
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the physiochemical aspects of the matrix in improving sulfur-loading for room-temperature sodium–sulfur batteries
Sungjemmenla
,
Chhail Bihari
Soni
,
S. K.
Vineeth
and
Vipin
Kumar
 *
*
Centre for Energy Studies (CES), Indian Institute of Technology Delhi, Hauz Khas, New Delhi, Delhi 110016, India. E-mail: vkumar@ces.iitd.ac.in
First published on 14th June 2021
Abstract
The sulfur cathode in Na/S batteries possesses a very high theoretical specific capacity of about 1675 mA h g−1 and specific energy of 1230 W h kg−1 (which is over five times that of the LiCoO2 cathode in Li-ion batteries), besides high abundance and cost-effectiveness of the electrode materials. The sulfur cathode in Na/S batteries undergoes various electrochemical processes, where a series of soluble sodium polysulfides are formed during the discharge reaction, which adversely affects the operation of the cell. Furthermore, the viable application of RT-Na/S batteries is severely challenged by various obstacles, including their short-life and low-sulfur utilization, which become more serious when sulfur loading is increased to the practically acceptable level of over 5 mg cm−2. Thus, there have been innovative efforts in recent years to manipulate the physiochemistry of the matrix to overcome these barriers toward the practical application of RT-Na/S batteries with an improved sulfur loading close to practical limits. The rational design of the matrix (i.e., physicochemical aspects) with a high-sulfur utilization and long lifespan are two crucial challenges that Na/S batteries are experiencing currently and require immediate attention to be addressed. This review highlights the recent progress on tuning the physiochemistry of the matrix through chemical and physical means to realize an improved sulfur-loading. Particularly, basic insight into the chemical binding, strategies for mesoscale assembly, unique architectures, and configurational innovation in the cathode are the specific focus. Finally, novel strategies to improve sulfur-loading are proposed to guide the future development of high-sulfur loading RT-Na/S batteries.
1. Introduction
There is growing interest in high-energy rechargeable batteries for large-scale stationary energy storage.1–3 Rechargeable lithium-ion batteries with an energy density of about 180 W h kg−1 are still the first choice for portable and mobile applications, but the high cost of Li-ion batteries per kWh limits their use in stationary storage applications.3–8 Besides, the performance of Li-ion batteries has plateaued due to the fundamental limitations of the electrode materials.Recently, interest in sodium–sulfur batteries has been revived due to their unique attributes, including high theoretical specific capacity and energy density, high natural abundance and long-term sustainability.9 In addition, sodium–sulfur electrochemistry offers several other advantages, such as (1) stable-operation, (2) minimal self-discharge, (3) low-cost per cycle, (4) high energy efficiency, and (5) non-toxicity.10 Significant progress has been made in high-temperature sodium–sulfur (HT-Na/S) battery technology owing to its high theoretical energy density (760 W h kg−1) and power capability with excellent durability for over 15 years or 2500 cycles.11 HT-Na/S batteries as cutting-edge technology have found applications in space, electric vehicles grid storage, and aeronautics.12,13 Attempts have been made to utilize HT-Na/S batteries for large-scale grid storage applications. For instance, Kawakami and coworkers stabilized the fluctuating wind power of about 51 MW using a 34 MW HT-Na/S battery system in Japan.14 Similarly, other groups used HT-Na/S batteries for load-leveling in wind-farms.15 HT-Na/S battery technology has matured over time and been well constructed and developed mainly for large-scale storage applications. However, despite these achievements, HT-Na/S is severely compromised regarding safety due to its high operating temperatures (>300 °C), which are required to achieve the desired ionic conductivity of the solid–electrolyte. In addition, the reaction between molten sulfur and sodium can liberate a high enthalpy of about −420 kJ mol−1, which may further contribute to an increase in the cell temperature.16 Thus, the high operating temperature (300–350 °C), high initial cost, corrosivity and low Coulombic efficiency of HT-Na/S batteries challenge their long-term viability.
In contrast to the HT-Na/S battery, the room-temperature sodium–sulfur (RT-Na/S) battery offers a safe and reliable operation with a low operating cost,17–19 delivering a remarkably high theoretical specific energy of ∼1230 W h kg−1.20 Park and co-workers proposed the idea of developing a room-temperature analogue instead, which consists of a solid form of both sodium and sulfur electrodes.21 This battery delivered an initial discharge capacity of about 489 mA h g−1; however, it could only be cycled for 20 cycles with a drastic decrease (nearly tenfold) in its reversible capacity. Wang et al.22 investigated for the first time the use of a conductive polymer-PAN/S as the cathode material for RT-Na/S to enhance both the cycling stability and performance of the cell. The hybrid composite sulfur cathode exhibited an improved performance of about 655 mA h g−1; however, the cell was plagued with a low cycle-life (only for 18 cycles), while it retained a reversible capacity of about 500 mA h g−1. Subsequently, significant progress has been made in the development of state-of-the-art composite cathodes with remarkable progress in the rate capability and ultra-long cycle-life.23,24 Recently, attempts have been made to understand the charging/discharging mechanism in Na/S batteries via both experiment and theoretical modeling. Although the intermediate reactions are very complex, the generally accepted discharging reaction in the RT-Na/S battery follows S8 → Na2S8 → Na2S6 → Na2S4 → Na2S2 → Na2S, where the long-chain polysulfides, i.e., Na2S8 and Na2S6, readily dissolve in the electrolyte.25 In most cases, the dissolved polysulfides move towards the anode (i.e., shutting effect) and hamper the anode operation permanently, leading to rapid capacity fading and poor cycling stability.26,27 Polysulfide cross-over or shuttling can be evaded partly through physical or chemical trapping by the matrix, i.e., carbon scaffold, in most cases.28 Extensive research efforts have been focused on finding better matrix materials that can offer great absorption sites for the long-chain polysulfides to bind without compromising with electronic conductivity of the electrode materials. However, the key challenge with this technology is the low sulfur loading, which lowers its capacity and the overall specific energy.
Although good progress in the electrochemistry of cathode composites has been achieved in recent years, the low sulfur loading, which is below 3 mg cm−2, eventually hinders the cell from reaching its unprecedented theoretical value. Furthermore, when attempts were made to improve the sulfur loading in the cathode, the performance of the cell was observed to deteriorate rapidly due to the lack of efficient binding sites, strong inter-particle interaction, and optimized ratio of electrolyte and sulfur (i.e., E/S).29 Thus, to achieve a high specific energy, it is urgent to increase the loading (>3 mg cm−2) and weight percentage of sulfur.30 Pope et al. reported different cathode architectures for Li/S batteries, where they analyzed the specific energy as a function of the sulfur loading.31 They identified that a higher specific energy of about 400 W h kg−1 could be achieved with sulfur loadings greater than 2 mg cm−2. It is evident that an increase in the sulfur loading has a profound impact on the practicality of metal–sulfur battery systems. Several research groups have presented impactful reviews on the Na–S battery;32–35 however, this mini-review introduces the key issues and different aspects of the matrix towards the development of a high loading sulfur cathode for RT-Na/S batteries. Particularly, the recent advances with a judicious combination of different composite materials and representative physical and chemical aspects of cathode engineering are summarized, as schematically depicted in Fig. 1. In addition, novel strategies to improve the sulfur loading are proposed to guide the future development of high-sulfur-loading sulfur cathodes for RT-Na/S batteries.
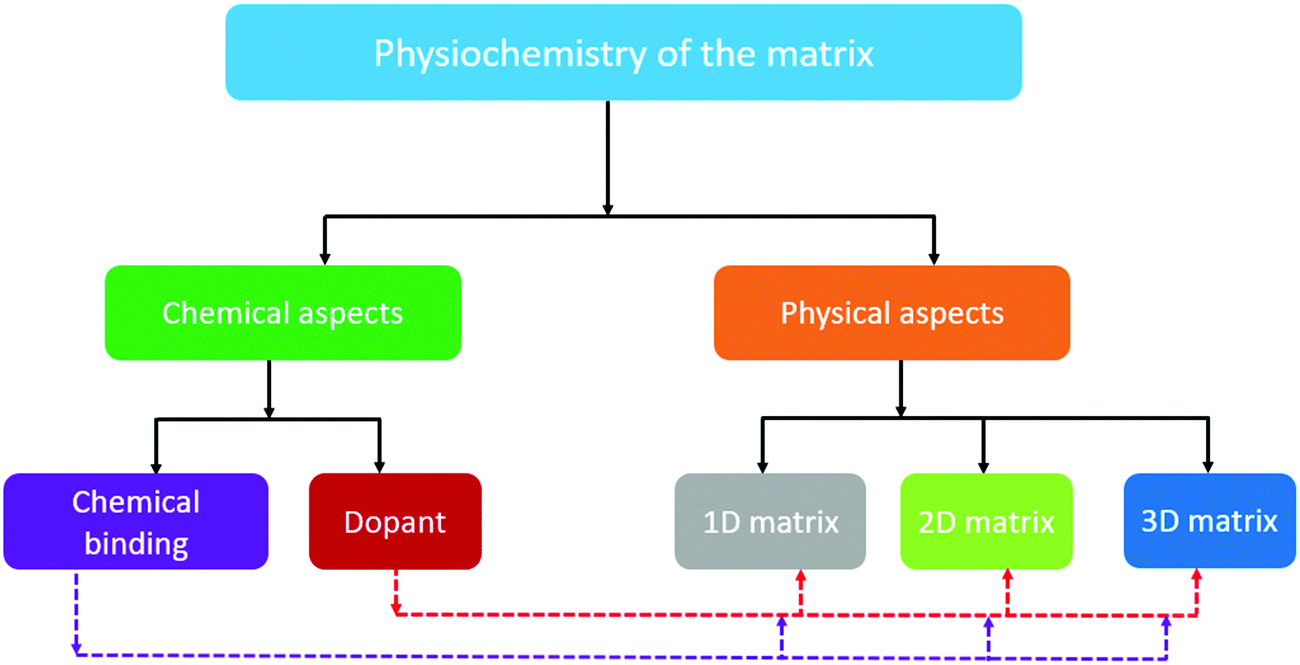 | ||
| Fig. 1 Schematic illustration of the rational design of the matrix for high-loading sulfur cathodes for RT/Na–S batteries. | ||
2. The physio-chemistry of the matrix for sulfur cathodes
A room-temperature Na–S battery is comprised of a sulfur cathode, sodium metal anode, and a separator soaked in a liquid electrolyte, as depicted schematically in Fig. 2. The sulfur cathode, which consists of elemental sulfur particles, binder, and conductive filler, undergoes severe chemical and structural changes during the charge/discharge reactions. The chemical and physical changes that occur in the matrix mainly dictate the stability of the sulfur cathode and kinetics of the conversion reactions. Although the charge/discharge reactions are very complicated, the intermediate reaction steps that occur during the reactions are presented below through a series of reactions (eqn (1)–(4)).34,36| S8 + 2Na+ + 2e− → Na2S8 (solid–liquid transition; ∼2.20 V vs. Na/Na+) | (1) |
| Na2S8 + 2Na+ + 2e− → 2Na2S4 (liquid–liquid transition; 2.20–1.65 V vs. Na/Na+) | (2) |
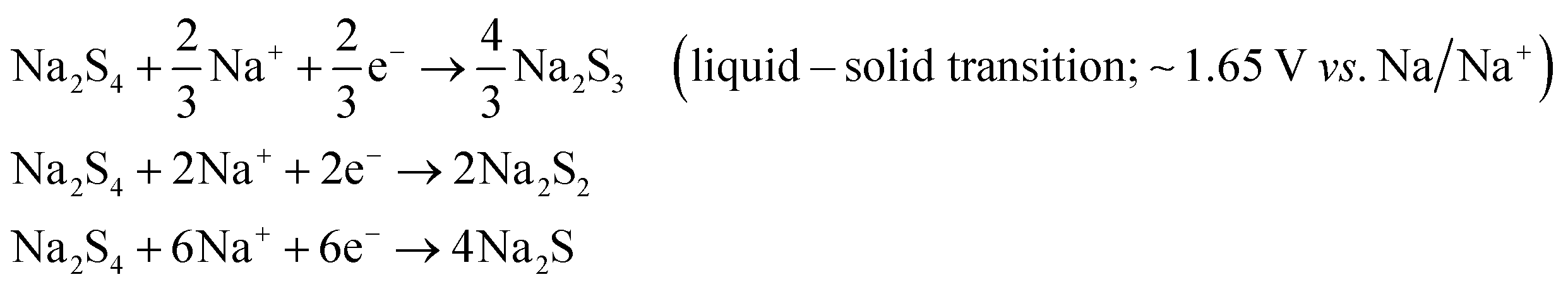 | (3) |
| Na2S2 + 2Na+ +2e− → 2Na2S (solid–solid transition; 1.65–1.20 V vs. Na/Na+) | (4) |
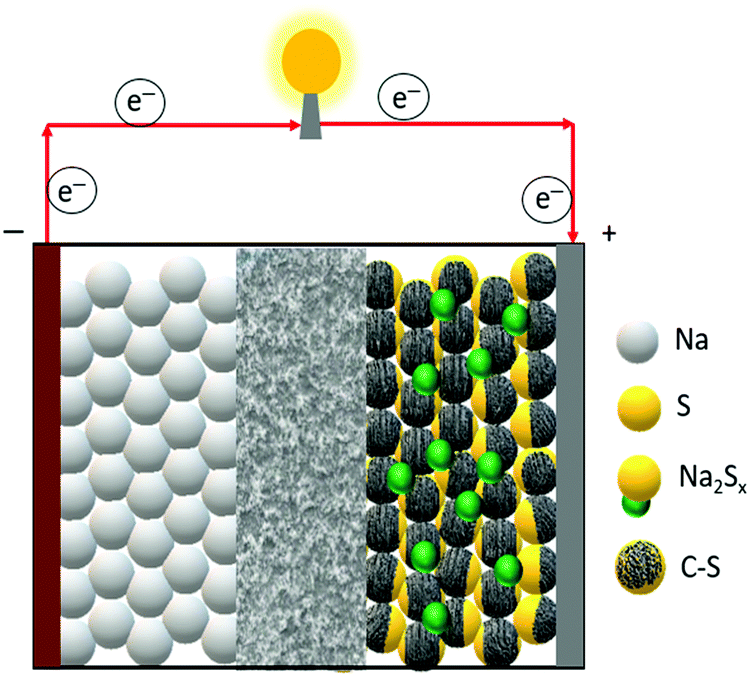 | ||
| Fig. 2 Schematic representation of an RT-Na/S battery. | ||
The overall reaction during the discharge process can be expressed as:
| Anode: 2Na → 2Na+ + 2e− |
| Cathode: 1/8S8 + 2Na+ +2e− → Na2S |
2.1 Chemical aspects
The sulfur cathode contributes two electrons to the reaction and offers a high theoretical capacity of about 1675 mA h g−1.43 However, it is immensely challenging to extract the theoretical capacity from sulfur cathode due to two major challenges. Firstly, the dissolution of high-order polysulfide intermediates (Na2S8–Na2S4, i.e., long-chain polysulfides) in organic electrolytes causes severe sulfur loss in the cathode, reducing the cycle-life of RT-Na/S. The dissolution of the high-order polysulfides also requires a flooded electrolyte (FE) to achieve a high power density, sacrificing the total energy density. Secondly, Na plating/stripping continuously consumes the Na-metal anode and electrolyte during cycling, which requires excess Na-metal and electrolyte, thus further reducing the Coulombic efficiency and energy density of RT-Na/S.To date, numerous strategies and different material chemistries have been developed to modify the matrix (i.e., sulfur host) and minimize dissolution of sodium polysulfides. To promote the kinetics of the conversion reactions, a chemically modified sulfur matrix has been demonstrated as a potential solution to enhance the performance of RT-Na/S batteries.
The other aspect that can be employed is to use binders to alter the chemical functionality of the matrix instead of polymeric coatings. The chemically altered binder approach greatly improves the structural integrity of the cathode.52–54 Although the binder accounts for only about 2 to 10 wt% of the overall electrode material, it plays a critical role in ensuring the stable performance of batteries. The primary function of a chemical binder is to provide (i) strong inter-particle adhesion and binding, (ii) abundant adsorption sites to adsorb or absorb the discharge products, and (iii) a robust and interconnected structure to prevent pulverization of the cathode material during volumetric contraction/expansion during the charge/discharge reactions.55–57 Traditionally, linear binders, such as poly(vinylidene-fluoride) (PVDF), have been used to form cathode films for RT-Na/S batteries. The polar groups (i.e., –CF2) of the PVDF binder molecules facilitate the adhesion among various components of the cathode, which include sulfur particles, conductive fillers, and the current collector. In addition, the van der Waals forces among the polymer chains provide good elasticity to accommodate the volume change upon repeated discharge/charge cycles. It is important to note that the volume change in the sulfur cathode is many times that of the conventional oxide cathode,40,58 and thus the PVDF binder encounters a significant challenge in ensuring long-term stability of the cathode, especially when the sulfur loading is high or close to the practical limits. Accordingly, by altering the molecular structure and surface chemistry of the binder molecules, researchers can explore alternative binders to promote better mechanical properties.59,60 Polar functional groups, for instance, –OH, and –COOH, have been incorporated to form covalent bonds with S–C composites and the substrate. Recently, Alex and co-workers tailored the binder interactions with the cathode materials through –COOH functional groups to gain a better perspective towards binder assembly.54 Carboxyl-containing binders were utilized as an alternative to the traditional PVDF binder due to the relatively stronger interaction of their polar groups with sodium polysulfide. Polyacrylic acid (PAA) binder, which behaves as a trap for the soluble sodium polysulfides during the sodiation cycles, can readily adsorb sodium polysulfide. The adsorption of sodium polysulfide was validated through theoretical calculations. For example, the binding energy of Na2S for PAA binder is about 2.02 eV, which is nearly 60% that of the PVDF binder (binding energy of about 1.20 eV). Scanning electron microscopy (SEM) images revealed the uniform particle distribution on the cathode surface for a PAA-based sulfur cathode, whereas the PVDF-based cathode contained large fractures and showed lower contrast due to the passivation layer on its surface (see Fig. 3a–d). As schematically illustrated in Fig. 3e, a conjugated pyridine-like backbone confirms the cyclization in forming strong interactions between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN bonds. The strong bonds formed with the carboxyl groups potentially act as a chemical trap if discharge products are inadvertently formed during sodiation. Consequently, due to these strong interactions, a sulfur content as high as 41 wt% could be achieved and the mass loading of the active material could reach about 1.8 mg cm−2. Due to the locally modified matrix structure, the rate capability tests showed high specific capacity values for the PAA-based cathodes of 1023 mA h g−1, 920 mA h g−1 and 800 mA h g−1 at 1C, 2C and 4C, respectively. In contrast, the PVDF based cathode exhibited a much lower specific capacity of 554 mA h g−1 at a C-rate of 4C.
C and CN bonds. The strong bonds formed with the carboxyl groups potentially act as a chemical trap if discharge products are inadvertently formed during sodiation. Consequently, due to these strong interactions, a sulfur content as high as 41 wt% could be achieved and the mass loading of the active material could reach about 1.8 mg cm−2. Due to the locally modified matrix structure, the rate capability tests showed high specific capacity values for the PAA-based cathodes of 1023 mA h g−1, 920 mA h g−1 and 800 mA h g−1 at 1C, 2C and 4C, respectively. In contrast, the PVDF based cathode exhibited a much lower specific capacity of 554 mA h g−1 at a C-rate of 4C.
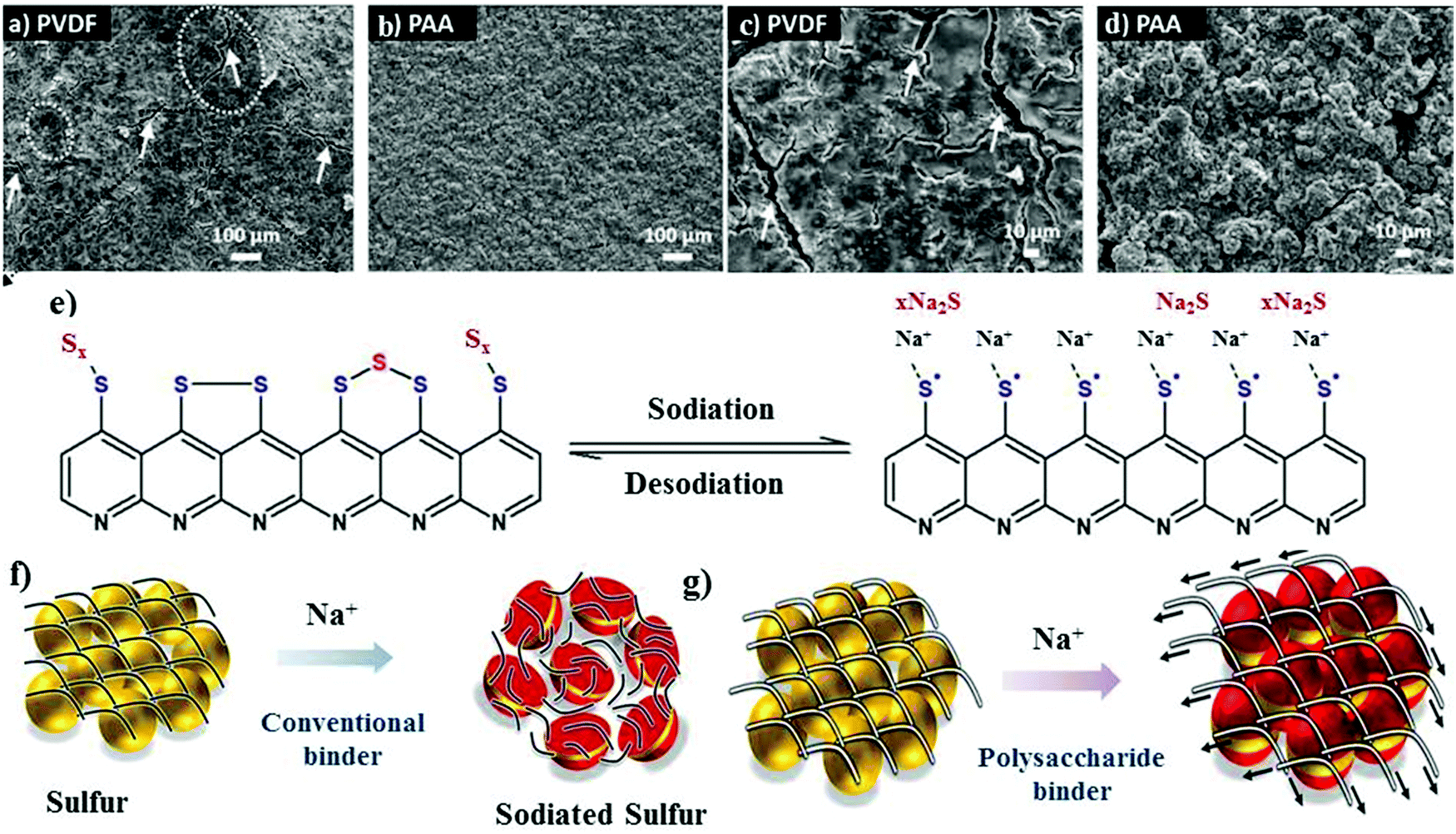 | ||
| Fig. 3 SEM images of (a and c) PVDF cathode and (b and d) PAA cathode at different magnifications. (e) Schematic illustration of the sulfur bonding modes in S-PAN with the corresponding sodiation/de-sodiation reaction. Reproduced with permission.54 Copyright 2020, The Royal Society of Chemistry. (f and g) Schematic of the proposed role of the conventional and polysaccharide binder to counteract the volume expansion of sulfur during sodium-ion insertion. Reproduced with permission.61 Copyright 2019, the American Chemical Society. | ||
Besides PAA, a hybrid sodium alginate–polyaniline matrix binder was also developed and observed to play a crucial role due to its inherent stiffness and swelling properties.61 In contrast to the conventional binder (see Fig. 3f), sodium alginate could preserve the structural integrity of the sulfur cathode during the sodium insertion and extraction process, as shown in Fig. 3g. In addition, the hybrid binder of sodium alginate–polyaniline could serve as a conductive matrix to facilitate the movement of ions and electrons and ensured a sulfur loading of about 2.05 mg cm−2.
Another broad approach of covalent fixing can reinforce the strong chemical binding interactions between the sulfur chains and the host molecules. Consequently, Arnab et al.30 reported a sulfur-embedded polymer matrix as the host for the cathode material for RT-Na/S batteries with a high sulfur loading of about 90 wt%. The sulfur co-polymer was obtained via a thermal ring-opening polymerization strategy using cardonol-based benzoxazine as the co-monomer (CS-90) (see Fig. 4a), which was then incorporated with reduced graphene oxide (CS-90/rGO). Besides the high reactivity of the co-monomer, the presence of multiple active sites prompted the co-monomer to anchor the polysulfane chains, resulting in an average S-loading of about 2.14 mg cm−2. The high binding effect of the covalently bonded active sulfur with the organic moiety units alleviated the dissolution effect of the sulfur species during cycling. However, the cathode delivered a capacity of 285 mA h g−1 after 100 cycles at a current density of 0.6C. Similarly, Wu and co-workers proposed a covalently bonded sulfur–carbon composite with benzenedisulfonic acid (SC-BDSA) with –SO3H and SO4− as the sulfur source in an RT-Na/S battery system, which ensured an active mass loading of ∼3 mg cm−2.62Fig. 4b schematically illustrates (1) a short-chain sulfur–carbon composite with a sulfur content of 8.53 wt%, (2) replacing benzenesulfonic acid with di-substituted benzenedisulfonic acid (BDSA) with an increased sulfur content of 18.33 wt% and (3) replacing the salt template with potassium sulfate, resulting in a high S-content of 40.07 wt%. Due to the O–S/C–S bridge bonds present in the sulfur species, high conductivity can be maintained to provide excellent interfacial contact among the particles in SC-BDSA. Consequently, the RS2O2− units formed on the surface of the complex matrix can act as an internal mediator to strongly bind the long-chain sodium polysulfides in the electrolyte. This results in catenating polythionates, which trigger their conversion to short-chain sodium polysulfides, as depicted in the following reaction.
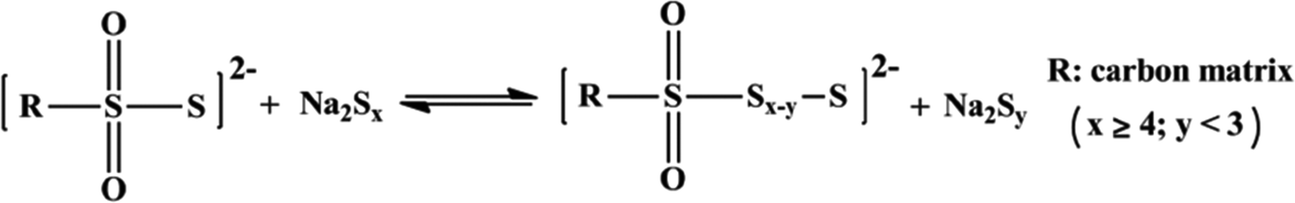
 | ||
| Fig. 4 (a) Chemical structure of copolymer by reaction of Ca monomer with sulfur. Reproduced with permission.30 Copyright 2017, the American Chemical Society. (b) Schematic representation of the synthetic processes of C-BSA, C-BDSA, and SC-BDSA. Reproduced with permission.62 Copyright 2019, John Wiley and Sons. (c) Schematic illustration of the synthesis of SC. Reproduced with permission.63 Copyright 2019, The Royal Society of Chemistry. (d) Synthetic route of a covalent sulfur–carbon composite via the solvothermal strategy. Reproduced with permission.64 Copyright 2020, the American Chemical Society. | ||
The cell exhibited high cyclic stability of 1000 cycles with a minimal decay rate of 0.035% per cycle with a reversible capacity of 452 mA h g−1 at 2500 mA g−1 and an initial discharge capacity of 696 mA h g−1.
Considering the covalent binding effect with sulfur, another group demonstrated the idea of confinement to boost the binding of sulfur with the matrix. Sulfur atoms were confined to a carbon matrix, which was named “thioether bond-functionalized carbon (SC)”.63 This can be an effective approach to mitigate the dissolution effect of soluble polysulfides. They investigated if the development of sulfur atoms bonded to carbon species can protect the system from reduced sulfur species of long chain-polysulfides owing to the generation of soluble sulfur species (S2− and S22−) in the routine carbonate electrolyte. SC, containing a thioether bond, can be untied through voltage scissors in the voltage range of 0.01–0.5 V vs. Na/Na+ and the cell can deliver a large reversible capacity for sodium storage. The insoluble reduced sodium species were then incorporated in the carbon defects induced by the cleaved sulfur (see Fig. 4c), overcoming the detrimental transformation to long-chain polysulfides. However, despite their effort to suppress the shuttle effect, the active material loading obtained was very low at 0.9–1.1 mg cm−2. Recently, Li et al.64 studied the in situ preparation of covalent configured carbon sulfur through the wet-chemical solvothermal strategy using carbon disulfide as the precursor in the presence of red phosphorus. S–C bonds were formed with the uniform distribution of sulfur on the periphery and interior of the carbon skeleton, as depicted in Fig. 4d. The ease of access of sodium ions between the carbon interlayer could activate the sulfur species electrochemically under 0.5 V. They claimed that the enlarged interlayer spacing of ∼0.4 nm allowed the free transportation of sodium ions into the matrix, which reacted with sulfur, allowing it to behave as a capacity donor in the subsequent cycles. The covalent cathode composite maintained a high reversible capacity of 811.4 mA h g−1 after 950 ultra-long cycles at 1.6C. Additionally, the covalent-S–C electrode delivered a remarkable capacity of 700 mA h g−1 at 8.1C.
Recently, Zhang et al.70 developed an elemental cobalt (i.e., atomic cobalt)-doped hollow carbon nanosphere matrix for the sulfur cathode, and reported an improved sulfur loading of about 5 mg cm−2. Due to the strong polar interaction between the Co and S atoms, the matrix could accommodate a high loading of sulfur with improved kinetics in the conversion reactions. Theoretical calculations were performed to understand the kinetically fast reaction of Na2S with the Co-doped matrix, as shown in Fig. 5a and b. The strong binding with the doped matrix allows a complete sodiation reaction, which effectively alleviates the shuttle effect and improves the cycling stability. The matrix doped with Co-atoms allows the facile adsorption of sodium polysulfides due to the relatively low or negative energy barrier for the adsorption reaction, as shown in Fig. 5c. Thus, this enabled stability for over 600 cycles at a current density of 100 mA g−1. The matrix without Co dopant could deliver an initial reversible of 1209 mA h g−1 with a rapid decay in the capacity of 271 mA h g−1 after 600 cycles. Significantly, the addition of cobalt clusters within the matrix enhanced the utilization of sulfur, where the sodium polysulfides confined within the shell of carbon could be catalytically reduced to Na2S. This attenuated the dissolution of polysulfides based on the polar–polar interactions, as illustrated in Fig. 5d and e.
 | ||
| Fig. 5 Density functional theory results and electrode reaction mechanism. (a) Optimized structures of Na2S4 cluster on carbon-supported Co6 cluster and (b) on carbon support. Purple: Na; yellow: S; blue: Co; gray: C; white: H. (c) Energy profiles of Na2S4 adsorption on carbon-supported Co6 cluster (in blue) and carbon support (in red). (d) Schematic illustration of the electrode reaction mechanism of atomic cobalt-decorated hollow carbon–sulfur host (S@Con-HC) and (e) hollow carbon hosting sulfur (S@HC), respectively. Reproduced with permission.70 Copyright 2018, Springer Nature. (f) Adsorption energies and (g) Gibbs free energies of NaPSs bound on the nitrogen-doped carbon surface and Au-decorated nitrogen-doped carbon. Reproduced with permission.71 Copyright 2020, The Royal Society of Chemistry. (h) Reaction pathway for Te0.04S0.96@pPAN and S@pPAN composites and hollow carbon hosting sulfur (S@HC), respectively. Reproduced with permission.72 Copyright 2019, the American Chemical Society. | ||
Zheng et al. studied “a highly feasible nano-copper-assisted immobilizing sulfur in high-surface-area mesoporous carbon (HSMC)” prepared via the multiple wetness impregnation and synchro-dry method with a S-content of 50 wt%.73 In addition to the physical encapsulation of sulfur in the mesoporous carbon, sulfur was also chemically stabilized with the copper nanoparticles, resulting in enhanced conductivity in the composite cathode. The HSMC–Cu–S cathode delivered a high discharge capacity of about 610 mA h g−1 at 0.03C after 110 cycles with a high Coulombic efficiency of about 100%. Despite the high initial discharge capacity of ∼1000 mA h g−1 and improved kinetics offered by the inclusion of the copper cluster in the matrix, the feasibility of the system was hindered due to its low sulfur loading (∼1 mg cm−2 with 50 wt% S-content). Similarly, Zhu et al.74 modified the carbon matrix with 0.14 at% iron atoms and nitrogen to develop a hybrid sulfur cathode. The nitrogen-doped carbon nanospheres can expedite the transport of ions and electrons, whereas the iron atoms serve as catalytic sites for stronger chemical interaction and conversion for the discharge products of sulfur. The hybrid cathode could deliver a reversible capacity of 359 mA h g−1 and 180 mA h g−1 at a current density of 0.1 A g−1 and 1 A g−1, respectively, with a high coulombic efficiency of about 100% after 200 cycles. Recently, Wang et al. introduced “an effective sulfiphilic matrix,” i.e., gold nanodot-decorated hierarchical N-doped carbon microspheres (CN/Au/S).71 Due to the high affinity of CN/Au towards sulfur atoms, a high sulfur content of about 56.5 wt% could be achieved. The matrix doped with gold nanodots (i.e., CN/Au/S) exhibited a higher binding energy for sulfur atoms compared to the matrix without dopant, indicating the strong interaction between the sulfur particles and gold nanodots, consequently trapping the sodium polysulfides during the cycling process. Apparently, the high binding energies of the elemental sulfur and sodium polysulfides upon the addition of gold clusters demonstrate their effectiveness in achieving a higher sulfur loading. The presence of gold nanodots enhanced the adsorption between the matrix and polysulfides (Na2Sn; 4 ≤ n ≤ 1), which helped alleviate the shuttle effect to a great extent. The as-modified matrix in the sulfur cathode exhibited long-term cycling stability, retaining a capacity of about 430 mA h g−1 after 1000 cycles, and a reversible capacity of about 369 mA h g−1 after 2000 cycles at a current density of 2 A g−1 and 10 A g−1, respectively. This is attributed to the fact that sulfur and its discharged species show higher absorption energies over CN/Au compared to the matrix without gold nanodots, as shown in Fig. 5f. In addition, the lower Gibbs free energy change for the gold doped matrix suggests the spontaneity of the reaction, resulting in enhanced adsorption capability, which efficiently mitigates the polysulfides shuttling, as shown in Fig. 5g.
Intrigued by the effect of elemental doping, Li et al.72 altered the SPAN matrix with tellurium atoms to form a sulfur cathode for RT-Na/S. The reaction kinetics were greatly improved with a high sulfur content of about 47.77 wt% of the active material. The tellurium-doped SPAN enhanced the reversible kinetics of the system (see Fig. 5h) with improved reversibility. Besides the fast reaction kinetics and the high electrochemical performance of Te0.04S0.96@pPAN, a high sodium ion diffusion coefficient and a higher electronic conductivity of at least 1.6 times that of pPAN@S could be achieved with Te0.04S0.96@pPAN. The cycle-life of over 600 cycles was attained with the Te0.04S0.96@pPAN cathode, retaining a discharge capacity of about 970 mA h g−1 at 0.5 A g−1 with a decay of about 0.015% per cycle. Due to the unique attributes of the Te0.04S0.96@pPAN cathode (i.e., high electronic and ionic conductivity, and fast conversion kinetics), it demonstrated an excellent rate capability at various current densities (0.1 A g−1 to 6 A g−1). It is apparent from the literature that the chemical modification of the matrix of the sulfur cathode significantly improves the battery performance, and it is anticipated that the role of chemical binding will create more opportunities to further improve the sulfur loading without compromising the fast reaction kinetics.
2.2 Physical aspects
The poor ionic conductivity of sulfur and relatively large size of Na+ ions result in extreme electronic and mechanical stress in the matrix.75,76 Therefore, it is desirable for the matrix to have a provision for altering the electronic conductivity to improve the electron transfer between the electrode/electrolyte interface without experiencing volume changes upon charge–discharge in the sulfur cathode. Due to limited availability of electronic and ionic sites in the conventional matrix, the RT-Na/S battery loses its capacity very fast.77 Thus, to promote the electrochemical stability, different strategies have been developed to alter the physical structure of the matrix (i.e., 1D, 2D and 3D structures) to concomitantly restrict polysulfides at the cathode and virtually eliminate their dissolution in the electrolyte.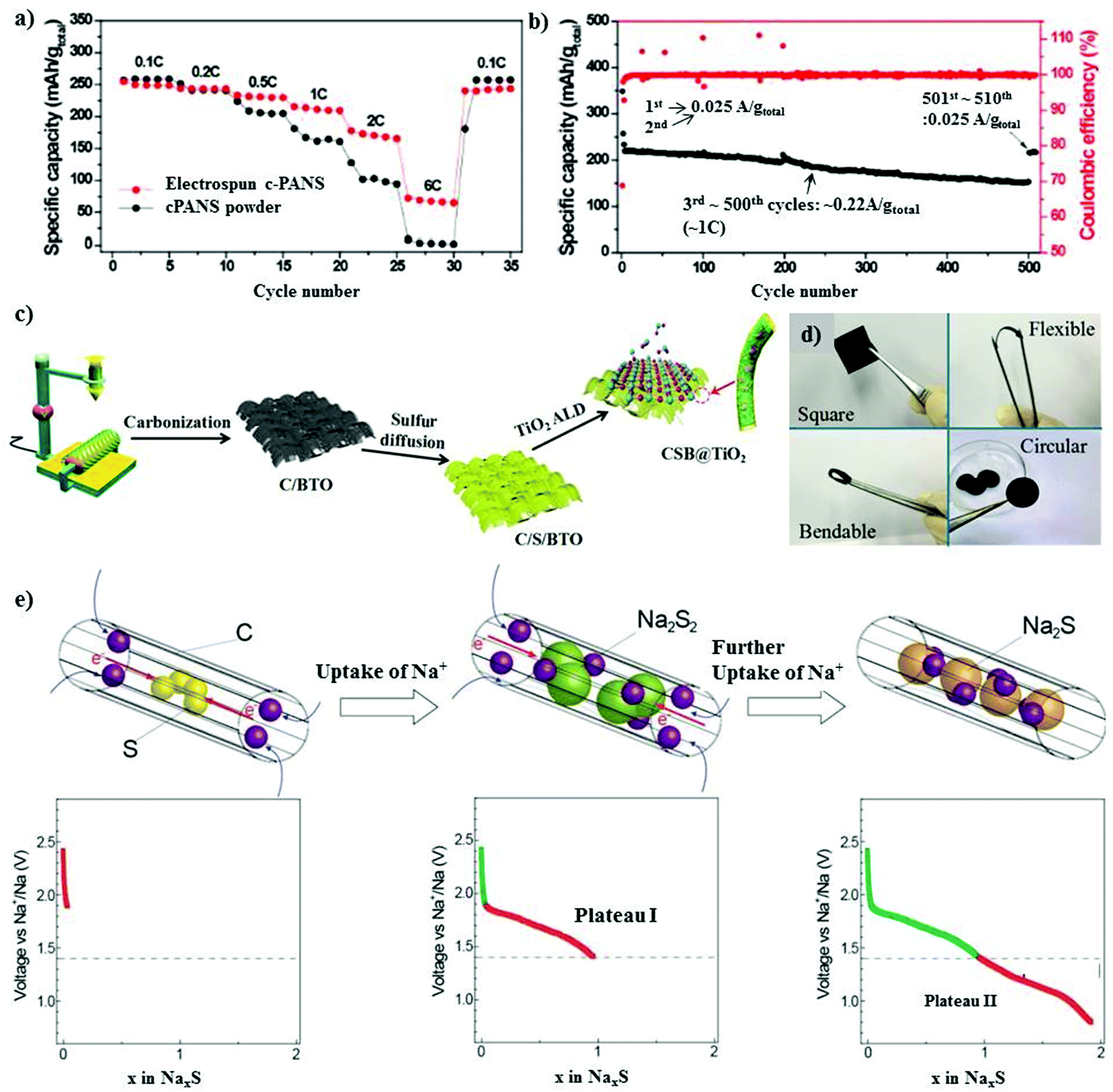 | ||
| Fig. 6 (a) Rate performances of the electrospun c-PANS NFs and c-PANS powder measured at various C-rates. The C-rates were the same for both charge and discharge in each cycle. (b) Capacity retention and Coulombic efficiencies of c-PANS NFs. The sample was measured at a low C-rate of 0.1C (0.025 A g−1 total) in the first two and final ten cycles, but at a higher C-rate of 1C (0.22 A g−1 total) in the cycle range of 3–500. Reproduced with permission.83 Copyright 2013, the American Chemical Society. (c) Schematic illustration of the preparation of the CSB@TiO2 electrode. (d) Photograph of the CSB@TiO2 free-standing electrode. Reproduced with permission.85 Copyright 2018, John Wiley and Sons. (e) Electrochemical reaction between S and Na+ ions during the discharge process in S/(CNT@MPC). Reproduced with permission.86 Copyright 2013, John Wiley and Sons. | ||
Generally, metal oxides are non-conductive with abundant active polar sites due to their oxygen ions. A judiciously designed metal oxide with polar sites may lead to a higher loading of sulfur, and simultaneously exhibit a high volumetric energy density with limited diffusion of polysulfides.87,88 Ma et al. implemented a new strategy for mitigating the shuttle effect by employing an amorphous structured transition metal oxide on a ferroelectric-encapsulated composite cathode to achieve high ionic transport across the electrode materials.85 The composite cathode was fabricated through a carbonization process and melt diffusion strategy with the active mass loading of about 1.2–1.4 mg cm−2, as shown in Fig. 6c and d. The electrochemical results for CSB@TiO2 were compared with that of pure C/S and C/S/BaTiO3, and it was found that the hybrid cathode CSB@TiO2 exhibited a superior performance. This can be attributed to the synergistic morphology of the atomic layer deposited layer on the surface of C/S/BaTiO3. The CSB@TiO2, C/S/BaTiO3, and C/S electrodes displayed a specific capacity of 1020 mA h g−1, 1101 mA h g−1 and 1120 mA h g−1, respectively, where a high reversible capacity of 611 mA h g−1 could be retained for the CSB@TiO2 cathode after 400 cycles. The cycling performance was evaluated at an even higher sulfur loading of about 3.3–3.5 mg cm−2, and the initial specific capacity at 0.5 A g−1 was 1055 mA h g−1 for C/S, 1040 mA h g−1 for C/S/BTO and 967 mA h g−1 for CSB@TiO2. Unconventional battery systems using cable-type configurations have been recently explored to provide an efficient capacity for battery devices.89 Xin et al. reported a composite cathode comprising co-axial cable-like carbon nanotubes with a mean diameter of 0.5 nm in a sulfur-infused microporous carbon matrix.86 S/CNT@MPC exhibited a sulfur content of about 40% with an overall active material loading of 1 mg cm−2. The cable-like structure provides a facile pathway for the movement of ions and electrons (Fig. 6e) without deteriorating the attributes of the 1D microporous matrix. The cathode with small sulfur molecules could deliver a high capacity of 1610 mA h g−1 in addition to displaying a high rate performance of 815 mA h g−1 even at a high C-rate of 2C. The high performance of S/CNT@MPC was attributed to the complete reduction of sodium polysulfides. An ultra-microporous carbon with a pore diameter of less than 0.7 nm can be more effective in providing better absorption sites for entrapping sodium polysulfides.90 The matrix was fabricated through an activation-free approach, and the elemental sulfur particles were infused under thermal treatment. Owing to the ultra-microporosity in the 3D matrix, the cell could deliver a reversible discharge capacity of 392 mA h g−1 after 200 cycles with a high Coulombic efficiency close to 100% at 1C.
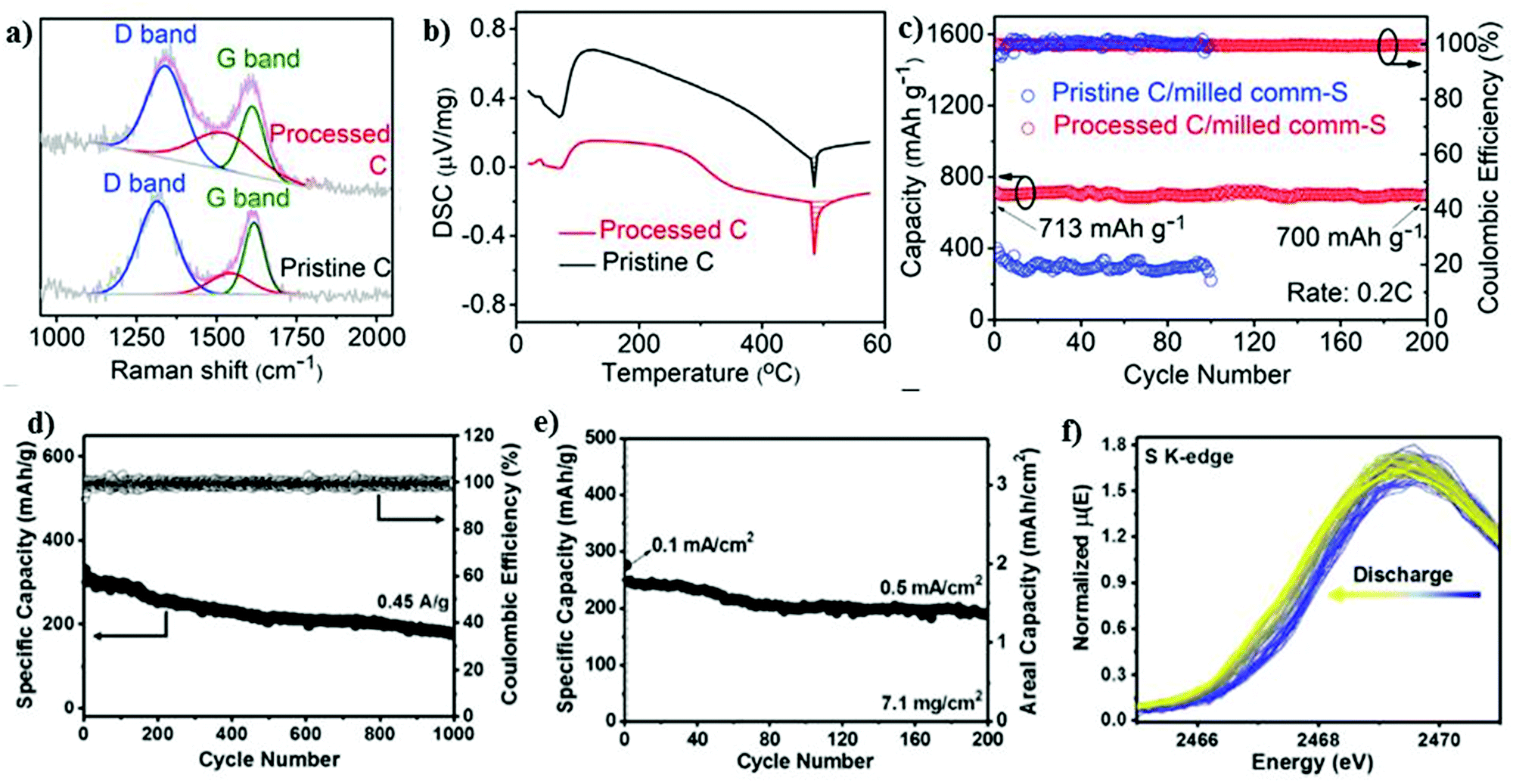 | ||
| Fig. 7 (a) Raman spectra and (b) DSC curves of pristine and processed C. (c) Cycling performance at 0.2C. Reproduced with permission.98 Copyright 2020, Elsevier. (d) Cycling stability and corresponding Coulombic efficiency at 0.45 A g−1. (e) Cycling stability of a high-loading electrode (7.1 mg cm−2) at 0.1 mA cm−2 for the first few cycles, and subsequently 0.5 mA cm−2. (f) Evolution of S K-edge XANES spectrum during sodiation. Reproduced with permission.99 Copyright 2017, National Academy of Sciences. | ||
Recently, an effective polysulfide anchoring composite was formulated by Sajjad and co-workers, where they studied 2D polar nitrogenated holey graphene (C2N) and non-polar polyaniline (C3N).100 The bilayer polar C2N was superior to the non-polar C3N in terms of its efficiency as an electrode enhancer for improving the conductivity of the sulfur cathode, thus attenuating the shuttle phenomena. A pure carbonaceous material system is relatively less or non-polar in nature, which impedes its practicality due to the weaker binding in confining the metal polysulfides.101–104 Recently, researchers have exploited metal-based composites as a possible matrix for the sulfur cathode owing to their highly polar nature and remarkable theoretical specific capacity values.105–108 Attributed to the highly polar sites within the 2D metal-based composite matrix, the additional benefits of strong “sulfiphilic” sites can be exploited.106 Consequently, an allotrope of molybdenum sulfide as the cathode has recently been examined with the aim of enhancing the loading of sulfur for the sulfur cathode in the RT-Na/S battery system. Ye et al. reported an amorphous structured MoS3 a “the sulfur-equivalent cathode material” for RT-Na/S batteries.99 A remarkable loading of about 7.1 mg cm−2 was achieved for the MoS3/S composite cathode, which is attributed to its unique atomic arrangement, where the Mo atoms were bridged between the sulfur and di-sulfur ligands. MoS3 as a cathode exhibited good electrochemical cycling stability for over 1000 cycles at 0.45 A g−1 with a Coulombic efficiency of ∼100%, as shown in Fig. 7d. At a current density of 0.5 A g−1, the cathode maintained the initial discharge capacity of ∼248 mA h g−1 with ∼76% capacity retention after 200 cycles, as illustrated in Fig. 7e. Operando X-ray absorption spectroscopy (XAS) was performed to observe the S K-edge XANES spectrum in the sulfur composite during the charge–discharge process. Profound changes were observed during the discharging of the sulfur cathode, which could be due to the strong interaction between the sulfur and sodium ions, as shown in Fig. 7f.
Kim et al. developed a flexible 2D matrix using a SPAN web/S composite as the cathode material for the RT-Na/S battery.82 The flexible web was prepared through a two-step electrospinning method followed by pyrolysis. The sulfur content was estimated to be about 41 wt%. The as-developed cathode exhibited good stability for over 200 cycles without any deterioration, dents, or cracks. The discharge capacity of about 343 mA h g−1, 257 mA h g−1, and 266 mA h g−1 could be achieved in the 1st, 2nd, and 200th cycles, respectively. Zhu et al. studied carbonized PAN (cPAN) with sulfur as a sulfur cathode for the Na/S battery system.109 The sulfur loading was estimated to be about 0.79 mg cm−2. The cell delivered a discharge capacity of about 311 mA h g−1, while maintaining a high Coulombic efficiency of ∼100% after 100 cycles.
Recently, Ma and co-workers examined a “carbon-wrapped nano-cobalt anchored graphene aerogel.”110 The matrix was fabricated through a heat-treatment method with Co/rGO and S in the weight ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, as illustrated in Fig. 8a–c. The framework of the graphene aerogel could provide a platform for successfully entrapping the sulfur particles, while the Co nanoparticles accelerated the kinetics of the conversion reactions, as shown in Fig. 8d–g. In addition, the nano-sized cobalt nanoparticles offered active sites for sulfur and its discharge products. The interconnected porous framework of graphene buffered the volume fluctuations during the cycling process. Consequently, the cathode delivered the initial discharge capacity of 572.8 mA h g−1 at 5C with a minimal decay rate of 0.01% per cycle.
:4, as illustrated in Fig. 8a–c. The framework of the graphene aerogel could provide a platform for successfully entrapping the sulfur particles, while the Co nanoparticles accelerated the kinetics of the conversion reactions, as shown in Fig. 8d–g. In addition, the nano-sized cobalt nanoparticles offered active sites for sulfur and its discharge products. The interconnected porous framework of graphene buffered the volume fluctuations during the cycling process. Consequently, the cathode delivered the initial discharge capacity of 572.8 mA h g−1 at 5C with a minimal decay rate of 0.01% per cycle.
 | ||
| Fig. 8 Schematic illustration of the S@Co/C/rGO electrode design. (a–c) Illustration of the synthesis. (d–g) Advantages of the S@Co/C/rGO composite. Reproduced with permission.110 Copyright 2020, Elsevier. | ||
3D infiltrated electrodes are promising matrix candidates, where sulfur can be infused into the 3D interconnected pores via different techniques, such as melt diffusion and vapor phase infiltration.117,118 The 3D infiltrated matrix offers alluring characteristics, which can be useful to improve the transportation kinetics, while increasing the thickness of the electrode materials.119,120 Unlike 3D assembled electrodes, microporous carbon with a size of <2 nm enhances the conductivity of the sulfur cathode, which can be designed to act as a strainer to successfully confine sulfur polysulfides.121 As a proof-of-concept, Carter et al. designed microporous carbon derived from sugar as a 3D matrix to support elemental sulfur.117 Sulfur particles were infused through a vapor infiltration chemical processing method, which could provide stable adsorption and successful confinement of the sulfur species, as schematically shown in Fig. 9a. The composite cathode demonstrated a stable performance for over 1500 cycles with the Coulombic efficiency of over 98%. The reversible capacity of about 300 mA h g−1 could be obtained at 1C, whereas the cell could deliver a discharge capacity of ∼700 mA h g−1 at 0.1C. The successful infiltration and confinement of sulfur in the microspheres alleviated the shuttle effect and maintained the stability of the cathode, but the sulfur content was about 35 wt%, which is much lower than the practical requirement. Despite the effective immobilization of sulfur offered by microporous carbon, its performance as a 3D matrix to hold sulfur is still impeded by its non-polar nature, uncontrollable porosity, and weak physio-sorption.122 Additionally, microporous carbon limits the loading of sulfur to under approximately 50 wt% due to its small size.123 Another potential solution to entrap or confine sulfur in a microporous 3D matrix can be achieved by engineering networks derived from metal organic frameworks (MOF).124–126 MOFs are a group of porous crystalline materials assembled by combining metal ions or clusters coordinated with organic linkers to form a highly ordered-porous spatial network.127–130 Wei's group pioneered the use of microporous carbon/sulfur as a 3D matrix for the sulfur cathode.131 The MOF-derived template (i.e., ZIF-8) was synthesized via a high temperature carbonization process with a loading of about 1 mg cm−2. By rationally tailoring the design, one can develop different MOF structures with prominent MOF matrices to encapsulate sulfur and physically entrap sodium polysulfides.
 | ||
| Fig. 9 (a) Schematic representation of the material processing steps using sucrose (sugar) to produce microporous sodium–sulfur battery cathodes. Reproduced with permission.117 Copyright 2017, the American Chemical Society. SEM images of (b) HCM, (c) HCMs-S, (d) PCM and (e) PCMs-S. Reproduced with permission.132 Copyright 2018, the American Chemical Society. (f) Schematic of the confinement in the S@iMCHS nanocomposite. Reproduced with permission.118 Copyright 2016, the American Chemical Society. (g) Procedure for the fabrication of c-ZIF-8/S. Reproduced with permission.133 Copyright 2016, The Royal Society of Chemistry. (h) Schematic illustration of the synthesis S@Con-HC. (i) Cycling performance of S@Con-HC and S@HC. Reproduced with permission.70 Copyright 2018, Springer Nature. | ||
Other possible configurations of the 3D matrix to enhance the sulfur loading and concomitantly inhibit polysulfide dissolution have also been developed. One of these configurations was proposed by Lee et al.,134 where they synthesized hollow carbon sphere/sulfur (HCS-S) with the sulfur content of about 56 wt% using a one-step hydrothermal method. The majority of the sulfur content was successfully impregnated within the ring-shaped hollow spheres. The cell could deliver a reversible capacity of 550 mA h g−1. Zhang et al. contributed towards improving the battery performance by assembling porous carbon microspheres (PCMs) as a 3D matrix to support sulfur particles.132 Multi-dimensional framework PCMs were constructed as a double-shell architecture with the outer carbon shell consisting of micronized-carbon shells and an inner carbon shell, with hollow carbon nanobeads. Elemental sulfur could be filled inside the porous carbon microspheres within the carbon shell, which was covered by the outermost part of the shell. Sulfur and its reaction intermediates were well-protected inside the double-shelled structure. There was about 28% sulfur embedded in the interconnected hollow carbon beads, and nearly 6% sulfur was inside the microspheres. Therefore, the double-shelled structure of the 3D matrix provided protection against direct contact with the electrolyte and mitigated polysulfide diffusion or dissolution in the electrolyte. A comparison was made with sulfur-filled hollow carbon microspheres (HCM), which could encapsulate about 44% sulfur in its hollow structure. The SEM micrographs of HCM and PCM are shown in Fig. 9b–e for comparison. Upon cycling, the PCM matrix agglomerated and became much denser with micronized particles. Consequently, PCMs as the host in the sulfur cathode showed an excellent rate performance at various current densities ranging from 100 mA g−1 to 2000 mA g−1. Owing to the double-carbon shell architecture, the cathode exhibited an initial discharge capacity of 1100 mA h g−1 and 795 mA h g−1 in the 2nd cycle and stabilized with the Coulombic efficiency of 63.6% in the 1st cycle and 83.5% in the subsequent cycles. Xia et al. designed a 3D matrix to improve the sulfur loading and provide exceptional adsorption capability for sodium polysulfides.135 A hierarchical 3D matrix of porous carbon nanofibres constructed together with carbon hollow nanobubbles (CHNBs@PCNFs) was obtained via an electrospinning method. To activate the formation of homogeneously distributed porosity in the matrix, metal azides were used as both a bubbling and porogen reagent. The CHNB@PCNF/S cathode for the RT-Na/S battery delivered the 1st discharge capacity of about 1214 mA h g−1 at 0.1C and retained a remarkable reversible capacity of 786 mA h g−1 after 50 cycles. The CHNB@PCNF/S cathode exhibited a high rate performance at various C-rates, including 0.1C, 0.2C, 0.5C, 1C, and 2C, which could be attributed to the high volume and surface area of PCNFs. CHNBs@PCNFs was regarded as an efficient building block to structurally integrate the 3D porous matrix. Efforts have been made to enhance the sulfur loading by exploring other structured matrices, e.g., mesoporous carbon. Wang et al. demonstrated an RT-Na/S battery using a mesoporous carbon hollow nanosphere/sulfur (iMCHS/S)-based sulfur cathode.118 The outer carbon shell could effectively trap sodium polysulfides, and the mesoporous shell could serve as a matrix to provide a conductive network for the sulfur particles. As depicted schematically in Fig. 9f, the interconnected mesoporous structure of the matrix could be loaded with a high amount of sulfur (about 4.1 mg cm−2). Due to the hollow interconnected mesoporous structures, sulfur could be encapsulated successfully. The as-developed cathode could deliver a reversible capacity of about 390 mA h g−1 and 127 mA h g−1 at a current density of 0.1 A g−1 and 5 A g−1, respectively. A reversible capacity of 292 mA h g−1 was retained even after 200 cycles of charge/discharge with about ∼88.8% capacity retention at a current density of 100 A g−1. Nonetheless, the cathode lacked stability in its structure and ability to confine the diffusion of polysulfides effectively.
With the assistance of certain dopants or metal clusters, the conductivity of materials can be greatly enhanced to offer strong confinement of sulfur species and sodium polysulfides.136–140 Nitrogen, as one of the best dopants, has been successfully used in Li–S batteries due to its excellent electron transfer properties and high electronegativity of 3.04.141–144 Chen's group synthesized a nitrogen-doped carbon composite matrix derived from zeolitic imidazolate framework-8 (ZIF-8). ZIF-8 represents a class of MOFs with a sodalite-type structure and regular mesoporous/microporous pore sizes. MOF-derived nitrogen-doped 3D matrices are of particular interest due to their simple fabrication process and reproducibility. Carbonized ZIF/S (cZIF/S) was fabricated via a melt diffusion method, resulting in the successful infusion of sulfur chains inside the nitrogen-doped cZIF-8 framework with 50% S-content, as shown in Fig. 9g. The cathode could be cycled for over 250 cycles with a reversible capacity of 500 mA h g−1 at 0.2C. The cZIF/S composite exhibited a good rate performance with C-rates ranging from 0.1C to 2C. The high rate performance could also be attributed to the addition of nitrogen dopant in the carbon framework, which facilitates: (1) the formation of covalent bonds with sulfur, (2) sulfur immobilization, (3) chemical bonding between sulfur and the oxygen functional groups in the carbon framework and (4) chemical adsorption of sulfur in the carbon matrix. The strong chemical bonding of sulfur with the oxygen functional groups of carbon was revealed using XPS. The sulfur loading achieved was about 0.7–0.9 mg cm−2, which is quite low and it may impede the system from reaching a high specific energy density.133 Exploiting the advantages of the porous structure of MOFs, in 2020, Xiao et al.145 proposed another MOF-based sulfur and nitrogen-doped porous carbon as a host. A Zn-derived MOF with 2,5-thiophenedicarboxylic acid and 1,4-bis(pyrid-4-yl)benzene as the ligands and a vapor-infiltration method were considered for efficiently infiltrating sulfur within the carbon matrix with strong covalent bonding. The composite cathode with a polydopamine coating and 37% S-content and 0.9–1.5 mg cm−2 S-loading could deliver an initial discharge capacity of 1003 mA h g−1 at 0.1 A g−1 and a reversible capacity of 680 mA h g−1 at 1A g−1 after 500 cycles. A 3D hierarchical morphology nanocomposite with different sulfur loadings of 25, 45, 65, and 86 wt% in nitrogen-doped graphene nanosheets was prepared recently.146 The 3D morphology of the composite resembled a voile structure, which confined the discharge products effectively to alleviate the shuttle effect. They concluded that the S-loading of 25 wt% displayed the best performance with an initial discharge capacity of 212 mA h g−1 and 136 mA h g−1 in the 10th cycle, which can be attributed to the ease of the intercalation of sodium ions. However, despite this, the partial dissolution of sodium polysulfides was observed during cycling even with the lowest loading of 25 wt%. The performance of the electrode material deteriorated with an increase in the sulfur loading; therefore, composites with a higher loading are needed given that they may enhance the energy density of the cell. Qiang et al. reported a nitrogen-doped porous carbon 3D matrix for the sulfur cathode to boost the cycle-life of RT-Na/S batteries.147 Doped-hierarchical-structured porous-based sulfur (N,S-HPC/S) was fabricated as a cathode material. The sulfur molecules could be effectively trapped within the porous carbon through a calcination process followed by melt diffusion. The sulfur loading of about 1.1 mg cm−2 could be achieved in this architecture. The presence of nitrogen improved the electrostatic interaction between the matrix and sodium polysulfides, which enhanced the entrapment of sodium polysulfides to inhibit side reactions. Guo et al. reported the synthesis of a unique 3D matrix of nickel concatenated by nitrogen doped on carbon fibres (NCFs) via an electrostatic spinning method.140 Each unit of nickel displayed a hollow morphology and could successfully buffer the volume changes during the charge–discharge cycles and also provide polar interactions bonds between Ni and S upon S-loading. Nickel, in combination with NCFs as the host for the S-cathode, exhibited a high rate capability ranging from 0.2C to 5C with a reversible capacity of 738.7 and 181.7 mA h g−1, respectively. In addition, the cathode could maintain the 1st discharge capacity of ∼431 mA h g−1 and ∼233 mA h g−1 after 270 cycles at 1C due to the high catalytic effect of the nickel units. However, despite the intriguing rate performance, the cathode could maintain an area loading of only about 0.5–0.7 mg cm−2. Another approach to enhance the loading of sulfur and prevent the dissolution of polysulfides was proposed by Zhang and co-workers.70 They used atomic cobalt as an effective electro-catalyst to accelerate the conversion reactions. As schematically illustrated in Fig. 9h, atomic cobalt and the elemental sulfur particles were efficiently encapsulated within the hollow framework of the porous carbon nanoparticles. A sulfur loading as high as 5 mg cm−2 could be achieved. Electron microscopy revealed uniformly dispersed atomic cobalt and sulfur particles within the carbon nanoparticles. The hollow carbon and atomic cobalt could effectively trap and bind with polysulfides, respectively, to improve the stability of the cell for over 600 cycles at a current density of 100 mA g−1, as shown in Fig. 9i.
To effectively harness the benefits of the transition metal sulfides to accelerate the reversible reactions, while controlling the diffusion of polysulfides in the electrolyte solution, Meyerson et al.148 reported a molybdenum sulfide-based sulfur cathode with an active mass loading of about 1.5–2.0 mg cm−2. SEM micrographs were captured for the as-developed molybdenum sulfide (i.e., MoS5.6) before cycling, showing angular-shaped particles of several micrometers. After ten cycles, the angular-shaped particles transformed into spherical structures particles with a reduced lateral size (see Fig. 10a–d). However, although the transformation of the angular-shaped particles into spherical particles increased the accessibility of sodium ions into the sodiation active sites, cracks were observed during long-term cycling. The MoS5.6 electrode showed a capacity of 537 mA h g−1 and 200 mA h g−1 at current densities of 50 mA g−1 and 1 A g−1, respectively.
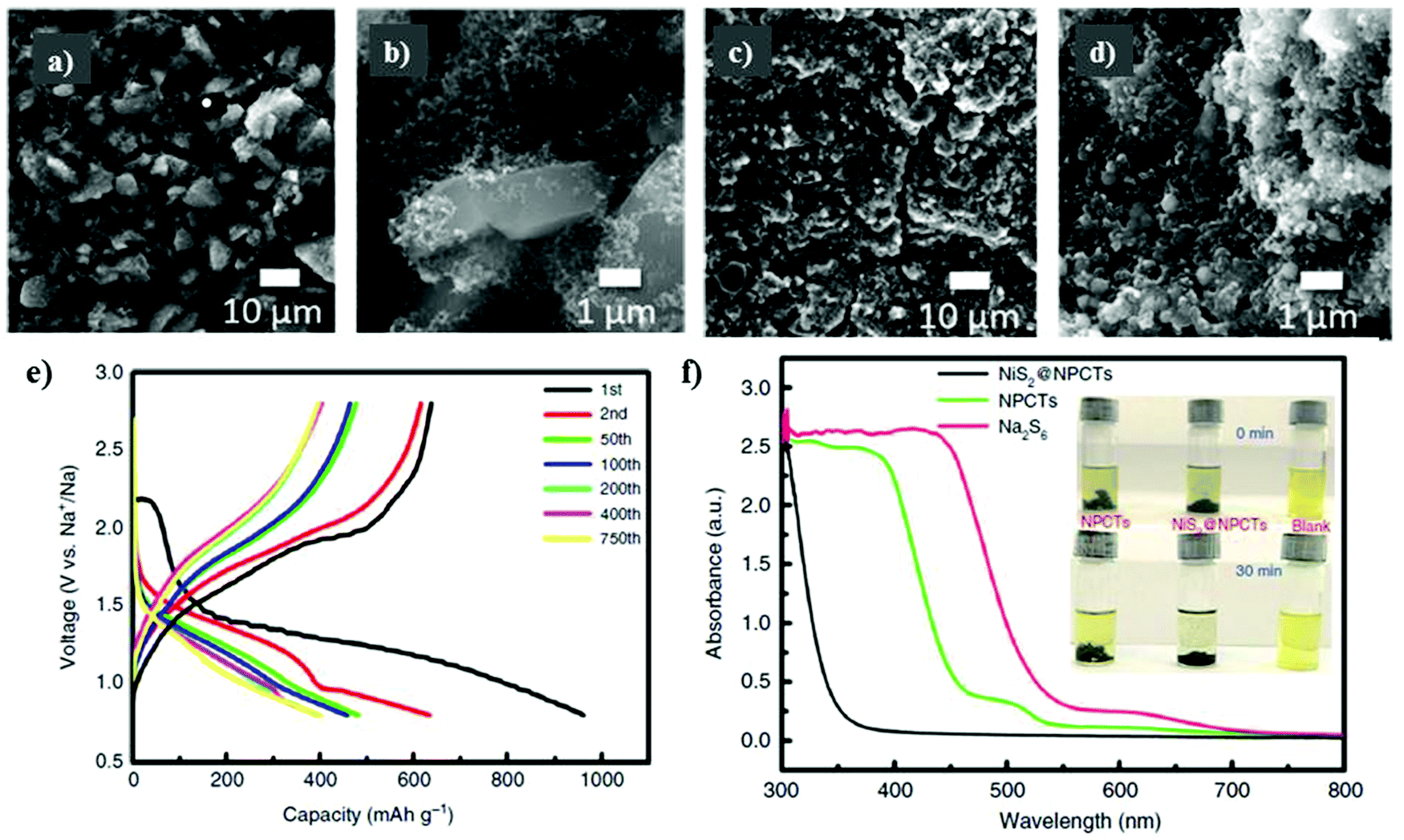 | ||
| Fig. 10 Scanning electron micrographs of the MoS5.6 electrode (a and b) before cycling and (c and d) after 10 cycles at 50 mA g−1. Reproduced with permission.148 Copyright 2020, the American Chemical Society. (e) Corresponding charge/discharge profiles of NiS2@NPCTs/S at different cycles. (f) Ultraviolet/visible (UV-vis) spectra and corresponding photographs (inset) of pure Na2S6 solution and the solution after exposure to NiS2@NPCTs and NPCTs. Reproduced with permission.139 Copyright 2019 Springer Nature. | ||
Recently, Yan et al.139 examined a nitrogen-doped porous carbon network to embed nickel sulfide nanocrystals as the sulfur cathode for RT-Na/S batteries. A high content of about 56% of sulfur in NiS2@NPCTs/S or a sulfur loading of about 2.5 mg cm−2 was achieved. The existence of two different types of sulfur atoms was confirmed by X-ray photoelectron spectroscopy (XPS). Due to the sulfur loading, NiS2@NPCTs/S as the cathode material exhibited distinct plateaus with a high capacity of 960 mA h g−1 in the 1st cycle, which remained stable at 410 mA h g−1 after 750 cycles, as shown in Fig. 10e. The high performance and increased sulfur loading were attributed to the synergistic effects of the transition metal sulfide with N-doped carbon network, which was confirmed by the UV visible spectra, as shown in Fig. 10f. However, although the cathode based on the N-doped 3D matrix could maintain an impressive cycling performance and relatively high sulfur loading, irreversible losses in the capacity were observed. Aslam and co-workers studied a polar metal chalcogenide as a matrix for the sulfur cathode in the RT-Na/S battery.116 A bipyramidal prism structure of cobalt sulfide was fabricated via simple reflux, as shown in Fig. 11a. A high sulfur content of about 64.5% or a sulfur loading of about 4.4 mg cm−2 could be obtained through this method. In contrast to the non-polar sulfur matrix, the polar matrix could physically block the outward diffusion of sodium polysulfides. The sulfur and its reduced products could be successfully entrapped through the polar sites and wide hollow cavity architecture, as depicted schematically in Fig. 11b and c. The CoS2/C (BPCS) cathode exhibited an interweaving hierarchical architecture with wide internal hollow spaces of about 376 nm to entrap the polysulfides. BPCS displayed a high discharge capacity of 1347 mA h g−1 in the 1st cycle, and 755 mA h g−1 in the 2nd cycle, and 701 mA h g−1 after 350 cycles with a high coulombic efficiency of 98.5%. The sulfur loadings of about 7.3 mg cm−2 and 9.1 mg cm−2 were also obtained, and the sulfur cathode was cycled at a rate of 0.5C, where the 1st areal capacity of about 6.24 mA h cm−2 and 8 mA h cm−2 was achieved, respectively. Simultaneously, they retained a capacity of 5.5 mA h cm−2 and 6.6 mA h cm−2 after ten cycles, respectively. The CoS2/C cathode presented a new reaction pathway to facilitate the reduction process from S to Na2S by mitigating the dissolution of sodium polysulfides in the electrolyte. Therefore, this may diminish the shuttle effect to provide better electrochemical performance even with a higher sulfur loading.
 | ||
| Fig. 11 (a) Synthesis of metal chalcogenide S@BPCX composites. (b) Diffusion of NaPSs in solid nonpolar host. (c) Suppression of NaPSs in hollow polar/catalytic host. Reproduced with permission.116 Copyright 2020, Springer Nature. (d) Cyclic voltammetry curves of the rGO/S/MnxOy@SA–PANI cathode at a scan rate of 20 μV s−1. (e) Schematic of the possible surface redox reaction between Na2S6 and MnxOy. Reproduced with permission.61 Copyright 2019, the American Chemical Society. | ||
One of the issues of increasing the thickness of the sulfur loading is the rapid dissolution of sulfur products due to the drop in the capillary pressure.119 In addition, a restricted reaction pathway for sodium ions and electrons, and dramatic volumetric changes are observed with an increased loading. Thus, rationally reinforcing different composites of reduced graphene oxide (rGO) and manganese oxide (MnxOy) with Na alginate/polyaniline hybrid binder to form a 3D matrix may be another effective approach to mitigate these issues.61 To design this type of interconnected 3D matrix, Ghosh and co-workers designed a rGO/S/MnxOy@SA–PANI composite cathode via a vacuum filtration method, which resulted in a sulfur loading of about 2.05 mg cm−2. Owing to the good electrical conductivity of rGO (4.8 × 103 S m−1) and cubic lattice structure of the MnxOy nanoparticles, the as-synthesized 3D conductive matrix with porous interconnected morphology could accommodate volume fluctuations during the reversible sodiation and desodiation reactions. Cyclic voltammetry was performed using the composite cathode, as shown in Fig. 11d, showing a series of cathodic peaks in the range of 1.2–2.8 V vs. Na/Na+, corresponding to: (1) the conversion of solid-phase S8 to liquid-phase long-chain polysulfides, (2) long-chain polysulfides to liquid-phase middle-chain polysulfides, and (3) middle-order polysulfides to insoluble short-chain polysulfides, respectively. The charge/discharge curve for the composite cathode showed an enhanced performance in the 1st, 2nd and 5th cycles at a current density of 0.2 A g−1. As a result of the enhanced ionic-electron distribution efficiency, the diffusion pathways of sodium ions and electrons could be shortened, contributing to the improved conversion reactions with minimal dissolution of polysulfides. In addition, MnxOy nanoparticles could act as a reducing agent for polysulfides, resulting in a high affinity of MnxOy nanoparticles (30–50 nm) towards Na2S6, which is evident in Fig. 11e. The sulfur cathode comprised of rGO/S/MnxOy@SA–PANI could deliver a remarkable energy density of 737 W h kg−1. The high performance of the cathode was attributed to the electrocatalytic activity of the MnxOy nanoparticles and the conductive nature of the rGO/S/MnxOy@SA–PANI composites. However, with an increase in the number of cycles, a gradual decay was observed in the reversible capacities, which could be due to the formation of a thick SEI on the surface of the anode, affecting the dissolution and deposition of sodium ions.
The inhomogeneous distribution of sulfur within the matrix often leads to the low utilization of sulfur, which has become a roadblock to reach the unprecedented specific capacity of the RT-Na/S battery.134,149,150 In the pursuit of developing an ideal host for elemental sulfur and confining the polysulfides, researchers have reported different composites in various structures, which have been found to be suitable for achieving a high sulfur loading cathode. One unique contribution was reported by Yu and Manthiram, where they explored different sodium/polysulfide batteries based on different composite materials.151 They designed a cathode composed of long-chain sodium polysulfides with high surface area multi-walled carbon nanotube (MWCNT) fabric, which served as the current collector for the sulfur cathode. The cycling performance of the dissolved liquid-phase NaPS catholyte/MWCNT with 60% sulfur content was investigated at a discharge cut-off voltage of 1.2 V for 30 cycles, which maintained a capacity of about 400 mA h g−1. NaPSs/MWCNT exhibited a sharp decay in capacity after the first few cycles, which could be due to the sluggish reversibility of the sodium polysulfides after the charge/discharge cycles. By varying the concentration of the sodium polysulfide catholyte in the composite material, the sulfur loading could be further enhanced. Therefore, Yu et al. performed further studies, but this time with short-chain polysulfides (Na2S) (80%), which were spread on MWCNT (20%) to enhance the conductivity of the insulated Na2S.152 Na2S, as they claimed, possesses “high electronic resistivity and low sodium ion diffusivity,” behaving as a special cathode with a high anchoring effect that is enough to encapsulate the charge/discharge products during the cycling process. The cell with the Na2S composite cathode was tested at two different C-rates with a discharge capacity of 660 mA h g−1 at C/10 and 540 mA h g−1 at C/3. The cell exhibited a cycle life of about 50 cycles, retaining capacities of 560 mA h g−1 and 380 mA h g−1, and a Coulombic efficiency of ∼90% after the subsequent cycles. As a result of its unique architecture with good electron-ionic transport, the Na2S/MWCNT electrode exhibited an energy density of ∼250 W h kg−1. However, the cell experienced high polarization with an increase in rate due to the inert nature of Na2S. In another contribution, they proposed a conductive matrix of carbon nanofibres anchored with high surface area activated carbon.153 The sulfur content of about 40–50 wt% could be accommodated within this matrix. CNF/AC was utilized as the matrix for the sodium polysulfide cathode to enhance the utilization of sulfur in the polysulfides. Recently, Bloi and co-workers revisited this configuration using nanostructured Na2S (90 wt%) with conductive carbon as the matrix for the sulfur cathode.154 The Na2S/C hybrid cathode, which was synthesized via carbothermal reduction at different temperatures, showed improved cathode utilization with enhanced stability and an initial discharge capacity of about 740 mA h g−1. One of the methods to achieve a higher and stronger adsorption of polysulfides can be realized by incorporating Na2S6 catholyte into a carbon cloth/MnO2 nanoarray.155 Carbon cloth/MnO2 showed a strong anchoring effect towards higher-order polysulfides, alleviating the shuttle effect and accelerating the reversibility and reaction kinetics. A sulfur loading of about 1.7 mg cm−2 was attained. The CC/MnO2@Na2S6 cathode displayed an initial specific capacity of ∼938 mA h g−1 with 67% retention after 500 cycles. Table 1 comprehensively summarizes the efforts made hitherto to enhance the sulfur loading and the cell performance. Despite the advantages offered by the judicious combination of different composite materials, dedicated efforts are still required to engineer matrix materials for the sulfur cathode.
| Matrix physiochemistry | Cathode system | Synthesis route | Active mass-loading (mg cm−2) | Initial discharge (mA h g−1) | Capacity (mA h g−1) after n cycles | Advantages | Disadvantages | |
|---|---|---|---|---|---|---|---|---|
| Chemical aspects | Chemical binding | Polyacrylic acid-based SPAN54 | Heat treatment | 1.1–1.8 | 1195@0.5C | 1000 at 1000 cycles@0.5C | Accommodate volumetric fluctuations due to strong binding | Low mass loading of sulfur |
| Sodium alginate based rGO/S/MnxOy61 | Vacuum filtration | 2.05 | >700@0.12C (0.2 A g−1) | 535 after 50 cycles@0.12C (0.2 A g−1) | Facilitated ion-diffusion process with cycle life up to 400 cycles | Low current densities | ||
| Carbon–sulfur90/rGO30 | Thermal ring-opening polymerisation | 2.14 | ∼500@0.6C (1 A g−1) | 285 after 100 cycles@0.6C (1 A g−1) | Large S-confinement and improved rate capability | Limited current densities | ||
| Covalent sulfur–carbon complex (SC-BDSA)62 | Calcination and thermal treatment | 3 | >900@0.06C (0.1 A g−1) | 590 after 200 cycles@0.06C (0.1 A g−1) | High anchoring ability to form insoluble intermediates | Low sulfur content | ||
| Sulfur into carbon matrix (SC)63 | Carbonized crosslinking polymerisation | 0.9–1.1 | ∼1070@0.06C (0.1 A g−1) | 500 after 100 cycles@0.06C (0.1 A g−1) | Fast kinetic conversion to short-chain polysulfides | Very low S-content (21.5%) and loading with capacity decay in initial few cycles | ||
| Dopant | Atomic cobalt-hollow carbon nanospheres70 | Sol–gel process | 5 | 1081@0.06C (0.1 A g−1) | 508 after 600 cycles@0.06C (0.1 A g−1) | Improved sulfur reactivity, high sulfur utilisation & alleviated polysulfide diffusion due to polar interactions | Partial reversibility of Na2S product | |
| High surface area mesoporous carbon–copper/sulfur73 | Solution impregnation strategy | 1.0 | 1000@0.03C | 610 after 110 cycles@0.03C | Stable reversible capacities with good rate capability | Low term cyclic performance with low loading of active material | ||
| Nitrogen-doped carbon nanospheres loaded with iron74 | Carbothermal reduction followed by calcination process | — | >350@0.6C (1 A g−1) | 180 after 200 cycles@1 A g−1 | Accelerated conversion reaction and shorter diffusion path for ions and electrons | Lower capacity attained for low values of current density and side reactions between solvents and polysulfides. | ||
| Gold nanodots on hierarchical N-doped carbon microspheres/sulfur71 | HCl etching process | — | 1967@0.06C (0.1 A g−1) | 701 after 110 cycles@0.1 A g−1 | Enhanced sulfur utilization & adsorption capability | Decay in capacity with an increase in cycle number | ||
| Tellurium-doped/SPAN72 | Heat treatment method | 1.0–1.2 | ∼1500@0.3C (0.5 A g−1) | 970 after 600 cycles@0.3C (0.5 A g−1) | Faster kinetic reactions & good performance in both ether and carbonate-based electrolyte | Very low active mass loadings and low current densities | ||
| Physical aspect | 1D matrix | cPANS83 | Electrospinning method | 1.0–1.2 | 364@0.1C | 153 after 500 cycles@1C | Stable cycling test, improved kinetics for de-sodiation & high rate capability up to 6C | Low reversible capacities with increased C-rates |
| Pyrolyzed PAN/selenium disulfide84 | Electrospinning method & heat treatment | 2 | 1043@0.6C (1 A g−1) | 800 after 400 cycles@1 A g−1 | Extreme stable cyclic life | Low voltage plateau & limited intrinsic reactivity | ||
| C/S/BaTiO3@titanium dioxide85 | Electrospinning technique and atomic layer deposition | 3.3–3.5 | 952@0.6C (1 A g−1) | 524.8 after 1400 cycles@0.6C (1 A g−1) | Ultra-long term cyclic stability & stable ionic transport interface | Low mass loadings | ||
| 2D matrix | Nanocarbon-promoted comm-S98 | Ball milling process | 0.8–2.0 | 713@0.2C | 700 after 200 cycles@0.2C (for 1.2 mg cm−2) | High binding affinity towards sulfur and its discharge products and cost-efficient | Low loading of sulfur with low C-rate | |
| SPAN82 | Pyrolysis method | — | 342@0.1C | 266 after 200 cycles@0.1C | High flexibility, rollability & bendability to 180° without any fracture | A continuous voltage decay in the first discharge | ||
| S/CPAN109 | Heat treatment | ∼0.79 | ∼310@0.06C (0.1 A g−1) | 251 after 100 cycles@0.06C (0.1 A g−1) | Enhanced ionic conductivity | Sharp decay in initial cycle and low areal loadings | ||
| Carbon wrapped cobalt nanoparticles on graphene aerogel110 | Thermal treatment | 572.8@5C | 286.5 after 1000 cycles@5C | Improved S-reactivity even at high C-rate and accommodation of fast volume fluctuations | Low S-content of only about 37.5% | |||
| 3D matrix | Carbon fibre cloth/sulfur111 | Solution impregnation | 1 | 390@0.1C | 120 after 300 cycles@0.1C (for 2 mg cm−2) | High electrolyte absorbability & flexibility | Increased polarization with increased loading | |
| Sugar-derived microporous carbon/sulfur117 | Isothermal vapor phase infiltration | — | ∼1524@0.1C | 300 after 1500 cycles@1C | Stable SEI layer & strong affinity for sulfur species | Low energy density possible | ||
| Ultra-microporous carbon/sulfur90 | Single step polymer carbonisation | 3 | >900@0.1C | 392 after 200 cycles@1C | Effective confinement between carbon and sulfur molecules | Sluggish kinetics of sodium ions | ||
| Porous carbon microspheres/sulfur132 | Chemical vapour deposition | — | 1100@0.06C (0.1 A g−1) | 290 after 350 cycles@0.1 A g−1 | Enhanced porosity to accommodate volume fluctuations & improved structured stability | Capacity loss | ||
| Carbon hollow nanobubbles@Porous carbon nanofibres/sulfur135 | Electrospinning technique | 1.4 | 1214@0.1C | 256 after 400 cycles@2C | Enhanced adsorption capability, high sulfur utilization with high reversibility | Low S-loading | ||
| Mesoporous carbon hollow nanospheres/sulfur118 | Sol–gel method | 3.2–4.1 | 1215@0.06C (0.1 A g−1) | 292 after 200 cycles@0.06C (0.1 A g−1) | Improved conductivity & high S electroactivity | Slow kinetics and serious polarization with increased number of cycles | ||
| MOF-derived S,N-doped porous carbon145 | Vapor-infiltration method | 0.9–1.5 | 1003@0.06C (0.1 A g−1) | 680 after 500 cycles@0.6C (1 A g−1) | Strong covalent bonding and reduced adsorption energy of NaPSs | Low content of sulfur (37 wt%) | ||
| Nitrogen-doped graphene nanosheets/sulfur146 | Chemical reaction deposition & low temperature heat treatment | — | 212@0.05C | 48 after 300 cycles@0.1C | Enhanced transport of sodium ions & increased S-loading achieved up to 86 wt% | Low confinement of the composite to sulfur species, leading to a decay in capacity with increased cycling and lower electrochemical performance with increase in sulfur wt% | ||
| N,S doped-hierarchical porous carbon147 | Customised roll-to-roll line strategies | 1.0–1.1 | ∼430@0.14C (0.23 A g−1) | 378 after 350 cycles@0.14C (0.23 A g−1) | Inhibits reaction of sodium polysulfides with the electrolyte & improved wettability | Limited current rate and lower loadings of active material | ||
| Nickel–nitrogen-doped carbon fibres140 | Electrostatic spinning process | 0.5–0.7 | ∼431@1C | ∼233 after 270 cycles@1C | Good catalytic effect of Ni on the conversion of NaPSs | Low electrochemical performance and low S-loading | ||
| Atomic cobalt-hollow carbon nanospheres70 | Sol–gel process | 5 | 1081@0.06C (0.1 A g−1) | 508 after 600 cycles@0.06C (0.1 A g−1) | Improved sulfur reactivity, high sulfur utilisation & alleviated polysulfide diffusion due to polar interactions | Partial reversibility of Na2S product | ||
| Molybdenum sulfide148 | Chemical oxidation | 1.5–2 | 400@0.12C (0.2 A g−1) | 300 after 100 cycles@0.12C (0.2 A g−1) | Decreased size of composite material allowing faster rate transport of ions | Capacity decay and low current density values | ||
| Nickel sulfide@nitrogen-doped porous carbon network139 | Freeze-drying followed by heat treatment | 2.5 | 960@0.6C (1 A g−1) | 401 after 750 cycles@0.6C (1 A g−1) | Effective electrocatalytic active sites with improved kinetics to promote fast conversion | Degraded capacity values with increase in current density | ||
| CoS2C@sulfur116 | Simple reflux method | 4.4 | 1347@0.5C | 675 after 800 cycles@0.5C (for 4.4 mg cm−2) | Successful entrapment of sodium polysulfides with high adsorption capability | Limited performance with increase in loading | ||
| rGO/S/MnxOy@SA–PANI61 | Vacuum filtration | 2.05 | >700@0.12C (0.2 A g−1) | 535 after 50 cycles@0.12C (0.2 A g−1) | Facilitated ion-diffusion process with cycle life up to 400 cycles | Low current densities | ||
3. Status of Na metal anode and electrolyte engineering
Owing to its high theoretical capacity (1166 mA h g−1), low electrochemical potential (−2.71 V) with respect to the standard hydrogen electrode, high abundance and cost effectiveness, the sodium anode is considered an appealing candidate as an anode for RT-Na/S batteries.156,157 However, there are numerous challenges that need to be addressed urgently, which include, but are not limited to the following;(1) Rapid chemical reactivity of Na metal with electrolyte.158 This leads to the decomposition of the electrolyte on the metal surface, which creates a layer of by-products, and the instability of this layer (solid electrolyte interphase) ultimately results in a decay in performance.
(2) Growth of dendrites.159,160 They are mostly caused because of surface protrusions, uneven distribution of current density and non-uniform deposition of Na, which result in the continuous growth of metal dendrites, eventually, short circuiting the system.
(3) Gas evolution.161 The formation of dendrites is one of the main reasons for gas evolution, which accumulates gradually upon the reaction between electrolyte and metal dendrites.
(4) Volume fluctuations.162,163 This can occur because of the increase or decrease in the thickness of the deposits, resulting in increased pressure and mechanical stresses.
Thus far, various strategies have been employed to stabilize the Na metal anode. For instance, Gu et al.164 reported an electrochemical surface polishing approach to achieve an ultra-smooth and ultra-thin SEI for an Na metal anode with an organic/inorganic rich multilayer structure. Jiao et al.165 proposed an implantable artificial protective layer to stabilize an Na metal anode obtained via the doctor-blade coating layer technique, where NaF particles were added to an N-methyl pyrrolidone (NMP) and polyvinylidene fluoride (PVDF) solution, giving a thickness of 20 μm. Zhao's group166 reported the rational design of carbon paper (CP) with N-doped carbon nanotubes (CNTs) for Na deposition as a 3D host that can accommodate Na ions. Owing to the good conductivity and high surface area of CP, it was used as a skeleton and the N-doped CNTs effectively reduced the contact angle between the Na metal and host. Thus far, electrolyte engineering is the simplest technique to form a nearly ideal artificial solid electrolyte interphase (SEI), where the reaction between the engineered electrolyte and Na metal plays a decisive role. An ideal electrolyte should possess some characteristic attributes such as: (i) good ionic conductivity (1–10 mS cm−1)167,168 to allow easy access to sodium ions, (ii) wide potential window (1.5–4.2 V vs. Na/Na+),169 (iii) not actively participate in the chemical reaction with a stabilized electrolyte-electrode interfacial contact, (iv) electrochemical compatibility with the components of the battery system and must be inflammable, and (v) cost-effective and environmental benignancy.33,170 Nevertheless, the aforementioned attributes can be challenging due to the instability of most of organic electrolytes in the presence of alkali metal anodes, resulting in the formation of a highly uncontrolled solid electrolyte interface (SEI).171 Thus, continuous research to engineer the electrolyte has been put forward to achieve most of the desirable properties. It should be noted that it is crucial to establish an electrolyte system with a suitable solvent, long-term stable sodium salts and additives to ensure the formation of a mechanically and chemically stable SEI.172,173 The progress made in electrolyte engineering is still in its infancy and highly limited due to its high corrosivity, narrow thermal stability window of the additives and sodium salts, besides their thermodynamic instability in the presence of Na metal.
The concept of designing localized metal-alloy artificial interfaces is relatively new, which exhibits tremendous potential to boost the stability of alkali metal anodes. Recently, Zheng's group174 devised a new strategy to intrinsically develop a localized metal-alloy interphase. The electrolyte was engineered by adding a controlled amount of additive such as SnCl2 to achieve the in situ emergence of an Na–Sn alloy interphase directly over the Na metal anode, as shown in Fig. 12a. The interphase was considered to result from the electrochemical reactions between SnCl2 and Na metal, where SnCl2 is reduced to Sn and forms an alloy with Na.177,178 A dendrite free anode and stable cycling were achieved for 500 h with 50 mM SnCl2 added to the EC/PC electrolyte, showing an improved performance compared to the Na3V2(PO4)3 (NVP) cathode. Although the stability of the anode improved, the deposition morphology appeared to be highly non-uniform. Thus, to design and develop a controlled and highly uniform metal-alloy interphase, an innovative methodology was proposed recently.175 Generally, Na deposition can allow equilibrium and controlled growth at low current densities in contrast to high current densities, which involve non-equilibrium and uncontrolled growth.179,180 For a perfect interphase, enhanced critical strain and high Young's modulus are essential to allow effective functioning at both low and high current densities. Non-uniform and uncontrolled growth due to volume expansion and contraction can be handled by high critical strain (ductile). Conversely, controlled and equilibrium growth can be obtained via a high Young's modulus (stiff), alleviating the generation of large dendrites.181 For rapid and smooth Na ion transport across the interphase, a low ionic diffusion barrier can be a crucial factor. For instance, existing SEI products such as NaF possess relatively large ionic diffusion barriers and Na2O is stiff but a very brittle ceramic product, whereas organic compounds possess high porosity but less stiffness.182,183 Consequently, the current Na anodes can only show stability at low current densities (0.25 to 1 mA cm−2).160,184,185 Thus to circumvent this issue, Kumar et al.186 demonstrated a biphasic interphase (BPI) that was grown by reacting aqueous ammonia vapour with an Na metal anode, consisting of two chemically different phases (NaNH2 and NaOH). NaNH2 shows high ductility with high critical strain and NaOH possesses good stiffness with a high Young's modulus. Consequently, the BPI showed a smooth track with a low ion diffusion barrier, guiding the ions to dissolve and deposit uniformly. As a result, highly stable Na plating/stripping characteristics at high current densities (1–50 mA cm−2) and high areal capacities (1–10 mA h cm−2) were obtained. Unlike the solid–liquid approach, a solid–vapor approach was utilized to realize the formation of highly controlled metal-alloy interphases directly over the Na metal anode. The direct reaction of tin-tetrachloride (SnCl4) vapor with Na metal led to the formation of localized metal-alloy interphases instantly. Due to the strong adhesion and enhanced Young's modulus of elasticity, the interphase could accommodate volume fluctuations. Conversely, a highly compact and uniform layer of metal-alloy ensures effective screening of the metal anode, while it provides improved ionic conductivity to the electrolyte ions. Besides exploring Sn-based alloys, it has been demonstrated that the reduction potential of the metal cation is a decisive factor in guaranteeing the formation of a metal alloy interphase. The reduction of the metal cation and further formation of an MAI with the Na anode due to the spontaneity of the reaction (ΔG < 0) is supported by a high positive reduction potential with respect to Na. Another strategy to form a sodiophilic metal alloy interphase was proposed by Tang et al.176 to enable efficient Na deposition and stripping. They compared the sodiophilicity of Au, Mo, Cr, Cu, Sn and Sb by electrochemical measurements and reported that only Sb, Au and Sn formed an alloy with Na metal.187–189 They further highlighted the continuous alloying–dealloying resulted in pulverisation of the sodiophilic metal alloys, leading to the disappearance of the sodiophilic interphase. A technique for controlling the cut-off potential was employed and the Na-metal alloy interphase was bound by binders, as shown in Fig. 12c, resulting in an enhanced performance. Therefore, the formation of metal-alloy interphases can be helpful in improving the performance of alkali-metal anodes.
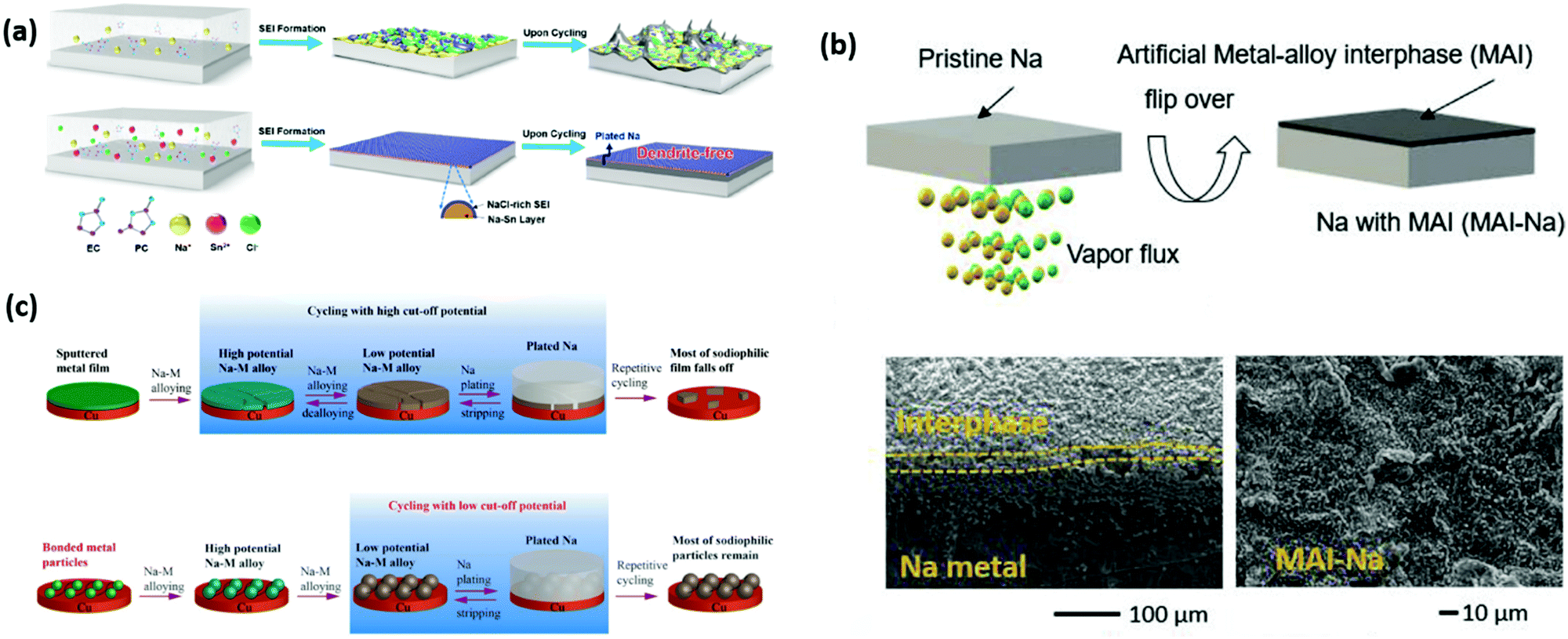 | ||
| Fig. 12 (a) Schematic of SEI generation on Na metal working in a traditional carbonate-based electrolyte, leading to dendrite growth and in situ-formed NaCl-rich SEI with Na–Sn alloy layer, resulting in uniform ion transport and dendrite-free cycling. Reproduced with permission.174 Copyright 2019, the American Chemical Society. (b) Schematic representation of process involved in forming an artificial metal alloy interphase and corresponding SEM images. Reproduced with permission.175 Copyright 2020, Elsevier. (c) Cycling with high cut-off potential, leading to most of the sodiophilic films falling off and cycling with low cut-off potential leads to the retention of most of the sodiophilic particles. Reproduced with permission.176 Copyright 2019, John Wiley and Sons. | ||
4. Conclusion and future outlook
The physiochemical engineering of the matrix is one of the key factors for the development of a stable and high sulfur-loaded sodium–sulfur battery system. Considering the fact that Na+ ions are relatively bigger and sodium polysulfide are comparatively more corrosive than their Li analogue, the RT-Na/S battery is expected to suffer more severely than that of Li–S battery system during its operation. With an increase sulfur loading or increased thickness of the cathode layer, several issues may become prominent, for instance, (1) a greater loading of active material in the electrode can result in material peel-off due to weak interparticle interaction, (2) as the thickness increases, the resistance increases due to the insulating nature of sulfur and (3) limited accessibility of electrolyte ions.190,191 Thus, various strategies have been developed to enhance the sulfur loading and mitigate polysulfide shuttling without adversely affecting the cell performance, and some of the important approaches were highlighted in the present review. Particularly, basic insight into the chemical binding, strategies for mesoscale assembly, unique architectures, and configurational innovation in the cathode were specific concerns. It is apparent from the literature that an improved sulfur loading can be realized in two ways. Firstly, through increased utilization of the sulfur cathode and mitigating the dissolution of the soluble discharge products during cycling. Secondly, infusing more and more sulfur into the matrix, and therefore, the matrix plays an important role in controlling the stability of RT-Na/S batteries. The primary focus of this review was to summarize the literature presenting novel ideas to alter the physiochemical properties of the matrix to enhance the sulfur loading.192 Although the fundamental properties of Na are largely different from Li, the development of RT-Na/S batteries has also been assisted by the progress made in Li–S batteries, and particularly the rational design of electrode materials.Under the premise of a high S-loading, a few strategies can be proposed to ensure a high specific energy density: (1) use of modified and polar separators, (2) highly porous current collector architecture, (3) employing a compatible electrolyte system, (4) developing different functional moieties and judicious combination of different binder systems and (5) replacing sulfur with sulfur-rich composites, for example transition metal sulfides. These developments have been reported for different battery systems to enhance their performance and energy density, and it is likely that their contribution in RT-Na/S will also result in positive effects.
In addition to fabricating suitable electrode architectures, an indirect approach is to develop modified separators for inhibiting the dissolution and simultaneously resolving the issue of the loss of the active materials. Lin et al.193 studied nickel cobaltite introduced into carbon nanofibres (NiCo2O4/CNF) as a modified separator to anchor the polysulfides and accelerate the conversion of long-chain to short-chain polysulfides through the Ni–S and Co–S bonding structure in an Li/S battery system. The modified separator demonstrated a well-defined interface with strong affinity towards polysulfides and rich polar nucleation sites, and thus could act as an accelerator and catalyze the conversion reactions. NiCo2O4/CNF could provide pathway for a fast ionic diffusion and electron mobility, resulting in a high S-loading of up to 7.9 mg cm−2.
Given that the design of a compatible electrolyte system is also important for achieving a stable system with higher safety, gel electrolytes are trailblazing technology for achieving a high S-content in cathode composites. To reach a S-loading of about 14 mg cm−2, a state-of-the art design of a highly elastic 3D-gel cathode was developed, which comprised an interconnected co-polymer skeleton (achieved through phase inversion strategy).194 The phase inversion strategy is a well-known universal technique for constructing a porous interconnected-polymeric network, and it can open up new pathways for the matrix to accommodate a high sulfur loading.195 During this process, three continuous phases merge, i.e., the S/C composite, the pores phase and a polymeric phase are formed in one-pot simultaneously, leading to the development of an electronically conductive scaffold to promote conductive pathways. The tri-continuous structured matrix with an electron conductive network consists of inter-penetrable macropores, which can favor electrolyte permeation and a high loading of sulfur. Gel electrolytes have also been fabricated in a similar fashion with an amorphous fluorinated co-polymer matrix with high ionic conductivity, rendering high flexibility and suppression of dendritic growth.
The binder is another important component, which is indispensable for achieving greater thickness of sulfur in the electrode material, and hence a high sulfur loading. However, the conventional way of using binders can limit the diffusion of electrolyte given that the binder can fill the spaces in the carbon matrix, thus resulting in a reduced sulfur loading in the electrode material.196 The premise of a high specific density can be ensured with a high sulfur loading. Recently, Shaibani and co-workers197 reported an expansion-tolerant architecture with a new approach to augment the number of active sites to accommodate a large fraction of active materials. Applying the bridging architecture mechanism,198 dispersion of a high modulus Na–CMC binder could be achieved by forming web-like bridging bonds between the neighbouring particles (without filling the voids of the matrix). A sulfur cathode with a sulfur loading as high as 15 mg cm−2 could be achieved through the “bridging architecture technique.” A new strategy of developing a high-loaded sulfur cathode system was recently proposed by Chen et al.56 A 3D multifunctional flexible network composed of highly polar groups was developed as a binder for the elemental sulfur powder. The amino group-functionalized network encapsulated sulfur particles (AFG)@S and allowed a high sulfur loading of about 8 mg cm−2 without deteriorating the stability and cycle-life of the cell. Theoretical calculation predicted the high affinity of the –NH2 groups towards sulfur particles and dissolved lithium polysulfide. In addition, the 3D structure of the network offers abundant absorption or adsorption sites for polysulfides. Considering that sodium polysulfides are more soluble than lithium polysulfides, it is expected that they may access the absorption sites more easily compared to lithium polysulfides. Therefore, designing a functional multi-dimensional structure with highly polar adsorption sites can be pivotal in improving the utilization and loading of sulfur in sulfur cathodes for RT-Na/S batteries. Furthermore, Han and co-workers199 formulated the idea of a nucleophilic bi-functional binder with the ability to immobilize polysulfides within the carbon matrix for achieving a higher thickness of active materials. The combination of a maleate and amine group in a binder provides high efficacy for binding, besides chemical and mechanical stability. The bi-functional maleate poly-(ethylene glycol) (PEG) binder resulted in a high sulfur loading of about 12 mg cm−2 with a sulfur content of 80 wt%. Hencz et al.200 illustrated the importance of a polymeric binder matrix in housing sulfur for providing the necessary interfacial interactions with an efficient interlocking mechanism.
Significant progress in the field of RT/Na–S batteries has been made in recent years; however, the viability of this technology still requires in-depth research to unveil the versatile aspects of the electrode materials. The physiochemical structure–property relationship needs to be developed in a more comprehensive manner to address the key challenges. There still exists a huge vacuum in this technology, which requires a better understanding of the conversion mechanisms through advanced characterization tools. Thus, continuous and innovative efforts are still required to further improve the performance of RT-Na/S batteries.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Sungjemmenla, Chhail Bihari Soni, and Vineeth S. K., acknowledge the scholarship awarded by the Indian Institute of Technology Delhi (IIT Delhi). This work was financially supported by the IIT Delhi.References
- T. Kousksou, P. Bruel, A. Jamil, T. El Rhafiki and Y. Zeraouli, Sol. Energy Mater. Sol. Cells, 2014, 120, 59–80 Search PubMed.
- S. Choudhury, S. Wei, Y. Ozhabes, D. Gunceler, M. J. Zachman, Z. Tu, J. H. Shin, P. Nath, A. Agrawal and L. F. Kourkoutis, Nat. Commun., 2017, 8, 1–10 Search PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 Search PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 Search PubMed.
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292–308 Search PubMed.
- J. Liu, J. G. Zhang, Z. Yang, J. P. Lemmon, C. Imhoff, G. L. Graff, L. Li, J. Hu, C. Wang and J. Xiao, Adv. Funct. Mater., 2013, 23, 929–946 Search PubMed.
- F. Schipper, E. M. Erickson, C. Erk, J.-Y. Shin, F. F. Chesneau and D. Aurbach, J. Electrochem. Soc., 2016, 164, A6220 Search PubMed.
- A. Manthiram, J. Phys. Chem. Lett., 2011, 2, 176–184 Search PubMed.
- Y. Wang, D. Zhou, V. Palomares, D. Shanmukaraj, B. Sun, X. Tang, C. Wang, M. Armand, T. Rojo and G. Wang, Energy Environ. Sci., 2020, 13, 3848–3879 Search PubMed.
- R. De Silva, M. Jayaweera, V. Perera, I. Jayarathna and S. Rosa, Sri Lankan J. Phys., 2014, 15, 19–29 Search PubMed.
- Z. Wen, J. Cao, Z. Gu, X. Xu, F. Zhang and Z. Lin, Solid State Ionics, 2008, 179, 1697–1701 Search PubMed.
- K. Jung, H.-J. Heo, J.-H. Lee, Y.-C. Park and C.-Y. Kang, Corros. Sci., 2015, 98, 748–757 Search PubMed.
- J. T. Kummer and N. Weber, SAE Trans., 1968, 76, 1003–1028 Search PubMed.
- N. Kawakami, Y. Iijima, M. Fukuhara, M. Bando, Y. Sakanaka, K. Ogawa and T. Matsuda, Development and field experiences of stabilization system using 34MW NAS batteries for a 51MW wind farm, IEEE International Symposium on Industrial Electronics (ISIE2010), 2010, pp. 2371–2376 Search PubMed.
- S. Tewari and N. Mohan, IEEE Trans. Power Syst., 2012, 28, 532–541 Search PubMed.
- G. Nikiforidis, M. Van de Sanden and M. N. Tsampas, RSC Adv., 2019, 9, 5649–5673 Search PubMed.
- V. Kumar, Y. Wang, A. Y. S. Eng, M.-F. Ng and Z. W. Seh, Cell Rep. Phys. Sci., 2020, 1, 100044 Search PubMed.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, ACS Cent. Sci., 2015, 1, 449–455 Search PubMed.
- H. Ryu, T. Kim, K. Kim, J.-H. Ahn, T. Nam, G. Wang and H.-J. Ahn, J. Power Sources, 2011, 196, 5186–5190 Search PubMed.
- X. Yu and A. Manthiram, Chem. – Eur. J., 2015, 21, 4233–4237 Search PubMed.
- C.-W. Park, J.-H. Ahn, H.-S. Ryu, K.-W. Kim and H.-J. Ahn, Electrochem. Solid-State Lett., 2006, 9, A123 Search PubMed.
- J. Wang, J. Yang, Y. Nuli and R. Holze, Electrochem. Commun., 2007, 9, 31–34 Search PubMed.
- S. H. Chung and A. Manthiram, Adv. Mater., 2019, 31, 1901125 Search PubMed.
- D. Liu, Z. Li, X. Li, Z. Cheng, L. Yuan and Y. Huang, ChemPhysChem, 2019, 20, 3164–3176 Search PubMed.
- R. Dominko, A. Vizintin, G. Aquilanti, L. Stievano, M. J. Helen, A. R. Munnangi, M. Fichtner and I. Arcon, J. Electrochem. Soc., 2017, 165, A5014 Search PubMed.
- J. H. Lee, J. Kang, S.-W. Kim, W. Halim, M. W. Frey and Y. L. Joo, ACS Omega, 2018, 3, 16465–16471 Search PubMed.
- J. Liu, D. Lu, J. Zheng, P. Yan, B. Wang, X. Sun, Y. Shao, C. Wang, J. Xiao and J.-G. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 21965–21972 Search PubMed.
- S. Zhang, Y. Yao and Y. Yu, ACS Energy Lett., 2021, 6, 529–536 Search PubMed.
- X. Wang, T. Gao, F. Han, Z. Ma, Z. Zhang, J. Li and C. Wang, Nano Energy, 2016, 30, 700–708 Search PubMed.
- A. Ghosh, S. Shukla, M. Monisha, A. Kumar, B. Lochab and S. Mitra, ACS Energy Lett., 2017, 2, 2478–2485 Search PubMed.
- M. A. Pope and I. A. Aksay, Adv. Energy Mater., 2015, 5, 1500124 Search PubMed.
- D. Kumar, S. K. Rajouria, S. B. Kuhar and D. Kanchan, Solid State Ionics, 2017, 312, 8–16 Search PubMed.
- T. Li, J. Xu, C. Wang, W. Wu, D. Su and G. Wang, J. Alloys Compd., 2019, 792, 797–817 Search PubMed.
- Y. Wang, Y. Zhang, H. Cheng, Z. Ni, Y. Wang, G. Xia, X. Li and X. Zeng, Molecules, 2021, 26, 1535 Search PubMed.
- D. Kumar, D. Kanchan, S. Kumar and K. Mishra, Mater. Sci. Energy Technol., 2019, 2, 117–129 Search PubMed.
- N. Chawla and M. Safa, Electronics, 2019, 8, 1201 Search PubMed.
- H. B. Wu, S. Wei, L. Zhang, R. Xu, H. H. Hng and X. W. Lou, Chem. – Eur. J., 2013, 19, 10804–10808 Search PubMed.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969 Search PubMed.
- A. Manthiram, S. H. Chung and C. Zu, Adv. Mater., 2015, 27, 1980–2006 Search PubMed.
- P. Adelhelm, P. Hartmann, C. L. Bender, M. Busche, C. Eufinger and J. Janek, Beilstein J. Nanotechnol., 2015, 6, 1016–1055 Search PubMed.
- D. Liu, Z. Li, X. Li, Z. Cheng, L. Yuan and Y. Huang, ChemPhysChem, 2019, 20, 3164–3176 Search PubMed.
- Y. X. Wang, W. H. Lai, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Mater., 2020, 32, 1903952 Search PubMed.
- Sungjemmenla, C. B. Soni and V. Kumar, Nanoscale Adv., 2021, 3, 1569–1581 Search PubMed.
- L. Wen, X. Wang, G. Q. Liu, H. Z. Luo, J. Liang and S. X. Dou, Surf. Innovations, 2017, 6, 13–18 Search PubMed.
- Z. Chen, Y. Qin, K. Amine and Y.-K. Sun, J. Mater. Chem., 2010, 20, 7606–7612 Search PubMed.
- F. Lecomte, J. Siepmann, M. Walther, R. J. MacRae and R. Bodmeier, Pharm. Res., 2004, 21, 882–890 Search PubMed.
- X. Yu and A. Manthiram, Matter, 2019, 1, 439–451 Search PubMed.
- Z. Wen, Y. Hu, X. Wu, J. Han and Z. Gu, Adv. Funct. Mater., 2013, 23, 1005–1018 Search PubMed.
- S. Lim, R. Lilly Thankamony, T. Yim, H. Chu, Y.-J. Kim, J. Mun and T.-H. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 1401–1405 Search PubMed.
- W. Zhou, Y. Yu, H. Chen, F. J. DiSalvo and H. C. D. Abruña, J. Am. Chem. Soc., 2013, 135, 16736–16743 Search PubMed.
- Y. Fu and A. Manthiram, Chem. Mater., 2012, 24, 3081–3087 Search PubMed.
- H. Yi, T. Lan, Y. Yang, H. Zeng, T. Zhang, T. Tang, C. Wang and Y. Deng, Energy Storage Mater., 2019, 21, 61–68 Search PubMed.
- M. Ling, L. Zhang, T. Zheng, J. Feng, J. Guo, L. Mai and G. Liu, Nano Energy, 2017, 38, 82–90 Search PubMed.
- A. Y. S. Eng, D.-T. Nguyen, V. Kumar, G. S. Subramanian, M.-F. Ng and Z. W. Seh, J. Mater. Chem. A, 2020, 8, 22983–22997 Search PubMed.
- S.-L. Chou, Y. Pan, J.-Z. Wang, H.-K. Liu and S.-X. Dou, Phys. Chem. Chem. Phys., 2014, 16, 20347–20359 Search PubMed.
- W. Chen, T. Qian, J. Xiong, N. Xu, X. Liu, J. Liu, J. Zhou, X. Shen, T. Yang and Y. Chen, Adv. Mater., 2017, 29, 1605160 Search PubMed.
- J. Liu, D. G. Galpaya, L. Yan, M. Sun, Z. Lin, C. Yan, C. Liang and S. Zhang, Energy Environ. Sci., 2017, 10, 750–755 Search PubMed.
- S. Waluś, G. Offer, I. Hunt, Y. Patel, T. Stockley, J. Williams and R. Purkayastha, Energy Storage Mater., 2018, 10, 233–245 Search PubMed.
- G. Rong-Nan and H. Wei-Qiang, J. Inorg. Mater., 2019, 34, 1021–1029 Search PubMed.
- Y. Jiao, W. Chen, T. Lei, L. Dai, B. Chen, C. Wu and J. Xiong, Nanoscale Res. Lett., 2017, 12, 1–8 Search PubMed.
- A. Ghosh, A. Kumar, A. Roy, M. R. Panda, M. Kar, D. R. MacFarlane and S. Mitra, ACS Appl. Mater. Interfaces, 2019, 11, 14101–14109 Search PubMed.
- T. Wu, M. Jing, L. Yang, G. Zou, H. Hou, Y. Zhang, Y. Zhang, X. Cao and X. Ji, Adv. Energy Mater., 2019, 9, 1803478 Search PubMed.
- K. Chen, H. Li, Y. Xu, K. Liu, H. Li, X. Xu, X. Qiu and M. Liu, Nanoscale, 2019, 11, 5967–5973 Search PubMed.
- J. Yan, W. Li, R. Wang, P. Feng, M. Jiang, J. Han, S. Cao, Z. Zhang, K. Wang and K. Jiang, ACS Energy Lett., 2020, 5, 1307–1315 Search PubMed.
- C. Ling and F. Mizuno, Phys. Chem. Chem. Phys., 2014, 16, 10419–10424 Search PubMed.
- F. Wu, R. Dong, Y. Bai, Y. Li, G. Chen, Z. Wang and C. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 21335–21342 Search PubMed.
- X. Wang, S. Wang, K. Shen, S. He, X. Hou and F. Chen, J. Mater. Chem. A, 2020, 8, 4007–4016 Search PubMed.
- Y. Fang, X. Y. Yu and X. W. Lou, Angew. Chem., 2019, 131, 7826–7830 Search PubMed.
- K. Zhang, F. Zhang, H. Pan, J. Yu, L. Wang, D. Wang, L. Wang, G. Hu, J. Zhang and Y. Qian, Electrochim. Acta, 2020, 354, 136648 Search PubMed.
- B.-W. Zhang, T. Sheng, Y.-D. Liu, Y.-X. Wang, L. Zhang, W.-H. Lai, L. Wang, J. Yang, Q.-F. Gu and S.-L. Chou, Nat. Commun., 2018, 9, 1–11 Search PubMed.
- N. Wang, Y. Wang, Z. Bai, Z. Fang, X. Zhang, Z. Xu, Y. Ding, X. Xu, Y. Du and S. Dou, Energy Environ. Sci., 2020, 13, 562–570 Search PubMed.
- S. Li, Z. Zeng, J. Yang, Z. Han, W. Hu, L. Wang, J. Ma, B. Shan and J. Xie, ACS Appl. Energy Mater., 2019, 2, 2956–2964 Search PubMed.
- S. Zheng, P. Han, Z. Han, P. Li, H. Zhang and J. Yang, Adv. Energy Mater., 2014, 4, 1400226 Search PubMed.
- J. Zhu, A. Abdelkader, D. Demko, L. Deng, P. Zhang, T. He, Y. Wang and L. Huang, Molecules, 2020, 25, 1585 Search PubMed.
- S. Wei, S. Xu, A. Agrawral, S. Choudhury, Y. Lu, Z. Tu, L. Ma and L. A. Archer, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- D. Kumar, D. Kanchan, S. Kumar and K. Mishra, Mater. Sci. Energy Technol., 2019, 2, 117–129 Search PubMed.
- X. Yu and A. Manthiram, J. Phys. Chem. Lett., 2014, 5, 1943–1947 Search PubMed.
- M. Wang, W. Wang, A. Wang, K. Yuan, L. Miao, X. Zhang, Y. Huang, Z. Yu and J. Qiu, Chem. Commun., 2013, 49, 10263–10265 Search PubMed.
- S. Wei, L. Ma, K. E. Hendrickson, Z. Tu and L. A. Archer, J. Am. Chem. Soc., 2015, 137, 12143–12152 Search PubMed.
- X. Wang, X. Hao, Y. Xia, Y. Liang, X. Xia and J. Tu, J. Membr. Sci., 2019, 582, 37–47 Search PubMed.
- J. Ye, F. He, J. Nie, Y. Cao, H. Yang and X. Ai, J. Mater. Chem. A, 2015, 3, 7406–7412 Search PubMed.
- I. Kim, C. H. Kim, S. H. Choi, J.-P. Ahn, J.-H. Ahn, K.-W. Kim, E. J. Cairns and H.-J. Ahn, J. Power Sources, 2016, 307, 31–37 Search PubMed.
- T. H. Hwang, D. S. Jung, J.-S. Kim, B. G. Kim and J. W. Choi, Nano Lett., 2013, 13, 4532–4538 Search PubMed.
- Z. Li, J. Zhang, Y. Lu and X. W. D. Lou, Sci. Adv., 2018, 4, eaat1687 Search PubMed.
- D. Ma, Y. Li, J. Yang, H. Mi, S. Luo, L. Deng, C. Yan, M. Rauf, P. Zhang and X. Sun, Adv. Funct. Mater., 2018, 28, 1705537 Search PubMed.
- S. Xin, Y. X. Yin, Y. G. Guo and L. J. Wan, Adv. Mater., 2014, 26, 1261–1265 Search PubMed.
- M. Shobana, J. Alloys Compd., 2019, 802, 477–487 Search PubMed.
- C. Ye, D. Chao, J. Shan, H. Li, K. Davey and S.-Z. Qiao, Matter, 2020, 2, 323–344 Search PubMed.
- W. G. Chong, J. Q. Huang, Z. L. Xu, X. Qin, X. Wang and J. K. Kim, Adv. Funct. Mater., 2017, 27, 1604815 Search PubMed.
- L. Hu, Y. Lu, T. Zhang, T. Huang, Y. Zhu and Y. Qian, ACS Appl. Mater. Interfaces, 2017, 9, 13813–13818 Search PubMed.
- Z. Zhang, L. L. Kong, S. Liu, G. R. Li and X. P. Gao, Adv. Energy Mater., 2017, 7, 1602543 Search PubMed.
- L. Qie and A. J. A. M. Manthiram, Adv. Mater., 2015, 27, 1694–1700 Search PubMed.
- Q. Pang, X. Liang, C. Y. Kwok, J. Kulisch and L. F. Nazar, Adv. Energy Mater., 2017, 7, 1601630 Search PubMed.
- Y. Ma, H. Zhang, B. Wu, M. Wang, X. Li and H. Zhang, Sci. Rep., 2015, 5, 14949 Search PubMed.
- G. Zhou, L. Li, C. Ma, S. Wang, Y. Shi, N. Koratkar, W. Ren, F. Li and H.-M. Cheng, Nano Energy, 2015, 11, 356–365 Search PubMed.
- M. Hagen, S. Dörfler, H. Althues, J. Tübke, M. Hoffmann, S. Kaskel and K. Pinkwart, J. Power Sources, 2012, 213, 239–248 Search PubMed.
- I. Kim, J.-Y. Park, C. H. Kim, J.-W. Park, J.-P. Ahn, J.-H. Ahn, K.-W. Kim and H.-J. Ahn, J. Power Sources, 2016, 301, 332–337 Search PubMed.
- X. Hu, Y. Ni, C. Wang, H. Wang, E. Matios, J. Chen and W. Li, Cell Rep. Phys. Sci., 2020, 1, 100015 Search PubMed.
- H. Ye, L. Ma, Y. Zhou, L. Wang, N. Han, F. Zhao, J. Deng, T. Wu, Y. Li and J. Lu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13091–13096 Search PubMed.
- M. Sajjad, T. Hussain, N. Singh and J. A. Larsson, Langmuir, 2020, 36, 13104–13111 Search PubMed.
- L. Sun, M. Li, Y. Jiang, W. Kong, K. Jiang, J. Wang and S. J. N. L. Fan, Nano Lett., 2014, 14, 4044–4049 Search PubMed.
- Z. Yuan, H.-J. Peng, J.-Q. Huang, X.-Y. Liu, D.-W. Wang, X.-B. Cheng and Q. Zhang, Adv. Funct. Mater., 2014, 24, 6105–6112 Search PubMed.
- X. Xu, D. Zhou, X. Qin, K. Lin, F. Kang, B. Li, D. Shanmukaraj, T. Rojo, M. Armand and G. Wang, Nat. Commun., 2018, 9, 3870 Search PubMed.
- D. Kumar, Solid State Ionics, 2018, 318, 65–70 Search PubMed.
- X. Bian, Y. Gao, Q. Fu, S. Indris, Y. Ju, Y. Meng, F. Du, N. Bramnik, H. Ehrenberg and Y. Wei, J. Mater. Chem. A, 2017, 5, 600–608 Search PubMed.
- X. Liu, J. Q. Huang, Q. Zhang and L. Mai, Adv. Mater., 2017, 29, 1601759 Search PubMed.
- Y. Xu, A. Sumboja, A. Groves, T. Ashton, Y. Zong and J. A. Darr, RSC Adv., 2020, 10, 41871–41882 Search PubMed.
- J. He, Y. Chen and A. Manthiram, iScience, 2018, 4, 36–43 Search PubMed.
- T. Zhu, X. Dong, Y. Liu, Y.-G. Wang, C. Wang and Y.-Y. Xia, ACS Appl. Energy Mater., 2019, 2, 5263–5271 Search PubMed.
- Q. Ma, G. Du, B. Guo, W. Tang, Y. Li, M. Xu and C. Li, Chem. Eng. J., 2020, 388, 124210 Search PubMed.
- Q. Lu, X. Wang, J. Cao, C. Chen, K. Chen, Z. Zhao, Z. Niu and J. Chen, Energy Storage Mater., 2017, 8, 77–84 Search PubMed.
- I. Kim, C. Kim, H. Kim, K.-W. Kim, J.-H. Ahn and H.-J. Ahn, J. Nanosci. Nanotechnol., 2018, 18, 6524–6527 Search PubMed.
- Q. Guo, S. Li, X. Liu, H. Lu, X. Chang, H. Zhang, X. Zhu, Q. Xia, C. Yan and H. Xia, Adv. Sci., 2020, 7, 1903246 Search PubMed.
- R. Fang, S. Zhao, P. Hou, M. Cheng, S. Wang, H. M. Cheng, C. Liu and F. Li, Adv. Mater., 2016, 28, 3374–3382 Search PubMed.
- H. Nara, T. Yokoshima, H. Mikuriya, S. Tsuda, T. Momma and T. Osaka, J. Electrochem. Soc., 2016, 164, A5026 Search PubMed.
- M. K. Aslam, I. D. Seymour, N. Katyal, S. Li, T. Yang, S.-J. Bao, G. Henkelman and M. Xu, Nat. Commun., 2020, 11, 1–11 Search PubMed.
- R. Carter, L. Oakes, A. Douglas, N. Muralidharan, A. P. Cohn and C. L. Pint, Nano Lett., 2017, 17, 1863–1869 Search PubMed.
- Y.-X. Wang, J. Yang, W. Lai, S.-L. Chou, Q.-F. Gu, H. K. Liu, D. Zhao and S. X. Dou, J. Am. Chem. Soc., 2016, 138, 16576–16579 Search PubMed.
- H. J. Peng, J. Q. Huang, X. B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 7, 1700260 Search PubMed.
- Y. Zhang, J. Ren, D. Wang, C. Zhang, F. Yin, A. Mukanova and Z. Bakenov, ChemElectroChem, 2018, 5, 1591–1598 Search PubMed.
- S. Rehman, K. Khan, Y. Zhao and Y. Hou, J. Mater. Chem. A, 2017, 5, 3014–3038 Search PubMed.
- S. Rehman, K. Khan, Y. Zhao and Y. Hou, J. Mater. Chem. A, 2017, 5, 3014–3038 Search PubMed.
- L. He, W. C. Li, S. Xu and A. H. Lu, Chem. – Eur. J., 2019, 25, 3209–3218 Search PubMed.
- R. Zhao, Z. Liang, R. Zou and Q. Xu, Joule, 2018, 2, 2235–2259 Search PubMed.
- X. Li, S. Zheng, L. Jin, Y. Li, P. Geng, H. Xue, H. Pang and Q. Xu, Adv. Energy Mater., 2018, 8, 1800716 Search PubMed.
- Z. Wang, H. Tao and Y. Yue, ChemElectroChem, 2019, 6, 5358–5374 Search PubMed.
- R. Chen, T. Zhao, T. Tian, S. Cao, P. R. Coxon, K. Xi, D. Fairen-Jimenez, R. Vasant Kumar and A. K. Cheetham, APL Mater., 2014, 2, 124109 Search PubMed.
- Q. Wu, X. Zhou, J. Xu, F. Cao and C. Li, J. Energy Chem., 2019, 38, 94–113 Search PubMed.
- J. Xu, W. Zhang, Y. Chen, H. Fan, D. Su and G. Wang, J. Mater. Chem. A, 2018, 6, 2797–2807 Search PubMed.
- A. E. Baumann, D. A. Burns, B. Liu and V. S. Thoi, Commun. Chem., 2019, 2, 1–14 Search PubMed.
- J. Zhou, R. Li, X. Fan, Y. Chen, R. Han, W. Li, J. Zheng, B. Wang and X. Li, Energy Environ. Sci., 2014, 7, 2715–2724 Search PubMed.
- L. Zhang, B. Zhang, Y. Dou, Y. Wang, M. Al-Mamun, X. Hu and H. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 20422–20428 Search PubMed.
- Y.-M. Chen, W. Liang, S. Li, F. Zou, S. M. Bhaway, Z. Qiang, M. Gao, B. D. Vogt and Y. Zhu, J. Mater. Chem. A, 2016, 4, 12471–12478 Search PubMed.
- D.-J. Lee, J.-W. Park, I. Hasa, Y.-K. Sun, B. Scrosati and J. Hassoun, J. Mater. Chem. A, 2013, 1, 5256–5261 Search PubMed.
- G. Xia, L. Zhang, X. Chen, Y. Huang, D. Sun, F. Fang, Z. Guo and X. Yu, Energy Storage Mater., 2018, 14, 314–323 Search PubMed.
- K. Chen, Z. Sun, R. Fang, Y. Shi, H. M. Cheng and F. Li, Adv. Funct. Mater., 2018, 28, 1707592 Search PubMed.
- M. Zhang, C. Yu, C. Zhao, X. Song, X. Han, S. Liu, C. Hao and J. Qiu, Energy Storage Mater., 2016, 5, 223–229 Search PubMed.
- R. Yan, M. Oschatz and F. Wu, Carbon, 2020, 161, 162–168 Search PubMed.
- Z. Yan, J. Xiao, W. Lai, L. Wang, F. Gebert, Y. Wang, Q. Gu, H. Liu, S.-L. Chou and H. Liu, Nat. Commun., 2019, 10, 1–8 Search PubMed.
- B. Guo, W. Du, T. Yang, J. Deng, D. Liu, Y. Qi, J. Jiang, S. J. Bao and M. Xu, Adv. Sci., 2020, 7, 1902617 Search PubMed.
- Y. L. Ding, P. Kopold, K. Hahn, P. A. van Aken, J. Maier and Y. Yu, Adv. Funct. Mater., 2016, 26, 1112–1119 Search PubMed.
- J. Song, M. L. Gordin, T. Xu, S. Chen, Z. Yu, H. Sohn, J. Lu, Y. Ren, Y. Duan and D. Wang, Angew. Chem., 2015, 127, 4399–4403 Search PubMed.
- W. Ren, W. Ma, S. Zhang and B. Tang, Chem. Eng. J., 2018, 341, 441–449 Search PubMed.
- Q. Li, J. Guo, J. Zhao, C. Wang and F. Yan, Nanoscale, 2019, 11, 647–655 Search PubMed.
- F. Xiao, X. Yang, H. Wang, J. Xu, Y. Liu, D. Y. Yu and A. L. Rogach, Adv. Energy Mater., 2020, 10, 2000931 Search PubMed.
- Y. Hao, X. Li, X. Sun and C. Wang, ChemistrySelect, 2017, 2, 9425–9432 Search PubMed.
- Z. Qiang, Y.-M. Chen, Y. Xia, W. Liang, Y. Zhu and B. D. Vogt, Nano Energy, 2017, 32, 59–66 Search PubMed.
- M. L. Meyerson, P. E. Papa, J. A. Weeks, A. G. Paul-Orecchio, A. Heller and C. B. Mullins, ACS Appl. Energy Mater., 2020, 3, 6121–6126 Search PubMed.
- I. Bauer, M. Kohl, H. Althues and S. Kaskel, Chem. Commun., 2014, 50, 3208–3210 Search PubMed.
- S. Wenzel, H. Metelmann, C. Raiß, A. K. Dürr, J. Janek and P. Adelhelm, J. Power Sources, 2013, 243, 758–765 Search PubMed.
- X. Yu and A. Manthiram, J. Phys. Chem. C, 2014, 118, 22952–22959 Search PubMed.
- X. Yu and A. Manthiram, Chem. – Eur. J., 2015, 21, 4233–4237 Search PubMed.
- X. Yu and A. Manthiram, Chem. Mater., 2016, 28, 896–905 Search PubMed.
- L. M. Bloi, J. Pampel, S. Dörfler, H. Althues and S. Kaskel, Adv. Energy Mater., 2020, 10, 1903245 Search PubMed.
- A. Kumar, A. Ghosh, A. Roy, M. R. Panda, M. Forsyth, D. R. MacFarlane and S. Mitra, Energy Storage Mater., 2019, 20, 196–202 Search PubMed.
- B. Sun, P. Xiong, U. Maitra, D. Langsdorf, K. Yan, C. Wang, J. Janek, D. Schröder and G. Wang, Adv. Mater., 2020, 32, 1903891 Search PubMed.
- Y. Zhao, K. R. Adair and X. Sun, Energy Environ. Sci., 2018, 11, 2673–2695 Search PubMed.
- X. Zheng, C. Bommier, W. Luo, L. Jiang, Y. Hao and Y. Huang, Energy Storage Mater., 2019, 16, 6–23 Search PubMed.
- X. Zheng, H. Fu, C. Hu, H. Xu, Y. Huang, J. Wen, H. Sun, W. Luo and Y. Huang, J. Phys. Chem. Lett., 2019, 10, 707–714 Search PubMed.
- H. Wang, C. Wang, E. Matios and W. Li, Nano Lett., 2017, 17, 6808–6815 Search PubMed.
- X. Chen, X. Shen, B. Li, H. J. Peng, X. B. Cheng, B. Q. Li, X. Q. Zhang, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 734–737 Search PubMed.
- J. Luo, C. Wang, H. Wang, X. Hu, E. Matios, X. Lu, W. Zhang, X. Tao and W. Li, Adv. Funct. Mater., 2019, 29, 1805946 Search PubMed.
- F. Wu, J. Zhou, R. Luo, Y. Huang, Y. Mei, M. Xie and R. Chen, Energy Storage Mater., 2019, 22, 376–383 Search PubMed.
- Y. Gu, W.-W. Wang, Y.-J. Li, Q.-H. Wu, S. Tang, J.-W. Yan, M.-S. Zheng, D.-Y. Wu, C.-H. Fan and W.-Q. Hu, Nat. Commun., 2018, 9, 1–9 Search PubMed.
- S. Wang, Y. Jie, Z. Sun, W. Cai, Y. Chen, F. Huang, Y. Liu, X. Li, R. Du and R. Cao, ACS Appl. Energy Mater., 2020, 3, 8688–8694 Search PubMed.
- Y. Zhao, X. Yang, L. Y. Kuo, P. Kaghazchi, Q. Sun, J. Liang, B. Wang, A. Lushington, R. Li and H. Zhang, Small, 2018, 14, 1703717 Search PubMed.
- K. Karuppasamy, J. Theerthagiri, D. Vikraman, C.-J. Yim, S. Hussain, R. Sharma, T. Maiyalagan, J. Qin and H.-S. Kim, Polymers, 2020, 12, 918 Search PubMed.
- S. S. Zhang, J. Electrochem. Soc., 2013, 160, A1421 Search PubMed.
- C. Vidal-Abarca, P. Lavela, J. L. Tirado, A. V. Chadwick, M. Alfredsson and E. Kelder, J. Power Sources, 2012, 197, 314–318 Search PubMed.
- L. Wang, T. Wang, L. Peng, Y. Wang, M. Zhang, J. Zhou, M. Chen, J. Cao, H. Fei and X. Duan, Natl. Sci. Rev., 2021 DOI:10.1093/nsr/nwab050.
- E. Matios, H. Wang, C. Wang and W. Li, Ind. Eng. Chem. Res., 2019, 58, 9758–9780 Search PubMed.
- M. S. Syali, D. Kumar, K. Mishra and D. Kanchan, Energy Storage Mater., 2020, 31, 352–372 Search PubMed.
- A. Y. S. Eng, V. Kumar, Y. Zhang, J. Luo, W. Wang, Y. Sun, W. Li and Z. W. Seh, Adv. Energy Mater., 2021, 2003493 Search PubMed.
- X. Zheng, H. Fu, C. Hu, H. Xu, Y. Huang, J. Wen, H. Sun, W. Luo and Y. Huang, J. Phys. Chem. Lett., 2019, 10, 707–714 Search PubMed.
- V. Kumar, A. Y. S. Eng, Y. Wang, D.-T. Nguyen, M.-F. Ng and Z. W. Seh, Energy Storage Mater., 2020, 29, 1–8 Search PubMed.
- S. Tang, Y. Y. Zhang, X. G. Zhang, J. T. Li, X. Y. Wang, J. W. Yan, D. Y. Wu, M. S. Zheng, Q. F. Dong and B. W. Mao, Adv. Mater., 2019, 31, 1807495 Search PubMed.
- B. Zhang, R. Dugas, G. Rousse, P. Rozier, A. M. Abakumov and J.-M. Tarascon, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- Z. Li, J. Ding and D. Mitlin, Acc. Chem. Res., 2015, 48, 1657–1665 Search PubMed.
- Y. Liu, Y. Zhu and Y. Cui, Nat. Energy, 2019, 4, 540–550 Search PubMed.
- B. L. Mehdi, J. Qian, E. Nasybulin, C. Park, D. A. Welch, R. Faller, H. Mehta, W. A. Henderson, W. Xu, C. M. Wang, J. E. Evans, J. Liu, J. G. Zhang, K. T. Mueller and N. D. Browning, Nano Lett., 2015, 15, 2168–2173 Search PubMed.
- H. Tian, Z. W. Seh, K. Yan, Z. Fu, P. Tang, Y. Lu, R. Zhang, D. Legut, Y. Cui and Q. Zhang, Adv. Energy Mater., 2017, 7, 1602528 Search PubMed.
- S. Choudhury, S. Wei, Y. Ozhabes, D. Gunceler, M. J. Zachman, Z. Tu, J. H. Shin, P. Nath, A. Agrawal, L. F. Kourkoutis, T. A. Arias and L. A. Archer, Nat. Commun., 2017, 8, 898 Search PubMed.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, ACS Cent. Sci., 2015, 1, 449–455 Search PubMed.
- Y.-J. Kim, H. Lee, H. Noh, J. Lee, S. Kim, M.-H. Ryou, Y. M. Lee and H.-T. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 6000–6006 Search PubMed.
- W. Luo, C. F. Lin, O. Zhao, M. Noked, Y. Zhang, G. W. Rubloff and L. Hu, Adv. Energy Mater., 2017, 7, 1601526 Search PubMed.
- V. Kumar, Y. Wang, A. Y. S. Eng, M.-F. Ng and Z. W. Seh, Cell Rep. Phys. Sci., 2020, 1, 100044 Search PubMed.
- W. Luo, F. Shen, C. Bommier, H. Zhu, X. Ji and L. Hu, Acc. Chem. Res., 2016, 49, 231–240 Search PubMed.
- M. Lao, Y. Zhang, W. Luo, Q. Yan, W. Sun and S. X. Dou, Adv. Mater., 2017, 29, 1700622 Search PubMed.
- J. M. Stratford, M. Mayo, P. K. Allan, O. Pecher, O. J. Borkiewicz, K. M. Wiaderek, K. W. Chapman, C. J. Pickard, A. J. Morris and C. P. Grey, J. Am. Chem. Soc., 2017, 139, 7273–7286 Search PubMed.
- Q. Lu, X. Wang, J. Cao, C. Chen, K. Chen, Z. Zhao, Z. Niu and J. Chen, Energy Storage Mater., 2017, 8, 77–84 Search PubMed.
- J. Brückner, S. Thieme, H. T. Grossmann, S. Dörfler, H. Althues and S. Kaskel, J. Power Sources, 2014, 268, 82–87 Search PubMed.
- L. Zhu, W. Zhu, X.-B. Cheng, J.-Q. Huang, H.-J. Peng, S.-H. Yang and Q. Zhang, Carbon, 2014, 75, 161–168 Search PubMed.
- J.-X. Lin, X.-M. Qu, X.-H. Wu, J. Peng, S.-Y. Zhou, J.-T. Li, Y. Zhou, Y.-X. Mo, M.-J. Ding and L. Huang, ACS Sustainable Chem. Eng., 2021, 9, 1804–1813 Search PubMed.
- W. Yan, J. Wei, T. Chen, L. Duan, L. Wang, X. Xue, R. Chen, W. Kong, H. Lin and C. Li, Nano Energy, 2021, 80, 105510 Search PubMed.
- X. Yang, Y. Chen, M. Wang, H. Zhang, X. Li and H. Zhang, Adv. Funct. Mater., 2016, 26, 8427–8434 Search PubMed.
- M. Hagen, D. Hanselmann, K. Ahlbrecht, R. Maça, D. Gerber and J. Tübke, Adv. Energy Mater., 2015, 5, 1401986 Search PubMed.
- M. Shaibani, M. S. Mirshekarloo, R. Singh, C. D. Easton, M. D. Cooray, N. Eshraghi, T. Abendroth, S. Dörfler, H. Althues and S. Kaskel, Sci. Adv., 2020, 6, eaay2757 Search PubMed.
- H. Kokubo, S. Nakamura and H. Sunada, Chem. Pharm. Bull., 1995, 43, 1402–1406 Search PubMed.
- P. Han, S.-H. Chung, C.-H. Chang and A. Manthiram, ACS Appl. Mater. Interfaces, 2019, 11, 17393–17399 Search PubMed.
- L. Hencz, H. Chen, H. Y. Ling, Y. Wang, C. Lai, H. Zhao and S. Zhang, Nanomicro Lett., 2019, 11, 1–44 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |