
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Are bis(pyridine)iodine(I) complexes applicable for asymmetric halogenation?†
Daniel
von der Heiden
 a,
Flóra Boróka
Németh
b,
Måns
Andreasson‡
c,
Daniel
Sethio
a,
Imre
Pápai
bd and
Mate
Erdelyi
*a
a,
Flóra Boróka
Németh
b,
Måns
Andreasson‡
c,
Daniel
Sethio
a,
Imre
Pápai
bd and
Mate
Erdelyi
*a
aDepartment of Chemistry – BMC, Uppsala University, SE-751 23 Uppsala, Sweden. E-mail: mate.erdelyi@kemi.uu.se
bInstitute of Organic Chemistry, Research Centre for Natural Sciences, H-1117 Budapest, Hungary
cDepartment of Chemistry and Molecular Biology, University of Gothenburg, SE-412 96 Gothenburg, Sweden
dDepartment of Chemistry, University J. Selyeho, 94505 Komárno, Slovakia
First published on 9th September 2021
Abstract
Enantiopure halogenated molecules are of tremendous importance as synthetic intermediates in the construction of pharmaceuticals, fragrances, flavours, natural products, pesticides, and functional materials. Enantioselective halofunctionalizations remain poorly understood and generally applicable procedures are lacking. The applicability of chiral trans-chelating bis(pyridine)iodine(I) complexes in the development of substrate independent, catalytic enantioselective halofunctionalization has been explored herein. Six novel chiral bidentate pyridine donor ligands have been designed, routes for their synthesis developed and their [N–I–N]+-type halogen bond complexes studied by 15N NMR and DFT. The chiral complexes encompassing a halogen bond stabilized iodenium ion are shown to be capable of efficient iodenium transfer to alkenes; however, without enantioselectivity. The lack of stereoselectivity is shown to originate from the availability of multiple ligand conformations of comparable energies and an insufficient steric influence by the chiral ligand. Substrate preorganization by the chiral catalyst appears a necessity for enantioselective halofunctionalization.
Introduction
Electrophilic halofunctionalization is a long-established, synthetic transformation that introduces two heteroatoms onto a carbon–carbon double bond.1,2 It is frequently applied in the synthesis of bioactive molecules and synthetic building blocks, whose halogen handle can easily be further manipulated. The basis of its understanding has been laid by stereochemical and kinetic studies performed in the early 20th century.3 According to the commonly accepted mechanism, electrophilic halofunctionalizations involve the formation of cyclic halonium ions that undergo ring-opening through backside attack by a nucleophile, yielding the anti-stereoisomers of the products selectively. An intermediate cyclic halonium ion was first proposed by Roberts and Kimball,1 and later NMR spectroscopically proven by Oláh.4–6 The formation of acyclic halocarbenium ions, particularly for smaller halogens, was also reported, and even a concerted mechanism has been put forward by Borhan, according to which the nucleophile's electron donation activates the alkene for electrophilic attack by a halenium ion.§7 Despite diastereoselectivity, there is generally no facial selectivity in the halenium addition to the olefin, and accordingly the addition results in a racemic product. Due to their high electron affinity, the reactivity of halenium ions is difficult to modulate. The monodentate coordination of halogens makes their positioning into a chiral environment challenging. In the past decade, vast efforts to develop an asymmetric variant of this reaction have been made, however, so far only with limited success whereas vast limitations remain.7–16A reactive halenium ion can be stabilized by a three-center, four-electron halogen bond in a [bis(pyridine)halogen(I)]-type complex,17 which allows the rational modulation of halenium ion reactivity.18 In such complexes, halonium ions,§ including even chloronium ions,19 are stable in solution and hence can be experimentally studied.17,20–22 [Bis(pyridine)iodine(I)] tetrafluoroborate was introduced as a mild halogen transfer and oxidation reagent by Barluenga,23 and the fundaments for the mechanistic understanding of its halenium transfer reaction were laid by Brown's reaction kinetic studies.24–26 [Bis(pyridine)halogen(I)] complexes easily dissociate in solution, leading to rapid ligand scrambling.27 To avoid the complications that are caused by such dynamic processes, following some initial studies22 the properties of halonium complexes have been primarily investigated using bidentate bis(pyridine)–type ligands,21 such as 1 shown in Fig. 1. The (1,2-bis(pyridine-2-ylethynyl)benzene) backbone28 promotes the formation of [N–X–N]+ complexes with co-planar pyridine rings, providing further helpful geometric control. Following extensive studies of the factors influencing the symmetry and stability of a three-center, four-electron halogen bond of halonium ions,18–22,29–31 this backbone also allowed the development of a stable asymmetric halonium complex.29 Building on the above studies, halogen bonded halonium ions have been made use of in building complex supramolecular systems,32–34 including also the first halonium ion-based halogen bonded molecular framework (XOF).35 Their potential applicability in molecular motors has also been recently explored.36 The interaction of halogen(I) with silver(I) ions when included into bis(pyridine)-type complexes have lately been reported.37,38
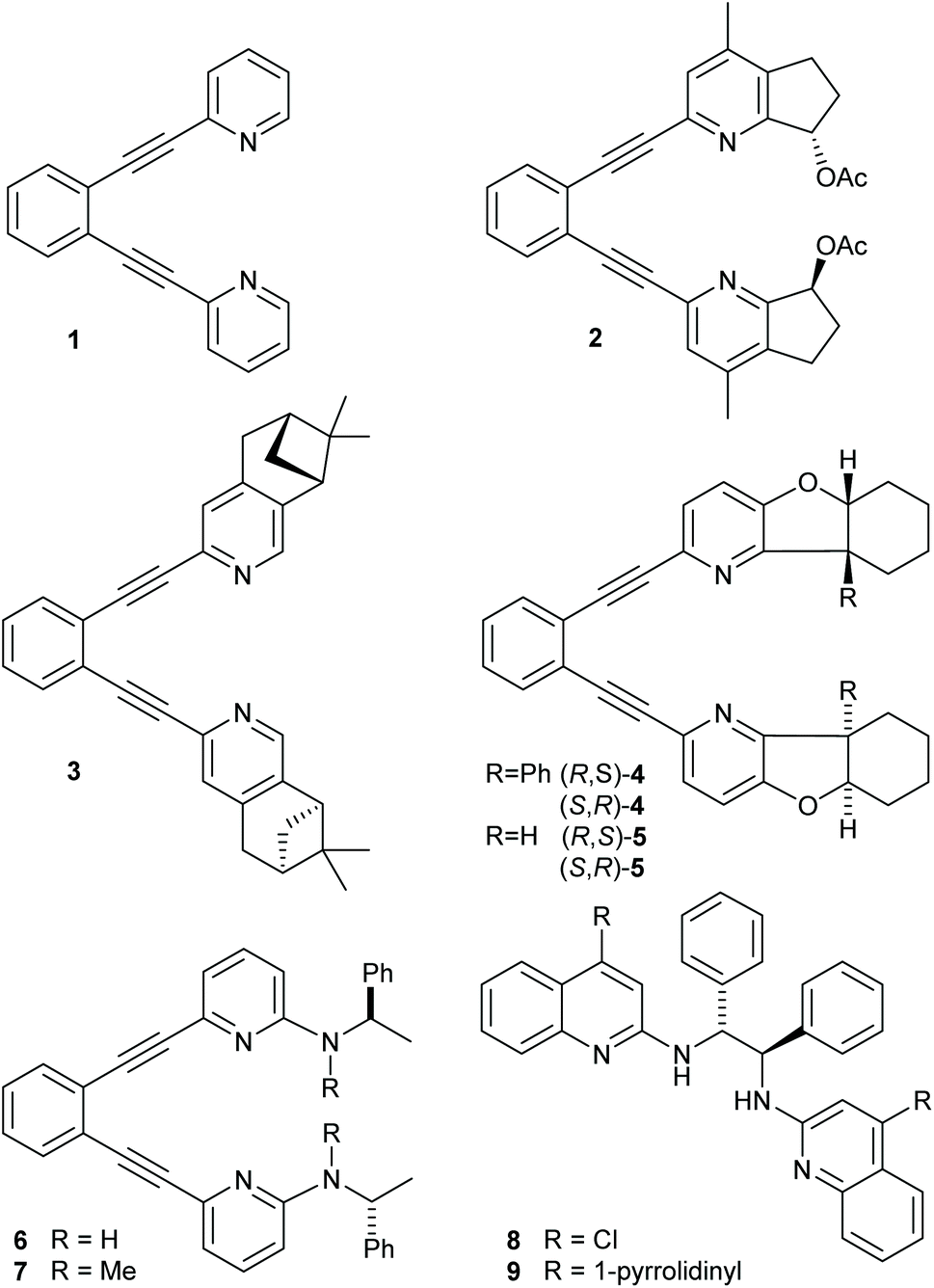 | ||
| Fig. 1 Ligands 1–9 used in iodine(I) transfer reactions. The corresponding iodine(I) complexes are denoted as 1-I, 2-I, … and 8-I. | ||
The use of a chiral [bis(2-menthylpyridine)bromine(I)] complex for asymmetric halocyclization has been explored by Brown. This yielded only negligible enantioselectivity (2.4–4.8% ee),25 likely due to the flexibility and the large distance of the applied 2-menthyl substituent from the reaction center. Herein, we explore whether more rigid (1,2-bis(pyridine-2-ylethynyl)-benzene)-type18,21 ligands that encapsulate a halonium ion into a chiral pocket could provide strong enough influence on the stereochemistry of halofunctionalization to make it enantioselective. Accordingly, we synthesized a series of chiral analogues of 1 (Fig. 1). These do not suffer from ligand scrambling,27 and may influence the halenium transfer process with both chiral pyridines even upon dissociation of the [N–I–N]+ bond. Whereas ligands 2 and 4 hold their chiral centers in a rigid framework and in the proximity of the pyridine nitrogen, 3 has its chiral centers further away, and 5 and 6 allow for geometric adjustments. Ligands 8–9 have originally been introduced by Johnston and coworkers,39 and were reported to provide enantioselectivity in halonium transfer when using N-iodosuccinimide as iodine-source in the presence of acid, and were here synthesized as positive controls.
Results and discussion
Synthesis
(1,2-Bis(pyridine-2-ylethynyl)benzene) (1) was prepared according to a previously established protocol,21 whereas ligands 2–7 were synthesized following the reaction routes shown in Schemes 1–4 (for details, see the Experimental section). Ligands 8 and 9 were prepared following Johnston's procedure.39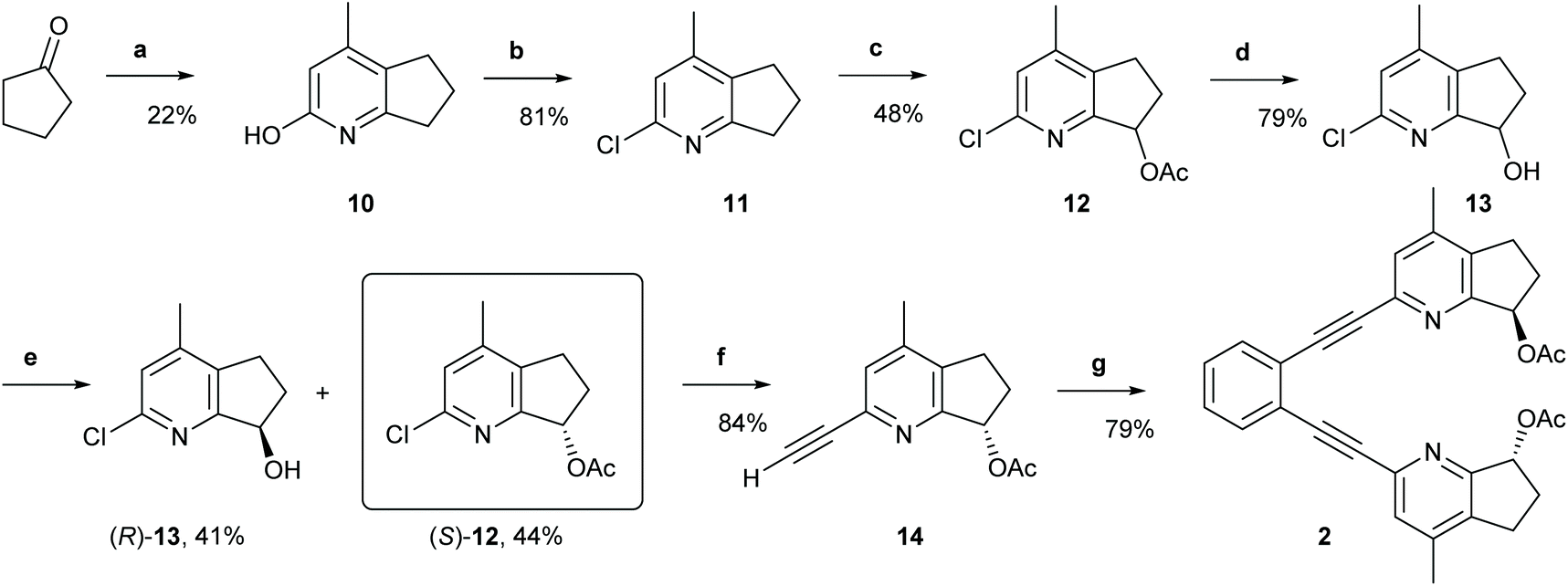 | ||
| Scheme 1 The synthetic route to ligand 2. Reagents and conditions: (a) Ethyl acetoacetate, NH4OAc, 135 °C, 20 h.; (b) Phenylphosphonic dichloride, 160 °C, 21 h; (c) (1) Glacial AcOH, H2O2, 80 °C to r.t., 22 h; (2) Ac2O, r.t. to 100 °C, 5 h; (d) LiOH, THF, H2O, r.t., 21 h; (e) Novozyme 435, vinyl acetate; r.t., 4 h; (f) (1) Triisopropylacetylene, trans-[PdCl2(CH3CN)2], XPhos, Cs2CO3, CH3CN, MW 110 °C, 20 min; (2) TBAF, THF, 0 °C, 2 h; (g) 1,2-diiodobenzene, Pd (PPh3)2Cl2, PPh3, CuI, Et2NH, MW 110 °C, 20 min. For details, see the ESI.† | ||
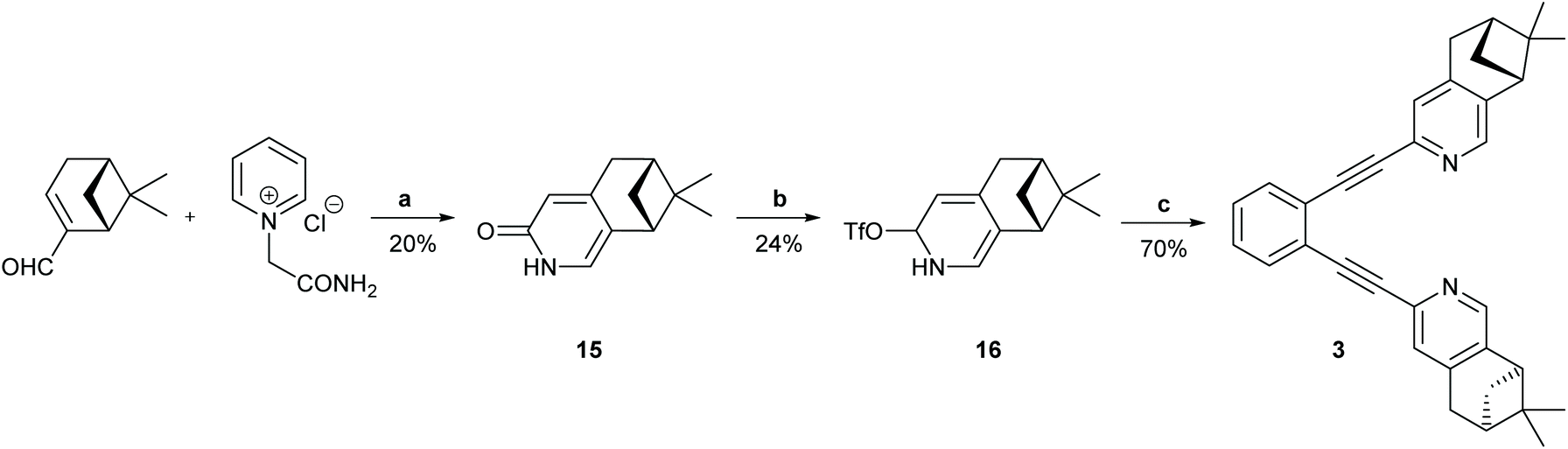 | ||
| Scheme 2 The synthetic route to ligand 3. Reagents and conditions: (a) 1. Piperidine, MeOH, reflux, Ar, 2 h. 2. HCONH2, AcOH, 200 °C, 1 h; (b) Tf2O, Et3N. CH2Cl2, −50 °C to r.t. 19 h; (c) 1,2-diethynylbenzene, Pd(PPh3)2Cl2, CuI, PPh3, DIEA, MW 110 °C, 15 min. For details, see the ESI.† | ||
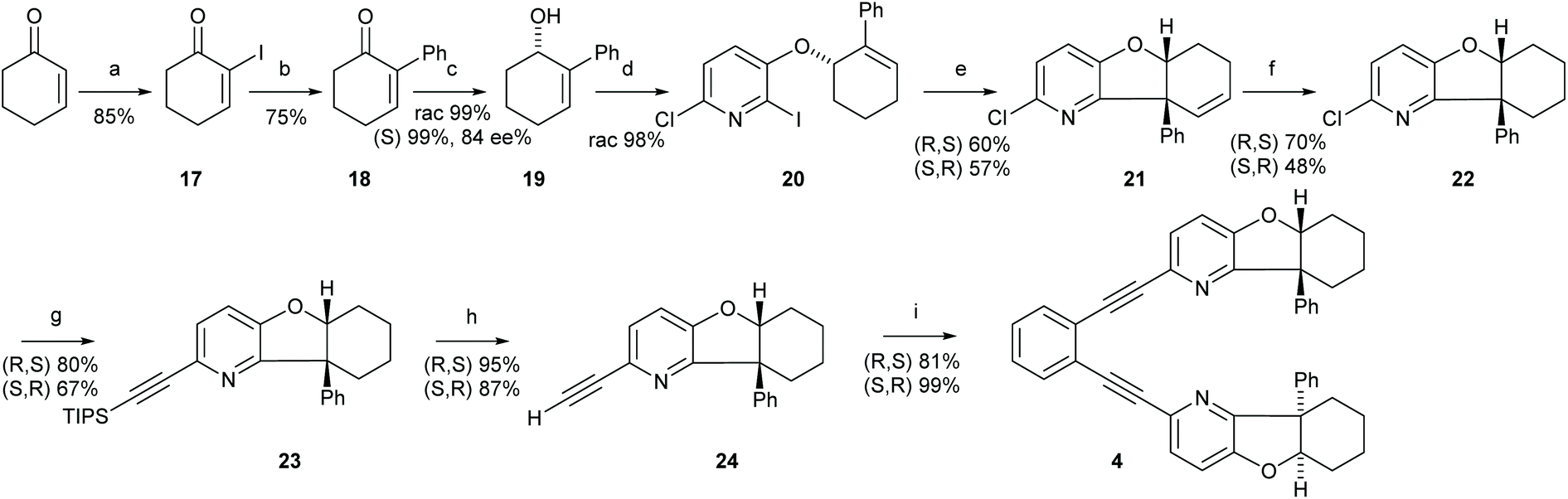 | ||
Scheme 3 The synthetic route to ligand 4. Reagents and conditions: (a) I2, DMAP, K2CO3, H2O/THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), r.t., 2 h.; (b) Pd/C, PhB(OH)2, Na2CO3, DME/H2O (1:1), r.t., 18 h; (c) CeCl3, NaBH4, MeOH, 0 °C to r.t. in 2 h; (d) 6-chloro-2-iodopyridine-3-ol, DIAD, PPh3, toluene, r.t., 3 days; (e) Pd(PPh3)2Cl2, Ag2CO3, NEt3, toluene, 110 °C, 24 h; (f) Rh/C, H2, MeOH; (g) trans-[PdCl2(CH3CN)2], XPhos, Cs2CO3, CH3CN, 85 °C, 8 h; (h) (1) NuB4F, THF, 0 °C, 2 h, then (2) 1,2-diiodobenzene, Pd2(dba)3, PPh3, CuI, NEt3, 45 °C, 20 h. For details, see the ESI.† :1), r.t., 2 h.; (b) Pd/C, PhB(OH)2, Na2CO3, DME/H2O (1:1), r.t., 18 h; (c) CeCl3, NaBH4, MeOH, 0 °C to r.t. in 2 h; (d) 6-chloro-2-iodopyridine-3-ol, DIAD, PPh3, toluene, r.t., 3 days; (e) Pd(PPh3)2Cl2, Ag2CO3, NEt3, toluene, 110 °C, 24 h; (f) Rh/C, H2, MeOH; (g) trans-[PdCl2(CH3CN)2], XPhos, Cs2CO3, CH3CN, 85 °C, 8 h; (h) (1) NuB4F, THF, 0 °C, 2 h, then (2) 1,2-diiodobenzene, Pd2(dba)3, PPh3, CuI, NEt3, 45 °C, 20 h. For details, see the ESI.† | ||
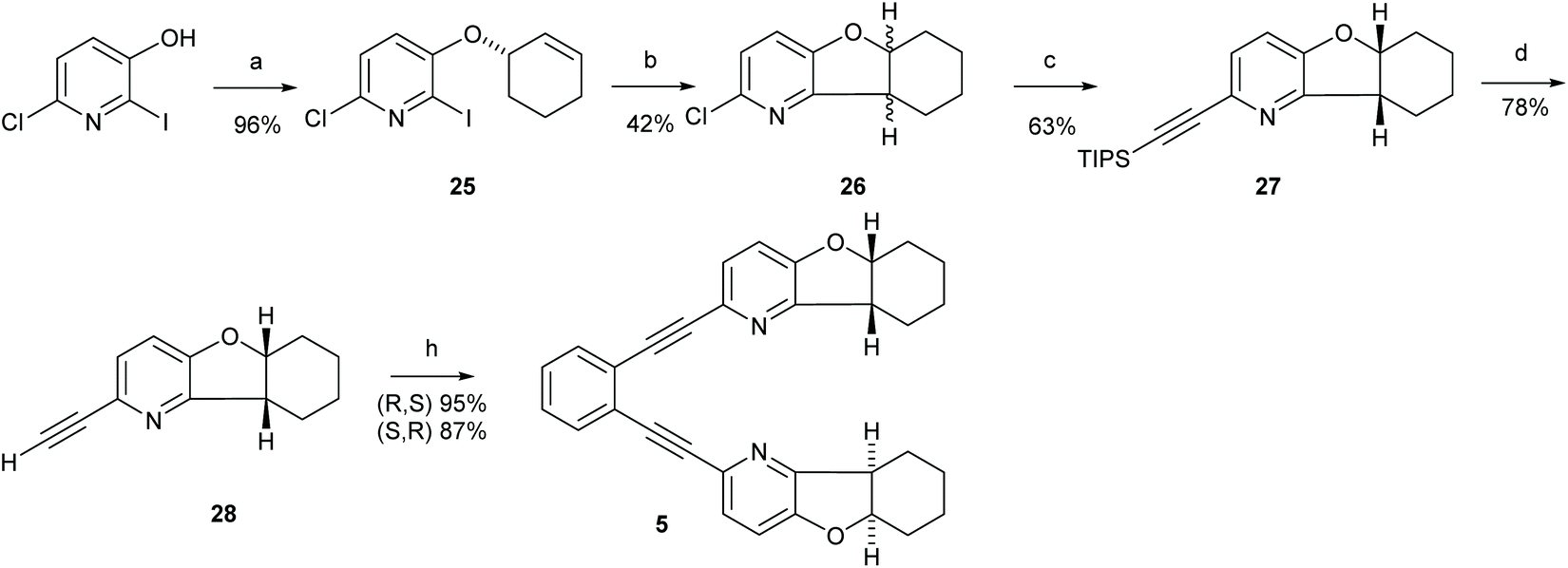 | ||
| Scheme 4 The synthetic route to ligand 5. Reagents and conditions: (a) Bromocylohex-1-ene, K2CO3, DMF, r.t. 17 h; (b) SmI2, Et3N, THF, r.t, 1 h; (c) triisopropylacetylene, trans-[PdCl2(CH3CN)2], XPhos, Cs2CO3, CH3CN, 90 °C, 18 h; (d) TBAF, THF, 0 °C, 2 h; (e) 1,2-diiodobenzene, Pd(PPh3)2Cl2, PPh3, CuI, Et2NH, 60 °C, 19 h. For details, see the ESI.† | ||
Stability
The iodine(I) complexes of 1–6 were achieved by mixing the free ligands with AgBF4 in dichloromethane, followed by the addition of I2, and removal of the AgI precipitate by centrifugation. The formation of the bis(pyridine)iodine(I) complexes was confirmed by observation of 15N NMR chemical shifts (Table 1) characteristic for such complexes.17,18,21,29 The iodine(I) complexes of 1–5 are stable at room temperature, whereas those of 6 and 7 can be studied at −5 °C and −35 °C, respectively. Ligands 8–9 do not form stable iodine(I) complexes, but undergo self-iodination.39 The stability of the [N−I−N]+ complexes remarkably correlates to the steric demand of the pyridine ortho-substituent. Hence, 1-I, the iodine(I) complex of 1 that possesses a sterically undemanding alkyne is stable at room temperature in solution, and so is its literature known 2,6-dialkyne analogue.32 The iodine(I) complexes 2-I–5-I that have sterically demanding substituents orienting away from the plane of the [N−I−N]+ halogen bond are also stable for several hours in solution at room temperature. In contrast, 6-I that has an sp2-hybridized ortho-pyridine substituent is less stable (Table 1), whereas 7-I was not stable in solution at −35 °C, expectably due to its even more sterically demanding ortho-substituent. To confirm our hypothesis on the importance of steric crowding, we performed geometry optimization of complexes 4, 6–7 and visualized the non-covalent interactions (Fig. 2, for details see section 4.3 in the ESI†). It should be noted here that 5-I is stabilized by an intramolecular hydrogen bond, where a filled p-orbital of iodine(I) acts as hydrogen bond acceptor and the amide proton as hydrogen bond donor, analogous to previously reported systems encompassing hydrogen bond-enhanced halogen bonds.40,41 In contrast, 7-I is destabilized by the repulsion between the N-methyl amine substituent and iodine. No significant steric crowding was predicted for complex 4-I that orients its sterically demanding phenyl group out of the plane of the three-center halogen bond.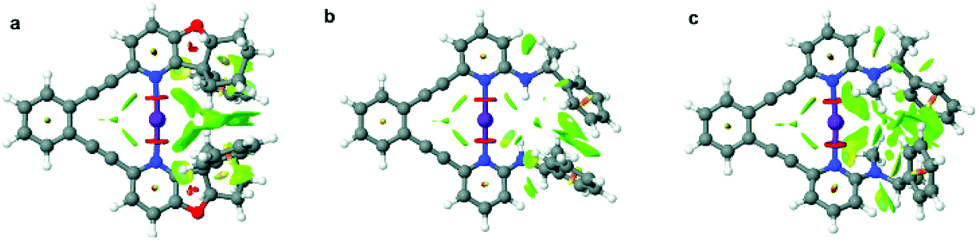 | ||
| Fig. 2 Non-covalent interaction (NCI) plots of 4-I (a), 6-I (b), and 7-I (c), calculated at the ωB97X-D/aug-cc-pVTZ level of theory, with strong repulsions being shown in red, weak repulsion in yellow, weak attraction in green, and strong attraction in blue. | ||
| Ligand | δ 15Nlig | δ 15NAg(I) | δ 15NI(I) | Stability |
|---|---|---|---|---|
| a The 15N NMR chemical shift of the free ligand is denoted as δ15Nlig, that of its silver(I) complex as δ15NAg(I), of its iodine(I) complex as δ15NI(I), and the stability of the iodine(I) complex at the conditions under which it is stable for at least 1 hour with < 5% decomposition. n.d. – not determined. | ||||
| 1 | −6518 | n.d. | −16618 | r.t.18 |
| (S)- 2 | −80 | n.d. | n.d. | n.d. |
| (S,S)- 3 | −84 | −29 | −171 | r.t. |
| (S,R)- 4 | −73 | −127 | −170 | r.t. |
| (S,R)- 5 | −74 | n.d. | −187.6 | n.d. |
| (R,R)- 5 | −115 (−285) | −163 (−283) | −199 (−273) | <−5 °C |
| (R,R)- 6 | −116 (−296) | −149 (−298) | n.d. | <−35 °C |
Halogen(I) transfer reaction
The non-chiral 1-I and its derivatives are mild halonium transfer agents.17,18 They react via an initial dissociation of one of the pyridines (Scheme 6), followed by coordination of the nucleophilic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond to the reactive single coordinated [pyridine-iodine(I)]+ species, yielding a three membered cyclic iodonium intermediate. The latter undergoes ring opening typically assisted by a nucleophile, which in halocyclizations is present intramolecularly in the substrate. The mechanism of iodine(I) transfer from bis(pyridine) complexes, such as 1, has been recently described based on UV-kinetic and computational data.31 Herein we assess whether chiral analogues of 1 presenting chiral functionalities near the coordinating nitrogen may influence the enantioselectivity of iodocyclisation.
C double bond to the reactive single coordinated [pyridine-iodine(I)]+ species, yielding a three membered cyclic iodonium intermediate. The latter undergoes ring opening typically assisted by a nucleophile, which in halocyclizations is present intramolecularly in the substrate. The mechanism of iodine(I) transfer from bis(pyridine) complexes, such as 1, has been recently described based on UV-kinetic and computational data.31 Herein we assess whether chiral analogues of 1 presenting chiral functionalities near the coordinating nitrogen may influence the enantioselectivity of iodocyclisation.
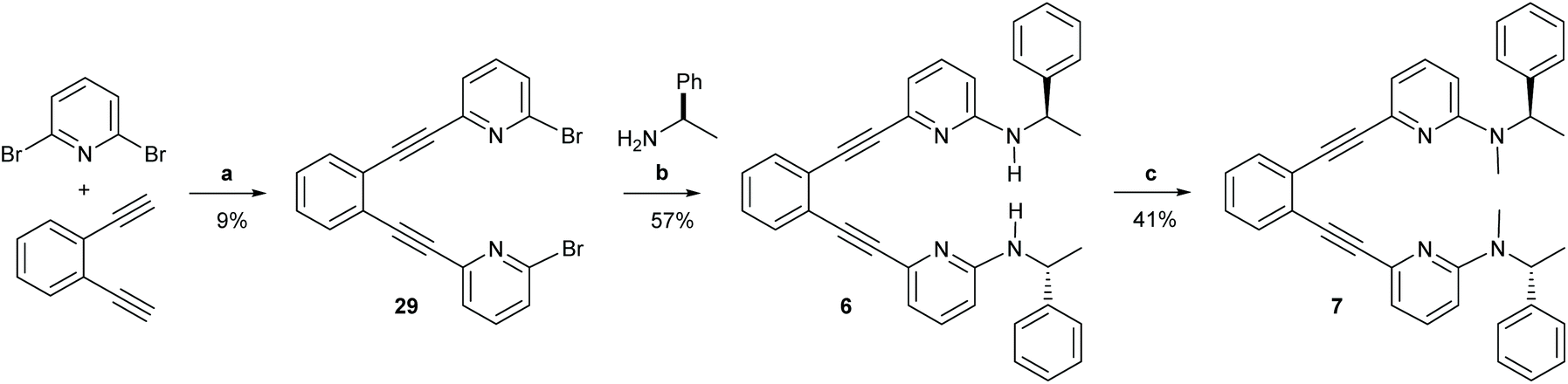 | ||
| Scheme 5 The synthetic route to ligands 6 and 7. Reagents and conditions: (a) Pd(PPh3)2Cl2, CuI NEt3, 110 °C, o.n.; (b) Pd2(dba)3, 1,3-bis(diphenylphosphino)propane, NaOtBu, (R)-(+)-1-phenzlethylamine, toluene, 90 °C, 3 h; (c) 1. NaH, DMF, r.t., 1 h, 2. MeI, DMF, r.t., 1 h, 3. NaH, DMF, r.t., 1 h, 4. MeI, DMF, r.t, 1 h. For details, see the ESI.† | ||
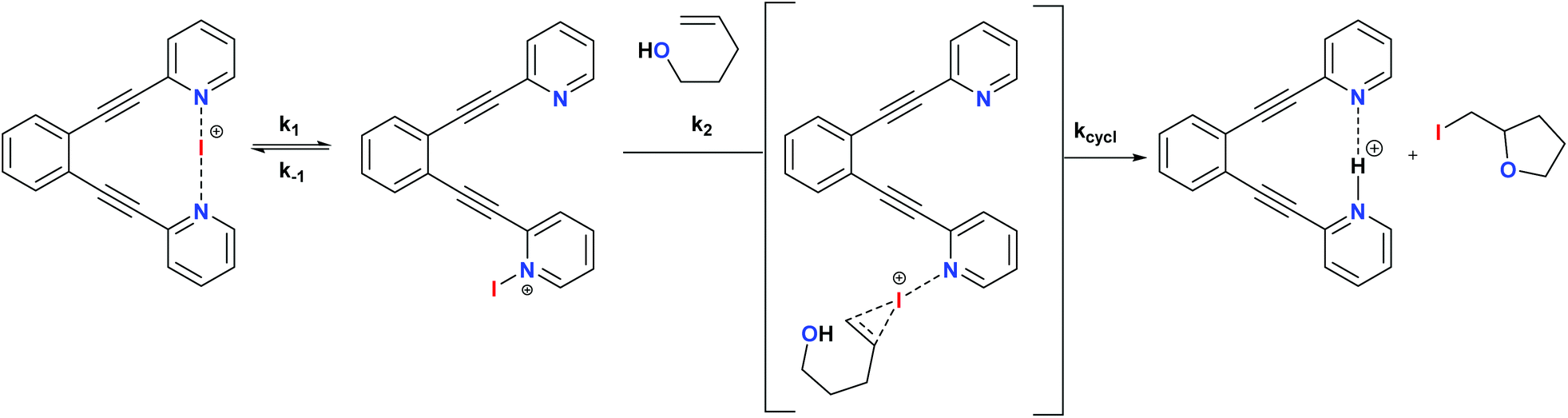 | ||
| Scheme 6 The simplified general mechanism of iodine(I) transfer from [(1,2-bis(pyridine-2-ylethynyl)benzene)iodineI()] (1)-type reagents. A detailed DFT-based description of the mechanism is given in ref. 29. The possible chiral influence of ligands 2–7 on the outcome of the iodocylization of alkenes is assessed herein.† | ||
To explore whether its chiral analogues 2–7 provide stereoinduction in halenium transfer reactions, we used iodocyclization as a model reaction. The iodine(I) complex of (R,S)-4-I was generated in situ by addition of stoichiometric I2 to a mixture of (R,S)-4-Ag and trans-styrylacetic acid at −20 °C, providing halocyclization with the full conversion of the iodine source in toluene, dichloromethane, acetonitrile or tetrahydrofuran as solvents (for details, see the ESI Table S3†). Despite being chiral, 2–5 did not induce significant enantiomeric excess (ee) in the halocyclization of trans-styrylacetic acid. As a positive control for iodocyclization, the pyridinium/N-iodosuccinimide mediated iodination of 5-phenylhex-5-enoic acid to 6-(iodomethyl)-6-phenyltetrahydro-2H-pyran-2-one using 8 and 9 as chiral catalysts, as introduced by Dobisch and Johnston,39 was used (Table 2. These provided measurable enantiomeric excesses (ees) confirming our chromatographic method's ability to detect ee (for details see ESI section 1 and 2.1–2.3†). No enantioselectivity was obtained using 2–7 as chiral catalysts.
|
|
||
|---|---|---|
| Catalyst | Acid | ee |
| a The ees were determined using chiral analytical HPLC using a Lux i-Amylose column (for further details, see Table S12 in section 2.3 in the ESI†). b The iodine(I) complex of (S)-2 was generated in situ by addition of I2 to its silver(I) complex. | ||
| (S)- 2 | — | 0%b |
| (S,S)- 3 | — | 0%b |
| (R,S)- 4 | HNTf2 | < 7% |
| (R,R)- 5 | — | 0%b |
| (R,R)- 6 | HOTf | 3% |
| 8 | HNTf2 | 61% |
| 9 | HNTf2 | 97%39 |
| 9 | HOTf | 93% |
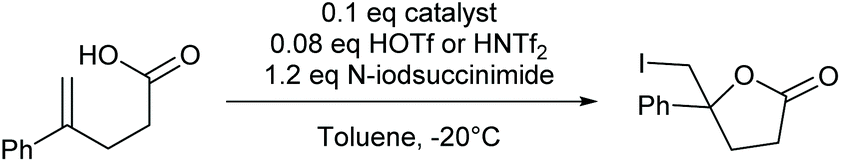
The enantioselectivity of the reactions of 8 and 9 has originally been proposed39 to originate from Brønsted acid activation of both N-iodosuccinimide and the substrate. The bifunctional catalysts 8 and 9 are supposed to interact with both N-iodosuccinimide and the substrate prior to the iodine(I) transfer. Using 1H NMR detection, we observed no decomposition when mixing 4 with N-iodsuccinimide to generate 4-I, whereas decomposition took place when, 4-H (protonated 4) was used instead (for details, see section 1.10 in the ESI†). Without contradicting the previous findings, this control experiment suggests that protonation is essential for the iodine(I) transfer from N-iodosuccinimide to the pyridine–nitrogen. The reactive N-iodopyridinium ion is formed upon transferring iodine(I) from N-iodosuccinimide to the pyridinium salt via I+/H+ exchange.
The chiral ligands 2–7 are similar to 8–9 bifunctional molecules, have two pyridine Lewis bases, and allow substrate coordination, and hence could be expected to induce enantioselectivity in halocyclizations. Enantioselectivity may originate from the irreversible addition of iodine(I) to the alkene via a halogen-bonded iodine(I) prereactive complex.42 In this, the iodine(I) simultaneously coordinates a chiral pyridine and the double bond, providing a chiral bidentate complex of iodine(I). Bidentate iodine(I) complexes are literature known.17,20 Alternatively, if the halogen addition step is reversible, the intramolecular nucleophilic attack of the carboxylate oxygen may provide enantioselectivity if the ligand remains coordinated at the time point of the nucleophilic attack. To assess the intermediate that undergoes the nucleophilic attack, we used 5-norbornene-2,3-dimethanol as a substrate (Scheme 7) as for this molecule enantioselectivity in product formation is determined at the time point of the nucleophilic attack, instead of the coordination of iodine(I) to the double bond. In case the chiral pyridine ligand remains coordinated to iodine(I) at the time point of the nucleophilic ring closure, enantioselectivity in the product formation is expected, presuming that the chiral information provided by the bis(pyridine)-ligand is close enough to the reaction centre. If the chiral ligand is dissociated from iodine(I) when the nucleophilic ring closure occurs, a racemic outcome is expected. Using (R,S)-4, no significant ee (2%) in the haloetherification of 5-norbornene-2,3-dimethanol was observed (Scheme 5). This suggests that pyridines do not form stable enough bis-coordinate iodine(I) complexes involving an alkene to induce enantioselectivity, or that the chiral ligand used in this experiment did not provide sufficient enough energy difference of the diaestereotopic transition states.
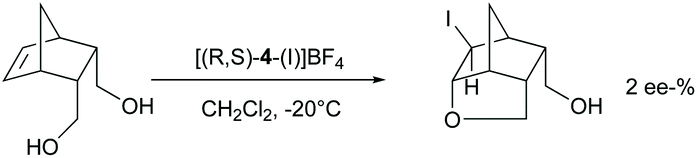 | ||
| Scheme 7 Haloetherification of 5-norbornene-2,3-dimethanol was used to selectively study enantioselectivity of the cyclization step. The enantiomeric excess (ee-%) has been determined following benzylation of the product using benzylanhydride, diisopropylethylamine in CH2Cl2, at 30 °C for 3 days. | ||
Iodocyclization reactions induced by 1–7-I are assumed to proceed via their singly coordinated open forms, which allow the interaction of the electrophilic iodine(I) with the substrate's olefinic double bond prior to iodocyclization. DFT calculations (see the Experimental section and section 4.1 in the ESI†) carried out for 4-I indicate that the ground state of this complex is the symmetric structure A (Fig. 3), which involves a strong three-center, four-electron [N–I–N]+ bond.20 The open forms B and C are by at least 13 kcal mol−1 less stable and may adopt several conformations, such as B1–C3 (Fig. 3) In the most favoured open forms B1 and B2, the pyridine-bound iodine(I) is in close contact either with the phenyl substituent or with the pyridine ring of the second arm of the ligand. Conformers C1, C2 and C3 lack such stabilizing interactions making the iodine(I) readily available for binding the olefinic bond of the ligand. Even if these conformers are considerably less stable than B1 and B2, they can be regarded as the reactive forms of the 4-I complex towards iodocyclization. Our structural analysis of these energetically close-lying reactive forms suggests that the chiral environment provided by ligand 4 is vaguely defined. The phenyl substituent of the chiral fused ring fragment of the dissociated arm of the ligand is in close vicinity to the iodine(I) providing partial steric shielding, whereas the rest of the fused ring system of the dissociated arm are remotely displaced. Most likely due to the weakness of the steric influence and to the availability of multiple ligand conformations, sufficient stereospecificity in substrate binding is not induced, explaining the inefficiency of 4 to induce enantiomeric excess.
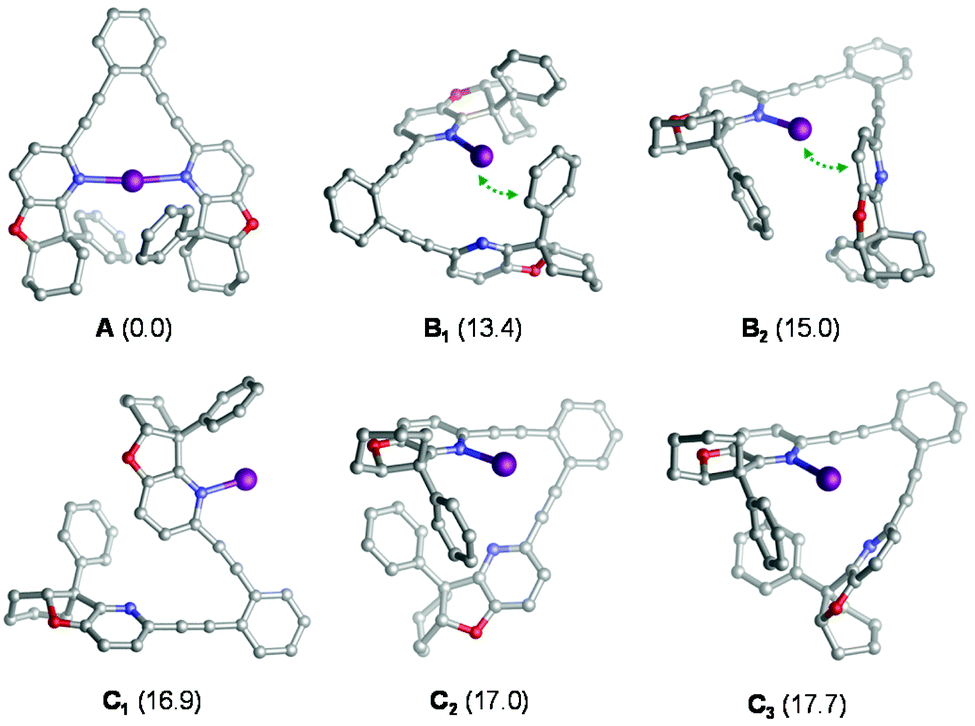 | ||
| Fig. 3 Various forms of complex [2-I]+ as obtained from DFT calculations. Relative stabilities are given in kcal mol−1 with respect to the symmetric chelating form. Hydrogen atoms are omitted for clarity. Close I+−π contacts in B1 and B2 are highlighted with green dotted arrows. | ||
Conclusions
We report the design and synthesis of six novel, bidentate and chiral bis(pyridine)-type ligands (2–7) and their assessment for enantioselective iodine(I) transfer. The three-center, four-electron [N–I–N]+ halogen bond complexes of the ligands were formed through two alternative pathways, either by conversion of the silver(I) complex of the ligands using iodine21,22 or by iodine transfer from N-iodosuccinimide in the presence of triflic acid.39 The stability of bidentate complexes was observed to be limited by the bulkiness of the substituent in the ortho position to the pyridine nitrogen, and thus we found that a hydrogen or alkyne (1) was well tolerated, whereas a substituted ortho-quarternary carbon (4) or an ortho-secondary amide (5) destabilized the iodine(I) complex. The ortho tertiary amide of 6 made the complex so unstable that it could not be studied by 15N NMR at −35 °C, but only generated in situ.The chiral complexes 2–7-I straightforwardly transferred iodenium ions to a model alkene; however, in our hands without enantioselectivity. We hypothesize that this is due to insufficient substrate preorganization by the chiral catalyst, which appears necessary for the induction of enantioselectivity.39 Our control experiments using 5-norbornene-2,3-dimethanol as substrate suggests the irreversible addition of the pyridine bound iodine(I) to the alkene along with rapid ligand dissociation, which prevents stereocontrol by the chiral bis(pyridine) ligands in the nucleophilic ring closure step. Alternatively, the lack of enantioselectivity of the iodine(I) transfer may be explained by the chiral information not being close enough to the reaction centre. Whereas a substrate-independent, catalytic enantioselective halofunctionalization protocol has been a long-sought target of the field, so far all known catalytic enantioselective halogenation protocols are substrate-dependent and require the pre-orientation of the substrate by the catalysts.7–14,16,39,43
This study not just provides some guidance for the further development of enantioselective halenium transfer reagents, but also presents a set of chiral bidentate ligands that may find applications in other fields, such as transition metal catalysis. Structurally closely related trans-chelating bis(pyridine)-ligands have been applied for complexation of copper,44–46 silver,28,47,48 iron,49 palladium,28,47,50–56 mercury,57 gold,58 and even of carbenes59 with several of these complexes having shown synthetically useful catalytic activities.45,51,54–56,58
Experimental section
General methods and materials
CH2Cl2 was dried by distillation over CaH2, and n-hexane by distillation over Na, benzophenone and tetraglyme. For NMR, deuterated solvents were dried by adding 3 Å molecular sieves to freshly opened bottles. All dry solvents were stored over 3 Å molecular sieves in a glovebox. Pyridine was redistilled prior to use. All other chemicals were used without further purification. For all synthesis performed in a glovebox, glassware had been dried at 150 °C in an oven, or in vacuo, at least overnight. NMR spectra were recorded on a Bruker Avance Neo 500 MHz spectrometer equipped with a TXO cryogenic probe, or an Agilent MR-400 equipped with an OneNMR probe. Chemical shifts are reported on the δ scale (ppm), with the residual solvent signal as an internal reference; CD2Cl2 (δH 5.32, δC 53.84), CDCl3 (δH 7.26, δC 77.16). Nitromethane (δN 0.0 ppm) was used as an external standard for 15N. To assign the 1H NMR resonances chemical shift (δ), multiplicity, coupling constants (J Hz) and number of hydrogens were considered. 2D spectra (1H,15N HMBC, 1H,13C HSQC, 1H,13C HMBC, TOCSY, and COSY) also aided correct assignment. Multiplicities are denoted as s (singlet), d (doublet), t (triplet), q (quartet), h (heptet), and m (multiplet). MestReNova 12.0.2. was used to process the NMR spectra.(1,2-Bis(pyridine-2-ylethynyl)benzene) (1) was prepared according to a previously established protocol21
:EtOAc (6:4) eluent. Compound 2 was obtained as an orange solid (61 mg, 67% yield). 1H NMR (500 MHz, CDCl3) δ 7.63 (AA′ part of AA′BB′, 2H, H-14), 7.39 (s, 1H, H-3), 7.39–7.42 (2 × s, 1H, H-3) 7.34 (BB′ part of AA′BB′, 2H, H-13), 6.06 (dd, J = 7.4, 4.1 Hz, 1H, H-7), 3.05–2.98 (m, 1H, H-5), 2.88–2.79 (m, 1H, H-5), 2.70–2.60 (m, J, 1H, H-6), 2.26 (s, 3H, Me-4), 2.14–2.07 (m, 4H, Me–9 and H-6). 13C NMR (126 MHz, CDCl3) δ 170.9 (C-8), 160.5 (C-7a), 144.2 (C-4), 142.8 (C-2), 137.2 (C-4a), 132.4 (C-13), 128.7 (C-14), 128.6 (C-3), 125.6 (C-12), 93.3 (C-11), 93.2 (C-10), 87.8 (C-7), 30.6 (C-6), 26.8 (C-5), 21.5 (Me–9), 18.6 (Me–4). 15N NMR (CDCl3) δ –80.2.
:pentane (7:3) as eluent, and further purified using ethyl/buthyl phophonic acid silica (Sigma-Aldrich) with MeOH as eluent. Compound 3 was obtained as an orange foam (265 mg, 76%). 1H NMR (500 MHz, CD2Cl2) δ 8.16 (s, 1H, H1), 7.63 (1H, H13), 7.59 (t, J = 0.9 Hz, 1H, H4), 7.38 (1H, H14), 2.97 (d, J = 2.8 Hz, 2H, H5), 2.85 (t, J = 5.5 Hz, 1H, H8), 2.72 (dt, J = 9.6, 5.5 Hz, 1H, H9′), 2.31 (tt, J = 5.5, 2.8 Hz, 1H, H6), 1.42 (s, 3H, CH37′), 1.21 (d, J = 9.6 Hz, 1H, H16′), 0.64 (s, 3H, CH37′′). 13C NMR (126 MHz, CD2Cl2) δ 147.1 (CH, C1), 145.3 (C, C8a), 143.3 (C, C4a), 141.6 (C, C3), 132.5 (CH, C13), 129.1 (CH, C14), 127.8 (CH, C4), 126.2 (C, C12), 94.3 (C, C10), 86.8 (C, C11), 45.2 (CH, C8), 40.7 (CH, C6), 39.7 (C, C7), 33.1 (CH2, C5), 32.2 (CH2, C9), 26.3 (CH3, C7′′), 21.7 (CH3, C7′). HRMS (MALDI TOF) calcd for C34H32N2 469.2643 [M + H]+, found m/z 469.2612. [α]20D −36.5 (c 0.135, CH2Cl2).
:80 v/v, 3 runs per PTLC) to give (5aS,9aR)-4 (370 mg, 0.592 mmol, 81%. Alternatively, (5aR,9aS)-4 was synthesized (340 mg, 0.544 mmol, 99%. Analytical HPLC was performed on a Lux® 5 μm Amylose-1 column (250 × 4.6 mm) with H2O/CH3CN 30:70 eluent at 1 ml min−1 flow rate, giving (5aS,9aR)-4 at 18.02 min, and (5aR,9aS)-4 at 17.65 min.
(5aS,9aR)-41H NMR (500 MHz, CDCl3): δ 7.61–7.57 (m, 2H, 3-H, 6-H), 7.49 (d, J = 8.3 Hz, 2H, 3′′-H), 7.42–7.37 (m, 2H, 2′′′-H, 6′′′-H), 7.33–7.27 (m, 6H, 3′′′-H, 5′′′-H, 4-H, 5-H), 7.23–7.19 (m, 2H, 4′′′-H), 6.82 (d, J = 8.3 Hz, 2H, 4′′-H), 5.08 (t, J = 5.0 Hz, 2H, 5a′′-H), 2.29–2.19 (m, 4H, 9′′-H), 2.05–1.97 (m, 2H, 6′′-Ha), 1.94–1.84 (m, 2H, 6′′-Hb), 1.69–1.50 (m, 2H, 7′′-Ha, 7′′-Hb, 8′′-Ha), 1.48–1.37 (m, 2H, 8′′-Hb). 13C NMR (126 MHz, CDCl3): δ 157.5 (C-10), 152.9 (C-5′′), 144.4 (C-1′′′), 135.6 (C-2′′), 131.9 (C-3, C-6), 128.6 (C-3f′′′, C-5′′′), 128.2 & 128.1 (C-3′′, C-4, C-5), 127.3 (C-2′′′, C-6′′′), 126.8 (C-4′′′), 125.9 (C-1, C-2), 116.6 (C-4′′), 93.8 (C-1′), 89.5 (C-5a′′), 86.5 (C-2′), 51.8 (C-9a′′), 33.2 (C-9′′), 27.2 (C-6′′), 21.1 (C-8′′), 19.3 (C-7′′). 15N NMR (51 MHz, CD2Cl2): δ–72.8 ppm (CH3NO2 as 0 ppm). LC-MS (ESI), [m/z]: 313.3 [M + 2H]2+, 625.4 [M + H]+.
(5aR,9aS)-41H NMR (500 MHz, CDCl3): δ 7.61–7.56 (m, 2H, 3-H, 6-H), 7.49 (d, J = 8.3 Hz, 2H, 3′′-H), 7.42–7.36 (m, 4H, 2′′′-H, 6′′′-H), 7.34–7.24 (m, 6H, 3′′′-H, 5′′′-H, 4-H, 5-H), 7.24–7.19 (t, J = 7.3, 1.6 Hz, 2H, 4′′′-H), 6.82 (d, J = 8.2 Hz, 2H, 4′′-H), 5.08 (t, J = 5.0 Hz, 2H, 5a-H), 2.29–2.20 (m, 4H, 9′′-H), 2.05–1.98 (m, 2H, 6′′-Ha), 1.94–1.85 (m, 2H, 6′′-Hb), 1.68–1.51 (m, 6H, 7′′-Ha, 7′′-Hb, 8′′-Ha), 1.48–1.37 (m, 2H, 8′′-Hb). 13C NMR (126 MHz, CDCl3): δ 157.5 (C-10), 152.8 (C-5′′), 144.4 (C-1′′′), 135.5 (C-2′′), 131.9 (C-3, C-6), 128.6 (C-3′′′, C-5′′′), 128.2 and 128.1 (C-3′′, C-4, C-5), 127.3 (C-2′′′, C-6′′′), 126.8 (C-4′′′), 125.8 (C-1, C-2), 116.6 (C-4′′), 93.8 (C-1′), 89.5 (C-5a′′), 86.5 (C-2′), 51.8 (C-9a′′), 33.2 (C-9′′), 27.2 (C-6′′), 21.1 (C-8′′), 19.3 (C-7′′). 15N NMR (51 MHz, CD2Cl2): δ −72.8 ppm. LC-MS (ESI), [m/z]: 313.3 [M + 2H]2+, 625.4 [M + H]+.
:Et2O (1:1) as eluent. Compound 5 was obtained as a yellow solid (17 mg, 47% yield) along with the monocoupled analogue. 1H NMR (400 MHz, CD2Cl2) δ 7.60 (m, 1H, H13), 7.51 (dt, J = 8.2, 0.7 Hz, 1H, H3), 7.35 (m, 1H, H14), 6.99 (d, J = 8.2 Hz, 1H, H4), 4.86 (dt, J = 7.2, 5.2 Hz, 1H, H5a), 3.30 (q, J = 7.2 Hz, 1H, H9a), 2.04–1.86 (m, 3H, C6 and C9), 1.79–1.66 (m, 1H, C9), 1.58–1.39 (m, 4H, C7 and C8). 13C NMR (101 MHz, CD2Cl2) δ 157.3 (C9b), 153.7 (C2), 135.0 (C4a), 132.3 (C13), 128.8 (C14), 127.8 (C3), 126.1 (C12), 116.3 (C4), 94.1 (C10), 86.3 (C11), 83.8 (C5a), 41.6 (C9a), 28.2 (C6), 26.7 (C9), 22.7 (C8), 20.8 (C7). 15N NMR (CD2Cl2) δ −71.25 (ppm). Enantiomer 5b [α]20D +130 (c 2 mg mL−1, CH2Cl2).
:0 to 96:4) provided pure 25 (104 mg, 0.201 mmol, 57%). 1H NMR (500 MHz, CDCl3): δ 7.53 (dd, J = 5.8, 3.3 Hz, 2H, H-3, H-6), 7.28–7.20 (m, 10H, H-4, H-5, H-5′′′, H-6′′′), 7.16–7.12 (m, 4H, H-4′′, H-7′′), 6.86 (dd, J = 7.3, 0.8 Hz, 2H, H-3′′), 6.02 (dd, J = 8.5, 0.8 Hz, 2H, 5′′-H), 5.02 (d, J = 6.2 Hz, 2H, NH), 4.56 (p, J = 6.5 Hz, 2H, H-1′′′), 1.44 (d, J = 6.7 Hz, 6H, H-2′′′). 13C NMR (126 MHz, CDCl3): δ 158.0 (C-6′′), 144.5 (C-3′′′), 141.6 (C-2′′), 137.7 (C-4′′), 132.3 (C-3, C-6), 128.9 (C-6′′′), 128.5 (4, 5), 127.3 (C-7′′′), 126.0 (C-5′′), 125.8 (C-1, C-2), 117.7 (C-3′′), 106.4 (C-5′′), 93.5 (C-2′), 86.7 (C-1′), 52.3 (C-1′′′), 24.7 (C-2′′′). 15N NMR (51 MHz, CDCl3): δ −285 (N-1′′′), −115 (N-1′′). HRMS (TurboSpray TOF) calcd for C36H30N4 519.2549, found m/z 519.2566.
:10, Rf = 0.41 product, Rf = 0.16 mono-methylated product) gave pure 26 (75 mg, 0.137 mmol, 41%). Analytical HPLC was performed using a Lux® 5 μm i-Amylose-1 column (250 × 4.6 mm) using hexane/iPrOH (90:10 to 20:80 [10 min], 20:80 [10 min] 0.75 ml min−1) providing (1R)-26 with 7.882 min retention time. 1H NMR (500 MHz, CD2Cl2) δ 7.67–7.60 (AA′BB′, 2H, H-3, H-6), 7.40–7.34 (AA′BB′, 2H, H-4, H-5), 7.35–7.23 (m, 12H, H-4′′, H-2′′′′, H-3′′′′, H-4′′′′), 6.96 (d, J = 7.2 Hz, 2H, H-3′′), 6.50 (d, J = 8.6 Hz, 2H, H-5′′), 6.19 (q, J = 7.0 Hz, 2H, H-1′′′), 2.73 (s, 6H, H-4′′′), 1.56 (d, J = 7.0 Hz, 6H, H-2′′′). 13C NMR (126 MHz, CD2Cl2) δ 159.2 (C-6′′), 143.0 (C-1′′′), 141.4 (C-2′′), 137.8 (C-4′′), 132.8 (C-6), 129.0 (C-5), 128.9 (C-3′′′′), 127.5 (C-2′′′′), 127.3 (C-4′′′′), 126.1 (C-1, C-2), 116.8 (C-3′′), 106.4 (C-5′′), 94.7 (C-2′), 86.1 (C-1′), 52.5 (C-1′′), 30.6 (C-4′′′), 16.6 (C-2′′′). 15N NMR (51 MHz, CD2Cl2): δ −296 (N3′′′), −116 (N1′′). HRMS (TurboSpray TOF) calcd for C38H34N4 [M + H]+ 547.2862, found m/z 547.2838.
(1R,2R)-N1,N2-Bis(4-chloroquinolin-2-yl)-1,2-diphenylethane-1,2-diamine (8) and (1R,2R)-1,2-diphenyl-N,1N2-bis(4-(pyrrolidin-1-yl)quinolin-2-yl)ethane-1,2-diamine (9) were prepared following the procedure of Johnston.39
Synthesis of (7R,7′R)-(1,2-phenylenebis(ethyne-2,1-diyl))bis(4-methyl-6,7-dihydro-5H-cyclopenta[b]pyridine-2,7-diyl) diacetate (2)
:MeOH 9:1) afforded compound 2 (455 mg, 81%) as a yellow oil that solidified upon standing in the fridge. The spectroscopic data was in agreement with that reported in the literature.611H NMR (500 MHz, CDCl3): δ 6.90 (s, 1H, H-3), 2.98 (dd, J = 7.5, 7.5 Hz, 2H, H-8), 2.82 (dd, J = 7.5, 7.5 Hz, 2H, H-6), 2.122 (s, 3H, CH3), 2.11 (m, 2H, CH2-5).
:Et2O (1:1) as eluent afforded compound 3 (615 mg, 48%) as a yellow oil. The spectroscopic data was in agreement with that reported in the literature.60,611H NMR (400 MHz, CDCl3): δ 9.8 (br s, 1H, OH), 7.24 (s, 1H, H-3), 3.22 (dd, J = 7.8, 7.8 Hz, 2H, H-8), 2.92 (dd, J = 7.8, 7.8 Hz, 2H, H-6), 2.22 (d, J = 0.9 Hz, 3H, CH3), 2.20 (m, 2H, CH2-5), 2.05 (s, 3H).
:H2O (3:1, 3 mL) was added lithium hydroxide monohydrate (220 mg, 5.2 mmol) and the resultant black solution was stirred at room temperature for 21 h. The reaction mixture was then diluted with HO and extracted with DCM. The combined organic phases were washed with sodium chloride aq. sol., dried over sodium sulfate and concentrated in vacuo to afford the corresponding crude product. Flash chromatography with hexane:EtOAc (1:1) as eluent afforded compound 13 (189 mg, 79%) as a white crystalline solid. The spectroscopic data was in agreement with that reported in the literature.621H NMR (400 MHz, CDCl3): δ 7.04 (s, 1H, H-3), 5.15 (m, 1H, H-8), 2.90–3.02 (m, 1H), 2.75–2.66 (m, 1H), 2.60–2.49 (m, 1H) 2.22 (s, 3H, CH3), 2.09–1.99 (m, 1H).
:EtOAC 1:1). Compound (R)-13 (100 mg, 44%) was obtained as a yellow oil and (S)-12 (75 mg, 41%) as a white crystalline solid. See NMR data given above.
:EtOAc (1:1) as eluent. Compound 14 was obtained as a brown oil, which was used in the next step without further purification. A solution of TBAF in THF (1.0 M, 0.5 mL, 0.5 mmol, 1.1 eq.) was added to a solution of the TIPs-protected alkyne in dry THF (4.4 mL). The reaction mixture was stirred at 0 °C for 2 h under N2 atmosphere. After being warmed to room temperature, the reaction mixture was diluted in Et2O and washed twice with brine. The organic phase was dried over Na2SO4 and the solution was subsequently concentrated under reduced pressure. The crude mixture was purified by column chromatography on silica gel with hexane:EtOAc (7:3) as eluent to give 1463 as a brown solid (80 mg, 84% (over two steps)). 1H NMR (500 MHz, CDCl3) δ 7.24 (s, 1H, H3), 6.03 (dd, J = 7.5, 4.2 Hz, 1H, H7), 3.10 (s, 1H, H12), 3.0–2.96 (m, 1H, H6), 2.86–2.77 (m, 1H, H6), 2.67–2.57 (m, 1H, H5), 2.28 (s, 3H, -Me4,), 2.10 (s,3H, -Me9), 2.08–2.03 (m, 1H, H5). 13C NMR (126 MHz, CDCl3) δ 170.9 (C8), 160.5 (C7a), 144.5 (C4), 141.6 (C2), 137.7 (C4a), 128.3 (C3), 83.1 (C7), 77.5 (C10), 76.5 (C12), 30.5 (C6), 26.7 (C5), 21.4 (-Me9), 18.6 (Me-4). LC-MS (ESI), [m/z]: 216.3 [M + H]+.
Synthesis of 1,2-bis(((6R,8R)-7,7-dimethyl-5,6,7,8-tetrahydro-6,8-methanoisoquinolin-3-yl)ethynyl)benzene (3)
:1) for the elution of secondary products, and with EtOAc/MeOH (9:1) to give 15 (500 mg, 20%) as a brown foam, which was used in the next step without further purification. The analytical data was in agreement with that reported.641H NMR (400 MHz, CDCl3) δ 6.88 (s, 1H), 6.39 (s, 1H), 2.89 (d, J = 1.5 Hz, 2H), 2.68–2.54 (m, 2H), 2.17 (tt, J = 5.8, 2.9 Hz, 1H), 1.34 (s, 1H), 1.17 (d, J = 9.6 Hz, 1H), 0.67 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 165.3, 152.4, 127.6, 126.4, 118.3, 43.9, 40.2, 39.5, 32.9, 32.4, 26.00, 21.6.
:1) to give compound 16 (1160 mg, 24%) as a yellow oil along, with recovered starting material 15 (1040 mg). 1H NMR (400 MHz, CDCl3) δ 7.88 (s, 1H, H-4), 6.95 (s, 1H, H-1), 3.03 (d, J = 3.0 Hz, 2H, H-5), 2.88 (t, J = 5.5 Hz, 1H, H-8), 2.72 (dt, J = 9.9, 5.7 Hz, 1H, H-9), 2.31 (tt, J = 5.7, 2.9 Hz, 1H, H-6), 1.41 (s, 3H, CH3), 1.19 (d, J = 9.9 Hz, 1H, H9), 0.62 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 154.6 (C, C-3), 150.7 (C, C-8a), 144.0 (CH, C-1), 143.7 (C, C-4a), 123.5 (C, OTf), 120.3 (C, OTf), 117.2 (C, OTf), 114.3 (CH, C-4), 114.0 (C, OTf), 44.1 (CH, C-8), 39.6 (CH, C-6), 39.3 (C, C-7), 33.2 (CH2, C-5), 31.6 (CH2, C-9), 25.9 (CH3), 21.4 (CH3). 19F NMR (376 MHz, CDCl3) δ −73.36. HRMS (MALDI TOF) calcd for C13H15F3NO3S 322.0724 [M + H]+, found m/z 3220716. [α]20D −36.5 (c 1.27, CDCl3).
:1, 30 mL) was treated with K2CO3 (800 mg, 5.8 mmol) at r.t. for 2.5 h. Following the removal of organic solvent, H2O was added and the aqueous solution was extracted with CH2Cl2 trice. The CH2Cl2 solution was dried over Na2SO4. After solvent evaporation, the residue was purified by column chromatography on silica gel with hexane as eluent to give 1,2-diethynylbenzene (270 g, 77% yield) as colorless oil, which solidified upon standing in the freezer. The spectroscopic data was in agreement with that previously reported.671H NMR (400 MHz, CDCl3) δ (ppm) δ 7.52 (dd, J = 5.7, 3.4 Hz, 1H), 7.31 (dd, J = 5.7, 3.4 Hz, 1H), 3.34 (s, 1H).
Synthesis of 1,2-bis-[((5aS,9aR)-9a-phenyl-5a,6,7,8,9,9a-hexahydrobenzofuro[3,2-b]pyridin-2-yl)-ethinyl]benzene (4)
:1 (250 mL) for 5 min. The solution was cooled to 0 °C. The cooling bath was removed and cyclohex-2-enone (5.40 mL, 55.8 mmol, 1.00 eq.) was added, by fast addition of the first 2 mL and the remaining volume added dropwise. The mixture was stirred for 1.5 h, complete conversion was confirmed by NMR of the reactions mixture. The mixture was cooled to 0 °C and was neutralized with aqueous 2 M HCl solution, extracted with Et2O (4 times of the THF volume), and the organic layer was 5 times carefully (!) washed with 1 M HCl to remove DMAP (causes deiodination). Washing with brine and finally with Na2S2O3 solution removes Iodine and the organic layer was dried over Na2SO4. Removal of the solvent gave a pure product; if necessary purification can be performed by column chromatography (silica gel ∼3 g mmol−1, n-hexane/EtOAc (10:90) to (30:70) [10CV], then (30:70) [5 CV]) to yield a yellow solid (10.6 g, 47.6 mmol, 85%). The product is volatile and should not be dried over vacuum or be exposed to heat. It was stored at −20 °C and under N2 atmosphere. On long time storage, C-2 homo-coupling was observed. The analytical data was in agreement with that reported.681H NMR (400 MHz, CDCl3): δ 7.76 (q, 3JHH = 4.3 Hz, 1H, H-3), 2.72–2.60 (m, 2H, H-6), 2.49–2.38 (m, 2H, H-4), 2.15–1.99 (m, 2H, H-5). 13C NMR (100 MHz, CDCl3): δ 192.3 (C-1), 159.6 (C-3), 104.0 (C-2), 37.4 (C-6), 30.1 (C-4), 23.0 (C-5).
:1 v/v) was added. PhB(OH)2 (4.49 g, 36.8 mmol, 1.50 eq.) and 2-iodocyclohex-2-en-1-one (17) (5.45 g, 24.6 mmol, 1.00 eq.) were added, and the mixture was stirred for 18 h at room temperature. It was filtered twice by vacuum filtration through a P2 frit, and the resulting dark red solution was extracted with Petroleum ether (4 × 100 mL). The combined organic layer was washed with brine (3 × 100 mL), dried over MgSO4, and the solvent was removed in vacuum. Flash column chromatography (50 g silica /3 g crude, n-hexane/EtOAc 10:90 to 20:80 [5CV], then 20:80 [5CV]) gave the desired product as a white solid (3.17 g, 18.4 mmol, 75%). The analytical data were in agreement with that reported in the literature.691H NMR (400 MHz, CDCl3): δ 7.38–7.27 (m, 5H, 8-H, 9-H, 10-H, 11-H, 12-H), 7.03 (t, 3JHH = 4.3 Hz, 1H, 3-H), 2.63–2.58 (m, 2H, 5-H), 2.54 (td, 3JHH = 6.0, 4.3 Hz, 2H, 4-H), 2.12 (p, 3JHH = 6.2 Hz, 2H, 5-H). 13C NMR (101 MHz, CDCl3): δ 198.1 (C-1), 148.1 (C-3), 140.6 (C-7), 136.7 (C-2), 128.8 and 128.1 (C-8,9,11,12), 127.7 (C-10), 39.2 (C-6), 26.8 (C-4), 23.1 (C-5). LC-MS (ESI), [m/z]: 173.3 [M + H]+
:40 to 70:30 in 30 min), 1 ml min−1, retention time of (S)-19 = 18.6 min, and of (R)-19 = 9.3 min. The enantiomers can be separated by preparative HPLC on a Lux® 5 μm Amylose-1 column (50 × 21.2 mm), with H2O/CH3CN (20:80) eluent mixture at 20 ml min−1 flow rate (loading: ∼200 mg per run in 2 mL CH3CN). The spectroscopic data of the enantiomers was in agreement with that previously reported.701H NMR (400 MHz, CD2Cl2): δ 7.46 (d, 3JHH = 7.4 Hz, 2H, 8-H, 12-H), 7.33 (t, 3JHH = 7.6 Hz, 2H, 9-H, 11-H), 7.27–7.22 (m, 1H, 10-H), 6.16 (t, 3JHH = 4.1 Hz, 1H, 3-H), 4.68 (br. q, 3JHH = 4.6 Hz, 1H, 1-H), 2.32–2.10 (m, 2H, 4-H), 1.96–1.63 (m, 4H, 5-H, 6-H), 1.61 (d, 3JHH = 5.92 Hz, 1H, O–H). 13C NMR (101 MHz, CD2Cl2): δ 141.1 (C-7), 139.7 (C-2), 129.2(C-3), 129.0 (C-8, C-12), 127.5 (C-10), 126.5 (C9, C-11), 65.9 (C-1), 32.4 (C-6), 26.6 (C-4), 17.9 (C-5). LC-MS (ESI), [m/z]: 157.2 [M + H-H2O]+.
:80) and 20 ml min−1 flow rate, with 200 mg per run dissolved in 2 mL CH3CN for loading. Analytical HPLC using a Lux® 5 μm Amylose-1 column (250 × 4.6 mm), H2O/CH3CN (60:40 to 70:30 in 30 min), 1 ml min−1, retention time (S)-20 = 16.8 min, (R)-20 = 9.3 min 1H NMR (400 MHz, CDCl3): δ 7.47 (d, 3JHH = 7.7 Hz, 2H, 8-H, 12-H), 7.34 (t, 3JHH = 7.5 Hz, 2H, 9-H, 11-H), 7.27 (d, 3JHH = 7.1 Hz, 1H, 10-H), 6.17 (t, 3JHH = 4.0 Hz, 1H, 3-H), 4.72 (q, 3JHH = 4.6 Hz, 1H, 1-H), 2.34–2.11 (m, 2H, 4-H), 2.03–1.65 (m, 4H, 5-H, 6-H), 1.64–1.59 (m, 1H, 13-H). 13C NMR (101 MHz, CDCl3): δ 140.3 (C-7), 139.2 (C-2), 128.8 (C-3), 128.7 (C-9, C-11), 127.2 (C-10), 126.1 (C-8, C-12), 65.6 (C-1), 31.7 (C-6), 26.2 (C-4), 17.5 (C-5). HRMS (MALDI TOF) calcd for C11H11ClINO 335.9652 [M + H]+, found m/z 335.9648.
:5 v/v [3CV], then 95:5 to 93:7 over [5CV], then 93:7 [5CV]) gave pure 21. Rac-21 was obtained as a colourless oil (3.3 g, 8.02 mmol, 98%). When enantiopure (R)-2-phenyl-cyclohex-2-enol (20) was used as starting material, (S)-6-chloro-2-iodo-3-((2-phenylcyclohex-2-ene-1-oxy)pyridine was observed in a mixture with its R-enantiomer. Thus ee is lost under the reaction conditions. Separation of the enantiomers was performed by preparative HPLC using a Lux® 5 μm Amylose-1 column (250 × 21.2 mm). Optimized conditions for the separation of (S)-21: H2O/CH3CN 30:70, 22 ml min−1 flow; loading: ∼100 mg per run sample in 0.5 mL CH3CN. Optimized conditions for (R)-21 (major product from (R)-Me-CBS): H2O/CH3CN 30:70, 22 mL min−1 flow rate; loading: ∼200 mg per run sample in 1 mL CH3CN. Analytical HPLC using a Lux® 5 μm Amylose-1 column (250 × 4.6 mm), with H2O/CH3CN (60:40 to 70:30 in 30 min), eluent at 1 ml min−1, eluting (S)-21 at 18.3 min, and (R)-21 at 21.6 min retention time. 1H NMR (400 MHz, CDCl3): δ 7.34–7.20 (m, 5H, 8′-H, 9′-H, 10′-H, 11′-H, 12′-H), 7.13 (d, 3JHH = 8.4 Hz, 1H, 5-H), 7.00 (d, 3JHH = 8.5 Hz, 1H, 4-H), 6.34 (dd, 3JHH = 5.1, 2.9 Hz, 1H, 3′-H), 5.13 (d, 3JHH = 3.7 Hz, 1H, 1′-H), 2.52–2.36 (m, 1H, 4′-Ha), 2.33–2.17 (m, 1H, 4′-Hb), 2.16–2.07 (m, 1H, 6′-Hb), 2.07–1.92 (m, 1H, 5′-Ha), 1.81 (dt, J = 13.4, 3.4 Hz, 1H, 6′-Ha), 1.77–1.69 (m, 1H, 5′-Hb). 13C NMR (101 MHz, CDCl3): δ 153.8 (C′–3), 141.5 (C′–6), 141.0 (C′–7′), 135.5 (C′–2′), 132.3 (C′–3′), 128.6 (C′–9′, C′–11′), 127.3 (C′–10′), 126.2 (C′–8′, C-12′), 123.6 (C′–5), 122.3 (C′–4), 111.4 (C′–2), 75.1 (C′–1′), 27.7 (C′–6′), 25.9 (C′–4′), 17.1 (C′–5′). LC-MS (ESI), [m/z]: 414.1 [M(37Cl) + H]+, 412.1 [M(35Cl) + H]+, 258.1 [M(37Cl)-C12H13]+, 256.1 [M(35Cl)-C12H13]+.
:3 to 90:10 v/v [10CV]) yielding (5aS,9aR)-mix of regioisomers of 21 (940 mg, 3.31 mmol, 60%) (5aR,9aS)-mix of regioisomers of 21 (740 mg, 2.61 mmol, 57%). All fractions that showed absorption at 300 nm were collected and used for the next step. (Note: finish the reaction after 4–8 h to prevent diasteroisomerisation by double bond migration. Desired product fractions can be identified by the phenyl UV absorption at 300 nm.) The mixed fractions containing 21 were dissolved in MeOH (1 mL/10 mg crude material) and Rh on carbon (5 mg/10 mg crude material, 5% metal loading) was added. 5 bar H2 was applied and the mixture was stirred until completion of the reaction was indicated by LC-MS. The product was purified by column chromatography (silica, ∼10 g mmol−1 starting material, 60 mL min−1, n-hexane/EtOAc 90:10 v/v [1CV] then gradient 90:10 to 70:30 v/v [10CV]) yielding (5aS,9aR)-2-chloro-9a-phenyl-5a,6,7,8,9,9a-hexahydrobenzofuro[3,2-b]pyridine, 660 mg, 2.31 mmol, 70%) or (5aR,9aS)-2-chloro-9a-phenyl-5a,6,7,8,9,9a-hexahydrobenzofuro[3,2-b]pyridine, 360 mg, 1.26 mmol, (48%). (Note: the starting material and the product are difficult to separate, and so complete conversion is necessary. Longer reaction times cause hydration of the Ph ring to Cy which is easier to separate.) Analytical HPLC (Lux® 5 μm Amylose-1 column (250 × 4.6 mm), H2O/CH3CN (50:50) with 1 ml min−1 flow rate eluting (5aS,9aR)-22 at 10.82 min, and (5aR,9aS)-22 at 11.46 min retention time. (5aR,9aS)-221H NMR (500 MHz, CDCl3): δ 7.42–7.38 (m, 2H, 2′-H, 6′-H), 7.33 (t, J = 7.6 Hz, 2H, 3′-H, 5′-H), 7.25–7.21 (tt, J = 7.3, 1.3 Hz, 1H, 4′-H), 7.06 (AB-spin system of higher order, 2H, 3-H, 4-H), 5.10 (t, J = 4.8 Hz, 1H, 5a-H), 2.26–2.19 (ddd, J = 14.0, 8.5, 3.6 Hz, 1H, 9-Ha), 2.17–2.10 (m, 1H, 9-Hb), 2.00–1.91 (m, 2H, 6-H), 1.68–1.55 (m, 3H, 7-Ha, 8-Ha, 8-Hb), 1.49–1.40 (m, 1H 7-Hb). 13C NMR (126 MHz, CDCl3): δ 157.7 (C-10), 152.3 (C-5), 143.6 (C-1′), 142.6 (C-2), 128.6 (C-3′, C-5′), 127.2 (C-2′, C-6′), 126.9 (C-4′), 122.8 and 119.5 (C-3, C-4), 89.5 (C-5a), 51.6 (C-9a), 33.3 (C-9), 27.0 (C-6), 20.9 (C-7), 19.3 (C-8).
:2 v/v [10CV]) gave the pure product ((5aR,9aS)-23 (590 mg, 1.37 mmol, 80%) Alternatively, (5aS,9aR)-23 was synthesized in (494 mg, 1.14 mmol, 67%). Analytical HPLC was performed on a Lux® 5 μm Amylose-1 column (250 × 4.6 mm), using H2O/CH3CN (30:70) as eluent with 1 ml min−1 flow rate, eluting the 5aS,9aR-23 at 11.29 min, and (5aR,9aS)-22 at 10.94 min retention times.
:5 v/v [10CV]) gave pure (5aS,9aR)-24 (488 mg, 1.69 mmol, 95%), or the corresponding procedure (5aR,9aS)-24 (330 mg, 1.20 mmol, 88%). Analytical HPLC was performed on a Lux® 5 μm Amylose-1 column (250 × 4.6 mm) using H2O/CH3CN (60:40) eluent at 1 ml min−1 flow rate, giving (5aS,9aR)-24 at 20.40 min, and (5aR,9aS)-24 at 21.19 min retention time. (5aS,9aR)-24. 1H NMR (500 MHz, CDCl3): δ 7.42–7.37 (m, 2H, 2′-H, 6′-H), 7.33–7.29 (m, 2H, 3′-H, 5′-H), 7.27 (d, J = 12.4 Hz, 1H, 3-H), 7.21 (tt, J = 7.3, 1.1 Hz, 1H, 4′-H), 7.02 (d, J = 8.3 Hz, 1H, 4-H), 5.11 (t, J = 5.0 Hz, 1H, 5a-H), 3.06 (s, 1H, 2′′-H), 2.21 (t, J = 6.0 Hz, 2H, 9-H), 2.04–1.94 (m, 1H, 6-Ha), 1.94–1.85 (m, 1H, 6-Hb), 1.66–1.49 (m, 3H, 7-Ha, 7-Hb, 8-Ha), 1.47–1.37 (m, 1H, 8-Hb). 13C NMR (126 MHz, CDCl3): δ 158.0 (C-10), 153.1 (C-5), 143.8 (C-1′), 134.1 (C-2), 128.4 (C-3′, C-5′), 127.5 (C-3), 127.2 (C-2′, C-6′), 126.7 (C-4′), 116.4 (C-4), 89.3 (C-5a), 83.4 (C-1′′), 75.5 (C-2′′), 51.5 (C-9a), 33.1 (C-9), 27.0 (C-6), 20.9 (C-8), 19.3 (C-7). LC-MS (ESI), [m/z]: 276.3 [M + H]+.
Synthesis of 1,2-bis(((5aS,9aS)-5a,6,7,8,9,9a-hexahydrobenzofuro[3,2-b]pyridin-2-yl)ethynyl)benzene (5)
:1) yielded compound 25 (600 mg, 92%) as a yellow oil. 1H NMR (800 MHz, CDCl3) δ 7.17 (d, J = 8.4 Hz, 1H, H5), 7.02 (d, J = 8.4 Hz, 1H, H-4), 6.07–6.00 (m, 1H, H-9), 5.83 (m, 1H, H-8), 4.74 (m, 1H, H-7), 2.18 (m, 1H, H-10), 2.10–2.01 (m, 1H, H-10), 1.98–1.88 (m, 3H, H-12 and H-11), 1.72–1.63 (m, 1H, H-11). 13C NMR (201 MHz, CDCl3) δ 153.8 (C-3), 141.5 (C-6), 134.0 (C-9), 124.5 (C-8), 123.6 (C-5), 122.5 (C-4), 111.3 (C-2), 73.9 (C-7), 28.3 (C-12), 25.1 (C-10), 18.8 (C-11). HRMS (MALDI TOF) calcd for C11H11ClINO 335.9652 [M + H]+, found m/z 335.9652.
:1) yielded 26 (50 mg, 40% yield) along with the 2-chloro-5-(cyclohex-2-en-1-yloxy)pyridine, that is deiodinated 25 (30 mg, 24% yield) and 25 (10 mg). The two enantiomers of 26 were separated on HPLC. Analytical HPLC was performed using a Lux 5μm Amylose-1 column (50 × 4.6 mm) using i-PrOH:CH3CN 4:1 with 1 mg mL−1 flow rate. Preparative HPLC was performed on a Lux 5μm Amylose-1 column (250 × 21.6 mm) using i-PrOH:CH3CN (40:30) as eluent with 15 ml min−1 (4 min) and subsequently 21 ml min−1 flow rate. Enantiomer 26a [α]20D −110 (c 9.9 mg mL−1, CH2Cl2), enantiomer 26b [α]20D +94 (c 9.8 mg mL−1, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.05–6.97 (m, 2H, H-3 and H-4), 4.81 (dt, J = 7.2, 4.9 Hz, 1H, H-5a), 3.23 (q, J = 7.2 Hz, 1H, H-9a), 2.01–1.92 (m, 2H, H9 and H-6), 1.91–1.82 (m, 1H, H-6), 1.69–1.58 (m, 1H, H-9), 1.56–1.46 (m, 3H, H-3, H-7 and H-8), 1.38 (m, 1H, H-8). 13C NMR (101 MHz, CDCl3) δ 156.5 (C-9b), 152.6 (C-9a), 142.2 (C-2), 122.4 (C-py), 119.0 (C-py), 83.5 (C-5a), 41.2 (C-9a), 27.6 (C-6), 26.5 (C-9), 21.9 (C-8), 20.2 (C-7). HRMS (MALDI TOF) calcd for C11H12ClNO 210.0686 [M + H]+, found m/z 210.0675.
:Et2O (90:10) as eluent. Compound 27 was obtained as a pale brown oil (75 mg, 68% yield). 1H NMR (400 MHz, CDCl3) δ 7.25 (d, J = 8.2 Hz, 1H, H-3), 6.94 (d, J = 8.2 Hz, 1H, H-4), 4.77 (dt, J = 7.0, 4.7 Hz, 1H, H-5a), 3.20 (q, J = 7.0 Hz, 1H, H-9a), 2.21–1.92 (m, 2H, H-9 and H-6), 1.93–1.65 (m, 1H, H6), 1.65–1.46 (m, 4H, H9 and H8 and H7), 1.45–1.29 (m, 1H, H-8), 1.12 (s, 21H, TIPS). 13C NMR (101 MHz, CDCl3) δ 156.9 (C-9b), 153.0 (C-4a), 135.1 (C-2), 127.8 (C-3), 115.1 (C-4), 106.6 (C10), 89.0 (C-11), 83.2 (C-5a), 41.2 (C-9a), 27.6 (C-6), 26.8 (C-9), 22.0 (C-8), 20.2 (C-7), 18.8 (CH3, TIPS), 11.5 (C, TIPS).
:2) to give 28 as crystalline brown solid (33 mg, 78% yield). Enantiomer 28a [α]20D −137 (c 2.7 mg mL−1, CH2Cl2). Enantiomer 28b [α]20D +125.5 (c 4.5 mg mL−1, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.25 (d, J = 8.2 Hz, 1H, H-3), 6.96 (d, J = 8.2 Hz, 1H, H-4), 4.82 (dt, J = 7.2, 5.0 Hz, 1H, H-5a), 3.24 (q, J = 7.3 Hz, 1H, H-9a), 3.05 (s, 1H, -C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, H-11), 2.06–1.82 (m, 3H), 1.76–1.58 (m, 1H), 1.57–1.45 (m, 3H), 1.45–1.32 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 157.0 (C-9b), 153.5 (C-4a), 133.7 (C-2), 127.1 (C-3), 116.0 (C-4), 83.3 (C-10), 83.3 (C-5a), 75.4 (-C11), 41.1 (C-9a), 27.6 (C-6), 26.4 (C-9), 21.9 (C-8), 20.2 (C-7). HRMS (MALDI TOF) calcd for C13H13NO 200.1075 [M + H]+, found m/z 200.1063.
CH, H-11), 2.06–1.82 (m, 3H), 1.76–1.58 (m, 1H), 1.57–1.45 (m, 3H), 1.45–1.32 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 157.0 (C-9b), 153.5 (C-4a), 133.7 (C-2), 127.1 (C-3), 116.0 (C-4), 83.3 (C-10), 83.3 (C-5a), 75.4 (-C11), 41.1 (C-9a), 27.6 (C-6), 26.4 (C-9), 21.9 (C-8), 20.2 (C-7). HRMS (MALDI TOF) calcd for C13H13NO 200.1075 [M + H]+, found m/z 200.1063.
Syntheses of 1,2-bis((6-(N-((1R)-1-phenylethyl)amino)pyridin-2-yl)ethinyl)benzene (5) and 1,2-bis((6-(N-methyl-N-((1R)-1-phenylethyl)amino)pyridin-2-yl)ethinyl)benzene (7)
:30, RF(product) = 0.21) gave pure 29 (155 mg, 0.354 mmol, 9%). 1H NMR (500 MHz, CDCl3): δ 7.88–7.82 (d, J = 8.0 Hz, 2H, H-3′′), 7.69–7.59 (m, 4H, H-4′′, H-3, H-6), 7.49–7.44 (d, J = 7.7 Hz, 2H, H-5′), 7.40 (dd, J = 5.8, 3.4 Hz, 2H, H-4, H-5). 13C NMR (126 MHz, CDCl3): δ [143.9 (C-2′′), 141.8 (C-6′′), 138.8 (C-4′′), 132.2 (C-5′′), 129.3 (C-4, C-5), 127.7 (C-1, C-2), 127.0 (C-3, C-6), 125.5 (C-3′′), 92.3 (C-2′), 89.2 (C-1′). LC-MS (ESI) [m/z]: [M + H]+ 437.0.
Computations
We applied density functional theory (DFT) in our present work to characterize the electronic structure of the investigated complexes. Truhlar's M06-2X hybrid-type exchange–correlation functional74 was used along with the Def2-SVP basis set75 for geometry optimizations and vibrational analysis. This choice for the approximated functional is justified by previous benchmark studies,76,77 which reported good performance of M06-2X for non-covalent and halogen bonding interactions, both important in our computational analysis. For each optimized structure, additional single-point energy calculations were performed with the larger Def2-TZVPP basis set.75 All these calculations (geometry optimizations and vibrational analysis as well) were carried out with the inclusion of solvent effects via the integral equation formalism of the polarizable continuum model (IEFPCM).78 The atomic radii and non-electrostatic terms in the IEFPCM calculations were those introduced by Truhlar and coworkers in terms of the SMD solvation model.79 For iodine, the atomic radius was set to 2.74 Å as suggested in the SMD18 refinement of the model.80 We used dichloromethane as a typical solvent utilized in our experiments.The thermal and entropic contributions to the Gibbs free energies were computed for 298.15 K and c = 1 mol dm−3 conditions and employing Grimme's quasi-RRHO approximation.81 This approach is expected to be more appropriate than the standard ideal gas RRHO (rigid rotor – harmonic oscillator) model, because the optimized structures of the present iodonium complexes have several low harmonic frequency modes (skeletal vibrations). The relative stabilities reported in our paper correspond to solution phase Gibbs free energies computed as G = E′0,sol + (G0,sol−E0,sol), where E′0,sol and E0,sol are solution phase electronic energies obtained at the M06-2X/Def2TZVPP and M06-2X/Def2SVP levels, respectively, and G0,sol is solution phase Gibbs free energy computed at M06-2X/Def2SVP level. All DFT calculations were carried out with the Gaussian16 software.82 The molecular structures were visualized with the CYLview program.83
The structure of the iodonium complex [4-I]+ examined computationally, particularly the singly coordinated open form, is conformationally complex, therefore, we used initial conformational screening to map possible structures. Our conformational analysis involved a Monte Carlo sampling using the OPLS_2005 force field as implemented in MacroModel,84 as well as the metadynamics sampling procedure provided by the CREST (Conformer–Rotamer Ensemble Sampling Tool)85 utility of the xtb program package.86 A large number of conformers were identified via this initial screening, and many of them (about 70 different structures) were subject to subsequent DFT calculations. We report the most stable and structurally distinct structures (ESI†).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Swedish Research Council (2016-03602, 2020-03431), FORMAS (2017-01173) and by Vinnova (2019-02160), H2020-MSCA-IF-2014 (657726) for financial support. We thank Inmaculada Rodriguez Lahoz (University of Gothenburg) for her contribution to the synthesis and characterisation of compounds 2–6. This project made use of the NMR Uppsala infrastructure, which is funded by the Department of Chemistry – BMC and the Disciplinary Domain of Medicine and Pharmacy. Part of the computations was performed on resources provided by Swedish National Infrastructure for Computing (SNIC) through National Supercomputer Center (NSC) at Linköping University under Project SNIC 2020/5-435. This work was also supported by the European Regional Development Fund via the “Support of research and development capacities in the area of nanochemical and supramolecular systems” project (code: 313011T583).Notes and references
- I. Roberts and G. E. Kimball, J. Am. Chem. Soc., 1937, 59, 947–948 CrossRef CAS.
- P. D. Barlett and D. S. Tarbell, J. Am. Chem. Soc., 1936, 58, 466–474 CrossRef.
- C. K. Ingold, Structure and Mechanism in Organic Chemistry, Cornell University Press, Ithaca, 1953 Search PubMed.
- G. A. Olah and J. M. Bollinger, J. Am. Chem. Soc., 1967, 89, 2993–2996 CrossRef CAS PubMed.
- G. A. Olah and J. M. Bollinger, J. Am. Chem. Soc., 1968, 90, 947–953 CrossRef CAS.
- G. A. Olah, J. M. Bollinger and J. Brinich, J. Am. Chem. Soc., 1968, 90, 2587–2594 CrossRef CAS.
- K. D. Ashtekar, M. Vetticatt, R. Yousefi, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2016, 138, 8114–8119 CrossRef CAS PubMed.
- A. Castellanos and S. P. Fletcher, Chem. – Eur. J., 2011, 17, 5766–5776 CrossRef CAS PubMed.
- Y. A. Cheng, W. Z. Yu and Y. Y. Yeung, Org. Biomol. Chem., 2014, 12, 2333–2343 RSC.
- A. J. Cresswell, S. T. Eey and S. E. Denmark, Angew. Chem., 2015, 54, 15642–15682 CrossRef CAS PubMed.
- S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., 2012, 51, 10938–10953 CrossRef CAS PubMed.
- C. K. Tan, L. Zhou and Y. Y. Yeung, Synlett, 2011, 1335–1339 CAS.
- U. Hennecke, Chem. – Asian J., 2012, 7, 456–465 CrossRef CAS PubMed.
- J. Chen and L. Zhou, Synthesis, 2014, 586–595 CAS.
- R. S. Brown, Acc. Chem. Res., 1997, 30, 131–137 CrossRef CAS.
- M. L. Landry and N. Z. Burns, Acc. Chem. Res., 2018, 51, 1260–1271 CrossRef CAS PubMed.
- L. Turunen and M. Erdelyi, Chem. Soc. Rev., 2020, 49, 2688–2700 RSC.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Grafenstein, K. Rissanen and M. Erdelyi, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed.
- A. Karim, M. Reitti, A.-C. C. Carlsson, J. Grafenstein and M. Erdelyi, Chem. Sci., 2014, 5, 3226–3233 RSC.
- A. C. Reiersolmoen, S. Battaglia, S. Oien-Odegaard, A. K. Gupta, A. Fiksdahl, R. Lindh and M. Erdelyi, Chem. Sci., 2020, 11, 7979–7990 RSC.
- A.-C. C. Carlsson, J. Grafenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdelyi, J. Am. Chem. Soc., 2012, 134, 5706–5715 CrossRef CAS PubMed.
- A.-C. C. Carlsson, J. Grafenstein, J. L. Laurila, J. Bergquist and M. Erdelyi, Chem. Commun., 2012, 48, 1458–1460 RSC.
- J. Barluenga, J. M. Gonzalez, P. J. Campos and G. Asensio, Angew. Chem., Int. Ed. Engl., 1985, 24, 319–320 CrossRef.
- A. A. Neverov, H. X. Feng, K. Hamilton and R. S. Brown, J. Org. Chem., 2003, 68, 3802–3810 CrossRef CAS PubMed.
- X. L. Cui and R. S. Brown, J. Org. Chem., 2000, 65, 5653–5658 CrossRef CAS PubMed.
- R. S. Brown, R. W. Nagorski, A. J. Bennet, R. E. D. Mcclung, G. H. M. Aarts, M. Klobukowski, R. Mcdonald and B. D. Santarsiero, J. Am. Chem. Soc., 1994, 116, 2448–2456 CrossRef CAS.
- D. von der Heiden, K. Rissanen and M. Erdelyi, Chem. Commun., 2020, 56, 14431–14434 RSC.
- E. Bosch and C. L. Barnes, Inorg. Chem., 2001, 40, 3097–3100 CrossRef CAS PubMed.
- S. Lindblad, K. Mehmeti, A. X. Veiga, B. Nekoueishahraki, J. Grafenstein and M. Erdelyi, J. Am. Chem. Soc., 2018, 140, 13503–13513 CrossRef CAS PubMed.
- A.-C. C. Carlsson, M. Uhrbom, A. Karim, U. Brath, J. Grafenstein and M. Erdelyi, CrystEngComm, 2013, 15, 3087–3092 RSC.
- S. Lindblad, F. B. Nemeth, T. Foldes, D. von der Heiden, H. G. Vang, Z. L. Driscoll, E. R. Gonnering, I. Papai, N. Bowling and M. Erdelyi, Chemistry, 2021 DOI:10.1002/chem.202102575.
- A. Vanderkooy, A. K. Gupta, T. Foldes, S. Lindblad, A. Orthaber, I. Papai and M. Erdelyi, Angew. Chem., Int. Ed., 2019, 58, 9012–9016 CrossRef CAS PubMed.
- U. Warzok, M. Marianski, W. Hoffmann, L. Turunen, K. Rissanen, K. Pagel and C. A. Schalley, Chem. Sci., 2018, 9, 8343–8351 RSC.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem, 2016, 55, 14033–14036 CrossRef CAS PubMed.
- G. Gong, S. Lv, J. Han, F. Xie, Q. Li, N. Xia, W. Zeng, Y. Chen, L. Wang, J. Wang and S. Chen, Angew. Chem., Int. Ed., 2021, 60, 14831–14835 CrossRef CAS PubMed.
- S. Lindblad, D. Sethio, O. B. Berryman and M. Erdelyi, Chem. Commun., 2021, 57, 6261–6263 RSC.
- J. S. Ward, A. Frontera and K. Rissanen, Chem. Commun., 2021, 57, 5094–5097 RSC.
- J. S. Ward, A. Frontera and K. Rissanen, Inorg. Chem., 2021, 60, 5383–5390 CrossRef CAS PubMed.
- M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 6068–6071 CrossRef CAS PubMed.
- D. A. Decato, A. M. S. Riel, J. H. May, V. S. Bryantsev and O. B. Berryman, Angew. Chem., Int. Ed., 2021, 60, 3685–3692 CrossRef CAS PubMed.
- A. M. S. Riel, R. K. Rowe, E. N. Ho, A. C. Carlsson, A. K. Rappe, O. B. Berryman and P. S. Ho, Acc. Chem. Res., 2019, 52, 2870–2880 CrossRef CAS PubMed.
- A. C. Legon, Angew. Chem., 1999, 38, 2686–2714 CrossRef.
- F. Romanov-Michailidis, L. Guenee and A. Alexakis, Org. Lett., 2013, 15, 5890–5893 CrossRef CAS PubMed.
- E. Bosch and C. L. Barnes, J. Coord. Chem., 2003, 56, 329–336 CrossRef CAS.
- T. Kawano, J. Kuwana, T. Shinomaru, C. X. Du and I. Ueda, Chem. Lett., 2001, 1230–1231 CrossRef CAS.
- T. Kawano, J. Kuwana and I. Ueda, Bull. Chem. Soc. Jpn., 2003, 76, 789–797 CrossRef CAS.
- N. Schultheiss, J. M. Ellsworth, E. Bosch and C. L. Barnes, J. Coord. Chem., 2008, 61, 322–327 CrossRef CAS.
- N. L. Brennan, C. L. Barnes and E. Bosch, Inorg. Chim. Acta, 2010, 363, 3987–3992 CrossRef CAS.
- S. Schonfeld, C. Lochenie, G. Horner and B. Weber, J. Phys.: Condens. Matter, 2019, 31, 504002 CrossRef PubMed.
- Y. Z. Hu, C. Chamchoumis, J. S. Grebowicz and R. P. Thummel, Inorg. Chem., 2002, 41, 2296–2300 CrossRef CAS PubMed.
- T. Kawano, T. Shinomaru and I. Ueda, Org. Lett., 2002, 4, 2545–2547 CrossRef CAS PubMed.
- M. S. Jung, Y. Suzaki, T. Saito, K. Shimada and K. Osakada, Polyhedron, 2012, 40, 168–174 CrossRef CAS.
- F. A. Pereira, T. Fallows, M. Frank, A. Q. Chen and G. H. Clever, Z. Anorg. Allg. Chem., 2013, 639, 1598–1605 CrossRef CAS.
- S. Atobe, M. Sonoda, Y. Suzuki, T. Yamamoto, H. Masuno, H. Shinohara and A. Ogawa, Res. Chem. Intermed., 2013, 39, 359–370 CrossRef CAS.
- S. Atobe, H. Masuno, M. Sonoda, Y. Suzuki, H. Shinohara, S. Shibata and A. Ogawa, Tetrahedron Lett., 2012, 53, 1764–1767 CrossRef CAS.
- Y. Suzaki, T. Saito and K. Osakada, J. Organomet. Chem., 2020, 910, 121088 CrossRef CAS.
- C. L. Barnes and E. Bosch, J. Chem. Crystallogr., 2006, 36, 563–566 CrossRef CAS.
- A. C. Reiersolmoen, D. Csokas, S. Oien-Odegaard, A. Vanderkooy, A. K. Gupta, A. C. Carlsson, A. Orthaber, A. Fiksdahl, I. Papai and M. Erdelyi, J. Am. Chem. Soc., 2020, 142, 6439–6446 CrossRef PubMed.
- A. Karim, N. Schulz, H. Andersson, B. Nekoueishahraki, A. C. Carlsson, D. Sarabi, A. Valkonen, K. Rissanen, J. Grafenstein, S. Keller and M. Erdelyi, J. Am. Chem. Soc., 2018, 140, 17571–17579 CrossRef CAS PubMed.
- A. Sakurai and H. Midorika, Bull. Chem. Soc. Jpn., 1968, 41, 165–167 CrossRef CAS.
- M. P. A. Lyle and P. D. Wilson, Org. Lett., 2004, 6, 855–857 CrossRef CAS PubMed.
- M. P. A. Lyle, A. A. Narine and P. D. Wilson, J. Org. Chem., 2004, 69, 5060–5064 CrossRef CAS PubMed.
- C. Schotes, M. Beller, K. Junge, W. Liu, J. Schneekoenig and T. Leischner, Germany Pat. WO2021058457A1, 2020.
- Y. Zhou, J. Dong, F. Zhang and Y. Gong, J. Org. Chem., 2011, 76, 588–600 CrossRef CAS PubMed.
- E. Anger, M. Rudolph, L. Norel, S. Zrig, C. Shen, N. Vanthuyne, L. Toupet, J. A. Williams, C. Roussel, J. Autschbach, J. Crassous and R. Reau, Chem. – Eur. J., 2011, 17, 14178–141798 CrossRef CAS PubMed.
- A. I. Aranda Perez, T. Biet, S. Graule, T. Agou, C. Lescop, N. R. Branda, J. Crassous and R. Reau, Chem. – Eur. J., 2011, 17, 1337–1351 CrossRef CAS PubMed.
- Q. Zhou, P. J. Carroll and T. M. Swager, J. Org. Chem., 1994, 59, 1294–1301 CrossRef CAS.
- M. E. Krafft and J. W. Cran, Synlett, 2005, 1263–1266 CrossRef CAS.
- F. X. Felpin, J. Org. Chem., 2005, 70, 8575–8578 CrossRef CAS PubMed.
- J. Hannedouche, J. A. Kenny, T. Walsgrove and M. Wills, Synlett, 2002, 263–266 CrossRef CAS.
- A. L. Gemal and J. L. Luche, J. Am. Chem. Soc., 1981, 103, 5454–5459 CrossRef CAS.
- J. L. Luche, J. Am. Chem. Soc., 1978, 100, 2226–2227 CrossRef CAS.
- A. Ford and S. Woodward, Angew. Chem., 1999, 38, 335–336 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- S. Kozuch and J. M. Martin, J. Chem. Theory Comput., 2013, 9, 1918–1931 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and E. Cances, J. Mol. Struct., 1999, 464, 211–226 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- E. Engelage, N. Schulz, F. Heinen, S. M. Huber, D. G. Truhlar and C. J. Cramer, Chem. - Eur. J., 2018, 24, 15983–15987 CrossRef CAS PubMed.
- S. Grimme, Phys. Chem. Eur. J., 2012, 18, 9955 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. Y. Legault, CYLview, v 1.0b, Universite de Sherbrooke, 2009 Search PubMed.
- L. Schrödinger, Macromodel, New York, USA, 2016 Search PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- XTB program package, https://xtb-docs.readthedocs.io/en/latest/contents.html#.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details, copies of NMR spectra and HRMS, HPLC chromatograms, details of computations. See DOI: 10.1039/d1ob01532j |
| ‡ Current address: Department of Chemistry, Umeå University, SE-901 87 Umeå, Sweden |
| § The hypocoordinate (6e) X+ is denoted as halenium, whereas the halogen of the hypercoordinate (10e) [N−X−N]+ complex as halonium ion. The former is formally a halogen bond donor (Lewis acid) that may simultaneously interact with two halogen bond acceptors (Lewis bases) and thereby form a 3-center, 4-electron halogen bonded complex. Both halenium and halonium ions are halogen(I) species. |
| This journal is © The Royal Society of Chemistry 2021 |