
Synthesis, structure, and luminescence properties of layered oxychloride Ba3Y2O5Cl2†
Yuki
Iwasa
 *a,
Yu
Su
a,
Yoshinori
Tsuchiya
a,
Makoto
Tatsuda
b,
Kohji
Kishio
ab,
Takayuki
Yanagida
c,
Fumi
Takada
d,
Taichiro
Nishio
d,
Yoshihiro
Tsujimoto
e,
Kotaro
Fujii
f,
Masatomo
Yashima
f and
Hiraku
Ogino
a
*a,
Yu
Su
a,
Yoshinori
Tsuchiya
a,
Makoto
Tatsuda
b,
Kohji
Kishio
ab,
Takayuki
Yanagida
c,
Fumi
Takada
d,
Taichiro
Nishio
d,
Yoshihiro
Tsujimoto
e,
Kotaro
Fujii
f,
Masatomo
Yashima
f and
Hiraku
Ogino
a
aNational Institute of Advanced Industrial Science and Technology, 1-1-1 Central 2, Umezono, Tsukuba, Ibaraki, 305-8568, Japan. E-mail: iwasa-y@aist.go.jp
bDepartment of Applied Chemistry, The University of Tokyo, 7-3-1 Hongo, Bunkyo, Tokyo 113-8656, Japan
cNara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara, 630-0192, Japan
dDepartment of Physics, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku, Tokyo 162-8601, Japan
eInternational Center for Materials Nanoarchitechtonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
fDepartment of Chemistry, School of Science, Tokyo Institute of Technology, Tokyo 152-8551, Japan
First published on 9th November 2020
Abstract
We report the synthesis, crystal structure, and optical properties of a new layered perovskite oxychloride, Ba3Y2O5Cl2. The structural and electronic properties of the compound were studied and compared with those of related compounds, Ba3RE2O5Cl2 (RE = Sc, Gd, and Lu). Furthermore, the luminescence properties due to Eu-doping were investigated. Polycrystalline Ba3Y2O5Cl2 samples were successfully synthesized by a solid-state reaction. The space group of the compound was I4/mmm and the lattice constants were a = 4.3971(8) Å and c = 24.848(5) Å. The Y atom in this compound shifted toward the apical oxygen site owing to the asymmetric coordination of O and Cl. Moreover, large anisotropic displacements were observed at the O sites. The direct bandgap of the compound was 5.4 eV. As Y3+ ions with a distorted octahedron have an asymmetric coordination environment, both magnetic and electric dipole transitions were observed by doping Eu3+ to Y3+ sites. Ba3Y2O5Cl2:Eu 4% also showed a high internal quantum yield (IQY) (>70%). Therefore, considering the photoluminescence spectra and high IQY, this compound is a potential host lattice for optical materials.
Introduction
In recent years, mixed-anion compounds have garnered much attention owing to the various functionalities that originate from their special structural and electronic features.1 The asymmetric local coordination environment of cations can be formed by utilizing two or more anions in the compound; this, in turn, causes a unique crystal field splitting and/or changes the transition probability of the luminescence centers. Controlling the coordinating environment of active cations in phosphor materials is a key method in inducing their luminescence properties as they are particularly sensitive to their coordinating anion environment.2–5 For example, Eu3+ ions show strong luminescence due to the 4f–4f transition of 5D0 → 7FJ (total angular momentum quantum number, J = 0, 1,…, 6). The magnetic dipole (MD) transitions are allowed in the case of J = 0, 1, 3, and 5, whereas the electric dipole (ED) transitions of J = 2, 4, and 6 are forbidden. Moreover, the ED transitions are allowed only when the Eu site has no inversion symmetry. Similarly, the charge transfer (CT) transitions, which occur upon the excitation of Eu3+ ions, are also affected by the local environment. Most highly efficient Eu3+ phosphors exhibit a narrow emission spectrum with a hyper-sensitive ED transition. However, considering the color rendering of white light illumination, a broad emission peak is suitable.The coordination environment of the luminescence center is determined by the structure of the host lattice. For example, the Y sites in Y2O3:Eu3+ and Y3Al5O12:Eu3+ are surrounded by six and eight oxygen ions, respectively, with rather high distortion. The asymmetric environment of rare-earth (RE) sites enables them to exhibit an ED transition. A greater variety of coordination environments can be realized for mixed anion compounds. For example, Y2O2S:Eu3+ is a well-known red phosphor, in which the Y site is surrounded by four oxygen and three sulfur ions. Strong ED transitions are observed because of their asymmetric environment. In addition, the excitation band appears at a longer wavelength than simple oxides, owing to the lower electronegativity of sulfur.6
In perovskite-related oxides, such as ABO3 and An+1BnO3n+1, there are two cation sites, the A-site (coordination number (CN) = 12) and the B-site (CN = 6); depending on the cation combinations, Eu3+ can be substituted at either of these sites. The A- and B-sites have different coordination environments. The difference in coordination results in a variety of luminescence properties.7,8 In the case of A-site substitution, the luminescence properties of Eu3+ can be fine-tuned by changing the local environment of the A-site.9–11 However, for instances of B-site substitutions, MD transitions are dominant as the local environment of the B-site is almost symmetric and luminescence properties cannot be controlled as it is not easy to change the environment of B-sites.12–14 In addition, if Eu3+ is located at a simple octahedral site, only the luminescence from MD transitions is observed.15 Furthermore, for mixed-anion compounds, there is a series of Ruddlesden–Popper-type perovskite compounds, with the chemical formula An+1BnO3n−1Cl2 (A: alkaline earth metals (AE), B: transition metals or RE elements, and n: number of perovskite blocks), such as A2BO2Cl2 (n = 1),16–18 A3B2O5Cl2 (n = 2)19–23 and A4B3O7.5Cl2 (n = 3).21,24 In these compounds, the local environment of the B-site is asymmetric because it is surrounded by both oxygen and chlorine. In addition, relatively large cations, such as RE elements, can occupy the B-site of these compounds, for example, Ba3Gd2O5Cl2.22 However, the photoluminescence properties of these compounds have not been reported thus far.
In this paper, we report the synthesis and crystal structure of a new layered oxychloride, Ba3Y2O5Cl2. It is interesting to study the luminescence properties of these compounds because of the specific local environment of the Y site. Optical properties such as absorption and photoluminescence on Eu-activation were studied and compared with those of related oxychlorides.
Experimental
Synthesis
Polycrystalline samples of Ba3Y2O5Cl2 and Ba(Y1−xEux)2O5Cl2 (Ba3Y2O5Cl2:Eu3+, x = 0–0.10) were synthesized via a conventional solid-state reaction with stoichiometric quantities of BaCO3 (3N), BaCl2 (3N), and Y2O3 (3N). Powder mixtures were pelletized and heated several times with intermittent grindings at 1173–1273 K in air for 24 h. The same procedure was followed for the synthesis of Ba3RE2O5Cl2 (RE = Gd, Ho, Er, and Lu). The chemical compositions were analyzed using energy-dispersive X-ray spectroscopy (EDX, Oxford SwiftED3000) equipped with a scanning electron microscope (SEM, Hitachi TM3000). Commercial Y2O3:Eu3+ powders (Nemoto & Co., Ltd) were used for the optical measurements.X-ray diffraction and structural refinement
Sintered samples were measured by powder X-ray diffraction (PXRD) with an Ultima-IV diffractometer (Rigaku). Using Cu Kα radiation, data were collected from 5° ≤ 2θ ≤ 80° with a 0.02° step size. The lattice parameters were refined by PXRD patterns using an Si internal standard.Transparent single crystals were found in the pellets synthesized by a normal solid-state reaction. Single crystal X-ray diffraction (SCXRD) data were collected using a Rigaku XtaLAB Pro diffractometer (Mo Kα radiation) at 300 K on a single crystal of 0.015 mm × 0.010 mm × 0.005 mm. The structural parameters were refined by the full-matrix least-squares method using SHELXL-2016.25 The details of SCXRD are shown in Table S1 (ESI†). When the occupancy factors were refined, the factors of all atoms except for Cl exceeded 1. The factor for Cl was refined to 0.962(13). These factors approached unity within 3σ. Therefore, the occupancy of all ions was fixed at 1.
DFT calculations
The density functional theory (DFT)-based calculations were performed for Ba3Y2O5Cl2 by the Vienna ab initio simulation package (VASP) code26 using the Perdew–Burke–Ernzerhof generalized gradient approximation (PBE-GGA) for the exchange and correlation functionals. Projector augmented-wave (PAW) potentials were used for Ba, Y, O, and Cl atoms.27,28 A plane-wave basis set with a cutoff of 500 eV was used (k-point mesh 5 × 5 × 5). The self-consistent field (SCF) tolerance was 1.0 × 10−7 eV·atom−1 and the cell parameters and atom positions were relaxed until the maximum force on each atom was less than 0.02 eV·Å−1.Optical properties
Diffuse reflectance was measured using a spectrophotometer (Shimadzu, UV-2600) equipped with an ISR-2600Plus integration sphere. The photoluminescence spectra and lifetime of luminescence were recorded using a spectrofluorometer (JASCO, FP-8500) and the internal quantum yield (IQY) was measured using an intensified multichannel spectrometer (Otsuka Electronics, MCPD-7000). Internal quantum yield (IQY) Φint is defined as follows: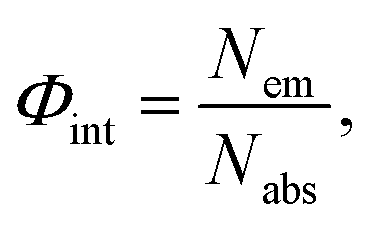 | (1) |
Results and discussion
Ba3Y2O5Cl2 samples were successfully prepared by solid-state synthesis. Plate-like particles were found on the surface of the pellet (Fig. S1, ESI†). The grain sizes ranged from a few tens to a few hundred micrometers. The atomic ratio was evaluated as Ba![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Y:Cl = 3:2.1:1.8 by EDX measurement, which is within the error range from the nominal composition of Ba3Y2O5Cl2. Fig. 1(a) shows the experimental and simulated PXRD patterns of Ba3Y2O5Cl2. The simulation pattern was determined from the results of the structural analysis. Except for a small amount of Y2O3 impurity, a single-phase was obtained. All peaks were assigned to the tetragonal cell with the space group I4/mmm. The lattice parameters obtained were a = 4.3971(8) Å and c = 24.848(5) Å. The intensities of the 00l reflections were larger than the simulation pattern because of the grain orientation. Furthermore, the phase was stable under air and no degradation was observed after a few days of exposure.
:Y:Cl = 3:2.1:1.8 by EDX measurement, which is within the error range from the nominal composition of Ba3Y2O5Cl2. Fig. 1(a) shows the experimental and simulated PXRD patterns of Ba3Y2O5Cl2. The simulation pattern was determined from the results of the structural analysis. Except for a small amount of Y2O3 impurity, a single-phase was obtained. All peaks were assigned to the tetragonal cell with the space group I4/mmm. The lattice parameters obtained were a = 4.3971(8) Å and c = 24.848(5) Å. The intensities of the 00l reflections were larger than the simulation pattern because of the grain orientation. Furthermore, the phase was stable under air and no degradation was observed after a few days of exposure.
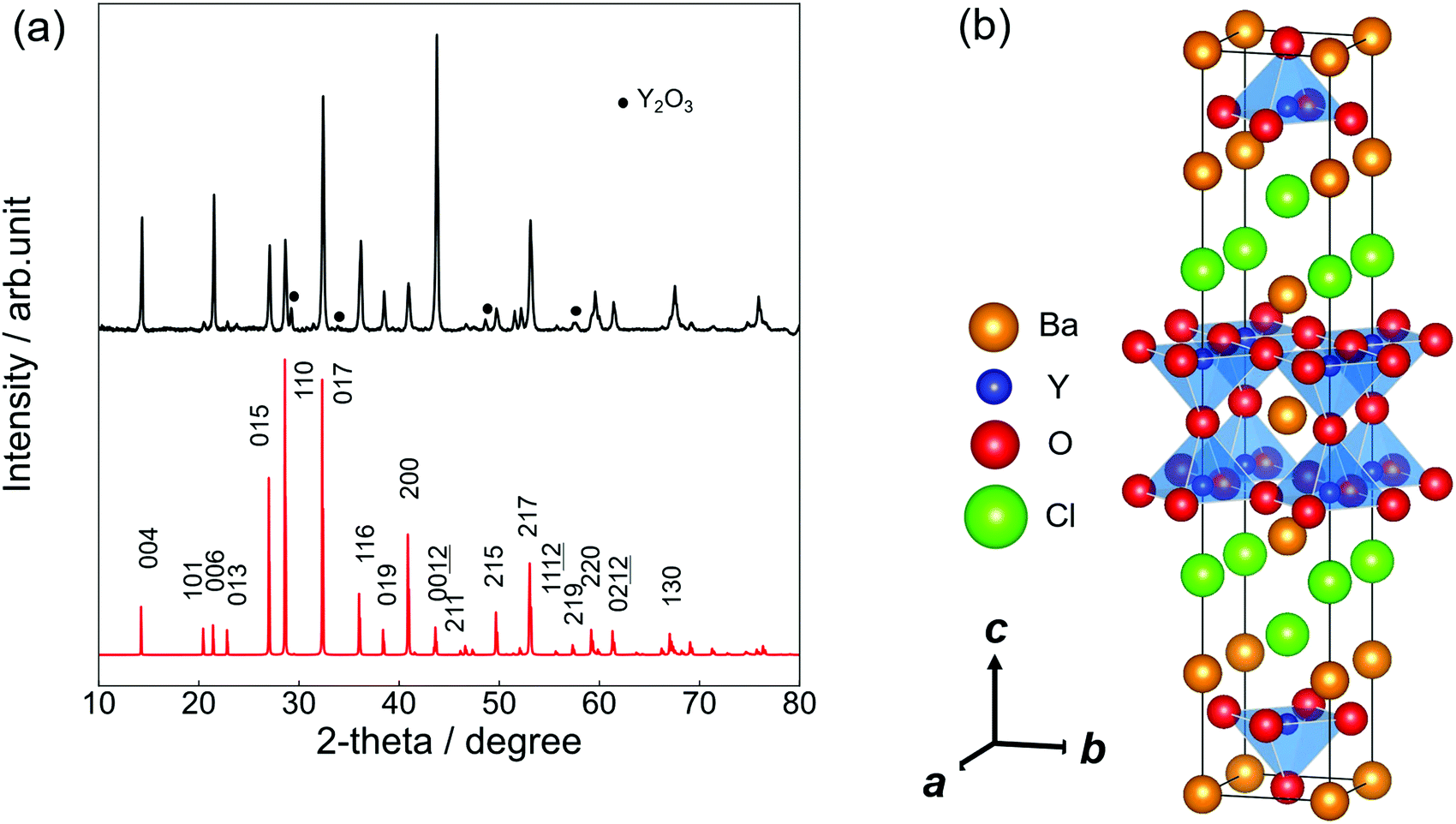 | ||
| Fig. 1 (a) PXRD patterns of Ba3Y2O5Cl2. The top pattern (black) shows the experimental result and the bottom pattern (red) shows a simulated pattern. (b) Crystal structure of Ba3Y2O5Cl2. The orange, blue, red and green balls represent Ba, Y, O and Cl ions, respectively. The blue pyramids show the polyhedra of YO5. | ||
Table 1 presents the results of the single-crystal structure analysis. The lattice parameters evaluated from SCXRD were a = 4.4120(2) Å and c = 24.8781(15) Å, which are nearly consistent with the lattice parameters evaluated by PXRD. Moreover, the calculated profiles were in good agreement (within 1.5%) with those optimized by the DFT calculations.
| Atom | x | y | z | Occ. | U iso/Å2 | U 11/Å2 | U 22/Å2 | U 33/Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I4/mmm (no. 139). a = 4.4120(2) Å, and c = 24.8781(15) Å. U12 = U13 = U23 = 0 for all sites. Rint = 0.059, and R1(I > 2σ(I)) = 0.020, wR2 = 0.040, and S = 1.26. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ba1 | 0 | 0 | 0 | 1.0 | 0.0243(3) | 0.0206(4) | 0.0206(4) | 0.0318(6) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ba2 | 0 | 0 | 0.16244(2) | 1.0 | 0.0100(2) | 0.0079(2) | 0.0079(2) | 0.0143(3) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Y | 1/2 | 1/2 | 0.08512(4) | 1.0 | 0.0079(3) | 0.0065(3) | 0.0065(3) | 0.0106(5) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oeq | 1/2 | 0 | 0.0969(2) | 1.0 | 0.0226(13) | 0.016(3) | 0.007(3) | 0.045(3) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oap | 1/2 | 1/2 | 0 | 1.0 | 0.051(5) | 0.071(7) | 0.071(7) | 0.012(6) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cl | 1/2 | 1/2 | 0.20543(10) | 1.0 | 0.0193(6) | 0.0208(9) | 0.0208(9) | 0.0163(14) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Fig. 1(b) illustrates the crystal structure of Ba3Y2O5Cl2 using the VESTA.29 The structure was isostructural to that of Ba3RE2O5Cl2 (RE = Sc, and Gd–Lu) (RE = Sc, and Gd–Lu)22,23 and comprised a double-perovskite-like Ba–Y–O layer and Ba–Cl rock-salt layer. In the perovskite layer, the double YO5Cl octahedra were connected by corner-shared apical oxygen (Oap) sites and the anion sites in the rock-salt layer were fully occupied by chlorine. Therefore, the structure obtained the stacking sequence, –(BaO)–(YO2)–(BaCl)2–(YO2)–.
According to the ionic radii reported by Shannon et al.,30 the ionic radius of Y3+ (0.900 Å, CN = 6) was close to that of Ho3+ (0.901 Å, CN = 6) and Er3+ (0.890 Å, CN = 6). Therefore, the lattice parameters of Ba3Y2O5Cl2 were expected to be similar to those of Ba3Ho2O5Cl2 and Ba3Er2O5Cl2. This was because Besara et al. reported lattice constants that were measured at 200 K, synthesized Ba3Ho2O5Cl2 and Ba3Er2O5Cl2via solid-state synthesis and evaluated the lattice parameters by PXRD at room temperature.22
The lattice constants of Ba3Ho2O5Cl2 were a = 4.408(11) Å, and c = 24.822(7) Å, and those of Ba3Er2O5Cl2 were a = 4.3969(7) Å, and c = 24.779(5) Å. Taking into account the difference between the lattice constants measured at 200 K and at room temperature, these parameters were quite consistent with the reported values.20,21 Therefore, the lattice constants of Ba3Y2O5Cl2 were larger than those of the other two compounds. In chlorides, such as YCl3 and HoCl3, the Y–Cl distance is usually longer than the Ho–Cl or Er–Cl distance;31 therefore, the larger lattice constants in Ba3Y2O5Cl2, especially for the c-axis, can be explained by this tendency.
Fig. 2 shows a schematic view of the local coordination environments of Ba1, Ba2, and Y. The CNs of Ba1, Ba2, and Y were 12, 9, and 6, respectively. Ba1 was coordinated to eight equatorial oxygen (Oeq) atoms, whereas four Oap and Ba2 atoms were coordinated by five Cl and four Oeq atoms. The Y site was coordinated by one Oap, four Oeq, and one Cl, and the local structure of the Y site was symmetric along the ab plane and asymmetric along the c-axis. The bond lengths of Ba1–Oeq and Ba1–Oap are 3.268(4) Å and 3.11976(15) Å, respectively. These bond lengths, especially Ba1–Oeq, were longer than the sum of the ionic radii of Ba2+ and O2− (3.01 Å). Owing to the large bonding lengths, the bond valence sums (BVSs) of Ba1 were estimated to be 0.9809, which were much smaller than the expected value (∼2) approximated from the formal valence of Ba. The bond lengths of Ba2–Cl were 3.287(3) Å and 3.2980(9) Å, and that of Ba2–Oeq was 2.743(3) Å. The Ba2–Cl bond length was almost equal to the sum of the ionic radii (3.28 Å) but the bond length of Ba2–Oeq was slightly smaller than the expected value (2.87 Å). The BVS of Ba2 was estimated to be 2.132. Y–Oap, Y–Oeq and the Y–Cl distances were 2.1176(11) Å, 2.2254(7) Å, and 2.993(3) Å, respectively. The Y–Oap distance was ∼10% shorter, while the Y–Cl distance was ∼10% larger than the sum of the ionic radii and the position of Y shifted toward Oap. This trend is explained by the difference in electronegativity between O2− and Cl− ions. Therefore, Y shifted from the center of the octahedron, creating an asymmetric coordination environment at the B-site.
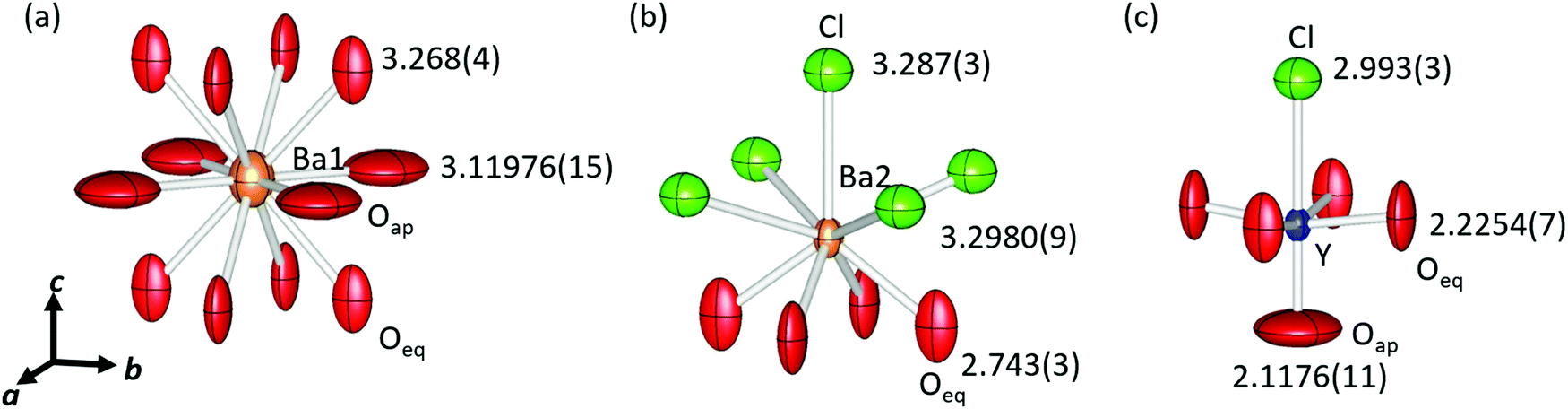 | ||
| Fig. 2 Schematic view of the crystal structure and local coordination environments of (a) Ba1, (b) Ba2 and (c) Y, respectively. The orange, blue, red and green ellipsoids show Ba, Y, O and Cl, respectively. Displacement ellipsoids are shown at the 99% level. Cation–anion bond length values are described on anions. | ||
There were large anisotropic displacements at the Oeq and Oap sites, as shown in Fig. 2. The thermal ellipsoids of Oeq were elongated along the c-axis and those of Oap were elongated along the ab plane. Su et al. reported that such elongation of thermal ellipsoids occurred because of the mismatch in compensation of each layer.23 The ideal sum of the radii of Y–O (2.30 Å) was greater than half the lattice constant (2.204 Å). Therefore, the Y–Oeq plane was compressed along the ab plane. In contrast, the bond length of Ba1–Oap (equal to a/√2), 3.11976(15) Å, was much larger than the ideal sum of radii for Ba–O, 3.01 Å; hence, the Ba1–Oap plane expanded along the ab plane.
Fig. 3 shows the band structure and partial density of state (PDOS) of Ba3Y2O5Cl2 obtained by the DFT calculations. The calculated band structure suggested that this compound had a direct bandgap of ∼3.6 eV, in contrast to the indirect bandgap of Ba3Sc2O5Cl2. The valence band maximum (VBM) was formed by the Oeq 2p orbital and the Oap 2p orbital was located lower than the Oeq 2p orbital. Cl 3p was located at a lower energy than the two O 2p orbitals. While the 3p orbital of Cl occupies a higher energy than the O 2p band in usual oxychlorides, the Cl 3p orbital in this compound did not contribute to the VBM, which was similar to that of the related Ba3RE2O5Cl2 compounds.22,23 The conduction band minimum (CBM) of Ba3Y2O5Cl2 was formed by the unoccupied 5d orbital of Ba1. Conversely, the Sc 3d orbital in Ba3Sc2O5Cl2 contributed to the CBM. This difference may explain the variations in the direct/indirect bandgap nature of these compounds. Ba2 5d and Y 4d orbitals were located at higher energies than the Ba1 5d orbitals. Therefore, the bandgap of Ba3Y2O5Cl2 consisted of Ba1 and Oeq in the perovskite-like layer and the Ba2 and Cl ions in the rock-salt layer did not contribute to the bandgap in the compound.
 | ||
| Fig. 3 (a) The band structure and (b) partial density of states (PDOS) of Ba3Y2O5Cl2. | ||
Fig. 4 shows the Tauc plots of Ba3Y2O5Cl2, as well as those of the related compounds, Ba3Lu2O5Cl2 and Ba3Gd2O5Cl2. Bandgaps were estimated using the following equation:
| (F(R)hν)2 = A(hν − Eg), | (2) |
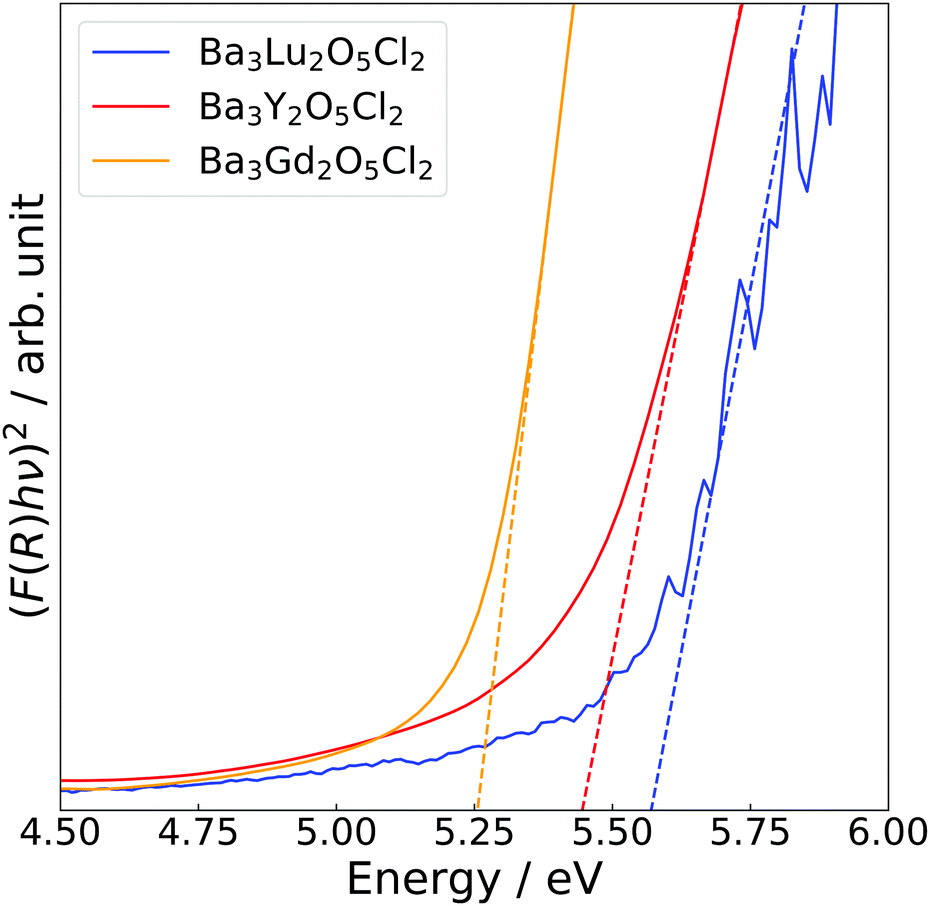 | ||
| Fig. 4 Tauc plots of Ba3RE2O5Cl2 (RE = Y, Gd and Lu) measured at room temperature. The solid lines show Kubelka–Munk transformed diffuse reflectance spectra and the dashed lines show the extrapolation of the linear portion at the band edge. The blue, red and orange colours represent Ba3Lu2O5Cl2, Ba3Y2O5Cl2, and Ba3Gd2O5Cl2, respectively. | ||
Fig. 5 shows the excitation and emission spectra and an image of luminescence under a mercury lamp (254 nm excitation) of Ba3Y2O5Cl2:Eu3+ 1% and commercial Y2O3:Eu3+, which was used as a reference. In the excitation spectra, both samples showed excitation peaks that are characteristic of the CT transition of Eu3+. In the case of Y2O3:Eu3+, there were two peaks at ∼230 mm and ∼250 nm, whereas for Ba3Y2O5Cl2:Eu3+, there was only one peak at ∼250 nm. Unlike some mixed-anion compounds, such as Y2O2S, a large redshift of the excitation peak was not observed in Ba3Y2O5Cl2. This was in agreement with the DFT calculations, in which the Cl 3p orbital was located at a lower energy than the O 2p orbital in the compound.
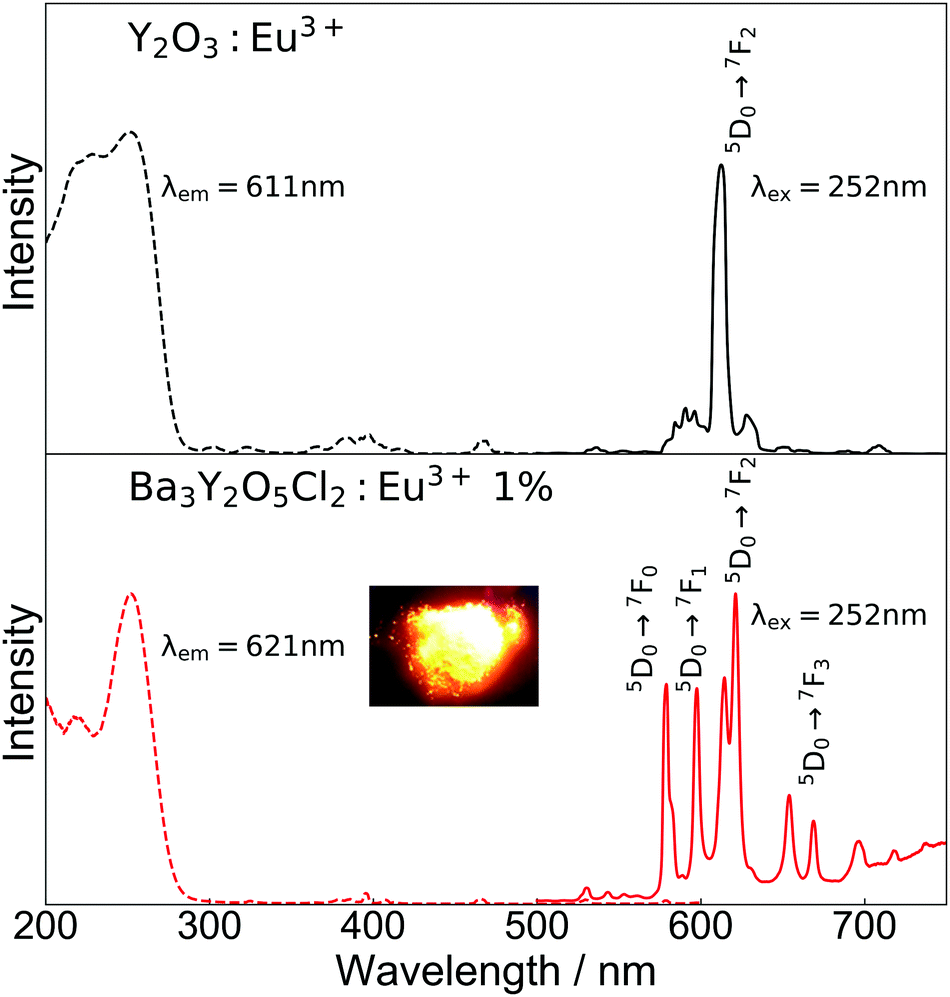 | ||
| Fig. 5 Photoluminescence of Y2O3:Eu3+ (black) and Ba3Y2O5Cl2:Eu3+ 1% (red) measured at room temperature. The dashed line shows the excitation spectrum, and the solid line shows the emission line. The inset image shows the emission of Ba3Y2O5Cl2:Eu3+ 1% under excitation. | ||
In the emission spectra, photoluminescence from the Eu3+ 4f–4f transitions was observed in both samples. A strong single peak of ED transition was observed for Y2O3:Eu3+ at 611 nm, which is in good agreement with the reported value. For Ba3Y2O5Cl2:Eu3+, emission from both ED and MD transition peaks was observed at 575–583 nm, 597–600 nm, 614–625 nm, 653–673 nm, and 693–725 nm. The emission color was orange, as shown in the inset image in Fig. 5. The aforementioned small amount of Y2O3 impurity present in the sample had no clear contribution to the luminescence. The intensities of the ED and MD transition peaks in this compound were almost similar, which has not been reported thus far. Owing to this, the compounds show a broad spectra in the red region. Moreover, this is contrary to the ED transition peak of a simple oxide perovskite with a B-site Eu3+, which is much lower than the MD transition peaks.12–14 As described above, the characteristic ED/MD transition ratio of the compound originates from the local structure of the Y site.
The lifetime of the luminescence at the 5D0 → 7F2 transition in Ba3Y2O5Cl2:Eu 4% is shown in Fig. 6. All emission peaks show almost similar decay curves with CT-state excitation. The decay curve I(t) was fitted by the following function:
 | (3) |
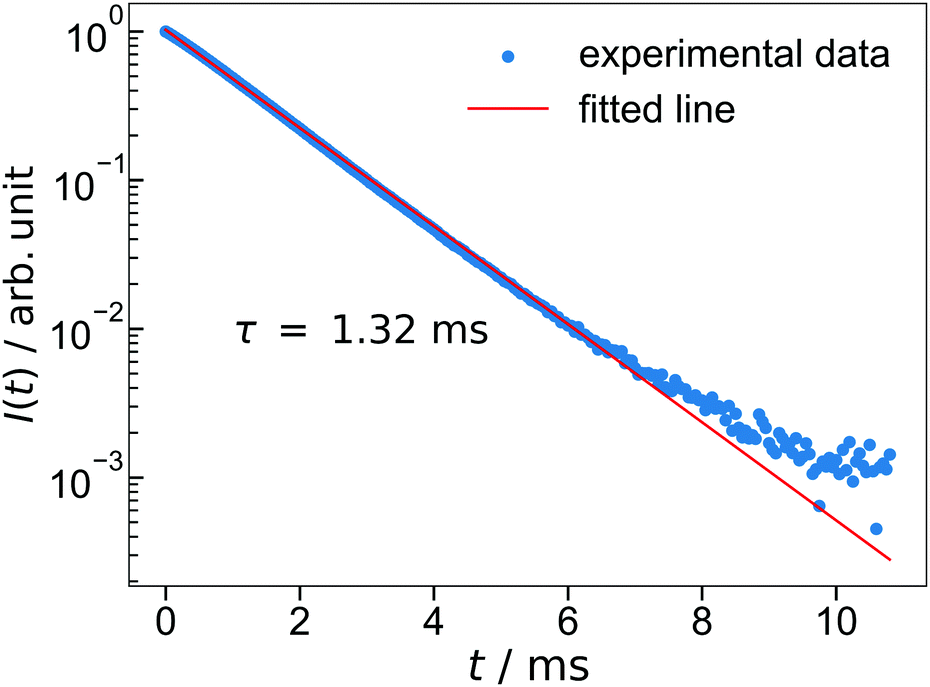 | ||
| Fig. 6 The luminescence decay of Ba3Y2O5Cl2:Eu 4%. The excitation wavelength is 252 nm and monitoring wavelength is 621 nm. | ||
The IQYs of Ba3Y2O5Cl2:Eu3+ with different Eu concentrations are shown in Fig. 7. The highest quantum yield was achieved at x = 0.04, with IQY of over 70%; this is larger than that of YOF:Eu3+.32 The IQY is in good agreement with the calculated quantum yield from the lifetime (Table S2, ESI†). Considering the high IQY, characteristic ED/MD transition ratio, and stability in air, Ba3Y2O5Cl2 can be a promising candidate for several phosphor materials, i.e., not only for Eu3+ but also for other RE elements.
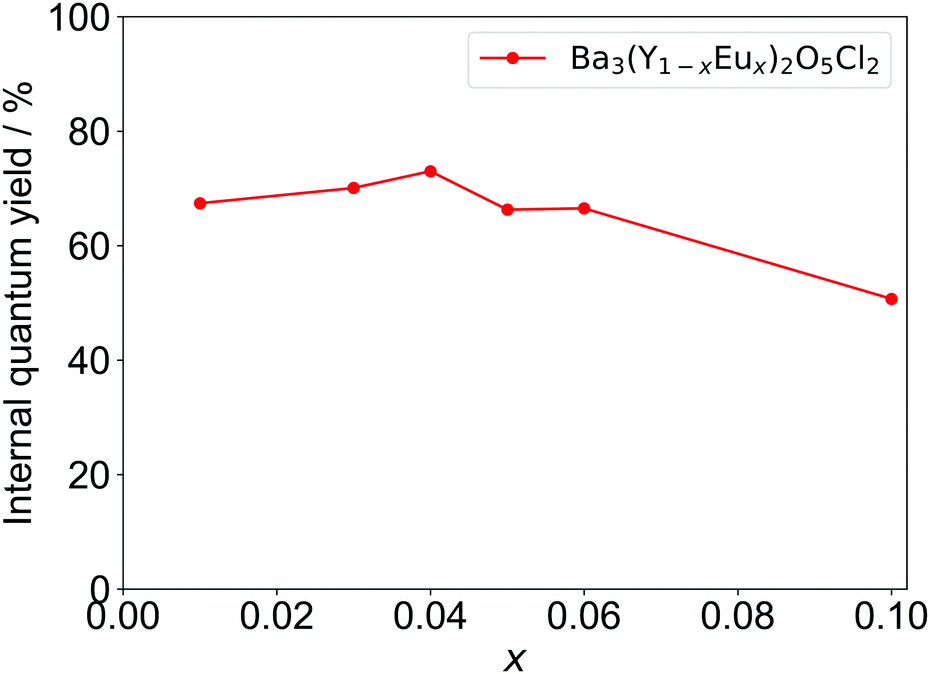 | ||
| Fig. 7 Internal quantum efficiencies (IQYs) of Ba3Y2O5Cl2 with different Eu concentrations. | ||
Conclusions
We successfully synthesized new layered oxychlorides, Ba3Y2O5Cl2, by a solid-state reaction. The space group of the compound was I4/mmm, and the lattice constants were a = 4.3971(8) Å and c = 24.848(5) Å. Y ions were located toward Oap in the YO5Cl octahedron because of the difference in electronegativity between O2− and Cl− ions. A direct bandgap was suggested by the DFT calculations, and VBM and CBM were formed by Oeq 2p and Ba 5d orbitals, respectively. The bandgap of Ba3Y2O5Cl2 was estimated to be 5.4 eV by diffuse reflectance measurement. In Ba3Y2O5Cl2:Eu3+, both MD and ED transitions were observed with similar intensities, and for 4% Eu-doping, IQY was as high as 70%.Author contributions
M. T., K. K., and H. O. designed and explored the new compound. Y. I., Y. S., Y. Tsuc., F. T., T. N., Y. Tsuj., and H. O. synthesized and measured the optical properties. Y. I., Y. S., T. Y., and H. O. measured luminescence properties. K. F. and M. Y. analyzed the crystal structure and performed the DFT calculations. All the authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS Grant-in-Aid for Scientific Research on Innovative Areas “Mixed Anion” (Grant Number JP16H06439, 16H06440, and 19H04711) and Grant-in-Aid No. 18J01627 and 20K15032. We thank Prof. T. Takeda and N. Hirosaki in NIMS for assistance with the IQY measurements.Notes and references
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 772 CrossRef.
- H. Kato, Y. Takeda, M. Kobayashi, H. Kobayashi and M. Kakihana, Front. Chem., 2018, 6, 1–7 CrossRef.
- M. Kobayashi, T. Yasunaga, H. Sato, H. Kato, K. Fujii, M. Yashima and M. Kakihana, Chem. Lett., 2017, 46, 795–797 CrossRef CAS.
- Y. Masubuchi, M. Tadaki and S. Kikkawa, Chem. Lett., 2018, 47, 31–33 CrossRef CAS.
- Y. Kitagawa, J. Ueda, M. G. Brik and S. Tanabe, Opt. Mater., 2018, 83, 111–117 CrossRef CAS.
- B.-M. Cheng, C.-K. Duan and P. A. Tanner, Opt. Mater., 2009, 31, 902–904 CrossRef CAS.
- M. K. Mahata, T. Koppe, T. Mondal, C. Brüsewitz, K. Kumar, V. Kumar Rai, H. Hofsäss and U. Vetter, Phys. Chem. Chem. Phys., 2015, 17, 20741–20753 RSC.
- M. K. Mahata, T. Koppe, K. Kumar, H. Hofsäss and U. Vetter, Sci. Rep., 2020, 10, 8775 CrossRef CAS.
- L. Zhang, B. Sun, Q. Liu, N. Ding, H. Yang, L. Wang and Q. Zhang, J. Alloys Compd., 2016, 657, 27–31 CrossRef CAS.
- L. Li, W. Chang, W. Chen, Z. Feng, C. Zhao, P. Jiang, Y. Wang, X. Zhou and A. Suchocki, Ceram. Int., 2017, 43, 2720–2729 CrossRef CAS.
- F. Fujishiro, T. Arakawa and T. Hashimoto, Mater. Lett., 2011, 65, 1819–1821 CrossRef CAS.
- H. Zhang, X. Fu, S. Niu and Q. Xin, J. Alloys Compd., 2008, 459, 103–106 CrossRef CAS.
- K. Ueda, S. Tanaka, T. Yoshino, Y. Shimizu and T. Honma, Inorg. Chem., 2019, 58, 10890–10897 CrossRef CAS.
- A. K. Kunti, N. Patra, R. A. Harris, S. K. Sharma, D. Bhattacharyya, S. N. Jha and H. C. Swart, Inorg. Chem., 2019, 58, 3073–3089 CrossRef CAS.
- F. Fujishiro, M. Murakami, T. Hashimoto and M. Takahashi, J. Ceram. Soc. Jpn., 2010, 118, 1217–1220 CrossRef CAS.
- Y. Kohsaka, M. Azuma, I. Yamada, T. Sasagawa, T. Hanaguri, M. Takano and H. Takagi, J. Am. Chem. Soc., 2002, 124, 12275–12278 CrossRef CAS.
- C. S. Knee and M. T. Weller, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 1–7 CrossRef.
- Y. Su, Y. Tsujimoto, A. Miura, S. Asai, M. Avdeev, H. Ogino, M. Ako, A. A. Belik, T. Masuda, T. Uchikoshi and K. Yamaura, Chem. Commun., 2017, 53, 3826–3829 RSC.
- J. F. Ackerman, J. Solid State Chem., 1991, 92, 496–513 CrossRef CAS.
- N. McGlothlin, D. Ho and R. J. Cava, Mater. Res. Bull., 2000, 35, 1035–1043 CrossRef CAS.
- S. M. Loureiro, C. Felser, Q. Huang and R. J. Cava, Chem. Mater., 2000, 12, 3181–3185 CrossRef CAS.
- T. Besara, D. C. Ramirez, J. Sun, N. W. Falb, W. Lan, J. N. Neu, J. B. Whalen, D. J. Singh and T. Siegrist, Inorg. Chem., 2018, 57, 1727–1734 CrossRef CAS.
- Y. Su, Y. Tsujimoto, K. Fujii, M. Tatsuta, K. Oka, M. Yashima, H. Ogino and K. Yamaura, Inorg. Chem., 2018, 57, 5615–5623 CrossRef CAS.
- C. S. Knee, A. A. Zhukov and M. T. Weller, Chem. Mater., 2002, 14, 4249–4255 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- D. H. Templeton and G. F. Carter, J. Phys. Chem., 1954, 58, 940–944 CrossRef CAS.
- T. Grzyb, M. Węcławiak, T. Pędziński and S. Lis, Opt. Mater., 2013, 35, 2226–2233 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc04415f |
| This journal is © The Royal Society of Chemistry 2020 |