
Symmetric cytotoxic trimeric and dimeric indole alkaloids isolated from Bousigonia angustifolia†
Bao-Bao
Shi
ab,
Jing-Song
Lu
c,
Jing
Wu
ab,
Mei-Fen
Bao
ab and
Xiang-Hai
Cai
 *a
*a
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China. E-mail: xhcai@mail.kib.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cDongzhimen Hospital, Beijing University of Chinese Medicine, Beijing 100700, China
First published on 16th March 2021
Abstract
A symmetric monoterpenoid indole alkaloid trimer, bousangustine A, is reported for the first time, as well as the dimeric alkaloids bousangustines B and C; these were isolated from the trunks of Bousigonia angustifolia. The structures, which feature a symmetric 6/9/5/6 ring system, were elucidated using comprehensive spectroscopic analysis. Their absolute configurations were determined via X-ray crystal diffraction, as well as computational chemistry. They can be constructed through Friedel–Crafts and free radical reactions, respectively. These compounds exhibited significant cytotoxicity against tumor cells.
Introduction
Since the development of bisindole vinblastine (VBL) and vincristine (VCR) into drugs, dimeric monoterpenoid indole alkaloids (MIAs) have attracted extensive attention. Owing to the inactive units of velbenamine and vindoline in VBL and VCR, the polymerization of indoles appears to be a crucial approach to obtain structural complexity, as well as bioactive diversity.1 There are two forms of polymerization of MIAs (Fig. S1B in ESI†). One is through a direct connection, such as asymmetric voacandimine A,2 coryzeylamine,3 deformylcoryzeylamine,3 suadimins A–C,4 and symmetric geleganidine B.5 The other is through an indirect connection, such as symmetric pleiokomenines A,6 meloyines III,7 melofusine I,8 melomorsine I,8 and voacinol2 using methylene, as well as geleganidine C through a carbonyl.5 Additionally, trimeric MIAs are unusual, for example, psychotripine from Psychotria pilifera,9 ervadivamines A/B from Ervatamia divaricata,10 and 3-hydroxy-14′-(3α′′-tabersonyl)voafrine B and 14-(3α′′′-tabersony1)-voafrine B in Catharanthus roseus,11 bousigonine B in Bousigonia mekongensis have been previously reported.12 These trimers are assembled by a single bond in sequence and are asymmetric structures. As part of our recent studies on dimeric7,8,13,14 and trimeric alkaloids,15 we have phytochemically researched plants of the genus Bousigonia, which contain bisindoles and dimers.16 This paper describes the isolation, structural determination, and potential bioactivity of three newly isolated MIAs (1–3) together with three known MIAs (4–6) (Fig. 1) obtained from the trunks of B. angustifolia Pierre.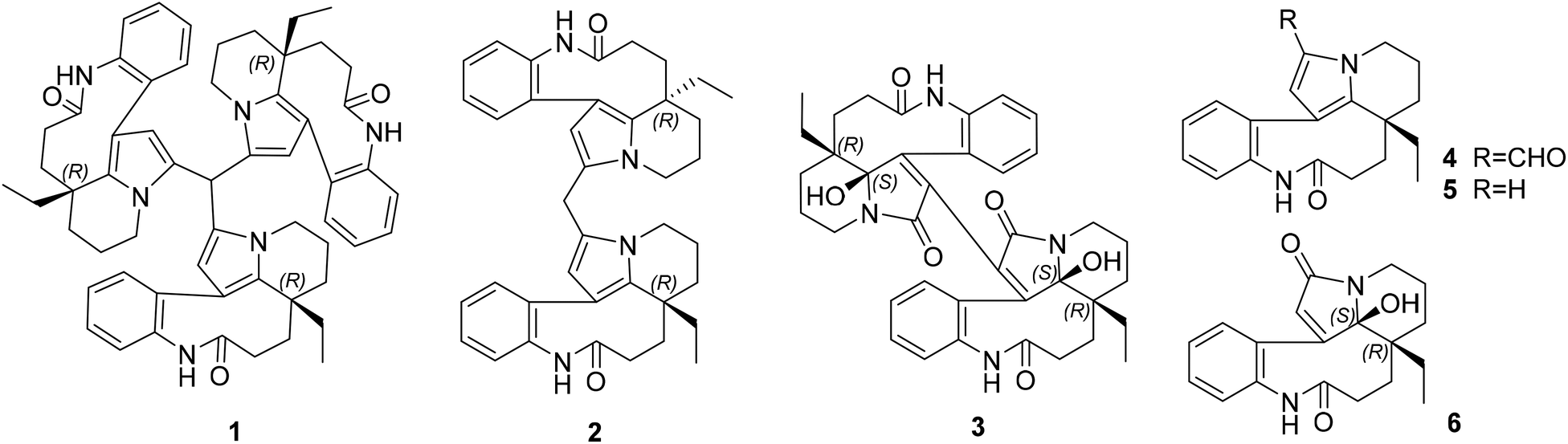 | ||
| Fig. 1 Chemical structures of bousangustines A–C (1–3) and the known compounds 4–6. | ||
Results and discussion
Compounds 1–3 were obtained as an amorphous powder and exhibited a positive reaction to Dragendorff's reagent. The 1H NMR spectrum of 1 revealed signals for four coupled indole protons (δH 7.10, 7.26, 7.18, 7.26), one olefinic proton (δH 5.06), one methyl (δH 0.60), one methine (δH 5.20, s), and six methylene protons (δH 1.39, 1.40, 1.80, 1.81, 2.07, 2.33, 2.34, each 1H, m; δH 1.38, 1H, d; δH 3.29 and 3.87, each 1H, dd; δH 1.67, 1H, td; and δH 1.13, 1H, dq) (Table 1). The 20 carbon signals in the 13C NMR and DEPT spectra of 1 could be classified as seven quaternary carbons (including one carbonyl at δC 179.9 and five olefinic ones at δC 118.1, 131.1, 131.3, 139.5 and 141.6), six methines (including five olefinic ones at δC 111.1, 127.2, 127.9, 128.9 and 132.5), six methylenes (δC 20.3, 29.2, 31.1, 33.7, 37.9 and 44.4) and one methyl at δC 8.5 (Table 1). The molecular formula C58H64N6O3 was established using the 13C NMR and high-resolution electron ionization mass spectrometry (HREIMS) data (m/z 915.4937 [M + Na] +, calculated for C58H64N6O3Na, 915.4932), which indicated 30 indices of hydrogen deficiency. However, the above described NMR data for 1 showed 20 carbon and 22 proton signals, only accounting for around one-third of the signals compared with the molecular formula. Therefore, compound 1 has a highly symmetrical structure. The planar structure was further elucidated by 2D NMR experiments. Analysis of the 1H–1H correlation spectroscopy (COSY) and heteronuclear multiple quantum coherence (HMQC) spectra revealed the presence of three molecular fragments: fragment A (C3–C14–C15), fragment B (C16–C17), fragment C (C18–C19) and an indole ring (Fig. 2). The heteronuclear multiple bond correlation (HMBC) correlations from H-3 to C-5 (δC 131.1) and C-21, from H-6 to C-7 (δC 118.1), C-8 (δC 141.6) and C-21, and from H-9 to C-7 indicated the presence of a pyrrole ring. Further analysis of the NMR spectrum suggested that 1 was closely related to known compounds 4 and 5.17 A careful comparison of the NMR data for 1 and 4 (Fig. S26 and S27†) disclosed the presence of an aldehyde group in 4, and a methine (δC 35.1 and δH 5.20) appeared in 1. The HMBC correlations from H-15 to C-17 (δC 37.9), C-19 (δC 31.1) and C-21 (δC 131.3), from H-19 to C-15 (δC 33.7), C-17 and C-21, and from H-17 to C-15, C-19 and C-21 further supported this similarity between 1 and 4. The methine was connected with C-5 by the key HMBC correlations from its proton signal δH 5.20 to C-5 and C-6 (δC 111.1), and from H-6 (δH 5.06) to C-5, C-8, C-21 and its carbon signal (δC 35.1). Furthermore, taking the molecular formula into consideration, this methine should connect to another two of the same units. Thus, the complete planar structure of 1 was established.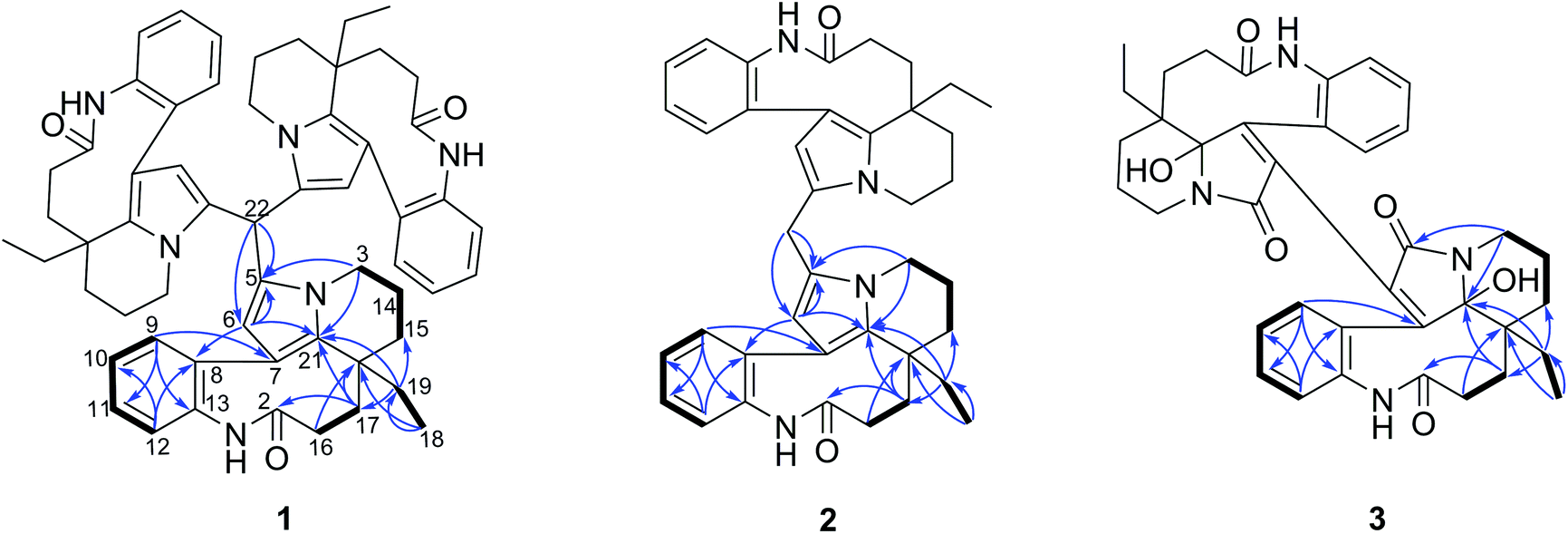 | ||
| Fig. 2 Key HMBC and COSY correlations for compounds 1–3. | ||
| No. | δ C (1)b | δ H (1)a | δ C (2)b | δ H (2)a | δ C (3)c | δ H (3)a |
|---|---|---|---|---|---|---|
| a Recorded at: 400 MHz. b Recorded at: 125 MHz. c Recorded at: 150 MHz. | ||||||
| 2/2′/2′′ | 179.9 s | 180.1 s | 177.4 s | |||
| 3/3′/3′′ | 44.4 t | 3.87 dd (12.4, 4.6) | 44.4 t | 3.99 dd (12.2, 4.6) | 37.4 t | 4.19 d (13.7) |
| 3.29 dd (12.4, 4.0) | 3.50 td (12.2, 4.9) | 3.02 m | ||||
| 5/5′/5′′ | 131.1 s | 129.2 s | 165.8 s | |||
| 6/6′/6′′ | 111.1 d | 5.06 s | 110.1 d | 5.38 s | 129.9 s | |
| 7/7′/7′′ | 118.1 s | 118.2 s | 154.5 s | |||
| 8/8′/8′′ | 141.6 s | 141.6 s | 133.6 s | |||
| 9/9′/9′′ | 132.5 d | 7.26 dd (7.7, 1.4) | 132.4 d | 7.36 dd (7.6, 1.7) | 130.3 d | 6.80 d (7.3) |
| 10/10′/10′′ | 127.9 d | 7.18 td (7.7, 1.5) | 128.1 d | 7.28 td (7.6, 1.5) | 128.8 d | 6.80 t (7.3) |
| 11/11′/11′′ | 128.9 d | 7.26 td (7.7, 1.4) | 128.9 d | 7.34 td (7.6, 1.7) | 130.4 d | 7.19 t (7.3) |
| 12/12′/12′′ | 127.2 d | 7.10 dd (7.7, 1.5) | 127.4 d | 7.18 dd (7.6, 1.5) | 127.1 d | 7.00 d (7.3) |
| 13/13′/13′′ | 139.5 s | 139.6 s | 135.3 s | |||
| 14/14′/14′′ | 20.3 t | 2.07 m | 20.3 t | 2.15 m | 21.1 t | 1.55 m |
| 1.81 m | 1.90 m | |||||
| 15/15′/15′′ | 33.7 t | 1.67 td (13.5, 3.1) | 33.7 t | 1.72 td (13.5, 3.1) | 34.5 t | 1.79 m |
| 1.39 m | 1.46 m | 1.67 d (14.1) | ||||
| 16/16′/16′′ | 29.2 t | 2.34 m | 29.2 t | 2.40 m | 28.2 t | 1.96 m |
| 1.80 m | 1.86 t (7.8) | |||||
| 17/17′/7′′ | 37.9 t | 2.33 m | 38.1 t | 2.40 m | 26.6 t | 1.79 m |
| 1.40 m | 1.46 m | 1.52 t (4.6) | ||||
| 18/18′/18′′ | 8.5 q | 0.60 t (7.4) | 8.5 q | 0.69 t (7.3) | 7.5 q | 0.48 t (7.7) |
| 19/19′/19′′ | 31.1 t | 1.13 dq (14.6, 7.4) | 31.2 t | 1.19 dq (14.6, 7.3) | 25.9 t | 1.04 dq (15.2, 7.7) |
| 1.38 dq (14.6, 7.4) | 1.46 dq (14.6, 7.3) | 1.45 dq (15.2, 7.7) | ||||
| 20/20′/20′′ | 40.3 s | 40.2 s | 45.5 s | |||
| 21/21′/21′′ | 131.3 s | 130.7 s | 93.8 s | |||
| 22 | 35.1 d | 5.20 s | 25.0 t | 3.74 s | ||
The molecular formula C39H44N4O2 with 20 degrees of unsaturation for alkaloid 2 was established by using the positive high resolution electrospray ionisation mass spectrometry (HRESIMS) ion at m/z 623.3355 ([M + Na]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +, calculated. 623.3356). The 13C NMR signals were only observed for 20 carbon atoms, indicating that 2 also has a symmetrical structure. The 1H and 13C NMR data of 2 were almost identical to those of compound 1 except for the methine group (CH-22) in 1, there is a methylene group (δC 25.0) in 2 instead. This presumption was supported by the HMBC correlations from H-22 (δH 3.74, 2H) to C-5 (δC 129.2) and C-6 (δC 110.1), and from H-6 to C-5, C-21 (δC 130.7) and C-22 (δC 25.0). However, 2 had the molecular formula C39H44N4O2 according to the results from the HRESIMS, C19H20N2O less than compound 1. This difference suggests that 2 is a symmetric dimer of 4 through a methylene bridge. Therefore, the planar structure of 2 was completely established.
+, calculated. 623.3356). The 13C NMR signals were only observed for 20 carbon atoms, indicating that 2 also has a symmetrical structure. The 1H and 13C NMR data of 2 were almost identical to those of compound 1 except for the methine group (CH-22) in 1, there is a methylene group (δC 25.0) in 2 instead. This presumption was supported by the HMBC correlations from H-22 (δH 3.74, 2H) to C-5 (δC 129.2) and C-6 (δC 110.1), and from H-6 to C-5, C-21 (δC 130.7) and C-22 (δC 25.0). However, 2 had the molecular formula C39H44N4O2 according to the results from the HRESIMS, C19H20N2O less than compound 1. This difference suggests that 2 is a symmetric dimer of 4 through a methylene bridge. Therefore, the planar structure of 2 was completely established.
The molecular formula C38H42N4O6 with 20 degrees of unsaturation of alkaloid 3 was established by the positive HRESIMS ion at m/z 673.2992 ([M + Na]+, calculated. 673.2997). The NMR data (Table 1) showed signals for 19 carbons and 21 protons, only accounting for half the number expected from the molecular formula. This indicated that 3 should be a symmetric dimer. The 1H NMR spectrum also revealed the same indole A ring (δH 6.80, 7.00, 6.80, 7.19), one methyl (δH 0.48, 3H), and six methylene protons. The 19 carbon signals shown in the 13C NMR and DEPT spectra of 3 could be classified as eight quaternary carbons (including two carbonyls at δC 165.8 and 177.4; four olefinic ones at δC 129.9, 133.6, 135.3 and 154.5, and one heteroatom-bearing at δC 93.8), four indole methines (δC 127.1, 128.8, 130.3 and 130.4), six methylenes (δC 21.1, 25.9, 26.6, 28.2, 34.5 and 37.4), and one methyl (δC 7.5 q). The above described data for 1 was similar to that obtained for the known leuconolam (6),18 except for the absence of the olefinic methine signal at δC 128.1 and the presence of a quaternary carbon signal at δC 129.9 in 1. For compound 3, the absence of a signal for H-6 (δH 5.79 in 6), together with the continuous correlations of H-9 (δH 6.80)/H-10 (δH 6.80)/H-11 (δH 7.19)/H-12 (δH 7.00) in the COSY spectrum (Fig. 2), indicated that both units must be directly connected through C-6/C-6′.
Unfortunately, attempts to prepare single crystals of 1–3 were not successful. However, a single crystal of alkaloid 4, and units of 1 and 2, were obtained. The X-ray diffraction analysis with Cu Kα radiation resulted in a Flack parameter of −0.01(6), giving the absolute configuration of alkaloid 4 (Fig. 3) as 20R. With regards to the biogenetic relationship between 1, 2, 4 and 5, the stereochemical structures of the four compounds were predicted to be identical. Therefore, the absolute configurations of 1 and 2 were determined to be 20R,20′R,20′′R and 20R,20′R, respectively. Furthermore, the calculated electronic circular dichromism (ECD) spectra were used to support this prediction. Although the Cotton effects of 1 and 2 were stronger and slightly shifted compared to those observed for 4 and 5 (Fig. 4a), the tendencies of the CD curves of the four compounds in the range of 200 to 350 nm were relatively consistent, supporting their identical configurations. In addition, the identity of the measured CD and calculated ECD spectrum of 1 and 2 (Fig. 4a) further confirmed this conclusion. Similar to 1 and 2, 4 and 5, 3 and 6 were predicted to have the same absolute configurations. A further search of the previously published literature revealed leuconolam as an authentic sample of a natural product, and its structure was unambiguously determined using X-ray analysis.18 Further comparison of the experimental chemical shifts and the calculated and experimental ECD spectra of 6 (Table S5 and Fig. S43†) revealed that 6 was leuconolam. Therefore, the stereoconfiguration of 3 was temporary deduced as 20R,21S,20′R,21′S. This assumption was confirmed by comparative analysis of the calculated and experimental ECD spectra. The identity of the measured circular dichromism (CD) and the calculated ECD spectra of 3 (Fig. 4b) supported this prediction. It should be mentioned that compound 3 possessed fast axial rotation rates in the order of seconds or faster and exhibited no axial chirality based on the correlation of the calculated energy barriers and rotation rates (ΔErot <20 kcal mol−1) (Fig. S42†).19
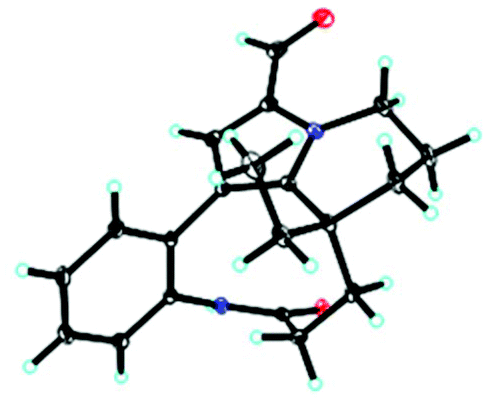 | ||
| Fig. 3 The X-ray structure of compound 4. | ||
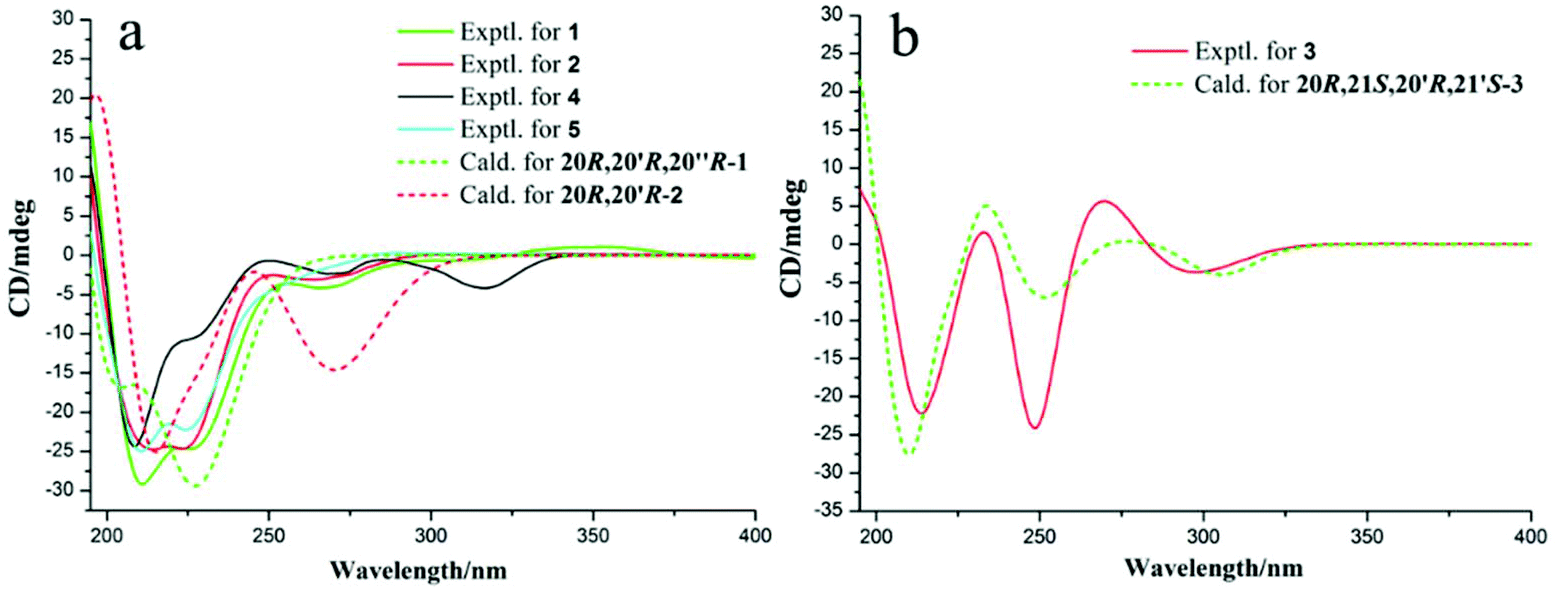 | ||
| Fig. 4 (a) Experimental ECD spectra and calculated ECD spectra for 1–2 and 4–5; and (b) experimental ECD spectra and calculated ECD spectra of 3 at the M06-2X/Def2SVP level in methanol. | ||
To determine whether the compounds occurred naturally in vivo, an UPLC-MS/MS analysis was performed. The dimeric alkaloids 2–3 could be detected as trace compounds in the total alkaloid fraction, which means that these alkaloids were probably natural products (Fig. 5).
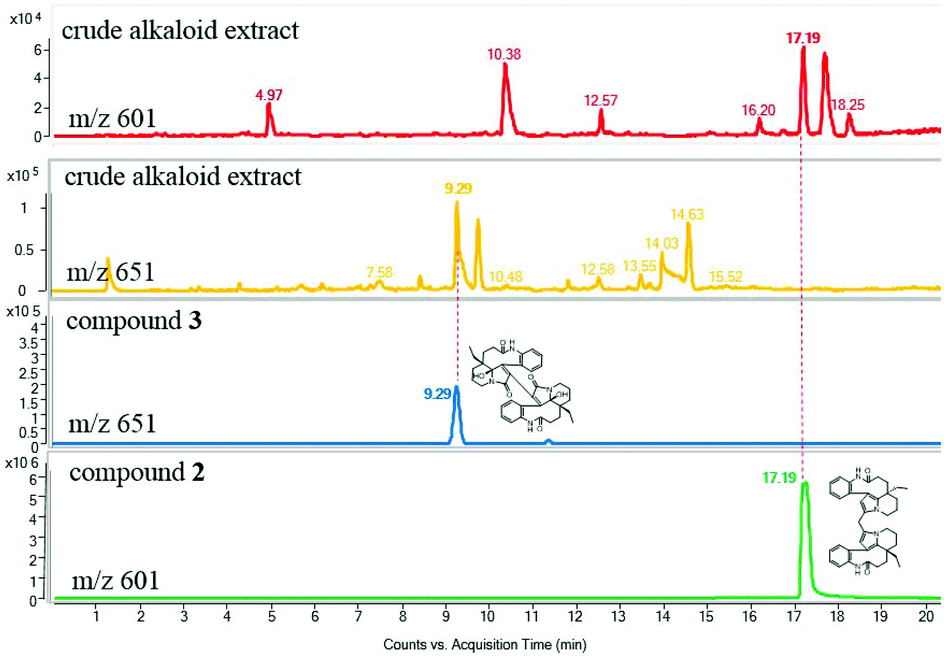 | ||
| Fig. 5 Positive-ion mode LC-MS analysis of the crude alkaloid extract and polymeric alkaloids (extracted ion chromatograms at m/z 601 and 651; conditions: YMC-pack ODS-A C18: 4.6 × 150 mm, flow rate: 1 mL min−1, a linear gradient of 10–100% CH3CN containing 0.01% NH4OH over 20 min, then eluting with 100% CH3CN for an additional 10 min). | ||
All of the isolates were evaluated in vitro against SMMC-7721, HeLa, HepG2, and A549 tumor cell lines. Compound 4 exhibited significant cytotoxicity against the SMMC-7721, HeLa and HepG2 cell lines with IC50 values of 0.52 ± 0.04, 0.60 ± 0.03 and 0.76 ± 0.05 μM, respectively. Compounds 1 and 2 showed moderate cytotoxic activities against SMMC-7721, HeLa, HepG2, and A549 cells, with IC50 values ranging from 4.31 to 16.93 μM. Compounds 3 and 6 showed no cytotoxicity at 40 μM (Table 2).
| No. | SMMC-7721 | HeLa | HepG2 | A549 |
|---|---|---|---|---|
| 1 | 10.99 ± 0.34 | 12.50 ± 1.11 | 16.3 ± 0.15 | 16.93 ± 0.33 |
| 2 | 4.31 ± 0.06 | 4.40 ± 0.30 | 7.02 ± 0.55 | 16.21 ± 0.01 |
| 3 | >40 | >40 | >40 | >40 |
| 4 | 0.52 ± 0.04 | 0.60 ± 0.03 | 0.76 ± 0.05 | >40 |
| 5 | 1.89 ± 0.22 | 1.76 ± 0.02 | 3.46 ± 0.22 | >40 |
| 6 | >40 | >40 | >40 | >40 |
| Vinorelbine | 3.02 ± 0.21 | 0.60 ± 0.03 | 0.76 ± 0.05 | 2.23 ± 0.06 |
Bousangustines A–C (1–3) are novel symmetrical MIAs trimer and dimers. Together with these, three known MIAs, rhazinal (4), rhazinilam (5), and leuconolam (6), were simultaneously isolated. Hence, we propose a plausible biosynthetic pathway for alkaloids 1–3 (Scheme 1). The pathway for 1–2 could start from 4 and 5 through a Friedel–Crafts reaction under acid conditions, generating the hydroxy intermediate I. I is then dehydrated to produce intermediate II. II is further coupled with 5, forming trimer 1 (Scheme 1, route A). On the other hand, II is reduced and gives dimer 2 (route B). Unlike the formation of 1 and 2, 6 is oxidized to generate the free radical intermediate III. The coupling of both units III gives 3 after isomerization (route C).
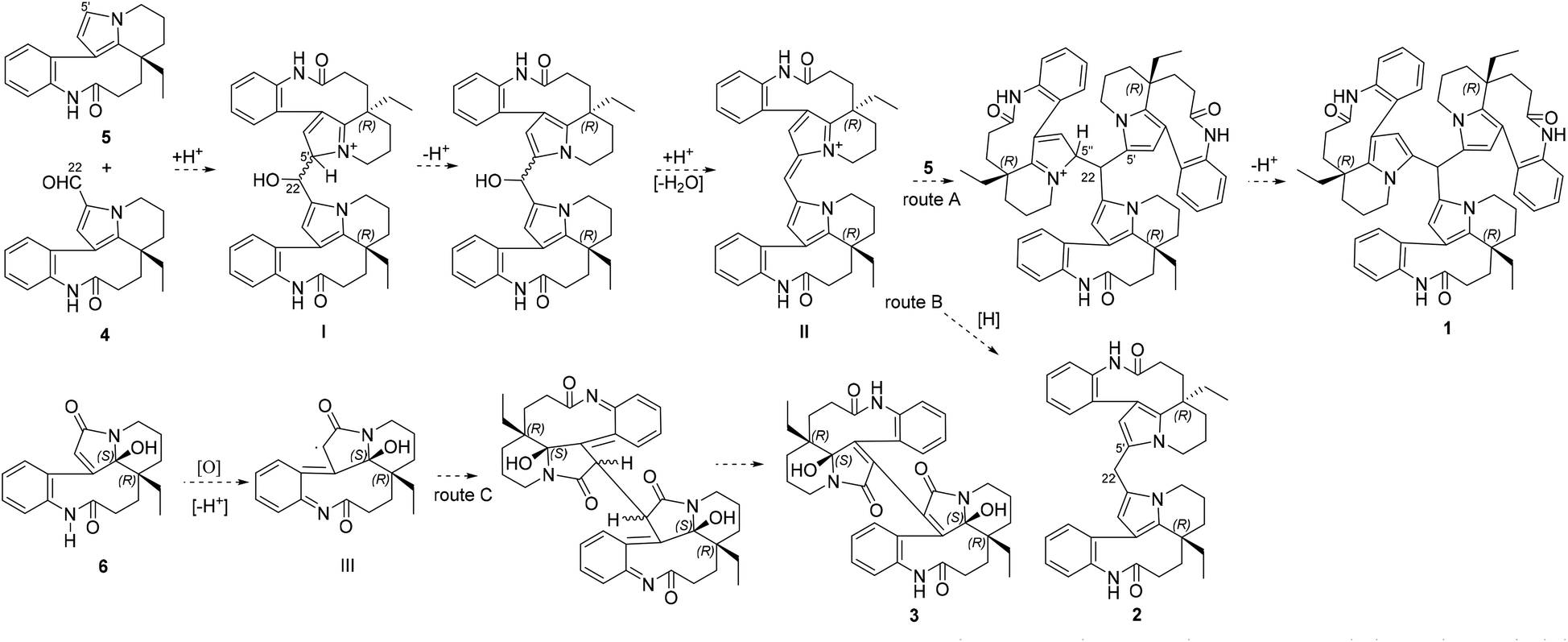 | ||
| Scheme 1 Proposed pathways to the dimeric and trimeric MIAs. | ||
Conclusions
In summary, three previously unreported bousangustines A–C were isolated from B. angustifolia. Bousangustine A is the first reported symmetric MIA trimer and bousangustines B–C are symmetric dimeric MIAs. Their absolute configurations were determined using X-ray crystal diffraction and ECD calculations. Bousangustines A and B may be constructed through a Friedel–Crafts reaction, while bousangustine C could be formed through free radical coupling from simultaneously isolated monomers. The liquid chromatography mass spectrometry (LC-MS) detection results of these novel isolates disclosed their natural properties. Furthermore, these macrocyclic compounds exhibited excellent cytotoxic activities. These findings enrich our knowledge of the chemical diversity of MIAs and will attract the interest of both chemical synthetic and pharmaceutical chemists.Experimental section
General experimental procedures
The optical rotations were measured using a Jasco P-1020 digital polarimeter (Jasco International Co., Tokyo, Japan). UV spectra were recorded on a Shimadzu 2401PC spectrophotometer (Shimadzu Corp., Kyoto, Japan). CD spectra were obtained on a Chirascan V100 circular dichroism spectrometer (Applied Photophysics, Surrey, UK). The MS data were recorded on an UPLC-IT-TOF MS (Shimadzu Corp., Kyoto, Japan) or Agilent G6230 TOF MS (Applied Biosystems, Ltd, Warrington, UK). 1H, 13C and 2D NMR spectra were obtained on Bruker AVANCE III-600, AVANCE III-500 and AVANCE III-400 MHz spectrometers (Bruker BioSpin GmBH, Rheinstetten, Germany) with SiMe4 as an internal standard. The chemical shifts (δ) are expressed in ppm with reference to the solvent signals. X-ray crystallographic analysis using Cu Kα radiation was performed on a Bruker D8 QUEST instrument (Bruker, Karlsruher, Germany). Column chromatography (CC) was performed on either silica gel (200–300 mesh, Qingdao Marine Chemical Co., Ltd, Qingdao, China) or RP-18 silica gel (20–45 μm, Fuji Silysia Chemical Ltd, Japan). Fractions were monitored by TLC on silica gel plates (GF254, Qingdao Marine Chemical Co., Ltd, Qingdao, China), and spots were visualized with Dragendorff's reagent spray. Medium pressure liquid chromatography (MPLC) was performed using a Buchi pump system coupled with RP-18 silica gel-packed glass columns (15 × 230 and 26 × 460 mm, respectively). High performance liquid chromatography (HPLC) was performed using Waters 1525E pumps (Waters Corp., Milford, MA, USA) coupled with analytical semi-preparative or preparative XBridge C18 columns (4.6 × 150, 10 × 150, and 19 × 250 mm, respectively). The HPLC system employed a Waters 2998 photodiode array detector and a Waters fraction collector III (Waters Corp., Milford, MA, USA).Plant material
Trunks of Bousigonia angustifolia Pierre were collected in Dec 2018 in areas along the Mengla County, Yunnan Province, P. R. China, and identified by Dr Jie Cai. A voucher specimen (No. Cai20181228) was deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.Extraction and separation
The dried and powdered trunks of B. angustifolia (25 kg) were extracted with MeOH (60 L × 3) at room temperature, these were then filtered and the solvent was evaporated in vacuo. The extract was dissolved in 0.5% aqueous HCl solution and extracted with ethyl acetate (EtOAc) three times. The remaining acidic aqueous solution was basified with 10% NH4OH to pH 7–8, and subsequently extracted with EtOAc. This afforded 210 g of the crude alkaloidal fraction. This EtOAc extract (210 g) was subjected to column chromatography (CC) over silica gel and eluted with gradient CHCl3-MeOH (1:0–0:1, v/v) to afford ten fractions (I-X). Fraction I (17.4 g) was chromatographed on a C18 MPLC column eluted with a gradient of MeOH–H2O (10:90–100:0, v/v) to give the eighteen subfractions I-1–I-18. Fraction I-12 (2.2g) was separated using a Sephadex LH-20 column eluted with MeOH. Six subfractions (I-12-1–I-12-6) were collected. I-12-6 (0.4 g) was separated using a C18 MPLC column with a gradient of MeOH–H2O (60:40–90:10, v/v) to afford three subfractions (I-12-6-1–I-12-6-3). Fraction I-12-6-1 (0.11 g) was purified using a preparative C18 HPLC column with a gradient of MeCN–H2O (30:70–45:55, v/v) to obtain 5 (12.5 mg, 33.5 min). Fraction I-12-6-2 (0.15 g) was purified using a preparative C18 HPLC column with a gradient of MeCN–H2O (40:60–55:45, v/v) to obtain 1 (2.4 mg, 69.3 min), 2 (9.1 mg, 50.2min) and 4 (9.8 mg, 27.4 min). Fraction I-18 (1.1g) was separated using a Sephadex LH-20 column eluted with MeOH. Five subfractions (I-18-1–I-18-5) were collected. Fraction I-18-3 (0.12g) was purified using a preparative C18 HPLC column with a gradient of MeCN–H2O (40:60–55:45, v/v) to obtain 3 (9.1 mg, 34.8 min). Fraction II (21.5 g) was chromatographed on a C18 MPLC column eluted with a gradient of MeOH–H2O (10:90–100:0, v/v) to give the twelve subfractions II-1–II-12. Fraction II-7 (3.3g) was separated using a Sephadex LH-20 column eluted with MeOH. Eight subfractions (II-7-1–II-7-8) were collected. Fraction II-7-7 (0.09 g) was purified using a preparative C18 HPLC column with a gradient of MeCN–H2O (25:75–40:60, v/v) to obtain 6 (8.7 mg, 24.6 min).
Bousangustine A (1): yellowish amorphous powder; C58H64N6O3; [α]23D −669.3 (c, 0.10, CH3OH); UV (CH3OH) λmax (logε) 195 (4.20), 348 (2.96) nm; 1H (400 MHz) and 13C (125 MHz) NMR data (methanol-d4) (Table 1); positive ESIMS m/z 915 [M + Na]+. HRESIMS (m/z 915.4937 [M + Na]+, calculated for C58H64N6O3Na 915.4932).
Bousangustine B (2): yellowish amorphous powder; C39H44N4O2; [α]23D −347.84 (c, 0.14, CH3OH); UV (CH3OH) λmax (logε) 195 (4.24) nm; 1H (400 MHz) and 13C (125 MHz) NMR data (methanol-d4) (Table 1); positive ESIMS m/z 623 [M + Na]+. HRESIMS (m/z 623.3355 [M + Na]+, calculated for C39H44N4O2Na 623.3356).
Bousangustine C (3): yellowish amorphous powder; C38H42N4O6; [α]23D −201.5 (c, 0.08, CH3OH); UV (CH3OH) λmax (logε) 195 (3.81) nm; 1H (400 MHz) and 13C (150 MHz) NMR data(methanol-d4) (Table 1); positive ESIMS m/z 673 [M + Na]+. HRESIMS (m/z 673.2992 [M + Na]+, calculated for C38H42N4O6Na 673.2997).
Bioassays
The cytotoxicity of compounds 1–6 was tested using the MTS assay. The cells were seeded into 96-well tissue culture dishes at 4 × 103 cells per well for HeLa and 5 × 103 cells per well for SMMC-7721, HepG2, and A-549 and cultured overnight at 37 °C in a 5% CO2 incubator for cell adhesion. Cells were then incubated in culture medium with each compound for 48 h. The MTS-reducing activity was evaluated by measuring the absorbance at 490 nm using a Cell Titer 96 A Queous One Solution Cell Proliferation Assay kit (Promega, USA) and an Infinite M200 Pro (Tecan, Austria) microplate reader. IC50 values were calculated using the Reed–Muench method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported in part by the Applied Basic Research Key Project of Yunnan (No. 2016FA030) and the National Natural Science Foundation of China (31872677). Computational resources used in this work were supported in part by SciGrid, Chinese Academy of Sciences.Notes and references
- A. Keglevich, A. Szigetvari, M. Dekany, C. Szantay, P. Keglevich and L. Hazai, Synthesis and in vitro antitumor effect of new vindoline Derivatives Coupled with triphenylphosphine, Curr. Org. Chem., 2019, 23, 852–858 CrossRef CAS.
- M. Kitajima, M. Iwai, N. Kogure, R. Kikura-Hanajiri, Y. Goda and H. Takayama, Aspidosperma-aspidosperma-type bisindole alkaloids from Voacanga africana, Tetrahedron, 2013, 69, 796–801 CrossRef CAS.
- H. Takayama, S. Subhadhirasakul, J. Mizuki, M. Kitajima, N. Aimi, D. Ponglux and S. Sakai, A new class of two dimeric indole alkaloids, coryzeylamine and deformylcoryzeylamine, from the leaves of Hunteria zeylanica in Thailand, Chem. Pharm. Bull., 1994, 42, 1957–1959 CrossRef CAS.
- X. H. Gao, Y. Y. Fan, Q. F. Liu, S. H. Cho, G. F. Pauli, S. N. Chen and J. M. Yue, Suadimins A–C, unprecedented dimeric quinoline alkaloids with antimycobacterial activity from Melodinus suaveolens, Org. Lett., 2019, 21, 7065–7068 CrossRef CAS PubMed.
- W. Zhang, X. J. Huang, S. Y. Zhang, D. M. Zhang, R. W. Jiang, J. Y. Hu, X. Q. Zhang, L. Wang and W. C. Ye, Geleganidines A-C, unusual monoterpenoid indole alkaloids from Gelsemium elegans, J. Nat. Prod., 2015, 78, 2036–2044 CrossRef CAS PubMed.
- E. O. N. Obiang, G. Genta-Jouve, J. F. Gallard, B. Kumulungui, E. Mouray, P. Grellier, L. Evanno, E. Poupon, P. Champy and M. A. Beniddir, Pleiokomenines A and B: dimeric aspidofractinine alkaloids tethered with a methylene group, Org. Lett., 2017, 19, 6180–6183 CrossRef PubMed.
- B. J. Zhang, C. Liu, M. F. Bao, X. H. Zhong, L. Ni, J. Wu and X. H. Cai, Novel monoterpenoid indole alkaloids from Melodinus yunnanensis, Tetrahedron, 2017, 73, 5821–5826 CrossRef CAS.
- X. H. Cai, H. Jiang, Y. Li, G. G. Cheng, Y. P. Liu, T. Feng and X. D. Luo, Cytotoxic indole alkaloids from Melodinus fusiformis and M. morsei, Chin. J. Nat. Med., 2011, 9, 259–263 CAS.
- X. N. Li, Y. Zhang, X. H. Cai, T. Feng, Y. P. Liu, Y. Li, J. Ren, H. J. Zhu and X. D. Luo, Psychotripine: a new trimeric pyrroloindoline derivative from Psychotria pilifera, Org. Lett., 2011, 13, 5896–5899 CrossRef CAS PubMed.
- Z. W. Liu, J. Zhang, S. T. Li, M. Q. Liu, X. J. Huang, Y. L. Ao, C. L. Fan, D. M. Zhang, Q. W. Zhang, W. C. Ye and X. Q. Zhang, Ervadivamines A and B, two unusual trimeric monoterpenoid indole alkaloids from Ervatamia divaricata, J. Org. Chem., 2018, 83, 10613–10618 CrossRef CAS PubMed.
- H. Schubel, W. Fahn and J. Stockigt, Formation of the first trimeric monoterpenoid indole alkaloids, Helv. Chim. Acta, 1989, 72, 147–150 CrossRef.
- Y. L. Wang, C. R. Guo, Y. Mu, Y. L. Lin, H. J. Yan, Z. W. Wang and X. J. Wang, Bousigonine A and B, bis- and tri-indole alkaloids from Bousigonia mekongensis and their preventing high glucose-induced podocyte injury activity, Tetrahedron Lett., 2019, 60, 151042 CrossRef CAS.
- B. J. Zhang, B. Wu, M. F. Bao, L. Ni and X. H. Cai, New dimeric and trimeric Erythrina alkaloids from Erythrina variegata, RSC Adv., 2016, 6, 87863–87868 RSC.
- Y. Yu, S. M. Zhao, M. F. Bao and X. H. Cai, Aspidosperma-type alkaloid dimer from Tabernaemontana bovina as a candidate for inhibition of microglial activation, Org. Chem. Front., 2020, 11, 1365–1373 RSC.
- B. J. Zhang, M. F. Bao, C. X. Zeng, X. H. Zhong, L. Ni, Y. Zeng and X. H. Cai, Dimeric Erythrina alkaloids from the flower of Erythrina variegata, Org. Lett., 2014, 16, 6400–6403 CrossRef CAS PubMed.
- (a) Y. H. Fu, H. P. He, Y. T. Di, S. L. Li, Y. Zhang and X. J. Hao, Mekongenines A and B, two new alkaloids from Bousigonia mekongensis, Tetrahedron Lett., 2012, 53, 3642–3646 CrossRef CAS; (b) Y. H. Fu, S. L. Li, S. F. Li, H. P. He, Y. T. Di, Y. Zhang and X. J. Hao, Cytotoxic eburnamine-aspidospermine type bisindole alkaloids from Bousigonia mekongensis, Fitoterapia, 2014, 98, 45–52 CrossRef CAS PubMed; (c) Y. H. Fu, Y. T. Di, H. P. He, S. L. Li, Y. Zhang and X. J. Hao, Angustifonines A and B, cytotoxic bisindole alkaloids from Bousigonia angustifolia, J. Nat. Prod., 2014, 77, 57–62 CrossRef CAS PubMed.
- T. S. Kam, Y. M. Tee and G. Subramaniam, Rhazinal, a formylrhazinilam derivative from a Malayan Kopsia, Nat. Prod. Lett., 1998, 12, 307–310 CrossRef CAS.
- S. H. Goh, A. R. M. Ali and W. H. Wong, Alkaloids of Leuconotis griffithii and L. eugenifolia (Apocynaceae), Tetrahedron, 1989, 45, 7899–7902 CrossRef CAS.
- S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, Revealing atropisomer axial chirality in drug discovery, ChemMedChem, 2011, 6, 505–513 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2048765. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qo01565b |
| This journal is © the Partner Organisations 2021 |