
A computational study of the reactivity of rare-earth/phosphorus Lewis pairs toward polymerization of conjugated polar alkenes†
Yanan
Zhao
 a,
Gen
Luo
ab,
Xin
Xu
c,
Zhaomin
Hou
ad and
Yi
Luo
*a
a,
Gen
Luo
ab,
Xin
Xu
c,
Zhaomin
Hou
ad and
Yi
Luo
*a
aState Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China. E-mail: luoyi@dlut.edu.cn
bInstitutes of Physical Science and Information Technology, Anhui University, Hefei 230601, China
cKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China
dOrganometallic Chemistry Laboratory, RIKEN Cluster for Pioneering Research, and Advanced Catalysis Research Group, RIKEN Center for Sustainable Resource Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
First published on 6th October 2020
Abstract
The polymerization mechanism of methyl methacrylate (MMA) catalyzed by rare-earth/phosphorus (RE/P) Lewis pairs has been systematically studied through density functional theory (DFT) calculations. Having achieved an agreement between theory and experiment, it is found that the polymerization of MMA mediated by intermolecular RE/P Lewis pairs mainly follows the bimetallic mechanism, while the monometallic pathway could not be excluded in the case of a La analogue. In comparison with phenyl phosphorus as a Lewis base, the higher activity of cyclohexyl phosphorus toward MMA polymerization could be ascribed to the electron-donation ability, rendering more electron flow in the addition reaction. Besides, a computational modelling of analogous intramolecular RE/P systems indicates that the size of the central metal and the length of the chain connecting Lewis pairs play an important role in the catalytic activity.
Introduction
Polymers derived from conjugated polar monomers have attracted much attention because an unsaturated bond and a polar functional group remain in the polymer chain and thus are capable of meeting specific applications or undergoing further functionalization. Besides the radical and anionic polymerization methods,1,2 the catalytic coordination insertion3 and conjugate addition such as group-transfer polymerization (GTP)4 and Lewis pair polymerization (LPP)5 are also useful for the synthesis of such polymers. The former is usually problematic in controllability, and transition-metal catalyzed coordination insertion is known to suffer from poisoning by the functional group.3 In the context of conjugate addition polymerization, LPP has attracted recent interest, thanks to the cooperation between the Lewis acid and base in chain initiation, propagation, and termination. In view of the cooperative behavior in LPP, the polymerization activity could be regulated by both the Lewis acid and Lewis base and thus showed rich chemistry, as demonstrated by previous works.6 Since the first LPP work reported by Chen and coworkers in 2010,5a significant progress has been made in this field.5b–h In Chen's primary work, Al(C6F5)3-based Lewis pairs were demonstrated to catalyze the polymerization of conjugated polar alkenes and a high molecular weight polymerization product was obtained. Hong7 and Zhang6,8 reported a series of main-group FLP catalysts for the polymerization of a wide range of conjugated polar monomers, some of which showed living and controllable features. In spite of group 4 metal involved ion-pair polymerization,9 transition-metal containing LPP is not well established.10,11 Therefore, a highly active transition-metal containing LPP system is still a worthy challenge, which could be improved by modulation of Lewis acids and bases.It is known that rare-earth metal complexes were reported to not only mediate the synthesis of small molecules12,13b but also catalyze the polymerizations of non-polar or polar olefins.13 To regulate the acidity of Lewis acids in LPP, Xu et al. developed intramolecular and intermolecular rare-earth/phosphorus (RE/P) Lewis pairs (Chart 1) for polymerizing conjugated polar monomers such as methyl methacrylate (MMA).14 Their works demonstrated that the polymerization activity is significantly affected by rare-earth metals and Lewis bases. The authors observed that La complex 3 showed higher activity than Sc analogue 2 and Lewis base PCy3 exhibited higher activity compared with PPh3. However, the origin of such activity discrepancy remained unclear.
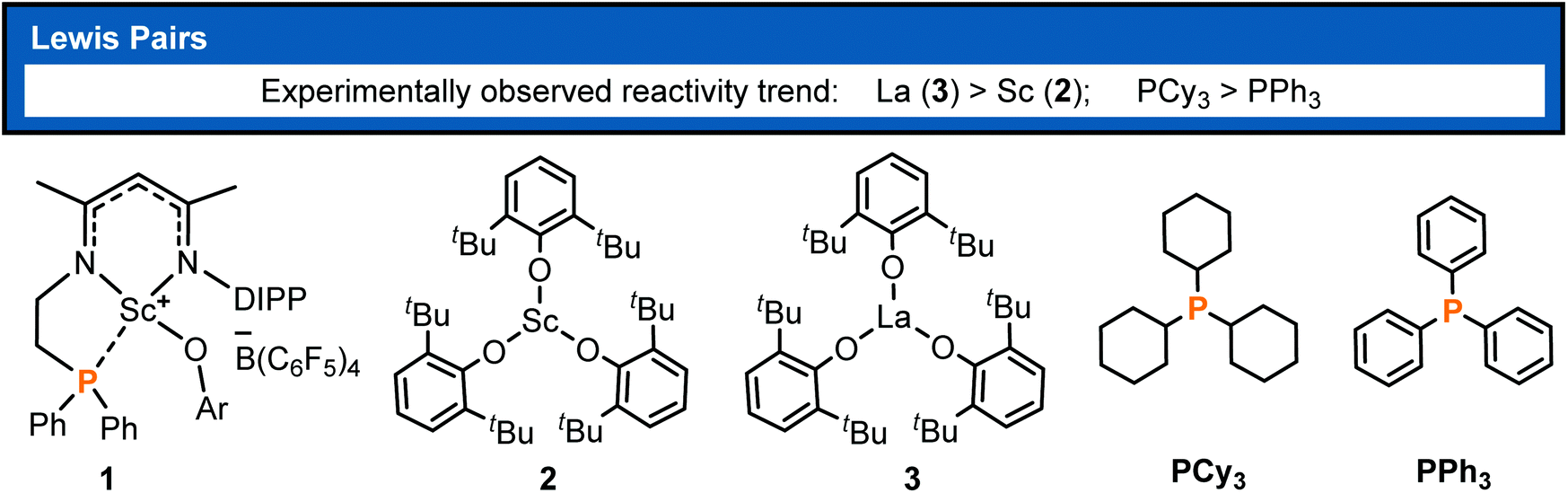 | ||
| Chart 1 Rare-earth metal Lewis pairs reported for LPP.14 | ||
A few theoretical mechanistic works on the mechanism of MMA polymerization by main-group FLPs have been documented.15–17 Chen and Cavallo at el. reported the calculations on the formation of the zwitterionic active species in LPP of MMA and found that NHC-based zwitterions were more stable compared with PR3-based analogues and the MMA addition favorably followed the bimetallic pathway.15 They also conducted mechanistic studies on the chain propagation and termination of LPP of conjugated polar monomers catalyzed by Al-based FLPs and found that intramolecular backbiting cyclization might account for the chain-termination,16 being in line with separate works by Lu et al.18 Taton and coworkers computationally demonstrated a cooperative mechanism in MMA polymerization catalyzed by a PR3/Me3SiNTf2 Lewis pair.17 In spite of these mechanistic studies on the main-group LPP, the theoretical study of such 1,4-conjugate addition polymerization catalyzed by RE/P Lewis pairs, to the best our knowledge, has not been reported to date.
Stimulated by our previous theoretical works on rare-earth metal catalyzed polymerizations,19 we are interested in the molecular-level factors governing the polymerization activity observed in the RE/P systems, with the purpose of elucidating the related reaction mechanism. It is generally considered that, in the LPP, stronger acidity of the Lewis acid could induce higher activity. Our primary calculation based on the HSAB (hard and soft acids bases) principle and DFT indicates that 2 has higher chemical hardness and thus stronger Lewis acidity in comparison with 3 (0.20 vs. 0.16 eV).19c,20 However, as aforementioned, the activity of 3 is higher than that of 2,14b which drove us to explore the origin of such activity discrepancy. In the present work, DFT calculations have been conducted for the intramolecular and intermolecular RE/P Lewis pairs (Chart 1) toward the polymerization of MMA, which is a commercially important and scientifically interesting commodity acrylic monomer. On the basis of the clarification of the related polymerization mechanism, the origin of different activity between Sc and La analogues and the effects of different Lewis bases have been elucidated, which could provide a piece of information for the development of LPP systems.
Results and discussion
Polymerization mechanism
It is generally considered that FLP (frustrated Lewis pair) mediated polymerization of conjugated polar alkenes follows Michael addition to grow a polymer chain rather than coordination insertion or single electron transfer.15,16 In the case of intramolecular Lewis pairs such as 1+ (Chart 1), as shown in Fig. 1, the reaction starts with the coordination of monomer MMA and then 1,4-addition (1acoor → 1aTS → 1a) could occur to provide 1a with a newly formed P–C bond. While the incoming monomer approaches the metal center of the active species 1a, the 1,4-addition reaction repeatedly takes place to form C–C bonds and the polymerization could smoothly occur. Such a C–C formation process has a moderate energy barrier (19.6 kcal mol−1) and is exergonic (Fig. 1), suggesting a feasible polymerization event.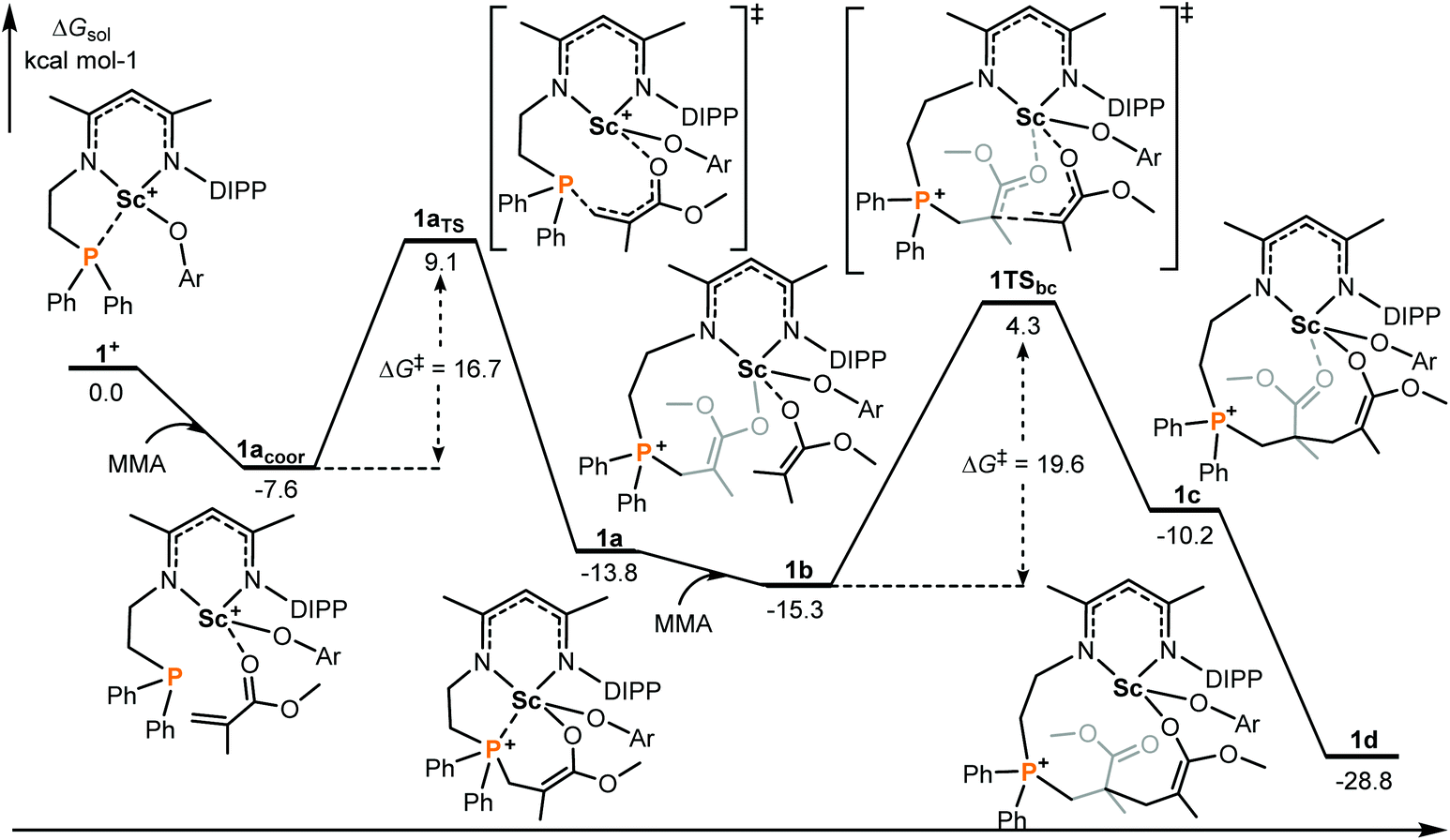 | ||
| Fig. 1 Computed energy profiles (M06(SMD)/6-311G**∩SDD//B3PW91/6-31G*∩SDD), see the ESI† for more details for 1+ mediated polymerization of MMA (intramolecular RE/P systems). Free energies (kcal mol−1) are relative to isolated reactants. | ||
In the case of the intermolecular RE/P Lewis pair (Chart 1), the monometallic and bimetallic mechanisms have been considered, respectively. As shown in Fig. 2, starting with 2C, the energy barrier for the monometallic mechanism (2TSde, 34.9 kcal mol−1, see Fig. S2 in the ESI† for the complete monometallic pathway) is significantly higher than that for the bimetallic pathway (2TSDE, 20.2 kcal mol−1), showing a preference for the bimetallic reaction manner. Although the enchainment of the second MMA is slightly endergonic (conversion of 2C to 2F), the addition of the third MMA is exergonic (2F to 2H), suggesting an energetically favorable enchainment process.
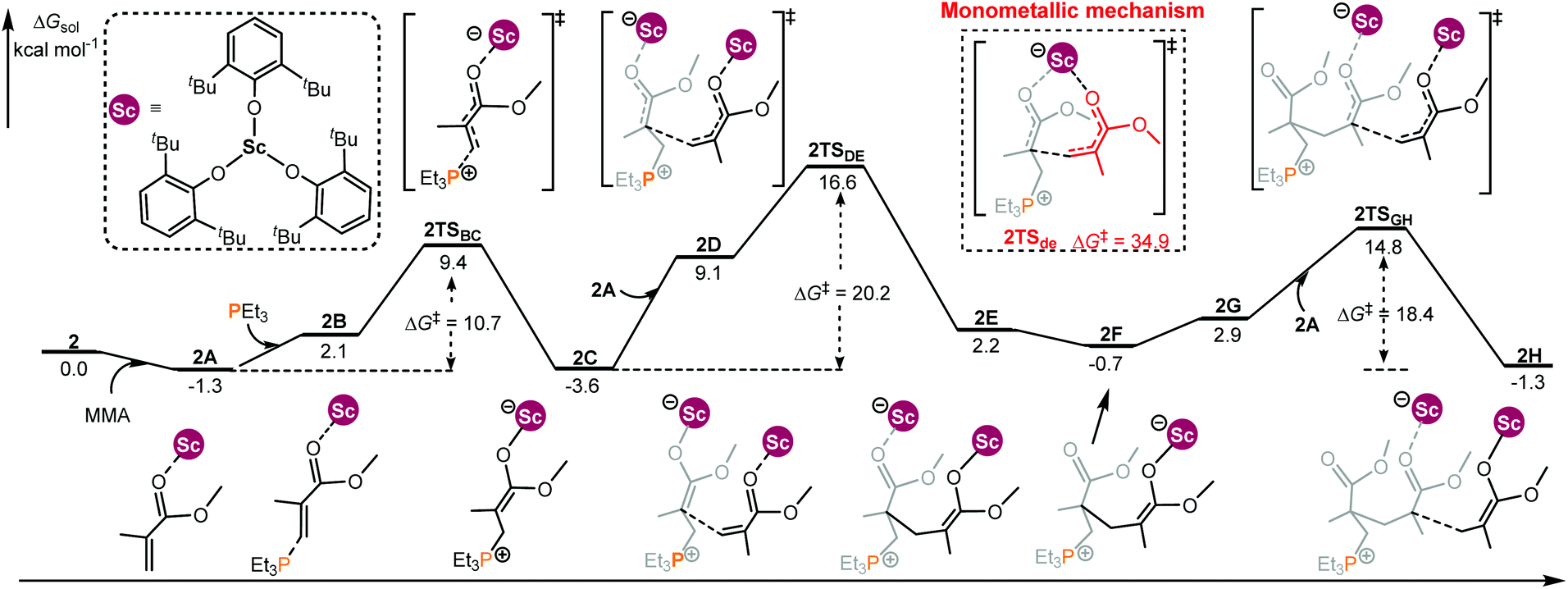 | ||
| Fig. 2 Computed energy profiles for 2/PEt3 mediated (intermolecular RE/P systems) bimetallic enchainment of MMA. Free energies (kcal mol−1) are relative to isolated reactants. | ||
To obtain better understanding of the priority of the bimetallic mechanism, the distortion/interaction analyses21 have been comparatively performed for the key transition states 2TSde and 2TSDE. During such analyses, the energies of the monomer moiety and the remaining metal complex (two fragments) in the TS geometries were evaluated via single-point calculations. Such single-point energies of the fragments and the energy of the TS were used to estimate the interaction energy ΔEint. These energies, together with the energies of the respective fragments in their optimal geometry, allow for the estimation of the deformation energies of the two fragments, ΔEdef(cat.) and ΔEdef(mono.). As the energy of the TS, ΔETS, is evaluated with respect to the energy of the two separated fragments, the relation ΔETS = ΔEint + ΔEdef(cat.) + ΔEdef(mono.) holds. As shown in Fig. 3, the total deformation energy ΔEdef in 2TSde is 60.7 (12.8 + 47.9) kcal mol−1, which could be hardly balanced out by its ΔEint (−12.3 kcal mol−1), leading to high ΔETS of 48.4 kcal mol−1. However, the ΔEdef in 2TSDE (31.9 kcal mol−1) is compensated by an interaction energy of −9.2 kcal mol−1, leading to ΔETS = 22.7 kcal mol−1, which is lower than that for 2TSde. As a consequence, the less steric hindrance inducing smaller deformations of the metal complexes could account for the higher stability of 2TSDE. A comparison of the structures of the two transition states indicates that the C1–C2 distance in 2TSde is obviously shorter than that in 2TSDE (2.09 vs. 2.17 Å, Fig. 3), suggesting a more crowded environment at the reaction center in 2TSde. This is also consistent with the greater deformations in the 2TSde case.
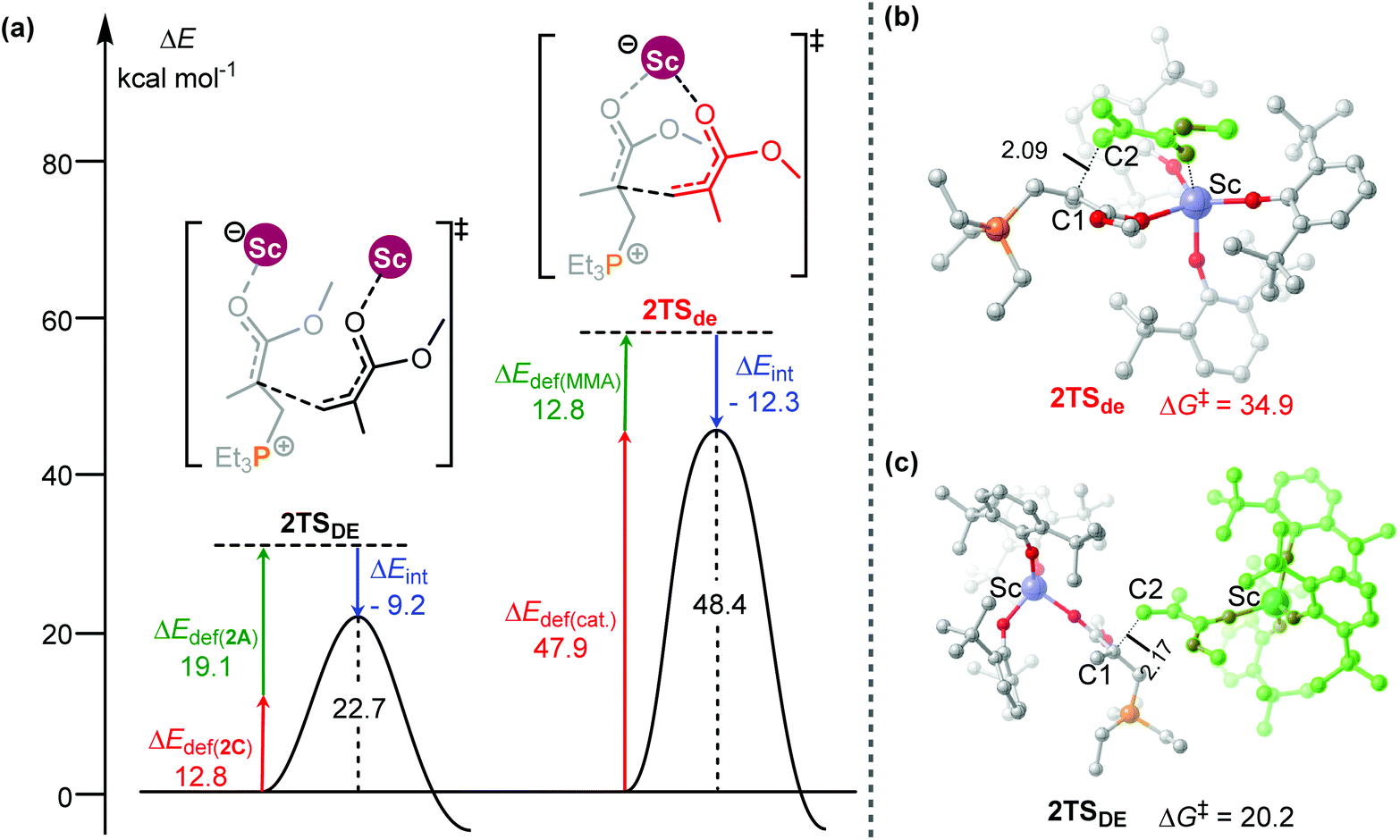 | ||
| Fig. 3 (a) Distortion/interaction analyses (kcal mol−1) and geometric parameters (distances in Å) of (b) 2TSde and (c) 2TSDE. | ||
Metal effect on catalyst activity
It was reported that different metal centers led to a significant activity difference in the intermolecular FLP system.14b This drove us to computationally compare the MMA addition mediated by an analogous complex such as 3 (La complex) with the same ligand as that in 2 (Sc complex, Chart 1). Like the 2/PEt3 system, it is also found that, in the case of 3/PEt3, the bimetallic mechanism is both kinetically and thermodynamically more favorable than the monometallic pathway (Fig. 4 and S3†). However, the overall energy barrier of the monometallic pathway is 16.1 kcal mol−1, which is not totally insurmountable under experimental conditions (room temperature). Besides, the monometal-mediated insertion process of MMA tends to be exergonic in chain propagation (3f → 3i, Fig. S3†). In view of this and the limited catalyst concentration, the monometallic mechanisms could not be excluded in the 3/PEt3 system. However, in the case of 2, the calculated energy barrier for the monometallic mechanism is too high (34.9 kcal mol−1) to be overcome under the experimental conditions. Therefore, only the bimetallic mechanism works in the 2 involved system. On the other hand, in the favorable bimetallic pathway, the overall energy barrier in the 3 catalyzed system is significantly lower than that in the 2 mediated reaction (10.9 vs. 20.2 kcal mol−1, Fig. 4 and 2). This is in line with the experimentally observed higher activity of 3 compared with 2.14b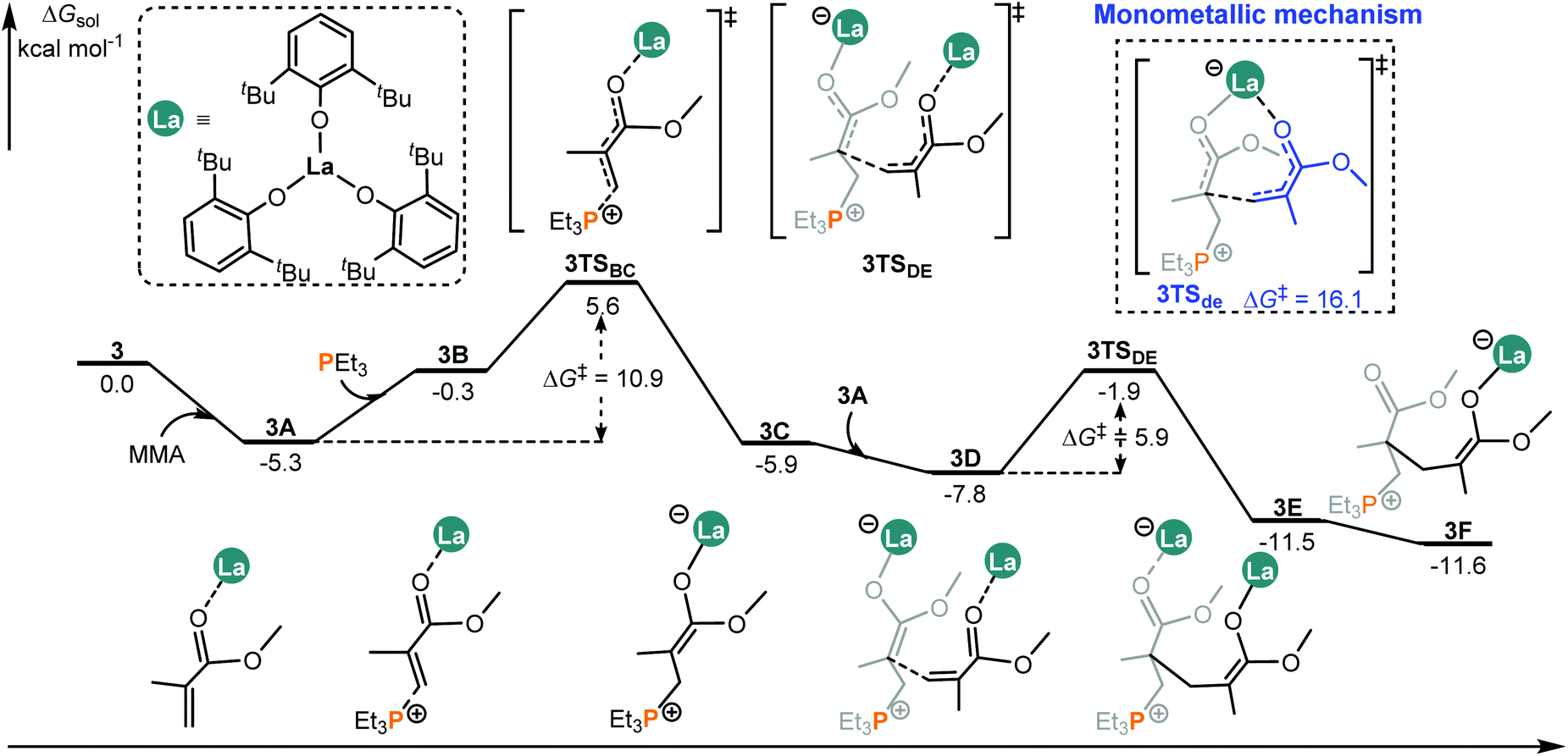 | ||
| Fig. 4 Computed energy profiles for the 3/PEt3 mediated (intermolecular RE/P systems) bimetallic pathway for MMA enchainment. Free energies (kcal mol−1) are relative to isolated reactants. | ||
To gain deeper insights into this activity difference between 2 and 3 in the bimetal mediated chain propagations, the similar distortion/interaction analyses21 of the two C–C bond forming transition states (2TSDE and 3TSDE, Fig. 2 and 4) were performed. As shown in Fig. 5, although the interaction between the two fragments in 3TSDE is weaker than that in 2TSDE (−8.7 vs. −9.2 kcal mol−1), the total deformation energy of the former (13.0 + 6.2 = 19.2 kcal mol−1) is significantly smaller than that of the latter (19.1 + 12.8 = 31.9 kcal mol−1), which compensates for the less negative interaction energy in 3TSDE, and eventually makes this TS more stable (free energy barrier of 5.9 vs. 20.2 kcal mol−1). These results suggest that the steric factor (geometrical deformation) accounts for the lower stability of 2TSDE. In order to compare the sterics around the metal center in 2 and 3, the topographic steric maps were generated using the SambVca 2.0 web tool.22 When submitting an optimized structure on the web page of SambVca 2.0 and selecting the center site, the buried volume can be therefore calculated, simultaneously generating a topographic steric map. As expected, 3 had a lower percentage of buried volume in total (%VBur, 52.0 vs. 61.5, Fig. 5b) because of the larger radius of La; thus the relatively open enchainment environment in 3 could account for its higher activity. On the other hand, the torsion angle difference Δ∠C1C2C3P = 22.4° and Δ∠O5C6C5C4 = 14.2° in 2TSDE is significantly larger than those in 3TSDE (Δ∠C1C2C3P = 7.4° and Δ∠O5C6C5C4 = 2.1°, Fig. S4†). Such a greater geometrical deformation in 2TSDE is also in line with the result of interaction/distortion analysis, resulting in a higher energy barrier and thus lower activity of the Sc complex.
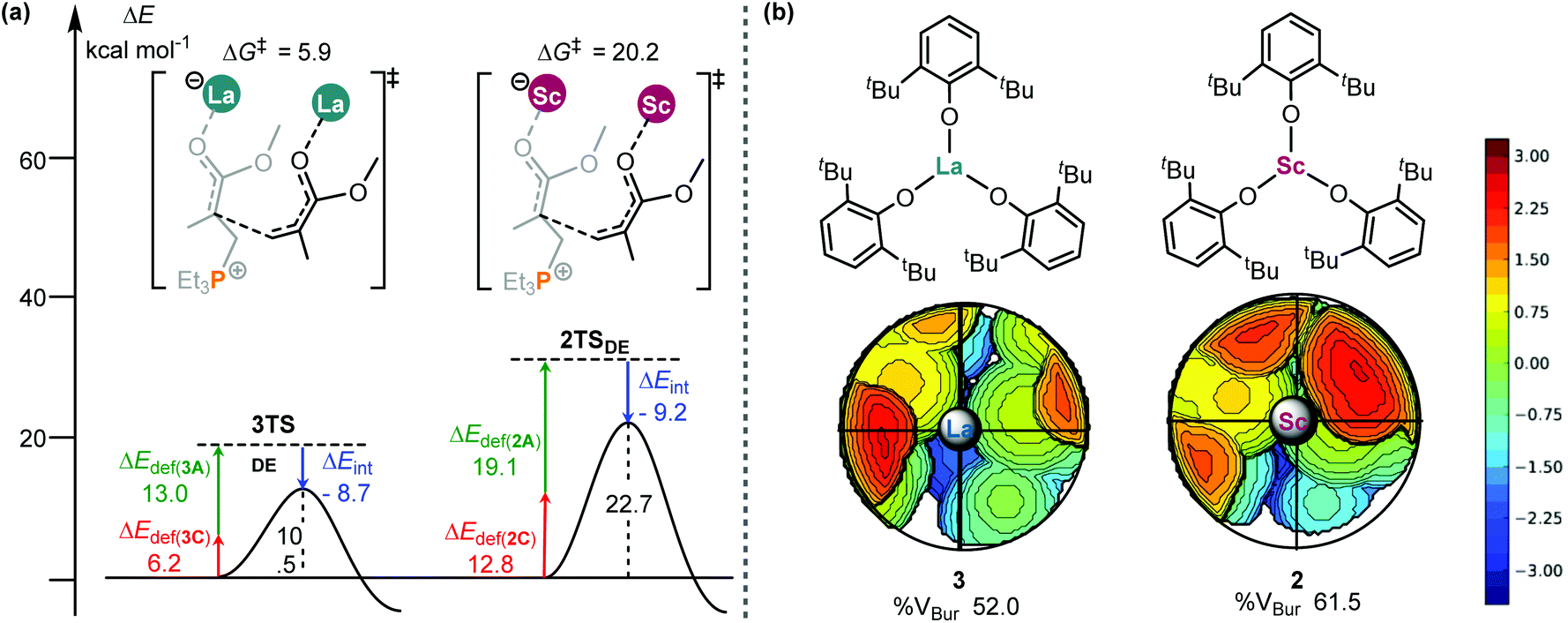 | ||
| Fig. 5 (a) Distortion/interaction analyses (kcal mol−1) of 3TSDE and 2TSDE; (b) topographical steric maps of catalysts 3 and 2. | ||
Lewis base effect on polymerization activity
An experimental observation was noted that changing the substituents on the P atom in the Lewis base can also alter polymerization activities.14b For instance, the replacement of PEt3 with triphenylphosphine (PPh3) noticeably decreased the activity. To understand the origin of such substituent effects, the energy profiles for MMA polymerization initiated by 2 and different bases R3P (R = Cy and Ph) were calculated. Following the bimetallic mechanism, the effect of the Lewis base on the polymerization activity is mainly reflected in the chain initiation step; thus only the chain initiation process is considered here.The computational results indicate that PCy3 as the Lewis base induced a lower energy barrier in comparison with PPh3 (Fig. 6). In addition, PCy3 involved zwitterionic species beyond the C–P bond formation transition state is more stable than the case of PPh3 (PCy3: −5.3 kcal mol−1vs. PPh3: 1.8 kcal mol−1). This result is in agreement with the activity trend experimentally observed. However, it remains a challenge to provide a chemically meaningful explanation for the origin of such a discrepancy in the activity. To obtain a piece of information on such differences, similar distortion/interaction analyses were performed for the key transition states TS2Cy and TS2Ph.
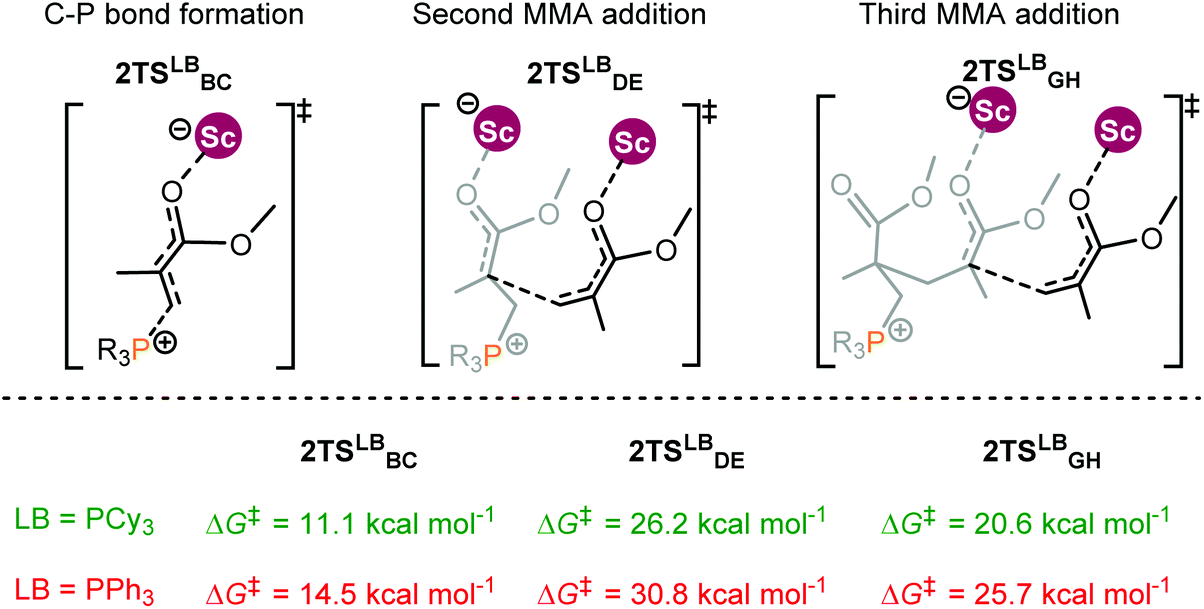 | ||
| Fig. 6 Transition states for MMA polymerization initiated by the 2/R3P pair (R = Cy and Ph). Free energies (kcal mol−1) are relative to isolated reactants. | ||
As shown in Fig. 7a, the two fragments (2C and 2A) are highlighted in black and green, respectively. The activation energy of each transition state (ΔETS) is decomposed into the distortion energy (ΔEdef) of the two reactive fragments and the interaction energy (ΔEint) between the deformed two fragments. In TS2Ph, the total distortion energy ΔEdef is 54.6 kcal mol−1, which could be partially balanced out by its ΔEint (−6.9 kcal mol−1) resulting in a ΔETS of 47.7 (54.6–6.9 = 47.7) kcal mol−1. However, the total distortion energy (ΔEdef = 55.9 kcal mol−1) in TS2Cy can be largely compensated by an interaction energy of −9.2 kcal mol−1, thus giving a lower ΔETS (46.7 kcal mol−1). The distortion/interaction model analysis indicates that higher interaction energy could account for the stability of TS2Cy in comparison with TS2Ph. To have a better understanding of such an interaction accounting for the relative stability of these two transition states, the ETS-NOCV23 (extended transition state and natural orbitals for chemical valence) calculations were performed by using the Amsterdam density functional (ADF) package24 to dissect the interaction energy (Fig. 7b). The interaction energy (ΔE′int) between the deformed 2A and 2CCy can be further dissected into Pauli repulsion (ΔEPauli), electrostatic interactions (ΔEelstat), orbital interactions (ΔEorbital), and dispersion interactions (ΔEdisp) (see computational methods in the ESI† for details). Of these interactions, the electrostatic (ΔEelstat), orbital (ΔEorbital), and dispersion (ΔEdisp) interactions are the stabilizing factors, while the Pauli repulsion (ΔEPauli) is the destabilizing factor. As shown in Fig. 7b, although the TS2Cy is disfavored by Pauli repulsion compared with 2TSPh (ΔΔEPauli = 112.3–98.9 = +13.4 kcal mol−1), it has stronger attractive orbital interactions (ΔΔEorbital = −7.9 kcal mol−1) and electrostatic interaction (ΔΔEelstat = −7.3 kcal mol−1) between the 2A and 2C moieties. These results indicate that the electronic properties of the Lewis base (PPh3 and PCy3) can alter the electron-density of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in the 2CCy and 2CPh, as also suggested by the CHelpG atomic charge25 (charges from the electrostatic potential using a grid-based method). As shown in Fig. 8a, the unsigned values of charge on C1 and C3 in 2CPh are smaller than those in 2CCy, suggesting an inductive effect of the Ph groups in 2CPh. This could account for the lower reactivity of PPh3 as a Lewis base because of the lower electron density on C1 atoms in comparison with PCy3 (−0.19 vs. −0.45), rendering more electron flow during the 1,4-addition in the case of PCy3. On the other hand, to gain a deeper understanding of the orbital interaction, orbital analysis based on the NOCV approach was performed, which could give useful information about the reorganization of the electronic density in the transition states through the electronic deformation density. Fig. 8b shows the leading electronic deformation densities and the corresponding energies for TS2Cy and 2TSPh. It is indicated that the ΔEorbital energy of TS2Cy (−47.8 kcal mol−1) is more negative than that of 2TSPh (−40.2 kcal mol−1). The charge flow from 2CCy (red region) to 2A (blue region) in TS2Cy is also more evident than that in 2TSPh. Therefore, different Lewis bases could influence the electronic population in the intermediate such as 2C and further affect the stability of the addition reaction transition state. In order to further explore the regulation effect of the Lewis base, the hydroxyl (electron-donating group) substituted phenyls have been modelled (Fig. 8a). As expected, the calculated CHelpG charge on C1 changed from −0.19 to −0.32 and the addition reaction energy barrier reduced from 30.8 to 26.5 kcal mol−1.
C bond in the 2CCy and 2CPh, as also suggested by the CHelpG atomic charge25 (charges from the electrostatic potential using a grid-based method). As shown in Fig. 8a, the unsigned values of charge on C1 and C3 in 2CPh are smaller than those in 2CCy, suggesting an inductive effect of the Ph groups in 2CPh. This could account for the lower reactivity of PPh3 as a Lewis base because of the lower electron density on C1 atoms in comparison with PCy3 (−0.19 vs. −0.45), rendering more electron flow during the 1,4-addition in the case of PCy3. On the other hand, to gain a deeper understanding of the orbital interaction, orbital analysis based on the NOCV approach was performed, which could give useful information about the reorganization of the electronic density in the transition states through the electronic deformation density. Fig. 8b shows the leading electronic deformation densities and the corresponding energies for TS2Cy and 2TSPh. It is indicated that the ΔEorbital energy of TS2Cy (−47.8 kcal mol−1) is more negative than that of 2TSPh (−40.2 kcal mol−1). The charge flow from 2CCy (red region) to 2A (blue region) in TS2Cy is also more evident than that in 2TSPh. Therefore, different Lewis bases could influence the electronic population in the intermediate such as 2C and further affect the stability of the addition reaction transition state. In order to further explore the regulation effect of the Lewis base, the hydroxyl (electron-donating group) substituted phenyls have been modelled (Fig. 8a). As expected, the calculated CHelpG charge on C1 changed from −0.19 to −0.32 and the addition reaction energy barrier reduced from 30.8 to 26.5 kcal mol−1.
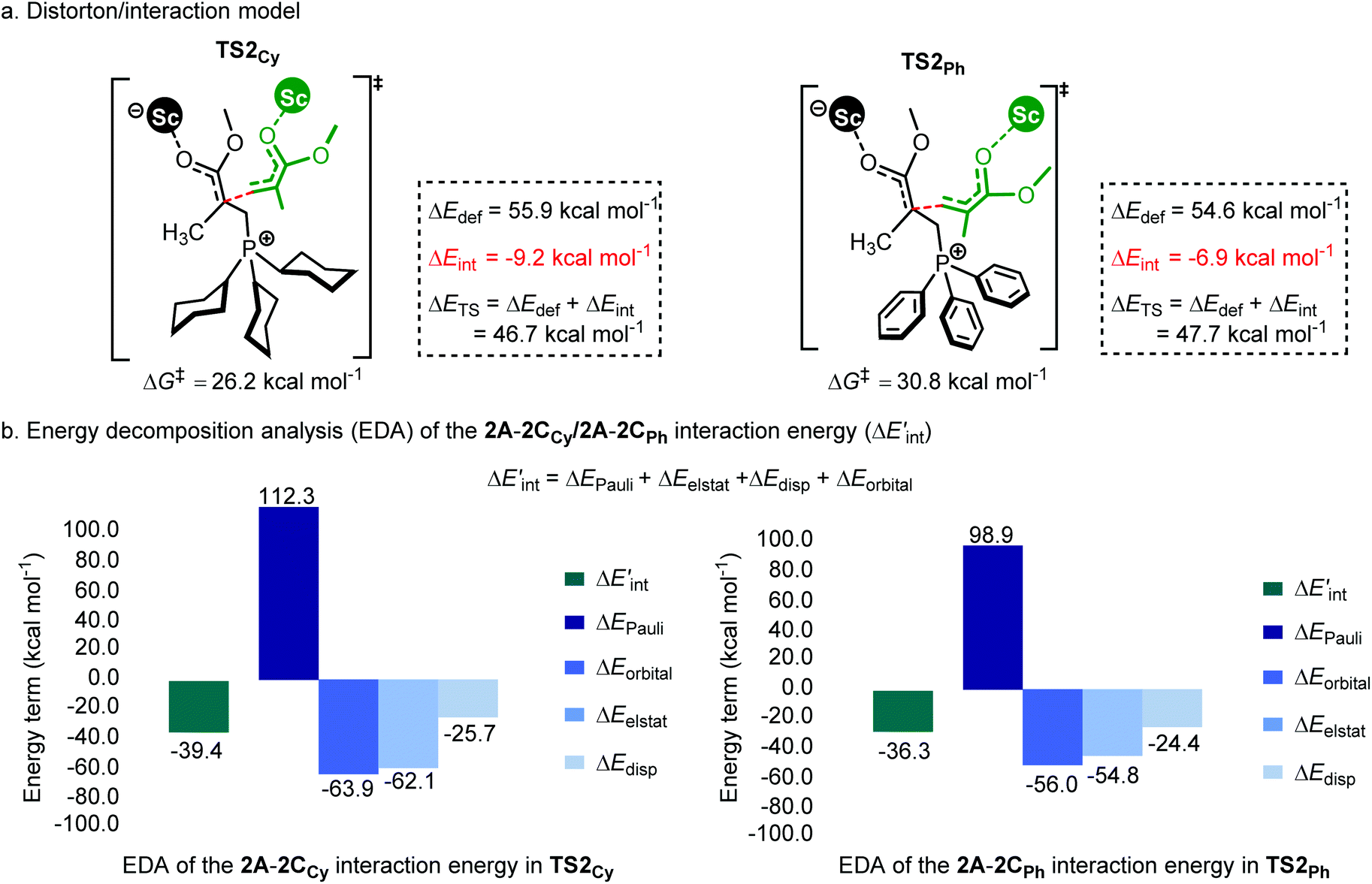 | ||
| Fig. 7 (a) Distortion/interaction model (kcal mol−1) and (b) energy decomposition analysis of TS2Cy and 2TSPh. ΔG‡ and ΔETS is calculated at the level of B3PW91/6-31G(d) planted in the Gaussian 16 program.26 ΔE′int is dissected into chemically meaningful terms according to the ETS-NOCV calculations at the level of B3LYP-D3/DZP planted in the Amsterdam density functional (ADF) package.24 | ||
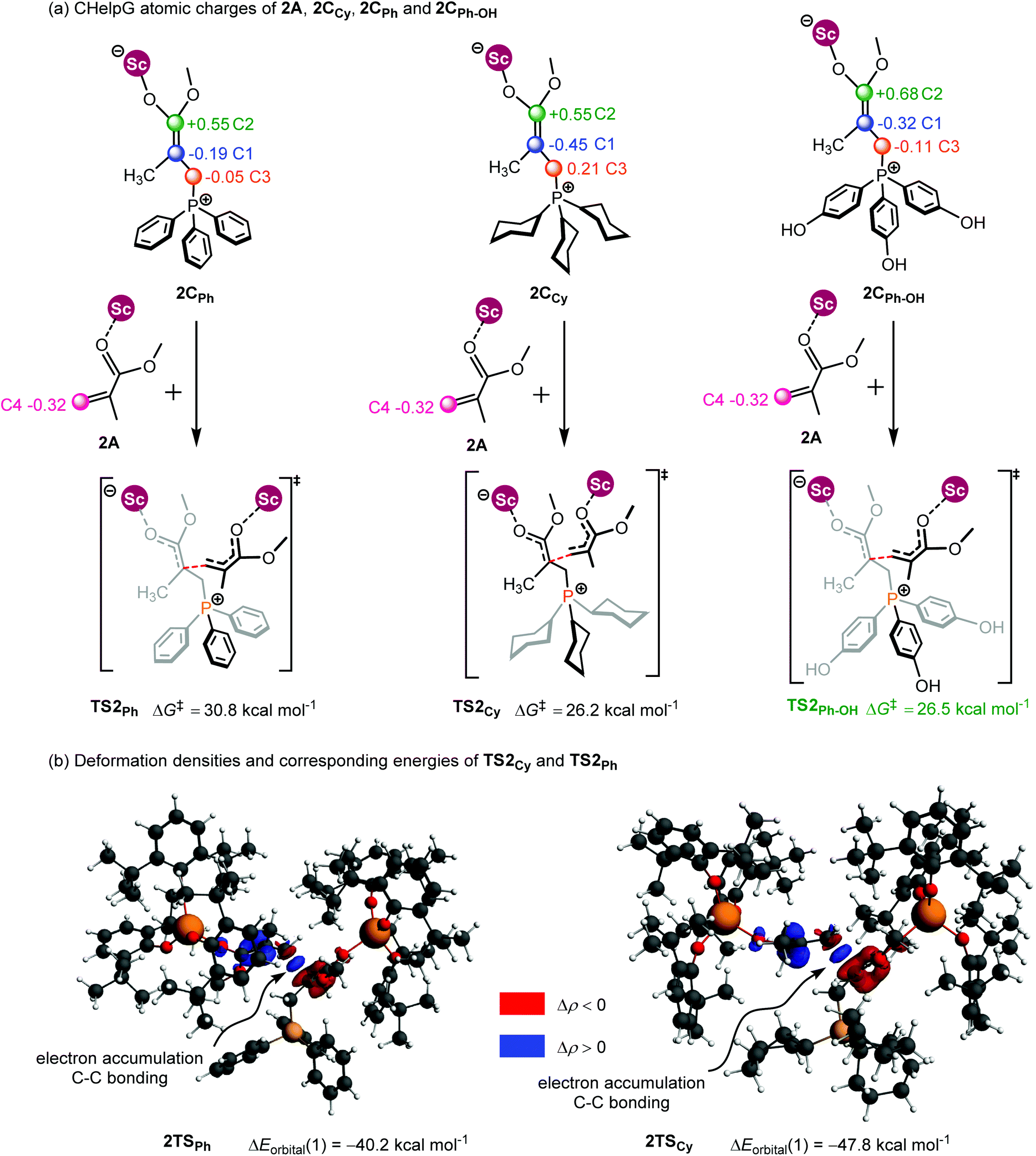 | ||
| Fig. 8 (a) CHelpG atomic charge25 in 2A, 2CPh, 2CCy and 2CPh–OH; (b) leading deformation densities and corresponding energies for TS2Ph and 2TSCy. The red color denotes charge depletion and the blue color represents charge accumulation (|Δρ| = 0.004 a.u. for all the density surfaces). | ||
Catalyst modification
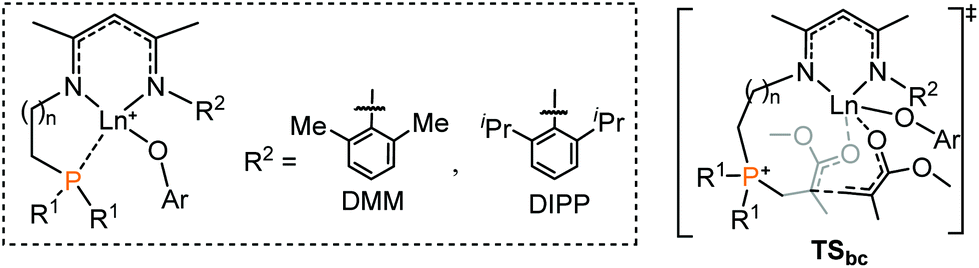
Inspired by the aforementioned calculation results, we speculated that it should be possible to improve the activity by modifying ligands in the intramolecular RE/P system.14a As aforementioned, the P–C bond formation is not the rate determining step (Fig. 1), which was not considered here. As shown in Table 1, on the basis of original catalyst 1, appropriate substituent alteration has no significant effect on the MMA enchainment energy barrier (ΔG‡TSbc, cat. 1, 4–7). Interestingly, when the number of bridging methylenes increased to 2, the energy barrier decreased (ΔG‡1 = 19.6 vs. ΔG‡8 = 17.2 kcal mol−1). Besides, when the metal Sc is changed to La, the energy barrier obviously decreased by 6.3 kcal mol−1 (ΔG‡1 = 19.6 vs. ΔG‡9 = 13.3 kcal mol−1). These results suggest that the proper flexibility of the arm bridging intramolecular Lewis pair and larger-sized metal could be beneficial to the addition of MMA.|
|
|||||
|---|---|---|---|---|---|
| Cat. | Ln | R1 | R2 | n | ΔG‡TSbc (kcal mol−1) |
| 1 | Sc | Ph | DIPP | 1 | 19.6 |
| 4 | Sc | Me | DIPP | 1 | 21.4 |
| 5 | Sc | Et | DIPP | 1 | 21.3 |
| 6 | Sc | Ph | DMM | 1 | 20.4 |
| 7 | Sc | Me | DMM | 1 | 23.1 |
| 8 | Sc | Ph | DIPP | 2 | 17.2 |
| 9 | La | Ph | DIPP | 2 | 13.3 |
Conclusions
In summary, the origin of activity difference in the polymerization of methyl methacrylate (MMA) catalyzed by various rare-earth/phosphorus Lewis pairs (RE/P), via Michael addition, has been computationally elucidated. The calculated energy landscapes indicate that the intramolecular RE/P mediated MMA polymerization is a significant exergonic process and features a moderate energy barrier. In the case of intermolecular RE/P, the bimetallic mechanism is kinetically preferable over the monometallic pathway. Unlike the Sc(OAr)3/PEt3 (Ar = 2,6-tBu2-C6H3) Lewis pair, however, the La analogue may also follow a monometallic mechanism in view of limited catalyst concentration and surmountable enchainment energy barrier. The detailed structure and energy analyses disclosed that geometrical deformation required for achieving the enchainment transition state mainly accounts for the activity discrepancy of the rare-earth metal complex as a Lewis acid in the intermolecular RE/P system. Meanwhile, given the same Lewis acid, the observed activity difference of various PR3 Lewis bases (R = Cy and Ph) could be ascribed to the electronic induction effect of the R group. Namely, the R group with stronger electron donation ability could increase the electron density on the reactive vinyl group and render more electron flow during the Michael addition, resulting in higher activity in comparison with the electron withdrawing group, which is in line with the experimentally observed activity trend. Besides, further computational modellings on a series of intramolecular RE/P catalysts indicate that increasing the radius of the central metal or the flexibility of the bridge connecting Lewis pair in the catalyst is beneficial to improving catalytic activity. These theoretical results are expected to be useful for the design of a more efficient RE/P polymerization system.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was partly supported by the NSFC (U1862115 and 21674014). The authors also thank the RICC (RIKEN Integrated Cluster of Clusters) and the Network and Information Center of the Dalian University of Technology for part of the computational resources. G. L. thanks the Innovation and Entrepreneurship Project of Overseas Returnees in Anhui Province (2020LCX006).References
- (a) F. Sugiyama, K. Satoh and M. Kamigaito, Regiospecific Radical Polymerization of Vinyl Methacrylate in the Presence of Lewis Acids into Soluble Polymers with Pendent Vinyl Ester Substituents, Macromolecules, 2008, 41, 3042–3048 CrossRef CAS; (b) J. Ma, C. Cheng, G. Sun and K. L. Wooley, Well-Defined Polymers Bearing Pendent Alkene Functionalities via Selective RAFT Polymerization, Macromolecules, 2008, 41, 9080–9089 CrossRef CAS.
- (a) Y. Murali Mohan, V. Raghunadh, S. Sivaram and D. Baskaran, Reactive Polymers Bearing Styrene Pendants through Selective Anionic Polymerization of 4-Vinylbenzyl Methacrylate, Macromolecules, 2012, 45, 3387–3393 CrossRef CAS; (b) S. Tanaka, R. Goseki, T. Ishizone and A. Hirao, Synthesis of Well-Defined Novel Reactive Block Polymers Containing a Poly(1,4-divinylbenzene) Segment by Living Anionic Polymerization, Macromolecules, 2014, 47, 2333–2339 CrossRef CAS.
- For reviewers, see: (a) C. Chen, Designing Catalysts for Olefin Polymerization and Copolymerization: Beyond Electronic and Steric Tuning, Nat. Rev. Chem., 2018, 2, 6–14 CrossRef CAS; (b) M. Chen and C. Chen, Polar and Functionalized Polyolefins: New Catalysts, New Modulation Strategies and New Materials, Acta Polym. Sin., 2018, 11, 1372–1384 Search PubMed; (c) D. J. Walsh, M. G. Hyatt, S. A. Miller and D. Guironnet, Recent Trends in Catalytic Polymerizations, ACS Catal., 2019, 9, 11153–11188 CrossRef CAS; (d) A. Nakamura, S. Ito and K. Nozaki, Coordination-Insertion Copolymerization of Fundamental Polar Monomers, Chem. Rev., 2009, 109, 5215–5244 CrossRef CAS; (e) B. Carrow and K. Nozaki, Transition-Metal-Catalyzed Functional Polyolefin Synthesis: Effecting Control through Chelating Ancillary Ligand Design and Mechanistic Insights, Macromolecules, 2014, 47, 2541–2555 CrossRef CAS; (f) L. Boffa and B. Novak, Copolymerization of Polar Monomers with Olefins Using Transition-Metal Complexes, Chem. Rev., 2000, 100, 1479–1493 CrossRef CAS.
- For recent reviews on the group-transfer polymerization, see: (a) E. Chen, Coordination Polymerization of Polar Vinyl Monomers by Single-Site Metal Catalysts, Chem. Rev., 2009, 109, 5157–5214 CrossRef CAS; (b) B. Soller, S. Salzinger and B. Rieger, Rare Earth Metal-Mediated Precision Polymerization of Vinylphosphonates and Conjugated Nitrogen-Containing Vinyl Monomers, Chem. Rev., 2016, 116, 1993–2022 CrossRef CAS.
- (a) Y. Zhang, G. Miyake and E. Chen, Alane-Based Classical and Frustrated Lewis Pairs in Polymer Synthesis: Rapid Polymerization of MMA and Naturally Renewable Methylene Butyrolactones into High-Molecular-Weight Polymers, Angew. Chem., Int. Ed., 2010, 49, 10158–10162 CrossRef CAS; (b) M. Hong, J. Chen and E. Chen, Polymerization of Polar Monomers Mediated by Main-Group Lewis Acid–Base Pairs, Chem. Rev., 2018, 118, 10551–10616 CrossRef CAS; (c) W. Zhao, J. He and Y. Zhang, Lewis Pairs Polymerization of Polar Vinyl Monomers, Sci. Bull., 2019, 64, 1830–1840 CrossRef CAS; (d) Y. Jia, Y. Wang, W. Ren, T. Xu, J. Wang and X. Lu, Mechanistic Aspects of Initiation and Deactivation in N-Heterocyclic Olefin Mediated Polymerization of Acrylates with Alane as Activator, Macromolecules, 2014, 47, 1966–1972 CrossRef CAS; (e) H. Wang, Q. Wang, J. He and Y. Zhang, Living Polymerization of Acrylamides Catalysed by N-heterocyclic Olefin-based Lewis Pairs, Polym. Chem., 2019, 10, 3597–3603 RSC; (f) Y. Bai, J. He and Y. Zhang, Ultra-High-Molecular-Weight Polymers Produced by the Immortal Phosphine-Based Catalyst System, Angew. Chem., Int. Ed., 2018, 57, 17230–17234 CrossRef CAS; (g) Y. Bai, H. Wang, J. He and Y. Zhang, Rapid and Scalable Access to Sequence-Controlled DHDM Multiblock Copolymers by FLP Polymerization, Angew. Chem., Int. Ed., 2020, 59, 11613–11619 CrossRef CAS; (h) M. McGraw, R. Clarke and E. Chen, Compounded Sequence Control in Polymerization of One-Pot Mixtures of Highly Reactive Acrylates by Differentiating Lewis Pairs, J. Am. Chem. Soc., 2020, 142, 5969–5973 CrossRef.
- (a) Q. Wang, W. Zhao, S. Zhang, J. He, Y. Zhang and E. Chen, Living Polymerization of Conjugated Polar Alkenes Catalyzed by N-Heterocyclic Olefin-Based Frustrated Lewis Pairs, ACS Catal., 2018, 8, 3571–3578 CrossRef CAS; (b) T. Xu and E. Chen, Probing Site Cooperativity of Frustrated Phosphine/Borane Lewis Pairs by a Polymerization Study, J. Am. Chem. Soc., 2014, 136, 1774–1777 CrossRef CAS; (c) M. Knaus, M. Giuman, A. Pöthig and B. Rieger, End of Frustration: Catalytic Precision Polymerization with Highly Interacting Lewis Pairs, J. Am. Chem. Soc., 2016, 138, 7776–7781 CrossRef CAS.
- (a) X. Wang, Y. Zhang and M. Hong, Controlled and Efficient Polymerization of Conjugated Polar Alkenes by Lewis Pairs Based on Sterically Hindered Aryloxide Substituted Alkylaluminum, Molecules, 2018, 23, 442–453 CrossRef; (b) X. Wang and M. Hong, Precise Control of Molecular Weight and Stereospecificity in Lewis Pair Polymerization of Semifluorinated Methacrylates: Mechanistic Studies and Stereocomplex Formation, Macromolecules, 2020, 53, 4659–4669 CrossRef CAS.
- (a) W. Zhao, Q. Wang, J. He and Y. Zhang, Chemoselective and living/controlled polymerization of polar divinyl monomers by N-heterocyclic olefin based classical and frustrated Lewis pairs, Polym. Chem., 2019, 10, 4328–4335 RSC; (b) Y. Bai and Y. Zhang, Polymerization of Polar Vinyl Monomers Catalyzed by Lewis Pairs, Acta Polym. Sin., 2019, 50, 233–246 Search PubMed.
- (a) E. Chen and M. Cooney, Amphicatalytic Polymerization: Synthesis of Stereomultiblock Poly(methyl methacrylate) with Diastereospecific Ion Pairs, J. Am. Chem. Soc., 2003, 125, 7150–7151 CrossRef CAS; (b) Y. Ning and E. Chen, Diastereospecific Ion-Pairing Polymerization of Functionalized Alkenes by Metallocene/Lewis Acid Hybrid Catalysts, Macromolecules, 2006, 39, 7204–7215 CrossRef CAS.
- (a) H. Ji, B. Wang, L. Pan and Y. Li, Lewis Pairs for Ring-Opening Alternating Copolymerization of Cyclic Anhydrides and Epoxides, Green Chem., 2018, 20, 641–648 RSC; (b) X. Li, B. Wang, H. Jia and Y. Li, Insights into the Mechanism for Ring-Opening Polymerization of Lactide Catalyzed by Zn(C6F5)2/Organic Superbase Lewis Pairs, Catal. Sci. Technol., 2016, 6, 7763–7772 RSC.
- A. Londaitsbehere, M. Sudupe, M. Mosquera, T. Cuenca and J. Cano, MMA Polymerization with Group 4 Alkyl-Free 14-Electron d0 Species, Eur. J. Inorg. Chem., 2020, 2020, 1589–1595 CrossRef CAS.
- (a) W. Mao, L. Xiang, C. Lamsfus, L. Maron, X. Leng and Y. Chen, Are Sc–C and Sc–P Bonds Reactive in Scandium Phosphinoalkylidene Complex? Insights on a Versatile Reactivity, Chin. J. Chem., 2018, 36, 904–908 CrossRef CAS; (b) C. Xue, Y. Luo, H. Teng, Y. Ma, M. Nishiura and Z. Hou, Ortho-Selective C–H Borylation of Aromatic Ethers with Pinacolborane by Organo Rare-Earth Catalysts, ACS Catal., 2018, 8, 5017–5022 CrossRef CAS; (c) Y. Luo, H. Teng, C. Xue, M. Nishiura and Z. Hou, Yttrium-Catalyzed Regioselective α-C–H Silylation of Methyl Sulfides with Hydrosilanes, ACS Catal., 2018, 8, 8027–8032 CrossRef CAS.
- (a) M. Nishiura and Z. Hou, Novel Polymerization Catalysts and Hydride Clusters from Rare-earth Metal Dialkyls, Nat. Chem., 2010, 2, 257–268 CrossRef CAS; (b) M. Nishiura, F. Guo and Z. Hou, Half-Sandwich Rare-Earth-Catalyzed Olefin Polymerization, Carbometalation, and Hydroarylation, Acc. Chem. Res., 2015, 48, 2209–2220 CrossRef CAS; (c) D. Cui, Studies on Homo- and Co-polymerizations of Polar and Non-polar Monomers Using Rare-earth Metal Catalysts, Acta Polym. Sin., 2020, 51, 12–29 Search PubMed; (d) C. Wang, G. Luo, M. Nishiura, G. Song, A. Yamamoto, Y. Luo and Z. Hou, Heteroatom-Assisted Olefin Polymerization by Rare-earth Metal Catalysts, Sci. Adv., 2017, 3, e1701011 CrossRef.
- (a) P. Xu, Y. Yao and X. Xu, Frustrated Lewis Pairs Like Reactivity of Rare-Earth Metal Complexes: 1,4-Addition Reactions and Polymerizations of Conjugated Polar Alkenes, Chem. – Eur. J., 2017, 23, 1263–1267 CrossRef CAS; (b) P. Xu and X. Xu, Homoleptic Rare-Earth Aryloxide Based Lewis Pairs for Polymerization of Conjugated Polar Alkenes, ACS Catal., 2018, 8, 198–202 CrossRef CAS; (c) P. Xu, L. Wu, L. Dong and X. Xu, Chemoselective Polymerization of Polar Divinyl Monomers with Rare-Earth/Phosphine Lewis Pairs, Molecules, 2018, 23, 360 CrossRef.
- Y. Zhang, G. Miyake, M. John, L. Falivene, L. Caporaso, L. Cavallo and E. Chen, Lewis Pair Polymerization by Classical and Frustrated Lewis Pairs: Acid, Base and Monomer Scope and Polymerization Mechanism, Dalton Trans., 2012, 41, 9119–9134 RSC.
- J. He, Y. Zhang, L. Falivene, L. Caporaso, L. Cavallo and E. Chen, Chain Propagation and Termination Mechanisms for Polymerization of Conjugated Polar Alkenes by [Al]-Based Frustrated Lewis Pairs, Macromolecules, 2014, 47, 7765–7774 CrossRef CAS.
- W. Ottou, E. Conde-Mendizabal, A. Pascual, A. Wirotius, D. Bourichon, J. Vignolle, F. Robert, Y. Landais, J. Sotiropoulos, K. Miqueu and D. Taton, Organic Lewis Pairs Based on Phosphine and Electrophilic Silane for the Direct and Controlled Polymerization of Methyl Methacrylate: Experimental and Theoretical Investigations, Macromolecules, 2017, 50, 762–774 CrossRef.
- Y. Jia, W. Ren, S. Liu, T. Xu, Y. Wang and X. Lu, Controlled Divinyl Monomer Polymerization Mediated by Lewis Pairs: A Powerful Synthetic Strategy for Functional Polymers, ACS Macro Lett., 2014, 3, 896–899 CrossRef CAS.
- (a) Y. Luo, Y. Luo, J. Qu and Z. Hou, QM/MM Studies on Scandium-Catalyzed Syndiospecific Copolymerization of Styrene and Ethylene, Organometallics, 2011, 30, 2908–2919 CrossRef CAS; (b) Y. Zhao, G. Luo, X. Kang, F. Guo, X. Zhu, R. Zheng, Z. Hou and Y. Luo, “C–H⋯π Interaction” Regulates the Stereoselectivity in Olefin Polymerization, Chem. Commun., 2019, 55, 6689–6692 RSC; (c) X. Kang, Y. Song, Y. Luo, G. Li and Z. Hou, Computational Studies on Isospecific Polymerization of 1-Hexene Catalyzed by Cationic Rare Earth Metal Alkyl Complex Bearing a C3iPr-trisox Ligand, Macromolecules, 2012, 45, 640–651 CrossRef CAS; (d) X. Kang, Y. Luo and Z. Hou, Theoretical Insights into Olefin Polymerization Catalyzed by Cationic Organo Rare-Earth Metal Complexes, in Computational Quantum Chemistry: Insights into Polymerization Reactions, ed. M. Soroush, Elsevier, Amsterdam, The Netherlands, 2019, pp. 327–356 Search PubMed.
- R. G. Pearson, Proc. Nati. Acad. Sci. US, 1986, 83, 8440–8441 CrossRef CAS.
- (a) F. Bickelhaupt and K. Houk, Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS; (b) I. Fernández and F. Bickelhaupt, Bioorthogonal Cycloadditions: Computational Analysis with the Distortion/Interaction Model and Predictions of Reactivities, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC; (c) F. Liu, Y. Liang and K. Houk, Bioorthogonal Cycloadditions: Computational Analysis with the Distortion/Interaction Model and Predictions of Reactivities, Acc. Chem. Res., 2017, 50, 2297–2308 CrossRef CAS; (d) K. Kitaura and K. Morokuma, A New Energy Decomposition Scheme for Molecular Interactions within the Hartree-Fock Approximation, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- (a) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, SambVca: A Web Application for the Calculation of the Buried Volume of N-Heterocyclic Carbene Ligands, Eur. J. Inorg. Chem., 2009, 2009, 1759–1766 CrossRef; (b) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps, Organometallics, 2016, 35, 2286–2293 CrossRef CAS . See also https://www.molnac.unisa.it/OMtools/sambvca2.0/.
- M. Mitoraj, A. Michalak and T. Ziegler, A Combined Charge and Energy Decomposition Scheme for Bond Analysis, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS.
- (a) G. te Velde, F. Bickelhaupt, E. Baerends, C. Fonseca Guerra, S. van Gisbergen, J. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS; (b) E. Baerends, J. Autschbach, D. Bashford, A. Bérces, F. Bickelhaupt, C. Bo, P. Boerrigter, L. Cavallo, D. Chong, L. Deng, R. Dickson, D. Ellis, M. van Faassen, L. Fan, T. Fischer, C. Fonseca Guerra, A. Ghysels, A. Giammona, S. van Gisbergen, A. Götz, J. Groeneveld, O. Gritsenko, M. Grüning, F. Harris, P. van den Hoek, C. Jacob, H. Jacobsen, L. Jensen, G. van Kessel, F. Kootstra, M. Krykunov, E. van Lenthe, D. McCormack, A. Michalak, M. Mitoraj, J. Neugebauer, V. Nicu, L. Noodleman, V. Osinga, S. Patchkovskii, P. Philipsen, D. Post, C. Pye, W. Ravenek, J. Rodríguez, P. Ros, P. Schipper, G. Schreckenbach, M. Seth, J. Snijders, M. Solà, M. Swart, D. Swerhone, G. te Velde, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang, T. Wesolowski, E. van Wezenbeek, G. Wiesenekker, S. Wolff, T. Woo, A. Yakovlev and T. Ziegler, ADF2009.01, Theoretical Chemistry, Vrije Universiteit, Amsterdam Search PubMed.
- C. Breneman and K. Wiberg, Determining Atom-Centered Monopoles from Molecular Electrostatic Potentials. The Need for High Sampling Density in Formamide Conformational Analysis, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, figures showing the monometallic pathway and geometrical parameters of key stationary points, and optimized Cartesian coordinates of all stationary points together with their single-point energies (a.u.) in solution and the imaginary frequencies (cm−1) of transition states (XYZ file). See DOI: 10.1039/d0qi01067g |
| This journal is © the Partner Organisations 2020 |