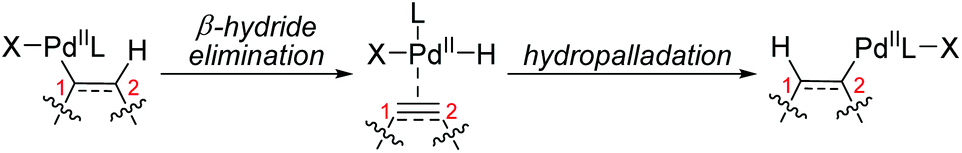DOI:
10.1039/D0QO00558D
(Review Article)
Org. Chem. Front., 2020,
7, 3530-3556
Recent advances in transition metal migration involving reactions
Received
12th May 2020
, Accepted 27th August 2020
First published on 29th August 2020
Abstract
Numerous elegant methodologies involving transition-metal migration processes have received increasing attention and they have gradually developed into significant synthetic tools in the past few decades. These strategies provide novel ways to build a carbon–metal bond at a new position, where it may not be facile or straightforward to introduce a metal atom. Hence, metal-migration reactions exhibit attractive practicability in synthetic organic chemistry. This review gives an up-to-date overview of catalytic methodologies involving metal-migration processes, which are sorted into categories such as 1,2- to 1,5- and 1,7-Pd migration, 1,3- to 1,5-Rh migration, 1,4- and 1,5-Fe migration, and so on. For most of these transformations, plausible mechanisms are demonstrated elaborately. Clarification of these processes is a key point for understanding metal-migration involving reactions and developing new high-performance methodologies.
1 Introduction
Transition-metal-catalyzed reactions have emerged as powerful alternatives to versatile functionalized molecules. Most importantly, significant progress has been witnessed in reactions for the construction of carbon–carbon and carbon–heteroatom bonds, exhibiting advantages of improved atom- and step-economy compared to conventional methods.1 Generally, carbon–metal bonds can be generated via oxidative addition, transmetalation, insertion into unsaturated bonds, C–H bond activation, and so on. However, it is not easy to introduce a metal moiety into a specific position of an organic molecular skeleton. Recently, the metal-migration process has shown attractive applications to form unique carbon–metal bonds, and they could be transformed into valuable compounds that otherwise would be difficult to achieve via classic processes. Palladium catalysis, as one of the most important synthetic tools in modern organic synthesis, shows a special application in the synthesis of complicated molecules via a controllable migration and remote C–H activation strategy. In 2005, Ma et al. contributed a prospective review on the elementary art of including 1,4-migration of palladium.2 In 2010, the Larock group summarized the reactions of 1,4 and 1,5-migration of palladium.3 Very recently, Gu et al. provided a comprehensive review on 1,4-migration of transition metals in organic synthesis, including palladium-catalyzed strategies.4 With the development of palladium chemistry, palladium migration became a quite unique tool in organic synthesis based on ready design and control. More and more novel transformations, involving 1,2- to 1,5- and 1,7-palladium migration, have emerged dramatically. Additionally, Rh, Fe, Co, Ir, Cr, Ni, and Pt also demonstrate appealing migratory behaviors in synthetic chemistry. Herein, this review mainly summarizes the metal-migration reactions in the past nearly 20 years, including elaborate mechanism presentation, hoping to give a preliminary understanding of this field for the chemists in need.
2 Palladium migration
2.1 1,2-Palladium migration
As illustrated in Scheme 1, 1,2-palladium migration occurs via sequential β-H elimination and hydropalladation processes in most cases. This elimination/reinsertion process frequently occurs in Heck type reactions, especially the migration of olefins, and chemists differ in their attitudes towards the process of reversible β-hydride elimination/reinsertion, which might not be considered and classified as a 1,2-palladium migration process. Hence in this section, only a few typical examples, classified as 1,2-Pd migration by the original authors, are exhibited to give a preliminary understanding for those unfamiliar with this field. More examples toward elimination/reinsertion processes can be found in metal chain walking research.5
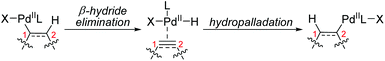 |
| | Scheme 1 Proposed process of 1,2-palladium migration. | |
The affecting factors and dynamics of 1,2-palladium migration are mainly associated with steric hindrance and the stability of the intermediate. The details will be described in the following reaction examples.
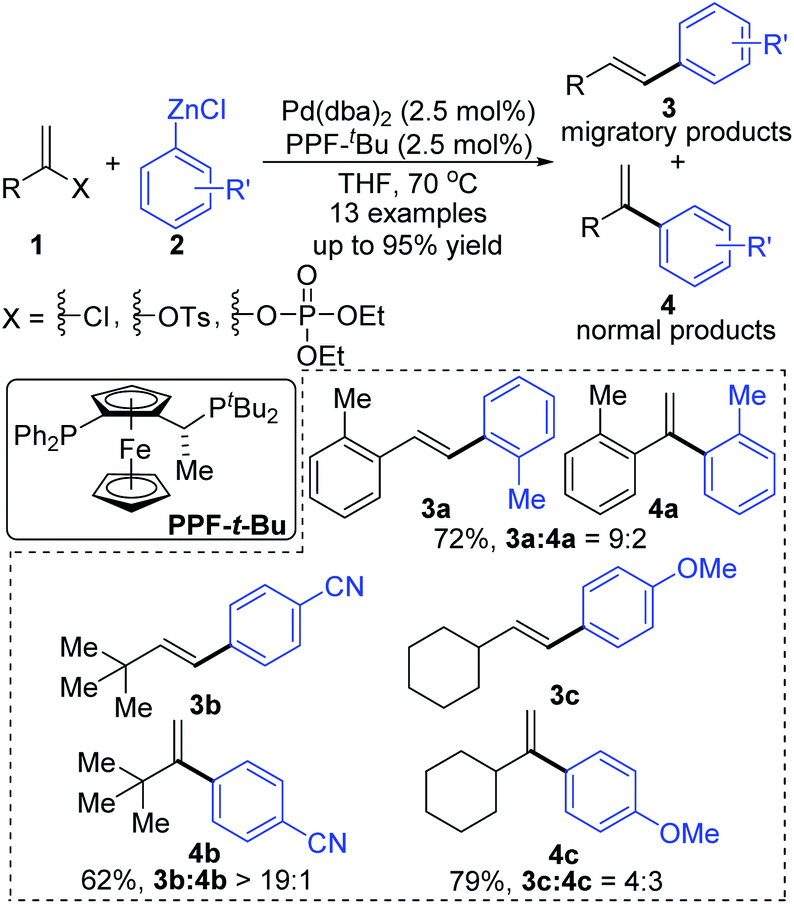
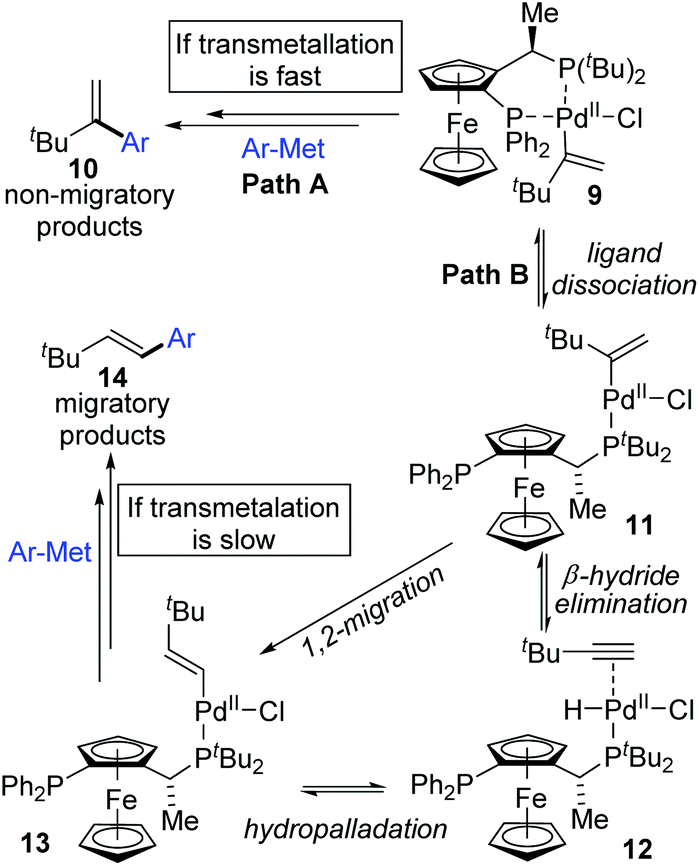
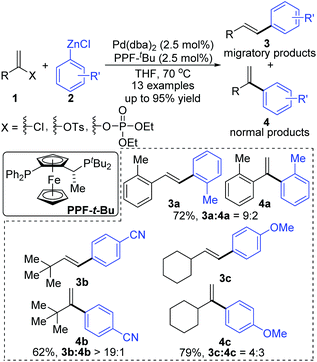
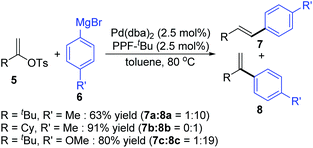
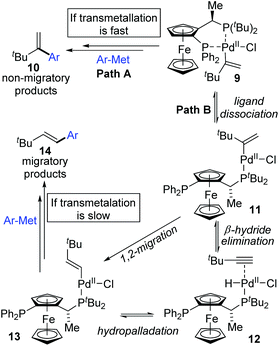
In 2006, a 1,2-migration involved palladium-catalyzed Negishi coupling was described by the Skrydstrup group (Scheme 2).6 The highest selectivity of 1,2-migration was achieved via employing Josiphos ligands with neither electron-donating nor -withdrawing substituents on the phenyl ring. The ratio of rearranged/normal products was up to 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with the combination of Pd(dba)2 and PPF-t-Bu. The employment of electron-deficient aryl zinc reagent seemed to favor this migratory process, such as 3b. Further exploration experiments toward more nucleophilic aryl Grignard reagents revealed that the nucleophilicity of the organometallic reagents might influence the competition between the transmetalation step and the intermediate-hydride elimination of the alkenyl Pd(II) species (Scheme 3). The pathways illustrated in Scheme 4 better visualize this process. The ligand based Pd(II) intermediate 9 would furnish the non-migratory products 10 in cases where the transmetalation was fast (path A), otherwise a ligand dissociation might take place to form a trigonally coordinated palladium(II) complex 11 possessing a vacant site (path B). Sequential β-hydride elimination in vinyl (12) and hydropalladation afforded the migrated vinylic Pd(II) compound 13, which finally provided the migratory products 14.
:1 with the combination of Pd(dba)2 and PPF-t-Bu. The employment of electron-deficient aryl zinc reagent seemed to favor this migratory process, such as 3b. Further exploration experiments toward more nucleophilic aryl Grignard reagents revealed that the nucleophilicity of the organometallic reagents might influence the competition between the transmetalation step and the intermediate-hydride elimination of the alkenyl Pd(II) species (Scheme 3). The pathways illustrated in Scheme 4 better visualize this process. The ligand based Pd(II) intermediate 9 would furnish the non-migratory products 10 in cases where the transmetalation was fast (path A), otherwise a ligand dissociation might take place to form a trigonally coordinated palladium(II) complex 11 possessing a vacant site (path B). Sequential β-hydride elimination in vinyl (12) and hydropalladation afforded the migrated vinylic Pd(II) compound 13, which finally provided the migratory products 14.
 |
| | Scheme 2 Skrydstrup's Negishi couplings involving a 1,2-palladium migration process. | |
 |
| | Scheme 3 Exploration experiments involving Grignard reagents with 1,2-migration. | |
 |
| | Scheme 4 Different reaction pathways based on the nucleophilicity of the organometallic reagents. | |
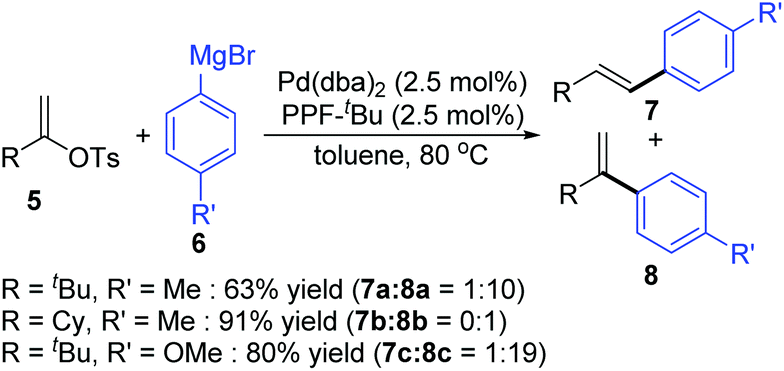
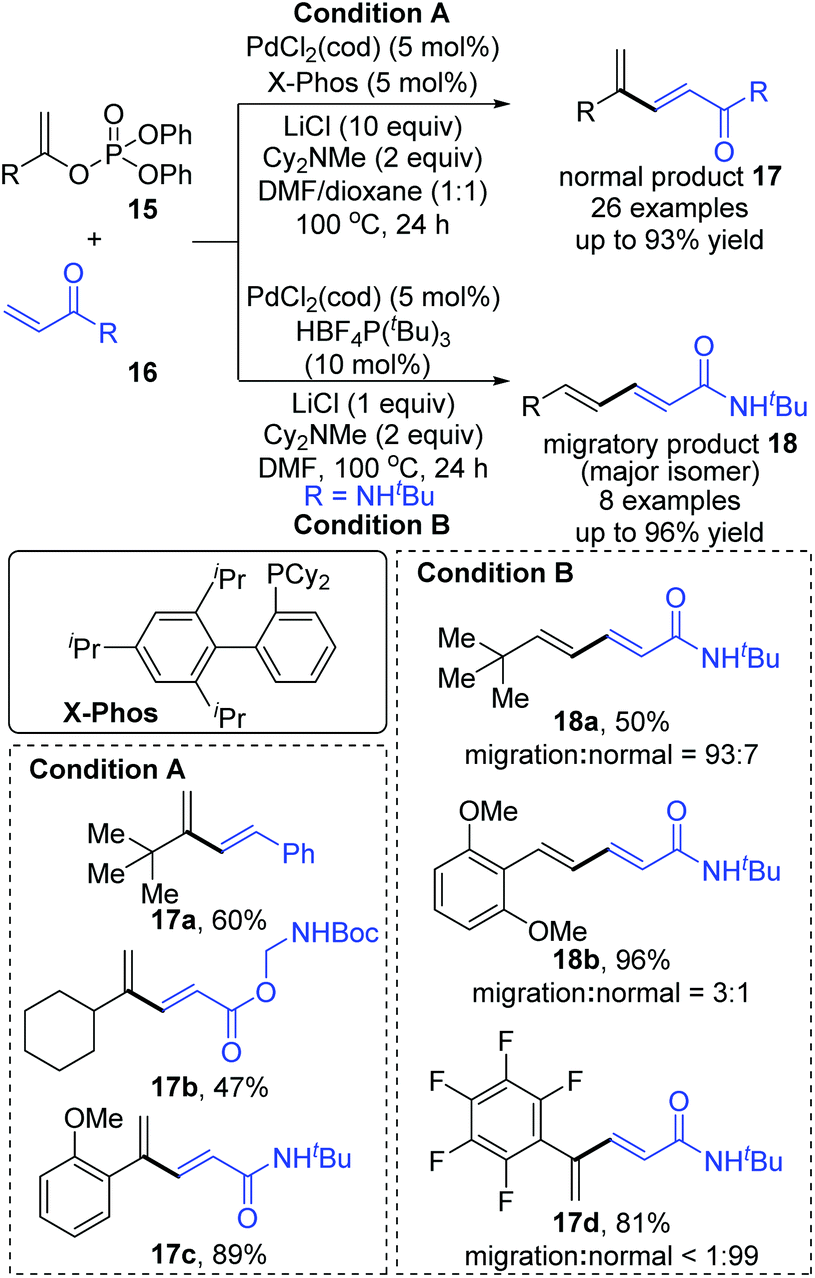
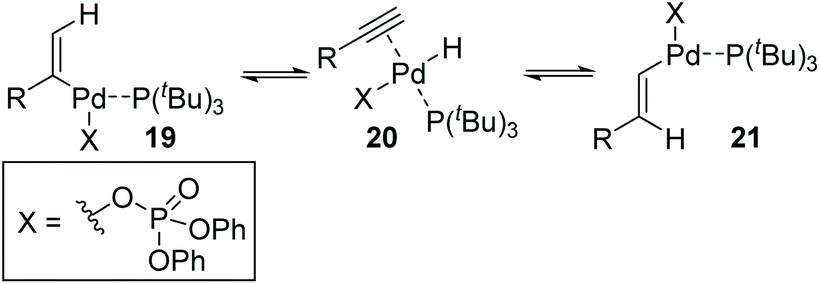
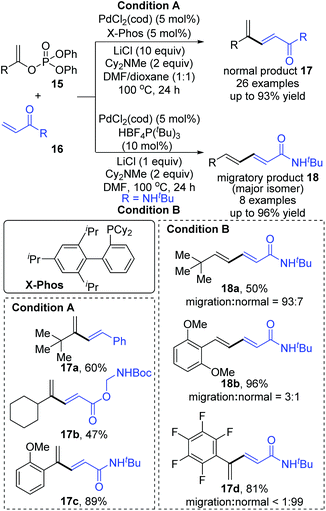
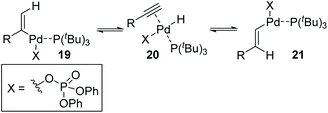
Then, Skrydstrup et al. further reported 1,2-palladium migration involved studies toward Heck reaction with alkenyl phosphates 15 (Scheme 5).7 The employment of X-Phos and excessive LiCl (10 equiv.) could suppress 1,2-migration efficiently to give the normal products 17, while ligand HBF4P(t-Bu)3 tended to encourage the migratory products 18. Studies also showed that the 1,2-migration was largely dependent on the C1-substituent, and high migratory selectivity was observed for substrates bearing a C1-alkyl quaternary carbon, such as product 18a. However, the perfluorophenyl vinyl phosphate afforded the non-migratory product 17d in a high yield under condition B, indicating that electronic factors held important implications for this transformation. The possible 1,2-palladium migration was revealed in Scheme 6. Initial oxidative addition of Pd(0) afforded a tri-coordinated Pd(II) intermediate 19 possessing a free site on the palladium for the following β-elimination step. And this elimination step was sensitive to the steric hindrance, which could prevent interaction of the metal center with the vinylic hydrogen. Just as condition A offered the non-migratory products exclusively by using the sterically hindered ligand X-Phos, with condition B, the hydropalladation of intermediate 20 allowed the Pd(II) group to move to the uncrowded vinyl-terminal and form the migratory Pd(II) complex 21.
 |
| | Scheme 5 Skrydstrup's Heck-type reactions involving 1,2-migration. | |
 |
| | Scheme 6 Possible mechanism for 1,2-Pd migration in Heck reactions. | |
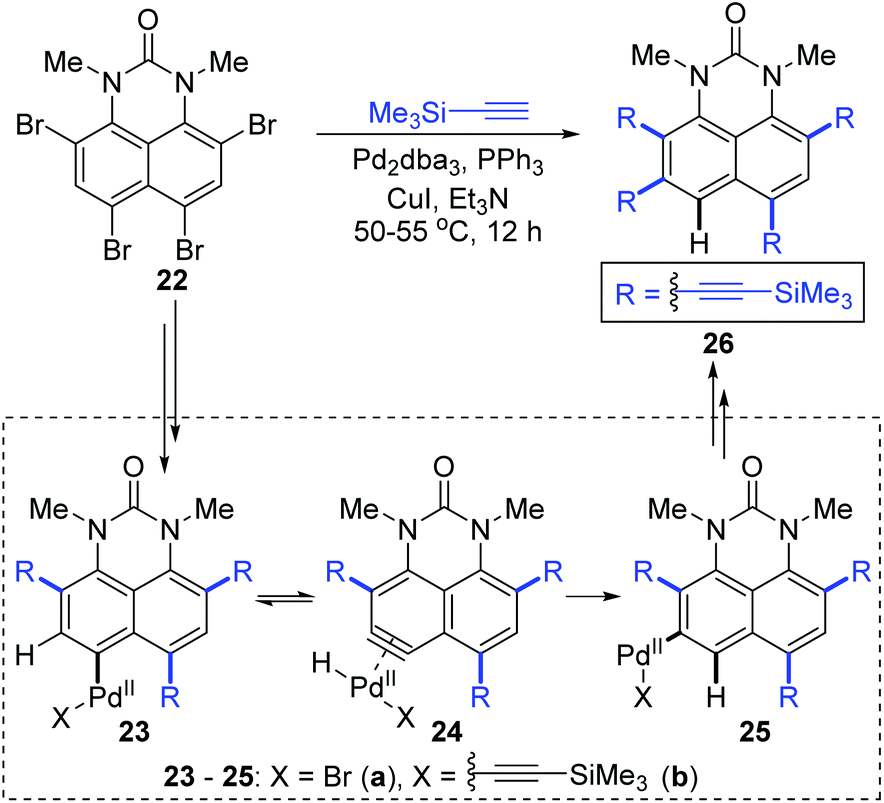
In 2016, Gulevskaya and co-workers reported a palladium-catalyzed reaction to synthesize a series of alkynyl derivatives of 1,3-dialkylperimidones.8 A previously unknown 1,2-migration of the Pd(II) group on the phenyl ring was observed when they dealt with the coupling of tetrabromide 22 with trimethylsilylacetylene (Scheme 7). Presumably, the formation of 26 originated from the 1,2-shift of the Pd(II) group in either intermediate 23a or 23b. And the motive force for such a migration should come from the harsh steric tension; therefore the PdBr group looked more likely to go through this migration. Inspired by 1,2-migrations in the Pd(II) alkenyl complexes,7a an aryne intermediate 24a was supposed to be generated via β-hydride elimination. The subsequent regioselective hydropalladation would furnish the migratory aryl palladium species 25a, which afforded the final product 26via Sonogashira coupling reaction.
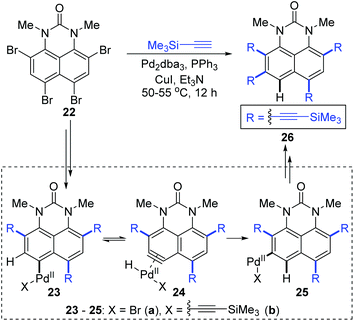 |
| | Scheme 7 An example of 1,2-Pd migration in Sonogashira coupling. | |
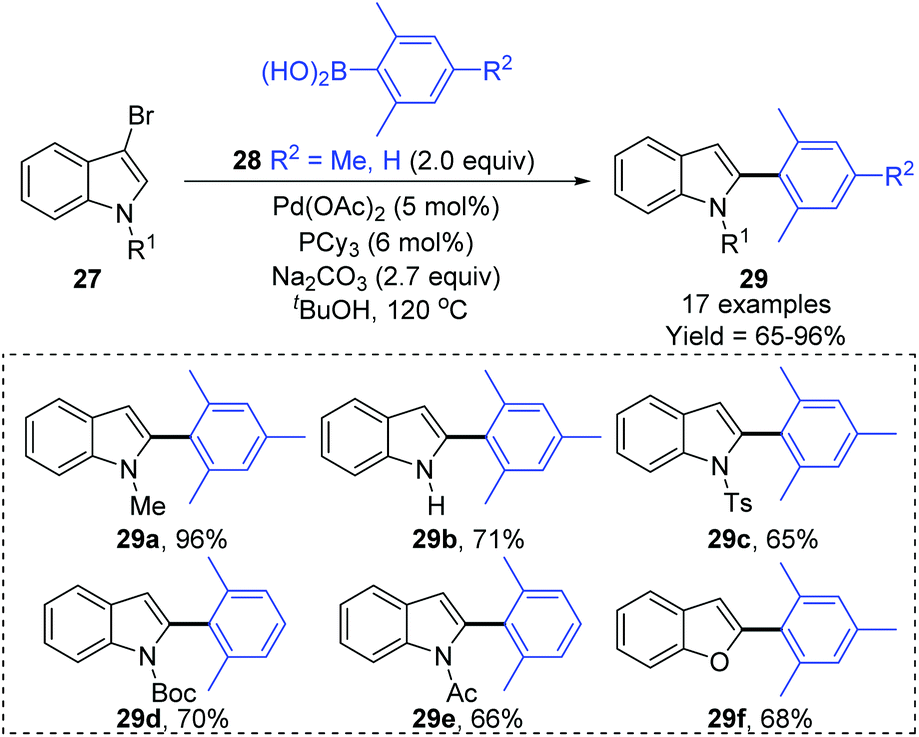
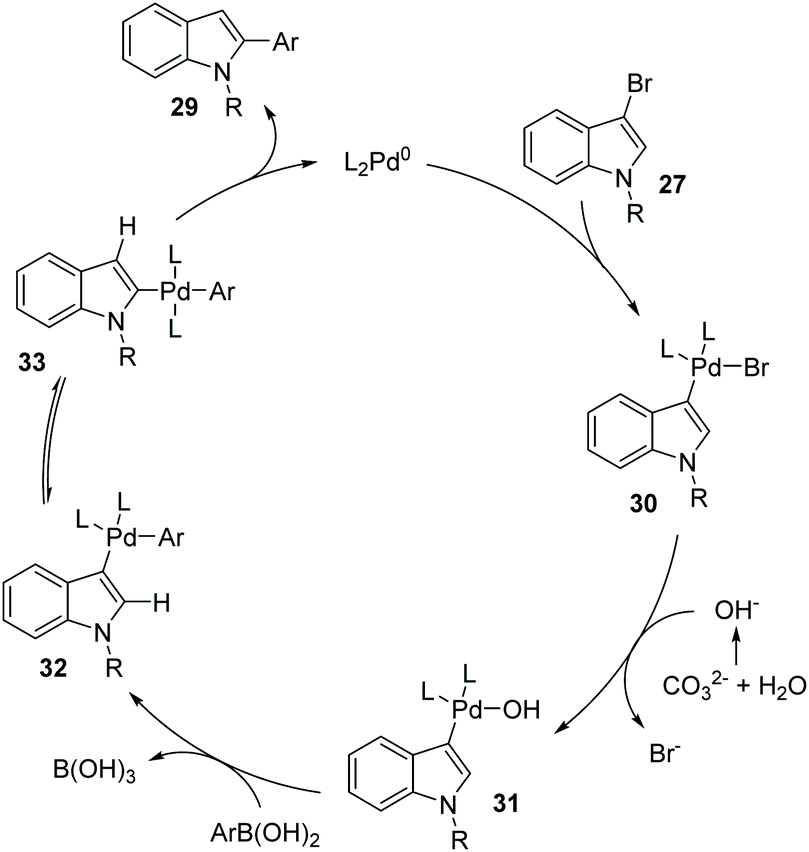
Later, a synthetic method for the preparation of 2-arylindoles involving a 1,2-palladium migration process was reported by the Yue group (Scheme 8).9 A series of 2-arylindoles 29 were obtained in moderate to high yields via Suzuki coupling reaction of 3-bromoindoles 27 with hindered boronic acid 28 catalyzed by Pd(OAc)2/PCy3. The steric hinderance of mesitylboronic acids was very crucial for this 1,2-migration, and no migratory product was observed in the presence of less hindered boronic acids. The basicity of the base also significantly affected the selectivity and yield, and weaker base gave a better performance. The proposed catalytic cycle is shown in Scheme 9. Initially, an aryl-palladium intermediate 30 was generated in situ via the oxidative addition of 27 to Pd(0). The sequential “oxo-palladium” pathway and transmetalation delivered the intermediate 32, which was compelled to undergo 1,2-palladium migration due to the effect of the aryl steric hindrance and the ligand. Finally, the reductive elimination of the thermodynamically more stable 2-palladium species 33 afforded the 1,2-migrated products 29.
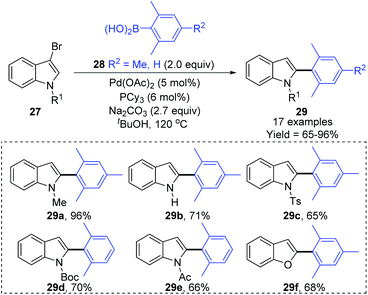 |
| | Scheme 8 Yue's 1,2-Pd migration strategy in the Suzuki coupling of indoles. | |
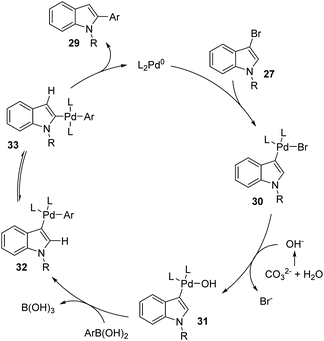 |
| | Scheme 9 Proposed mechanism for the Suzuki coupling of indoles via 1,2-Pd migration. | |
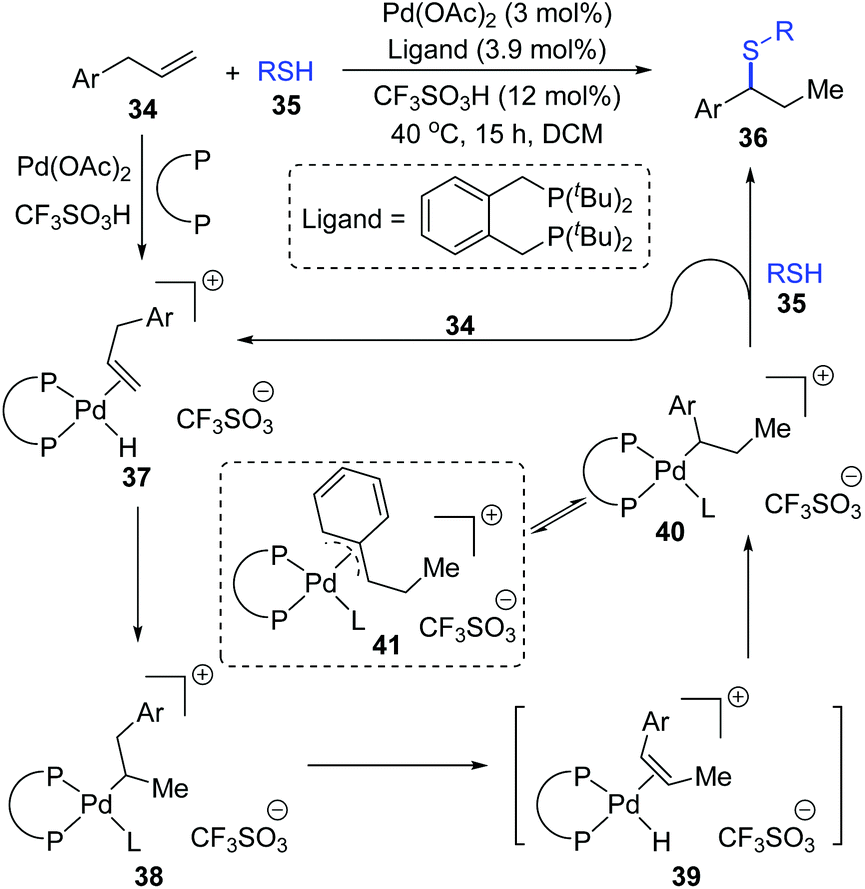
Moreover, a possible 1,2-palladium migration is also observed in the work of the Fleischer group.10 As shown in Scheme 10, a tandem olefin migration/hydrothiolation of allyl benzenes was achieved, which was facilitated by an in situ generated palladium hydride 37. In the proposed catalytic cycle, a 1,2-palladium migration could take place from intermediate 38 to 40via the elimination/coordination/insertion sequence. The more stable η3-coordination mode of species 41 might be responsible for the migratory motivation. In this procedure, coordination of thiol to a palladium atom (L in 38, 40, and 41) might occur in advance to account for the final formation of a C–S bond; alternatively, direct substitution between 40 and 35 was also feasible.
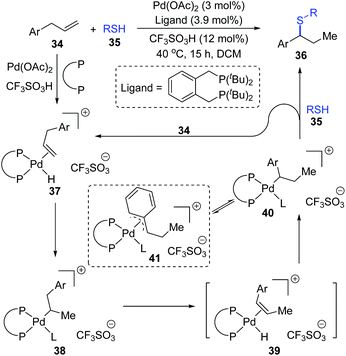 |
| | Scheme 10 Hydrothiolation of allyl benzenes via 1,2-palladium migration. | |
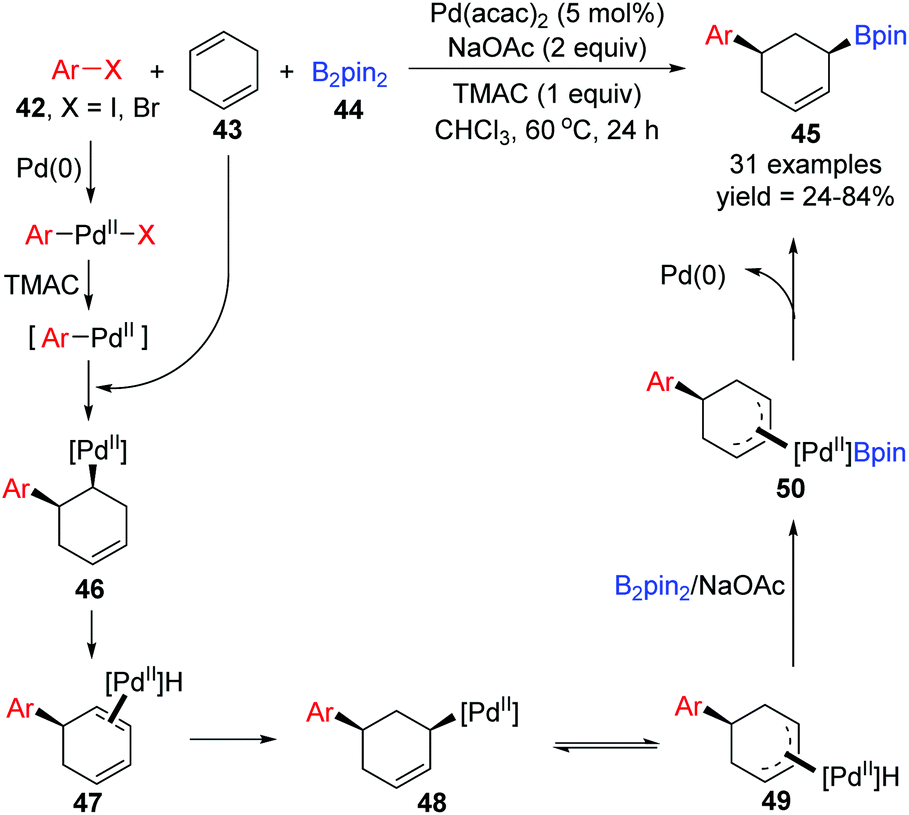
Recently, the Yin group achieved a novel palladium-catalyzed arylboration of 1,4-cyclohexadienes 43, which allowed expedited access to diverse functionalized 1,3-disubstituted cyclohexanes 45 from readily available starting materials (Scheme 11).11 Notably, this three-component reaction did not rely on the application of dative ligands, but a cheap ammonium chloride salt (TMAC) instead, and the 1,3-difunctional product 45 prepared via this protocol possessed exclusive cis-diastereoselectivity. In the proposed catalytic cycle, the intermediate Ar–PdII–X, generated from oxidative addition of aryl halides 42 with Pd(0), would further transform into a new Pd(II) species Ar–PdII in the presence of TMAC, which was inserted by 1,4-cyclohexadienes 43 to afford intermediate 46. Then the intermediate 46 rapidly underwent β-H elimination and migratory insertion. The excellent stereoselectivity observed in the products revealed that the Pd–H did not dissociate from the substrate 47 in this process. It seemed that the migratory force could be explained by a more stable π-allylPd(II) intermediate 49. Finally, sequential transmetalation with B2pin2 and reductive elimination gave the target products 45.
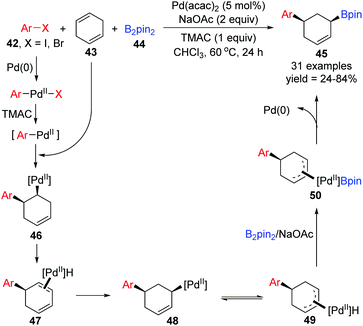 |
| | Scheme 11 Palladium-catalyzed 1,3-arylboration of unconjugated dienes. | |
2.2 1,3-Palladium migration
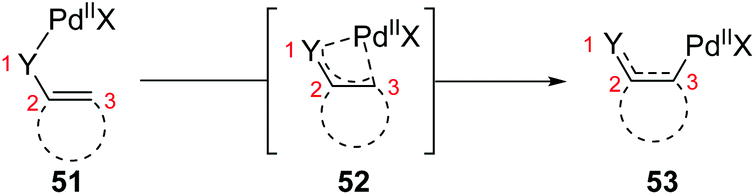
So far, transformation involving 1,3-palladium migration is just an emerging research project in the field of metal migration, and the relevant mechanism study is still in its infancy. Hence the elaborate mechanisms of 1,3-palladium migration are not well explored. Based on the current reports, structures of allyl palladium seem to be significant for the 1,3-migration, so it can be speculated that the Pd-migration may proceed via an intermediate 52 similar to π-allylpalladium (Scheme 12). However, there is no concrete evidence to prove this process, and further mechanism investigation experiments are essential. Moreover, several existing examples proposed as 1,3-Pd shifts seem to be indistinguishable with the π-allylic palladium chemistry, such as the following first example. Therefore, getting direct evidence for defining a 1,3-palladium migration involved strategy is another imperative in future research.
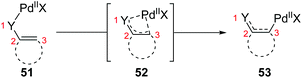 |
| | Scheme 12 Possible procedure of 1,3-palladium migration. | |
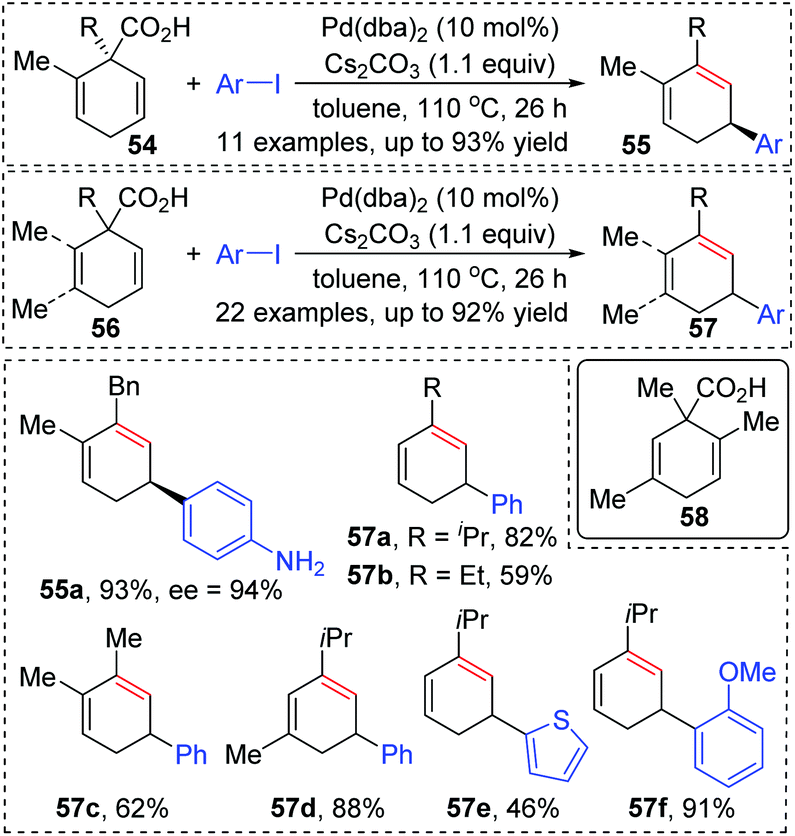
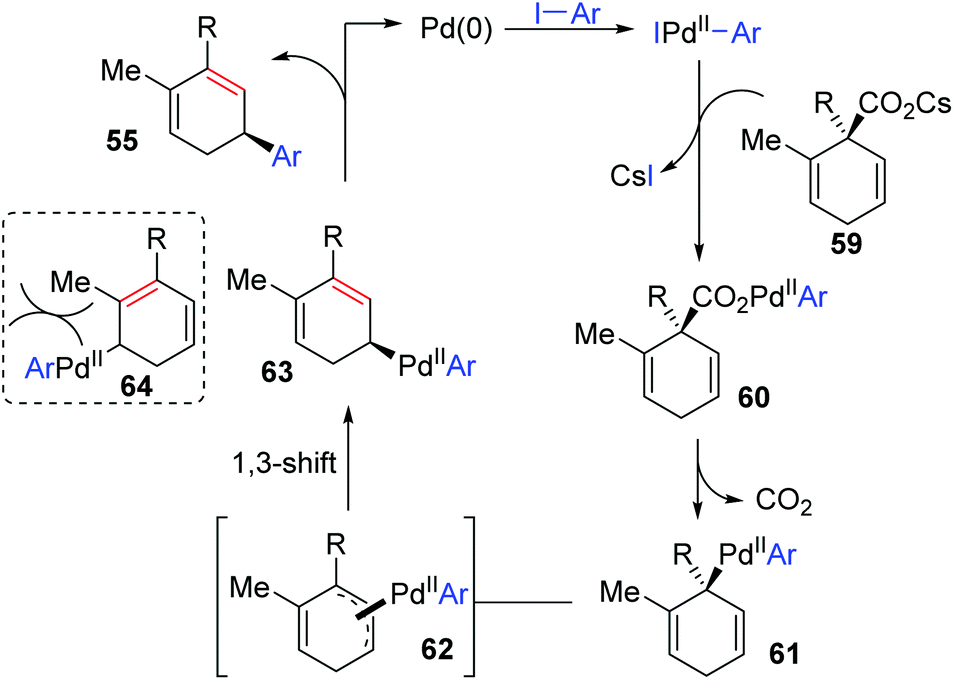
In 2011, Studer and co-workers developed a palladium-catalyzed coupling of 2,5-cyclohexadiene-1-carboxylic acid derivatives with aryl iodides, achieving stereoselective C(sp3)–C(sp2) bond formation (Scheme 13).12 Chiral substrates 54 were converted into the corresponding dienes 55 stereospecifically, while racemic products 57 were obtained from achiral substrates 56. The steric hindrance of the R group has a significant effect on this transformation. For instance, substrates possessing isopropyl groups (R = i-Pr) with larger steric hindrance than the ethyl group (R = Et) afforded the corresponding products in higher yield (57a and 57b). When an additional methyl group was installed at either the 2- or 3-position of the substrate 56, palladium migration took place preferentially on the side of the olefin without substituent (57c and 57d). However, the 2,5-dimethyl substrate 58 did not give the corresponding product due to steric reasons. The proposed catalytic cycle is illustrated in Scheme 14. Taking chiral substrates 54 as an example, I–Pd(II)–Ar species were generated via oxidative addition of aryl iodide to Pd(0). Then ligand exchange between I–Pd(II)–Ar and cesium salt 59 could form intermediate 60, which further provided 2,5-cyclohexadienyl palladium species 61 with retention of stereochemistry via a decarboxylation process. Stereospecific 1,3-Pd migration via a π-allylpalladium intermediate 62 delivered the migratory species 63. Final reductive elimination led to target chiral product 55 with the release of Pd(0). Additionally, the existence of regioisomeric intermediate 64 could not be entirely excluded; however, intramolecular tension makes it hard to compete with 63. Furthermore, even if the intermediate 64 is generated, it is more likely to reconvert into 63via a 1,3-shift process.
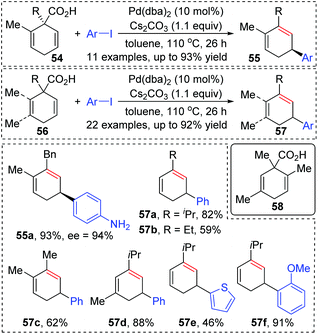 |
| | Scheme 13 Pd-Catalyzed coupling of 2,5-cyclohexadiene-1-carboxylic acid derivatives with aryl iodides. | |
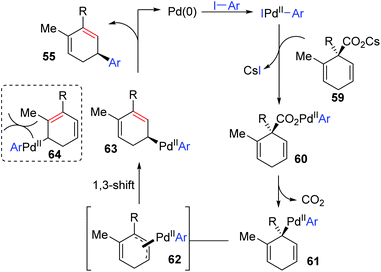 |
| | Scheme 14 Suggested mechanism for the reaction of 2,5-cyclohexadiene-1-carboxylic acids with aryl iodides. | |
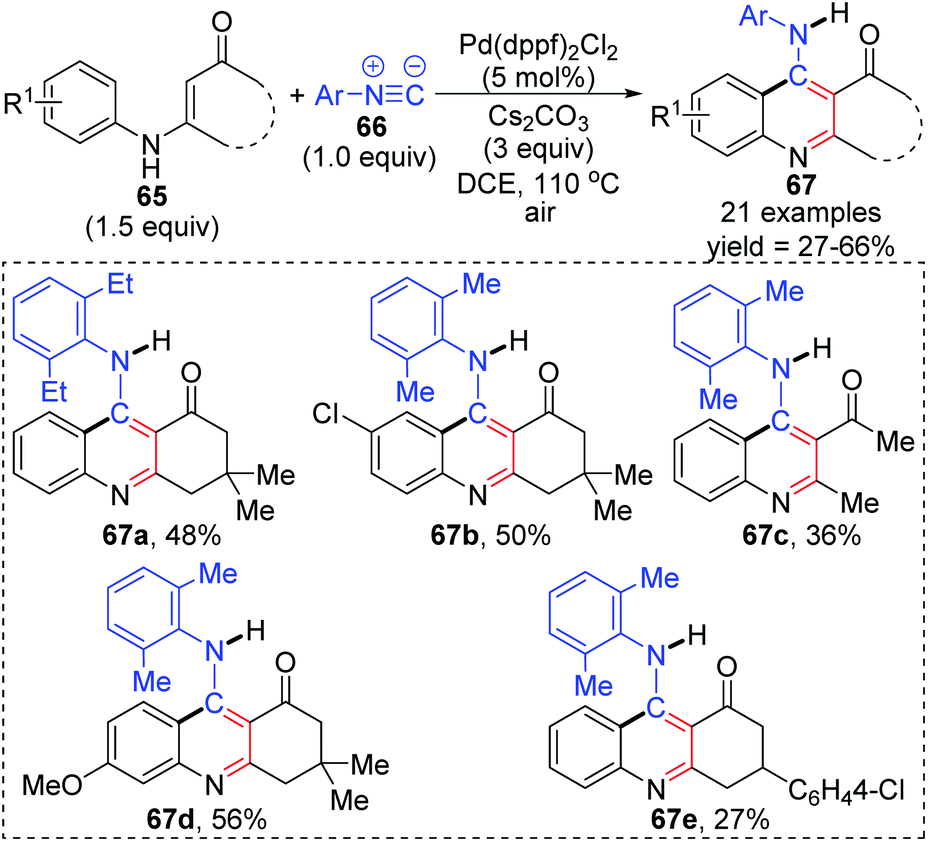
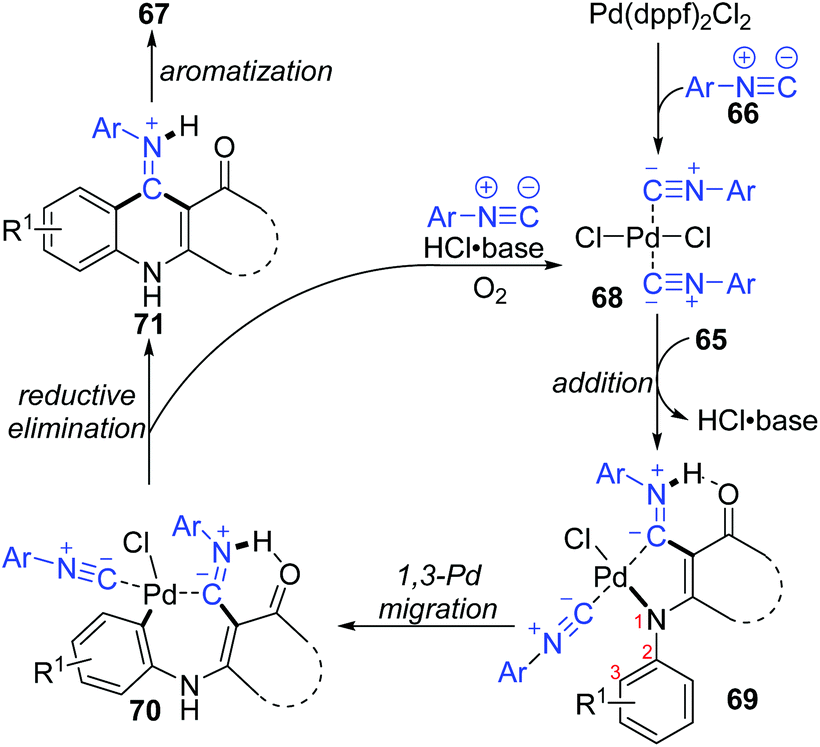
In 2015, a 1,3-palladium migration involved novel synthetic methodology for preparing 4-aminoquinoline derivatives 67 was reported by the Wang and Ji group (Scheme 15).13 This protocol employed diverse enaminones 65 and isocyanides 66 as the reaction substrates to give the corresponding products in moderate yields, while the annulation did not take place with enaminones bearing a strong electron-withdrawing group in the benzene ring, such as nitro. On the basis of exploration experiments, a plausible reaction mechanism was proposed for this 1,3-migration process (Scheme 16). Initial ligand exchange between Pd(dppf)2Cl2 and isocyanide 66 generated a bis(isocyanide)palladium(II) complex 68. Subsequent addition of enaminones 65 to complex 68 afforded the Pd(II) intermediate 69 with a five-membered ring transition structure. Then, a seven-membered palladacycle 70 was furnished via 1,3-Pd shift from the amine to the adjacent aryl C–H bond. And the results of the kinetic isotope effect revealed that the cleavage of the aryl C–H bond by 1,3-palladium migration is not the rate-determining step. Subsequent reductive elimination led to the acidic intermediate 71, which was unstable and converted into the desired cyclization product 67 rapidly under basic conditions via aromatization. The activated Pd(II) species 68 was regenerated by air oxidation in the presence of hydrochloride and isocyanide simultaneously.
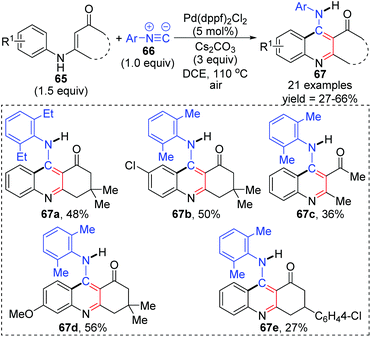 |
| | Scheme 15 Synthesis of 4-aminoquinoline derivatives via 1,3-palladium migration. | |
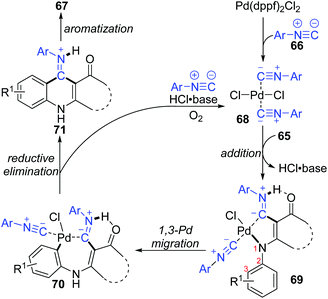 |
| | Scheme 16 Possible mechanism for preparing 4-aminoquinolines from enaminones and isocyanides. | |
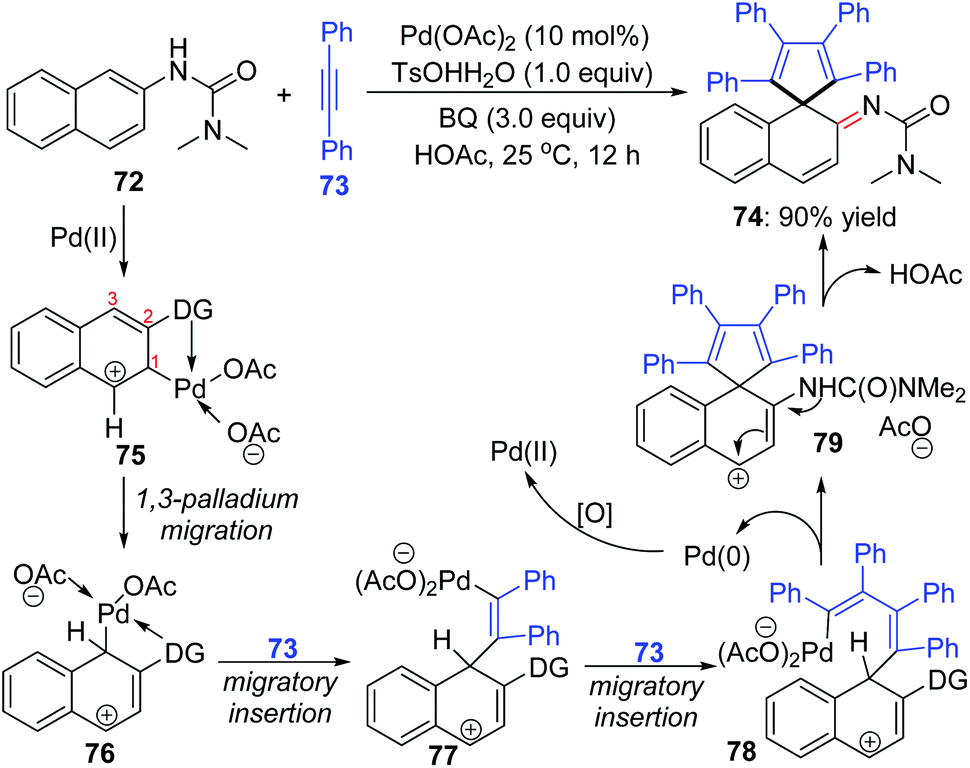
An interesting 1,3-palladium migration process was also observed in Wang's strategy of synthesizing imine derivatives via dearomatization reaction of N-aryl ureas with internal alkynes.14 The Pd-migration occurred when 2-naphthyl ureas 72 were employed as the substrates, and a proposed catalytic pathway to shed light on the migrated product formation is illustrated in Scheme 17. Based on the primary mechanism investigations, an intermediate 75 should be generated preferentially from electrophilic attack of the directing-group-coordinated Pd(OAc)2 to the 3-position of 2-naphthyl urea 72. The sequential 1,3-shift of the Pd center generated intermediate 76, which was probably thermodynamically more stable due to the stabilization of the C–Pd bond with an adjacent phenyl group. Intermediates 77 and 78 were afforded via migratory insertion of alkyne 73. Allyl sp3 C–H olefination of 78 delivered the spirocyclic compound 79, accompanied by Pd(0) release. The final migrated product 74 was generated by deprotonation of 79.
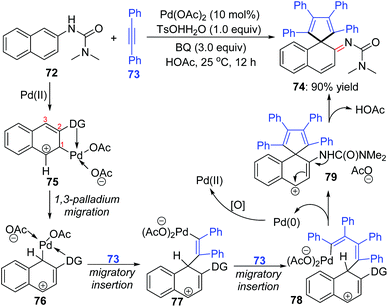 |
| | Scheme 17 1,3-Palladium migration in synthesizing imine derivatives. | |
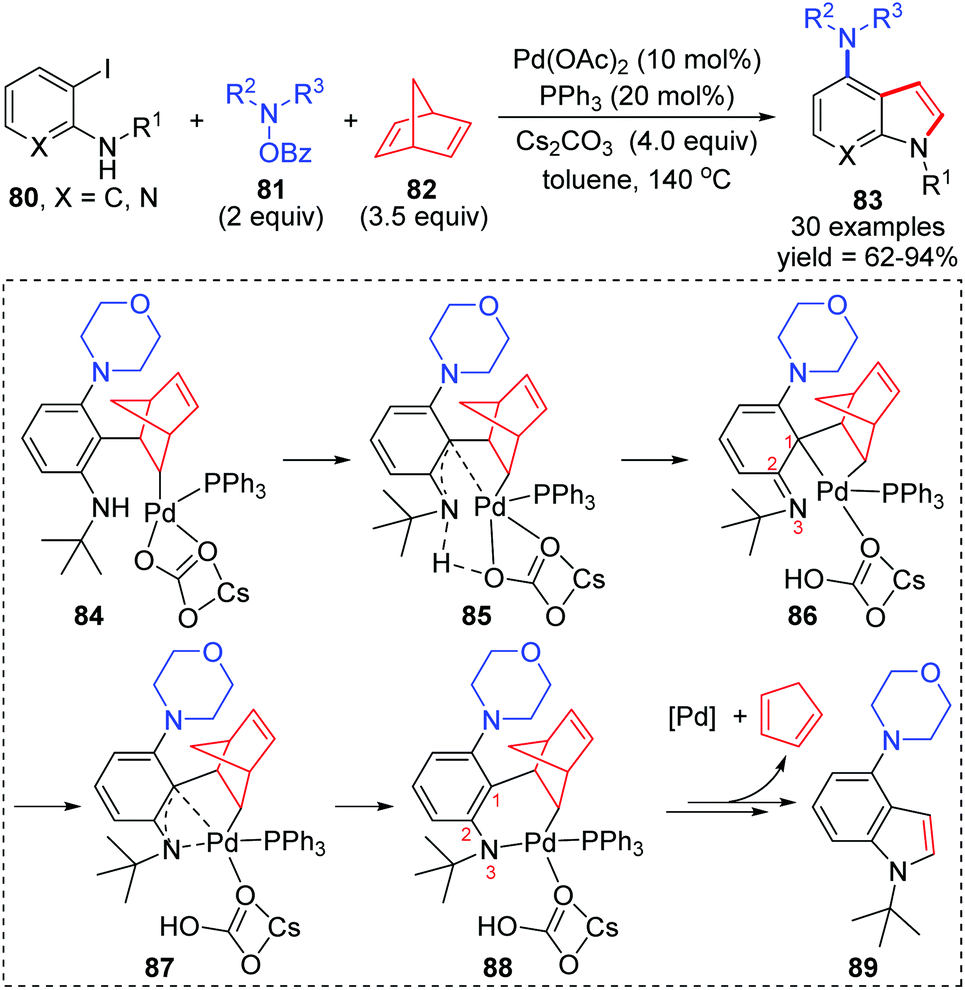
In 2019, the Liang group reported a three-component cross-coupling of o-iodoaniline 80, N-benzoyloxyamines 81, and norbornadiene 82 to access highly functionalized 4-aminoindoles 83 (Scheme 18).15 The Catellani and retro-Diels–Alder strategy was employed in this domino process. And this method also exhibited good industrialization potential by a gram-scale reaction investigation (79% yield of isolated product). On the basis of density functional theory calculations, the authors pointed out that a 1,3-palladium migration process should be involved in the intramolecular Buchwald coupling of this reaction. Details are also illustrated in Scheme 18. Firstly, the coordination of benzene with palladium might cause the carbonyl group of the carbonate to leave and generate an imine (from 84 to 86). Then, 1,3-Pd migration occurred to form intermediate 88via a π-allylicpalladium analogue 87, which further released the Pd catalyst and 1,3-cyclopentadiene to afford the target product 89. However, it is important to note that the process from intermediates 84 to 86 was merely proposed according to the computation studies, which was not supported by experimental results. Hence, the subsequent 1,3-Pd migration was only assumed by the authors to rationalize the calculations.
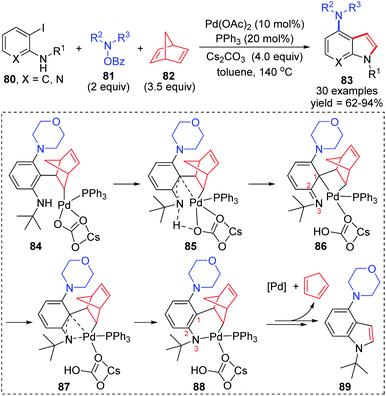 |
| | Scheme 18 1,3-Palladium migration that occurred in intramolecular Buchwald coupling. | |
2.3 1,4-Palladium migration
1,4-Palladium migration is the most widely investigated strategy among metal-migration involving reactions (Scheme 19). Due to its relatively stable transition state of a five-membered ring, such as species 91, 92, and 93, 1,4-palladium migration is quite common in organometallic chemistry compared with other migratory types. The strategy of 1,4-migration can be widely used to metalate a remote C–H bond, generating a new organometallic species that is difficult to achieve by other conventional approaches. Numerous elegant methodologies involving various types of 1,4-palladium migrations, such as aryl-to-vinyl, vinyl-to-aryl, aryl-to-aryl, aryl-to-benzyl etc., have been developed over the past decade. In this section, only complementary reports are summarized according to the previously reported reviews;2–4 more strategies toward 1,4-Pd migration can be found in the referenced literature.
 |
| | Scheme 19 Possible transition state of 1,4-palladium migration. | |
2.3.1 Aryl-to-vinyl 1,4-palladium migration.
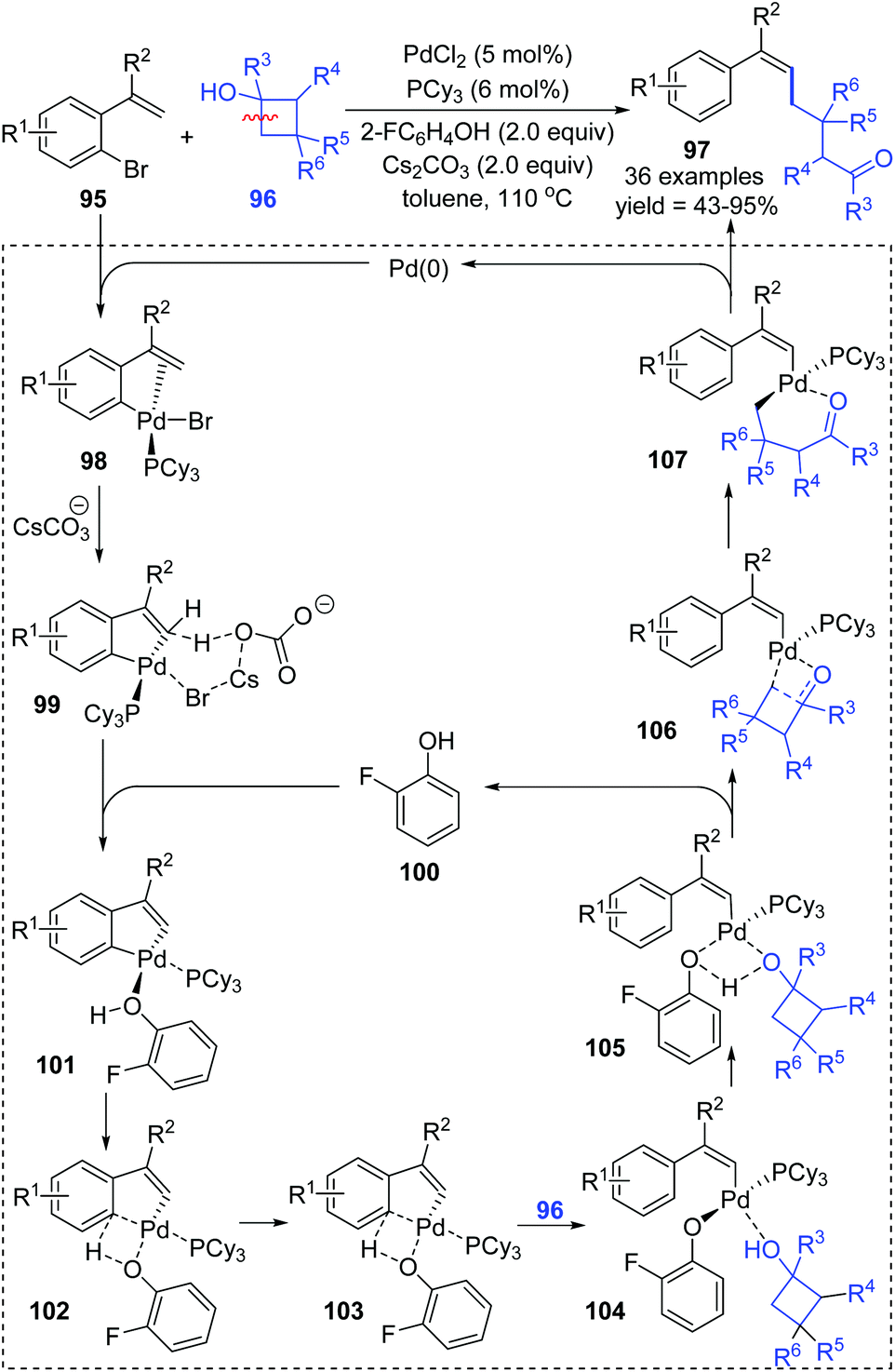
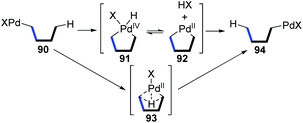
In early 2020, the Yu and Zhou group achieved a Pd-catalyzed C–H alkylation of gemdisubstituted ethylenes 95via an aryl to vinyl 1,4-palladium migration/ring-opening C–C cleavage process (Scheme 20).16 The trisubstituted alkenes 97 were afforded in high yields with good regioselectivity, broad substrate scopes, and good functional group tolerance. An elaborate catalytic cycle, proposed based on mechanistic study and DFT calculations, is illustrated in Scheme 28. After oxidative addition of Pd(0) to the C–Br bond (95), a subsequent 1,4-palladium migration process occurred from the aryl to vinyl position via an intermolecular hydrogen atom transfer (101 to 104). Coordination of cyclobutanol 104 to the palladium center caused its hydroxyl hydrogen to shift to the 2-F-phenoxy functionality, readily removing 2-FC6H4OH from the metal center (105 to 106). Then, a ring-opening C–C cleavage process of 106 afforded the thermally stable intermediate 107, which further delivered the final product 97 by reductive elimination.
 |
| | Scheme 20 Pd-Catalyzed coupling reaction via an aryl to vinyl 1,4-palladium migration/ring-opening C–C cleavage process. | |
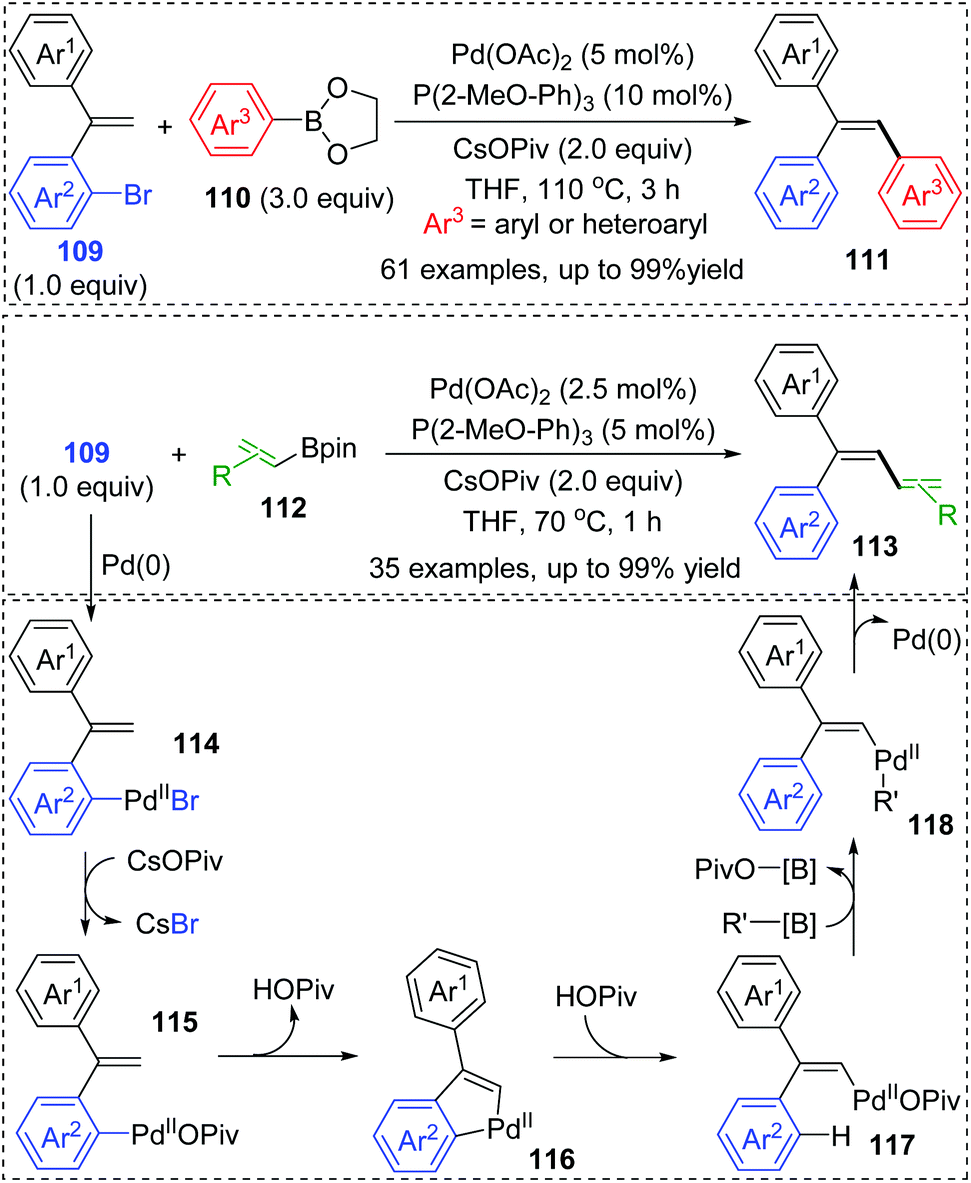
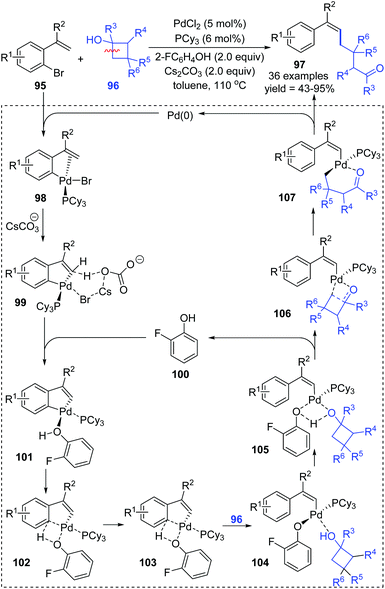
Very recently, a novel Suzuki–Miyaura coupling enabled by aryl to vinyl 1,4-palladium migration was achieved by the Feng and Lin group (Scheme 21).17 Diverse stereo-defined multisubstituted olefins 111 were synthesized from coupling of ortho-vinyl bromobenzene 109 with aryl/heteroaryl boronate esters 110. On the other hand, ortho-vinyl bromobenzene 109 also reacted with alkenyl boron reagents 112 to prepare a series of 1,3-dienes 113 efficiently. Good yields, broad substrate scope, and excellent functional-group tolerance were observed in these transformations. The tentative catalytic cycle is proposed in Scheme 29. The exchange of the bromide anion with PivO− occurred to aryl-Pd(II)Br intermediate 114 to form a new Pd(II) species 115. Then, the following C–H activation energetically favored a five-membered palladacycle(II) 116via a concerted metalation–deprotonation mechanism. Subsequent proton transfer with the aid of HOPiv delivered the palladium-migratory species 117, which further underwent transmetalation with organoboron reagents to furnish intermediate 118. Final reductive elimination led to the target product 111 or 113 and regeneration of Pd(0).
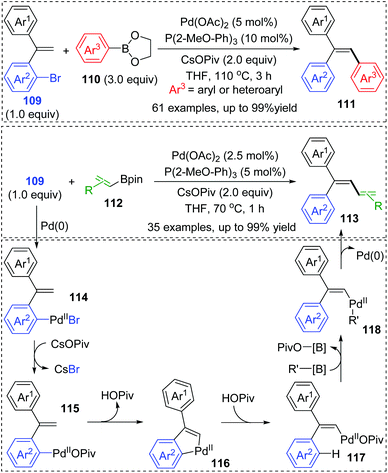 |
| | Scheme 21 Pd-Catalyzed Suzuki–Miyaura coupling via aryl to vinyl 1,4-palladium migration. | |
2.3.2 Vinyl-to-aryl 1,4-palladium migration.
According to the current reports, vinyl to aryl 1,4-Pd migration usually occurs to a vinyl palladium intermediate formed in situ via insertion of a C–Pd bond into an alkyne. Similar to other 1,4-Pd shifts, the metathesis between the C(sp2)–Pd bond of the vinyl and C(sp2)–H bond of the aryl also takes place via a five-membered transition state.
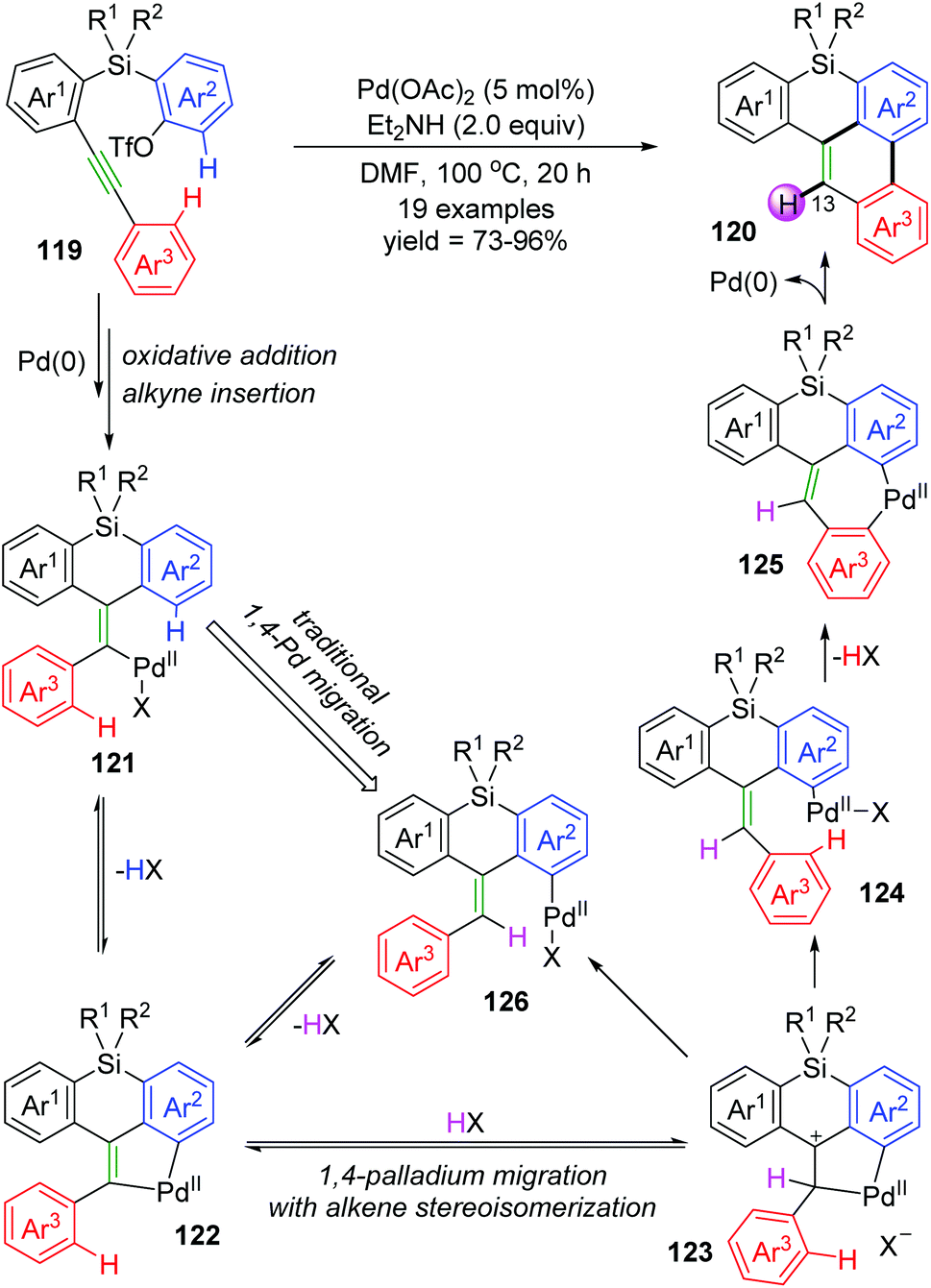
Recently, Shintani and co-workers developed a Pd-catalyzed intramolecular cyclization to prepare 8H-benzo[e]phenanthro[1,10-bc]silanes 120 from easily accessible 2-((2-(arylethynyl)aryl)silyl)aryl triflates 119 under simple reaction conditions.18 As shown in Scheme 22, two C–H bonds on Ar2 and Ar3 of substrate 119 were cleaved to form a C–C bond between the two rings, and a new C–H bond was generated at the 13-position of the resulting product 120. However, several deuterium-labelling experiments revealed that the H atom at the 13-position was derived from an external hydrogen donor (Et2NH and/or residual water in the solvent) instead of the Ar2 group. According to these results, a new mode of 1,4-palladium migration involving concomitant alkene stereoisomerization was preliminarily established as illustrated in Scheme 22. Initial successive oxidative addition of aryl triflate 119 to palladium(0) and insertion of alkyne generated the alkenylpalladium 121. Then, C–H bond activation on Ar2 led to a five-membered palladacycle 122, which underwent protonation by an external proton donor to form a cationic species 123. Subsequent cleavage of the alkyl–Pd bond with accompanying formation of a Z alkene afforded the key intermediate 124, which could further undergo C–H bond activation to generate a seven-membered palladacycle 125. Final reductive elimination gave the target product 120, and regenerated the Pd(0) catalyst. Nevertheless, the traditional 1,4-palladium migratory product 126 could not be excluded for the catalytic cycle.
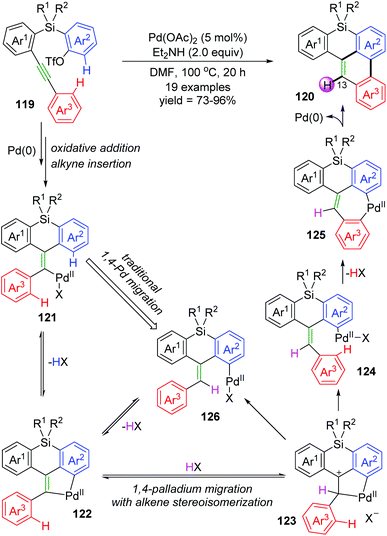 |
| | Scheme 22 Shintani's 1,4-Pd migration with concomitant alkene stereoisomerization. | |
2.3.3 Alkyl-to-aryl 1,4-palladium migration.
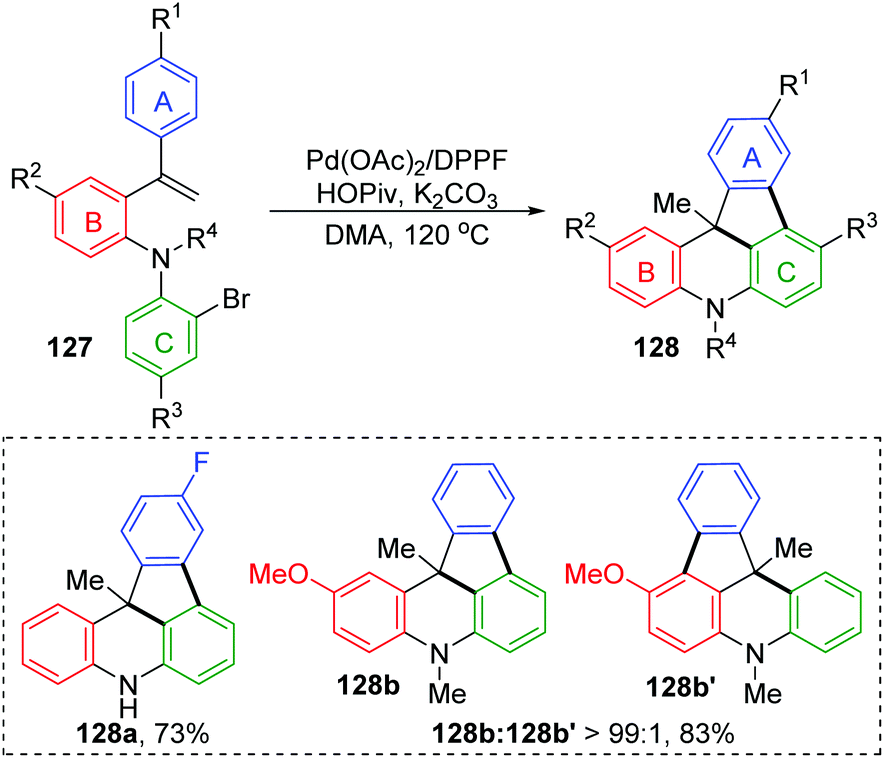
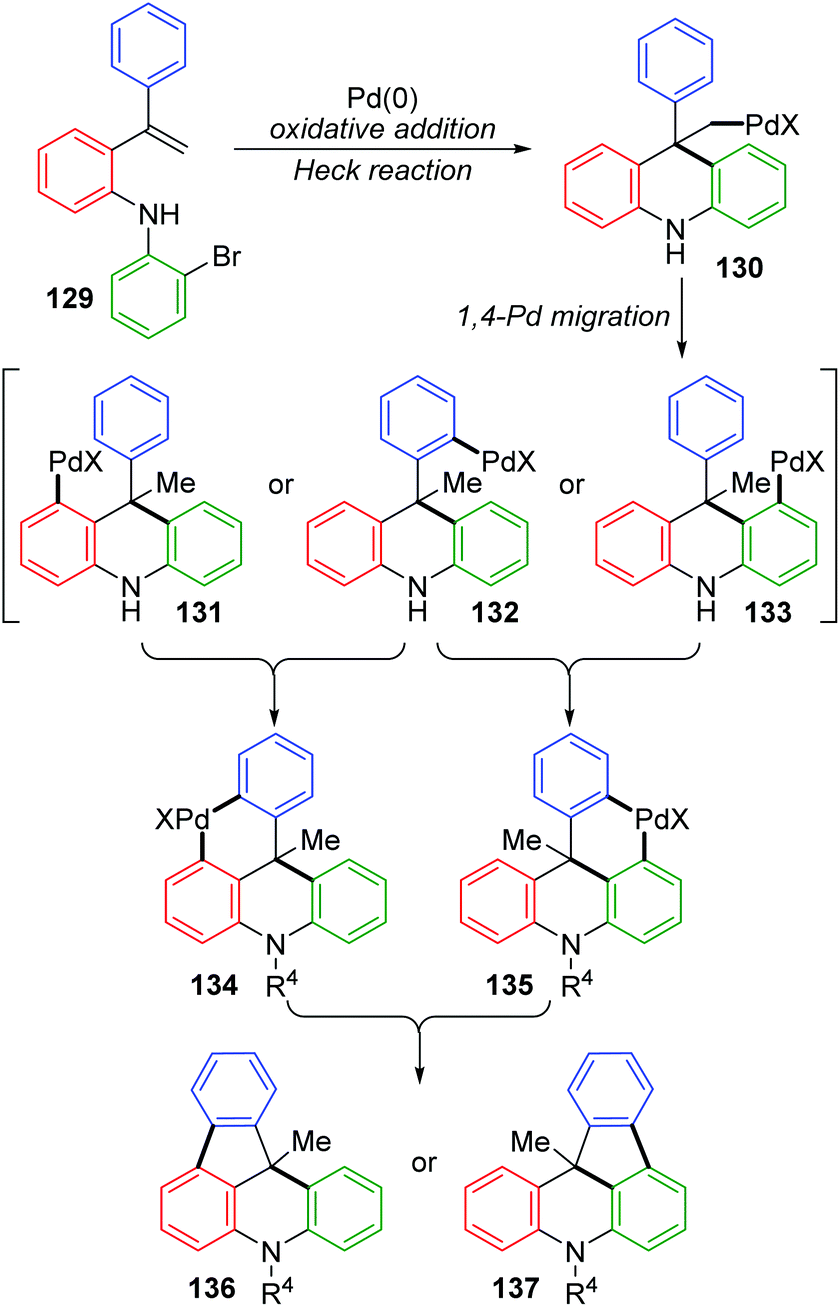
The Wang and Ji group achieved a palladium-catalyzed strategy for the synthesis of dihydroindeno [1,2,3-kl]acridine derivatives 128 (Scheme 23).19 The reaction afforded a five-membered ring and six-membered ring simultaneously via a 1,4-palladium migration process. Two regioisomers could be produced, while there were substituents on the rings B or C, such as 128b and 128b′. A possible reaction pathway is illustrated in Scheme 24. Successive oxidative addition of 129 to Pd(0) and intramolecular Heck addition afforded species 130, which could activate the ortho aromatic C–H bond on the A, B or C rings. Then, 1,4-palladium migration occurred to provide three regioisomers 131, 132 or 133. A six-membered palladacycle 134 or 135 was generated via intramolecular cyclization. Finally, the desired products were obtained as a result of reductive elimination, accompanied by reborn Pd(0) catalyst.
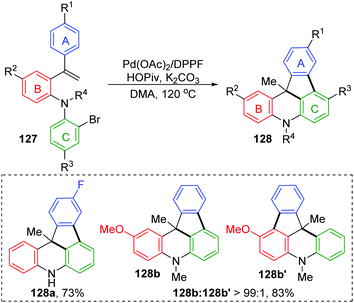 |
| | Scheme 23 Pd(0)-Catalyzed reaction for the synthesis of dihydroindeno[1,2,3-kl]acridines. | |
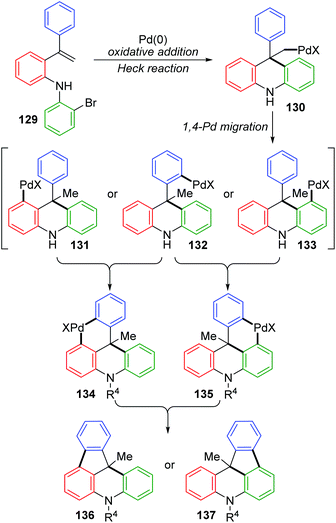 |
| | Scheme 24 Plausible mechanism for the Pd-catalyzed synthesis of dihydroindeno[1,2,3-kl]acridines. | |
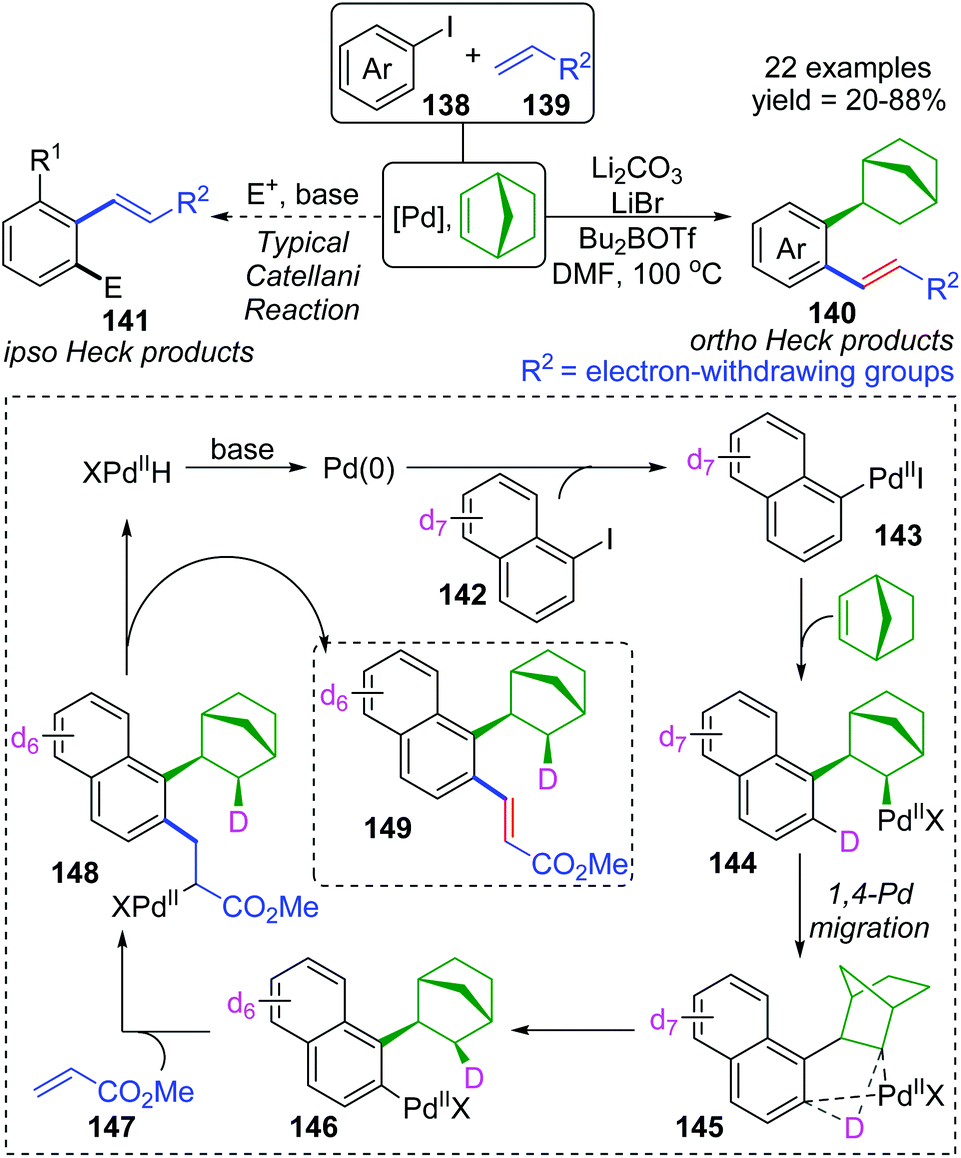
For the palladium/norbornene-catalyzed Catellani reaction,20 it is well-known that multiple types of vicinal functionalized arenes can be achieved via the aryl-norbornyl-palladacycle (ANP) intermediate. Recently, Rago and Dong discovered an unexpected ortho-Heck reaction via 1,4-palladium migration under Catellani reaction conditions (Scheme 25).21 In the presence of lithium salts, aryl iodide 138 reacted with electron-deficient olefins 139 to furnish the ortho Heck products 140 instead of commonly observed ipso Heck products 141 in Catellani-type reaction; meanwhile, a norbornyl group was introduced at the ipso position of the arene. A possible reaction pathway involving 1,4-Pd migration was proposed according to a series of deuterium labeling studies. Taking deuterated substrate 142 as an example, initial oxidative addition and subsequent norbornene insertion generated the alkyl palladium species 144, just like the classical Catellani process. However, the 1,4-palladiuim shift took place instead of forming an ANP intermediate to afford the aryl palladium species 146, accompanied by full transfer of the D atom to norbornene. Results of an isotope crossover experiment also revealed that the 1,4-shift was an intramolecular procedure (145), on account of no deuterium exchange between molecules. Finally, the ortho Heck product 149 was furnished through a typical Heck reaction pathway; cis-migratory insertion (146 to 148) and cis-β-H elimination (148 to 149).
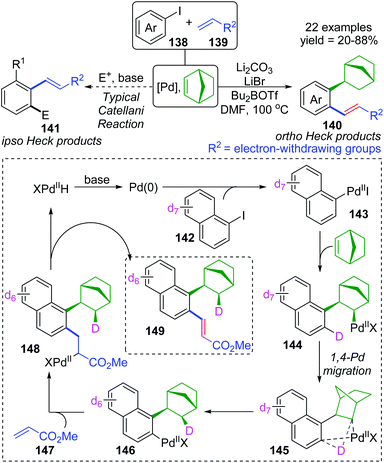 |
| | Scheme 25 Palladium/norbornene-catalyzed strategy toward an ortho-Heck reaction versus a typical Catellani reaction. | |
2.3.4 Aryl-to-aryl 1,4-palladium migration.
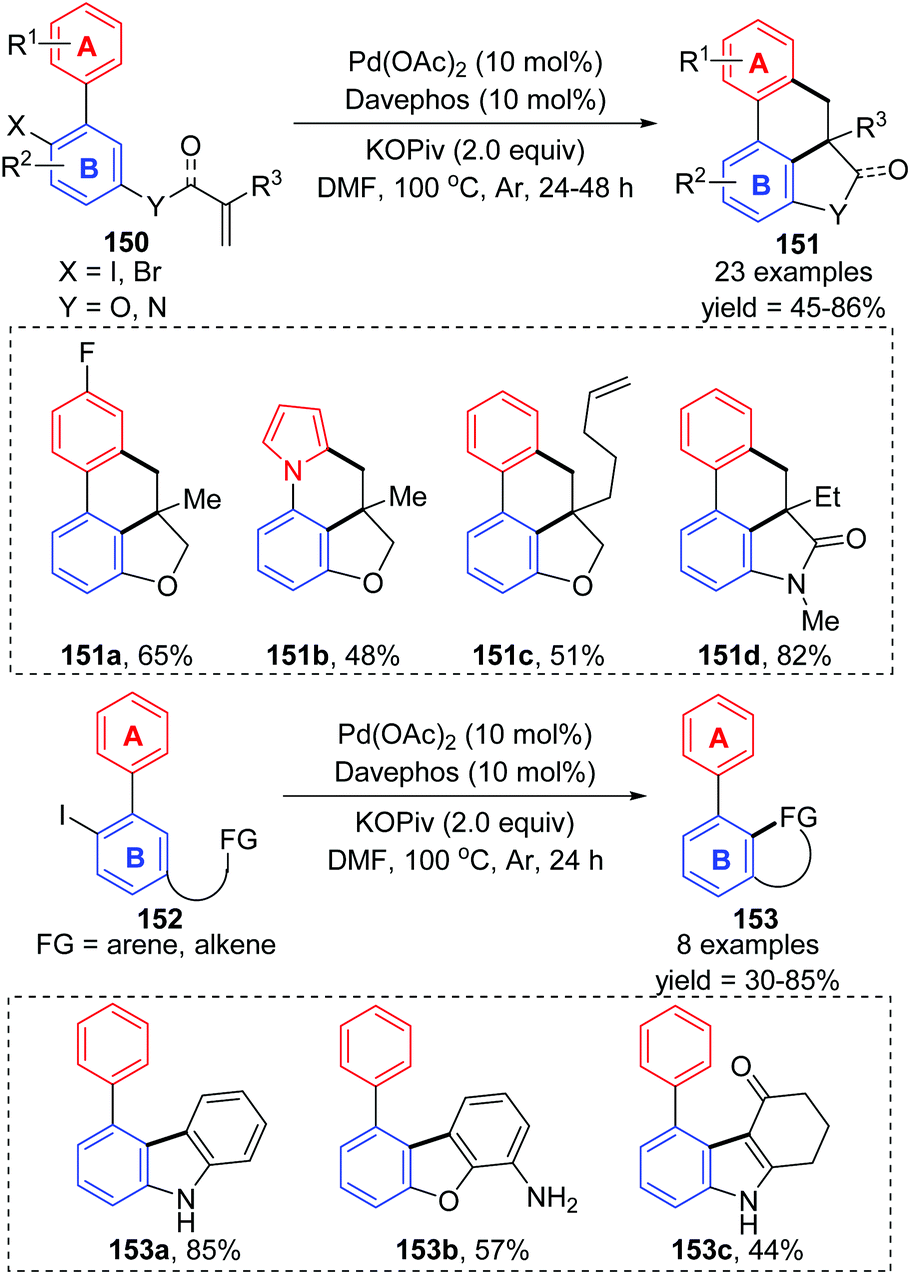
Recently, Lin and Yao et al. reported an intramolecular remote C–H activation via 1,4-palladium migration, delivering diverse polycyclic frameworks 151 and 153 in moderate to good yields (Scheme 26).22 The mechanism is similar to that described earlier in this section; successive oxidative addition, twice 1,4-palladium migration between ring A and ring B, alkene insertion, and a C–H activation process were responsible for the final product. Additionally, the authors also pointed out the reversibility of the 1,4-palladium migration between ring A and B.
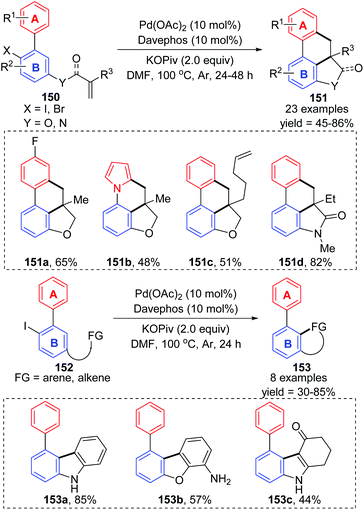 |
| | Scheme 26 Preparation of polycyclic frameworks via a 1,4-palladium migration pathway. | |
2.3.5 Aryl-to-acyl 1,4-palladium migration.
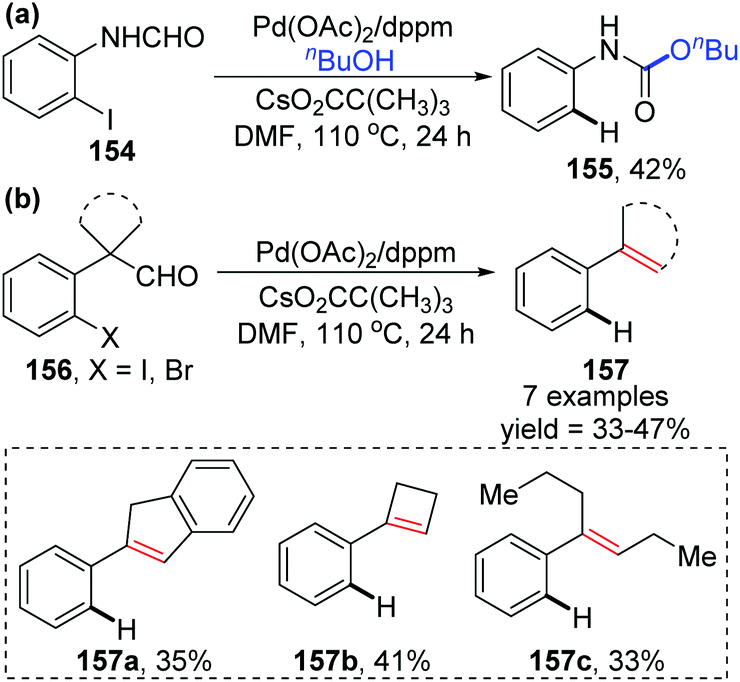
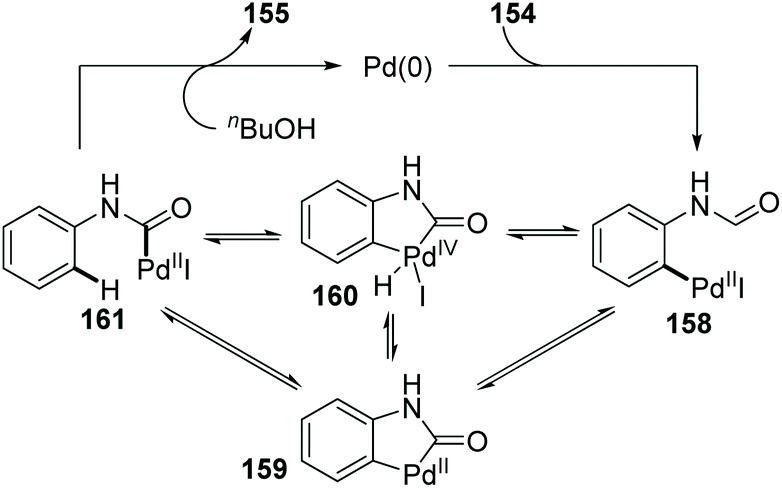
In 2009, the Larock group reported a novel strategy to activate an acyl C–H bond by employing different aldehydes as substrates, along with a simultaneous palladium shift from the aryl to acyl position (Scheme 27).23 This transformation offered a carbamate 155 in the presence of nucleophile butanol (Scheme 27a), or a decarbonylation/elimination product 157 in the absence of butanol (Scheme 27b). However, both of the two types of products were obtained in comparatively low yields. A possible reaction mechanism is described in Scheme 28. Taking the formamide 154 as an example, the initial oxidative addition of a carbon–halogen bond to Pd(0) formed an arylpalladium species 158, which could cyclize to Pd(II) intermediate 159 or further insert into the neighboring acyl C–H bond to afford Pd(IV) intermediate 160. The migrated acylpalladium species 161 could be generated from either 159 or 160, and then would be trapped by butanol to provide carbamate 155. Alternatively, in the absence of a nucleophile, the analogue of 161 would undergo decarbonylation and β-hydride elimination to deliver the Heck-type products 157.
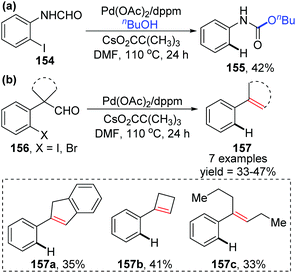 |
| | Scheme 27 Pd-Catalyzed strategy for the activation of an acyl C–H bond. | |
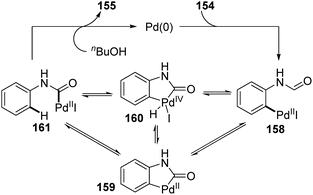 |
| | Scheme 28 Proposed catalytic cycle for the synthesis of carbamate. | |
2.3.6 Aryl-to-alkyl 1,4-palladium migration.
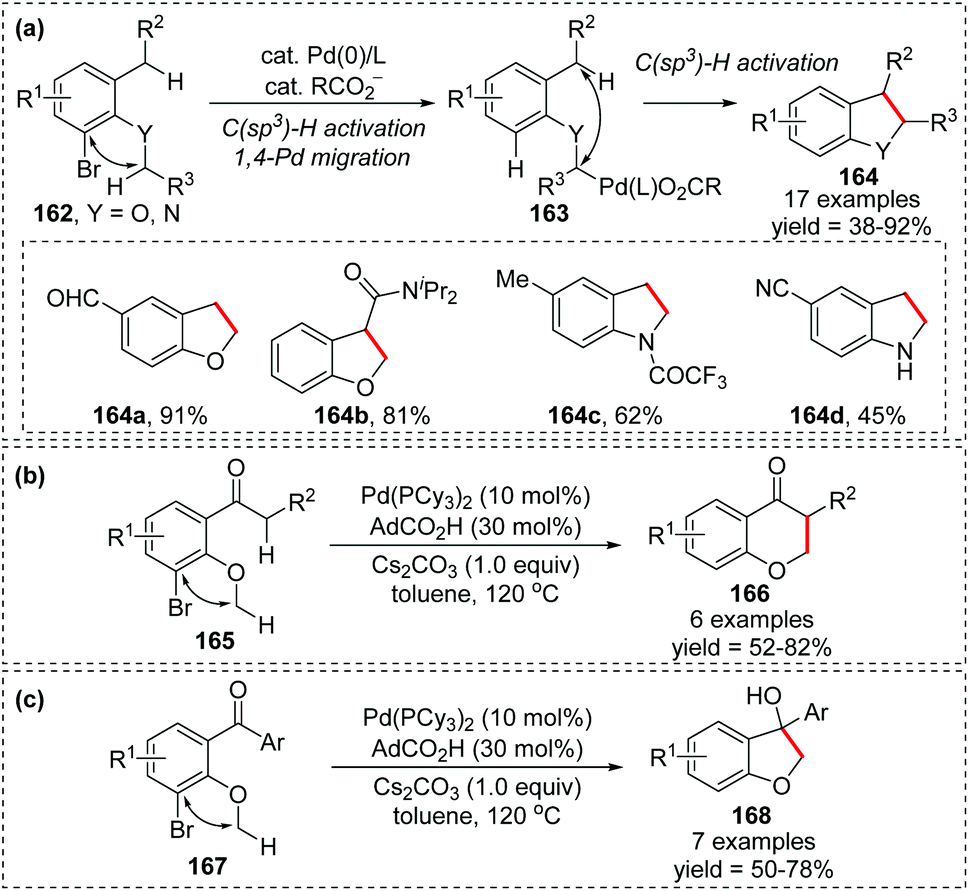
In 2019, a novel intramolecular coupling of two C(sp3)–H bonds via 1,4-Pd migration from aryl to alkyl was developed by Baudoin and co-workers (Scheme 29).24 This reaction proceeded under redox-neutral conditions, with the C–Br bond acting as an internal oxidant. Numerous 2,3-dihydrobenzofurans and indolines 164 were prepared in moderate to good yield (Scheme 29a), and a few 6-membered ring products 166 were also achieved as shown in Scheme 29b. Moreover, in the absence of an enolizable position, the migrated palladium intermediate could attack the ketone to afford a tertiary alcohol 168 (Scheme 29c). However, generally, this transformation is restricted to moderately acidic C–H bonds, adjacent to an oxygen or nitrogen atom on one side and benzylic or adjacent to a carbonyl group on the other side.
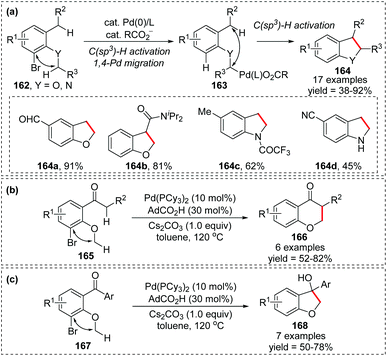 |
| | Scheme 29 1,4-Palladium migration assisted C(sp3)–H/C(sp3)–H coupling. | |
2.3.7 Alkyl-to-acyl 1,4-palladium migration.
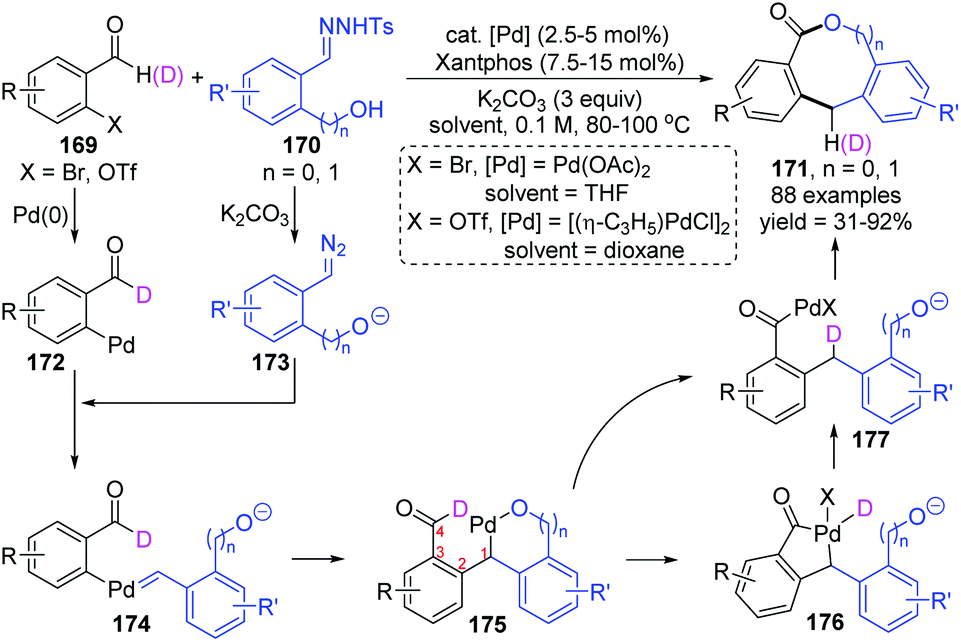
Recently, the Huang and Li group reported a novel strategy for synthesizing diverse lactones via a palladium carbene migratory insertion enabled 1,4-palladium migration process (Scheme 30).25 This protocol achieved a formal dimerization of two readily available reactants, 169 and 170, providing valuable coupling products medium-sized lactones 171, which are common structural motifs recurring in many biologically active compounds. Good yield, functional group tolerance, and broad substrate scope were observed in this transformation. A proposed catalytic cycle is also shown in Scheme 30; initial oxidative addition of Pd(0) to o-pseudo-halo benzaldehyde 169 formed a Pd(II) intermediate 172, which further acted as a receptor to react with diazo compound 173, affording the palladium-carbene intermediate 174. Subsequent intramolecular migratory insertion of 174 generated a new Pd(II) species 175. The 1,4-palladium shift from alkyl to acyl could occur directly or via a five-membered palladacycle 176, producing the Pd-migrated intermediate 177. Final ring closure of 177 generated the target medium-sized lactone 171.
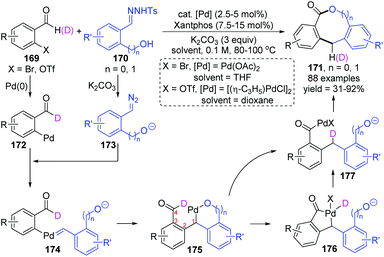 |
| | Scheme 30 Synthesis of medium-sized lactones via alkyl to acyl 1,4-Pd migration. | |
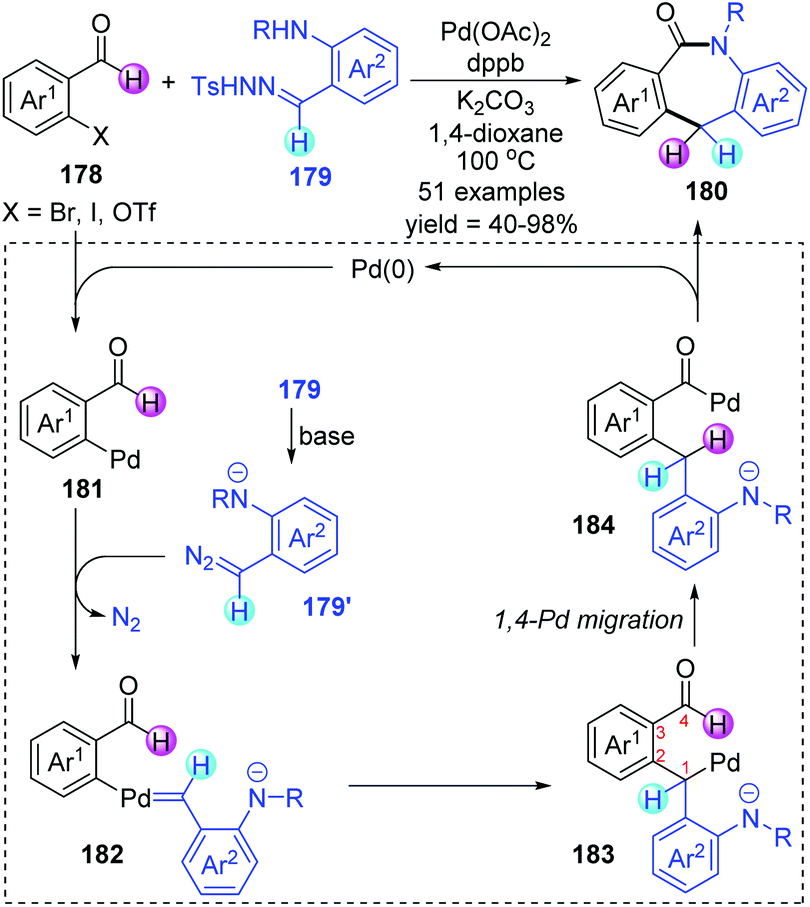
Shortly afterwards, Huang and co-workers further achieved a palladium-catalyzed formal [4 + 3] annulation for preparing valuable dibenzo-[b,e]azepin-6-ones 180 from easily accessible o-haloarylaldehydes 178 and N-tosylhydrazones 179 (Scheme 31).26 Broad substrate scope, excellent functional group tolerance, and feasible scale-up reactions gave this strategy practicality and synthetic potential. The transformation proceeded via an unconventional approach to functionalize the aldehyde C–H bond. A series of deuterium labelling experiments revealed the origin of hydrogen atoms on the central ring; a process of hydrogen atom migration from the acyl to the dibenzylic position should be involved in this conversion. Thus, a catalytic cycle involving 1,4-Pd migration was proposed as depicted. The initial oxidative addition between o-haloarylaldehydes 178 and a Pd(0) catalyst would generate Pd(II) intermediate 181, which subsequently reacted with the in situ generated diazo compound 179′ to afford a palladium carbene intermediate 182. Subsequent intramolecular metal carbene migratory insertion produced an alkyl palladium species 183. The crucial alkyl-to-acyl 1,4-palladium migration occurred to give a benzoyl palladium complex 184, which further underwent ring closure to furnish the target seven-membered N-heterocyclic products 180 and regenerate the Pd(0) catalyst.
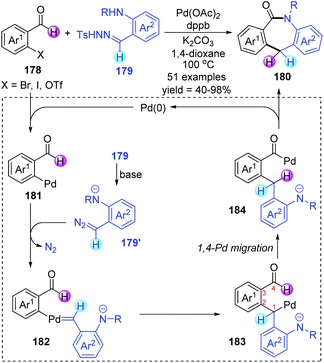 |
| | Scheme 31 Synthesis of dibenzo[b,e]azepin-6-one frameworks via two-component coupling. | |
2.4 1,5-Palladium migration
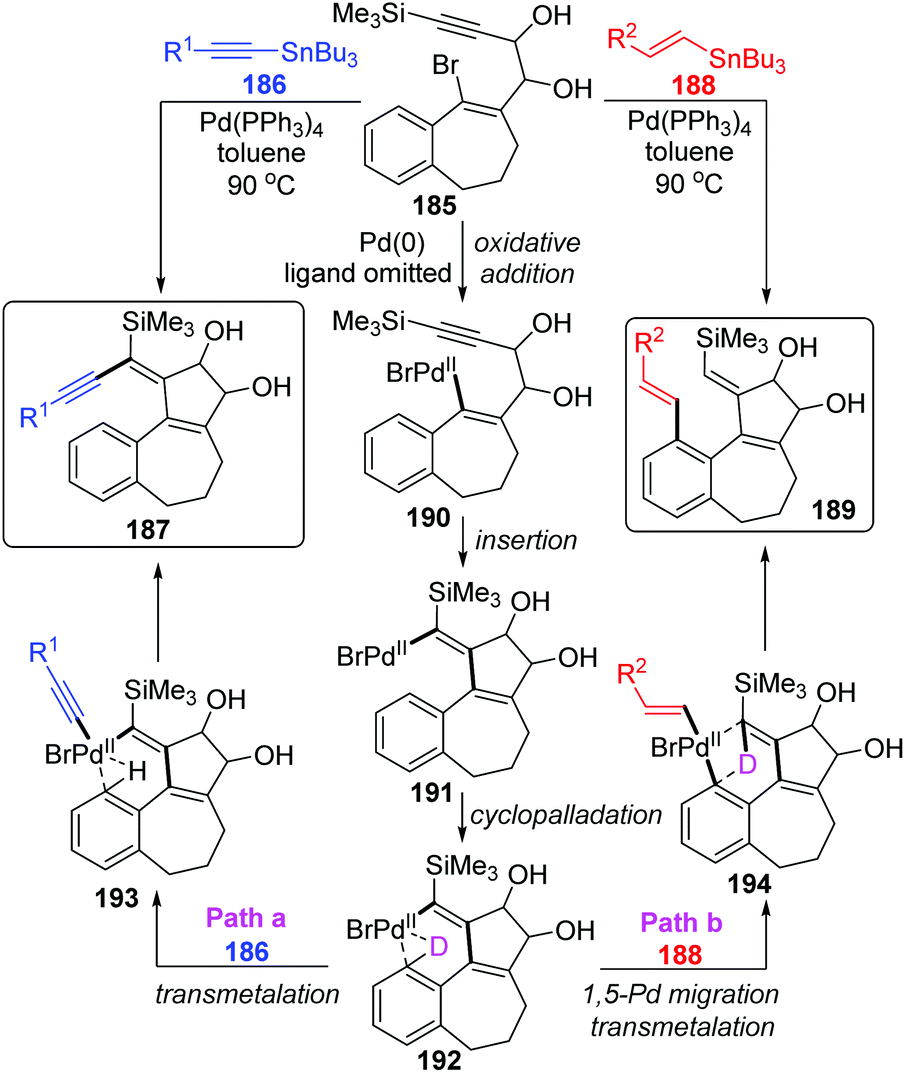
In 2004, Suffert and Bour described a new cyclization and C–H activation/Stille cross-coupling reaction of benzosuberone derivatives 185, which was regioselective and controlled by stannylated reagents (Scheme 32).27 In this study, a series of rearrangement products 189 were obtained in moderate yields by employing the vinylstannanes 188 as stannylated reagents. A 1,5-palladium migration was supposed to be responsible for this rearrangement; however, this migration process was totally suppressed and non-migratory product 187 was afforded when alkynylstannanes 186 were employed instead of 188. Observation of deuterium labelling experiments strongly supported that a location swap between the Pd-group and aryl D atom occurred through the coordination sphere of palladium, as shown in compound 192. A plausible mechanism is also illustrated in Scheme 32. Once the key intermediate 192 was generated via sequential oxidative addition of Pd(0) (185 to 190), insertion of a triple bond (190 to 191), and cyclopalladation via an agostic interaction (191 to 192), the transmetalation would take place directly between alkynylstannanes 186 and key intermediate 192 to furnish the non-migrated products 187 (path a). Alternatively, when vinylic reagents 188 were used instead (path b), the vinylic groups were preferentially installed on the aromatic position to provide rearrangement products 189via 1,5-palladium shift from vinyl to aryl. The stereoelectronic factors of stannylated reagents were potentially related to such regioselectivity.
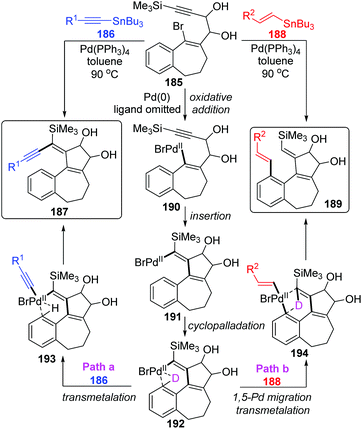 |
| | Scheme 32 Regioselective Stille cross-coupling reactions controlled by stannylated reagents. | |
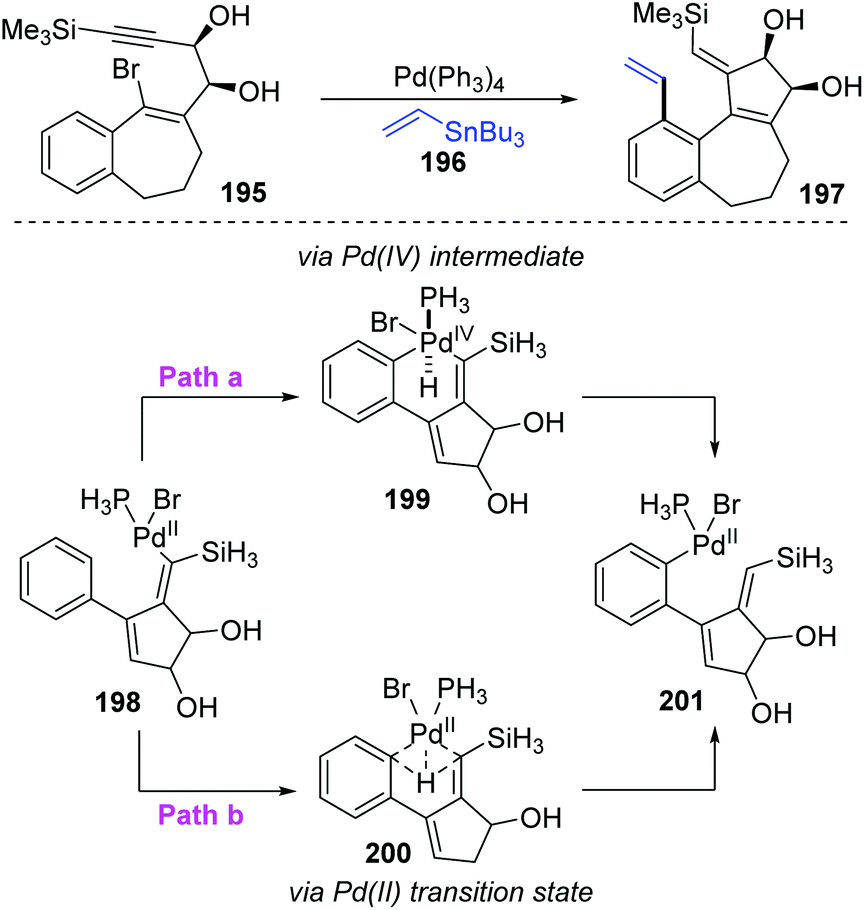
Later, the Dedieu group further carried out a DFT/B3LYP model study on this unusual 1,5 vinyl to aryl palladium shift that occurred in cyclocarbopalladation-Stille coupling tandem reaction (Scheme 33).28 Two possible pathways were proposed for this migration. As shown in path a, a hydridophenylvinyl Pd(IV) intermediate 199 was formed via the phenyl C–H oxidative addition on the palladium atom, which subsequently underwent C–H reductive elimination to provide the phenyl Pd(II) complex 201. Alternatively, in path b, a one-step process occurs, in which the bivalent oxidation state of the palladium atom is retained (species 200). The energy computation of the various transition states and intermediates revealed that the possible mechanism of this migration preferred a one-step hydrogen transfer between the two negatively charged carbon atoms of the vinyl and phenyl groups (path b). The two-step pathway involving a Pd(IV) intermediate (path a) is not likely to take place.
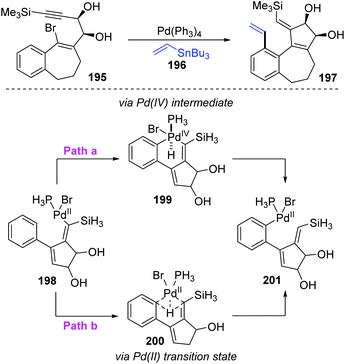 |
| | Scheme 33 1,5-Palladium migration involved cyclocarbopalladation-Stille coupling. | |
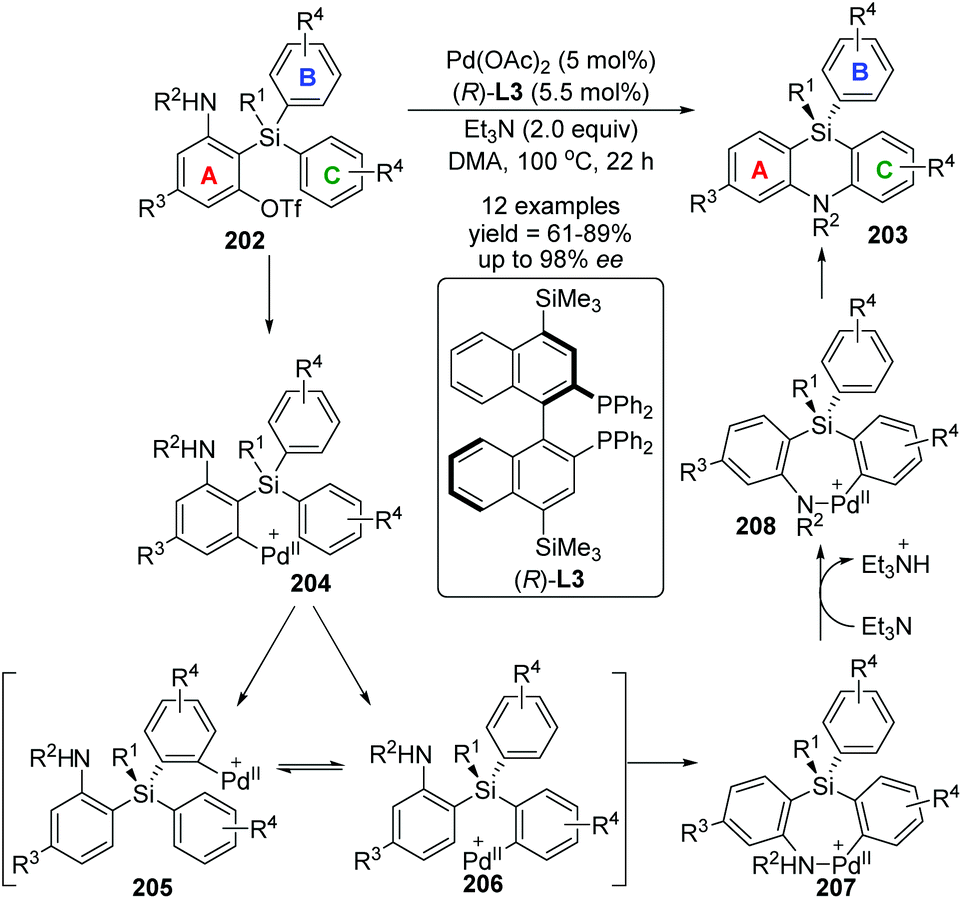
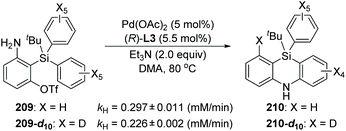
In 2017, Nozaki and Shintani et al. developed a palladium/(R)-L3-catalyzed asymmetric synthesis of silicon-stereogenic 5,10-dihydrophenazasilines 203 (Scheme 34).29 A series of mechanistic investigations were carried out, revealing that an enantioselective 1,5-palladium migration was involved in this conversion. And this migratory process also represented the first example of asymmetric induction at the step of metal migration itself. For this transformation, restriction of substrates should be noticed, and functional group R4 in the aromatic B and C must be kept in the same type and position, or a pair of isomers was obtained. The detailed catalytic mechanism is also shown in Scheme 34. The arylpalladium(II) compound 204 was generated via oxidative addition of 202 to Pd(0). Then, a 1,5-palladium migration occurred between two aryl groups to afford the Pd(II) species 205 or 206, which was followed by intramolecular coordination of the amino group to the palladium center. The resulting compound 207 further turned into palladacycle 208via Et3N-assisted deprotonation. Final reductive elimination and C–N bond-forming provided the desired product 203 along with regeneration of the active Pd(0) catalyst. Moreover, a KIE (Kinetic Isotope Effect) investigation, KIE value = 1.31, revealed that the 1,5-palladium migration is not the turnover-limiting step for this transformation (Scheme 35).
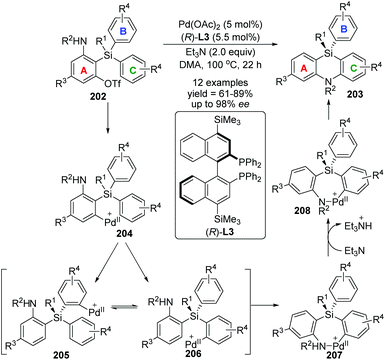 |
| | Scheme 34 Palladium-catalyzed asymmetric synthesis of 5,10-dihydrophenazasilines. | |
 |
| | Scheme 35 Kinetic isotope effect investigations during 1,5-palladium migration. | |
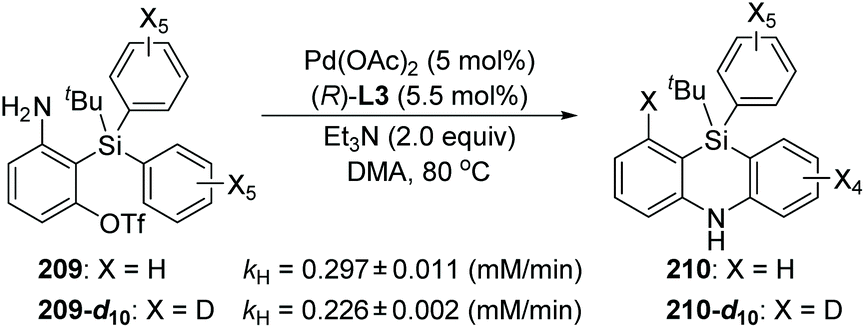
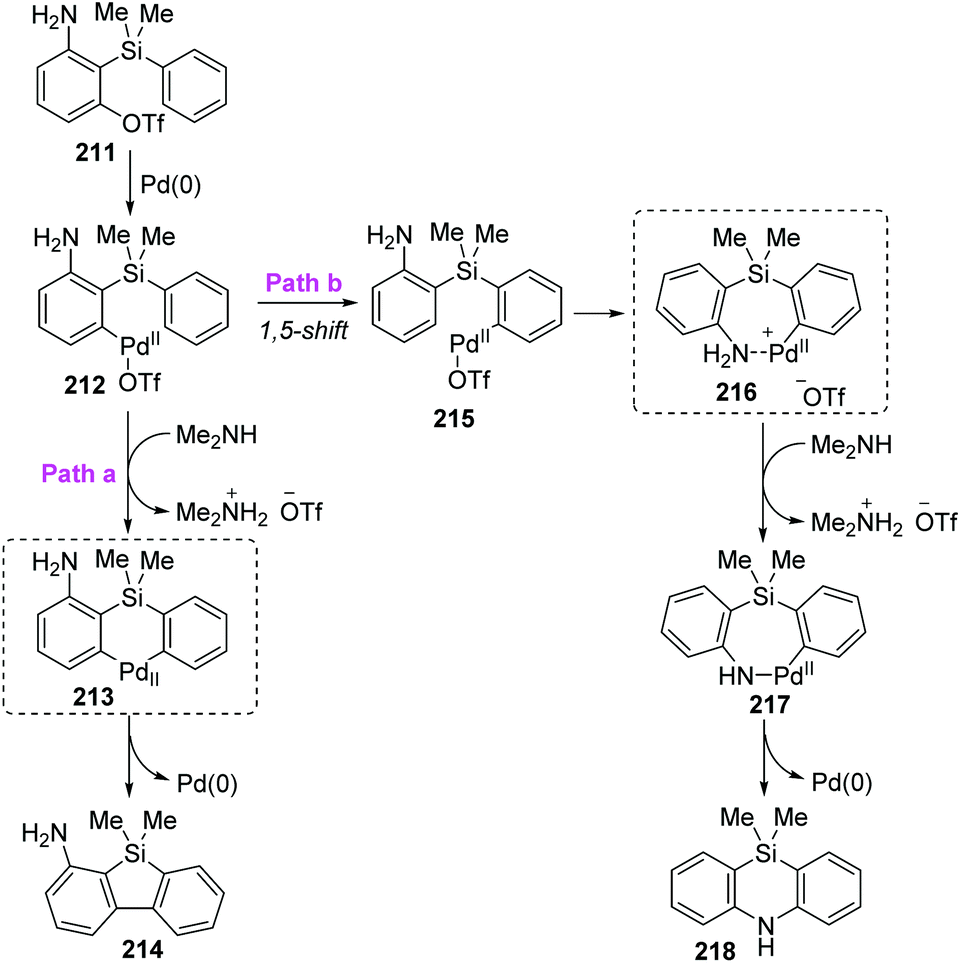
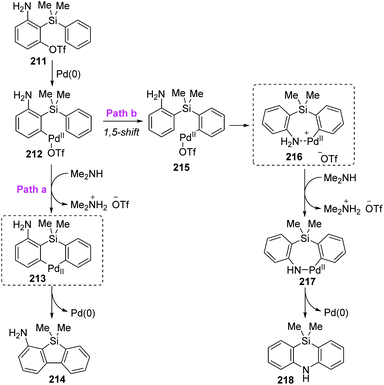
Taking the 2-(dimethylphenylsilyl)phenyl triflates 211 as a model substrate, Nozaki and co-workers further theoretically examined the palladium-catalyzed reaction as shown in Scheme 36.30 Computation pointed that the neutral diorganopalladium(II) intermediate 213 should be involved rather than a 1,5-palladium migration process when generating the dibenzosilole 214 (path a). On the other hand, in path b, a low-energy amine-coordinated palladacycle intermediate 216 should be present to drive the reaction toward 5,10-dihydrophenazasiline 218 instead of dibenzosilole 214.
 |
| | Scheme 36 Proposed catalytic cycle for the conversion of 211 to 218. | |
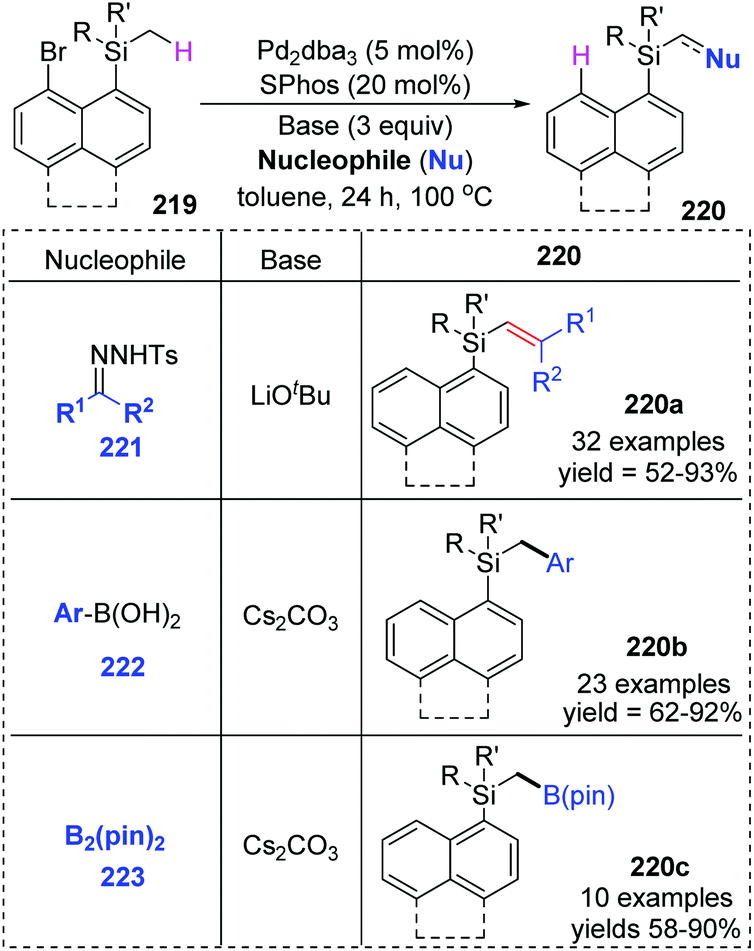
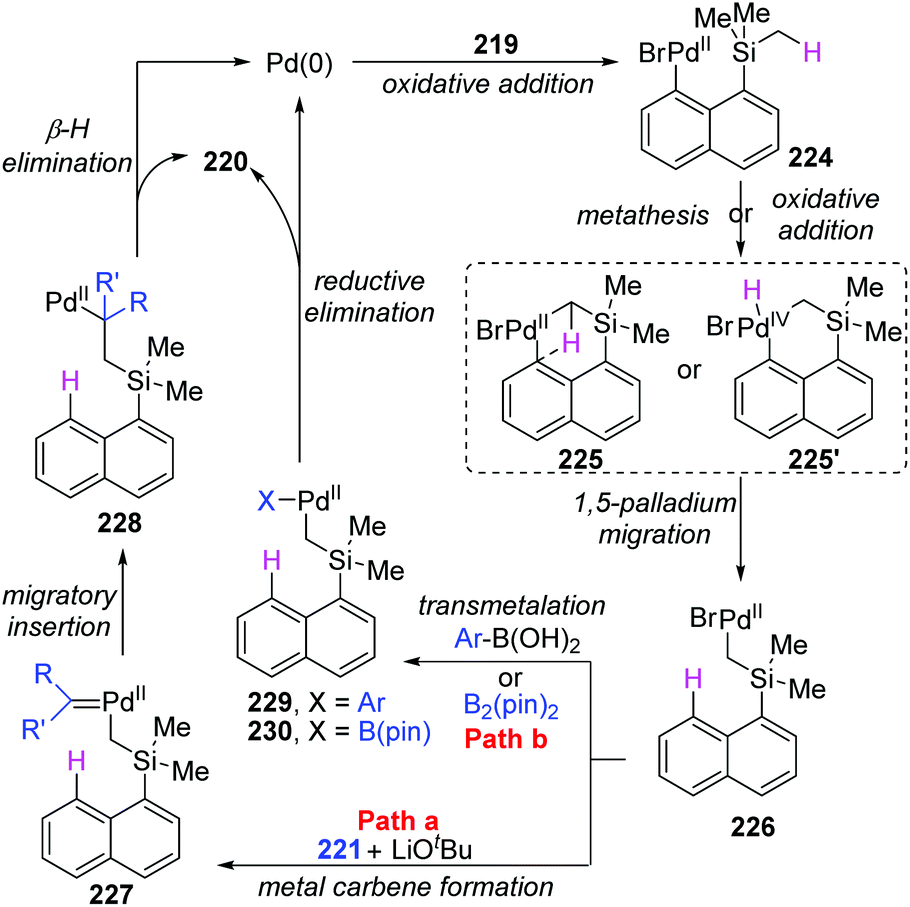
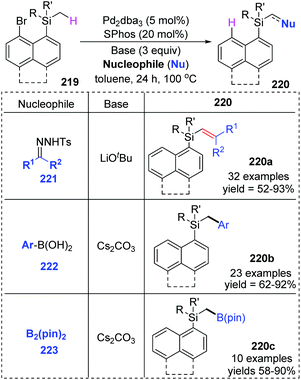
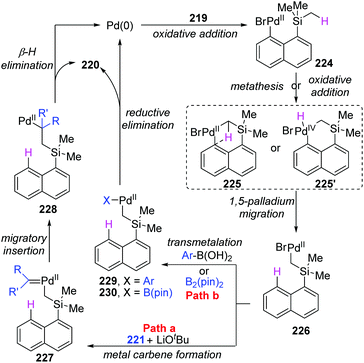
Early in this year, the Zhao group achieved an efficient remote C(sp3)–H functionalization of a methyl group on silicon by employing three different types of nucleophiles, including carbene coupling, Suzuki coupling, and Miyaura borylation reaction (Scheme 37).31 (8-Bromonaphthalen-1-yl)silane derivatives 219 could afford diverse vinyl (220a), benzyl (220b), and boryl silanes (220c) in moderate to excellent yields. This is the first example of a transformation involving 1,5-palladium migration from aryl to alkyl. The detailed catalytic cycle is illustrated in Scheme 38. Oxidative addition of 219 to Pd(0) could generate Pd(II) intermediate 224, which underwent a successive σ-bond metathesis between the C(sp2)–Pd bond and C(sp3)–H bond (225) to form Pd-migrated species 226. Alternatively, the formation of the Pd(IV)–H species 225′via oxidative addition of the C(sp3)–H bond to Pd(II) intermediate 224 and subsequent reductive elimination could also be responsible for 226. In path a, the reaction of intermediate 226 with the carbene precursor generated from 221 could give a Pd(II)-carbene complex 227. The following migratory insertion afforded the C(sp3)–Pd(II) species 228, and further β-hydride elimination delivered vinylsilane product 220a and regenerated the Pd(0) catalyst. In path b, transmetalation between Pd(II) intermediate 226 and aryl boronic acid (or B2pin2) led to either the Pd(II) species 229 or 230. Final reductive elimination resulted in the corresponding products and released Pd(0).
 |
| | Scheme 37 Palladium-catalyzed remote C(sp3)–H functionalization of the methyl group on silicon. | |
 |
| | Scheme 38 Proposed mechanism for the C(sp3)–H functionalization of the methyl group on silicon. | |
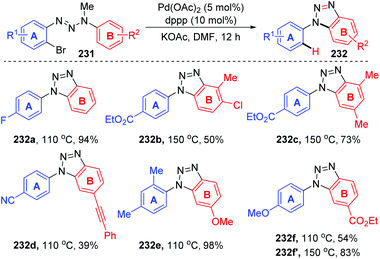
2.5 1,7-Palladium migration
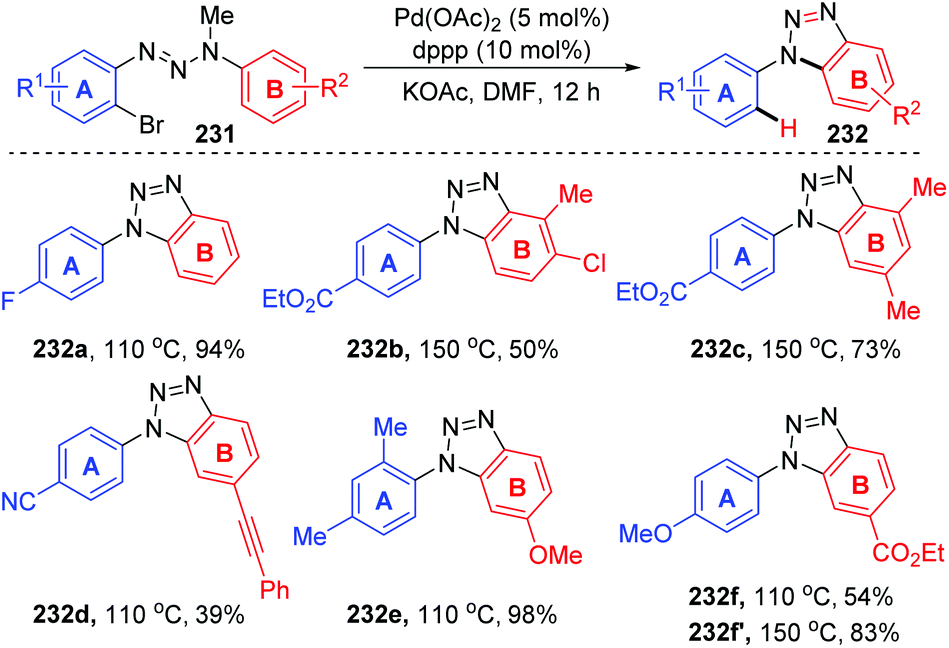
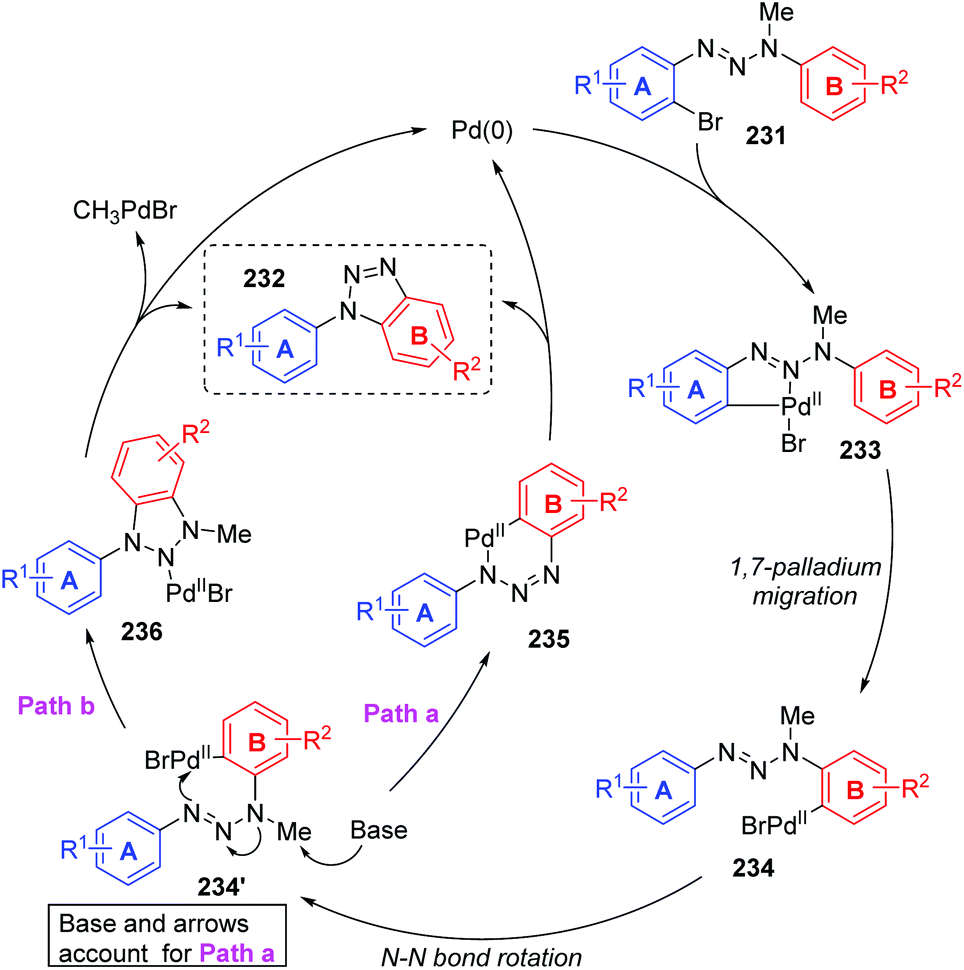
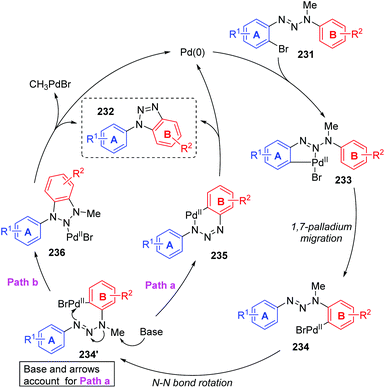
In 2011, Ren and co-workers reported a novel strategy for the regioselective synthesis of benzotriazoles 232via a sequential 1,7-palladium shift/cyclization/dealkylation process (Scheme 39).32 The transformation was performed at 110 °C or 150 °C to give the target products in excellent yields with high regioselectivity. A possible reaction mechanism for this transformation is depicted in Scheme 40. Initially, oxidative addition of Pd(0) to a C(sp2)–Br bond generated the intermediate 233. Then, the coordination of palladium with the middle nitrogen atom of the triazene moiety would make it close enough to the other aromatic ring, which allowed the 1,7-Pd migration to take place via C–H bond activation to give species 234 (an eight-membered-ring palladacycle intermediate33 might be involved). With the aid of a base, compound 234′ suffered intramolecular cyclization to afford the six-membered-ring palladacycle intermediate 235 (path a), which would provide the final product 232 and release the Pd(0) catalyst. An alternative reaction pathway (path b) cannot be excluded; 234′ could form a five-membered ring 236 by the insertion of an N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond into a C–Pd bond, and the final β-CH3 elimination would also be possible to furnish the target product 232.
N double bond into a C–Pd bond, and the final β-CH3 elimination would also be possible to furnish the target product 232.
 |
| | Scheme 39 Pd-Catalyzed intramolecular amination for the synthesis of benzotriazoles. | |
 |
| | Scheme 40 Proposed mechanism for the synthesis of benzotriazoles. | |
2.6 Other types of palladium migration
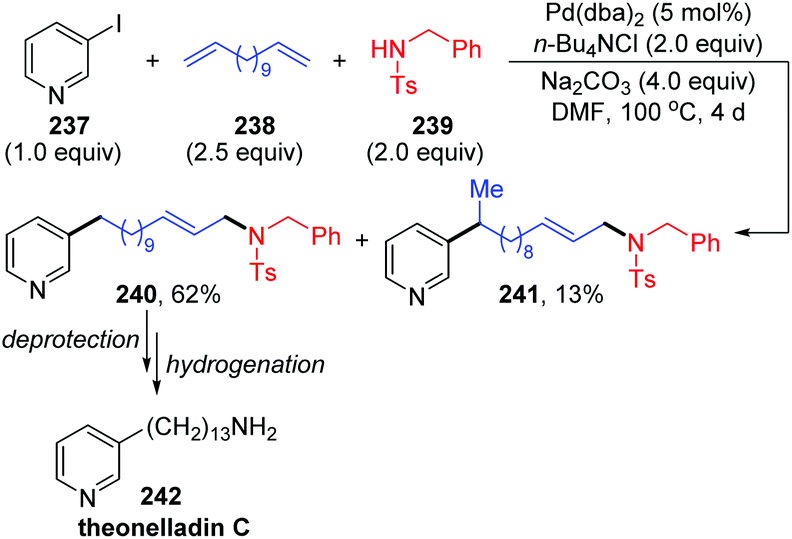
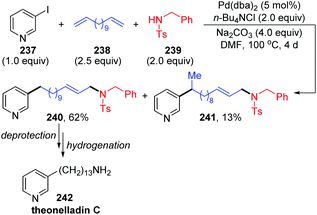
In 2002, Larock and co-workers reported a novel long-chain palladium migration strategy involving three-component coupling of 3-iodopyridine, long-chain nonconjugated dienes and appropriate nitrogen nucleophiles (Scheme 41).34 This transformation provided a convenient approach to prepare the pyridine alkaloids and related natural products, which contained a long chain with an aromatic or heteroaryl group on one end of the chain and diverse functional groups on the other end. For example, as shown in Scheme 41, employing 1,12-tridecadiene 238 as the long-chain dienes, followed by deprotection and hydrogenation, would afford the natural product theonelladin C 242 readily. However, there are still some shortcomings in this reaction. Ammonia and primary amines did not work well in this three-component coupling process. And the by-product isomer 241, generated via introducing a pyridylpalladium group to the more hindered internal carbon was inevitable.
 |
| | Scheme 41 Three-component coupling reaction via palladium long-distance migration. | |
3 Rhodium migration
3.1 1,3-Rhodium migration
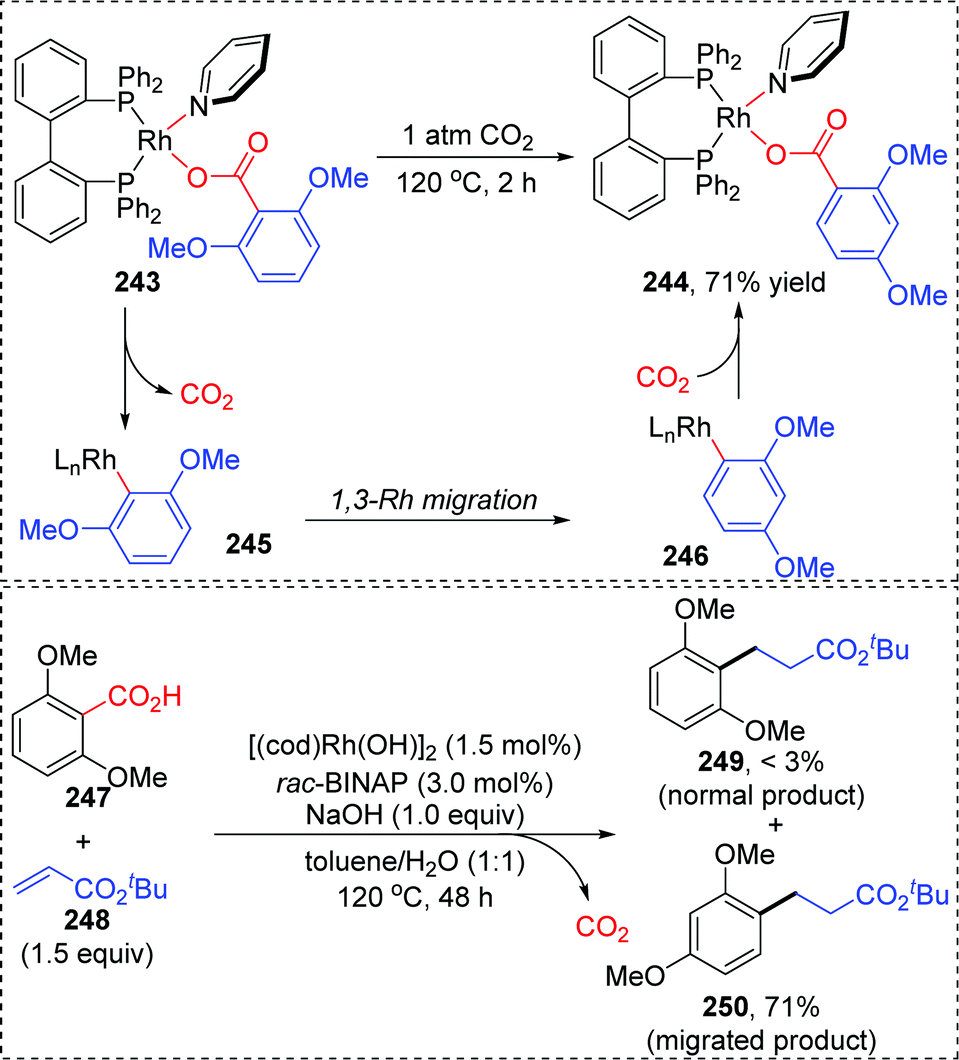
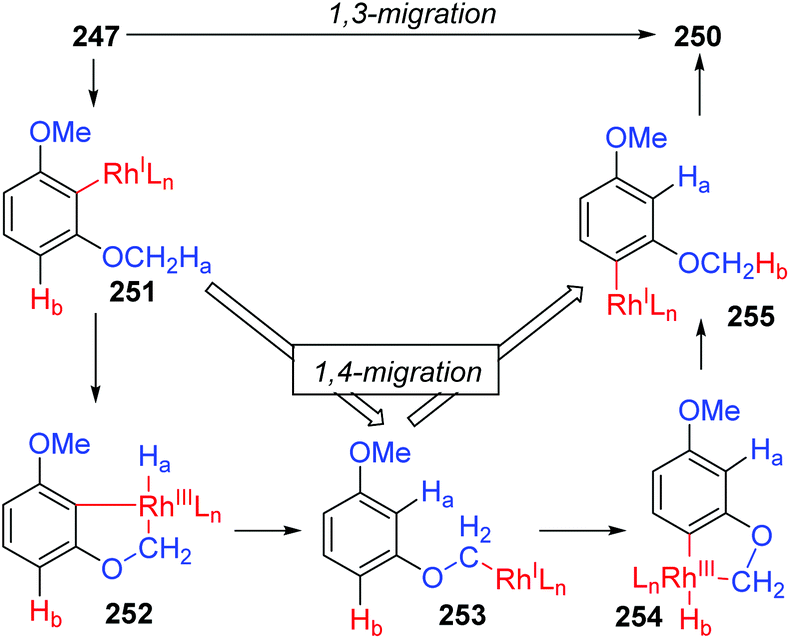
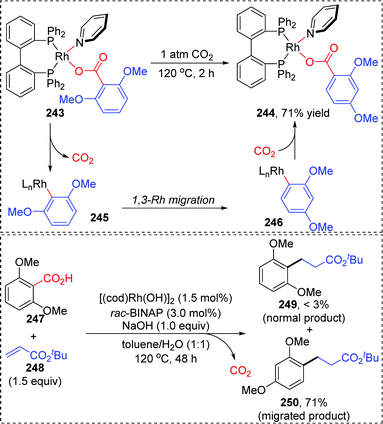
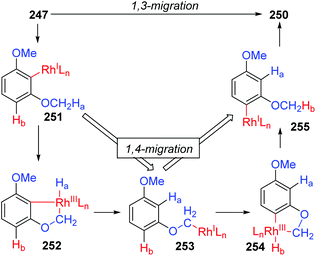
In 2013, the rearrangement of Rh(I) 2,6-dimethoxybenzoate complex 243via an unusual 1,3-Rh migration process was reported by the Zhao and Sun group (Scheme 42).35 2,4-Dimethoxybenzoate 244 was obtained in 71% yield under a CO2 atmosphere while only 34% yield was obtained under a N2 atmosphere, indicating that a decarboxylation/carboxylation process should be involved in this transformation. Decarboxylation of 243 generated a Rh(I) 2,6-dimethoxyphenyl intermediate 245, which then underwent 1,3-Rh/H shift to form the migrated Rh(I) 2,4-dimethoxybenzoate species 246 with reduced steric strain. Final reinsertion of CO2 into the Rh-aryl bond gave the more stable complex 244. This 1,3-Rh migration also took place in analogical transformation. For example, 2,6-dimethoxybenzoic acid 247 reacted with t-butyl acrylate 248 smoothly under a trace of rhodium catalyst to provide migrated product 250 in good yield and selectivity. A proposed pathway for this 1,3-Rh migration is illustrated in Scheme 43. A deuterium-labeling study revealed that two consecutive 1,4-migrations mediated by a methoxy group should be responsible for the resulting 1,3-Rh migration. The starting cyclometalation of aryl-Rh(I) 251 activated the Csp3–H bond of methoxy at the ortho position and generated a Rh(III) hydrido intermediate 252, followed by reductive elimination to afford a Rh(I) aryloxyalkyl 253. Further activation of the aromatic C–H bond at the less hindered meta position afforded another Rh(III) cyclometalated species 254, which underwent reductive elimination at the methoxy position to produce the final product 255.
 |
| | Scheme 42 Rearrangement of Rh(I) 2,6-dimethoxybenzoate derivatives via 1,3-Rh migration. | |
 |
| | Scheme 43 Proposed mechanism for 1,3-Rh migration in the rearrangement of the Rh(I) complex. | |
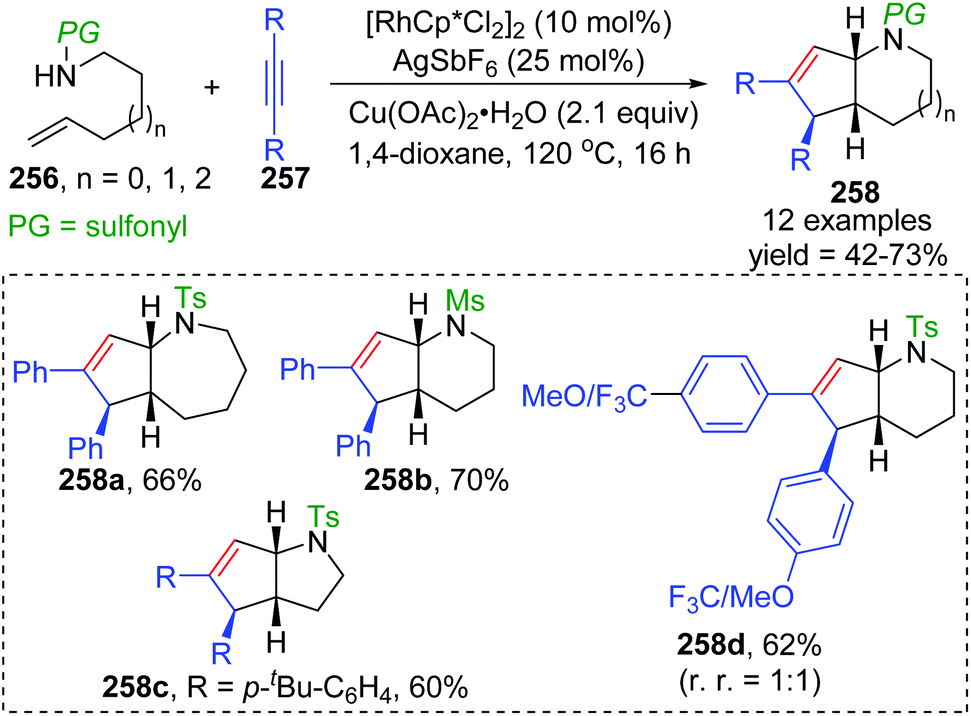
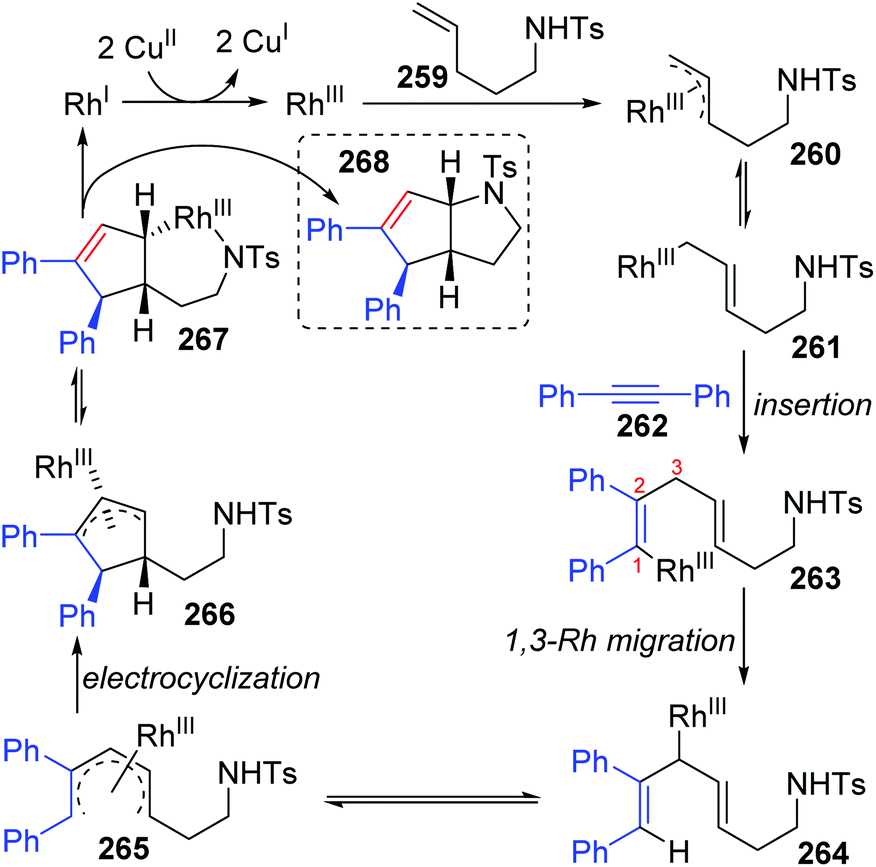
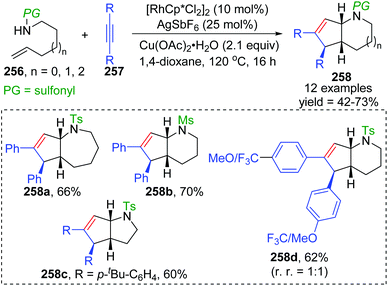
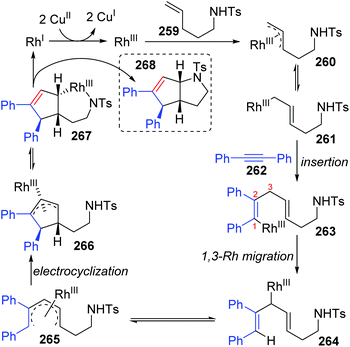
In 2015, synthesis of stereochemically complex azabicyclic structures via Rh(III)-catalyzed allylic C(sp3)–H activation/electrocyclization was achieved by Rovis and Archambeau (Scheme 44).36 Unsaturated N-tosylsulfonamide 256 reacted with diphenylacetylene 257 to afford the desired products 258 with complete control of diastereoselectivity, and the phenyl group was located on the exo-face of the molecule. Rh(III), Ag(I) and Cu(II) species, and high reaction temperature (120 °C) were all necessary for this conversion, while the reaction was shut down when the temperature decreased to 80 °C. Moreover, substituents on both sides of alkyne 257 should stay the same because the electronic properties of the alkyne did not influence the regioselectivity; for instance, an unsymmetrical alkyne led to a mixture of products 258d. The results of deuterium-labeling experiments strongly supported a 1,3-Rh migration process involved in the catalytic cycle as illustrated in Scheme 45. Initially, the allylic C(sp3)–H bond of 259 was activated by a Rh(III) catalyst, generating a π-allyl complex 260 and the homologous isomer 261. Subsequent migratory insertion of alkyne 262 to 261 led to the vinylrhodium(III) species 263, which underwent vinyl-to-allyl 1,3-Rh migration to afford the bis(allyl)rhodium(III) complex 264. Affected by steric resistance, the following 4π-electrocyclization preferred to form a π-allylrhodium(III) 266 with specific stereo-configuration. Successive N-metalation and reductive elimination gave the target dicyclo-product 268 and Rh(I) species. Final copper-mediated oxidation of the resulting Rh(I) species regenerated the active Rh(III) catalyst.
 |
| | Scheme 44 Rh-Catalyzed synthesis of azabicyclic derivatives from unsaturated N-sulfonamides. | |
 |
| | Scheme 45 Proposed mechanism for the synthesis of azabicyclic derivatives. | |
3.2 1,4-Rhodium migration
Among various types of metal-migration involved reactions, 1,4-rhodium migration is another well-studied subject no less than 1,4-palladium migration during the past twenty years. This chapter only summarizes several new reports published recently, and more information toward 1,4-rhodium migration can be found in Gu's review.4
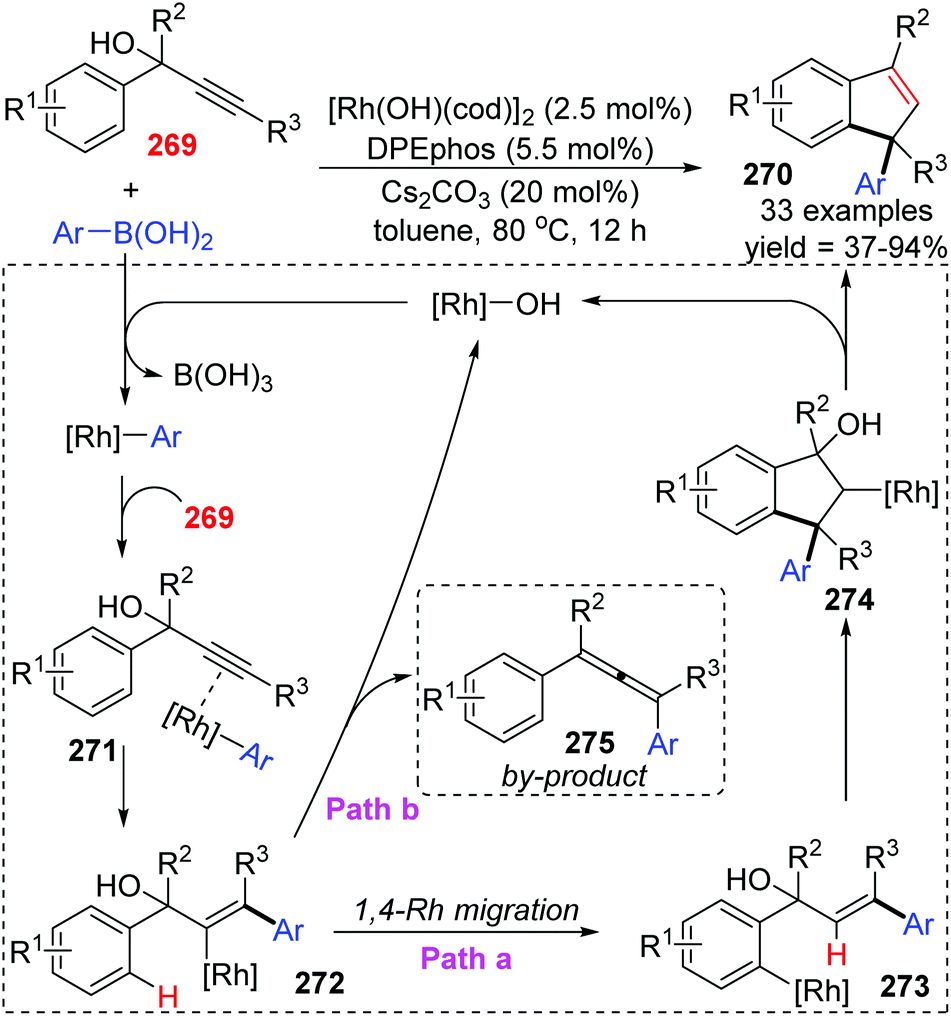
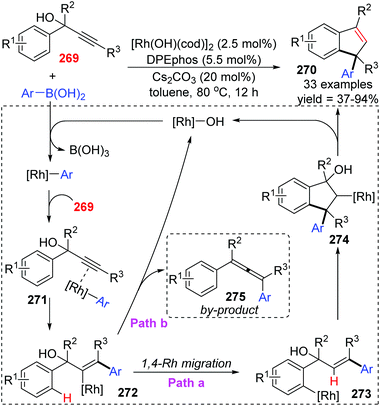
The Tian and Dou group achieved a rhodium-catalyzed expeditious synthesis of indenes 270 from propargyl alcohols 269 and arylboronic acids via the selective 1,4-rhodium migration over β-oxygen elimination (Scheme 46).37 Ligands played a crucial role in this transformation, and DPEphos was found to be uniquely effective for the high selectivity of migratory products. Results of density functional theory (DFT) calculations revealed that the employment of DPEPhos as the ligand lowered the relative energy toward key intermediates of the 1,4-migration path. As illustrated in Scheme 46, a plausible mechanism involving 1,4-Rh shift is responsible for the indene products. Initially, an arylrhodium intermediate was generated via transmetalation of the hydroxorhodium catalyst with arylboronic acids, which further coordinated with alkynyl to afford the vinylrhodium complex 272 by regioselective syn-arylrhodation. In path a, 1,4-rhodium migration from 272 gave an arylrhodium species 273, followed by a second intramolecular arylrhodation to form the key alkylrhodium intermediate 274. Ultimately β-oxygen elimination occurred to furnish the target indene derivatives 270 and regenerated the active Rh catalyst simultaneously. Alternatively, premature β-oxygen elimination of intermediate 272 would lead to the allene products 275 (path b).
 |
| | Scheme 46 Rhodium-catalyzed synthesis of indenes from propargyl alcohols and organoboronic acids. | |
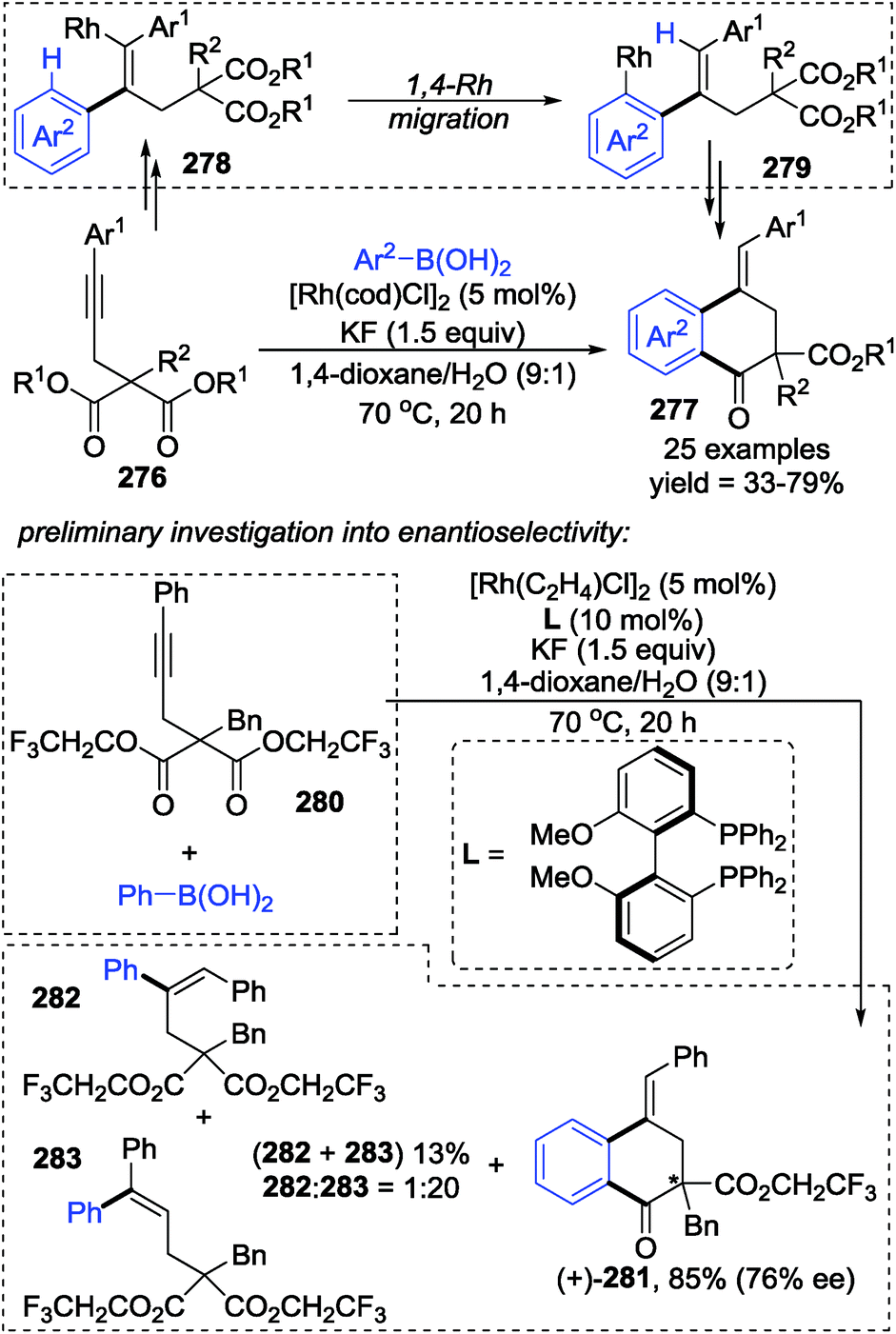
Lam and co-workers developed a rhodium-catalyzed arylative cyclization of alkynyl malonates 276 with arylboronic acids via 1,4-rhodium(I) migration to prepare functionalized 1-tetralones 277 in good yields (Scheme 47).38 This strategy performed excellently by using KF as a base in 1,4-dioxane/H2O (9:1) as the solvent. And additionally, the stereo-configuration of the products could be affected by the chirality of the ligands. Preliminary synthesis of chiral products (+)-281 (76% ee) along with an inseparable mixture of 282 and 283 was achieved by employing a chiral bisphosphine-rhodium complex as the precatalyst; however, no further investigation toward enantioselectivity has been conducted. Although the reaction mechanism was not proposed, an alkenyl-to-aryl 1,4-Rh(I) migration was considered to be the key step for the generation of a benzo six-membered heterocycle, as shown from 278 to 279.
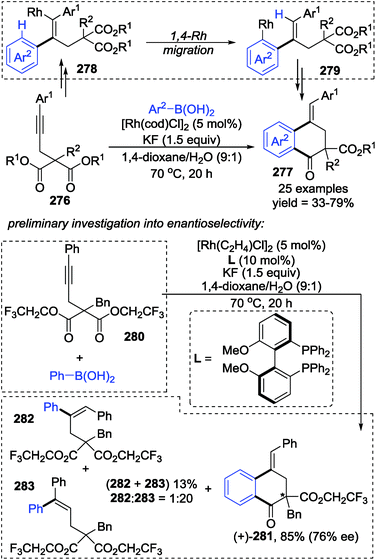 |
| | Scheme 47 Rh-Catalyzed arylative cyclization of alkynyl malonates with arylboronic acids. | |
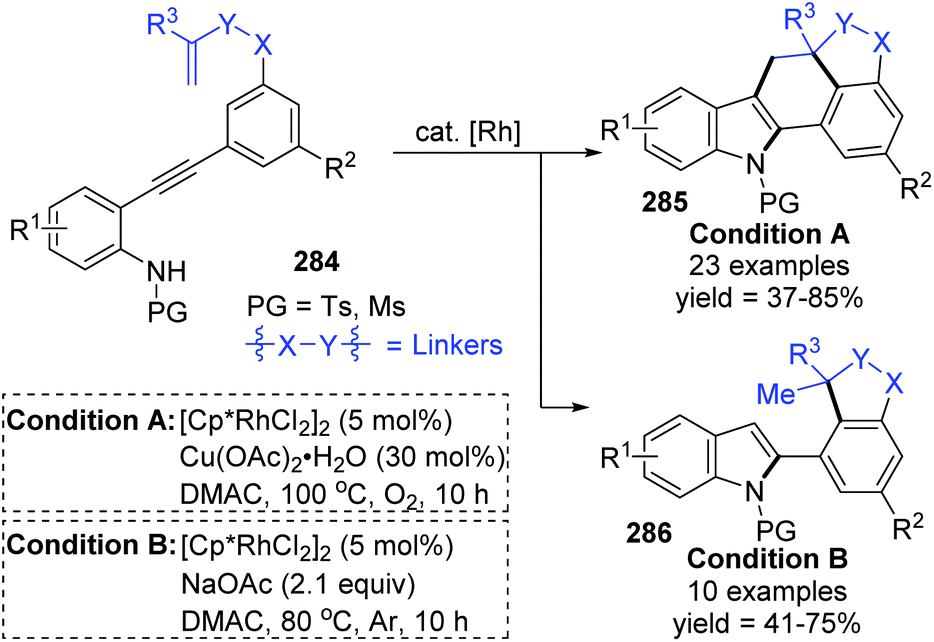
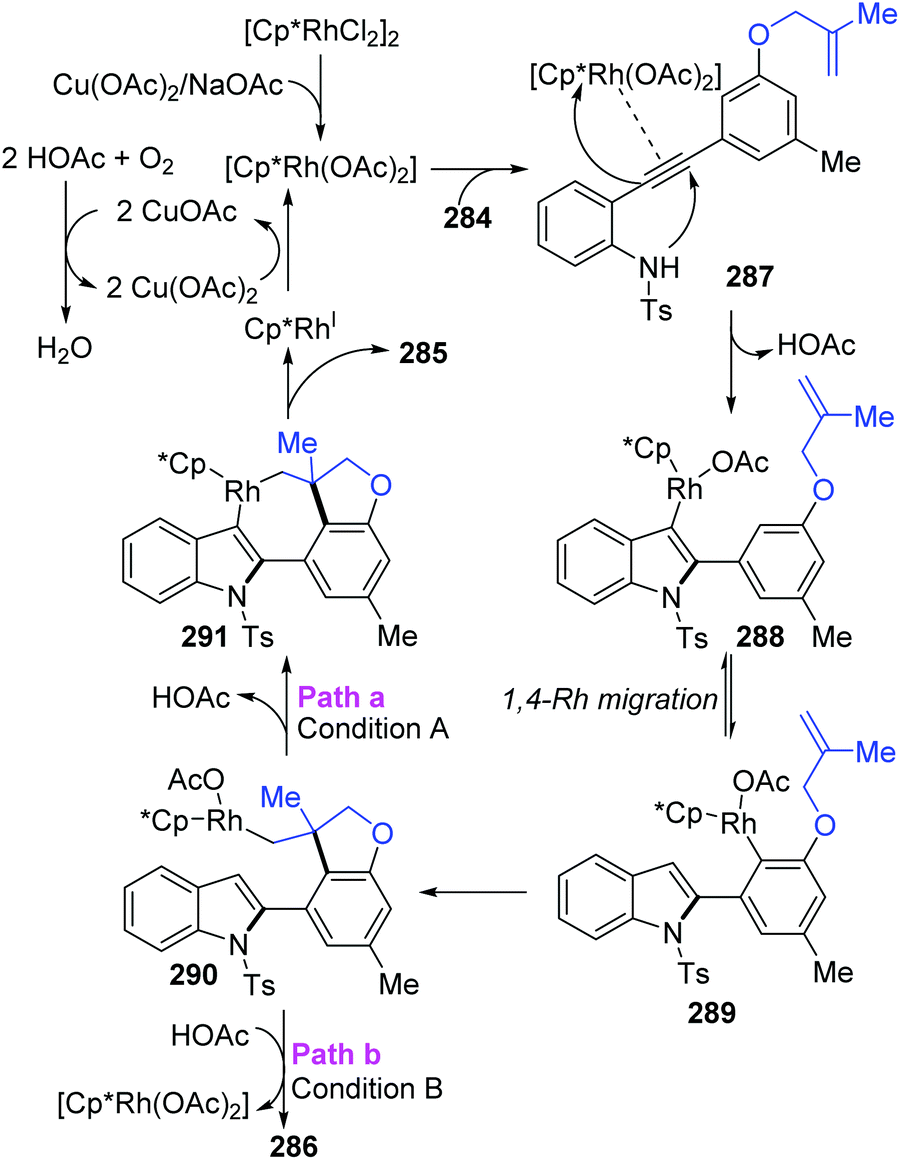
The Lin and Yao group reported a rhodium(III)-catalyzed strategy for the divergent construction of indole-fused polycyclic frameworks via an aryl-to-aryl 1,4-Rh migration process (Scheme 48).39 Different types of products were obtained by changing the reaction conditions. Carboannulation product 285 was afforded in the presence of Rh and Cu(OAc)2·H2O catalysts under an oxygen atmosphere, while hydroarylation product 286 was generated by employing a Rh catalyst and NaOAc under an argon atmosphere. A plausible pathway is illustrated in Scheme 49. Initial coordination of active Rh(III) to alkynyl and subsequent intramolecular electrophilic cyclization furnished the 3-indolyl rhodium species 288. Subsequently, C–H bond activation/aryl-to-aryl 1,4-rhodium migration took place to generate an aryl rhodium intermediate 289, which underwent intramolecular migratory insertion of the alkene to the Rh–C bond to give the alkyl rhodium intermediate 290. In path a (condition A), the indole C3 C–H activation offered the seven-membered rhodacycle complex 291. Final reductive elimination provided the target pentacyclic product 285, and the active Rh(III) catalyst was regenerated from Rh(I) via oxidation with the assistance of Cu(OAc)2/O2. Alternatively, in path b (condition B), the alkyl rhodium intermediate 290 underwent a direct protonation/demetalation process to afford 2-aryl indoles 286 and regenerate the Rh(III) catalyst.
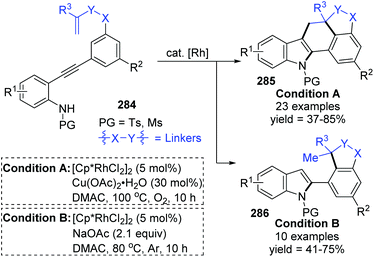 |
| | Scheme 48 Rh(III)-Catalyzed reaction to prepare diversified indole-fused polycycles. | |
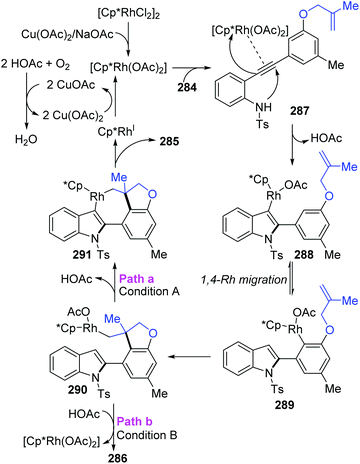 |
| | Scheme 49 Proposed catalytic pathway for the synthesis of indole-fused polycycles. | |
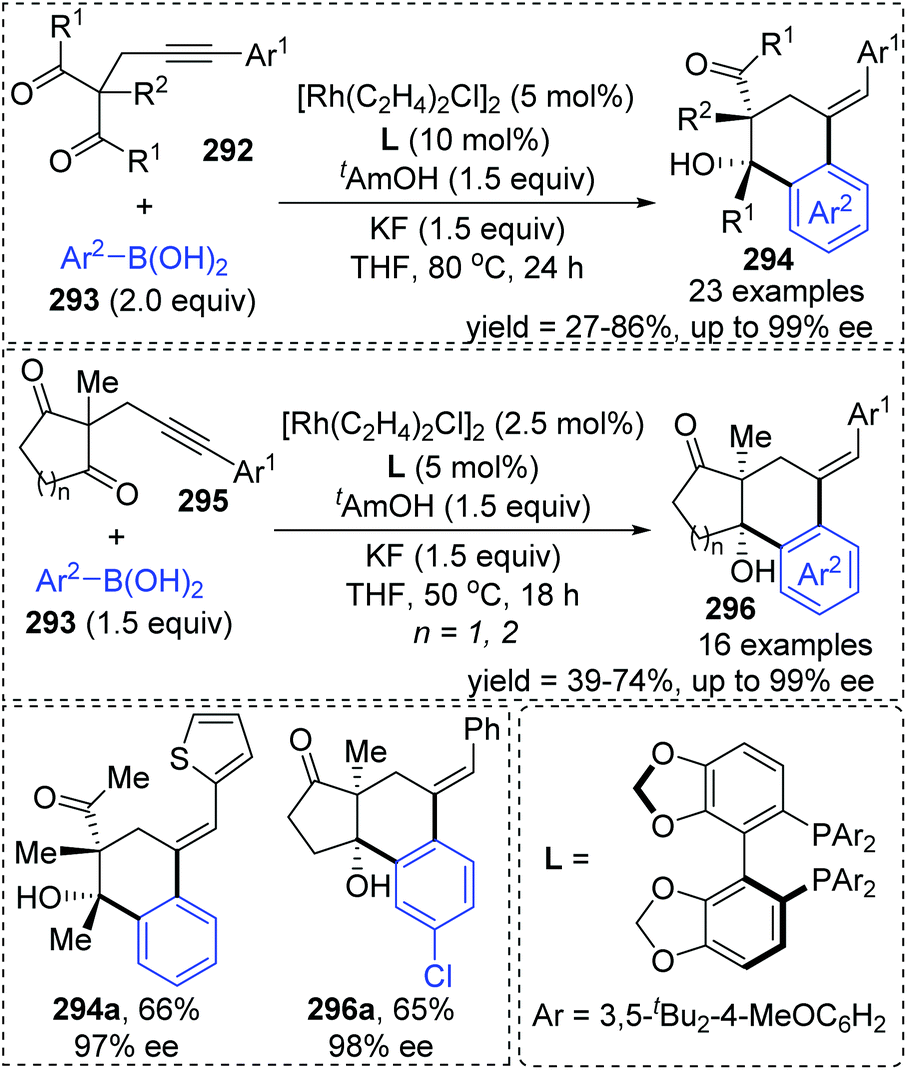
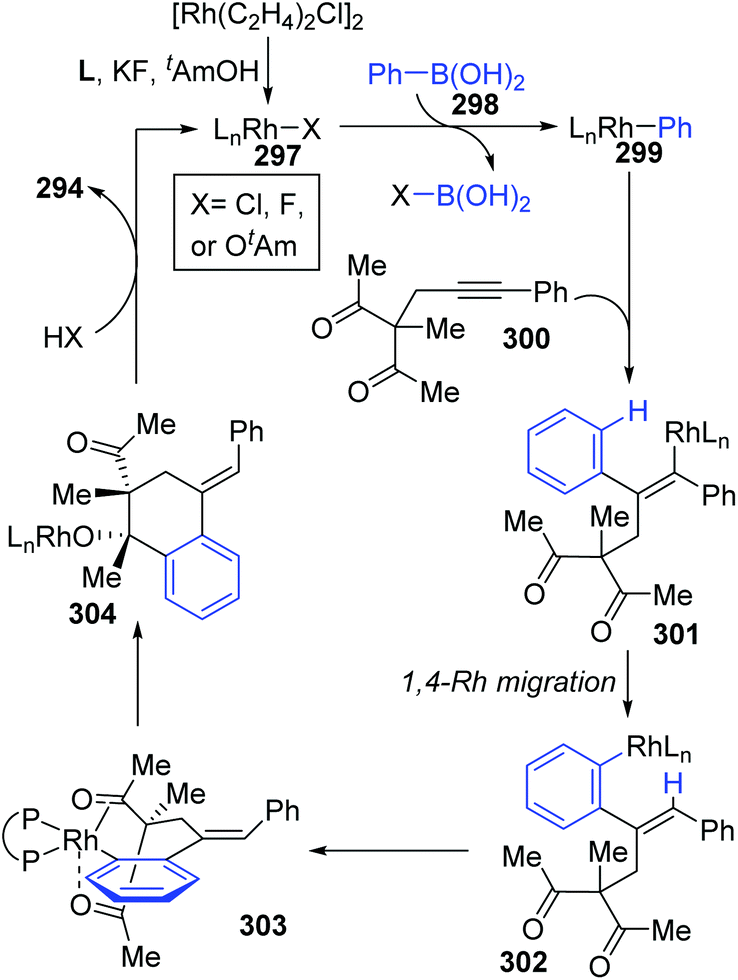
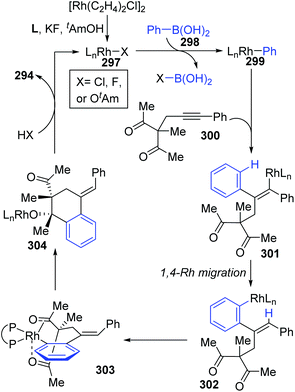
A chiral bisphosphine–rhodium complex promoted diastereo- and enantioselective reaction of arylboronic acids 293 with alkynyl 1,3-diketones was achieved by Lam and co-workers (Scheme 50).40 Both acyclic and cyclic 1,3-diketones (292 and 295) performed effectively to afford the corresponding arylative cyclization product 294 and 296 with high enantioselectivity in moderate to good yields. An alkenyl-to-aryl 1,4-Rh(I) migration process played a significant role in this transformation (Scheme 51). Initially, a chiral complex 297 consisting of one bisphosphine bound to one rhodium atom was formed from the mixture of [Rh(C2H4)Cl]2, L, KF, and t-AmOH, which further turned into an arylrhodium species 299via transmetalation with PhB(OH)2. Taking 300 as an example, the successive migratory insertion and 1,4-rhodium shift provided the key arylrhodium intermediate 302. The relative configuration of products 294 could be explained by an experimental stereochemical model where cyclization proceeds through a conformation similar to intermediate 303. Specific coordination of one ketone to the rhodium atom enabled the carbonyl group to be aligned with the arylrhodium bond, and the second ketone was coordinated to rhodium in an axial position. Depending on the geometric constraints, subsequent nucleophilic attack and migratory insertion formed the resulting configuration 304, which underwent protonation to afford target products 294.
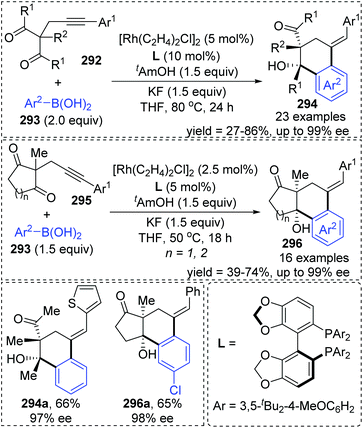 |
| | Scheme 50 Rhodium/chiral-bisphosphine complex catalyzed reaction of arylboronic acids with alkynyl 1,3-diketones. | |
 |
| | Scheme 51 Proposed catalytic mechanism for the reaction of arylboronic acids with alkynyl 1,3-diketones. | |
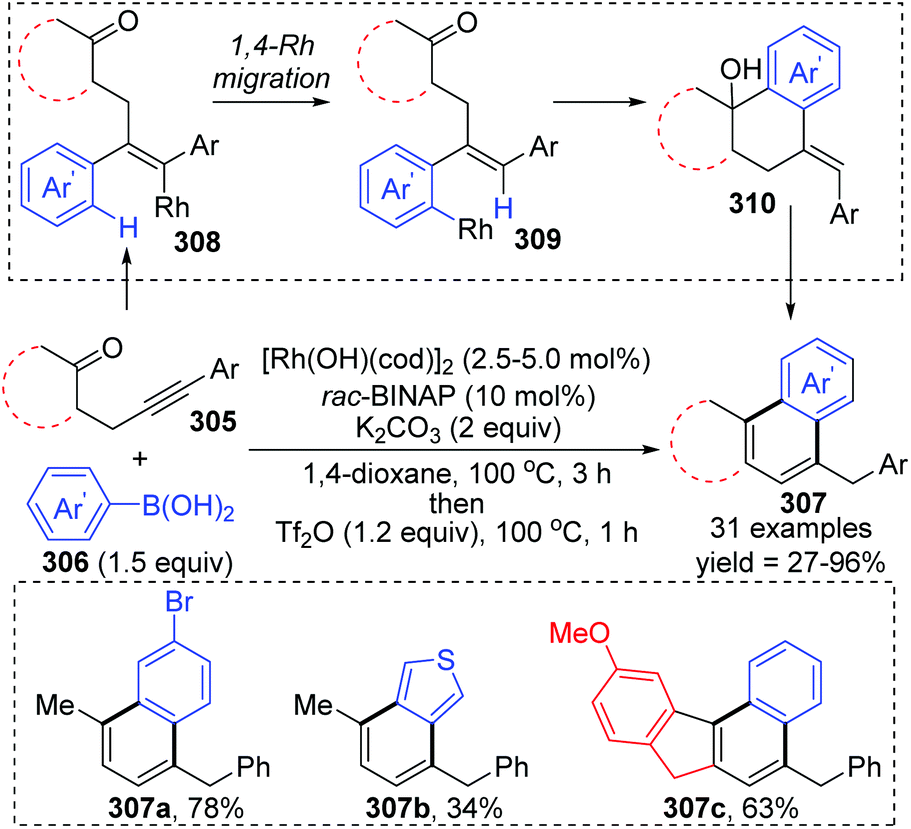
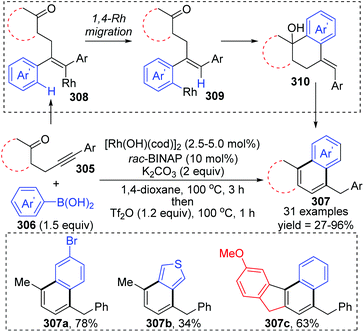
Matsuda and co-workers achieved a rhodium(I)-catalyzed addition of arylboronic acids 306 to β-(arylethynyl) ketones 305, involving an aryl-to-aryl 1,4-rhodium migration process (Scheme 52).41 The conversion started with the regioselective addition of an arylrhodium(I) species to the alkyne moiety of ketone 308, followed by 1,4-rhodium migration generating an arylrhodium(I) species 309. Intramolecular nucleophilic addition and subsequent protonation were responsible for the formation of the six-membered ring. The resulting tetralin alcohols 310 further underwent dehydration/aromatization upon treatment with Tf2O, providing access to various fused aromatic compounds 307.
 |
| | Scheme 52 Rhodium(I)-catalyzed reaction of β-(arylethynyl)ketones with boronic acids. | |
3.3 1,5-Rhodium migration
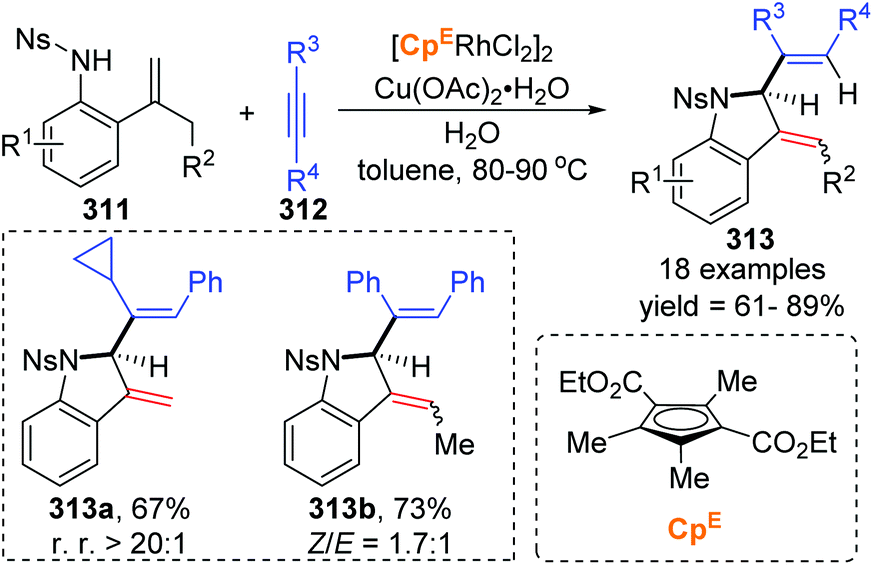
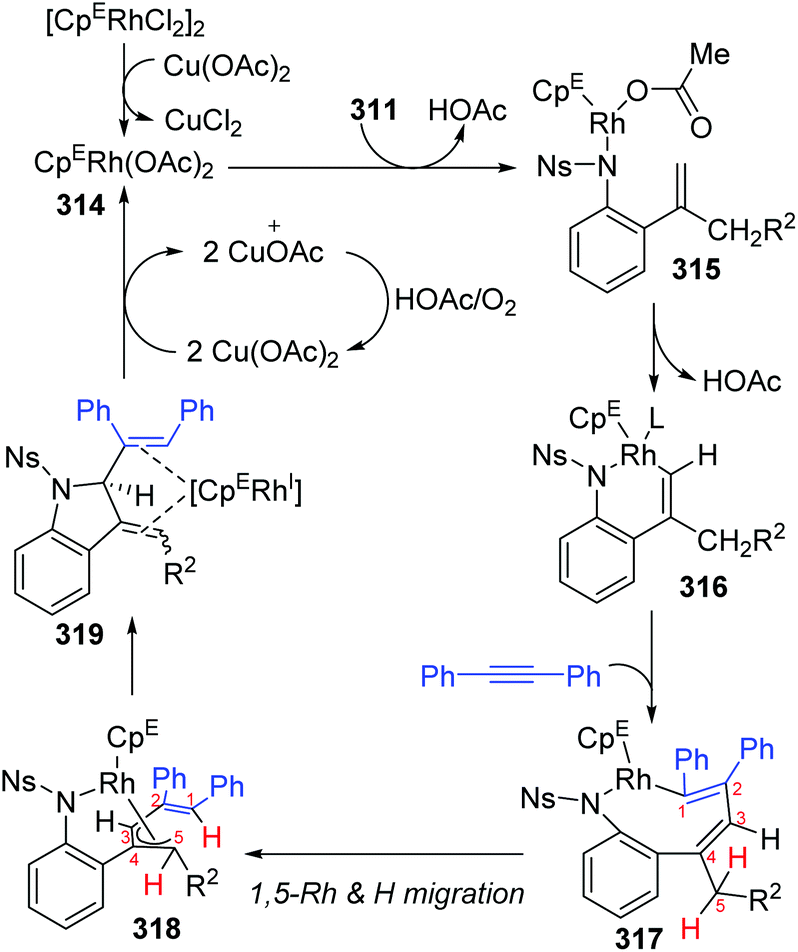
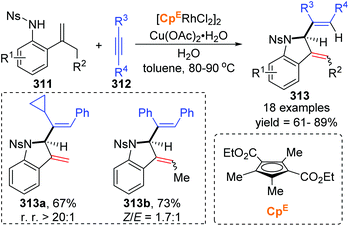
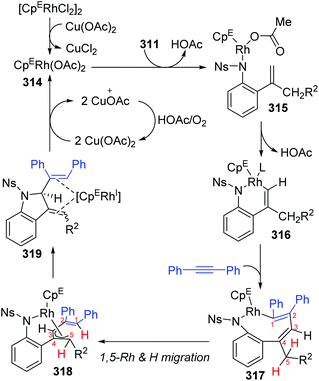
In 2018, a Rh(III)-catalyzed annulation of 2-alkenyl anilides 311 with alkynes 312via C–H activation was developed by the Mascareñas and Gulías group, providing a series of 2-substituted indoline derivatives 313 (Scheme 53).42 Various alkyne components were well tolerated, and symmetrical alkynes with electron-rich or deficient aromatic substituents performed smoothly to give the corresponding products in good yields. Additionally, high regioselectivity was observed when nonsymmetrical alkynes were employed, favoring products with the aryl substituent at the terminal position of the alkene, such as 313a (rr > 20:1). According to deuterium scrambling investigations, the possible reaction pathway is illustrated in Scheme 54. An active rhodium acetate species 314 was formed, which underwent ligand exchange of one of the acetates with the amide of 311 to generate a complex 315. Thereafter, activation of the alkenyl C–H bond and carbometalation took place to afford a rhodacycle 316, followed by migratory insertion of the 1,2-diphenylethyne to give a new rhodacycle intermediate 317. Successive internal 1,5-hydrogen/rhodium exchange and reductive elimination afforded the corresponding indoline product 313. Active Rh catalyst was regenerated with the assistance of Cu(OAc)2.
 |
| | Scheme 53 Rhodium(III)-catalyzed annulation of 2-alkenyl anilides with alkynes. | |
 |
| | Scheme 54 Possible mechanism for the cyclization of 2-alkenyl anilides. | |
4 Iron migration
4.1 1,4-Iron migration
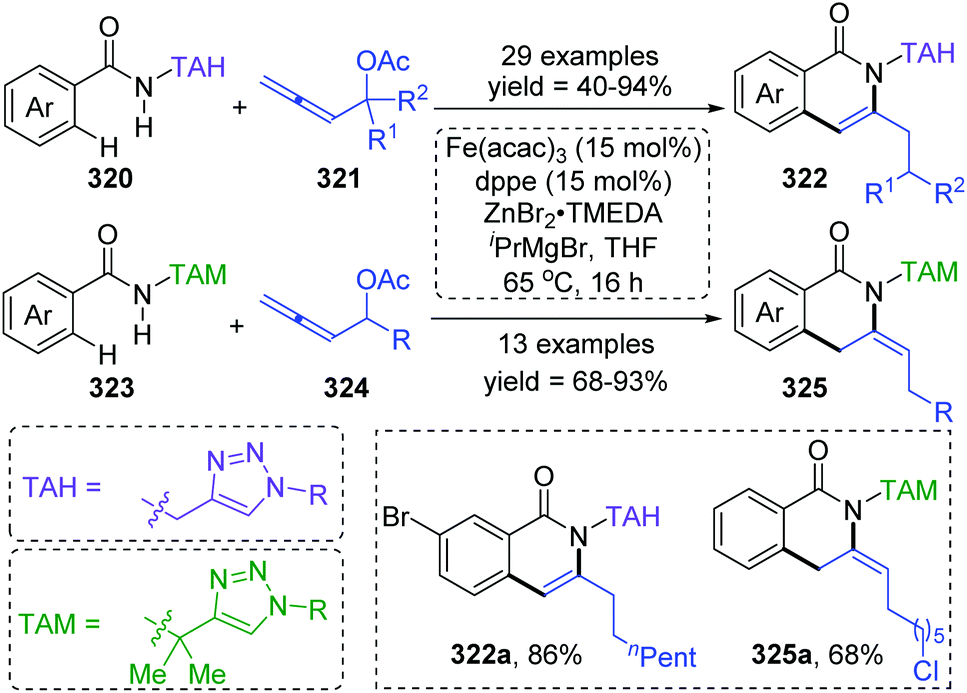
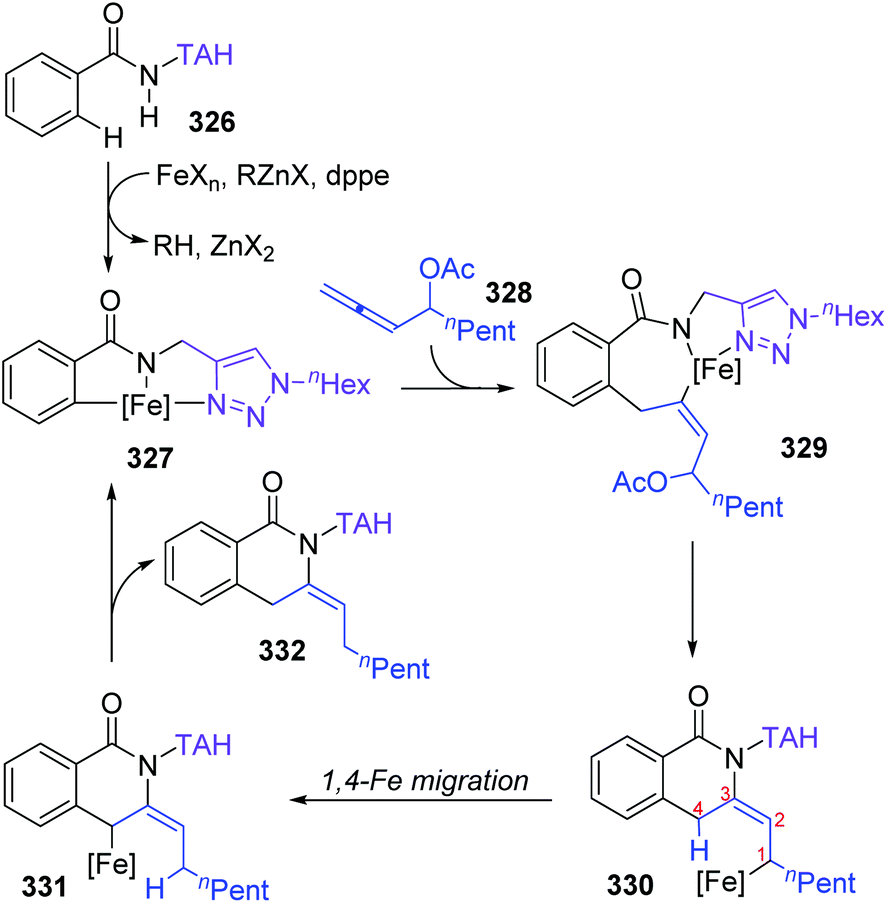
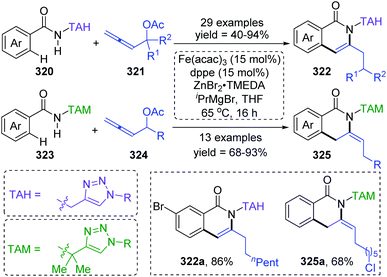
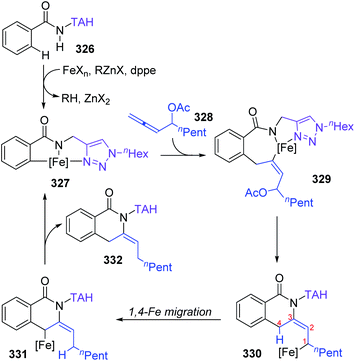
Formal iron migration reports emerged in 2018, Ackermann and co-workers described a strategy toward iron-catalyzed C–H/N–H/C–O/C–H activation and functionalization assisted by removable triazoles (Scheme 55).43 A variety of methylene-tethered triazoles 320 was converted into the corresponding isoquinolones 322 at room temperature, and further synthesis of nonaromatic exo-methylene dihydroisoquinolines 325 was also achieved by replacing the THA group with a dimethylmethylene TAM group. Various functional groups were tolerated in this transformation, for example, bromo substituents on arenes survived very well to provide target product 322a in high yields. Based on the results of preliminary mechanism investigations, a possible reaction pathway involving a formal 1,4-iron migration process is demonstrated in Scheme 56. Taking substrate 326 as an example, initial facile C–H metalation generated an aryliron complex 327, followed by allene migratory insertion to afford the intermediate 329. Then oxidation-induced reductive elimination of 329 formed an allyl iron species 330, which experienced 1,4-iron migration to give the stabilized allylic-benzylic iron intermediate 331. The process of 1,4-iron/hydrogen shift was strongly supported by deuterium-labeling experiments. Ultimate proto-demetalation with the amide motif of the substrate 331 furnished exo-methylene-3,4-dihydroisoquinoline 332, and further isomerization generated the corresponding isoquinolone products 322.
 |
| | Scheme 55 Ackermann's strategy of Fe-catalyzed C–H/N–H/C–O/C–H activation. | |
 |
| | Scheme 56 Possible reaction mechanism for the synthesis of isoquinolones and dihydroisoquinolines. | |
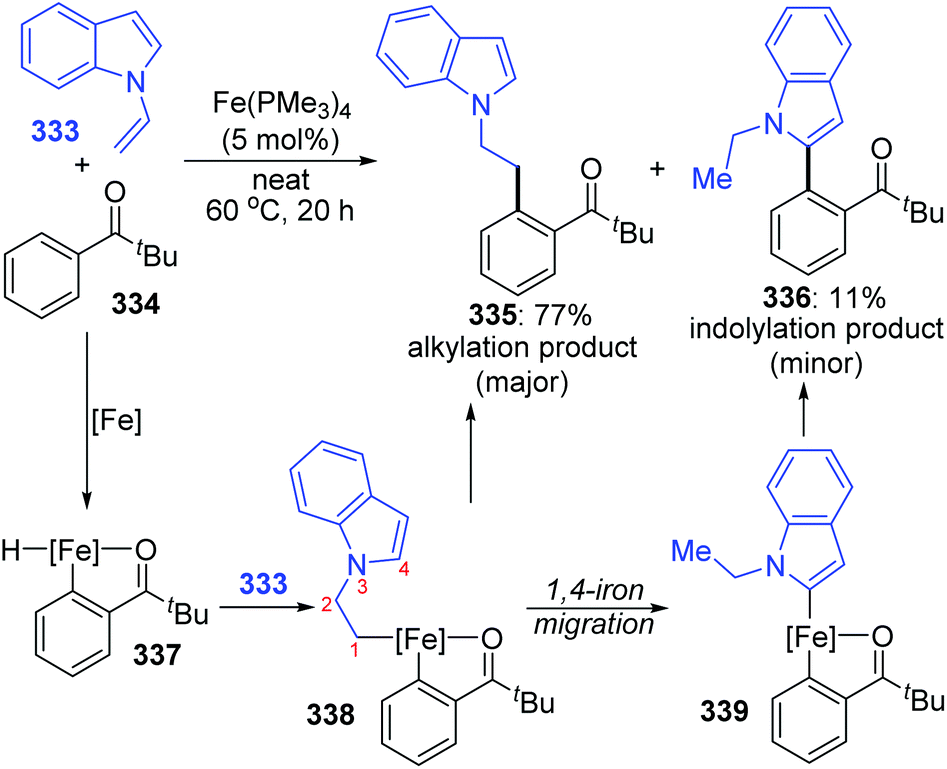
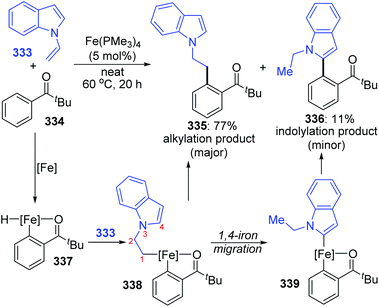
An iron-catalyzed ortho-selective C–H alkylation of aromatic ketones 334 with N-alkenylindoles 333 by employing a Fe(OMe3)4 catalyst was developed by Kakiuchi and co-workers (Scheme 57).44 A minor product indolylation species 336 was considered to occur via a 1,4-iron migration procedure. As shown, initial C–H oxidative addition of aromatic ketones 334 to the iron catalyst and subsequent hydrometalation of N-alkenylindoles 333 gave an organoiron complex 338. The uncommon alkyl-to-aryl 1,4-iron migration prior to reductive elimination took place to form an indolyliron intermediate 339, which was responsible for the minor indolylation product. Although the indolylation product 336 was obtained in very low yield, it offered a new perspective for direct introduction of an aryl group at the ortho position of aromatic ketones.
 |
| | Scheme 57 Iron-catalyzed ortho-selective C–H alkylation of aromatic ketones with N-alkenylindoles. | |
4.2 1,5-Iron migration
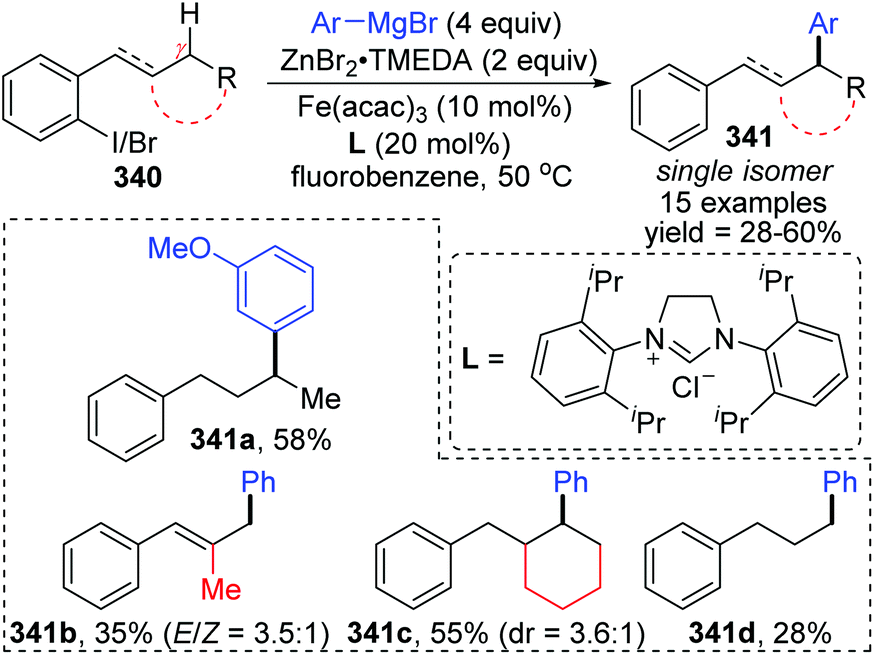
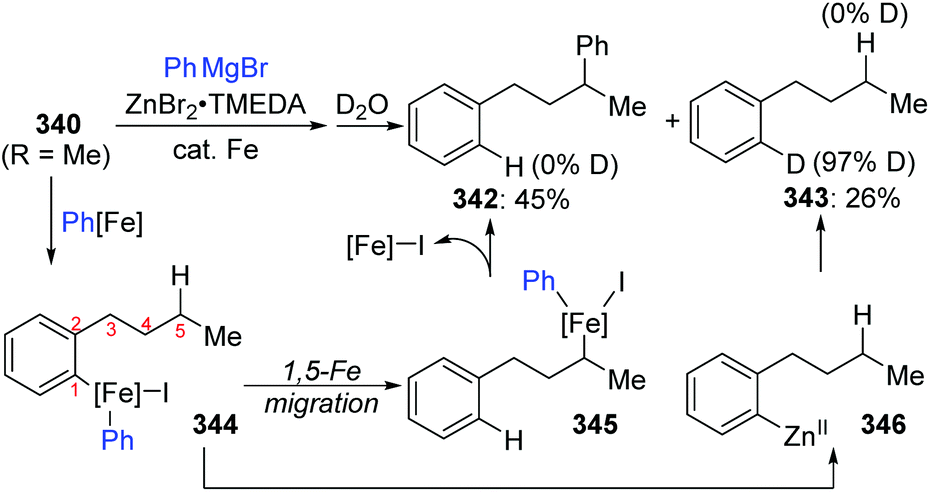
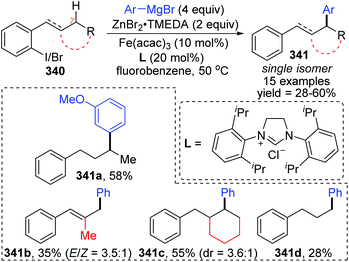
A novel 1,5-iron migration was observed in Fe-catalyzed arylation of the aliphatic C–H bonds of 2-iodoalkylarenes 340 by Nakamura and Ilies et al. (Scheme 58).45 Long alkyl chains were arylated with high regioselectivity at the γ-C–H bond by employing an iron(III) salt and a N-heterocyclic carbene ligand, providing diverse arylated alkanes 341 whose synthesis typically relied on transition-metal-catalyzed cross-coupling using alkyl halides that were difficult to access. Proposed key reaction intermediates are illustrated in Scheme 59. An organoiron complex Ph-[Fe] was envisioned to be generated from the iron salt and organometallic reagent, which might add to the aryl iodide 340via single electron transfer (SET) to generate an iron intermediate 344. Notably, unlike the classical type of metal migration discussed above, this iron migration was supposed to occur via a radical pathway. The preliminary suggestion was that 344 behaved in a radical manner and cleaved the aliphatic C–H bond via 1,5-hydrogen/iron shift to afford an alkyliron species 345, which then furnished the target product 342 through reductive elimination. However, no direct evidence supports this conclusion yet, and further investigations are imperative. Moreover, the reaction quenched with D2O afforded the phenylated product 342 with no deuterium incorporation, while the dehalogenated by-product 343 exhibited 97% deuterium incorporation on the carbon where the iodine atom was located. The existence of by-product 343 also should be responsible for the relatively low conversion yields.
 |
| | Scheme 58 Iron-catalyzed remote arylation of aliphatic C–H via a 1,5-Fe shift. | |
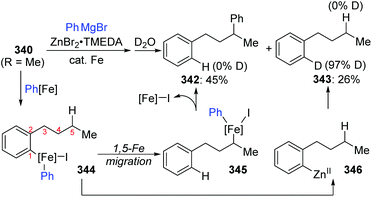 |
| | Scheme 59 Deuterium labeling experiment for Fe-catalyzed remote arylation. | |
5 Cobalt migration
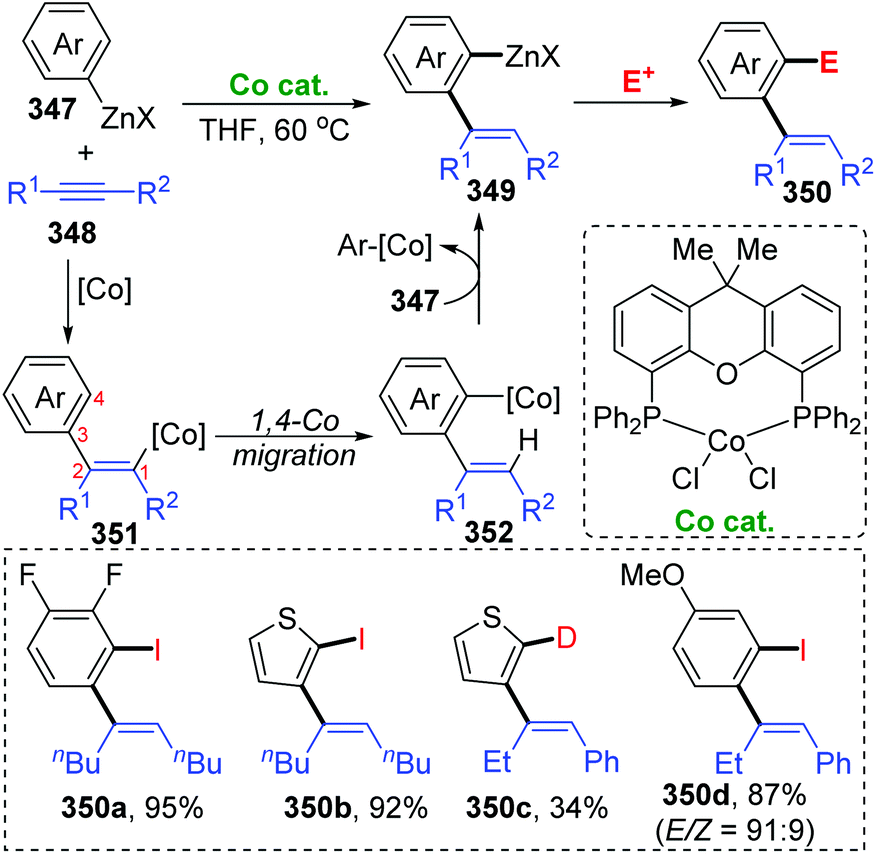
Until now, all three reports involving cobalt migration have come from Yoshikai's team. In 2012, the first example of cobalt-migration involving reactions was described by Yoshikai and co-workers.46 Diverse ortho-alkenylarylzinc species 349 were prepared by addition of arylzinc reagents 347 to alkynes 348 in the presence of Co-catalyst (Scheme 60). In the beginning, arylcobalt species were generated from the arylzinc reagents and active Co-catalyst, and the subsequent insertion into the alkyne formed an alkenylcobalt intermediate 351. Then, 1,4-cobalt migration via C–H bond activation took place to afford an ortho-alkenylarylcobalt species 352, which underwent transmetalation with the arylzinc reagent 347 to furnish a crucial ortho-alkenylarylzinc species 349 and regenerate the arylcobalt species. By employing different external electrophiles, the ortho-alkenylarylzinc species 349 could be easily converted into various corresponding products efficiently.
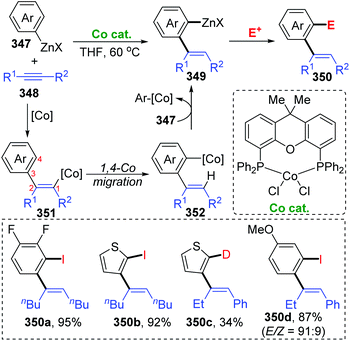 |
| | Scheme 60 Yoshikai's cobalt-catalyzed addition of arylzinc reagents to alkynes. | |
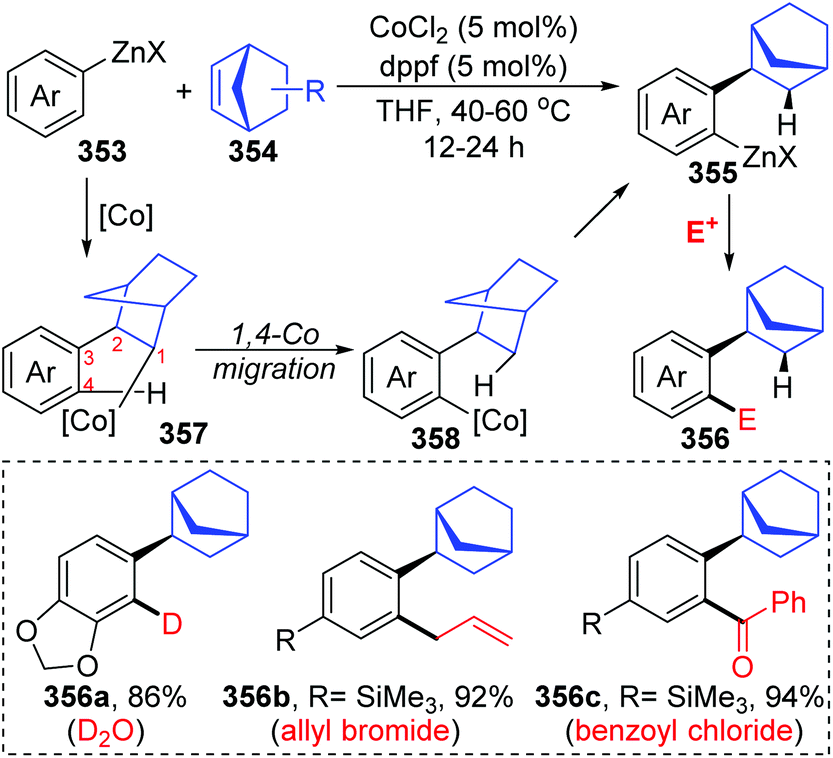
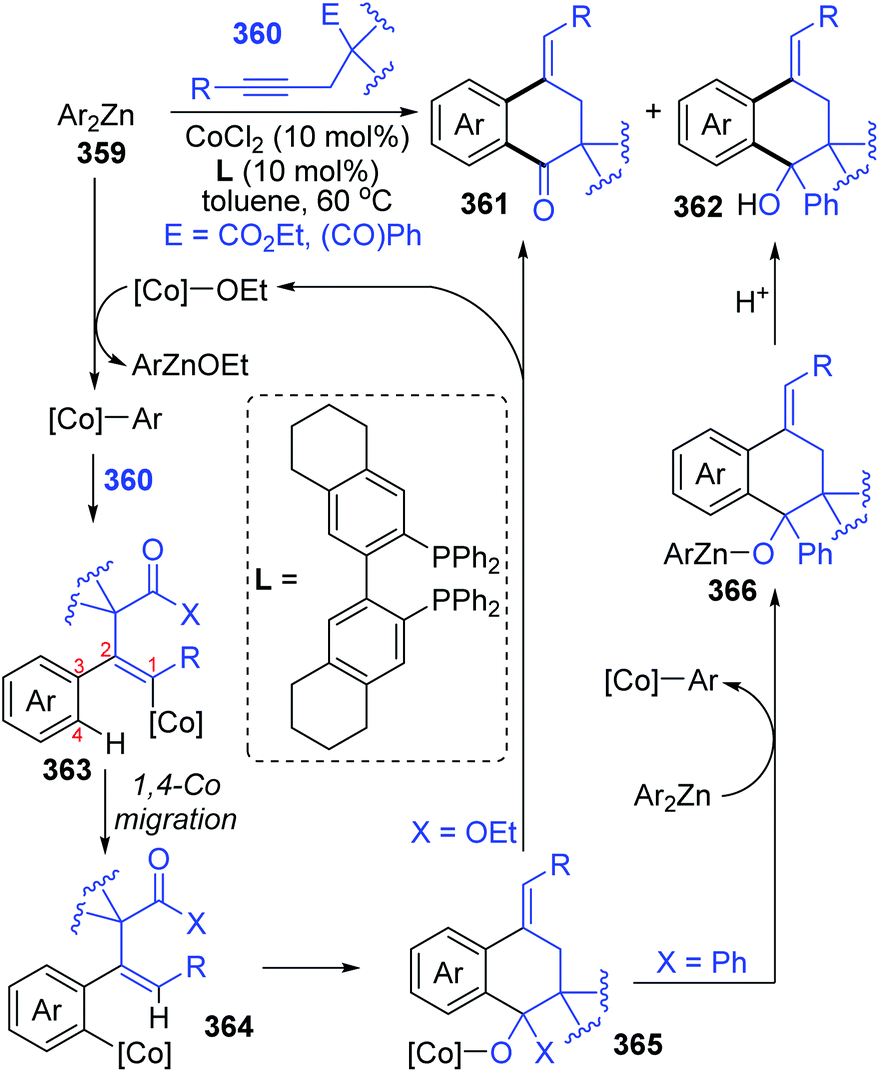
In the next four years, the Yoshikai group further developed two strategies involving 1,4-cobalt migration by using arylzinc reagents: (i) synthesis of o-(2-exo-norbornyl)arylzinc species 353via addition of arylzinc reagents to norbornene derivatives 354 (Scheme 61),47 and (ii) cyclization of acetylenic esters/ketones 360 with arylzinc reagents (Scheme 62).48 As shown in Scheme 61, the transformations of preparing o-(2-exo-norbornyl)arylzinc compounds 355 proceeded in a similar way as the first example described above; however, an alkyl-to-aryl 1,4-coblat migration occurred instead of the vinyl-to-aryl one. Then, common electrophilic trapping reactions under copper or palladium catalysis gave diverse products in excellent yields, such as 356b and 356c. The third case illustrated in Scheme 62 is slightly different from the first two cases; without extra electrophiles, a diarylzinc reagent was utilized to react with acetylenic esters/ketones 360 to prepare a series of benzo-fused cyclic ketone or alcohol products (361 and 362). Likewise, starting with the formation of arylcobalt species, successive insertion of alkynes and vinyl-to-aryl 1,4-coblat migration took place to form an ortho-alkenylarylcobalt complex 364. In the presence of an intramolecular carbonyl group, 6-exo-cyclization occurred readily to afford bicyclic skeleton intermediate 365. Subsequent β-alkoxy elimination furnished the benzo-fused cyclic ketones 361 (X = OEt) with a concomitant Co–OEt species, which further underwent transmetalation with the arylzinc reagent to regenerate the arylcobalt species. Alternatively, direct transmetalation of 365 with the arylzinc reagent could give a zinc alkoxide compound 366 and regenerate the arylcobalt species straight (X = Ph). The benzo-fused cyclic alcohol products 362 were obtained via protonation of zinc alkoxide compound 366.
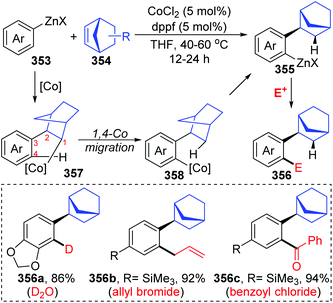 |
| | Scheme 61 Co-Catalyzed synthesis of o-(2-exo-norbornyl)arylzinc species from arylzinc reagents. | |
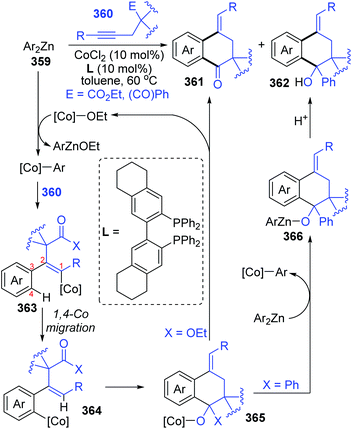 |
| | Scheme 62 Co-Catalyzed cyclization of acetylenic esters/ketones with arylzinc reagents. | |
6 Iridium migration
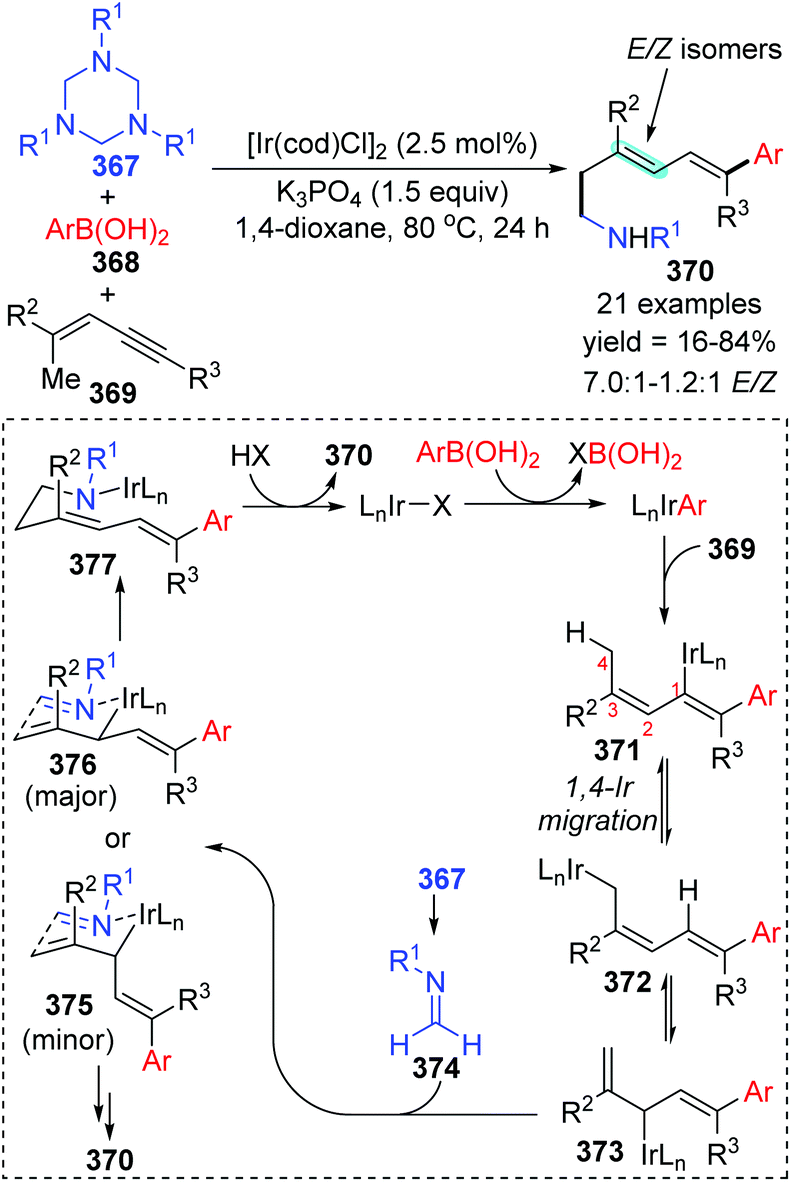
In 2014, a iridium-catalyzed three-component coupling reaction of 1,3-enynes with arylboronic acids and triazinanes was achieved by Lam and co-workers via alkenyl-to-allyl 1,4-iridium(I) migration, furnishing diverse 1,5-functionalized 1,3-dienes 370 (Scheme 63).49 The conversion started with transmetalation of an active iridium complex with arylboronic acid, which generated an aryliridium species. Further coordination and insertion of alkyne into the aryliridium species led to the alkenyliridium intermediate 371, which subsequently turned into an allyliridium species 372via alkenyl-to-allyl 1,4-iridium(I) migration. Then, the complex 373 was formed as a result of an σ–π–σ isomerization of species 372. Subsequent allylation of imine 374 with complex 373 through a chair-like conformation 376 (major transition state) afforded the iridium amide 377, which offered the final product 370 by protonation. Alternatively, the formation of conformation 375 (minor transition state) was responsible for the E/Z isomers of the target products.
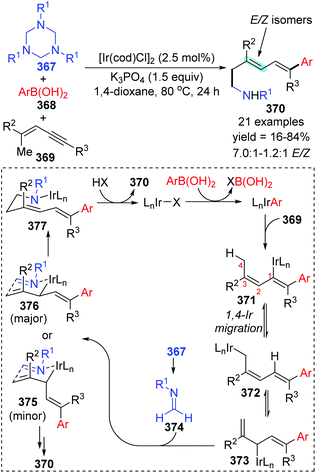 |
| | Scheme 63 Ir-Catalyzed 1,5-difunctionalization of 1,3-enynes with arylboronic acids and triazinanes. | |
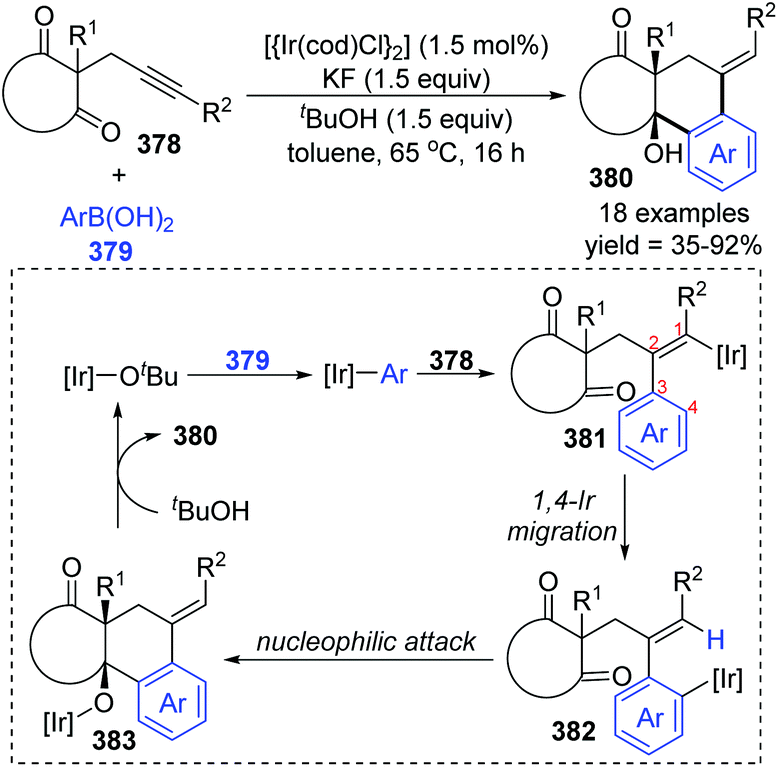
Later the same year, Lam et al. reported another case involving 1,4-iridium migration. Various complex polycycles 380 were prepared with good stereoselectivity via iridium-catalyzed arylative cyclization of alkynones 378 (Scheme 64).50 The transformation of 378 was conducted with PhB(OH)2 in the presence of [{Ir(cod)Cl}2] (1.5 mol%), KF (1.5 equiv.) as a mild base, and t-BuOH (1.5 equiv.) as a proton source. After the formation of [Ir]-Ot-Bu from a precatalyst, an aryliridium species was formed subsequently via transmetalation between the [Ir]-Ot-Bu and arylboronic acid. Migratory insertion of the alkyne 378 to aryliridium species took place to afford alkenyliridium complex 381, and then an alkenyl-to-aryl 1,4-iridium migration occurred to give the aryliridium intermediate 382. In the presence of an intramolecular carbonyl, the 6-exo cyclization triggered by nucleophilic attack generated a tertiary-alcohol-containing tricycle 383, followed by protonation to furnish the final product 380 and concomitant iridium butoxide.
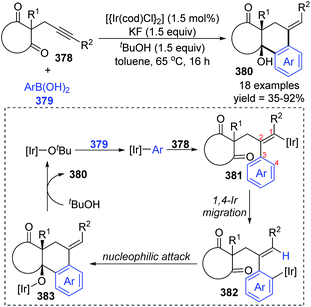 |
| | Scheme 64 Lam's strategy toward the iridium-catalyzed cyclization of alkynones. | |
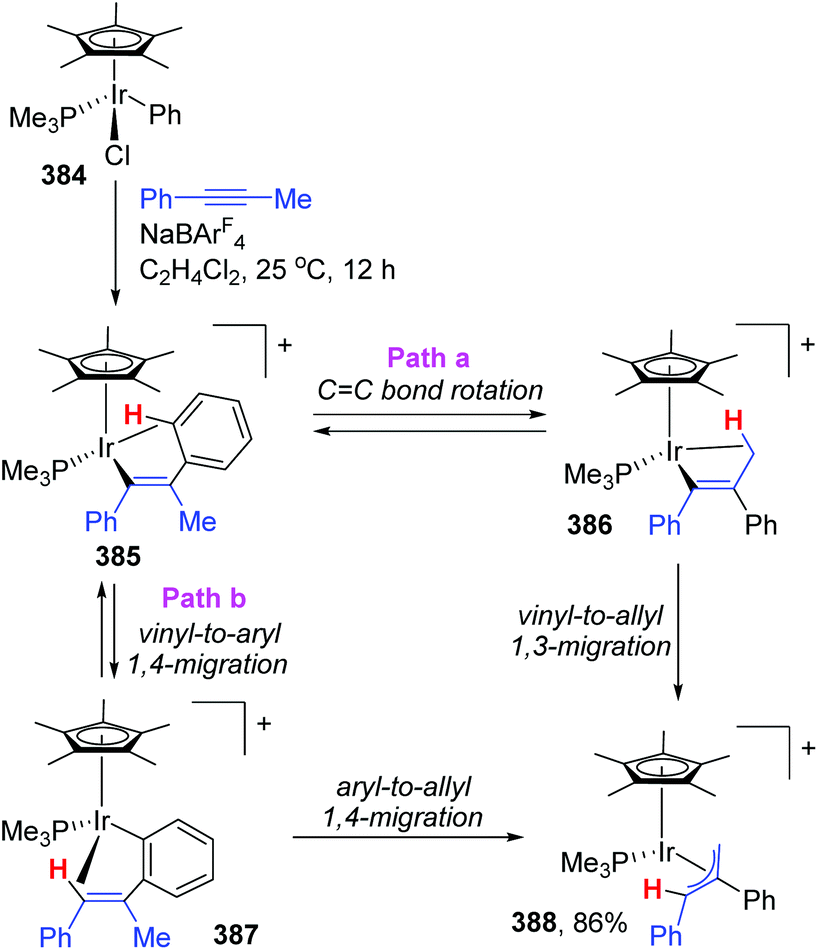
The appearance of iridium-migration was also observed in the reaction of phenyliridium(III) complex [Cp*IrCl-(Ph)(PMe3)] 384 with PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CMe in the presence of NaBArF4 at 25 °C, which furnished a π-allyl complex 388 as the end product in 86% yield (Scheme 65).51 Ishii and co-workers studied the intermediates of the reaction in depth by single-crystal X-ray and deuterium labeling experiments, and pointed out that both 1,3- and 1,4-iridium migration was involved in the formation of π-allyl complex 388. The procedure is also depicted in Scheme 65. This π-allyl complex 388 was produced via two distinct mechanisms, (i) CC bond rotation in 385 and subsequent direct vinyl-to-allyl 1,3-Ir migration (path a), and (ii) successive vinyl-to-aryl and aryl-to-allyl 1,4-Ir migrations (path b). Both of the pathways were strongly supported by deuterium labeling experiments. Overall, this reaction provided the first experimental evidence of formal 1,3-metal migration accompanied by C–H bond activation, which was of great significance for the future metal migration strategies.
CMe in the presence of NaBArF4 at 25 °C, which furnished a π-allyl complex 388 as the end product in 86% yield (Scheme 65).51 Ishii and co-workers studied the intermediates of the reaction in depth by single-crystal X-ray and deuterium labeling experiments, and pointed out that both 1,3- and 1,4-iridium migration was involved in the formation of π-allyl complex 388. The procedure is also depicted in Scheme 65. This π-allyl complex 388 was produced via two distinct mechanisms, (i) CC bond rotation in 385 and subsequent direct vinyl-to-allyl 1,3-Ir migration (path a), and (ii) successive vinyl-to-aryl and aryl-to-allyl 1,4-Ir migrations (path b). Both of the pathways were strongly supported by deuterium labeling experiments. Overall, this reaction provided the first experimental evidence of formal 1,3-metal migration accompanied by C–H bond activation, which was of great significance for the future metal migration strategies.
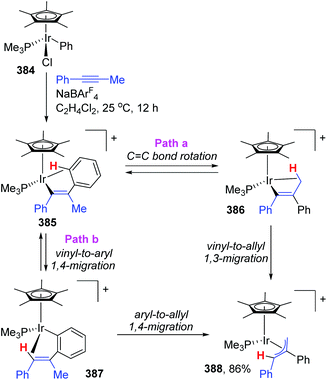 |
| | Scheme 65 1,3- and 1,4-Ir migrations in a Cp*Ir(III) complex. | |
7 Chromium migration
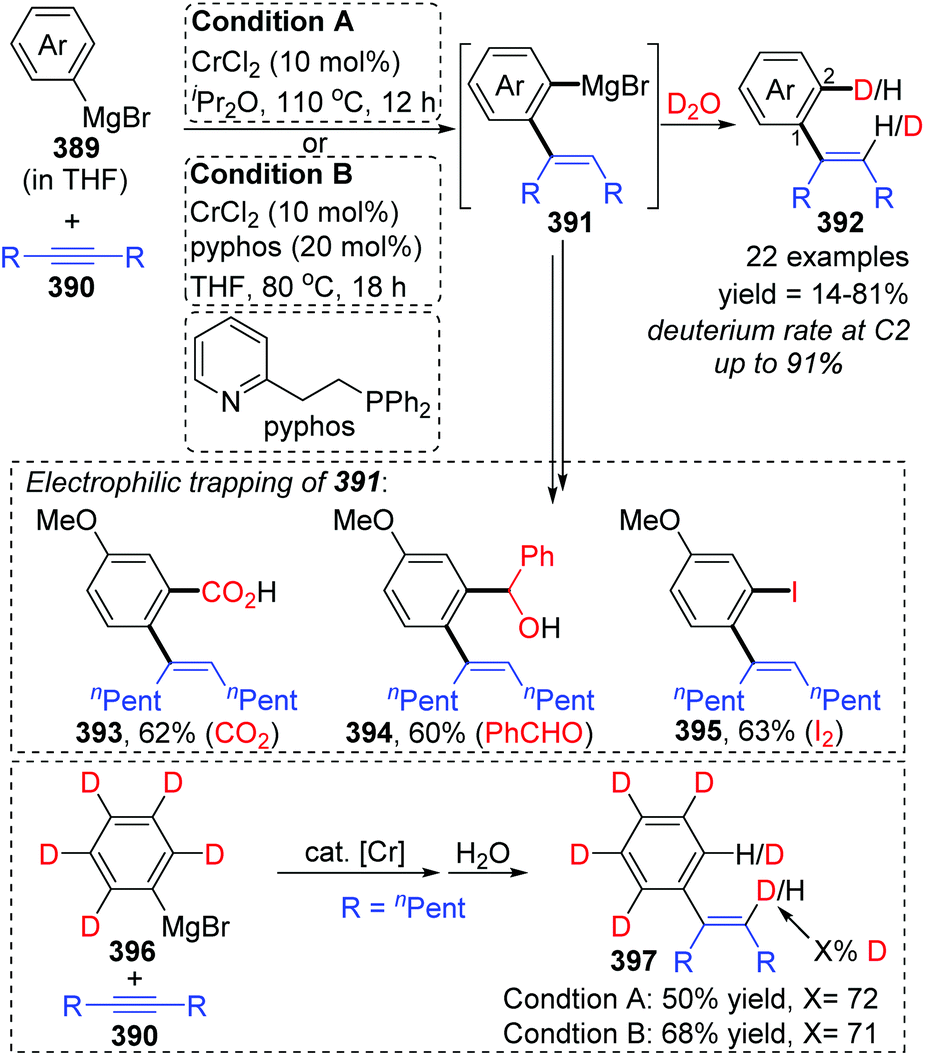
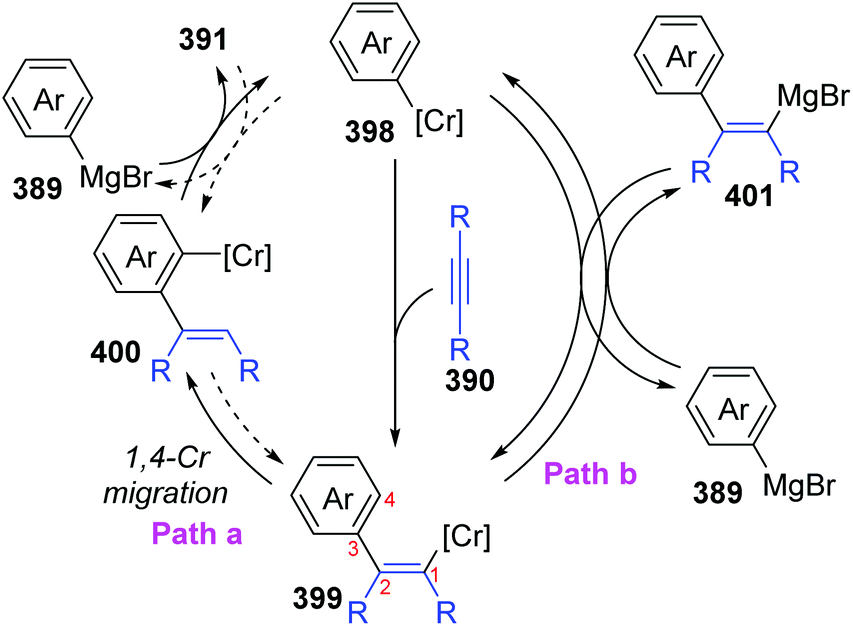
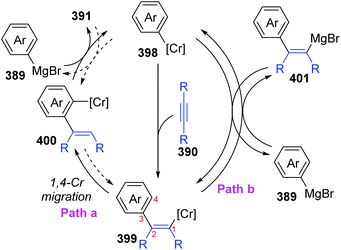
In 2017, Yan and Yoshikai described a chromium-catalyzed addition reaction of an arylmagnesium bromide 389 to an unactivated internal alkyne 390, affording the ortho-alkenylarylmagnesium bromide 391via alkenyl-to-aryl 1,4-chromium migration (Scheme 66).52 This transformation performed smoothly at almost the same level under the two reaction conditions, and the reaction was allowed to proceed at relatively low temperature (80 °C) by using the pyphos as a ligand (condition B), while the ligand-free condition was 110 °C (condition A). Reactions quenched with D2O provided various C2-deuterated adducts 392. Arylmagnesium bromides possessing an electron-donating group had good performance in this transformation, while employment of alkynes 390 with different substituents at both ends resulted in quite low yields. Furthermore, a series of electrophilic trapping regents, such as CO2 and PhCHO, worked well to provide the corresponding products in moderate yields (393 to 395). Pentadeuteriophenylmangesium bromide 396 underwent the standard reactions to give target product 397 with high deuteration rate at the vinyl portion, which should result from 1,4-chromium shift. The proposed reaction pathway is demonstrated in Scheme 67. This conversion started with the generation of an arylchromium species 398 from the chromium precatalyst and Grignard reagent. Subsequent insertion of the alkyne 390 formed an alkenylchromium complex 399, which further afforded an ortho-alkenylarylchromium species 400via 1,4-chromium migration (path a). Final transmetalation with the arylmagnesium bromide furnished the crucial ortho-alkenylarylmagnesium bromide 391 with concomitant regeneration of the arylchromium species 398. In consideration of incomplete 1,4-migration observed in product 392, the reversible process of direct transmetalation between alkenylchromium species 399 and Grignard reagent 389 (path b) should be present to compete with the 1,4-Cr shift, which accounted for the formation of normal products 401.
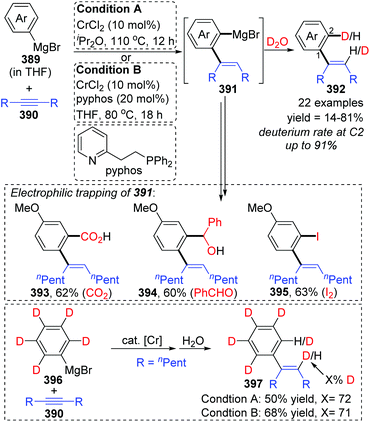 |
| | Scheme 66 Yoshikai's Cr-catalyzed reaction of arylmagnesium bromides with unactivated internal alkynes. | |
 |
| | Scheme 67 Possible reaction pathways for the reaction of arylmagnesium bromides with internal alkynes. | |
The above is the only example towards chromium migration; however, it revealed attractive resemblances compared with the 1,4-cobalt migration46 reported by the same team in 2012 (see the section on Cobalt migration). From these resemblances we can suppose that if metal-migration occurred in other Co-catalyzed reactions, it could also take place in Cr-catalyzed transformations, which needs to be verified in the future.
8 Nickel migration
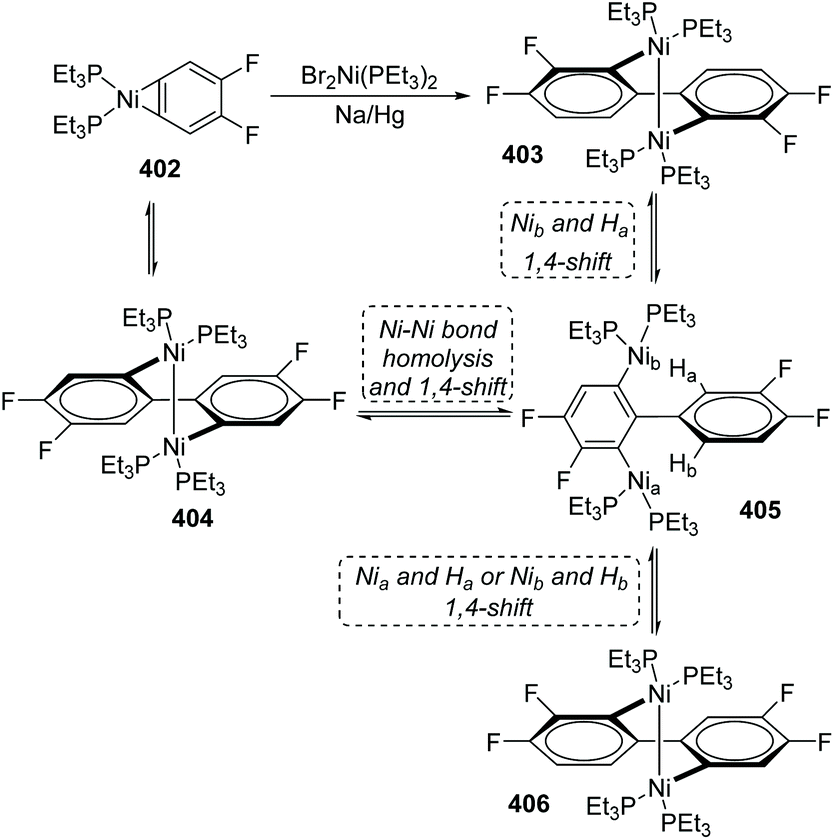
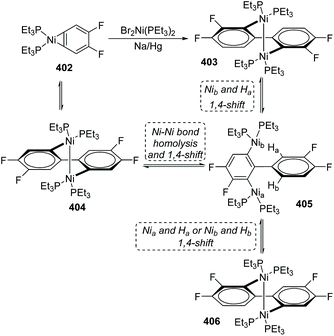
One case of nickel-migration was observed in isomerizing the aryne complex 402 into the biaryl complex 403 from Johnson's report.53 As shown in Scheme 68, the aryne complex 402 reacted with a catalytic amount of Br2Ni(PEt3)2 over 1% Na/Hg to afford the dinuclear Ni(I) biaryl complex 403via a combination of C–C bond formation and C–H bond rearrangement. Compound 404 was supposed to be formed initially from 402 under catalysis, which turned into a proposed intermediate 405 by Ni–Ni bond homolysis and 1,4-shift owing to the thermodynamic instability. Direct 1,4-shift of Nib and Ha could furnish the final biaryl complex 403 immediately; however, the concentration of an alternative product, 406, generated from 405via 1,4-shift of Nia and Ha (or Nib and Hb), was double that of 403 early in the reaction. Hence, complex 406 should be an intermediate corresponding to kinetics. The reversibility of the reactions allowed complex 406 to be converted into the more stable product 403 completely in approximately 48 h. The potential possibility of applying a nickel-migration strategy to practical synthetic reactions is revealed by Johnson's findings.
 |
| | Scheme 68 Johnson's synthesis of a dinuclear Ni(I) biaryl complex via a 1,4-Ni shift. | |
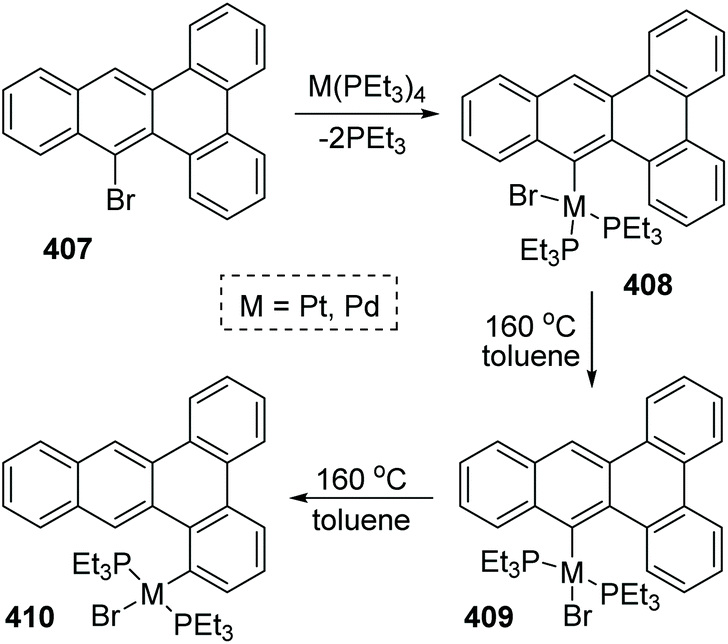
9 Platinum migration
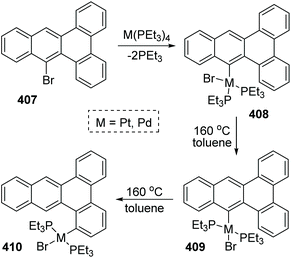
A 1,4-migration of platinum and palladium at the edge of dibenz[a,c]anthracene was reported by Sharp and Singh.54 The established process of metal migration by isolating metal complexes is illustrated in Scheme 69. When the intermediate 408 in toluene solution in a sealed-tube was heated at 160 °C, it would isomerize into trans complex 409. This transposition of a Br atom was supposed to alleviate the intense steric strain derived from the two PEt3 groups. After the isomerization was nearly completed, the concentration of 409 began to drop off, accompanied by the emergence and increase of a new migratory product 410. By comparing the structures of 409 and 410, it was revealed that the less constrained structure of 410 might account for the greater stability than 409, which was also responsible for the 1,4-migratory behavior of metal groups.
 |
| | Scheme 69 Metal-migration at the edge of dibenz[a,c]anthracene. | |
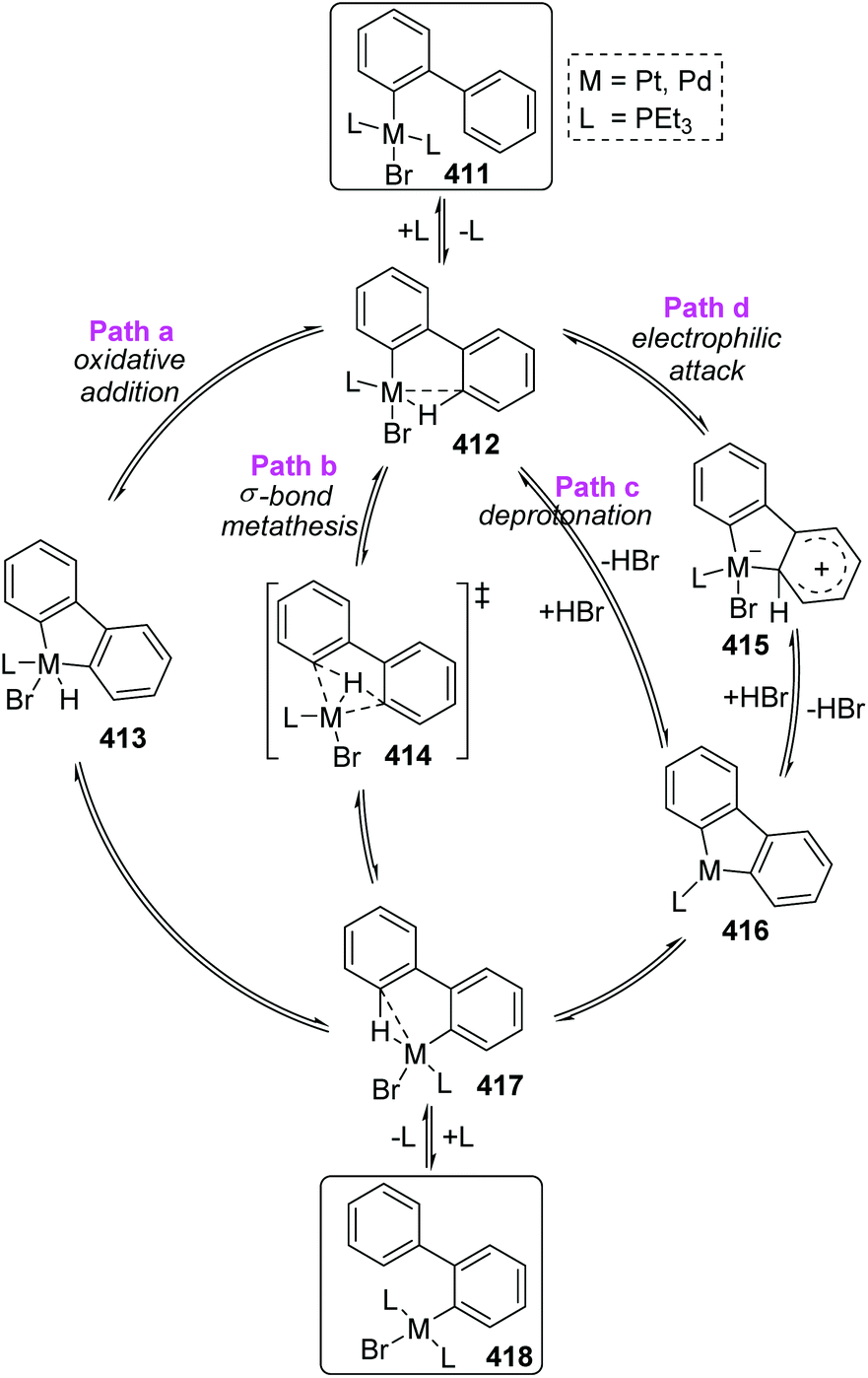
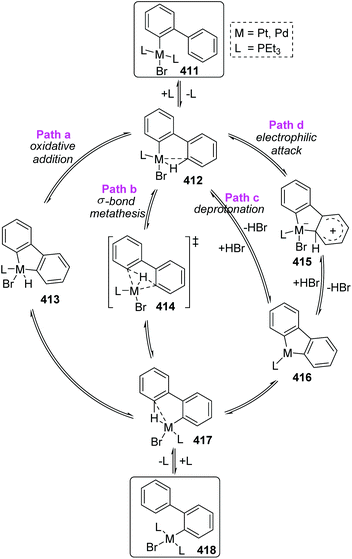
In Scheme 70, the abbreviated 411 was taken as an example to reveal the mechanistic pathways considered for 1,4-migration. The initial pre-agostic C–H of the phosphine ligand would change into a full-agostic and furnish the intermediate 412. There were several possible routes that could account for the final complex 418, such as oxidative addition of an aryl C–H bond to Pd(II) (path a), σ-bond metathesis (path b), deprotonation of an acidic agostic C–H (path c), or electrophilic attack of metal to aryl (path d). However, the deuterium scrambling experiments ruled out any possibility of path c and path d, because no deuteration was observed. Additionally, this migration process was promoted by polar solvents; therefore path b was the most possible reaction pathway and might have significant proton-transfer character.
 |
| | Scheme 70 Proposed mechanistic pathways for 1,4-migration on dibenz[a,c]anthracene. | |
10 Conclusions
This review has summarized the recent advances in transition-metal-migration involving reactions. To make the reader understand this field easily and deeply, most of the reaction mechanisms have been discussed in detail. Compared with relatively well-studied 1,4-palladium and 1,4-rhodium migration chemistry, other types of metal-migration reactions are just beginning to be studied, and most of the transition states have not been found or demonstrated. In addition, the migrations of nickel and platinum are only found in metal complexes and are not applied in practical methodologies. However, regularity of migration can still be observed from Pd- and Rh-migration strategies, providing theoretical help for other metal-migration protocols. In most cases, the metal atom coordinated with the ligands tends to shift to the position with less steric hindrance. Hence, the migration of transition-metals is more likely driven by intramolecular steric hindrance effects, for instance, the inherent spatial tension on a molecular level, or ligands with large steric hindrance may facilitate the migration process. Additionally, some experimental results involving Pd-migration strategies also revealed that the palladium atom tends to migrate to a more acidic position;55 in other words, the migration site is related to the acidity of the C–H bond. Metal-migration is considered a reversible process in many mechanistic diagrams, which is supported by extensive experimental evidence. Additionally, other competing paths, in particular irreversible β-X (X = O, N, F, etc.) elimination against 1,4-metal migration, should also be considered. Metal-migration chemistry would be promoted significantly if further migratory experimental data and details of transition states were obtained. Moreover, the application of metal-migration strategies in the total synthesis of natural products and pharmaceuticals is also worthy of further exploration. Overall, although many transition metals have only been used in metal migration strategies, the feasibility of using migration in synthetic chemistry has also been proved. Thus, the development and prospects of the metal migration field appear to be very promising, and we can expect to see many more reports involving new methodologies soon.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
H. L. is grateful for the support from the Shandong Provincial Natural Science Foundation (ZR2018MB008, ZR2018MB012), Youth Innovative Talents Attracting and Cultivating Plan of Colleges and Universities in Shandong Province, the National Natural Science Foundation of China (21671121), and the Shandong Provincial Key Research and Development program of China (2017GGX20131).
Notes and references
-
(a)
J.-Q. Yu, Catalytic Transformations via C−H activation, Science of Synthesis, Georg Thieme Verlag KG, Stuttgart, 2015, vol. 1 and 2 Search PubMed;
(b) J. F. Hartwig, Evolution of C–H bond functionalization from methane to methodology, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS.
- S. Ma and Z. Gu, 1,4-Migration of rhodium and palladium in catalytic organometallic reactions, Angew. Chem., Int. Ed., 2005, 44, 7512–7517 CrossRef CAS.
-
F. Shi and R. C. Larock, Remote C–H activation via through-space palladium and rhodium migrations, Springer, Berlin, Heidelberg, 2009, pp. 123–164 Search PubMed.
- A. Rahim, J. Feng and Z. Gu, 1,4-Migration of transition metals in organic synthesis, Chin. J. Chem., 2019, 37, 929–945 CrossRef CAS.
- T. Kochi, S. Kanno and F. Kakiuchi, Nondissociative chain walking as a strategy in catalytic organic synthesis, Tetrahedron Lett., 2019, 60, 150938 CrossRef.
- A. T. Lindhardt, T. M. Gøgsig and T. Skrydstrup, Studies on the 1,2-migrations in Pd-catalyzed Negishi couplings with JosiPhos ligands, J. Org. Chem., 2009, 74, 135–143 CrossRef CAS.
-
(a) J.-P. Ebran, A. L. Hansen, T. M. Gøgsig and T. Skrydstrup, Studies on the Heck reaction with alkenyl phosphates: can the 1,2-migration be controlled? Scope and limitations, J. Am. Chem. Soc., 2007, 129, 6931–6942 CrossRef CAS;
(b) A. L. Hansen, J.-P. Ebran, M. Ahlquist, P.-O. Norrby and T. Skrydstrup, Heck coupling with nonactivated alkenyl tosylates and phosphates: examples of effective 1,2-migrations of the alkenyl palladium(II) intermediates, Angew. Chem., Int. Ed., 2006, 45, 3349–3353 CrossRef CAS.
- E. A. Filatova, A. V. Gulevskaya, A. F. Pozharskii and V. A. Ozeryanskii, Synthesis and some properties of alkynyl derivatives of 1,3-dialkylperimidones. An example of the 1,2-palladium migration in the Sonogashira reaction, Tetrahedron, 2016, 72, 1547–1557 CrossRef CAS.
- G. Yue, Y. Wu, C. Wu, Z. Yin, H. Chen, X. Wang and Z. Zhang, Synthesis of 2-arylindoles by Suzuki coupling reaction of 3-bromoindoles with hindered benzoboronic acids, Tetrahedron Lett., 2017, 58, 666–669 CrossRef CAS.
- P. M. Kathe and I. Fleischer, Palladium-catalyzed tandem isomerization/hydrothiolation of allylarenes, Org. Lett., 2019, 21, 2213–2217 CrossRef CAS.
- H. Pang, D. Wu, H. Cong and G. Yin, Stereoselective palladium-catalyzed 1,3-arylboration of unconjugated dienes for expedient synthesis of 1,3-disubstituted cyclohexanes, ACS Catal., 2019, 9, 8555–8560 CrossRef CAS.
- C.-M. Chou, I. Chatterjee and A. Studer, Stereospecific palladium-catalyzed decarboxylative C(sp3)–C(sp2) coupling of 2,5-cyclohexadiene-1-carboxylic acid derivatives with aryl iodides, Angew. Chem., Int. Ed., 2011, 50, 8614–8617 CrossRef CAS.
- Z.-Y. Gu, X. Wang, J.-J. Cao, S.-Y. Wang and S.-J. Ji, Chemoselective Pd-catalyzed isocyanide insertion reaction of enaminones by C–H functionalization: hydrolysis or cyclization through 1,3-palladium migration, Eur. J. Org. Chem., 2015, 4699–4709 CrossRef CAS.
- P. Jiang, Y. Xu, F. Sun, X. Liu, F. Li, R. Yu, Y. Li and Q. Wang, Pd(II)-catalyzed ortho-C−H olefination/dearomatization of N-aryl ureas: an approach to imine derivatives, Org. Lett., 2016, 18, 1426–1429 CrossRef CAS.
- B.-S. Zhang, Y. Li, Z. Zhang, Y. An, Y.-H. Wen, X.-Y. Gou, S.-Q. Quan, X.-G. Wang and Y.-M. Liang, Synthesis of C4-aminated indoles via a catellani and retro-Diels−Alder strategy, J. Am. Chem. Soc., 2019, 141, 9731–9738 CrossRef CAS.
- Q. Wang, R. Chen, J. Lou, D. H. Zhang, Y.-G. Zhou and Z. Yu, Highly regioselective C−H alkylation of alkenes through an aryl to vinyl 1,4-palladium migration/C−C cleavage cascade, ACS Catal., 2019, 9, 11669–11675 CrossRef CAS.
- M.-Y. Li, P. Han, T.-J. Hu, D. Wei, G. Zhang, A. Qin, C.-G. Feng, B. Z. Tang and G.-Q. Lin, Suzuki-Miyaura coupling enabled by aryl to vinyl 1,4-palladium migration, iScience, 2020, 100966 CrossRef CAS.
- T. Tsuda, Y. Kawakami, S.-M. Choi and R. Shintani, Palladium-catalyzed synthesis of benzophenanthrosilines by C−H/C−H coupling through 1,4-palladium migration/alkene stereoisomerization, Angew. Chem., 2020, 59, 8057–8061 CrossRef CAS.
- Z.-Y. Gu, C.-G. Liu, S.-Y. Wang and S.-J. Ji, Pd-catalyzed intramolecular Heck reaction, C(sp2)−H activation, 1,4-Pd migration, and aminopalladation: chemoselective synthesis of dihydroindeno[1,2,3-kl]acridines and 3-arylindoles, Org. Lett., 2016, 18, 2379–2382 CrossRef CAS.
- M. Catellani, F. Frignani and A. Rangoni, A complex catalytic cycle leading to a regioselective synthesis of o,o’-disubstituted vinylarenes, Angew. Chem., Int. Ed. Engl., 1997, 36, 119–122 CrossRef CAS.
- A. J. Rago and G. Dong, Unexpected ortho-Heck reaction under the Catellani conditions, Org. Lett., 2020, 22, 3770–3774 CrossRef CAS.
- P. Li, Q. Li, H. Weng, J. Diao, H. Yao and A. Lin, Intramolecular remote C−H activation via sequential 1,4-palladium migration to access fused polycycles, Org. Lett., 2019, 21, 6765–6769 CrossRef CAS.
- T. Kesharwani, A. K. Verma, D. Emrich, J. A. Ward and R. C. Larock, Studies in acyl C−H activation via aryl and alkyl to acyl “through space” migration of palladium, Org. Lett., 2009, 11, 2591–2593 CrossRef CAS.
- R. Rocaboy, I. Anastasiou and O. Baudoin, Redox-neutral coupling between two C(sp3)−H bonds enabled by 1,4-palladium shift for the synthesis of fused heterocycles, Angew. Chem., Int. Ed., 2019, 58, 14625–14628 CrossRef CAS.
- Y. Yu, P. Chakraborty, J. Song, L. Zhu, C. Li and X. Huang, Easy access to medium-sized lactones through metal carbene migratory insertion enabled 1,4-palladium shift, Nat. Commun., 2020, 11, 461–469 CrossRef CAS.
- Y. Yu, L. Ma, J. Xia, L. Xin, L. Zhu and X. Huang, A modular approach to dibenzo-fused ε-lactams: palladium carbene bridging C−H activation and its synthetic application, Angew. Chem., Int. Ed. DOI:10.1002/anie.202007799.
- C. Bour and J. Suffert, Cyclocarbopalladation: sequential cyclization and C−H activation/Stille cross-coupling in the Pd-5-exo-dig reaction, Org. Lett., 2005, 7, 653–656 CrossRef CAS.
- A. J. Mota, A. Dedieu, C. Bour and J. Suffert, Cyclocarbopalladation involving an unusual 1,5-palladium vinyl to aryl shift as termination step: theoretical study of the mechanism, J. Am. Chem. Soc., 2005, 127, 7171–7182 CrossRef CAS.
- Y. Sato, C. Takagi, R. Shintani and K. Nozaki, Palladium-catalyzed asymmetric synthesis of silicon-stereogenic 5,10-dihydrophenazasilines via enantioselective 1,5-palladium migration, Angew. Chem., Int. Ed., 2017, 56, 9211–9216 CrossRef CAS.
- N. Misawa, T. Tsuda, R. Shintani, K. Yamashita and K. Nozaki, Palladium-catalyzed intramolecular C−H arylation versus 1,5-palladium migration: a theoretical investigation, Chem. – Asian J., 2018, 13, 2566–2572 CrossRef CAS.
- J.-L. Han, Y. Qin, C.-W. Ju and D. Zhao, Divergent synthesis of vinyl-, benzyl-, and borylsilanes: aryl to alkyl 1,5-palladium migration/coupling sequences, Angew. Chem., 2020, 59, 6555–6560 CrossRef CAS.
- J. Zhou, J. He, B. Wang, W. Yang and H. Ren, 1,7-Palladium migration via C−H activation, followed by intramolecular amination: regioselective synthesis of benzotriazoles, J. Am. Chem. Soc., 2011, 133, 6868–6870 CrossRef CAS.
-
(a) L.-C. Campeau, M. Parisien and K. Fagnou, Biaryl synthesis via direct arylation: establishment of an efficient catalyst for intramolecular processes, J. Am. Chem. Soc., 2004, 126, 9186–9187 CrossRef CAS;
(b) M. Leblanc and K. Fagnou, Allocolchicinoid synthesis via direct arylation, Org. Lett., 2005, 7, 2849–2852 CrossRef CAS;
(c) C. C. Hughes and D. Trauner, Concise total synthesis of (−)-frondosin B using a novel palladium-catalyzed cyclization, Angew. Chem., Int. Ed., 2002, 41, 1569–1572 CrossRef CAS.
- Y. Wang, X. Dong and R. C. Larock, Synthesis of naturally occurring pyridine alkaloids via palladium-catalyzed coupling/migration chemistry, J. Org. Chem., 2003, 68, 3090–3098 CrossRef CAS.
- J. Zhang, J.-F. Liu, A. Ugrinov, A. F. X. Pillai, Z.-M. Sun and P. Zhao, Methoxy-directed aryl-to-aryl 1,3-rhodium migration, J. Am. Chem. Soc., 2013, 135, 17270–17273 CrossRef CAS.
- A. Archambeau and T. Rovis, Rhodium(III)-catalyzed allylic C(sp3)−H activation of alkenyl sulfonamides: unexpected formation of azabicycles, Angew. Chem., Int. Ed., 2015, 54, 13337–13340 CrossRef CAS.
- N. Liu, J. Yao, L. Yin, T. Lu, Z. Tian and X. Dou, Rhodium-catalyzed expeditious synthesis of indenes from propargyl alcohols and organoboronic acids by selective 1,4-rhodium migration over β-oxygen elimination, ACS Catal., 2019, 9, 6857–6863 CrossRef CAS.
- L. O'Brien, S. N. Karad, W. Lewis and H. W. Lam, Rhodium-catalyzed arylative cyclization of alkynyl malonates by 1,4-rhodium(I) migration, Chem. Commun., 2019, 55, 11366–11369 RSC.
- S. Guo, R. Pan, Z. Guan, P. Li, L. Cai, S. Chen, A. Lin and H. Yao, Synthesis of indole-fused polycyclics via rhodium-catalyzed undirected C−H activation/alkene insertion, Org. Lett., 2019, 21, 6320–6324 CrossRef CAS.
- A. Groves, J. Sun, H. R. I. Parke, M. Callingham, S. P. Argent, L. J. Taylor and H. W. Lam, Catalytic enantioselective arylative cyclizations of alkynyl 1,3-diketones by 1,4-rhodium(I) migration, Chem. Sci., 2020, 11, 2759–2764 RSC.
- T. Matsuda, T. Izutsu and M. Hashimoto, Rhodium(I)-catalyzed arylative annulation of β-alkynyl ketones for preparation of fused aromatics, Eur. J. Org. Chem., 2020, 306–310 CrossRef CAS.
- M. Font, B. Cendón, A. Seoane, J. L. Mascareñas and M. Gulías, Rhodium(III)-catalyzed annulation of 2-alkenyl anilides with alkynes through C−H activation: direct access to 2-substituted indolines, Angew. Chem., Int. Ed., 2018, 57, 8255–8259 CrossRef CAS.
- J. Mo, T. Müller, J. C. A. Oliveira and L. Ackermann, 1,4-Iron migration for expedient allene annulations through iron-catalyzed C−H/N−H/C−O/C−H functionalizations, Angew. Chem., Int. Ed., 2018, 57, 7719–7723 CrossRef CAS.
- N. Kimura, T. Kochi and F. Kakiuchi, Iron-catalyzed ortho-selective C−H alkylation of aromatic ketones with N-alkenylindoles and partial indolylation via 1,4-iron migration, Asian J. Org. Chem., 2019, 8, 1115–1117 CrossRef CAS.
- B. Zhou, H. Sato, L. Ilies and E. Nakamura, Iron-catalyzed remote arylation of aliphatic C−H bond via 1,5-hydrogen shift, ACS Catal., 2018, 8, 8–11 CrossRef CAS.
- B.-H. Tan, J. Dong and N. Yoshikai, Cobalt-catalyzed addition of arylzinc reagents to alkynes to form ortho-alkenylarylzinc species through 1,4-cobalt migration, Angew. Chem., Int. Ed., 2012, 51, 9610–9614 CrossRef CAS.
- B.-H. Tan and N. Yoshikai, Cobalt-catalyzed addition of arylzinc reagents to norbornene derivatives through 1,4-cobalt migration, Org. Lett., 2014, 16, 3392–3395 CrossRef CAS.
- J. Yan and N. Yoshikai, Cobalt-catalyzed arylative cyclization of acetylenic esters and ketones with arylzinc reagents through 1,4-cobalt migration, ACS Catal., 2016, 6, 3738–3742 CrossRef CAS.
- R. E. Ruscoe, M. Callingham, J. A. Baker, S. E. Korkis and H. W. Lam, Iridium-catalyzed 1,5-(aryl)aminomethylation of 1,3-enynes by alkenyl-to-allyl 1,4-iridium(I) migration, Chem. Commun., 2019, 55, 838–841 RSC.
- B. M. Partridge, J. S. González and H. W. Lam, Iridium-catalyzed arylative cyclization of alkynones by 1,4-iridium migration, Angew. Chem., Int. Ed., 2014, 53, 6523–6527 CrossRef CAS.
- Y. Ikeda, K. Takano, S. Kodama and Y. Ishii, 1,4- and 1,3-Metal migration in a Cp*IrIII complex: experimental evidence of direct 1,3-metal migration, Organometallics, 2014, 33, 3998–4004 CrossRef CAS.
- J. Yan and N. Yoshikai, Chromium-catalyzed migratory arylmagnesiation of unactivated alkynes, Org. Chem. Front., 2017, 4, 1972–1975 RSC.
- A. L. Keen, M. Doster and S. A. Johnson, 1,4-Shifts in a dinuclear Ni(I) biarylyl complex: a mechanistic study of C-H bond activation by monovalent nickel, J. Am. Chem. Soc., 2007, 129, 810–819 CrossRef CAS.
- A. Singh and P. R. Sharp, Pt and Pd 1,4-shifts at the edge of dibenz[a,c]anthracene, J. Am. Chem. Soc., 2006, 128, 5998–5999 CrossRef CAS.
- M. A. Campo, H. Zhang, T. Yao, A. Ibdah, R. D. McCulla, Q. Huang, J. Zhao, W. S. Jenks and R. C. Larock, Aryl to aryl palladium migration in the Heck and Suzuki coupling of o-halobiaryls, J. Am. Chem. Soc., 2007, 129, 6298–6307 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a and
Fagang
Wang
*a
*a and
Fagang
Wang
*a