DOI:
10.1039/D1CE00807B
(Paper)
CrystEngComm, 2021,
23, 5531-5539
Slight ligand modifications within multitopic linear hydroxamates promotes connectivity differences in Cu(II) 1-D coordination polymers†
Received
18th June 2021
, Accepted 22nd July 2021
First published on 23rd July 2021
Abstract
The novel multitopic ligands N-hydroxy-4-((2-hydroxy-3-methoxybenzyl)amino)benzamide (L3H3) and N-hydroxy-4-((2-hydroxybenzyl)amino)benzamide (L4H3) have been synthesised through the Schiff base coupling and subsequent reduction of 4-aminophenylhydroxamic acid and either o-vanillin (to give L3H3) or 2-hydroxybenzaldehyde (to give L4H3). These linear multitopic ligands bind Cu(II) centres at both the hydroxamate and phenol ends to form the 1-D coordination polymers [Cu(II)(L3H2)2]n (1) and {[Cu(II)(L4H2)2]·2MeOH}n (2). Slight differences in the structures of L3H3 and L4H3 lead to significant extended connectivity changes upon Cu(II) metalation that are exampled by a 27% decrease in intra-chain Cu(II)⋯Cu(II) distance upon moving from 1 to 2. The significant conformation and metal binding differences shown by L3H2− and L4H2− in 1 and 2 respectively have been rationalised using density functional theory (DFT) calculations. Hirshfeld surface analysis has been employed to assess and visualise the intra- and intermolecular interactions in both complexes.
Introduction
Coordination polymers (CPs) are extended network materials comprising repeating coordination entities that propagate along one (1-D), two (2-D) or three (3-D) directions and are derived from a combination of metal ion nodes and divergent bridging ligands.1 Consequently, the resultant topology can often be tailored through careful node selection (metal ion geometry preferences)2 and ligand design (number of functional sites and/or shape).3 The ability to exercise synthetic and topological control over the assembly of a coordination polymer has enormous value to the synthetic chemist/materials scientist. Apart from the initial satisfaction it would derive, such undertakings give the protagonist more than a fighting chance of imparting the required functionality (or indeed multi-functionality) to the resultant material.2,3 Such applications may lie in one of a number of research fields that include molecular magnetism (such as spin-crossover behaviour (SCO);4 single-chain magnets (SCM)5 and single-ion magnetism (SIM)/single-molecule magnetism (SMM)6), electrical conductivity,7 luminescence8,13c,e and homo-/heterogeneous catalysis.9,10a The latter relies on the designer transmitting porosity to their extended architectures.10 Indeed, such porous materials are also of intense interest in areas such as gas storage and separation,11 drug delivery12 and sensor materials.4b,13
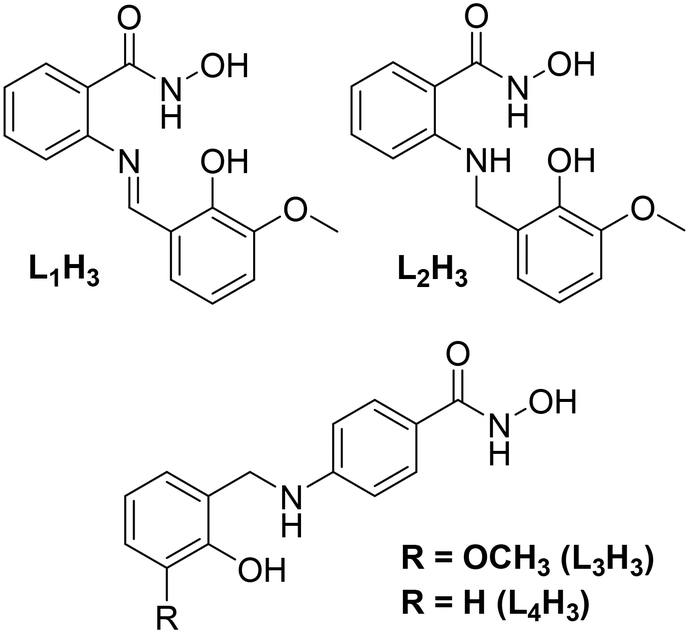
Previous work in our group has described the in situ formation (and Cu(II) ligation) of a series of ligands constructed from the Schiff base coupling of 2-amino-phenylhydroxamic acid and o-vanillin (and its analogues). The planarity of the resulting ligands (e.g. o-[(E)-(2-hydroxy-3-methoxyphenyl)methylideneamino]benzohydroxamic acid; L1H3 in Scheme 1), gave rise to a family of layered planar Cu(II) cages ranging in nuclearity from [Cu(II)10] to [Cu(II)30].14 We went on to show that the selective one-pot imine reduction (using sodium triacetoxyborohydride)15 of the o-[(E)-(2-hydroxy-3-methoxyphenyl)methylideneamino]benzohydroxamic acid ligand afforded the target ligand N-hydroxy-2-((2-hydroxy-3-methoxybenzyl)amino)benzamide (L2H3; Scheme 1). The introduction of a secondary amine group rendered the resultant ligand non-planar as illustrated upon subsequent Cu(II) ligation when forming the 12-MC-4Cu(II) metallacrown [Cu(II)5(L2H)4(MeOH)2](NO3)2·3H2O·4MeOH.16
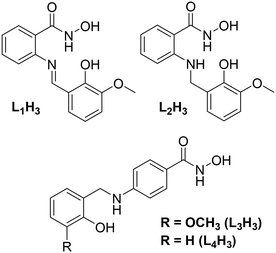 |
| | Scheme 1 (Top) ChemDraw representation of the ligands o-[(E)-(2-hydroxy-3-methoxyphenyl)methylideneamino]benzohydroxamic acid (left; L1H3) and N-hydroxy-2-((2-hydroxy-3-methoxybenzyl)amino)benzamide (right, L2H3) previously used in the production of a series of polynuclear Cu(II) complexes (see main text for details). (Bottom) ChemDraw representation of the ligands N-hydroxy-4-((2-hydroxy-3-methoxybenzyl)amino)benzamide (L3H3) and N-hydroxy-4-((2-hydroxybenzyl)amino)benzamide (L4H3) used in this work. | |
Results and discussion
In this work we describe the design, synthesis and Cu(II) ligation of the multitopic ligands N-hydroxy-4-((2-hydroxy-3-methoxybenzyl)amino)benzamide (L3H3) and N-hydroxy-4-((2-hydroxybenzyl)amino)benzamide (L4H3). Akin to ligands L1–2H3 (Scheme 1), ligands L3H3 and L4H3 are forged through the Schiff base coupling and subsequent imine reduction of 4-amino-phenylhydroxamic acid and either o-vanillin (L3H3) or salicylaldehyde (L4H3) and differ only in the coupling site (the 2-position in L1–2H3cf. 4-position in L3–4H3). The result is the formation of two linear multitopic ligands specifically designed to produce coordination polymers as demonstrated through the construction of the 1-D chains [Cu(II)(L3H2)2]n (1) and {[Cu(II)(L4H2)2]·2MeOH}n (2) as described below.
Structural descriptions
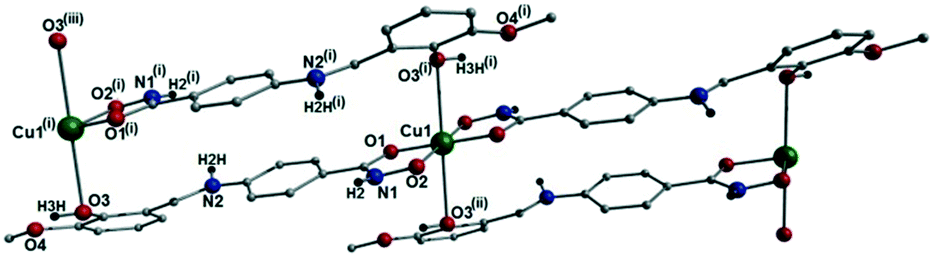
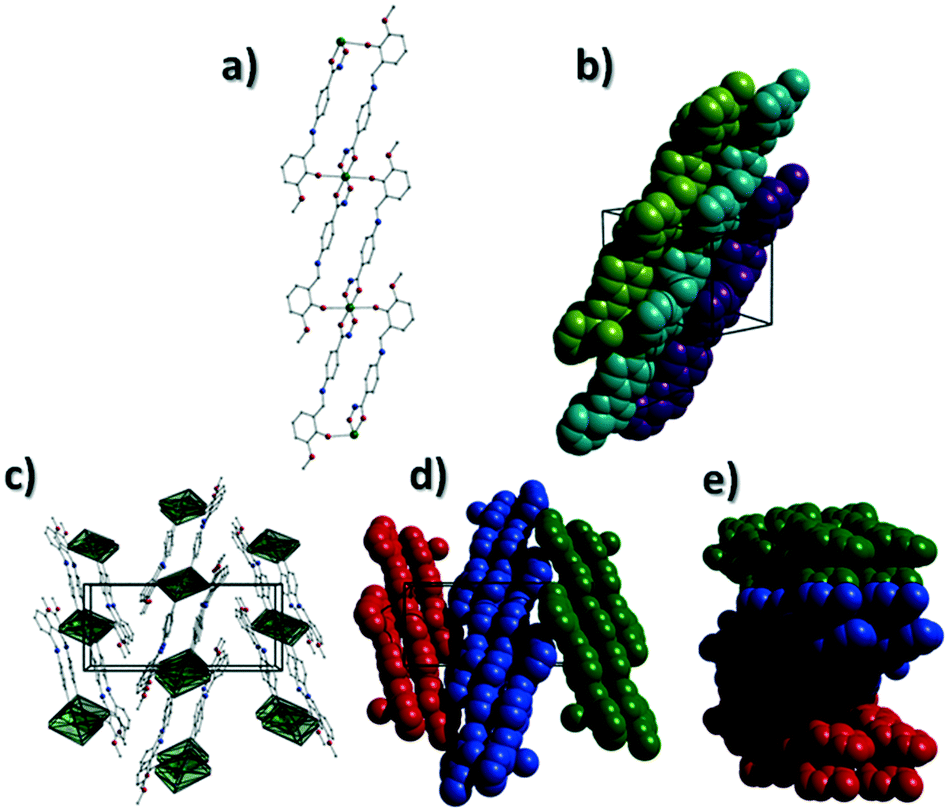
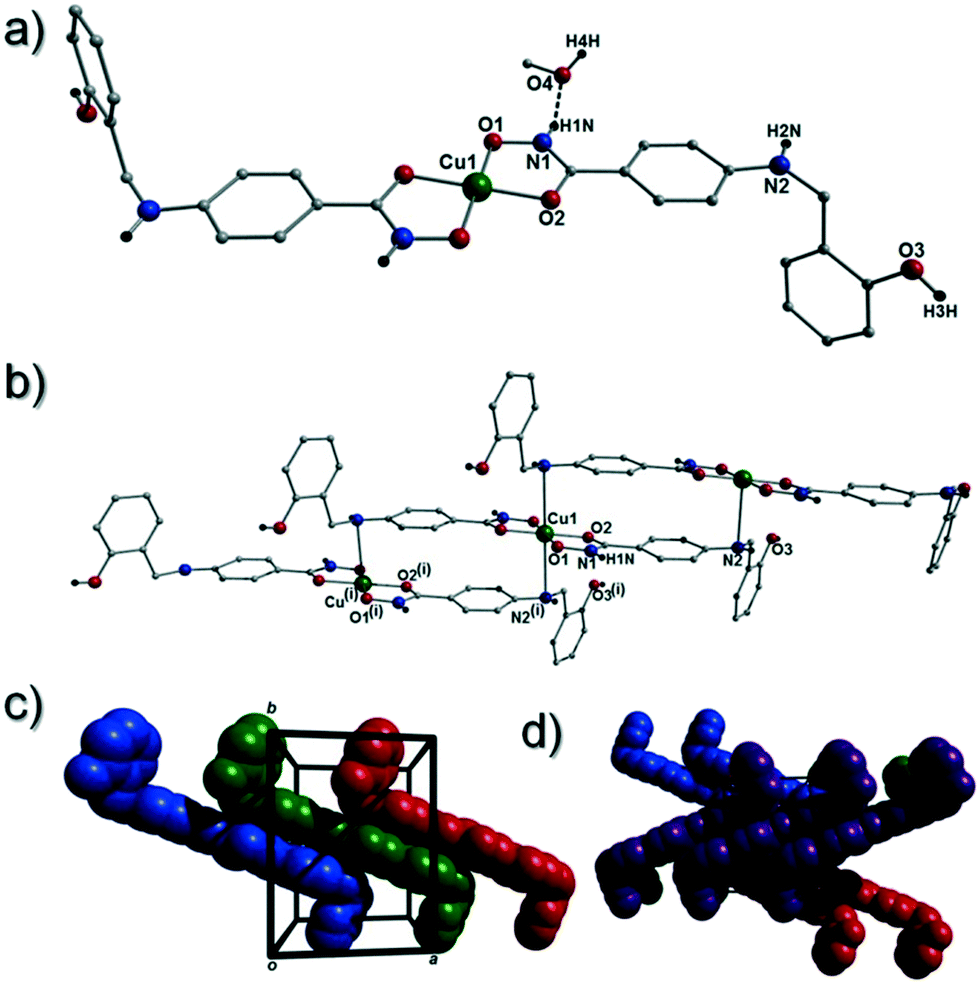
[Cu(II)(L3H2)2]n (1) crystallises in the monoclinic P21/c space group and the asymmetric unit comprises one Cu(II) centre (Cu1) and one L3H2− ligand. Each axially elongated J–T distorted octahedral Cu(II) centre in 1 is bound at the equatorial positions by two singly deprotonated L3H2− ligands that chelate through their hydroxamate (O2) and carbonyl (O1) oxygen atoms (Cu1–O1 = 1.93 Å, Cu1–O2 = 1.91 Å). The axial sites at each metal centre are occupied through long contacts with Ophen oxygen atoms (O3 and s.e.) belonging to neighbouring L3H2− ligands (Cu1–O3 = 2.74 Å and s.e.). Moreover, intra-ligand H-bonding interactions are observed between phenolic protons, H3H, and juxtaposed –OMe oxygen atoms (O4) (O3(H3H)⋯O4 = 2.16 Å). The multitopic nature of the L3H2− moieties in 1 results in the formation of the ribbon topology chains in 1 as shown in Fig. 1. The individual chains in 1 propagate in superimposable rows along the ac plane of the unit cell and produce an intra-chain Cu⋯Cu distance of 11.75 Å. The chains in 1 are stabilised by intra-chain π–π interactions between neighbouring hydroxamate phenyl rings giving a [C2–C6]centroid⋯[C2′–C6′]centroid distance of 3.83 Å. The individual chains in 1 stack on top of one another in a superimposable manner and are connected through H-bonding interactions (e.g. N1(H2)⋯O2′ = 2.03 Å, N2(H2H)⋯O2′ = 2.38 Å and N2(H2H)⋯O3′ = 2.95 Å) (Fig. 3). The resultant H-bonded stacks arrange themselves into the space efficient herring bone motif along the b direction of the unit cell and are also connected through a combination of H-bonding (C15(H15A)⋯O3′ = 2.83 Å) and C–H⋯π interactions ([C9–14]centroid⋯(H12′)C12 = 3.10 Å) (Fig. 2 and 3a).
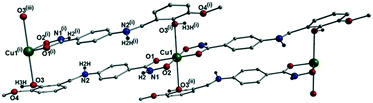 |
| | Fig. 1 Crystal structure of the coordination polymer in [Cu(II)(L3H2)2]n (1). Colour code as used throughout the text: green (Cu), grey (C), blue (N), red (O) and black (H). The majority of hydrogen atoms have been omitted for clarity. Symmetry codes: (i) 1 + x, y, −1 + z; (ii) −1 − x, 1 − y, 1 − z and (iii) 2 + x, y, −2 + z. | |
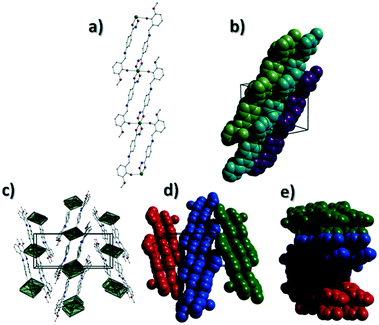 |
| | Fig. 2 (a) A polyhedral representation of a single chain in 1 highlighting the ribbon topology. All hydrogen atoms have been omitted for clarity. (b) Space-fill represented and colour coded H-bonded stacks of chains in 1 as viewed along the ab plane of the unit cell. Each colour represents a single chain. (c) Polyhedral representation of the packing observed in 1. (d and e) Space-fill and colour coded representation of H-bonded stacks comprising multiple 1D chains of 1 as viewed along the a (d) and c (e) unit cell direction. Note: figures c and d are equivalent and represent polyhedral (c) and space-fill (d) forms, respectively. | |
 |
| | Fig. 3 (a) Packing in 1 highlighting the individual 1-D chains stacking along the ac unit cell direction. (b) Two individual {Cu(II)(L3H2)2} chains connected through inter-chain complementary hydrogen bonding represented as dashed lines (N1(H2)⋯O2′ = 2.03 Å). | |
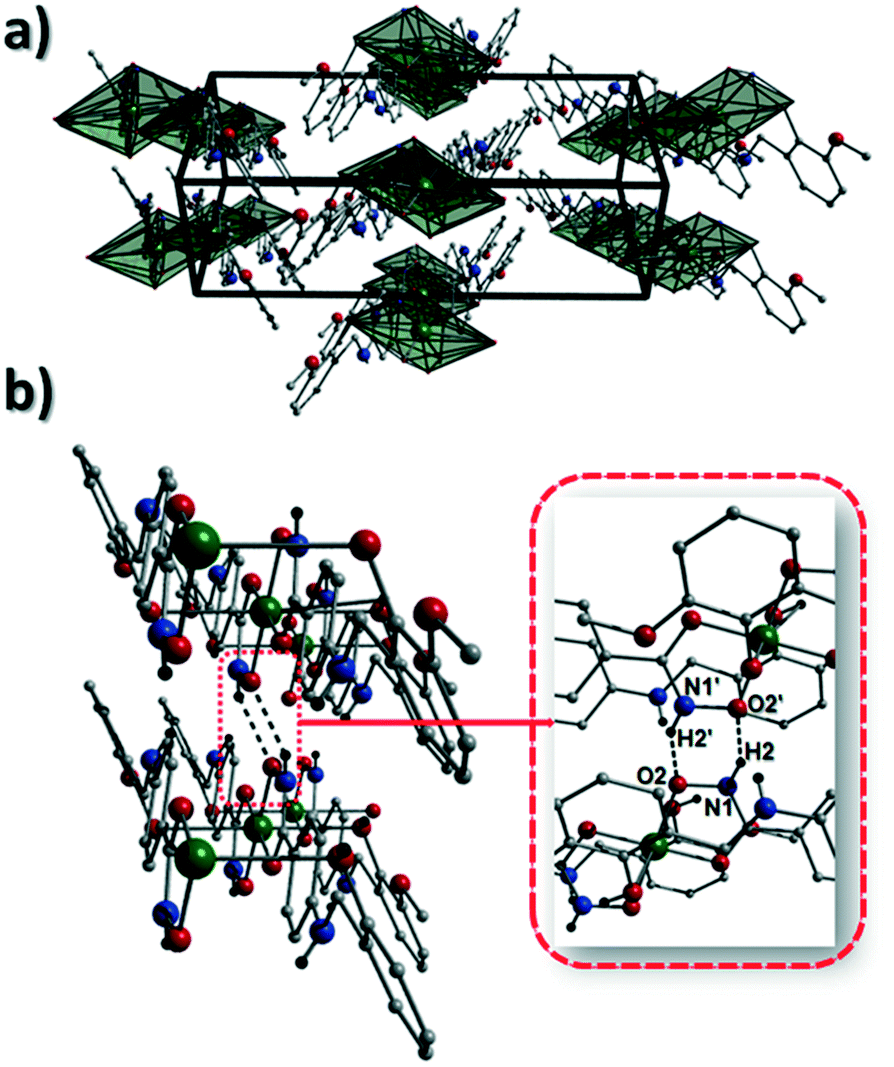
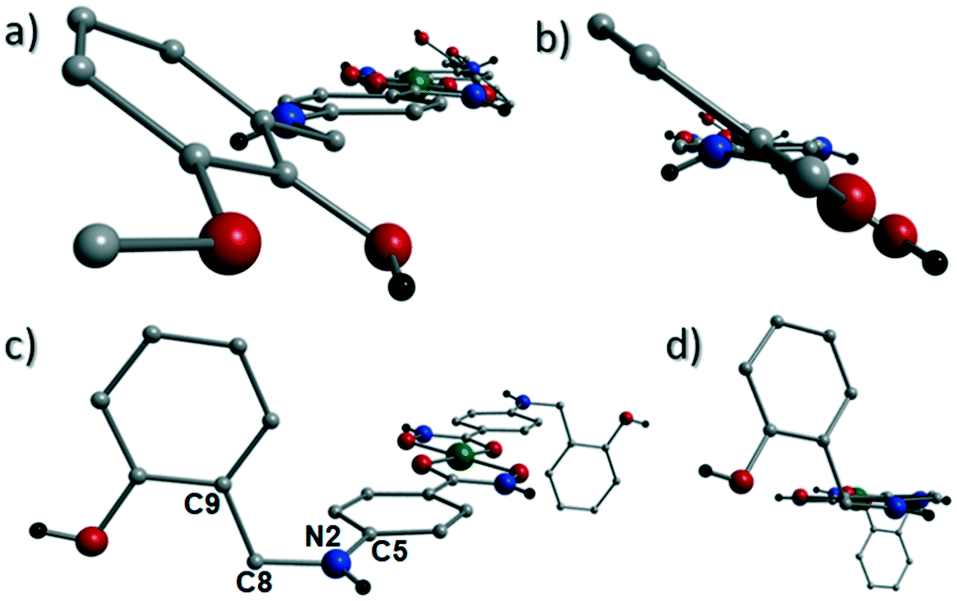
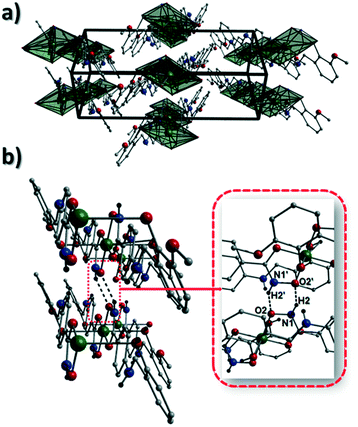
Akin to 1, the complex {[Cu(II)(L4H2)2]·2MeOH}n (2) crystallises in the monoclinic P21/c space group (Table 2). The asymmetric unit comprises an axially elongated Cu(II) centre, a single L4H2− ligand and a methanol solvent of crystallisation that sits at a H-bonding distance from the amide proton of the hydroxamate section of the ligand (N1(H1N)⋯O4 = 1.98 Å; Fig. 4a). Two L4H2− ligands chelate to the metal centre at distances of 1.91 Å (Cu1–O1) and 1.93 Å (Cu1–O2) to give the {Cu(II)(L4H2)2} chair shaped building block in 2 as opposed to the near planar {Cu(II)(L3H2)2} units in 1 (Fig. 4acf.1). The major difference between the structure in 1cf.2 lies in the axial connectivity at the Cu(II) centres in 2. Here, the 1-D chains in 2 are propagated by extremely long axial contacts between the metal centres and secondary amine N atoms (N2) located at the junction of the hydroxamate and phenolic units within each L4H2− ligand (Cu1–N2′ = 3.04 Å), as opposed to the Ophen oxygen donor atoms in 1 (Fig. 1cf.4b). Indeed, a Cu(II)–N distance of 3.04 Å is greater than the sum of their van der Waals radii (∼2.95 Å) and this interaction should be deemed weak, as corroborated using Hirshfeld surface analysis (vida infra).
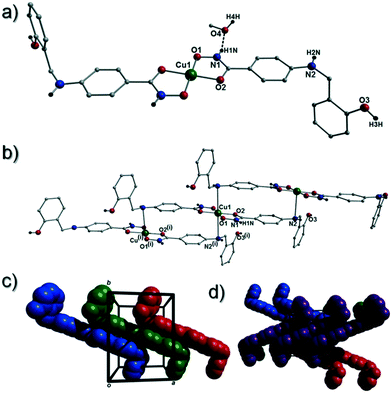 |
| | Fig. 4 (a) A single {[Cu(II)(L4H2)2]·2MeOH} unit in 2. The asymmetric unit has been labelled and only one MeOH solvent of crystallisation is shown. The dashes black line shows an inter-molecular H-bond at a distance of 1.98 Å (N1(H1N)⋯O4). The chain arrangement in 2 as viewed in normal (b) and space-fill mode (c), where each colour represents an independent {Cu(II)(L4H2)2} unit (symmetry code: (i) = 1 + x, y, z). (d) Space-fill representation of the packing in 2. Each colour represents an H-bonded stack of 1-D chains in 2 as viewed along the c unit cell direction. | |
The result is a much shorter intra-chain Cu1⋯Cu1′ distance of 8.62 Å in 2 (cf. 11.75 Å in 1) (Fig. 4b). More specifically, this ligand modification gives rise to a 27% decrease in the intra-nodal (Cu(II)⋯Cu(II)) distance on moving from 1 to 2. Interestingly, the deliberate omission of the –OMe group in L4H3 allows each ligand to distort to a much greater extent than observed in 1 (Fig. 5). More specifically, the phenolic aromatic rings in 2 twist away from their phenyl hydroxamate counterparts to produce a torsion angle of 75.1° (C5–N2–C8–C9) compared to the more co-planar value of 164.5° (C5–N2–C8–C9) exhibited by the L3H2− units in 1 (Fig. 5acf. c). These distortions can also be quantified by assessing the dihedral angles forged between the two aromatic rings belonging to each ligand (35.8° in 1 and 84.6° in 2) (Fig. 5). Computational studies have been carried in an attempt to rationalise these differences and are described later in this work. The individual chains in 2 propagate in a step-like manner along the a direction of the unit cell (Fig. 4b) and arrange themselves in space efficient stacks along the ab plane. These individual stacks pack along the c-direction in an alternating fashion, as highlighted in Fig. 4d. The methanol solvents of crystallisation (labelled C15–O4(H4H)) sit at H-bonding distance from L4H2− amide N atoms at a distance of 1.98 Å (N1(H1N)⋯O4) and act as molecular mortar by forming an O–H⋯π interactions with both nearby phenolic rings (O4(H4H)⋯[C9–C14]centroid = 2.54 Å) and Ophen oxygen donor atoms (O3) (O3⋯(H15C′)C15′ = 2.67 Å). The L3H2− secondary amine N atoms (N2 and s.e.) also partake in inter-chain H-bonding with neighbouring ligand Ophen aromatic rings (N2(H2N)⋯[C9′–C14′]centroid = 3.14 Å). The IR spectra of [Cu(II)(L3H2)2]n (1) and {[Cu(II)(L4H2)2]·2MeOH}n (2) show bands centred around 1608–1588 cm−1 in 1 and 1606–1592 cm−1 in 2 and are attributed to the ketonic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes associated with the hydroxamate L3H2− and L4H2− ligands, while resonances at 1064 cm−1 in 1 and 1079 cm−1 in 2 are assigned to N–O stretches and in combination corroborate the chelating nature of these hydroxamate ligands. Peaks at 1439 cm−1 and 1452 cm−1 in 1 and 1414 cm−1 and 1453 cm−1 in 2 are attributed to N–H deformation and C–N stretching frequencies, respectively.17
O stretching modes associated with the hydroxamate L3H2− and L4H2− ligands, while resonances at 1064 cm−1 in 1 and 1079 cm−1 in 2 are assigned to N–O stretches and in combination corroborate the chelating nature of these hydroxamate ligands. Peaks at 1439 cm−1 and 1452 cm−1 in 1 and 1414 cm−1 and 1453 cm−1 in 2 are attributed to N–H deformation and C–N stretching frequencies, respectively.17
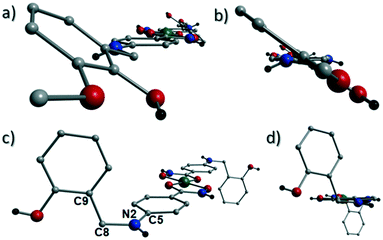 |
| | Fig. 5 The coordination polymers in 1 (a and b) and 2 (c and d) highlighting significant differences in phenolic ring positions in relation to their conjoined hydroxamate fragments. The planes of the phenolic and hydroxamate aromatic rings in 1 lie at an angle of 35.8° from one another as illustrated in figure b (the equivalent dihedral angle in 2 is 84.6°; fig. c). Figure c also highlights the C5–N2–C8–C9 torsion angle of 164.5° in 2. The equivalent torsion in 1 (also labelled C5–N2–C8–C9) provides an angle of 75.1° (a). | |
Hirshfeld surface studies
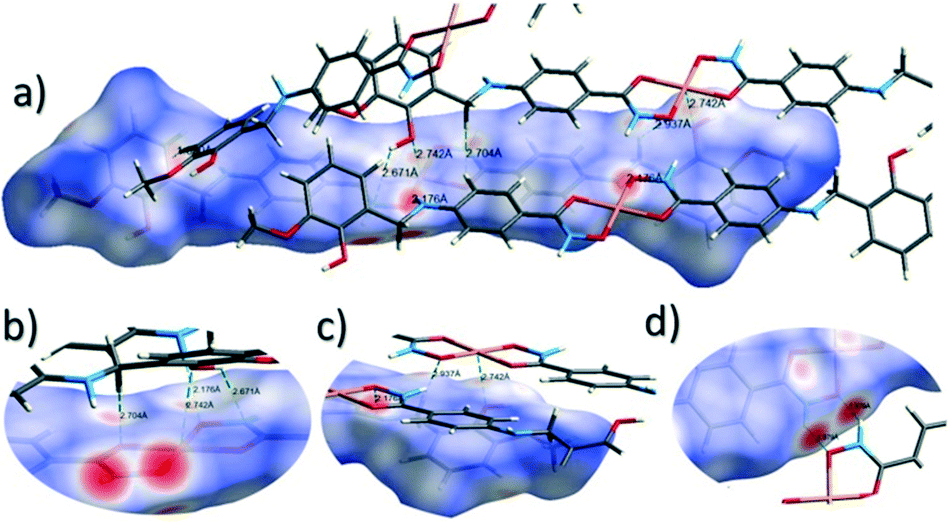
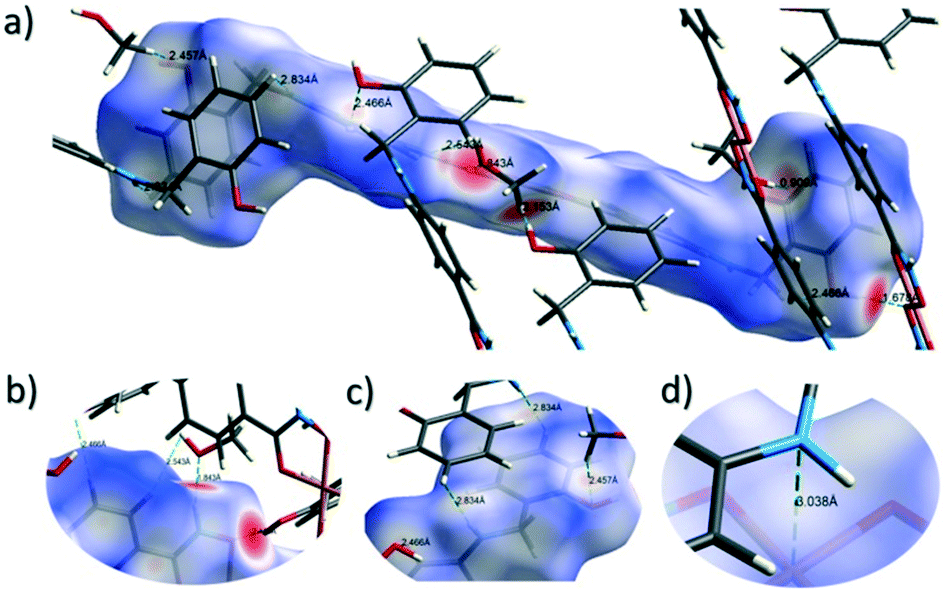
The close intermolecular interactions in 1 and 2 were further surveyed and visualised by carrying out a Hirshfeld surface (HS) analysis using the dnorm, curvedness and shape index mapping functions.18Fig. 6 and 8 depict the Hirshfeld surfaces (mapped over dnorm) for [Cu(II)(L3H2)2]n (1) and {[Cu(II)(L4H2)2]·2MeOH}n (2), respectively. In these plots, the red, blue and white regions represent interatomic interactions that are shorter (red), longer (blue) and commensurate (white) with vdW separations. Therefore, a close intermolecular interaction is envisaged when a particular vicinity of a molecules surface exhibits a red region of colouration. This is illustrated in Fig. 6 as it shows (for instance): 1) an inter-chain C–H⋯OC interaction at a distance of 2.7 Å (corresponding to the C8(H8A)⋯O1′ = 2.8 Å interaction observed in the crystal structure (Fig. 6a and b)) and 2) inter-chain complementary H-bonds between two adjacent hydroxamate functional groups at a distance of 1.88 Å (corresponding to the N1(H2)⋯O2′ = 2.03 Å interaction observed in the crystal structure) (Fig. 6d). The Cu1–O3′ long axial contact that allows chain propagation in 1 is also observed here at a distance of 2.74 Å (Fig. 6b and c). The inter-chain C–H⋯π interactions in 1 can be observed in the dnorm and shape index Hirshfeld surfaces given in Fig. 7. Likewise, the inter-chain C–H⋯π interactions observed in 2 (C11(H11)⋯[C2′–C7′]centroid = 3.44 Å and s.e.) are also corroborated using dnorm and shape index HS analysis (Fig. S10†).
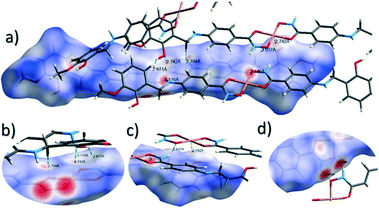 |
| | Fig. 6 Hirshfeld surface analysis of [Cu(II)(L3H2)2]n (1) mapped over the dnorm function (−0.56 to +1.37 a.u.−1) highlighting a number of intermolecular interactions observed in the crystal structure including the Cu–O3′ long contact at a distance of 2.74 Å (figures a–c). Figure d shows complementary hydrogen bonding between juxtaposed hydroxamate functional groups at a distance of 1.88 Å (N1(H2)⋯O2′). Note: the CrystalExplorer program normalises all X–H bond lengths to values obtained experimentally from neutron diffraction studies. The red and blue spots highlight long and short interatomic contacts, respectively. White regions represent interatomic distances commensurate with van der Waals separations. | |
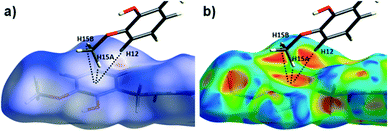 |
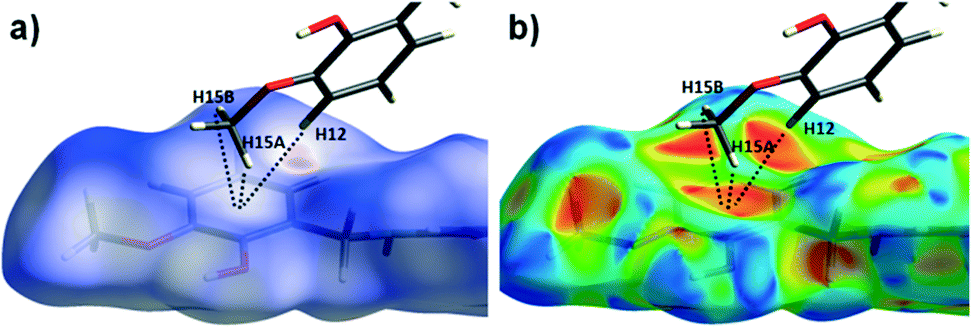
| | Fig. 7 The dnorm (a) and shape index (b) Hirshfeld surfaces for 1, highlighting the inter-chain C–H⋯π interactions: C12(H12)⋯[C9′–C14′]centroid = 3.10 Å; C15(H15A)⋯[C9′–C14′]centroid = 3.53 Å and C12(H15B)⋯[C9′–C14′]centroid = 3.48 Å (these are distances taken from the crystal structure data). | |
The Hirshfeld surface (dnorm) of {[Cu(II)(L4H2)2]·2MeOH}n (2) shows a number of intermolecular H-bonding interactions in the crystal. For instance, a short contact between Ophen protons (H3H and s.e.) and juxtaposed hydroxamate O donor atoms (O1 and s.e.) is highlighted in Fig. 8a, giving a distance of 1.68 Å. This interaction corresponds to the O3(H3H)⋯O1′ = 1.81 Å interaction observed in the crystal structure of 2. A significant H-bonding interaction is also observed (1.84 Å) between the MeOH solvent of crystallisation and a neighbouring hydroxamate N–H group and correlates with the N1(H1N)⋯O4 = 1.98 Å interaction observed in the crystal structure (Fig. 8b). As predicted, the HS plot centred on the Cu1–N2′ interaction in 2 is indeed indicative of a weak interaction (white colouration), with a distance at the very limit of the sum of their individual van der Waals radii (3.04 Å; Fig. 8d). The intermolecular interactions in 1 and 2 can also be visualised through their Hirshfeld surfaces mapped over both curvedness (Fig. S6 and S8†) and shape index (Fig. S7 and S9†). The curvedness plots in 1 and 2 each indicate flat regions around their aromatic rings (as expected), while the shape index surfaces for both complexes visualise the ‘bumps and hollows’ (shown as blue and red spots, respectively) associated with intermolecular interactions first highlighted via their dnorm surface plots in Fig. 6 and 8.
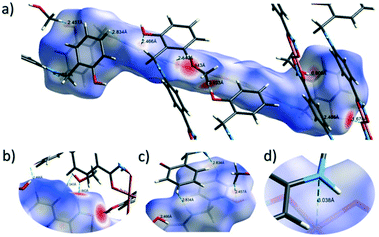 |
| | Fig. 8 Hirshfeld surface analysis of {[Cu(II)(L4H2)2]·2MeOH}n (2) using the dnorm function (−0.73 to +1.45 a.u.−1) and highlighting a number of inter-molecular interactions (a–c) including the very weak Cu1–N2′ = 3.04 Å long contact (d). | |
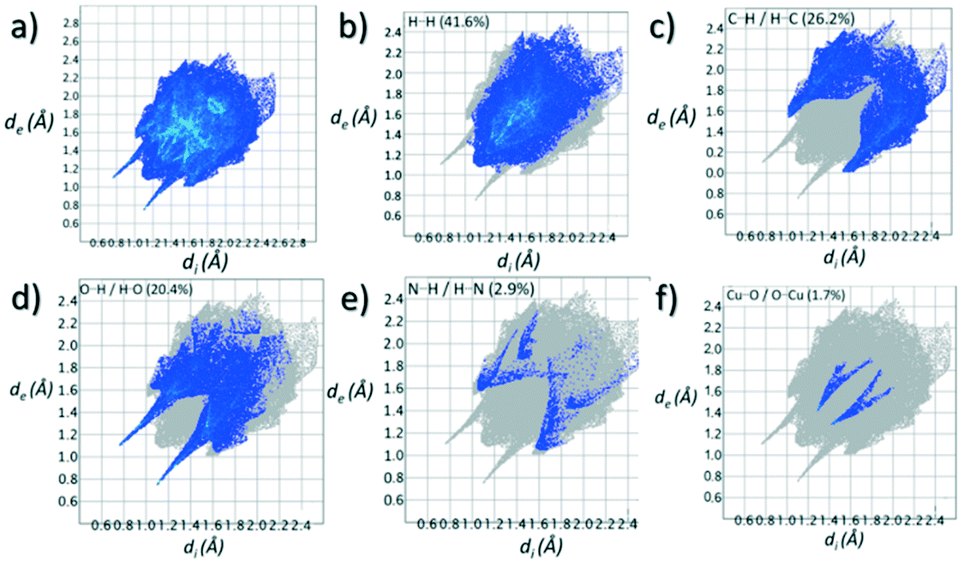
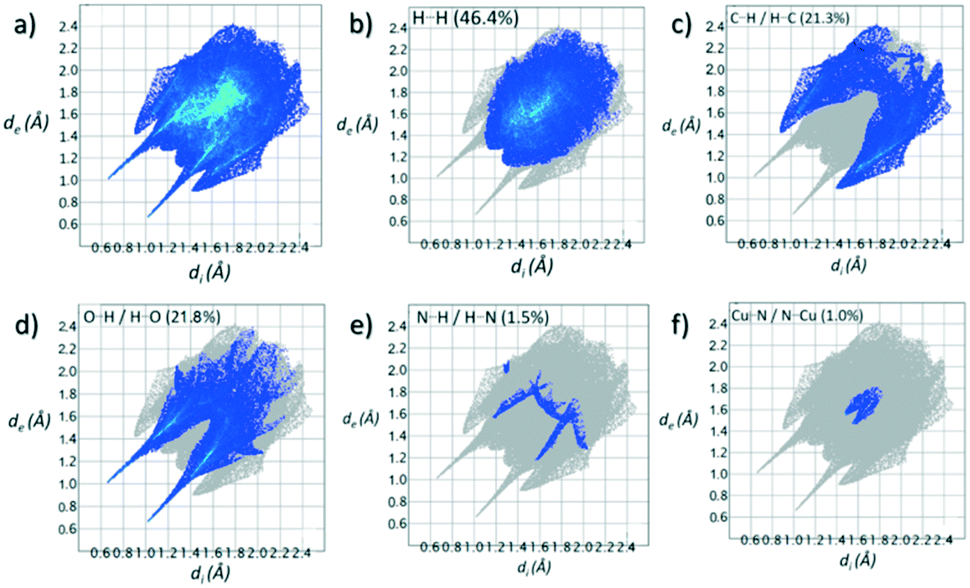
Contributions to the surface of the molecule from each atom in [Cu(II)(L3H2)2]n (1) and {[Cu(II)(L4H2)2]·2MeOH}n (2) shows that the majority of their intermolecular interactions come from H⋯H contacts (41.6% (1) and 46.4% (2)) (Table 1).19 Other significant contributions to the overall surface come from reciprocated C⋯H (26.2% in 1 and 21.3% in 2) and O⋯H (20.4% in 1 and 21.8% in 2) interactions. As is commonly observed, much smaller contributions are provided by N⋯H/H⋯N hydrogen bonding interactions (2.9% in 1 and 1.5% in 2). The Cu–O and Cu–N long contacts that effectively allow chain propagation in 1 and 2 provide 1.7% and 1.0% contributions to their molecular surfaces as shown in the 2-D fingerprint plots of Fig. 9f (1) and 10f (2), respectively.
Table 1 Relative percentage of close contact interactions contributing to the Hirshfeld surfaces in 1 and 2. For a full breakdown analysis see Tables S2 and S3†
| [Cu(II)(L3H2)2]n (1) |
| Contact |
Percentage contribution (%) |
| H⋯H |
41.6 |
| C⋯H/H⋯C |
26.2 |
| O⋯H/H⋯O |
20.4 |
| N⋯H/H⋯N |
2.9 |
| C⋯C |
2.9 |
| Cu⋯O/O⋯Cu |
1.7 |
| O⋯O |
1.3 |
| N⋯C/C⋯N |
0.4 |
| {[Cu(II)(L4H2)2]·2MeOH}n (2) |
| H⋯H |
46.4 |
| O⋯H/H⋯O |
21.8 |
| C⋯H/H⋯C |
21.3 |
| C⋯C |
3.0 |
| N⋯H/H⋯N |
1.5 |
| Cu⋯N/N⋯Cu |
1.0 |
| N⋯C/C⋯N |
0.9 |
| N⋯O/O⋯N |
0.2 |
Table 2 Selected crystal data obtained from 1 and 2
|
|
1
|
2·2MeOH |
|
Includes guest molecules.
Mo–Kα radiation, graphite monochromator.
wR2 = [∑w(|Fo2| − |Fc2|)2/∑w|Fo2|2]1/2.
For observed data.
R
1 = ∑||Fo| − |Fc||/∑|Fo|.
|
| Formulaa |
C30H30N4O8Cu1 |
C30H34N4O8Cu1 |
|
M
W
|
638.12 |
642.15 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
|
a/Å |
6.7098(2) |
8.61560(10) |
|
b/Å |
21.4689(8) |
11.31420(10) |
|
c/Å |
9.4280(2) |
14.8436(2) |
|
α/° |
90 |
90 |
|
β/° |
91.889(2) |
100.8870(10) |
|
γ/° |
90 |
90 |
|
V/Å3 |
1357.38(7) |
1420.89(3) |
|
Z
|
2 |
2 |
|
T/K |
100.0(2) |
100.0(2) |
|
λ
/Å |
0.71073 |
0.71073 |
|
D
c/g cm−3 |
1.561 |
1.501 |
|
μ(Mo-Kα)/mm−1 |
0.867 |
0.828 |
| Meas./indep. (Rint) refl. |
26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 867/9697 (0.0487) 867/9697 (0.0487) |
12946/11113 (0.0206) |
| Restraints, parameters |
0, 205 |
0, 199 |
| wR2 (all data)c |
0.1113 |
0.1406 |
|
R
1
,
|
0.0481 |
0.0424 |
| Goodness of fit on F2 |
1.303 |
1.138 |
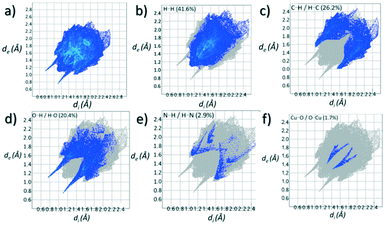 |
| | Fig. 9 (a) Full 2-D fingerprint surface interactions plot for [Cu(II)(L3H2)2]n (1) along with plots for H⋯H (b), C⋯H/H⋯C (c), O⋯H/H⋯O (d), N⋯H/H⋯N (e) and Cu⋯O/O⋯Cu (f) contacts (di = internal distance, de = external distance in Å). | |
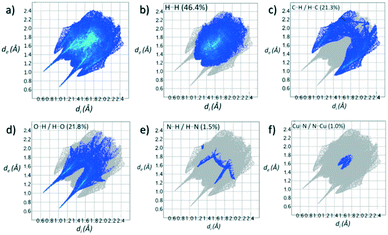 |
| | Fig. 10 (a) Full 2-D fingerprint surface interactions plot for {[Cu(II)(L4H2)2]·2MeOH}n (2) along with plots for H⋯H (b), C⋯H/H⋯C (c), O⋯H/H⋯O (d), N⋯H/H⋯N (e) and Cu⋯N/N⋯Cu (f) contacts (di = internal distance, de = external distance in Å). | |
Geometry optimisation studies
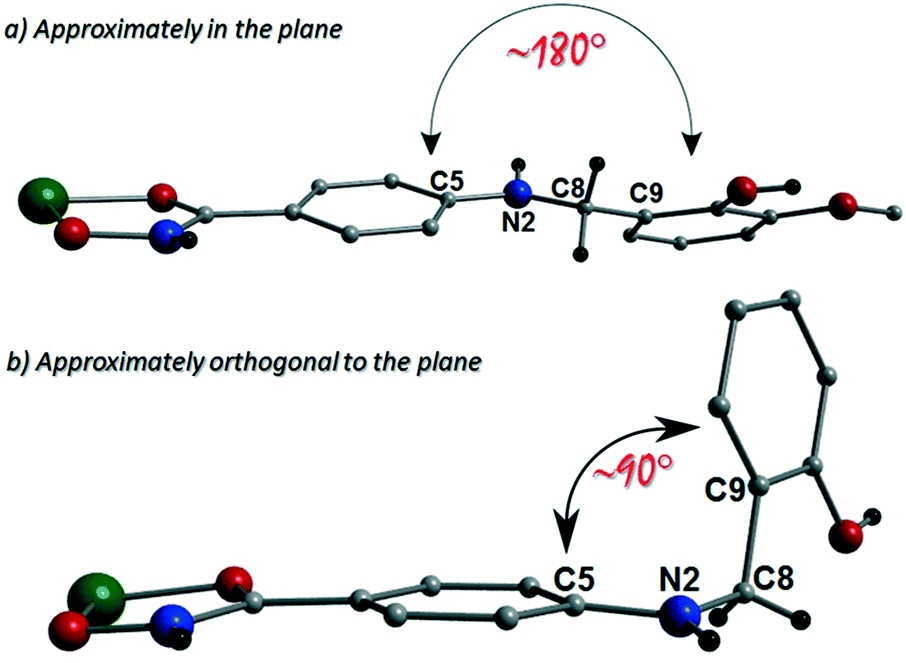
The more extreme distortion observed by the L4H2− units in 2 when compared to the L3H2− in 1 also give rise to differing Cu(II) binding sites at the phenolic sections of the ligands. We found it interesting that a metal binding site change is imposed through the omission of just one –OMe group. In order to gain insights into these observations, theoretical models of the two distinct geometries were created and geometry optimization computational studies were conducted using a cluster model derived from the two X-ray structures. Models of [Cu(II)(L)2] in the two configuration observed were fully optimized as described previously. The fundamental difference in the two conformations is the relative orientation of the two ligand phenyl rings as indicated in Fig. 11. In L3H2−, the rings are found to exist in the same plane (∼180°) while for L4H2−, they adopt a conformation orthogonal (∼90°) to each other. The calculations showed that the orthogonal conformation was energetically preferred for both ligands in 1 and 2. While this conformation is indeed observed in the experimental crystal structure of 2, the planar configuration is found for 1. Further analysis showed that the energy penalty for 2 to adopt the less preferential conformation is just 1.2 kcal mol−1, whereas it is considerably larger for 1, at 3.5 kcal mol−1. It would therefore appear that the conformation observed in 2 is a function of the lower energy penalty associated with the orthogonal geometry, while complex 1 (exhibiting a more planar geometry) gains additional stabilization from the axial Cu–Ophen interactions (strong electron donor ROH groups) observed with adjacent ligands in the crystal lattice, leading to the 1-D chain topology.
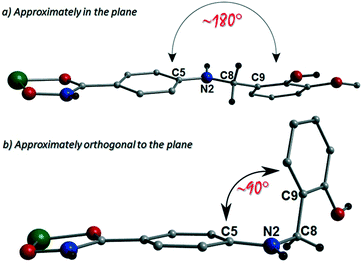 |
| | Fig. 11 Schematic using crystal data from [Cu(II)(L3H2)2] (1) (a) and {[Cu(II)(L4H2)2]·2MeOH} (2) (b) to highlight the approximately planar and orthogonal positions in relation to their respected ligand aromatic rings. | |
Powder X-ray diffraction studies on 1 and 2 were uses to confirm that their bulk samples were consistent with their single crystal data (Fig. S2 and S3†). This was carried out using simulations produced by the Mercury software package.20 Using a Johnson Matthey balance, the room temperature magnetic moment (μeff) of 1 (1.69 BM) and 2 (1.64 BM) was found to be consistent with that expected for a magnetically dilute Cu(II) chain (μS.O. = 1.73 BM) (Table S1†).‡
Conclusions
We have described in this work the design and synthesis of the novel linear multitopic ligands N-hydroxy-4-((2-hydroxy-3-methoxybenzyl)amino)benzamide (L3H3) and N-hydroxy-4-((2-hydroxybenzyl)amino)benzamide (L4H3). Upon Cu(II) ligation the self-assembly of the 1-D chains [Cu(II)(L3H2)2]n (1) and {[Cu(II)(L4H2)2]·2MeOH}n (2) is observed. Slight differences in the functionality of ligands L3H3vs. L4H3, namely the omission of an –OMe group in the latter, give rise to pertinent connectivity changes when closely inspecting chains 1 and 2. This is best exampled by noting that the intra-nodal Cu(II)⋯Cu(II) distance in 2 is significantly shorter than the corresponding length in 1 (11.75 Å (1) cf. 8.62 Å (2)). Although both these distances are too long for the possibility of magnetic exchange, such observations highlight the importance of ligand design and the potential ramifications associated with even slight modifications when designing magnetic coordination polymers. The coordination number and geometry flexibility of the Cu(II) ion no doubt promotes successful CP formation in this work. Indeed, this is highlighted further when we note that attempts to produce other 1st row transition metal analogues have so far been fruitless. Nevertheless, work is ongoing on the elucidation of viable synthetic pathways for further metal coordination of the novel ligands L3H3 and L4H3. Hirshfeld surface studies on both [Cu(II)(L3H2)2]n (1) and {[Cu(II)(L4H2)2]·2MeOH}n (2) have been employed to map and visualise all intermolecular interactions and to this end showed good alignment with contacts previously proposed upon close inspection of their crystal structures. Geometry optimisation computational studies were carried out on both complexes in order to probe the experimentally observed differences in their ligand conformations. It was shown that the ∼90° orthogonal geometry (with respect to the dihedral angle produces by their ligand aromatic rings), was preferred in both cases, although the observation was more pronounced in 2 over 1.
Experimental section
Materials
All solvent and chemicals were used as purchased. All chemicals were purchased from Sigma Aldrich.
Analytical methods
Infra-red spectra were recorded on a Perkin Elmer FT-IR Spectrum 100 spectrometer (School of Natural Sciences, Bangor University). 1H and 13C NMR spectra were obtained at room temperature (298 K) on a Bruker Ultrashield™ 400 Plus with Sample Xpress at 400 MHz. Chemical shifts are reported in ppm and referenced to DMSO (1H: 2.50 ppm, 13C: 39.52 ppm). Elemental analysis was carried out at OEA Laboratories (Kelly Bray, Cornwall). The room temperature magnetic moments (μeff) for 1 and 2 were obtained using a Johnson Matthey balance situated at the School of Natural Sciences, Bangor University (see ESI† for more details).
X-ray crystallography
Complexes 1 and 2 were collected on an Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ Super Bright molybdenum rotating anode generator with HF Varimax optics (100 m focus) (CCDC numbers: 1941525 (1) and 1941526 (2)). The cell determination and data collection of each complex was carried out using the CrystalClear-SM Expert package (Rigaku, 2012). Each data reduction, cell refinement and absorption correction were carried out using CrysAlisPro software (Rigaku OD, 2015),21 while all structures were solved and refined using SHELXT and SHELXL-2014 (ref. 22) within OLEX-2.23 Powder XRD was carried out using a PANalytical Philips X'Pert 3040/60 diffractometer at 45 kV and 35 mA between 5 and 60° 2θ using Ni-filtered Cu-Kα1 radiation (λ = 1.5405 Å) at the School of Natural Sciences, Bangor University.
Computational methodology
Computational models of complexes 1 and 2 were constructed from their experimental X-ray coordinates. Both models comprise of a Cu(II) metal centre coordinated to two linear hydroxamate ligands [Cu(II)(L3–4H2)2]. The complexes were fully optimized in Gaussian G16 (ref. 24) using the DFT M062x functional25 and the 6-311+G(d,p) basis set for H, N, C, O atoms and TZVP for Cu. Geometry optimizations were performed using default settings. Two additional models were generated by modifying the model of L3H2− into L4H2−, and vice versa. The relative energies associated with each configuration were then determined for both ligands. Hirshfeld surface analysis was carried out using the CrystalExplorer software.26
Preparation of ligands L3H3 and L4H3
The synthesis of ligands N-hydroxy-4-((2-hydroxy-3-methoxybenzyl)amino)benzamide (L3H3) and N-hydroxy-4-((2-hydroxybenzyl)amino)benzamide (L4H3) along with their precursors are described in the ESI.†
Preparation of complexes 1 and 2
All reactions were performed under aerobic conditions and all reagents and solvents were used as purchased. Caution: although no problems were encountered in this work, care should be taken when manipulating the potentially explosive nitrate salts.
Synthesis of [Cu(II)(L3H2)2]n (1)
Cu(NO3)2·3H2O (0.025 g, 0.10 mmol), 4-((2-hydroxy-3-methoxybenzyl)amino)-N-hydroxybenzamide (L3H3) (0.030 g, 0.10 mmol) and tetraethylammonium hydroxide (TEAOH) (0.015 g, 0.10 mmol) were dissolved in methanol (20 cm3) and the resultant solution stirred for 3 h at room temperature. The resultant yellow-green solution was then filtered and X-ray quality crystals of 1 were obtained upon slow evaporation of the mother liquor in 18% (11.5 mg; crystals) yield. Elemental analysis (%) calculated as 1·H2O (C30H32N4O9Cu1): C 54.92, H 4.92, N 8.54; found: C 54.25, H 4.83, N 8.76. FT-IR (cm−1): 3498 (m), 3313 (w), 3189 (w), 2955 (w), 2837 (w), 1608 (s, sh), 1588 (s), 1562 (m), 1543 (w), 1477 (s), 1452 (m), 1439 (m), 1393 (w), 1358 (m), 1335 (w), 1271 (m), 1257 (w), 1211 (m), 1188 (m), 1141 (s), 1130 (w), 1064 (s), 1021 (s), 915 (s, sh), 854 (m), 828 (s), 800 (m), 774 (s), 767 (s), 735 (s), 640 (m), 615 (m), 581 (m), 550(m), 503 (s), 453 (s).
Synthesis of {[Cu(II)(L4H2)2]·2MeOH}n (2)
Cu(NO3)2·3H2O (0.025 g, 0.10 mmol), N-hydroxy-4-((2-hydroxybenzyl)amino)benzamide (L4H3) (0.03 g, 0.11 mmol) and tetraethylammonium hydroxide (TEAOH) (0.015 g, 0.10 mmol) were dissolved in methanol (20 cm3) and stirred at room temperature for 3 h. The resultant yellowish green solution was then filtered and X-ray quality crystals of 2 were obtained upon slow evaporation of the mother liquor in 15% (9.1 mg; crystals) yield. Elemental analysis (%) calculated as 2 (C30H34N4O8Cu1): C 56.11, H 5.34, N 8.72; found: C 56.02, H 4.75, N 8.78. FT-IR (cm−1): 3624 (m), 3538 (s), 3391 (m), 3208 (w), 3132 (m), 3062 (w), 2940 (m), 2839 (w), 2723 (m), 2611 (m), 2233 (w), 2107 (w), 1899 (w), 1606 (s, sh), 1592 (s), 1533 (m), 1501 (s, sh), 1453 (s, sh), 1414 (w), 1395 (w), 1354 (w), 1333 (m), 1311 (w), 1273 (s), 1242 (s), 1195 (w), 1177 (s, sh), 1157 (w), 1110 (w), 1072 (s), 1033 (s, sh), 1013 (s), 920 (s, sh), 861 (w), 826 (s, sh), 762 (s, sh), 715 (m), 661 (s), 636 (s), 582 (s), 525 (w) 506 (s), 436 (s), 414 (s).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
BFM would like to thank the University of Maiduguri and the Petroleum Technology Development Fund (PTDF) for their award of an overseas fellowship. We would like to thank the EPSRC National Crystallographic Service at Southampton University for their continuous support. MPG would like to acknowledge support from KMITL (KREF046211). LFJ would like to thank the University of Wolverhampton for their support (JC).
Notes and references
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. Paik Suh and J. Reedjik, CrystEngComm, 2012, 14, 3002–3004 RSC.
-
(a) C.-T. Chen and K. S. Suslick, Coord. Chem. Rev., 1993, 128, 293–322 CrossRef CAS;
(b) C. Janiak, Dalton Trans., 2003, 2781–2804 RSC;
(c) S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 CrossRef CAS;
(d) J.-P. Zhang, Z.-C. Huang and X.-M. Chen, Chem. Soc. Rev., 2009, 38, 2385–2396 RSC;
(e) C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388 RSC;
(f) W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688–764 CrossRef CAS PubMed.
-
(a) F. Thebauldt, S. A. Barnett, A. J. Blake, C. Wilson, N. E. Champness and M. Schröder, Inorg. Chem., 2006, 45, 6179–6187 CrossRef PubMed;
(b) W. M. Bloch, C. J. Doonan and C. J. Sumby, CrystEngComm, 2013, 15, 9663–9671 RSC;
(c) M. Du, C.-P. Li, C.-S. Liu and S.-M. Fang, Coord. Chem. Rev., 2013, 257, 1282–1305 CrossRef CAS.
-
(a) V. Niel, J. M. Martinez-Agudo, M. Carmen Muñoz, A. B. Gaspar and J. Antonio Real, Inorg. Chem., 2001, 40, 3838–3839 CrossRef CAS;
(b) G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762–1765 CrossRef CAS PubMed;
(c) S. M. Neville, G. J. Halder, K. W. Chapman, M. B. Duriska, B. Moubaraki, K. S. Murray and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 12106–12108 CrossRef CAS PubMed;
(d) W. Liu, X. Bao, L.-L. Mao, J. Tucek, R. Zboril, J.-L. Liu, F.-S. Guo, Z.-P. Ni and M.-L. Tong, Chem. Commun., 2014, 50, 4059–4061 RSC;
(e) C. Bartual-Murgui, A. Akou, C. Thibault, G. Molnar, C. Vieu, L. Salmon and A. Bousseksou, J. Mater. Chem. C, 2015, 3, 1277–1285 RSC;
(f) Z.-P. Ni, J.-L. Liu, M. N. Hoque, W. Liu, J.-Y. Li, Y.-C. Chen and M.-L. Tong, Coord. Chem. Rev., 2017, 335, 28–43 CrossRef CAS;
(g) C. Pham and F. Paesani, Inorg. Chem., 2018, 57, 9839–9843 CrossRef CAS PubMed;
(h) A. Tissot, X. Keese, S. Giannopoulou, I. Stenger, L. Binet, E. Riviere and C. Serre, Chem. Commun., 2019, 55, 194–197 RSC.
-
(a)
C. Coulon, H. Miyasaka and R. Clerac, Struct. Bonding, Single-Molecule Magnets and Related Phenomena, Springer, 2006, pp. 163–206 Search PubMed;
(b) L. Bogani, A. Vindigni, R. Sessoli and D. Gatteschi, J. Mater. Chem., 2008, 18, 4750–4758 RSC;
(c) H.-L. Sun, Z.-M. Wang and S. Gao, Coord. Chem. Rev., 2010, 254, 1081–1100 CrossRef CAS.
-
(a)
A. D. Katsenis, E. K. Brechin and G. S. Papaefsathiou, Encyclopedia of Inorganic and Bioinorganic Chemistry, Wiley Publishers, 2014, pp. 245–258 Search PubMed;
(b) J. J. Baldoví, E. Coronado, A. Gaita-Ariño, C. Gamer, M. Giménez-Marqués and G. Mínguez Espallargas, Chem. – Eur. J., 2014, 20, 10695–10702 CrossRef PubMed;
(c) D. Aulakh, J. B. Pyser, X. Zhang, A. A. Yakovenko, K. R. Dunbar and M. Wreidt, J. Am. Chem. Soc., 2015, 137, 9254–9257 CrossRef CAS PubMed;
(d) X. Zhang, V. Vieru, X. Feng, J.-L. Liu, Z. Zhang, B. Na, W. Shi, B.-W. Wang, A. K. Powell, L. F. Chibotaru, S. Gao, P. Cheng and J. R. Long, Angew. Chem., Int. Ed., 2015, 54, 9861–9865 CrossRef CAS;
(e) C.-M. Liu, D.-Q. Zhang Hao and D.-B. Zhu, Sci. Rep., 2017, 7, 11156–11161 CrossRef PubMed.
-
(a) G. Givaja, P. Amo-Ochoa, C. J. Gómez-García and F. Zamora, Chem. Soc. Rev., 2012, 41, 115–147 RSC;
(b) C. H. Hendon, D. Tiana and A. Walsh, Phys. Chem. Chem. Phys., 2012, 14, 13120–13132 RSC;
(c) A. C. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66–69 CrossRef CAS PubMed;
(d) L. Sun, M. G. Campbell and M. Dinč, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS.
-
(a) L. Ma, O. R. Evans, B. M. Foxman and W. Lin, Inorg. Chem., 1999, 38, 5837–5840 CrossRef CAS;
(b) D. M. Ciurtun, N. G. Pschirer, M. D. Smith, U. H. F. Bunz and H.-C. zur Loye, Chem. Mater., 2001, 13(9), 2743–2745 CrossRef;
(c) T. H. Kim, Y. W. Shin, J. H. Jung, J. S. Kim and J. Kim, Angew. Chem., Int. Ed., 2008, 47, 685–688 CrossRef CAS PubMed;
(d) Y. Hasegawa and T. Nakinishi, RSC Adv., 2010, 5, 338–353 RSC;
(e) Y. Hasegawa, T. Nakanishi and Y. Kitagawa, RSC Adv., 2018, 347–370 Search PubMed;
(f) W. P. Lustig and J. Lin, Coord. Chem. Rev., 2018, 373, 116–147 CrossRef CAS.
-
(a) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC;
(b) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC;
(c) P. D. Tran, R. V. Tran, M. Orio, S. Torelli, Q. D. Truong, K. Nayuki, Y. Sasaki, S. Y. Chiam, R. Yi, I. Honma, J. Barber and V. Artero, Nat. Mater., 2016, 15, 640–646 CrossRef CAS PubMed;
(d) E. Loukopoulos and G. E. Kostakis, J. Coord. Chem., 2018, 71(3), 371–410 CrossRef CAS.
-
(a) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS;
(b) M. L. Foo, R. Matsuda and S. Kitagawa, Chem. Mater., 2014, 26, 310–322 CrossRef CAS.
-
(a)
X. Lin, N. R. Champness and M. Schröder, Top. Curr. Chem., Functional Metal-Organic Frameworks: Gas Storage, Separation and Catalysis, Springer, 2010, vol. 293, pp. 35–76 Search PubMed;
(b) S. Ma and H.-C. Zhou, Chem. Commun., 2010, 46, 44–53 RSC;
(c) B. Li, H.-M. Wen, W. Zhou and B. Chen, J. Phys. Chem. Lett., 2014, 5, 3468–3479 CrossRef CAS PubMed;
(d) J. Duan, W. Jin and R. Krishna, Inorg. Chem., 2015, 54, 4279–4284 CrossRef CAS PubMed;
(e) K. S. Song, D. Kim, K. Polychronopoulou and A. Coskun, ACS Appl. Mater. Interfaces, 2016, 8, 26860–26867 CrossRef CAS.
-
(a) P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 5974–5978 CrossRef CAS PubMed;
(b) P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. E. Eubank, D. Hertaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marseaud, P.-N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS;
(c) R. C. Huxford, J. Della Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef CAS PubMed;
(d) Z. Ma and B. Moulton, Coord. Chem. Rev., 2011, 255, 1623–1641 CrossRef CAS;
(e) I. Abánades Lázaro and R. S. Forgan, Coord. Chem. Rev., 2019, 380, 230–259 CrossRef.
-
(a) Z. Xie, L. Ma, K. E. deKrafft, A. Jin and W. Lin, J. Am. Chem. Soc., 2010, 132, 922–923 CrossRef CAS PubMed;
(b) H. Tan, B. Liu and Y. Chen, ACS Nano, 2012, 6(12), 10505–10511 CrossRef CAS PubMed;
(c) X. Zhang, X. Luo, N. Zhang, J. Wu and Y.-Q. Huang, Inorg. Chem. Front., 2017, 4, 1888–1894 RSC;
(d) C. Chen, X. Zhang, P. Gao and M. Hu, J. Solid State Chem., 2018, 258, 86–92 CrossRef CAS;
(e) M. Arici, New J. Chem., 2019, 43, 3690–3697 RSC.
- C. McDonald, D. W. Williams, P. Comar, S. J. Coles, T. D. Keene, M. B. Pitak, E. K. Brechin and L. F. Jones, Dalton Trans., 2015, 44, 13359–13368 RSC.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- M. B. Fugu, R. J. Ellaby, H. M. O'Connor, M. B. Pitak, W. Klooster, P. N. Horton, S. J. Coles, M. H. Al-mashhadani, I. F. Perepichka, E. K. Brechin and L. F. Jones, Dalton Trans., 2019, 48, 10180–10190 RSC.
-
(a) J. Yang, P. J. Bremer, I. L. Lamont and A. J. McQuillan, Langmuir, 2006, 22, 10109–10117 CrossRef CAS PubMed;
(b) B. Kurzak, H. Kozlowski and E. Farkas, Coord. Chem. Rev., 1992, 114, 169–200 CrossRef CAS.
-
(a) J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed;
(b) M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC;
(c) S. L. Tan, M. M. Jotani and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 308–318 CrossRef CAS.
- C. F. Matta, J. Hernández-Trujillo, T.-H. Tang and R. F. W. Bader, Chem. – Eur. J., 2003, 9, 1940–1951 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
-
Rigaku OD, CrysAlis PRO, Rigaku Oxford Diffraction Ltd, Yarnton, England, 2015 Search PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
-
M. J. Frisch, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120(1–3), 215–241 Search PubMed.
- D. Jayatilaka, S. K. Wolff, D. J. Grimwood, J. J. McKinnon and M. A. Spackman, Acta Crystallogr., Sect. A: Found. Crystallogr., 2006, 62, 90 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1941525 and 1941526. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce00807b |
| ‡ The powder spectrum obtained from {[Cu(II)(L4H2)2]·2MeOH}n (2) shows signs of crystallinity loss (peak broadening) presumably during sample preparation (e.g. loss of interstitial MeOH solvent molecules of crystallisation upon exposure to air and sample grinding). |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab