Cu | 58 |
| EC (77.3) | ||||||||||
List of PET radioisotopes
Halogens
Applications. Since the early days of nuclear medicine, several bromine isotopes have been considered for therapy (auger emitting 77Br), SPECT (80mBr), and for PET imaging (75Br and 76Br). With intermediary physicochemical properties between fluorine and iodine, radiobromines allow for fine-tuning molecular mass, volume, polarizability, and lipophilicity as alternatives to fluorine-18 (18F; t1/2: 109.8 min, 100% β+) or radioiodines (see next section). Of the two positron emitting bromine isotopes, 75Br (t1/2: 96.7 min, 75% β+, 25% EC), has the more favorable characteristics for PET applications with a larger yield of lower energy positrons and a lower number of other gamma-generating decay modes. While the long half-life daughter 75Se (t1/2: 119.8 d, 100.0% EC) warrants careful dose estimation, several 75Br-labeled agents have undergone preclinical and clinical studies including cardiac fatty acid metabolism,24 neuroreceptor mapping,25–28 and glucose analogues.29
The other positron-emitting isotope is 76Br (t1/2: 16.2 h, 55% β+, 45% EC). It has seen wider adoption primarily because it decays to stable 76Se1,30,31 and possesses a half-life well matched to protein pharmacokinetics as illustrated through 76Br-labeled antibodies and their fragments for preclinical tumor imaging.1 Beyond antibody radiolabeling, there are examples of 76Br-labeled neuroimaging tracers,32–34 peptides,35 reporter gene studies,36 and agents for studying sympathetic innervation.37 Despite this there are limitations to using 76Br-labelled tracers for PET imaging applications. Similar to radioiodine, it is prone to dehalogenation in vivo where it generally remains in blood circulation and slowly accumulates in the gastric mucosa.38 The high levels of blood retention negatively affects image quality, particularly in blood rich organs (i.e. the heart and lungs) hours after the tracer has been administered.1,38 Furthermore, bromide has a long biological half-life in human plasma (about 10 days) which makes elimination of labile [76Br]Br− post-imaging problematic.39 These concerns will surely be addressed as new 76Br-labelled tracers are developed, given an expanded role of this isotope with new β+ γ coincidence PET methodologies.40,41 A recent review of the chemistry and applications of radiobromines42 provides more insight into their applications.
Production. The primary nuclear reaction used to produce 75Br on solid-target medical cyclotrons is the 76Se(p,2n)75Br nuclear reaction.30,43–46 Although enriched elemental selenium has been used to produce solid targets for proton bombardment,30,44–46 its low melting point and low thermal conductivity necessitates current restrictions to maintain target integrity during bombardment. Alloyed selenides such as NiSe, Ag2Se, CuAgSe, Cu2Se and PbSe30,44,47 have been developed as suitable alternatives which permit irradiation at higher currents, significantly increasing yields. One drawback of selenium alloy targets is poor adherence to heat-conducting target backings.44 The beam energy used for the production of 75Br ranges from 24 to 21.5 MeV.43,44 For this energy range, the theoretical 75Br yields (assuming 76Se-enriched targets), ranged from 1184 to 1480 MBq (μA h)−1 (i.e. 32–40 mCi (μA h)−1),44 although production at higher energies (i.e. 28 to 22 MeV) provided yields up to three times greater.30 The major impurity produced was 76Br43–45 although trace amounts of 72As (t1/2: 26.0 h, 87.8% β+, 12.2% EC), 73As (t1/2: 80.3 d, 100.0% EC), 75Se (t1/2: 119.8 d, 100.0% EC) and 77Br (t1/2: 57.0 h, 0.7% β+, 99.3% EC) have been reported.44
There are several reactions that can be used to produce 76Br, however few can be carried out using lower-energy medical cyclotrons.46 Nevertheless, a feasible production route is the 76Se(p,n)76Br nuclear reaction, which occurs in the 16 to 8 MeV beam energy range.48 Like 75Br, the target was composed of an enriched selenium alloy, specifically Cu2Se48,49 or NiSe.47 Bombarding a Cu2Se target with beam energies ranging from 16 to 8 MeV, the yield of 76Br ranged from 65 to 70 MBq (μA h)−1 (i.e. 1.8 to 2.1 mCi (μA h)−1) at end of bombardment (EOB).48 The primary impurity produced for this production route was 77Br (2%).30,46
Product purification and target recovery. The primary method used to isolate bromine from irradiated selenium was thermochromatographic dry distillation.30,43,44,46,48 This procedure required heating the selenide targets in an enclosed, oxygen-free apparatus such as a furnace or dedicated distillation vessel. The distillation temperature depended on the type of target being processed, with 300 °C being ideal for distillation of bromine from elemental selenium targets43,44 and temperatures ranging from 1090 to 1100 °C being ideal for the selenide targets.30,48 To ensure an oxygen-free environment, an inert gas such as argon was continuously passed through the system. In addition to reducing oxygen exposure, the argon gas mobilizes vaporized bromine and selenium gases generated from the distillation out of the furnace and into a cold trap for collection. The vaporized selenium was the first to condense along the walls of the tubing at temperatures between 120 and 215 °C,44 while the bromine began to condense closer to room temperature. Any uncondensed bromine can be collected by bubbling the gas stream through water44 or a dilute NaOH solution.48 The condensed bromine can be removed by washing with pure water or absolute ethanol.30,44 Once cooled, the heated targets can be recycled and repeatedly irradiated, typically up to 20 times with <1% target material loss per run.30,48 The product separation yield for the process ranged from 65–75%.48
Summary. Table 2 below summarizes key production and purification parameters for 75Br and 76Br not hitherto mentioned in Table 1.
| Radioisotope | 75Br, 76Br |
|---|---|
| Target material | Elemental selenium, selenides (NiSe, Ag2Se, CuAgSe, Cu2Se, PbSe) |
| Product purity | — |
| Major impurities | 76Br, 72As, 73As, 75Se, 77Br |
| Separation method | Thermochromatographic dry distillation |
| (300–1100 °C in Ar; water or ethanol for precipitate dissolution; NaOH trap) | |
| Product separation yield | 65–75% |
| Target recovery yield | (100 − x)%, where ‘x’ is no. of times reused |
Applications. Apart from 99mTc, radioiodines are the most commonly used radionuclides in nuclear medicine. Complementary to the near ideal SPECT isotope 123I (t1/2: 13.2 h, 100.0% EC), the positron emitting 124I (t1/2: 4.2 d, 22.7% β+, 77.3% EC) has been used extensively in immunoPET imaging and other protein-based PET applications due to its well-understood radiochemistry and long half-life.1,50 Thus far, 124I has been used for cancer diagnosis, monitoring and treatment planning, as well as mechanistic, and pharmacokinetic studies.51–54 Beyond antibodies, 124I labelled peptides and small molecules targeting neurotransmitters systems, tumor hypoxia, prostate specific membrane antigen (PSMA), and neuroinflammation have also been investigated in humans.55–57 However, the use of 124I is not without its limitations, not the least of which is its comparatively low positron yield at a high end-point energy. This results in low spatial resolution58 which may be addressed through hybrid imaging with PET/MR instruments possessing strong magnetic fields.59 Another drawback is the large number of gammas released from electron capture, increasing patient radiation dose, and further limiting image resolution. These can be corrected for with special software1,3 and may be turned into an advantage using the aforementioned β+/γ coincidence tomography.41 To avoid high doses to the thyroid gland from the high energy positron and gammas, ideally the attachment of 124I to biomolecules is kinetically inert.60 Readers are referred to recent reviews on the radiochemistry and application of radioiodines for more information.23,42 Taking advantage of increased interest in theranostic radiopharmaceuticals, 124I may see a surge in upcoming years as a companion to 131I (t1/2: 8.02 d, β−: 971 keV) or used for therapy itself (70% absorbed dose of 131I.61
Production. While 124I can be produced through a number of different nuclear reaction pathways, however the most feasible production via low-energy proton cyclotron is through the 124,125Te(p,xn)124I nuclear reactions. The radioisotope can be produced through the irradiation of natural tellurium (isotopic composition: 0.09% 120Te, 2.55% 122Te, 0.89% 123Te, 4.74% 124Te, 7.07% 125Te, 18.84% 126Te, 31.74% 128Te, 34.08% 130Te) metalloid targets, >98% enriched 124,125Te metalloid targets, and enriched tellurium oxides electro-deposited or melted onto platinum or tantalum substrates. The optimum beam energy range used for irradiation can range from 14 to 5 MeV for 124Te-based targets, and 22 to 4 MeV for 125Te-based targets. The primary impurities produced during bombardment using 124Te-based targets were 123I (t1/2: 13.2 h, 100.0% EC), 125I (t1/2: 59.4 d, 100.0% EC), 126I (t1/2: 12.9 d, 1.0% β+, 47.3% β−, 51.7% EC) and 130I (t1/2: 12.4 h, 100.0% β−), while the primary impurities for 125Te-based targets are 123I and 125I.9,46,54,62,63 For the 124Te(p,n)124I nuclear reaction within the 14 to 7 MeV energy range, reported yields ranged from 5 to 21 MBq (μA h)−1 (i.e. 0.1 to 0.6 mCi (μA h)−1) with major impurities equaling less than 1%. For the 125Te(p,2n)124I nuclear reaction within the 22 to 4 MeV energy range, reported yields ranged from 43 to 111 MBq (μA h)−1 (i.e. 1.2 to 3.0 mCi (μA h)−1) with impurities comprising as much as 13% of the final product.54,62–64
Product purification and target recovery. The preferred method for separating the radioiodine from the post-bombarded tellurium target material is via dry (thermochromatographic) distillation,46,54,63 which is facilitated by the favorable melting and solidification properties of TeO2. Conventional dry distillation was carried out using a ventilated quartz tube where the irradiated TeO2 was heated to temperatures between 670 to 820 °C for periods ranging from 5 to 20 min in carrier gases of either air, argon, helium, or oxygen. This approach aimed to extract as much of the iodine product using a NaOH trap, however, any prematurely condensed iodine can be removed via dissolution using a weak buffer solution. To minimize tellurium evaporation, Al2O3/quartz wool was integrated into the distillation setup.63 The separation efficiency for the dry distillation stage ranged from 80% to 95%.54 Wet chemistry methods were also used to chemically isolate iodine from the tellurium targets, but these methods were less preferred due to product dilution in the final solution. The process was carried out in two stages, with the first being a target dissolution stage using an oxidizing alkali, and the second a reduction stage facilitated by the addition of aluminum powder.9
After the iodine has been extracted, the spent tellurium targets can be recycled. Metallic tellurium targets can be dissolved in a mixture of hydrogen peroxide and hydrochloric acid, and the resulting solution can subsequently be reduced using either hydrogen bromide or a mixture of hydrazine and sodium sulfite. Reduction via the former recovered tellurium in the form of tellurite, while the latter facilitated the recovery of metallic tellurium.9 For tellurium oxide targets, a multistage process involving vacuum distillation, acid dissolution and chemical precipitation was used to recover metallic tellurium.9 In general, the recovery yields for tellurium-based targets range from 60 to 90%.54 However, despite the innovations in tellurium recovery, 124I production is still negatively affected by high target material costs.62
Summary. Table 3 summarizes key production and purification parameters for 124I not previously mentioned in Table 1.
| Radioisotope | 124I |
|---|---|
| Target material | Natural and enriched tellurium, enriched tellurium oxide |
| Product purity | 87–>99% |
| Major impurities | 123I, 125I, 126I, 130I |
| Primary separation method | Thermochromatographic dry distillation |
| (670–820 °C in air, Ar, He or O2; weak buffer for precipitated dissolution; NaOH trap) | |
| Product separation yield | 80–95% |
| Target recovery yield | 60–90% |
Transition metals
Applications. Copper has many positron-producing radionuclides (i.e. 60Cu (t1/2: 23.7 min, 93.0% β+, 7.0% EC), 61Cu (t1/2: 3.3 h, 61.0% β+, 39.0% EC) and 64Cu (t1/2: 12.7 h, 17.6% β+, 38.5% β-, 43.9% EC)) that are finding use in PET applications primarily due to their well-established coordination chemistry.10 Although less frequently utilized, 60Cu and 61Cu have been used to label various bioactive molecules for hypoxia and blood flow studies.17,21,65
The most commonly used copper radioisotope is 64Cu, with the first-in-human use as [64Cu]ASTM for tumor hypoxia.66 There are several characteristics that make 64Cu well suited for PET imaging. Besides its well elucidated chemistry, its favorable half-life allows it to be used for radiolabeling various targeting vectors like peptides, antibodies, and nanoparticles.1,10 Additionally, the positron energy for 64Cu (mean: 278 keV, max: 653 keV) is comparable to 18F (mean: 250 keV, max: 634 keV), leading to similar positron range and excellent spatial resolution.67 With respect to use in immunoPET, there exists some dissonance between radiolabeling conditions and antibody stability. Moreover, some 64Cu radiotracers were found to be susceptible to metabolism in the liver, which permits transchelation of the radioisotope with other copper-binding proteins such as Cu/Zn superoxide dismutase (Cu/Zn SOD) in the liver and metallothionein in the blood.1,3,68 These limitations may be overcome with improved chelator technology.69,70 Lastly, it warrants mentioning the potential theranostic pairing with 67Cu (t1/2: 2.58 d, β−: 100%, avg. Eβ−: 141 keV) which makes it ideal for radioimmunotherapy monitoring.71
Production. As 60Cu is typically produced via the 60Ni(p,n)60Cu nuclear reaction, occurring with natural nickel (isotopic composition: 68.08% 58Ni, 26.22% 60Ni, 1.14% 61Ni, 3.63% 62Ni, 0.93% 64Ni) and enriched 60Ni targets when bombarded with a beam energy of 14.7 MeV.65 For the 14.7 MeV bombardment of >99% enriched 60Ni electroplated on a gold target backing, the yield at EOB was 2146 MBq (μA h)−1 (i.e. 58 mCi (μA h)−1) with the primary impurities produced to include 61Cu (t1/2: 3.3 h, 61.0% β+, 39.0% EC) (0.05%) and 57Co (t1/2: 271.7 d, 100.0% EC) (0.025%).65 When bombardment was carried out using natural nickel targets for similar conditions in the same study, EOB yields of 291 MBq (μA h)−1 (i.e. 7.9 mCi (μA h)−1) and 385 MBq (μA h)−1 (i.e. 10.4 mCi (μA h)−1) were reported along with the presence of 55Co (t1/2: 17.5 h, 76.0% β+, 24.0% EC) (0.65%) and 61Cu (0.60%) as the major impurities.65
Both the 61Ni(p,n)61Cu65,72 and 64Zn(p,α)61Cu17,72 nuclear reactions have been reported for the production of 61Cu. Production via 61Ni(p,n)61Cu required proton optimum beam energies ranging from 14.7 to 9 MeV and enriched 61Ni targets. For the 14.7 MeV bombardment of >99% enriched 61Ni electroplated onto a gold target substrate with a thickness of 118 μm and diameter of 5 mm, the 61Cu yield at EOB was 281 MBq (μA h)−1 (i.e. 7.6 mCi (μA h)−1) while the primary impurity produced was 58Co (t1/2: 70.9 d, 14.9% β+, 85.1% EC) (0.04%).65 In the same study, 60Ni(d,n)61Cu reactions were investigated but produced lower yields at higher per bombardment costs. Alternatively, 61Cu can be produced using the 64Zn(p,α)61Cu nuclear reaction. The reaction has been investigated with natural zinc (isotopic composition: 49.17% 64Zn, 27.73% 66Zn, 4.04% 67Zn, 18.45% 68Zn, 0.61% 70Zn) foils and enriched 64Zn electroplated on to gold, silver or aluminum substrates with beam energies ranging from 17.6 to 11.7 MeV (maximum production occurs circa 14.5 MeV).17 When >99% enriched 64Zn was bombarded with 14.5 MeV protons, the yield of 61Cu was 13 MBq (μA h)−1 (i.e. 0.4 mCi (μA h)−1) with a purity exceeding 95%.17 Despite having completed similar experiments using natural zinc foil targets in the same study, the results were too variable to draw valid conclusions. The primary impurities produced during the bombardment of zinc targets were 66Ga (t1/2: 9.5 h, 57.0% β+, 43.0% EC), 67Ga (t1/2: 3.3 d, 100.0% EC) and 68Ga (t1/2: 67.7 min, 88.9% β+, 11.1% EC).17 Based on the data generated, 61Cu production from the bombardment of nickel was found to be more efficient than production using zinc. However, the former production route can be expensive due to the high cost associated with enriched 61Ni. Since enriched 64Zn is significantly cheaper than enriched 61Ni, the production of 61Cu from zinc targets has found some consideration as an alternative route.17
64Cu is primarily produced via the 64Ni(p,n)64Cu nuclear reaction.9,62 The solid target used for the production is typically enriched 64Ni that has been electroplated onto a gold62,73–77 or rhodium10 target backing substrate. Although the threshold for the reaction is 2.5 MeV, production of 64Cu has been reported for beam energies ranging from 15 to 10 MeV since this range offers the highest yields.62 Numerous yields for the production of 64Cu have been reported in the literature. McCarthy et al.76 reported yields ranging from 85 to 185 MBq (μA h)−1 (i.e. 2.3 to 5 mCi (μA h)−1) when gold substrates electroplated with >95% 64Ni (thicknesses ranging from 132 to 311 μm) were bombarded with a 15.5 MeV proton beam, while McCarthy et al.65 reported yields of 296 MBq (μA h)−1 (i.e. 8 mCi (μA h)−1) for greater purity targets of similar design (i.e. >99% 64Ni electroplated onto a gold substrate with a thickness of 215 μm and diameter of 5–6 mm) irradiated at 14.7 MeV. Obata et al.77 reported yields ranging from 22 to >111 MBq (μA h)−1 (i.e. 0.6 to >3 mCi (μA h)−1) when they bombarded gold substrates that were electroplated with 94.8% 64Ni with a 12 MeV proton beam. More recently, a saturation yield of 5883 MBq (μA h)−1 (i.e. 159 mCi (μA h)−1) has been reported by Avila-Rodriguez et al. (2007) when they bombarded gold substrates electroplated with >95% enriched 64Ni with an 11.4 MeV proton beam. The primary impurity produced for this reaction is 55Co (t1/2: 17.5 h, 76.0% β+, 24.0% EC) (0.012%).65 However, other relevant impurities include 57Ni (t1/2: 35.6 h, 43.6% β+, 56.4% EC), 56Co (t1/2: 77.2 d, 19.7% β+, 80.3% EC), 57Co (t1/2: 271.7 d, 100.0% EC) and 58Co (t1/2: 70.9 d, 14.9% β+, 85.1% EC).73
Product purification and target recovery. Isolation of the copper product from the target material begins with target dissolution in strong acid (i.e. 6 M HNO3 or 6 N HCl at temperatures ranging from 90 to 100 °C (Jeffery et al. 2012). The acidic solution is passed through an anion exchange column filled with AG1-X8 resin which retains the copper product along with the nickel target material and any cobalt impurities present. The nickel target material is eluted first using 6 N HCl75,76 for HCl-dissolved targets, or 0.2 M HCl in 96% methanol74 for HNO3-dissolved targets. The cobalt impurity fraction retained by the column can be eluted with 4 M HCl75 for HCl-dissolved targets, or 0.3 M HCl in 72% ethanol74 for HNO3-dissolved targets. Finally, the copper product can be eluted using dilute acid (i.e. 0.1–0.5 M) or distilled water75,76 for HCl-dissolved targets, or 0.3 M HCl in 40% ethanol74 for HNO3-dissolved targets. Separation yields exceeding 95% have been reported for the process.75
Implementation of a dedicated recycling scheme for 60Ni recovery is not typical due to its low material cost,65 which stems from the large natural abundance of the isotope. However, recycling of enriched 64Ni is necessary to keep production costs down. Several methods have been developed for 64Ni recovery. The method described by Obata et al.77 made use of two heating stages conducted in series. In the first heating stage, the solution containing the eluted nickel was evaporated to dryness at 150 °C before it was rehydrated with distilled water. In the second heating stage, the rehydrated solution was heated at 900 °C for 24 h. This resulted in the formation of 64NiO which was then used for the preparation of future targets. The minimum recycling efficiency for this process was 94%. A second method was discussed by McCarthy et al.,76 and required several evaporation and acid dissolution steps be carried out in series. The first two acid dissolution steps were performed with HNO3 and the final acid dissolution step was performed with H2SO4. After H2SO4 dissolution, the solution was diluted and the pH was adjusted to 9 with the addition of concentrated NH4OH. The solution was available for electroplating after the addition of ammonium sulfate and a final dilution with deionized water. The overall efficiency of this recovery process exceeded 90%.
For the case of 61Cu production from zinc targets, Rowshanfarzad et al.72 and Asad et al.17 outlined a method which made use of multiple column separation stages in series. The zinc portion of the targets were dissolved in 10 M HCl at room temperature before passing through a cation exchange column filled with AG 50W-X8 resin (200–400 mesh). The column retained the copper product, the gallium impurities, and the unreacted zinc target material. The copper product and zinc target materials were eluted with 10 M HCl and the eluant was passed through an AG 1-X8 anion exchange column (200–400 mesh). Both the copper product and zinc target material were retained, and the copper product was selectively eluted with 2 M HCl. In order to recycle the zinc target material (as in the case of enriched targets), the column can be eluted with 0.5 M HCl.17 This solution was then evaporated to dryness before 6 M H2SO4 was added to digest any resin residues from the prior chromatography stages. The evaporation/digestion stage was repeated before the recovered zinc was finally reconstituted as an electroplating solution. The overall yield for the 64Zn recovery process was about 90%.17
Summary. Table 4 summarizes key production and purification parameters for 60Cu, 61Cu and 64Cu not mentioned in Table 1.
| Radioisotope | 60Cu, 61Cu, 64Cu | |
|---|---|---|
| Target material | Natural and enriched nickel (all), natural and enriched zinc (for 61Cu only) | |
| Product purity | 60Cu | ∼98.8 to ∼99.9% |
| Major impurities | 61Cu, 55Co, 57Co | |
| Product purity | 61Cu | ∼99.6% for 61Ni(p,n)61Cu nuclear reaction; >95% for 64Zn(p,α)61Cu nuclear reaction |
| Major impurities | 58Co for 61Ni(p,n)61Cu nuclear reaction; 66Ga, 67Ga, 68Ga for 64Zn(p,α)61Cu nuclear reaction | |
| Product purity | 64Cu | ∼95 to >99% |
| Major impurities | 55Co, 56Co, 57Co, 58Co, 57Ni | |
| Separation method | Ni | Anion exchange chromatography (AG 1-X8 resin) |
| - Target dissolution: (a) HNO3 or (b) strong HCl | ||
| - Ni target elution: (a) 0.2 N HCl in 96% methanol or (b) 6N HCl | ||
| - Co impurity elution: (a) 0.3% HCl in 72% ethanol or (b) 4 N HCl | ||
| - Copper elution: (a) 0.3 N HCl in 40% ethanol or (b) water | ||
| Product separation yield | 95% | |
| Target recovery yield | 90% | |
| Separation method | Zn | Ion exchange chromatography in series (AG 50W-X8 resin (cation) and AG 1-X8 resin (anion)) |
| - Target dissolution: 10 N HCl | ||
| - (Cation exchange) Cu and Zn elution: 10 N HCl | ||
| - (Anion exchange) Cu elution: 2 N HCl | ||
| - (Anion exchange) Zn target elution: 0.5 N HCl | ||
| Product separation yield | — | |
| Target recovery yield | 90% | |
Applications. Cobalt is a human micronutrient and component of the essential vitamin B12 (cobalamin). As a potential competitor radionuclide to 64Cu, 55Co (t1/2: 17.5 h, 76.0% β+, 24.0% EC) has been shown to mimic calcium intake in infarcted brain tissue. This property makes it useful in the imaging of recent cerebral brain damage resulting from cerebral tumors, stroke and other brain injuries.78–81 Recent reports have demonstrated high resolution PET images obtained with peptide and affibody chelates of 55Co.82,83 Ionic 55Co species are residualizing radiotracers. Significant amounts of 55Co can accumulate in organs such as the bladder, liver and kidney84 with the majority eliminated or decayed three days post-injection. 55Co has also shown great promise in the field of immunoPET imaging and has been used to label an anti-EGFR affibody82 as well as several peptides.83,85 Unfortunately, widespread clinical use of this isotope may be hampered as 55Co decays into 55Fe (t1/2: 2.73 years) while releasing large amounts of high energy gamma rays from electron capture in the process.1 Nonetheless, it remains a valuable tool for preclinical PET investigations.
Production. There are two major nuclear reactions available to produce 55Co on small medical cyclotrons by utilizing either the 56Fe(p,2n)55Co81,86,87 or the 58Ni(p,α)55Co.81,88,89 Production via 56Fe(p,2n)55Co involved irradiating 56Fe-enriched iron foils87 with a minimum proton beam energy of about 16 MeV,86,87 although the reaction peaks at an energy near 23.5 MeV.87 However, this production route was undesirable since significant amounts of the chemically-inseparable 56Co (t1/2: 77.2 d, 19.7% β+, 80.3% EC) impurity was also co-produced.81 The 58Ni(p,α)55Co production route can be carried out through the bombardment of natural nickel (isotopic composition: 68.08% 58Ni, 26.22% 60Ni, 1.14% 61Ni, 3.63% 62Ni, 0.93% 64Ni) foils89 or enriched 58Ni-electroplated onto silver88 or gold81 disk substrates. Irradiations have been carried out using proton beams with energies of about 16 MeV (ref. 88 and 90) to 15 MeV.81 The primary impurities produced during irradiation were 57Co (t1/2: 271.7 d, 100.0% EC), 58Co (t1/2: 70.9 d, 14.9% β+, 85.1% EC) and 57Ni (t1/2: 35.6 h, 43.6% β+, 56.4% EC), particularly when natural nickel targets were used for the production.89 The saturation yield of 55Co when natural nickel foils were bombarded with a beam energy of 16 MeV was 236 MBq (μA h)−1 (i.e. 6.3 mCi (μA h)−1) at EOB with purity exceeding 97%.89 55Co yields from 16 MeV proton beam bombardments of >99% 58Ni-electroplated gold disk substrates resulted in 9.3 MBq (μA h)−1 (i.e. 0.3 mCi (μA h)−1) with a purity exceeding 92%.88
Product purification and target recovery. Column-based separation techniques have been used to isolate cobalt from the nickel targets after heated dissolution with concentrated HCl.81,88,89 The first example used an AG1-X8 anion exchange resin81,89 which retained the cobalt product when the acidic target solution was passed through. Dilute HCl with concentrations ranging from 0.4–0.5 M (ref. 81 and 89) could be used to elute the cobalt product from the resin. Another stationary phase, DGA branched resin,88 was demonstrated to selectively release 55Co upon elution with 3 M HCl. The overall separation efficiency for the process generally exceeded 90%.88,89 If the method used for target preparation was the electroplating of enriched 58Ni on a metal disk substrate, 58Ni was recovered with a 94% efficiency from the spent target solution using a series of drying and chemical cleaning stages before finally being reconstituted as an electroplating solution.88
Summary. Table 5 summarizes key production and purification parameters for 55Co not mentioned in Table 1.
| Radioisotope | 55Co |
|---|---|
| Target material | Natural and enriched nickel |
| Product purity | 92 to >97% |
| Major impurities | 57Co, 58Co, 57Ni |
| Separation method | Anion exchange chromatography (AG1-X8 resin) |
| - Target dissolution: concentrated HCl | |
| - Co elution: 0.4–0.5 N HCl | |
| Product separation yield | >90% |
| Target recovery yield | 94% |
Applications. A considerable amount of interest in the use of 52gMn (t1/2: 5.6 d, 29.4% β+, 70.6% EC) in PET applications has developed since its first use as a myocardial perfusion PET agent in 1985.91 The radionuclide has been used in several preclinical imaging studies, including murine 4T1 xenografts, functional β-cells in type 1 and type 2 diabetes, and neural imaging in primates.22,92 The interest in 52gMn stems from its decay characteristics, ease of production, and its potential to act as a surrogate to non-invasively quantify MR contrast agent.93 The 52gMn radioisotope has a long half-life which facilitates late time-point studies up to 2 to 3 weeks following tracer administration, making it suitable for ImmunoPET applications.1,94 Additionally, positron decay occurs at a low positron energy (β+ Emax: 575 keV), promoting high resolution imaging that is comparable to 18F.93 Despite its numerous advantages, caution is exercised when using 52gMn for PET applications since it has several high energy gamma emissions (i.e. 744, 935, and 1434 keV) coupled with prolonged residence in critical organs which require careful dosimetry studies prior to translation.
Production. 52gMn is primarily produced using the 52Cr(p,n)52gMn nuclear reaction.92,94–96 The reaction takes place within the 20 to 10 MeV beam energy range.93 The main target material used is natural chromium (isotopic composition: 4.35% 50Cr, 83.79% 52Cr, 9.50% 53Cr, 2.37% 54Cr) which can be loaded on to metal target substrates such as copper95 and silver94 via electroplating or pressing.92,94,95 The natural chromium targets may also be a metal disk92,93 or foil.96 The primary impurity produced during the reaction was 54Mn (t1/2: 312.2 d, 100.0% EC) via the 54Cr(p,n)54Mn nuclear reaction,92,94 however this impurity can be greatly reduced if 52Cr-enriched targets are used. 52mMn (t1/2: 21.1 min, 96.6% β+, 1.6% EC, 1.8% IT) was also produced97 from the irradiation, however the radionuclide decays into 52gMn relatively quickly after its production. When chromium metal disks were bombarded with 16 MeV proton beams, a yield of 9.6 MBq (μA h)−1 (i.e. 0.26 mCi (μA h)−1) was reported at a purity level of 99.55%.93 For the composite, chromium/silver pressed targets bombarded at the same beam energy, a yield of 5 MBq (μA h)−1 (i.e. 0.1 mCi (μA h)−1) at a purity level of about 95% was reported.94 When natural chromium foils were irradiated with 12.5 MeV proton beams, a yield of 4.5 MBq (μA h)−1 (i.e. 0.12 mCi (μA h)−1) and a purity exceeding 99.0% was reported by Topping et al. (2013).
Product purification. The major method used to isolate manganese from the chromium targets is ion exchange chromatography. There are several different resins that can be used to carry out the isolation procedure. The most frequently used resin was the AG 1-X8 which required dissolution of the bombarded target in 11 to 12 M HCl.93,96–98 After dissolution, the target solution was passed through the resin which selectively retained the manganese product, and elution was carried out using 0.1 M HCl. The separation efficiency of the process exceeded 97%.97 Apart from the AG 1-X8 resin, the 50W-X8 resin from Dowex or AG was also used to perform the separation, as discussed by Chaple and Lapi92 and Wooten et al.95 Methods for recovering the chromium target material have not been discussed, likely because recovery schemes tend not to be feasible when the large natural abundance of 52Cr is considered.
Summary. Table 6 summarizes key production and purification parameters for 52gMn not mentioned in Table 1.
| Radioisotope | 52gMn |
|---|---|
| Target material | Natural chromium |
| Product purity | 95 to 99.55% |
| Major impurities | 54Mn, 52mMn |
| Separation method | Ion exchange chromatography (AG1-X8 resin or other resins) |
| - Target dissolution: 11 N HCl | |
| - Mn elution: 0.1 N HCl | |
| Product separation yield | 97% |
| Target recovery yield | n/a |
Applications. A relative newcomer PET radionuclide, 90Nb (t1/2: 14.6 h, 53.0% β+, 47.0% EC) is finding increasing consideration as a labelling agent in immunoPET applications due to close agreement between its half-life and the pharmacokinetics of peptides, antibody fragments and antibodies.99,100 Additionally, 90Nb has a high probability of positron decay at moderate emission energies (β+ Emax: 1.5 MeV), which facilitates high-resolution PET imaging.101 Protein labelling studies have been carried out as a preliminary step, including 90Nb-labelled monoclonal antibodies rituximab and bevacizumab in high yields.102–104 90Nb has also been used to label the peptide derivative DFO-succinyl-(D)Phe1-octreotide.105 Although easily produced using low-energy cyclotrons, efficient separation methods ensuring quick isolation of 90Nb from bombarded target materials have not yet been established.99,105
Production. 90Nb is primarily produced via the 90Zr(p,n)90Nb nuclear reaction within the 19 to 7 MeV beam energy range.99,105 However, 90Nb could also be produced via the 91Zr(p,2n)90Nb nuclear reaction when the incident beam energy exceeded 15 MeV.105 Maximum 90Nb production occurred at a beam energy of 13.5 MeV.105 The target material for 90Nb production was typically natural zirconium foils (isotopic composition: 51.45% 90Zr, 11.22% 91Zr, 17.15% 92Zr, 17.38% 94Zr, 2.80% 96Zr),99,101–103,105 although the use of this target produced a significant number of impurities, making it unsuitable for clinical trial applications.105 The primary impurities produced during the bombardment of natural zirconium were 89g/mNb (t1/2: 2.0 h, 75.0% β+, 25.0% EC/66.0 min, 81.0% β+, 19.0% EC), 91mNb (t1/2: 60.9 d, 96.6% IT, 3.4% EC), 92mNb (t1/2: 10.2 d, 0.1% β+, 99.9% EC), 95g/mNb (t1/2: 35.0 d, 100.0% β-/t1/2: 3.6 d, 5.6% β-, 94.4% IT), 96Nb (t1/2: 23.4 h, 100.0% β-), 87Y (t1/2: 79.8 h, 0.2% β+, 99.8% EC) and 89Zr (t1/2: 78.4 h, 22.7.0% β+, 77.3% EC).102,105 An alternative production route using ZrO2 from enriched 90Zr has been proposed by ref. 105, in which case the target preparation involved sedimentation of enriched ZrO2 powder onto a copper substrate. Production using the 90ZrO2 targets had up to two orders of magnitude lower impurity levels. When three, 0.25 mm thick natural zirconium foils were irradiated with a 17.5 MeV proton beam, a 90Nb yield of 145 MBq (μA h)−1 (i.e. 3.9 mCi (μA h)−1) was reported with a purity of 97% at EOB.102 A yield of 596 MBq (μA h)−1 (i.e. 16.1 mCi (μA h)−1) with purity exceeding 95% was reported following the bombardment of three 90ZrO2-on-copper foil targets with an incident proton beam energy of 11.9 MeV, 4 hours after EOB.105
Product purification. There are two established methods that have been used to isolate the niobium product from post-bombarded zirconium targets. The first method was described by Radchenko et al.100,103 and required serial ion exchange chromatography separation. Irradiated zirconium targets were first dissolved in 28 M hydrofluoric acid and diluted to 21 M before loading on to the first ion exchange column. Cation exchange was performed by passing the solution through a DOWEX 50 × 8 resin (200–400 mesh) to retain trace metal ion impurities (i.e. copper and iron), colloids and undissolved particulate, while allowing the dissolved target material and product to pass through. After washing with concentrated HF, the emerging solution from the cation exchange stage was transferred to the second ion exchange stage. Anion exchange was performed using an AG 1 × 8 resin (200–400 mesh) resin to retain the niobium product. To elute the niobium, a mixture of 1% H2O2 in 6 M HCl101 or 0.1 M oxalic acid103 was used. If the former was used for elution, the H2O2 should be removed by heating the eluate for 5 min at 120 °C. The overall efficiency of the extraction procedure ranged from 93 to 95%.
A second method was described by Radchenko et al.,101,102 which made use of a liquid–liquid extraction procedure. After placing into a small ice-water bath, the bombarded zirconium targets were dissolved in 48% HF (28 M). After complete target dissolution, 10 M HCl and saturated boric acid were added. More than 99% of the niobium product was then partitioned into chloroform using 0.02 M N-benzoyl-N-phenylhydroxylamine as a phase transfer catalyst. Liquid–liquid extraction was then performed with vigorous shaking for 20 min. After washing the organic phase with a mixture of 9 M HCl and 0.001 M HF and then 9 M HCl, respectively, the niobium product was re-extracted from the organic phase using aqua regia with an extraction rate ranging from 90–95%. To increase the purity of the obtained 90Nb product, the aqueous layer was subsequently passed through an anion exchange resin. The niobium containing solution was evaporated to dryness and the resulting residue was dissolved in a mixture of 0.25 M HCl and 0.1 M oxalic acid. Anion exchange was performed on the residue using a small Aminex A27 column, and the niobium product was eluted using a mixture of 6 M HCl and 0.01 M oxalic acid. The overall efficiency of the extraction procedure ranged from 76 to 81%.
In general, methods for recovering the zirconium target material have not been discussed, likely due to the lack of feasibility of any proposed recovery scheme given the relatively large natural abundance of 90Zr.
Summary. Table 7 summarizes key production and purification parameters for 90Nb not mentioned in Table 1.
| Radioisotope | 90Nb | |
|---|---|---|
| Target material | Natural and enriched zirconium oxide | |
| Product purity | >95 to 97% | |
| Major impurities | 89g/mNb, 91mNb, 92mNb, 95g/mNb, 96Nb, 87Y, 89Zr | |
| Separation method | 1 | Serial ion exchange chromatography (DOWEX 50 × 8 resin (cation), AG 1 × 8 resin (anion)) |
| - Target dissolution: 28 M HF | ||
| - (Cation exchange) 1st Nb elution: concentrated HF | ||
| - (Anion exchange) 2nd Nb elution: 1% H2O2 in 6 M HCl | ||
| Product separation yield | 93 to 95% | |
| Separation method | 2 | Liquid–liquid extraction |
| - Target dissolution: 48% HF | ||
| - Extraction solvents: 10 M HCl + saturated boric acid, 0.02 M N-benzoyl-N-phenylhydroxylamine in CHCl3, aqua regia | ||
| Product separation yield | 76 to 81% | |
| Target recovery yield | n/a | |
Applications. Favorable decay characteristics and the potential availability of a 44Ti/44Sc generator has brought 44gSc (t1/2: 4.0 h, 94.3% β+, 5.7% EC) into clinical focus over the past decade. Scandium-44 is a trivalent radionuclide that is readily conjugated to peptides and proteins using common macrocyclic chelators such as DOTA and NOTA. This facilitates the growing number of preclinical and clinical PET studies reported,106–110 and was exemplified by 44gSc-labelling of PSMA-617 (i.e. peptidomimetic inhibitors) as an alternative for 68Ga-labelling.111 Due to the four-fold longer half-life, dosimetry measurements could be obtained 19 h post-injection to better match the pharmacokinetics of the 177Lu-labelled therapeutic. Additionally, 44Sc forms a true theranostic pairing with 47Sc.112
Production. There are many methods that can be used to produce 44gSc, ranging from the 44Ti/44gSc generator62,111 to small medical cyclotrons.62,110,113,114 With respect to cyclotron production via solid targetry, one of the most common production routes is through the 44Ca(p,n)44g,mSc nuclear reaction. The process involves the use of solid targets formed from natural calcium (isotopic composition: 96.94% 40Ca, 0.65% 42Ca, 0.14% 43Ca, 2.09% 44Ca, >0.01% 46Ca, 0.19% 48Ca) and enriched 44Ca metal,113 as well as enriched [44Ca]CaCO3 powders.62,114 The nuclear reaction takes place over beam energies ranging from 18 to 6 MeV, with the reaction cross-section maximums between 13 and 10 MeV.114 The yield of 44gSc produced from the bombardment of >94% enriched [44Ca]CaCO3 powders at a beam energy of 11 MeV was 50 MBq (μA h)−1 (i.e. 1.4 mCi (μA h)−1),114 while that obtained from the bombardment of natural calcium at a beam energy of 15.6 MeV was 32 MBq (μA h)−1 (i.e. 0.9 mCi (μA h)−1) with a purity exceeding 95%.110 It should be pointed out that targets with high concentrations of 44Ca perform poorly as solid targets due to their low electrical and heat conductivity properties.110 The primary impurity produced during bombardment was 44mSc (t1/2: 58.6 h, 98.8% IT, 1.2% EC) which has a negligible effect on PET studies when produced in small amounts.110 44mSc has found new use as an in vivo generator in various pharmacokinetic studies62,113 due to its longer half-life and eventual decay into 44gSc. Other impurities produced during the proton irradiation of 44Ca include 47Sc (t1/2: 3.3 d, 100.0% β-) and 48Sc (t1/2: 43.7 h, 100.0% β-), which were formed from trace amounts of 48Ca via the 48Ca(p,2n)47Sc and 48Ca(p,n)48Sc nuclear reactions, respectively.110,114 Small amounts of 43Sc (t1/2: 4.0 h, 88.1% β+, 11.9% EC)110 and 46Sc (t1/2: 83.8 d, 100.0% β-)62 have also been reported.
Product purification and target recovery. After acid dissolution, the scandium product can be isolated from irradiated calcium targets through column chromatography. However, the specific column and resin used varied with the type of target bombarded. With 44Ca metal, the target was first dissolved in concentrated HCl and then a UTEVA extraction resin was used to retain both the scandium product and the target material.110 The bulk target material was eluted with the 10 M HCl, while the scandium product was eluted subsequently with water, recovering ∼80% of the 44gSc activity at EOB. When irradiating [44Ca]CaCO3, a chelating ion exchange resin (i.e. Chelex 100) was used114 after target dissolution in 0.1 M HCl. The resin retains both the scandium product and the target material, with the latter eluted first using 0.01 M HCl. The scandium product was removed using 1 M HCl with a separation efficiency exceeding 70%. A target-material recovery stage was further described in the work published by Krajewski et al.114 Briefly, the target-containing effluent was concentrated with ammonia. The new solution was subsequently heated to 170 °C to decompose NH4Cl formed from the ammonia addition, and to evaporate any remaining water. 44Ca was recovered as 44CaO with a recovery efficiency of 60%, although the authors believed the small batch sizes processed strongly affected the recovery efficiency. Regardless, the addition of a recovery stage was worthwhile given the cost of 44Ca.62
Summary. Table 8 summarizes key production and purification parameters for 44gSc not mentioned in Table 1.
| Radioisotope | 44gSc | |
|---|---|---|
| Target material | Natural and enriched calcium, enriched calcium carbonate | |
| Product purity | >95% | |
| Major impurities | 44mSc, 47Sc, 48Sc, 43Sc, 46Sc | |
| Separation method | 1 | Ion exchange chromatography (UTEVA extraction resin) |
| - Target dissolution: concentrated HCl | ||
| - Ca elution: 10 M HCl | ||
| - Sc elution: water | ||
| Product separation yield | 80% | |
| Target recovery yield | — | |
| Separation method | 2 | Ion exchange chromatography (Chelex 100 resin) |
| - Target dissolution: 0.1 M HCl | ||
| - Sc elution: 0.01 M HCl | ||
| - Ca elution: 1 M HCl | ||
| Product separation yield | 70% | |
| Target recovery yield | 60% | |
Applications. The workhorse of nuclear medicine is the SPECT isotope 99mTc.115 As a PET surrogate, 94mTc (t1/2: 52 min, 70.2% β+, 29.8% EC) has seen limited use. Efflux transporter activity has been measured with 94mTc-sestamibi. The tracer was used in a preclinical multi-drug resistance study involving P-glycoprotein knock-out and wild-type mice, and showed delayed tracer clearance in the liver and kidney of the former group.116 Other examples include examining cardiac function and estrogen receptor expression.117,118 A major application of 94mTc remains the quantitative assessment of novel 99mTc radiopharmaceuticals through in vivo PET studies.119 The limited adoption of 94mTc in PET can be attributed to several factors, with the most limiting being the prevalence of the high-energy gamma ray that is released during its decay.120 Clinical applications using 94mTc for testing 99mTc radiopharmaceuticals are also limited by the relatively short half-life of 94mTc,120 as well as the poor dosimetry of 94mTc which is seven-fold less favorable than that of 99mTc.121
Production. 94mTc can be produced using the 94Mo(p,n)94mTc nuclear reaction on small medical cyclotrons.11,119–121 The reaction takes place within the 13 to 6 MeV energy range,11,119 and has been carried out using both natural molybdenum foils (isotopic composition: 14.53% 92Mo, 9.15% 94Mo, 15.84% 95Mo, 16.67% 96Mo, 9.60% 97Mo, 24.39% 98Mo, 9.82% 100Mo) and 94Mo-enriched molybdenum oxide (i.e. MoO3) pellets and powders.120–122 When using 94Mo-enriched targets for production, the major impurity produced was 94gTc (t1/2: 293 min, 10.5% β+, 89.5% EC),119 especially when bombardment was conducted with beam energies equaling or exceeding 13 MeV.120 When natural molybdenum was used for production, impurities such as 95g/mTc (t1/2: 61.0 d, 0.4% β+, 96.1% EC, 3.9 IT/t1/2: 20.0 h, 100.0% EC), 96g/mTc (t1/2: 4.3 d, 100.0% EC/t1/2: 51.5 min, 98.0% IT, 2.0% EC) and 99mTc (t1/2: 6.0 h, 100.0% IT) were present in addition to 94gTc.120–122 Despite the myriad of impurities formed, 94mTc production from natural molybdenum has been considered suitable for human radiopharmaceutical studies.120 However, special attention should be given to 96mTc (which does not emit any imageable gamma rays)120 and 95Tc,123 which are both produced in significant amounts and have been shown to increase patient dose. When natural molybdenum foils were bombarded with an 11 MeV proton beam, a saturation yield of 111 MBq (μA h)−1 (i.e. 3 mCi (μA h)−1) was reported.121 However, a theoretical yield of 2000 MBq (μA h)−1 (i.e. 54 mCi (μA h)−1) has been calculated for thick targets of enriched 94Mo bombarded with beam energies ranging from 13 to 7 MeV.120,122
Product purification and target recovery. The primary method used to separate the technetium product from the metallic or oxide molybdenum target material was thermochromatographic dry distillation.120,121 This requires heating of the irradiated targets in moist air,120 oxygen or helium.121 For molybdenum oxide targets heated to temperatures below 800 °C, volatile technetium species such as [94mTc]HTcO4 and [94mTc]Tc2O7 sublime and were transported by the carrier gas until condensation temperatures (i.e. 250 to 350 °C) were reached. When heating was carried out at temperatures above 800 °C (i.e. 1090 °C (ref. 120)), both the technetium product and molybdenum target material sublimed. The molybdenum condenses first in the 600 to 800 °C temperature range, while the technetium product condenses at the cooler temperatures mentioned above. The condensed technetium product can be dissolved using hot NaOH and purified using a minimized alumina column. The entire separation process required approximately 25 minutes, and had an 80 to 85% product separation efficiency. The molybdenum target material was well isolated and could be easily recovered for immediate reuse with a recovery efficiency exceeding 95%.
Summary. Table 9 summarizes key production and purification parameters for 94mTc not mentioned in Table 1.
| Radioisotope | 94mTc |
|---|---|
| Target material | Natural molybdenum, enriched molybdenum oxides |
| Product purity | — |
| Major impurities | 94Tc, 95g/mTc, 96g/mTc, 99mTc |
| Separation method | Thermochromatographic dry distillation |
| (500 to 1090 °C in moist air, O2 or He; hot NaOH for precipitate dissolution) | |
| Product separation yield | 80 to 85% |
| Target recovery yield | >95% |
Applications. Favorable decay characteristics has garnered increase consideration of 45Ti (t1/2: 3.08 h, 84.8% β+, 15.2% EC) as a PET imaging agent. These include a reasonable half-life and decaying to a stable daughter isotope, both of which are desirable features for PET radionuclides.124 Additionally, 45Ti has a high probability of positron decay at a moderate maximum positron energy (β+ Emax: 1040).92,124 Most positron decay events proceed directly to the ground state, making positron annihilation the primary source of gamma rays in the system.124,125 These decay characteristics yield images that possess a high resolution.92 In addition to its favorable decay properties, 45Ti is also available to produce as discussed below. Despite its appeal, research on 45Ti radiolabeled tracers is limited as a result attributed to titanium's complicated chemistry and its high reactivity with oxygen.92,126 Regardless, a few studies using 45Ti-radiolabelled molecules exist. 45Ti has been used to label human serum albumin, phytate, DTPA and citrate for pre-clinical studies.125 A few bioconjugate studies have been completed including protein and peptide targeting vectors. Biodistributions studies in mice using [45Ti]transferrin observed high 45Ti uptake in the spleen and liver126 indicative of transchelation. Another recent example attached 45Ti to a PSMA-targeted peptidomimetic.126 In an interesting attempt to develop dual modality probes, chelate free mesoporous silica nanoparticles were labeled with 45Ti for non-invasive in vivo quantification.127
Production. 45Ti is produced via the 45Sc(p,n)45Ti nuclear reaction.128 The reaction primarily takes place within the 16 to 8 MeV energy range, with a threshold energy of 2.9 MeV.128 Regardless, a range restricted to 15 to 8 MeV was recommended to reduce the production of isotopic and non-isotopic impurities during bombardment.124 The primary impurities produced during bombardment were 44Ti (t1/2: 59.1 years, 100.0% EC) from the 45Sc(p,2n)44Ti nuclear reaction, and 44gSc (t1/2: 4.0 h, 94.3% β+, 5.7% EC) from the 45Sc(p,pn)44gSc and 45Sc(p,d)44gSc nuclear reactions.128 The target material was natural scandium (isotopic composition: 100.00% 45Sc) foils.92,124–126,128 A few values for 45Ti yields have been reported in the literature. Vavere and Welch126 reported yields of 422 MBq (μA h)−1 (i.e. 11.4 mCi (μA h)−1) after bombarding 0.25 mm thick natural scandium foils with a 14.7 MeV proton beam. However, yields as low as 230 MBq (μA h)−1 (i.e. 6.2 mCi (μA h)−1) for bombardments at 11.8 MeV have been reported.127
Product purification. Cation-exchange chromatography has been used to isolate the titanium product from the scandium targets, and the method has been extensively described by Vavere et al.125 and Vavere and Welch.126 Post-bombarded targets were first dissolved in 6 N HCl, and the final solution was added to a cation exchange column filled with AG 50W-X8 resin (100–200 mesh). 45Ti was then eluted with 6 N HCl, and this eluate was heated in the presence of a nitrogen stream to evaporate the HCl from the final product. Greater than 92% of the eluted 45Ti product was obtained with a radionuclide purity of 99.8%. Methods for recovering the scandium target material have not been discussed as such methods would likely not be feasible due to the naturally high abundance of 45Sc.
Summary. Table 10 summarizes key production and purification parameters for 45Ti not mentioned in Table 1.
| Radioisotope | 45Ti |
|---|---|
| Target material | Natural scandium |
| Product purity | >99% |
| Major impurities | 44Ti, 44gSc |
| Separation method | Cation exchange chromatography (AG 50W-X8 resin) |
| - Target dissolution: 6 M HCl | |
| - Ti elution: 6 M HCl | |
| Product separation yield | >92% |
| Target recovery yield | n/a |
Applications. As part of the original theranostic pairing,129 86gY (t1/2: 14.7 h, 31.9% β+, 68.2% EC) is a residualizing radioisotope that has promising applications in radioimmunotherapy as an imaging compliment to its chemically-identical therapeutic counterpart, 90Y. Owing to its well-elucidated chelation chemistry, 86gY has been used to label several different antibodies, although the produced radiotracers have been limited to use in preclinical studies1 due to a few limitations. 86gY decays primarily by electron capture, generating several gamma emissions in the process. This negatively affects imaging quality and necessitates the use of corrections.130 A second limitation is the accumulation of unbounded 86gY (due to conjugate dissociation or transchelation) in bone marrow, resulting in acute bone marrow toxicity. However, it should be mentioned that the risk of transchelation for ligands such as DOTA is low given the high thermodynamic and kinetic inertness of Y-DOTA chelates.131 Various peptides132,133 and antibodies134–137 have been radiolabeled with the intention of monitoring radioimmunotherapy. With respect to its utility as a theranostic pair, the half-life of 86gY is short compared to 90Y and may limit the flexibility of radioimmunotherapy applications.1,138 Beyond immunoPET, 86Y has potential for use as a surrogate for gadolinium for non-invasive quantification of MR contrast agents.139
Production. 86gY is primarily produced via the 86Sr(p,n)86gY nuclear reaction, which also produces significant amounts of 86mY (t1/2: 47.4 min, 99.3% IT, 0.4% β+, 0.3% EC), a shorter half-life impurity which quickly decays into 86gY.62 86gY has been produced from the bombardment of solid targets made from pressed, enriched 86SrCO3 (ref. 140–142) or 86SrO141 powders using proton beams of energies ranging from 15.1 to 11 MeV.140,142 Naturally, the yield of 86gY varied with the solid target material used as well as the specific irradiation conditions employed. When bombardments were carried out using 95.6% enriched 86SrCO3 solid target using a 15.1 MeV proton beam, an average 86gY yield of 48 MBq (μA h)−1 (i.e. 1.3 mCi (μA h)−1) with a purity exceeding 99% was obtained.62,140 Alternatively, bombardments carried out on 96.4% enriched 86SrO targets using a 14.5 MeV proton beam produced much higher 86gY yields of 166 MBq (μA h)−1 (i.e. 4.5 mCi (μA h)−1), with 86mY (220%), 87g/mY (t1/2: 79.8 h, 0.2% β+, 99.8% EC/t1/2: 13.3 h, 98.4% IT, 0.8 β+, 0.8% EC) (0.27%/0.43%) and 88Y (t1/2: 106.6 d, 0.2% β+, 99.8% EC) (0.024%) being the primary impurities.62,141
Product purification and target recovery. Multistage-electrolysis and filtration were two primary methods used to extract yttrium from post-bombardment strontium solid targets. The multistage-electrolysis procedure has been reported by Reischl et al.140 and Yoo et al.,141 and first required dissolution of the target material in 2.8 M HNO3. To displace any oxygen and carbon dioxide dissolved in solution, the solution is sparged with an inert gas (i.e. argon) for 10 to 15 min before electrolysis began and continued through two electrolysis stages. The first electrolysis stage was carried out using a platinum plate cathode and anode. Yttrium was deposited on the cathode, followed by transfer to a platinum wire during a second electrolysis stage, where it was later removed and used for downstream labelling procedures. A simpler filtration method required dissolution of the targets in 6 M HCl, followed by the subsequent addition of 1 M NH4OH.142 The resulting solution was then filtered under vacuum and washed with water to remove any residual strontium-containing liquid absorbed into the filter paper. Although the filtration-based separation technique was considerably faster than the multistage-electrolysis technique, filtration-based separation suffered from a lower efficiency which affected overall yttrium yields. Multistage-electrolysis boasts separation efficiencies of about 97%, while the separation yield for filtration was about 88%.62 The strontium remaining in solution after electrolysis and filtration was available to be recovered for reuse. Depending on the chemical form of the strontium, a solution of (NH4)2CO3 (ref. 140 and 142) or NH4OH followed by the addition of saturated ammonium carbonate solution141 was used to precipitate the strontium in the form of 86SrCO3 with a recovery efficiency exceeding 90%. The 86SrCO3 can be converted to 86SrO through thermal decomposition if desired.141
Summary. Table 11 summarizes key production and purification parameters for 86gY not mentioned in Table 1.
| Radioisotope | 86gY | |
|---|---|---|
| Target material | Strontium oxides | |
| Product purity | >99% | |
| Major impurities | 86mY, 87g/mY, 88Y | |
| Separation method | 1 | Multistage electrolysis |
| - Target dissolution: 2.8 M or 4% HNO3 | ||
| - Platinum plate cathode and anode | ||
| - Inert gas environment | ||
| Product separation yield | ∼97% | |
| Separation method | 2 | Filtration |
| - Target dissolution: 6 M HCl | ||
| - Y precipitator: 1 M NH4OH | ||
| Product separation yield | ∼88% | |
| Target recovery yield | >90% | |
Applications. Zinc is the second most abundant transition metal in the human body being essential to numerous somatic processes, ranging from enzymatic stability to DNA regulation.143–145 Consequently, zinc dysregulation has been associated with many diseases, such as diabetes, Alzheimer's disease, and various cancers including breast, pancreatic and prostate cancer.143,145,146 The critical role of zinc homeostasis in addition to its association to numerous diseases makes the development of non-invasive techniques for monitoring fluctuations in somatic zinc levels of great interest.143,144 Of the many zinc radioisotopes, only three are positron-emitters, with 63Zn (t1/2: 38.5 min, 92.7% β+, 7.3% EC) showing the greatest promise due to its high probability for positron decay and ease of production.143 For conjugation, zinc only has one oxidation state which makes designing inert chelates much simpler. Despite its attractive properties, 63Zn is limited by its comparatively short half-life, which restricts its viable application to about 2 hours.143 Regardless, 63Zn radiotracers are thought to be suitable for a variety of short investigative studies such as blood/tissue zinc transport, pancreas and prostate transport kinetics, and blood–brain barrier investigations.143 A few clinical and preclinical PET studies have used a [63Zn]zinc citrate radiotracer to monitor zinc transport.144 One preclinical study found that the radiotracer mainly accumulated in the gastrointestinal tract of mice, although a less significant accumulation in the brain of the animals was also reported. A clinical study comparing [63Zn]citrate uptake in brains of Alzheimer's disease patients and age-matched controls found no observed difference in uptake although the clearance of radioactivity was significantly slower in the patient group.147
Production. Although several nuclear reactions can be used to produce 63Zn, the only reaction suitable for production using small medical cyclotrons is the 63Cu(p,n)63Zn nuclear reaction.144 This reaction takes place within the 16 to 5 MeV range using enriched 63Cu as well as natural copper (isotopic composition: 69.17% 63Cu, 30.83% 65Cu). In both cases, 63Zn production was maximized in the 13 to 12 MeV energy range. The primary target material for the reactions were natural copper foils148 or natural copper that had been electroplated onto a suitable metal substrate such as gold.146 When a natural copper foil was bombarded with a 16 MeV proton beam, the reported 63Zn yield was 2470 MBq (μA h)−1 (i.e. 66.8 mCi (μA h)−1) with a purity exceeding 99.9%.148 The primary impurities generated during bombardment were 64Cu (t1/2: 12.7 h, 38.5% β−, 10.8% β+, 50.7% EC), 62Zn (t1/2: 9.2 h, 8.2% β+, 91.8% EC), and 65Zn (t1/2: 243.9 d, 1.4% β+, 98.6% EC).144,148 The presence of the 65Zn isotopic impurity is problematic for PET applications due to its long half-life. To limit the production of this impurity, bombardments at beam energies exceeding 11 MeV have been recommended.148
Product purification. A method for isolating zinc from the irradiated natural copper targets is described by ref. 148. The natural copper target was first dissolved in concentrated HNO3 before undergoing cation exchange with an AG 50W-X8 resin. The resin retained the zinc product, which can be eluted using a mixture of 0.05 N HCl and 85% acetone. The eluted zinc was obtained with a separation efficiency of 88.0%. Methods for recovering the copper target material have not been discussed as such methods would likely be impracticable due to the naturally high abundance of 63Cu.
Summary. Table 12 summarizes key production and purification parameters for 63Zn not mentioned in Table 1.
| Radioisotope | 63Zn |
|---|---|
| Target material | Natural copper |
| Product purity | >99.9% |
| Major impurities | 64Cu, 62Zn, 63Zn, 65Zn |
| Separation method | Cation exchange chromatography (AG 50-X8 resin) |
| - Target dissolution: concentrated HNO3 | |
| - Zn elution: 0.05 N HCl and 85% acetone | |
| Product separation yield | 88% |
| Target recovery yield | n/a |
Applications. The well matched half-life of 89Zr (t1/2: 78.4 h, 22.7% β+, 77.3% EC) with the pharmacokinetics of antibodies has led to more than 90 clinical studies, highlighted by the success of the ZEPHIR trial.4 These 89Zr-labelled antibodies are used to plan and monitor cancer treatments, visualize in vivo biodistribution, determine target expression, and conduct radiotherapy dosimetry. With the advancements in its production and labelling technology, 89Zr has been used to label numerous antibodies (i.e. HER1-targeted cetuximab, HER2-targeted trastuzumab, VEGF-targeted bevacizumab, CD20-targeted ibritumomab tiuxetan, transforming growth factor-β (TGF-β)-targeted fresolimumab, CD105-targeted TRC105 and PSMA-targeted 7E11) for both clinical and pre-clinical studies.1 Most of these examples employed desferoxime (DFO) chelator which is less than ideal for zirconium149 with significant concentrations of unbound 89Zr found with certain radiotracers such as 89Zr-fresolimumab and 89Zr-bevacizumab. This represents a major drawback of 89Zr as free radiometal will accumulate in the bone and may negatively affect bone marrow health.150,151 Ongoing efforts to develop improved 89Zr chelates may address many of these concerns.152–154
Production. Although several nuclear reactions can be used to produce 89Zr, only the 89Y(p,n)89Zr reaction can be feasibly carried out on small medical cyclotrons. Natural yttrium (isotopic composition: 100.00% 89Y) targets can be bombarded with beam energies between 14 to 9 MeV, although the actual range for reasonable, high purity production is somewhat narrower.62 Bombardments with beam energies exceeding 11.6 MeV lead to the production of the long half-life contaminant, 88Y (t1/2: 106.6 d, 0.2% β+, 99.8% EC) formed via the 89Y(p,pn)88Y reaction,155 while beam energies greater than 13 MeV resulted in the production of a second long half-life contaminant, 88Zr (t1/2: 83.4 d, 100.0% EC) which is inseparable from the final product and is formed via the 89Y(p,2n)88Zr reaction.62 Bombardments with beam energies below 10 MeV are not sufficient for 89Zr production.62 Many different forms of yttrium targets have been investigated to produce 89Zr. Foil and pressed pellet targets were produced entirely from natural yttrium. Alternatively, targets can be composites composed of yttrium or an yttrium oxide (Y2O3) layered onto a suitable metal substrate such as copper using sputtering and deposition techniques.156,157 A high purity yield of 58 MBq (μA h)−1 (i.e. 1.6 mCi (μA h)−1)64,158 has been reported in the literature for 89Zr production on yttrium metal targets in the aforementioned beam energy range. While the natural abundance of 89Y is monoisotopic it contains trace impurities of iron, titanium, and gadolinium. Proton interactions with those trace elements during bombardment form radionuclide impurities such as 48V (t1/2: 16.0 d, 49.9% β+, 50.1% EC), 156Tb (t1/2: 5.3 d, 100.0% EC), 65Zn (t1/2: 243.9 d, 1.4% β+, 98.6% EC) and 56Co (t1/2: 77.2 d, 19.7% β+, 80.3% EC) which negatively affect 89Zr antibody labelling and patient dosimetry.156
Product purification. To ensure high zirconium purity, column-based separation methods are generally utilized, with the most common being weak cation exchange.158,159 The irradiated yttrium targets were dissolved in strong (1 to 3 M) hydrochloric acid, and the solution was passed through a hydroxamate resin column. The hydroxamate column absorbed the zirconium ions in solution and later eluted using 1 M oxalic acid with a separation efficiency exceeding 70%. Depending on the application, a second column-based separation and washing stage could be used to eliminate the oxalic acid from the complex it forms with zirconium since oxalic acid is highly toxic to the human body. When performed, the result was an zirconium product with a final purity exceeding 99.9%.156–158 Methods for recovering the yttrium target material have not been discussed because recovery of natural Y targets is not practiced due to its economic characteristics vs. the purchase of new target material.
Summary. Table 13 summarizes key production and purification parameters for 89Zr not mentioned in Table 1.
| Radioisotope | 89Zr |
|---|---|
| Target material | Natural yttrium and its oxide |
| Product purity | >99.9% |
| Major impurities | 88Y, 88Zr, 48V, 156Tb, 65Zn, 56Co |
| Separation method | Cation exchange chromatography (hydroxamate resin) |
| - Target dissolution: 1 to 3 M HCl | |
| - Zr elution: 1 M oxalic acid | |
| Product separation yield | 70% |
| Target recovery yield | n/a |
Main group elements
Applications. Due to its comparatively long half-life, 66Ga (t1/2: 9.5 h, 57.0% β+, 43.0% EC) has received some consideration in the field of PET for longer tracking of radiogallium. However only a few examples of 66Ga PET radiotracers have been reported as the high energy emissions from its decay raises both patient and healthcare personnel safety concerns.1 In one study, 66Ga was used to label monoclonal antibodies (mAbs) to investigate tumor angiogenesis in animals. The study found that the radiotracer did not clear quickly from the bloodstream of the animals, resulting in moderate amounts of the radiotracer being found in the lungs, liver, heart, kidneys and spleen.160 A second animal study demonstrated that unbound 66Ga accumulated in the bone and liver, pointing to metabolism or transchelation.161 On the other hand, 68Ga (t1/2: 67.7 min, 88.9% β+, 11.1% EC) has found extensive use as a small-molecule imaging tool within the last decade.162–164 68Ga is particularly advantageous for the labelling of such compounds since its half-life is generally comparable to their pharmacokinetics. Another group of targeting molecules that can be radiolabeled using 68Ga are peptides. These radiotracers are easy and inexpensive to produce and show quick blood clearance and tissue diffusivity. Most of the peptides that have been labelled are analogous to somatostatin, a regulatory protein primarily produced in the nervous and digestive systems. These analogues are able to interact with somatostatin receptors found in normal human tissues (i.e. the gastrointestinal tract, kidney, thyroid, spleen, brain, pancreas) and in a variety of malignancies (i.e. neuroendocrine tumors, malignant lymphomas, renal cell carcinomas, prostate cancer, breast cancer and small cell lung cancer), thus facilitating PET investigations. 68Ga has also been used to label melanocortin analogues for investigating melanocortin-receptor containing tissues (i.e. fibroblasts, leukocytes, keratinocytes, melanocytes and endothelial and antigen-presenting cells), and bombesin analogues for investigating bombesin-receptor containing tissue found in tumors (i.e. breast and prostate cancers). It is evident that 68Ga peptide radiotracers have been used in numerous preclinical and clinical studies,165 including nearly 400 clinical investigations. The promising potential of 68Ga radiopharmaceuticals have found clinical utility through successful imaging of PSMA, somatostatin receptors, and FAPI.163,164,166
Production. 66Ga can be produced from the bombardment of both natural zinc (isotopic composition: 49.17% 64Zn, 27.73% 66Zn, 4.04% 67Zn, 18.45% 68Zn, 0.61% 70Zn) and 66Zn-enriched foils,167 or after electroplating onto gold or silver substrates.160 The radionuclide is primarily produced via the 66Zn(p,n)66Ga nuclear reaction at a minimum beam energy of 6 MeV,161 but irradiations are typically carried out at much higher energies (i.e. such as 9.8 MeV,167 13 MeV,160 14.5 MeV (ref. 167) and 15 MeV (ref. 161)). Yields of 511 MBq (μA h)−1 (i.e. 13.8 mCi (μA h)−1) with a purity of 99.96% have been achieved from the 14.5 MeV bombardment of 98.9% enriched 66Zn targets167 although higher yields of 700 MBq (μA h)−1 (i.e. 18.9 mCi (μA h)−1) have been reported elsewhere.64 The primary impurity produced during irradiation was 67Ga (t1/2: 3.3 d, 100.0% EC).161,167
While 68Ga has traditionally been produced with 68Ge/68Ga generators producing 1850 MBq and 3700 MBq of 68Ga, cyclotrons have alternatively been used for its production via the proton bombardment of 68Zn-enriched targets. Recent progress using liquid targets has demonstrated modest production yields above 68Ge/68Ga generators. However solid targets show significant potential for much higher capacity production.168 Solid targets may be foils or zinc-coated metal backings (i.e. aluminum, gold or silver) that are generally prepared via electroplating or sputtering.169–171 The primary reaction governing the production of 68Ga is the 68Zn(p,n)68Ga nuclear reaction, which takes place within the 14 to 11 MeV beam energy range.62 Researchers have reported approximate yields of 1240 MBq (μA h)−1 (i.e. 33.5 mCi (μA h)−1) from the bombardment of natural zinc targets at a beam energy of 12.5 MeV,171 and yields of 1380 MBq (μA h)−1 (i.e. 37.3 mCi (μA h)−1) for the bombardment of enriched >99% 68Zn electroplated silver targets with a beam energy of 12.5 MeV.169 A novel enriched zinc target design (backed with silver) irradiated at 13 MeV demonstrated a final yield of 1213 MBq (μA h)−1 (i.e. 32.8 mCi (μA h)−1) at end of separation (EOS) with a purity exceeding 99%.172 The major impurities produced from the reaction were 66Ga (t1/2: 9.5 h, 57.0% β+, 43.0% EC) and 67Ga (t1/2: 3.3 d, 100.0% EC), and they are inseparable from the desired isotope.62 Measures can be taken to reduce 66/67Ga production during target irradiation including: (1) reduce the amounts of 66Zn and 67Zn in the targets by using highly enriched 68Zn to limit the 66Zn(p,n)66Ga and 67Zn(p,n)67Ga nuclear reactions, and (2) limit the maximum beam energy to 12 MeV to reduce the 68Zn(p,2n)67Ga nuclear reaction62 respectively.169
Product purification and target recovery. Gallium can be extracted from the zinc targets through two major methods. The first method made use of cation exchange chromatography where irradiated targets were dissolved in 10 to 12 M HCl and passed through a cation exchange resin, such as the AG 50W-X8 or AG 50W-X2, which retained both the zinc target material and gallium product. The zinc target material was then eluted using 10 M HCl, and was recovered with efficiencies ranging from 76 to 80%. The gallium product was subsequently eluted with 4 M HCl with an overall separation efficiency as high as 90%.160,167,170 To increase the purity of the obtained gallium, a washing stage using 7 N HCl was carried out immediately before eluting the gallium product.170 Alternatively, this procedure was carried out in a fully automated way using commercially available modules with separation efficiencies up to 80%.169,171–173 Unreacted zinc target material was recovered from the 10 M HCl solution through electrolysis using a voltage of 5 V and a current of 1 A with efficiencies exceeding 76%.169
The second method for isolating gallium is liquid–liquid extraction. Preparation steps for this procedure involved dissolving the irradiated zinc targets in 12 M acid and diluting the resulting solution to 7 M with water. The dissolution was then subjected to a series of extraction and back-extraction stages using various solvents (i.e. diisopropyl ether, 7 M HCl and water) until gallium was obtained with the desired purity.167 Maximum separation efficiency for the gallium product was 79% and the recovery efficiency for the zinc target material exceeded 99%.
Summary. Table 14 summarizes key production and purification parameters for 66Ga and 68Ga not mentioned in Table 1.
| Radioisotope | 66Ga, 68Ga | |
|---|---|---|
| Target material | 66Ga | Natural and enriched zinc |
| Product purity | >99.9% | |
| Major impurities | 67Ga | |
| Target material | 68Ga | Enriched zinc |
| Product purity | >99% | |
| Major impurities | 66Ga, 67Ga | |
| Separation method | 1 | Cation exchange chromatography (AG 50W-X8 and AG 50W-X2 resins) |
| - Target dissolution: 10 to 12 M HCl | ||
| - Zn elution: 10 M HCl | ||
| - Ga elution: 4 M HCl | ||
| Product separation yield | 90% | |
| Target recovery yield | 76 to 80% | |
| Separation method | 2 | Liquid–liquid extraction |
| - Target dissolution: 12 M HCl | ||
| - Extraction solvents: Diisopropyl ether, 7 M HCl and water | ||
| Product separation yield | 79% | |
| Target recovery yield | >99% | |
Applications. 72As (t1/2: 26.0 h, 87.8% β+, 12.2% EC) is considered a promising PET radionuclide for several reasons including its long nuclear half-life which permits the radiolabeling of biomolecules with longer biological half-lives.174,175 The resulting radiotracers can be used in several extended biochemical and physiological process studies such as immunoimaging and receptor mapping. A second favorable characteristic arises from the unique soft Lewis acid properties of arsenic to covalently bond to thiol (or sulfhydryl) groups.1,174,175 Theoretically, this allows arsenic's radioisotopes to directly label numerous biomolecules with little to no chemical modification.1,175 Several radioarsenic labeled antibodies and nanoparticles have been reported, although none have entered clinical use.176–178 Finally, the positron emitting 72As provides a matched pair with 77As (t1/2: 38.8 h, 100% β−) which will behave identically in vivo for imaging and radiotherapy, respectively.179 The theranostic advantage of the pairing makes up for the poor image resolution resulting from arsenic's large positron range (i.e. 3.6 mm in soft tissue).58 Despite its favorable qualities however, research on the PET applications of 72As has been slow as a result of the limited supply of isotopically-enriched germanium, the primary material for the production of various arsenic radioisotopes,1,175 although selenium has been used on occasion.180 The limited supply of isotopically-enriched germanium increases target costs which translates into an overall increase in 72As production costs.175
Production. 72As can be produced using a 72Se/72As generator, or using a solid target cyclotron.1 With respect to the latter, 72As can be produced using a variety of nuclear reactions such as 72Ge(p,n)72As, 73Ge(p,2n)72As, 74Ge(p,3n)72As and 76Se (p,x)72As.180,181 Of the listed reactions, the most commonly used was the 72Ge(p,n)72As nuclear reaction which took place within the 18 to 8 MeV range,174 noting that 72As production at beam energies exceeding 15 MeV contained a greater number of impurities in the final product.174 The targets used for 72As production were typically 72Ge-enriched germanium disks178 and 72Ge-enriched germanium oxide that had been electroplated onto substrates made of copper174 and niobium.175 Additionally, natural germanium oxide174 or natural germanium (isotopic composition: 20.57% 70Ge, 27.45% 72Ge, 7.75% 73Ge, 36.50% 74Ge, 7.73% 76Ge) foil182 have also been used to produce 72As in other studies. The primary impurities produced during bombardment were 71As (t1/2: 65.3 h, 28.3% β+, 71.7% EC), 74As (t1/2: 17.8 d, 34.0% β−, 29.0% β+, 37.09% EC), 76As (t1/2: 26.2 h, 100.0% β−),69Ge(t1/2: 39.1 h, 24.0% β+, 76.0% EC) and 67Ga (t1/2: 3.3 d, 100.0% EC) (175,178,182). The 72As production yield for >96% enriched germanium oxide electroplated onto a niobium substrate and bombarded with 16.1 MeV protons was 90 MBq (μA h)−1 (i.e. 2.4 mCi (μA h)−1), with purity exceeding 99%.178 The theoretical 72As yield for natural germanium oxide electroplated onto a copper substrate and bombarded with a proton beam energy of 15 MeV to 8 MeV was 60 MBq (μA h)−1 (i.e. 1.6 mCi (μA h)−1).174
Product purification and target recovery. Methods for isolating arsenic from the germanium targets have been described extensively in Enferadi et al.,174 Ellison et al.175,178 and Guin et al.183 In general, the methods required dissolution of the germanium targets using solutions of 6 M NaOH or hot aqua regia, followed by the addition of 6 to 10 N HCl. The addition of the HCl was necessary to convert the germanium metal to GeCl4, which is significantly more volatile than the metallic arsenic product. Thirty percent hydrogen peroxide was then added to oxidize the arsenic to the As(V) state. The final dissolution was evaporated to near dryness, after which distillation was performed on the resulting residue. About 97% of the 72As was left behind as residue from the distillation, and then further purified through ion exchange chromatography after dissolution in 10 M HCl. Anion exchange was carried out by passing the arsenic solution through a Bio-Rad AG 1 × 8 resin (200–400 mesh), which retained the arsenic product as well as any residual germanium and gallium impurities. The arsenic could be collected by eluting with 10 M HCl, and the procedure had a separation efficiency of 93%, resulting in an overall As(V) separation efficiency of about 90%. However, if it is desirous to have the arsenic in the As(III) state instead of its As(V) state, SPE can be carried out subsequently. The SPE procedure had an efficiency of 57% and after its application, the overall separation efficiency of the As(III) product was 51%. Recovery of the germanium target material could be achieved by treating the acidic condensate from the dissolution stage with 6 M NaOH. This neutralized the condensate, and converted GeCl4 to GeO which could be easily reduced to metallic germanium when sufficiently heated. The overall recovery efficiency reported by Ellison et al.178 was about 60%, however it should be emphasized that higher recovery efficiencies could be achieved if the recovery process was optimized.
Summary. Table 15 summarizes key production and purification parameters for 72As not mentioned in Table 1.
| Radioisotope | 72As |
|---|---|
| Target material | Natural and enriched germanium and their oxides |
| Product purity | >99% |
| Major impurities | 71As, 74As, 76As, 67Ga, 69Ge |
| Separation method | Distillation, Anion exchange chromatography and SPE |
| - (Distillation) target dissolution: 6 M NaOH or hot aqua regia + 6 to 10 N HCl + 30% H2O2 | |
| - (Chromatography) target distillation: 10 M HCl | |
| - As elution: 10 M HCl | |
| Product separation yield | >90% As(V), ∼51% As(III) |
| Target recovery yield | ∼60% |
Conclusion
Given the surge in clinical use and interest in several metallic radioisotopes, solid target systems for small (≤24 MeV) medical cyclotrons are rapidly evolving to enable large-scale production with consistent yield, composition and quality. With this, many factors need to be considered when choosing to develop in-house radiometal production capabilities, including (i) the variability in medical cyclotron design (i.e. beam energy, current and machine configuration); (ii) target isotope availability; (iii) quality/quantity variations when compared to alternative, non-cyclotron production routes, and (iv) the intended application of the radioisotope. While many of the metallic radioisotopes discussed herein are being developed as imaging agents unto themselves, several have the potential to be theragnostic partners to a range of chemically diverse therapeutic radioisotopes, and require considerations in chemical compatibility and physical/biological half-life. Of those diagnostic radioisotopes with theragnostic applications, the community will likely see near-term adoption of cyclotron-produced 68Ga and 89Zr, and a resurgence of interest in 64Cu. Radioisotopes with element-identical theragnostic application such as 86Y, 44Sc and 124I may encounter increases in use as long as their production and/or recycling methodologies can be sufficiently optimized. Yet others, such as cyclotron-produced 99mTc, may emerge as important enablers for those regions in which supply via non-cyclotron-based alternatives remain problematic.Nomenclature and units
| α | Alpha particle |
| β− | Beta emission |
| β+ | Positron emission |
| d | Days |
| EC | Electron capture |
| Emax | The endpoint energy (i.e. maximum energy) of an emitted positron |
| EOB | End of bombardment |
| h | Hours |
| IT | Isomeric transition |
| keV | Kiloelectron volts |
| MBq | Megabecquerels |
| MeV | Megaelectron volts |
| μA | Microamperes |
| mCi | Millicuries |
| min | Minutes |
| M | Molar (i.e. mols of acid per liter) |
| n | Neutron |
| N | Normal (i.e. moles of dissociated H+ ions per liter) |
| °C | Degrees celsius |
| PET | Positron emission tomography |
| p | Positron |
| SPECT | Single photon computed tomography |
| SPE | Solid phase extraction |
| t1/2 | Half-life |
| γ | Gamma ray |
Conflicts of interest
There are no conflicts of interest to declare.References
- Y. Zhou, K. E. Baidoo and M. W. Brechbiel, Mapping Biological Behaviors by Application of Longer-Lived Positron Emitting Radionuclides, Adv. Drug Delivery Rev., 2013, 65(8), 1098–1111 CrossRef CAS PubMed.
- A. Boschi, P. Martini, V. Costa, A. Pagnoni and L. Uccelli, Interdisciplinary tasks in the cyclotron production of radiometals for medical applications - The case of 47Sc as example, Molecules, 2019, 24(3), 444 CrossRef PubMed.
- S. Reddy and M. Robinson, ImmunoPET In Cancer Models, Semin. Nucl. Med., 2010, 40(3), 182–189 CrossRef PubMed.
- E. Boros and J. Holland, Chemical aspects of metal ion chelation in the synthesis and application antibody-based radiotracers, J. Labelled Compd. Radiopharm., 2018, 61(9), 652–671 CrossRef CAS PubMed.
- G. Stöcklin and V. W. Pike, Radiopharmaceuticals for Positron Emission Tomography-Methodological Aspects, Springer Science & Business Media, Vol. 24, 1993 Search PubMed.
- M. Sajjad and R. M. Lambrecht, Cyclotron targetry for medical radioisotope production, Nucl. Instrum. Methods Phys. Res., Sect. B, 1989, 40, 11001104 Search PubMed.
- H. F. Valdovinos, S. Graves, P. Ellison, T. Barnhart and R. J. Nickles, Earth, air, fire and water: A targetry quartet, AIP Conf. Proc., 2017, 1845 Search PubMed.
- S. M. Qaim, Target development for medical radioisotope production at a cyclotron, Nucl. Instrum. Methods Phys. Res., 1989, 282(1), 289–295 CrossRef.
- International Atomic Energy Agency, Cyclotron produced radionuclides: physical characteristics and production methods International Atomic Energy Agency, Vienna, Austria, 2009 Search PubMed.
- C. J. Anderson and R. Ferdani, Copper-64 radiopharmaceuticals for PET imaging of cancer: Advances in preclinical and clinical research, Cancer Biother.Radiopharm., 2009, 24(4), 379–393 CrossRef CAS PubMed.
- M. Fassbender, A. F. Novgorodov, F. Rösch and S. M. Qaim, Excitation Functions of 93Nb(3He,xn)93m'g,94m,g,95m,gTc-Processes from Threshold up to 35 MeV: Possibility of Production of 94mTc in High Radiochemical Purity using a Thermochromatographic Separation Technique, Radiochim. Acta, 1994, 65(4), 215–222 CrossRef CAS.
- H. Büyükuslu, A. Kaplan, G. Yıldırım, A. Aydin, E. Tel and M. Bölükdemir, Production cross sections of medical 110,111 In radionuclides, Kerntechnik, 2010, 73(3), 103–108 CrossRef.
- T. Kostelnik and C. Orvig, Radioactive main group and rare earth metals for imaging and therapy, Chem. Rev., 2018, 119(2), 902–956 CrossRef PubMed.
- A. Rahman and A. Awal, Production of 149Tb, 152Tb, 155Tb and 161Tb from gadolinium using different light-particle beams, J. Radioanal. Nucl. Chem., 2020, 323(2), 731–740 CrossRef CAS.
- A. Klein, F. Rösch and S. Qaim, Investigation of 50Cr(d,n)51Mn and natCr(p, x)51Mn processes with respect to the production of the positron emitter 51Mn, Radiochim. Acta, 2000, 88(5), 253–264 CrossRef CAS.
- E. Boros and A. Packard, Radioactive transition metals for imaging and therapy, Chem. Rev., 2018, 119(2), 870–901 CrossRef PubMed.
- A. H. Asad, S. V. Smith, S. Chan, C. M. Jeffery, L. Morandeau and R. I. Price, Cyclotron production of 61Cu using natural Zn & enriched 64Zn targets, in AIP Conference Proceedings, 2012, pp. 91–5 Search PubMed.
- T. Z. Wong, J. L. Lacy, N. A. Petry, T. C. Hawk, T. A. Sporn and M. W. Dewhirst, et al., PET of hypoxia and perfusion with 62Cu-ATSM and 62Cu-PTSM using a 62Zn/62Cu generator, Am. J. Roentgenol., 2008, 190(2), 427–432 CrossRef PubMed.
- E. Aluicio-Sarduy, N. Thiele, K. Martin, B. Vaughn, J. Devaraj and A. Olson, et al., Establishing radiolanthanum chemistry for targeted nuclear medicine applications, Chemistry, 2020, 26(6), 1238 CrossRef CAS PubMed.
- E. Aluicio-Sarduy, T. Barnhart, J. Weichert, R. Hernandez and J. Engle, Cyclotron produced 132La as a PET imaging surrogate of therapeutic 225Ac, J. Nucl. Med., 2021, 62(7), 1012–1015 CrossRef PubMed.
- E. Boros and A. B. Packard, Radioactive Transition Metals for Imaging and Therapy, Chem. Rev., 2018, 119(2), 870–901 CrossRef PubMed.
- E. Aluicio-Sarduy, P. Ellison, T. Barnhart, W. Cai, R. Nickles and J. Engle, PET radiometals for antibody labeling, J. Labelled Compd. Radiopharm., 2018, 61(9), 636–651 CrossRef CAS PubMed.
- H. Coenen and J. Ermert, Expanding PET-applications in life sciences with positron emitters beyond fluorine-18, Nucl. Med. Biol., 2021, 92, 241–269 CrossRef CAS PubMed.
- H. Coenen, M. Harmand, G. Kloster and G. Stöcklin, 15-(p-[75Br] bromophenyl) pentadecanoic Acid: Pharmacokinetics and Potential as Heart Agent, J. Nucl. Med., 1981, 22(10), 891–896 CAS.
- O. De Jesus, G. Van Hoffaert, D. Glock, L. Goldber and A. Friedman, Synthesis of a radiobrominated analog of SCH 23390, a selfctive dopamine D1/DA1 antagonist, J. Labelled Compd. Radiopharm., 1986, 23(9), 919–925 CrossRef CAS.
- S. Moerlein and G. Stoecklin, Synthesis of high specific activity [75Br]-and [77Br] Bromperidol and tissue distribution studies in the rat. Journal of medicinal chemistry, J. Med. Chem., 1985, 28(9), 1319–1324 CrossRef CAS PubMed.
- S. Moerlein, P. Laufer, G. Stöcklin, G. Pawlik, K. Wienhard and W. Heiss, Evaluation of 75Br-labelled butyrophenone neuroleptics for imaging cerebral dopaminergic receptor areas using positron emission tomography, Eur. J. Nucl. Med., 1986, 12(4), 211–216 CrossRef CAS PubMed.
- J. Rice, N. Hussain and E. Lavoie, Synthesis of [methoxy-14C] eugenol, J. Labelled Compd. Radiopharm., 1987, 24(9), 1043–1049 CrossRef CAS.
- G. Kloster, P. Laufer, W. Wutz and G. Stöcklin, 75,77Br-and 123 I-analogues of D-glucose as potential tracers for glucose utilisation in heart and brain, Eur. J. Nucl. Med., 1983, 8(6), 237–241 CrossRef CAS PubMed.
- D. J. Rowland, T. J. McCarthy and M. J. Welch, Radiobromine for imaging and therapy, in Handbook of Radiopharmaceuticals: Radiochemistry and Applications, 2002, pp. 441–465 Search PubMed.
- P. McQuade, D. J. Rowland, J. S. Lewis and M. J. Welch, Positron-Emitting Isotopes Produced on Biomedical Cyclotrons, Curr. Med. Chem., 2005, 12(7), 807–818 CrossRef CAS PubMed.
- M. Kassiou, C. Loc’h, M. Bottlaender, K. Mardon, M. Ottaviani and C. Coulon, et al., (+)-[76Br] A-69024: a non-benzazepine radioligand for studies of dopamine D1 receptors using PET, Nucl. Med. Biol., 2002, 29(3), 295–302 CrossRef CAS PubMed.
- C. Foged, C. Halldin, C. Loc’h, B. Mazière, P. Karlsson and M. Mazière, et al., 11C-and 76Br-labelled NNC 22-0010, selective dopamine D1 receptor radioligands for PET, Nucl. Med. Biol., 1996, 23(6), 837–844 CrossRef CAS PubMed.
- C. Lundkvist, C. Loc’h, C. Halldin, M. Bottlaender, M. Ottaviani and C. Coulon, et al., Characterization of bromine-76-labelled 5-bromo-6-nitroquipazine for PET studies of the serotonin transporter, Nucl. Med. Biol., 1999, 26(5), 501–507 CrossRef CAS PubMed.
- L. Lang, W. Li, H. Jia, D. Fang, S. Zhang and X. Sun, et al., New methods for labeling RGD peptides with bromine-76, Theranostics, 2011, 1, 341 CrossRef CAS PubMed.
- S. Cho, L. Ravasi, L. Szajek, J. Seidel, M. Green and H. Fine, et al., Evaluation of 76Br-FBAU as a PET reporter probe for HSV1-tk gene expression imaging using mouse models of human glioma, J. Nucl. Med., 2005, 46(11), 1923–1930 CAS.
- S. Watanabe, H. Hanaoka, J. Liang, Y. Iida, K. Endo and N. Ishioka, PET Imaging of Norepinephrine Transporter–Expressing Tumors Using 76Br-meta-Bromobenzylguanidine, J. Nucl. Med., 2010, 51(9), 1472–1479 CrossRef CAS PubMed.
- R. Rossin, D. Berndorff, M. Friebe, L. M. Dinkelborg and M. J. Welch, Small-animal PET of tumor angiogenesis using a 76Br-labeled human recombinant antibody fragment to the ED-B domain of fibronectin, J. Nucl. Med., 2007, 48(7), 1172–1179 CrossRef CAS PubMed.
- N. Vaiseman, G. Koren and P. Pencharz, Pharmacokinetics of oral and intravenous bromide in normal volunteers, J. Toxicol. Clin. Toxicol., 1986, 24(5), 403–413 CrossRef CAS PubMed.
- C. Lang, D. Habs, K. Parodi and P. Thirolf, Sub-millimeter nuclear medical imaging with high sensitivity in positron emission tomography using β+ γ coincidences, J. Instrum., 2014, 9(1), P01008 CrossRef.
- M. Sitarz, J. Cussonneau, T. Matulewicz and F. Haddad, Radionuclide candidates for β+ γ coincidence PET: an overview, Appl. Radiat. Isot., 2020, 155, 108898 CrossRef CAS PubMed.
- D. Wilbur and M. Adam, Radiobromine and radioiodine for medical applications, Radiochim. Acta, 2019, 107(9–11), 1033–1063 CrossRef CAS.
- Progress in Radiopharmacy. Progress in Radiopharmacy. ed. P Cox, S Mather, C Sampson and C Lazarus, Springer Science & Business Media; 2012 Search PubMed.
- Z. Kovács, G. Blessing, S. M. Qaim and G. Stöcklin, Production of 75Br via the 76Se(p,2n)75Br reaction at a compact cyclotron, Int. J. Appl. Radiat. Isot., 1985, 36(8), 635–642 CrossRef.
- S. M. Qaim, Recent developments in the production of 18F, 75,76,77Br and 123I, Int. J. Radiat. Appl. Instrum. Appl. Radiat. Isot., 1986, 37(8), 803–810 CrossRef CAS.
- D. J. Schlyer, Production of Radionuclides in Acclerators, in Handbook of radiopharmaceuticals, radiochemistry and applications, 2003. pp. 1–70 Search PubMed.
- K. Breunig, I. Spahn, S. Spellerberg and H. H. Coenen, Production of no-carrier-added radiobromine: New nickel selenide target and optimized separation by dry distillation, Radiochim. Acta, 2015, 103(5), 397–402 CrossRef CAS.
- V. Tolmachev, A. Lövqvist, L. Einarsson, J. Schultz and H. Lundqvist, Production of 76Br by a low-energy cyclotron, Appl. Radiat. Isot., 1998, 49(12), 1537–1540 CrossRef CAS.
- A. Bruskin, I. Sivaev, M. Persson, H. Lundqvist, J. Carlssona and S. Sjoberg, et al., Radiobromination of monoclonal antibody using potassium [76Br](4 isothiocyanatobenzyl-ammonio)-bromo-decahydro-closo-dodecaborate (Bromo-DABI), Nucl. Med. Biol., 2004, 31(2), 205–211 CrossRef CAS PubMed.
- S. Mahajan and C. Divgi, The role of iodine-124 positron emission tomography in molecular imaging, Clin. Transl. Imaging, 2016, 4(4), 297–306 CrossRef.
- J. A. O'Donoghue, P. M. Smith-Jones, J. L. Humm, S. Ruan, D. A. Pryma and A. A. Jungbluth, et al., 124I-huA33 antibody uptake is driven by A33 antigen concentration in tissues from colorectal cancer patients imaged by immuno-PET, J. Nucl. Med., 2011, 52(12), 1878–1885 CrossRef PubMed.
- I. Verel, G. W. M. Visser, O. C. Boerman, J. E. M. Van Eerd, R. Finn and R. Boellaard, et al., Long-lived positron emitters zirconium-89 and iodine-124 for scouting of therapeutic radioimmunoconjugates with PET, Cancer Biother.Radiopharm., 2003, 18(4), 655–661 CrossRef CAS PubMed.
- G. C. Jayson, J. Zweit, A. Jackson, C. Mulatero, P. Julyan and M. Ranson, et al., Molecular imaging and biological evaluation of HuMV833 anti-VEGF antibody: Implications for trial design of antiangiogenic antibodies, J. Natl. Cancer Inst., 2002, 94(19), 1484–1493 CrossRef CAS PubMed.
- A. M. S. Braghirolli, W. Waissmann, J. B. Da Silva and G. R. Dos Santos, Production of iodine-124 and its applications in nuclear medicine, Appl. Radiat. Isot., 2014, 90, 138–148 CrossRef CAS PubMed.
- H. Coenen, K. Dutschka, S. Müller, L. Geworski, J. Farahati and C. Reiners, Nca radiosynthesis of [123,124 I] β-CIT, plasma analysis and pharmacokinetic studies with SPECT and PET, Nucl. Med. Biol., 1995, 22(8), 977–984 CrossRef CAS PubMed.
- C. Zechmann, A. Afshar-Oromieh, T. Armor, J. Stubbs, W. Mier and B. Hadaschik, et al., Radiation dosimetry and first therapy results with a 124 I/131I-labeled small molecule (MIP-1095) targeting PSMA for prostate cancer therapy, Eur. J. Nucl. Med. Mol. Imag., 2014, 41(7), 1280–1292 CrossRef CAS PubMed.
- C. Foss, D. Plyku, A. Ordonez, J. Sanchez-Bautista, H. Rosenthal and I. Minn, et al., Biodistribution and radiation dosimetry of 124I-DPA-713, a PET radiotracer for macrophage-associated inflammation, J. Nucl. Med., 2018, 59(11), 1751–1756 CrossRef CAS PubMed.
- L. Carter, A. Kesner, E. Pratt, V. Sanders, A. Massicano and C. Cutler, et al., The impact of positron range on PET resolution, evaluated with phantoms and PHITS Monte Carlo simulations for conventional and non-conventional radionuclides, Mol. Imag. Biol., 2020, 22(1), 73–84 CrossRef CAS PubMed.
- N. Shah, H. Herzog, C. Weirich, L. Tellmann, J. Kaffanke and L. Caldeira, et al., Effects of magnetic fields of up to 9.4 T on resolution and contrast of PET images as measured with an MR-BrainPET, PLoS One, 2014, 4, e95250 CrossRef PubMed.
- S. D. Wilbur, S. W. Hadley, M. D. Hylarides, P. G. Abrams, P. A. Beaumier and A. C. Morgan, et al., Development of a stable radioiodinating reagent to label monoclonal antibodies for radiotherapy of cancer, J. Nucl. Med., 1989, 30(2), 216–226 Search PubMed.
- M. Berman, L. Braverman, J. Burke, L. De Groot, K. McCormack and T. Oddie, et al., Summary of Current Radiation Dose Estimates to Humans from 123I, 124I, 125I, 126I, 130I, 131I, and 132I as Sodium Iodide, J. Nucl. Med., 1975, 16(12), 1214–1217 Search PubMed.
- M. A. Synowiecki, L. R. Perk and J. F. W. Nijsen, Production of novel diagnostic radionuclides in small medical cyclotrons, EJNMMI Radiopharm. Chem, 2018, 3(1), 3 CrossRef PubMed.
- L. Koehler, K. Gagnon, S. McQuarrie and F. Wuest, Iodine-124: A promising positron emitter for organic PET chemistry, Molecules, 2010, 15(4), 2686–2718 CrossRef CAS PubMed.
- S. M. Qaim, Nuclear data for production and medical application of radionuclides: Present status and future needs, Nucl. Med. Biol., 2017, 44, 31–49 CrossRef CAS PubMed.
- D. W. McCarthy, L. A. Bass, P. D. Cutler, R. E. Shefer, R. E. Klinkowstein and P. Herrero, et al., High purity production and potential applications of copper-60 and copper-61, Nucl. Med. Biol., 1999, 26(4), 351–358 CrossRef CAS PubMed.
- S. Lapi, J. Lewis and F. Dehdashti, Evaluation of hypoxia with copper-labeled diacetyl-bis (N-methylthiosemicarbazone), Semin. Nucl. Med., 2015, 45(2), 177–185 CrossRef PubMed.
- M. Conti and E. Lars, Physics of pure and non-pure positron emitters for PET: a review and a discussion, EJNMMI Phys, 2016, 3(1), 1–7 CrossRef PubMed.
- W. Ping Li, L. A. Meyer, D. A. Capretto, C. D. Sherman and C. J. Anderson, Receptor-binding, biodistribution, and metabolism studies of 64Cu-DOTA-cetuximab, a PET-imaging agent for epidermal growth-factor receptor-positive tumors, Cancer Biother.Radiopharm., 2008, 23(2), 158–171 CrossRef PubMed.
- M. Lewis, C. Boswell, R. Laforest, T. Buettner, D. Ye and J. Connett, et al., Conjugation of monoclonal antibodies with TETA using activated esters: biological comparison of 64Cu-TETA-1A3 with 64Cu-BAT-2IT-1A3, Cancer Biother.Radiopharm., 2001, 16(6), 483–494 CrossRef CAS PubMed.
- B. Gutfilen, S. Souza and G. Valentini, Copper-64: a real theranostic agent, Drug Des., Dev. Ther., 2018, 12, 3235 CrossRef CAS PubMed.
- N. Smith, D. Bowers and D. Ehst, he production, separation, and use of 67Cu for radioimmunotherapy: a review, Appl. Radiat. Isot., 2012, 70(10), 2377–2383 CrossRef CAS PubMed.
- P. Rowshanfarzad, M. Sabet, A. Reza Jalilian and M. Kamalidehghan, An overview of copper radionuclides and production of 61Cu by proton irradiation of natZn at a medical cyclotron, Appl. Radiat. Isot., 2006, 64(12), 1563–1573 CrossRef CAS PubMed.
- P. Rajec, V. Csiba, M. Leporis, M. Štefečka, E. L. Pataky and M. Reich, et al., Preparation and characterization of nickel targets for cyclotron production of 64Cu, J. Radioanal. Nucl. Chem., 2010, 286(3), 665–670 CrossRef CAS.
- C. M. Jeffery, S. V. Smith, A. H. Asad, S. Chan and R. I. Price, Routine production of copper-64 using 11.7MeV protons, AIP Conf. Proc., 2012, 1509(1), 84–90 CrossRef CAS.
- M. A. Avila-Rodriguez, J. A. Nye and R. J. Nickles, Simultaneous production of high specific activity 64Cu and 61Co with 11.4 MeV protons on enriched 64Ni nuclei, Appl. Radiat. Isot., 2007, 65(10), 1115–1120 CrossRef CAS PubMed.
- D. W. McCarthy, R. E. Shefer, R. E. Klinkowstien, L. A. Bass, W. H. Margeneau and C. S. Cutler, et al., Efficient production of high specific activity 64Cu using a biomedical cyclotron, Nucl. Med. Biol., 1997, 24(1), 35–43 CrossRef CAS.
- A. Obata, S. Kasamatsu, D. W. McCarthy, M. J. Welch, H. Saji and Y. Yonekura, et al., Production of therapeutic quantities of 64Cu using a 12 MeV cyclotron, Nucl. Med. Biol., 2003, 30(5), 535–539 CrossRef CAS PubMed.
- J. De Reuck, K. Paemeleire, P. Santens, K. Strijckmans and I. Lemahieu, Cobalt-55 positron emission tomography in symptomatic atherosclerotic carotid artery disease: Borderzone versus territorial infarcts, Clin. Neurol. Neurosurg., 2004, 106(2), 77–81 CrossRef PubMed.
- J. De Reuck, P. Santens, J. Keppens, J. De Bleecker, K. Strijckmans and P. Goethals, et al., Cobalt-55 positron emission tomography in recurrent ischaemic stroke, Clin. Neurol. Neurosurg., 1999, 101(1), 15–18 CrossRef CAS.
- H. Jansen, R. Dierckx, J. Hew, A. Paans, J. Minderhoud and J. Korf, Positron emission tomography in primary brain tumours using cobalt-55, Nucl. Med. Commun., 1997, 18(8), 734–740 CrossRef CAS PubMed.
- T. Mastren, B. Marquez, D. Sultan, E. Bollinger, P. Eisenbeis and T. Voller, et al., Cyclotron production of high - Specific activity 55Co and in vivo evaluation of the stability of 55Co metal-chelate-peptide complexes, Mol. Imag., 2015, 14(10), 7290 CrossRef PubMed.
- J. Garousi, K. Andersson, J. Dam, B. Olsen, B. Mitran and A. Orlova, et al., The use of radiocobalt as a label improves imaging of EGFR using DOTA-conjugated Affibody molecule, Sci. Rep., 2017, 7(1), 1–10 CrossRef PubMed.
- J. Dam, B. Olsen, C. Baun, P. Høilund-Carlsen and H. Thisgaard, Vivo Evaluation of a bombesin analogue labeled with Ga-68 and Co-55/57, Mol. Imag. Biol., 2016, 18(3), 368–376 CrossRef CAS PubMed.
- P. Goethals, A. Volkaert, C. Vandewielle, R. Dierckx and N. Lameire, 55Co-EDTA for renal imaging using positron emission tomography (PET): a feasibility study, Nucl. Med. Biol., 2000, 27(1), 77–81 CrossRef CAS.
- B. Mitran, H. Thisgaard, U. Rosenström, J. Dam, M. Larhed and V. Tolmachev, et al., High contrast PET imaging of GRPR expression in prostate cancer using cobalt-labeled bombesin antagonist RM26, Contrast Media Mol. Imaging, 2017, 1–10 Search PubMed.
- M. Al-abyad, M. N. H. Comsan and S. M. Qaim, Excitation functions of proton-induced reactions on with particular reference to the production of 57Co, Appl. Radiat. Isot., 2009, 67(1), 122–128 CrossRef CAS PubMed.
- A. G. Wain and I. L. Jenkins, Functions for the bombardment, J. Inorg. Nucl. Chem., 1970, 32(1970), 1419–1425 Search PubMed.
- H. F. Valdovinos, R. Hernandez, S. Graves, P. A. Ellison, T. E. Barnhart and C. P. Theuer, et al., Cyclotron production and radiochemical separation of 55Co and 58mCo from 54Fe, 58Ni and 57Fe targets, Appl. Radiat. Isot., 2017, 130, 90–101 CrossRef CAS PubMed.
- H. F. Valdovinos, S. Graves, T. Barnhart and R. J. Nickles, 55Co separation from proton irradiated metallic nickel, AIP Conf. Proc., 2014, 1626(1), 217–220 CrossRef.
- P. Goethals, A. Volkaert, C. Vandewielle, R. Dierckx and N. Lameire, 55Co-EDTA for renal imaging using positron emission tomography (PET): A feasibility study, Nucl. Med. Biol., 2000, 27(1), 77–81 CrossRef CAS PubMed.
- M. Daube and R. Nickles, Development of myocardial perfusion tracers for positron emission tomography, Int. J. Nucl. Med. Biol., 1985, 12(4), 303–314 CrossRef CAS.
- I. F. Chaple and S. E. Lapi, Production and Use of the First-Row Transition Metal PET Radionuclides 43,44Sc, 52Mn, and 45Ti, J. Nucl. Med., 2018, 59(11), 1655–1659 CrossRef CAS PubMed.
- C. M. Lewis, S. A. Graves, R. Hernandez, H. F. Valdovinos, T. E. Barnhart and W. Cai, et al., 52Mn production for PET/MRI tracking of human stem cells expressing divalent metal transporter 1 (DMT1), Theranostics, 2015, 5(3), 227–239 CrossRef CAS PubMed.
- S. A. Graves, R. Hernandez, J. Fonslet, C. G. England, H. F. Valdovinos and P. A. Ellison, et al., Novel Preparation Methods of 52Mn for ImmunoPET Imaging, Physiol. Behav., 2015, 26(10), 2118–2124 CAS.
- A. L. Wooten, B. C. Lewis, R. Laforest, S. V. Smith and S. E. Lapi. Cyclotron Production and PET/MR Imaging of 52Mn, in Proceedings of the 15th International Workshop on Targetry and Target Chemistry. 2014 Search PubMed.
- G. J. Topping, P. Schaffer, C. Hoehr, T. J. Ruth and V. Sossi, Manganese-52 positron emission tomography tracer characterization and initial results in phantoms and in vivo, Med. Phys., 2013, 40(4), 042502-2–042502-8 CrossRef PubMed.
- J. Fonslet, S. Tietze, A. I. Jensen, S. A. Graves and G. W. Severin, Optimized procedures for manganese-52: Production, separation and radiolabeling, Appl. Radiat. Isot., 2017, 121, 38–43 CrossRef CAS PubMed.
- A. I. Jensen, G. W. Severin, A. E. Hansen, F. P. Fliedner, R. Eliasen and L. Parhamifar, et al., Remote-loading of liposomes with manganese-52 and in vivo evaluation of the stabilities of 52Mn-DOTA and 64Cu-DOTA using radiolabelled liposomes and PET imaging, J. Controlled Release, 2018, 269, 100–109 CrossRef CAS PubMed.
- S. Busse, J. Brockmann and F. Rösch, Radiochemical separation of no-carrier-added radioniobium from zirconium targets for application of 90Nb-labelled compounds, Radiochim. Acta, 2002, 90(7), 411–416 CrossRef CAS.
- V. Radchenko, S. Busse and F. Roesch, Desferrioxamine as an appropriate chelator for 90Nb: Comparison of its complexation properties for M-Df-Octreotide (M=Nb, Fe, Ga, Zr), Nucl. Med. Biol., 2014, 41(9), 721–727 CrossRef CAS PubMed.
- V. Radchenko, D. V. Filosofov, O. K. Bochko, N. A. Lebedev, A. V. Rakhimov and H. Hauser, et al., Separation of 90Nb from zirconium target for application in immuno-pet, Radiochim. Acta, 2014, 102(5), 433–442 CrossRef CAS.
- V. Radchenko, H. Hauser, M. Eisenhut, D. J. Vugts, G. A. Van Dongen and F. Roesch, 90Nb - A potential PET nuclide: Production and labeling of monoclonal antibodies, Radiochim. Acta, 2012, 100(11), 857–864 CrossRef CAS.
- V. Radchenko, D. V. Filosofov, J. Dadakhanov, D. V. Karaivanov, A. Marinova and A. Baimukhanova, et al., Direct flow separation strategy, to isolate no-carrier-added 90Nb from irradiated Mo or Zr targets, Radiochim. Acta, 2016, 104(9), 625–634 CrossRef CAS.
- A. De La Fuente, V. Radchenko, T. Tsotakos, C. Tsoukalas, M. Paravatou-Petsotas and A. Harris, et al., Conjugation, labelling and in vitro/in vivo assessment of an anti-VEGF monoclonal antibody labelled with niobium isotopes, J. Radioanal. Nucl. Chem., 2018, 318(3), 1991–1997 CrossRef CAS.
- S. Busse, F. Rösch and S. M. Qaim, Cross section data for the production of the positron emitting niobium isotope 90Nb via the 90Zr(p,n)-reaction, Radiochim. Acta, 2002, 90(1), 1–5 CrossRef CAS.
- E. Koumarianou, N. Loktionova, M. Fellner, F. Roesch, O. Thews and D. Pawlak, et al., 44Sc-DOTA-BN [2-14] NH2 in comparison to 68Ga-DOTA-BN [2-14] NH2 in pre-clinical investigation. Is 44Sc a potential radionuclide for PET?, Appl. Radiat. Isot., 2012, 70(12), 2669–2676 CrossRef CAS PubMed.
- H. Honarvar, C. Müller, S. Cohrs, S. Haller, K. Westerlund and A. Karlström, et al., Evaluation of the first 44Sc-labeled Affibody molecule for imaging of HER2-expressing tumors, Nucl. Med. Biol., 2017, 45, 15–21 CrossRef CAS PubMed.
- A. Khawar, E. Eppard, J. Sinnes, F. Roesch, H. Ahmadzadehfar and S. Kürpig, et al., [44Sc] Sc-PSMA-617 biodistribution and dosimetry in patients with metastatic castration-resistant prostate carcinoma, Clin. Nucl. Med., 2018, 43(5), 323–330 CrossRef PubMed.
- G. Orteca, J. Sinnes, S. Rubagotti, M. Iori, P. Capponi and M. Piel, et al., Gallium-68 and scandium-44 labelled radiotracers based on curcumin structure linked to bifunctional chelators: Synthesis and characterization of potential PET radiotracers, J. Inorg. Biochem., 2020, 204, 110954 CrossRef CAS PubMed.
- R. Hernandez, H. F. Valdovinos, Y. Yang, R. Chakravarty, H. Hong and T. E. Barnhart, et al., 44Sc: An attractive isotope for peptide-based PET imaging, Mol. Pharm., 2014, 11(8), 2954–2961 CrossRef CAS PubMed.
- E. Eppard, A. de la Fuente, M. Benešová, A. Khawar, R. A. Bundschuh and F. C. Gärtner, et al., Clinical translation and first in-human use of [44Sc]Sc-PSMA-617 for pet imaging of metastasized castrate-resistant prostate cancer, Theranostics, 2017, 7(18), 4359 CrossRef CAS PubMed.
- K. Siwowska, P. Guzik, K. Domnanich, J. Monné Rodríguez, P. Bernhardt and B. Ponsard, et al., Therapeutic potential of 47Sc in comparison to 177Lu and 90Y: Preclinical investigations, Pharmaceutics, 2019, 11(8), 424 CrossRef CAS PubMed.
- V. Radchenko, C. A. Meyer, J. W. Engle, C. M. Naranjo, G. A. Unc and T. Mastren, et al., Separation of 44Ti from proton irradiated scandium by using solid-phase extraction chromatography and design of 44Ti/44Sc generator system, J. Chromatogr. A, 2016, 1477, 39–46 CrossRef CAS PubMed.
- S. Krajewski, I. Cydzik, K. Abbas, A. Bulgheroni, F. Simonelli and U. Holzwarth, et al., Cyclotron production of 44Sc for clinical application, Radiochim. Acta, 2013, 101(5), 333–338 CrossRef CAS.
- S. Banerjee, M. Pillai and N. Ramamoorthy, Evolution of Tc-99m in diagnostic radiopharmaceuticals, Semin. Nucl. Med., 2001, 31(4), 260–277 CrossRef CAS PubMed.
- H. Bigott, D. McCarthy, F. Wüst, J. Dahlheimer, D. R. Piwnica-Worms and M. Welch, Production, processing and uses of 94mTc. Journal of Labelled Compounds and Radiopharmaceuticals, J. Labelled Compd. Radiopharm., 2001, 44(S1), S119–S121 CrossRef.
- C. Stone, B. Christian, R. Nickles and S. Perlman, Technetium 94m-labeled methoxyisobutyl isonitrile: dosimetry and resting cardiac imaging with positron emission tomography, J. Nucl. Cardiol., 1994, 1(5), 425–433 CrossRef CAS PubMed.
- H. Bigott, E. Parent, L. Luyt, J. Katzenellenbogen and M. Welch, Design and synthesis of functionalized cyclopentadienyl tricarbonylmetal complexes for technetium-94m PET imaging of estrogen receptors, Bioconjugate Chem., 2005, 16(2), 255–264 CrossRef CAS PubMed.
- C. D. Illan and B. W. Wieland, Evaluation of a recoil-escape fiber target using 94Mo (p,n) 94mTc to produce 94TcO4-precursor for radiolabeled compounds useful in positron emission tomography, Duke University Medical Center, Durham NC, 2001 Search PubMed.
- S. M. Qaim, Production of high purity 94mTc for positron emission tomography studies, Nucl. Med. Biol., 2000, 27(4), 323–328 CrossRef CAS PubMed.
- B. T. Christian, R. J. Nickles, C. K. Stone, T. L. Mulnix and J. Clark, Improving the radionuclidic purity of 94mTc for PET imaging, Appl. Radiat. Isot., 1995, 46(2), 69–73 CrossRef CAS.
- F. Rösch and S. M. Qaim, Nuclear Data Relevant to the Production of the Positron Emitting Technetium Isotope 94mTc via the 94Mo(p,n)-reaction, Radiochim. Acta, 1993, 62(3), 115–122 CrossRef.
- X. Hou, J. Tanguay, K. Buckley, P. Schaffer, F. Bénard and T. J. Ruth, et al., Molybdenum target specifications for cyclotron production of 99mTc based on patient dose estimates, Phys. Med. Biol., 2015, 61(2), 542–553 CrossRef PubMed.
- M. Sadeghi, M. Enferadi and H. Nadi, 45Ti, a candidate for positron emission tomography: Study of the cyclotron production, Radiochemistry, 2011, 53(4), 411–414 CrossRef CAS.
- A. L. Vavere, R. Laforest and M. J. Welch, Production, processing and small animal PET imaging of titanium-45, Nucl. Med. Biol., 2005, 32(2), 117–122 CrossRef CAS PubMed.
- A. Vavere and M. Welch, Preparation, biodistribution, and small animal PET of 45Ti-transferrin, J. Nucl. Med., 2005, 46(4), 683–690 CAS.
- F. Chen, H. F. Valdovinos, R. Hernandez, S. Goel, T. E. Barnhart and W. Cai, Intrinsic radiolabeling of Titanium-45 using mesoporous silica nanoparticles, Acta Pharmacol. Sin., 2017, 38(6), 907–913 CrossRef CAS PubMed.
- S. Kuhn, I. Spahn, B. Scholten and H. H. Coenen, Positron and γ-ray intensities in the decay of 45Ti, Radiochim. Acta, 2015, 103(6), 403–409 CrossRef CAS.
- F. Rösch, S. Qaim and G. Stöcklin, Production of the positron emitting radioisotope 86Y for nuclear medical application, Appl. Radiat. Isot., 1993, 44(4), 677–681 CrossRef.
- T. K. Nayak, K. Garmestani, K. E. Baidoo, D. E. Milenic and M. W. Brechbiel, PET imaging of tumor angiogenesis in mice with VEGF-A-targeted 86Y-CHX-A′′′-DTPA-bevacizumab, Int. J. Cancer, 2011, 128(4), 920–926 CrossRef CAS PubMed.
- T. Wadas, E. Wong, G. Weisman and C. Anderson, Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease, Chem. Rev., 2010, 110(5), 2858–2902 CrossRef CAS PubMed.
- T. Clifford, C. Boswell, G. Biddlecombe, J. Lewis and M. Brechbiel, Validation of a Novel CHX-A ‘‘Derivative Suitable for Peptide Conjugation: Small Animal PET/CT Imaging Using Yttrium-86-CHX-A ‘‘-Octreotide, J. Med. Chem., 2006, 49(14), 4297–4304 CrossRef CAS PubMed.
- L. Wei, X. Zhang, F. Gallazzi, Y. Miao, X. Jin and M. Brechbiel, et al., Melanoma imaging using 111In-, 86Y-and 68Ga-labeled CHX-A ′′-Re (Arg11) CCMSH, Nucl. Med. Biol., 2009, 36(4), 345–354 CrossRef CAS PubMed.
- H. Chong, K. Garmestani, D. Ma, D. Milenic, T. Overstreet and M. Brechbiel, Synthesis and Biological Evaluation of Novel Macrocyclic Ligands with Pendent Donor Groups as Potential Yttrium Chelators for Radioimmunotherapy with Improved Complex Formation Kinetics, J. Med. Chem., 2002, 45(46), 3458–3464 CrossRef CAS PubMed.
- C. Kang, X. Sun, F. Jia, H. Song, Y. Chen and M. Lewis, et al., Synthesis and preclinical evaluation of bifunctional ligands for improved chelation chemistry of 90Y and 177Lu for targeted radioimmunotherapy, Bioconjugate Chem., 2012, 23(9), 1775–1782 CrossRef CAS PubMed.
- L. Koi, R. Bergmann, K. Brüchner, J. Pietzsch, H. Pietzsch and M. Krause, et al., Radiolabeled anti-EGFR-antibody improves local tumor control after external beam radiotherapy and offers theragnostic potential, Radiother. Oncol., 2014, 110(2), 362–369 CrossRef CAS PubMed.
- T. Nayak, K. Garmestani, K. Baidoo, D. Milenic and M. Brechbiel, Preparation, biological evaluation, and pharmacokinetics of the human anti-HER1 monoclonal antibody panitumumab labeled with 86Y for quantitative PET of carcinoma, J. Nucl. Med., 2010, 51(6), 942–950 CrossRef CAS PubMed.
- S. Walrand, R. Barone, S. Pauwels and F. Jamar, Experimental facts supporting a red marrow uptake due to radiometal transchelation in 90Y-DOTATOC therapy and relationship to the decrease of platelet counts, Eur. J. Nucl. Med. Mol. Imag., 2011, 38(7), 1270–1280 CrossRef CAS PubMed.
- M. Le Fur, N. Rotile, C. Correcher, V. Clavijo Jordan, A. Ross and C. Catana, et al., Yttrium-86 Is a Positron Emitting Surrogate of Gadolinium for Noninvasive Quantification of Whole-Body Distribution of Gadolinium-Based Contrast Agents, Angew. Chem. Int. Ed., 2020, 59(4), 1474–1478 CrossRef CAS PubMed.
- G. Reischl, F. Rösch and H. J. Machulla, Electrochemical separation and purification of yttrium-86, Radiochim. Acta, 2002, 90(4), 225–228 CrossRef CAS.
- J. Yoo, L. Tang, T. A. Perkins, D. J. Rowland, R. Laforest and J. S. Lewis, et al., Preparation of high specific activity 86Y using a small biomedical cyclotron, Nucl. Med. Biol., 2005, 32(8), 891–897 CrossRef CAS PubMed.
- M. A. Avila-Rodriguez, J. A. Nye and R. A. Nickles, Production and separation of non-carrier-added 86Y from enriched 86Sr targets, Appl. Radiat. Isot., 2008, 66(1), 9–13 CrossRef CAS PubMed.
- T. R. DeGrado, M. K. Pandey, J. F. Byrne, H. P. Engelbrecht, H. Jiang and A. B. Packard, et al., Preparation and preliminary evaluation of 63Zn-zinc citrate as a novel PET imaging biomarker for zinc, J. Nucl. Med., 2014, 55(8), 1348–1354 CrossRef CAS PubMed.
- M. Rostampour, M. Aboudzadeh, M. Sadeghi and S. Hamidi, Theoretical assessment of production routes for 63Zn by cyclotron, J. Radioanal. Nucl. Chem., 2016, 309(2), 677–684 CAS.
- T. R. DeGrado, B. J. Kemp, M. K. Pandey, H. Jiang, T. M. Gunderson and L. R. Linscheid, et al., First PET imaging studies with 63Zn-zinc citrate in healthy human participants and patients with Alzheimer disease, Mol. Imag., 2016, 15, 1–10 CrossRef CAS PubMed.
- M. Rostampour, M. Sadeghi, M. Aboudzadeh, K. Yousefi and S. Hamidi, Investigation of the thermal performance of natCu target for 63Zn production, Appl. Radiat. Isot., 2018, 141, 1–4 CrossRef CAS PubMed.
- T. DeGrado, B. Kemp, M. Pandey, H. Jiang, T. Gunderson and L. Linscheid, et al., First PET imaging studies with 63Zn-zinc citrate in healthy human participants and patients with Alzheimer disease, Mol. Imag., 2016, 15, 1536012116673793 CrossRef PubMed.
- M. Rostampour, M. Sadeghi, M. Aboudzadeh, S. Hamidi, N. Soltani and F. B. Novin, et al., Experimental study and simulation of 63Zn production via proton induce reaction, Appl. Radiat. Isot., 2018, 136, 32–36 CrossRef CAS PubMed.
- S. Heskamp, R. Raavé, O. Boerman, M. Rijpkema, V. Goncalves and F. Denat, 89Zr-immuno-positron emission tomography in oncology: state-of-the-art 89Zr radiochemistry, Bioconjugate Chem., 2017, 28(9), 2211–2223 CrossRef CAS PubMed.
- T. H. Oude Munnink, M. E. A. Arjaans, H. Timmer-Bosscha, C. P. Schro
![[d with combining umlaut]](https://www.rsc.org/images/entities/char_0064_0308.gif) er, J. W. Hesselink and S. R. Vedelaar, et al., PET with the 89Zr-labeled transforming growth factor-β antibody fresolimumab in tumor models, J. Nucl. Med., 2011, 52(12), 2001–2008 CrossRef CAS PubMed.
er, J. W. Hesselink and S. R. Vedelaar, et al., PET with the 89Zr-labeled transforming growth factor-β antibody fresolimumab in tumor models, J. Nucl. Med., 2011, 52(12), 2001–2008 CrossRef CAS PubMed. - W. B. Nagengast, E. G. De Vries, G. A. Hospers, N. H. Mulder, J. R. De Jong and H. Hollema, et al., In vivo VEGF imaging with radiolabeled bevacizumab in a human ovarian tumor xenograft, J. Nucl. Med., 2007, 48(8), 1313–1319 CrossRef CAS PubMed.
- J. Yoon, B. Park, E. Ryu, Y. An and S. Lee, Current Perspectives on 89Zr-PET Imaging, Int. J. Mol. Sci., 2020, 21(12), 4309 CrossRef CAS PubMed.
- N. Bhatt, D. Pandya and T. Wadas, Recent advances in zirconium-89 chelator development. Molecules, Molecules, 2018, 23(3), 638 CrossRef PubMed.
- J. Holland, Predicting the thermodynamic stability of zirconium radiotracers, Inorg. Chem., 2020, 59(3), 2070–2082 CrossRef CAS PubMed.
- A. M. Dabkowski, S. J. Paisey, M. Talboys and C. Marshall, Optimization of cyclotron production for radiometal of zirconium 89, Acta Phys. Pol., A, 2015, 127(5), 1479–1482 CrossRef.
- A. Kasbollah, P. Eu, S. Cowell and P. Deb, Review on production of 89Zr in a medical cyclotron for PET radiopharmaceuticals, J. Nucl. Med. Technol., 2013, 41(1), 35–41 CrossRef PubMed.
- M. J. O'Hara, N. J. Murray, J. C. Carter and S. S. Morrison, Optimized anion exchange column isolation of zirconium-89 (89Zr) from yttrium cyclotron target: Method development and implementation on an automated fluidic platform, J. Chromatogr. A, 2018, 1545, 48–58 CrossRef PubMed.
- G. Severin, J. Engle, T. Barnhart and R. Nickles, 89Zr Radiochemistry for Positron Emission Tomography, Med. Chem., 2011, 7(5), 389–394 CrossRef CAS PubMed.
- W. E. Meijs, J. D. M. Herscheid, H. J. Haisma, R. Wijbrandts, F. van Langevelde and P. J. Van Leuffen, et al., Production of highly pure no-carrier added 89Zr for the labelling of antibodies with a positron emitter, Appl. Radiat. Isot., 1994, 45(12), 1143–1147 CrossRef CAS.
- J. W. Engle, H. Hong, Y. Zhang, H. F. Valdovinos, D. V. Myklejord and T. E. Barnhart, et al., Positron emission tomography imaging of tumor angiogenesis with a 66Ga-labeled monoclonal antibody, Mol. Pharm., 2012, 9(5), 1441–1448 CrossRef CAS PubMed.
- Ö. Ugur, P. J. Kothari, R. D. Finn, P. Zanzonico, S. Ruan and I. Guenther, et al., Ga-66 labeled somatostatin analogue DOTA-DPhe1-Tyr3-octreotide as a potential agent for positron emission tomography imaging and receptor mediated internal radiotherapy of somatostatin receptor positive tumors, Nucl. Med. Biol., 2002, 29(2), 147–157 CrossRef.
- M. Khan, S. Khan, S. El-Refaie, Z. Win, D. Rubello and A. Al-Nahhas, Clinical indications for gallium-68 positron emission tomography imaging, Eur. J. Surg. Oncol., 2009, 35(6), 561–567 CrossRef CAS PubMed.
- M. Perera, N. Papa, M. Roberts, M. Williams, C. Udovicich and I. Vela, et al., Gallium-68 prostate-specific membrane antigen positron emission tomography in advanced prostate cancer—updated diagnostic utility, sensitivity, specificity, and distribution of prostate-specific membrane antigen-avid lesions: a systematic review and meta-, Eur. Urol., 2020, 77(4), 403–417 CrossRef PubMed.
- C. Kratochwil, P. Flechsig, T. Lindner, L. Abderrahim, A. Altmann and W. Mier, et al., 68Ga-FAPI PET/CT: tracer uptake in 28 different kinds of cancer, J. Nucl. Med., 2019, 60(6), 801–805 CrossRef CAS PubMed.
- F. Giesel, C. Kratochwil, T. Lindner, M. Marschalek, A. Loktev and W. Lehnert, et al., 68Ga-FAPI PET/CT: biodistribution and preliminary dosimetry estimate of 2 DOTA-containing FAP-targeting agents in patients with various cancers, J. Nucl. Med., 2019, 60(3), 386–392 CrossRef CAS PubMed.
- U. Hennrich and M. Benešová, [68Ga] Ga-DOTA-TOC: The First FDA-Approved 68Ga-Radiopharmaceutical for PET Imaging, Pharmaceuticals, 2020, 13(3), 38 CrossRef CAS PubMed.
- M. R. Lewis, D. E. Reichert, R. Laforest, W. H. Margenau, R. E. Shefer and R. E. Klinkowstein, et al., Production and purification of gallium-66 for preparation of tumor-targeting radiopharmaceuticals, Nucl. Med. Biol., 2002, 29(6), 701–706 CrossRef CAS PubMed.
- M. E. Rodnick, C. Sollert, D. Stark, M. Clark, A. Katsifis and B. G. Hockley, et al., Cyclotron-Based Production of 68Ga, [68Ga]GaCl3, and [68Ga]Ga-PSMA-11 from a Liquid Target, EJNMMI Radiopharm. Chem, 2020, 5(1), 1–18 CrossRef PubMed.
- B. J. B. Nelson, J. Wilson, S. Richter, M. J. M. Duke, M. Wuest and F. Wuest, Taking cyclotron 68Ga production to the next level: Expeditious solid target production of 68Ga for preparation of radiotracers, Nucl. Med. Biol., 2020, 80, 24–31 CrossRef PubMed.
- J. W. Engle, V. Lopez-Rodriguez, R. E. Gaspar-Carcamo, H. F. Valdovinos, M. Valle-Gonzalez and F. Trejo-Ballado, et al., Very high specific activity 66/68Ga from zinc targets for PET, Appl. Radiat. Isot., 2012, 70(8), 1792–1796 CrossRef CAS PubMed.
- S. Zeisler, A. Limoges, J. Kumlin, J. Siikanen and C. Hoehr, Fused Zinc Target for the Production of Gallium Radioisotopes, Instruments, 2019, 3(1), 10 CrossRef CAS.
- H. Thisgaard, J. Kumlin, N. Langkjær, J. Chua, B. Hook and M. Jensen, et al., Multi-curie production of gallium-68 on a biomedical cyclotron and automated radiolabelling of PSMA-11 and DOTATATE, EJNMMI Radiopharm. Chem, 2021, 6(1), 1–11 CrossRef PubMed.
- J. Kumlin, J. H. Dam, C. J. Chua, S. Borjian, A. Kassaian and B. Hook, et al., Multi-Curie Production of Gallium-68 on a Biomedical Cyclotron, Eur. J. Nucl. Med. Mol. Imag., 2019, 46(suppl. 1, 1, SI), S39 Search PubMed.
- M. Enferadi, M. Sadeghi and H. Nadi, 72As, a powerful positron emitter for immunoimaging and receptor mapping: Study of the cyclotron production, Radiochemistry, 2011, 53(4), 407–410 CrossRef CAS.
- P. A. Ellison, F. Chen, T. E. Barnhart, R. J. Nickles, W. Cai and O. T. Dejesus, Production and isolation of 72As from proton irradiation of enriched 72GeO2 for the development of targeted PET/MRI agents, in: Proceedings of the 15th International Workshop on Targetry and Target Chemistry. 2015 Search PubMed.
- M. Jennewein, M. Lewis, D. Zhao, E. Tsyganov, N. Slavine and J. He, et al., Vascular imaging of solid tumors in rats with a radioactive arsenic-labeled antibody that binds exposed phosphatidylserine, Clin. Cancer Res., 2008, 14(5), 1377–1385 CrossRef CAS PubMed.
- P. Ellison, F. Chen, S. Goel, T. Barnhart, R. Nickles and O. DeJesus, et al., Intrinsic and stable conjugation of thiolated mesoporous silica nanoparticles with radioarsenic, ACS Appl. Mater. Interfaces, 2017, 9(8), 6772–6781 CrossRef CAS PubMed.
- P. A. Ellison, T. E. Barnhart, F. Chen, H. Hong, Y. Zhang and C. P. Theuer, et al., High Yield Production and Radiochemical Isolation of Isotopically Pure Arsenic-72 and Novel Radioarsenic Labeling Strategies for the Development of Theranostic Radiopharmaceuticals, Bioconjugate Chem., 2016, 27(1), 179–188 CrossRef CAS PubMed.
- A. DeGraffenreid, Y. Feng, D. Wycoff, R. Morrow, M. Phipps and C. Cutler, et al., Dithiol aryl arsenic compounds as potential diagnostic and therapeutic radiopharmaceuticals, Inorg. Chem., 2016, 55(16), 8091–8098 CrossRef CAS PubMed.
- W. Ali. Evaluation of Nuclear Reaction Cross Sections Relevant to the Production of Emerging Diagnostic Radioisotopes Fe-52 and As-72. Government College University, Lahore, 2012 Search PubMed.
- I. Spahn, G. F. Steyn, F. M. Nortier, H. H. Coenen and S. M. Qaim, Excitation functions of natGe(p,xn)71,72,73,74As reactions up to 100 MeV with a focus on the production of 72As for medical and 73As for environmental studies, Appl. Radiat. Isot., 2007, 65(9), 1057–1064 CrossRef CAS PubMed.
- Y. Li, S. F. Shen, J. H. Gu, S. H. Shi and J. Y. Liu, A study on the decay of 72As, Chin. Phys., 2005, 14, 487–493 CrossRef CAS.
- R. Guin, S. K. Das and S. K. Saha, Separation of carrier-free arsenic from germanium, J. Radioanal. Nucl. Chem., 1998, 227(1–2), 181–183 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |