
Electrochemical degradation of perfluoroalkyl acids by titanium suboxide anodes†
Yaye
Wang‡
a,
Randall “David”
Pierce‡
Jr.
a,
Huanhuan
Shi
ab,
Chenguang
Li
ab and
Qingguo
Huang
 *a
*a
aCollege of Agricultural and Environmental Sciences, Department of Crop and Soil Sciences, University of Georgia, Griffin, GA 30223, USA. E-mail: qhuang@uga.edu
bState Key Laboratory of Pollution Control and Resources Reuses, School of the Environment, Nanjing University, Nanjing 210023, P. R. China
First published on 18th November 2019
Abstract
Our recent findings indicate effective electrochemical degradation of perfluoroalkyl acids (PFAAs) in aqueous solutions using novel titanium suboxide (TSO) anodes. This provides a potentially promising technology to treat per- and polyfluoroalkyl substances (PFASs) in legacy AFFF stockpiles or in wastewater and contaminated groundwater. The degradation of PFAAs in TSO-based electrooxidation was evaluated with solutions of different compositions containing eight PFAAs either individually or in a mixture and under a range of operation conditions. Surface area normalized pseudo-first order rate constants were obtained and interpreted in terms of PFAA degradation behaviors and mechanisms. The PFAA degradation rates were further correlated to the molecular and electronic descriptors of the chemicals calculated based on density functional theory. The results of this study further the understanding of PFAS degradation in TSO-based anodic oxidation and provide a basis for process optimization, design and scaling.
Water impactWith the development of PFASs as a major water contaminant concern, treatment technologies have been needed for these highly stable compounds. Our work herein presents a promising treatment method by electrooxidation using titanium suboxide anodes for the degradation of multiple PFAS species. |
1. Introduction
Per- and polyfluoroalkyl substances (PFASs) have been used in industrial applications since the latter half of the 20th century primarily as surfactants suited for harsh conditions because of their extreme chemical and thermal stability. These compounds obtain their high stability largely due to C–F bonds that replace the typical C–H bonds in organic molecules.1 PFASs have emerged as a public concern because of their persistence in the environment, bioaccumulation tendency and risks to human and other biological populations.2 In particular, perfluoroalkyl acids (PFAAs) pose a unique threat as degradation of many PFASs become persistent PFAA by-products during many technological and biological degradation processes.3,4 PFAAs comprise some of the most well-known PFASs such as perfluorooctanesulfonate (PFOS) and perfluorooctanoic acid (PFOA).Wildlife and human monitoring services have identified PFAAs in mammal populations spanning the entire planet with PFAA levels in human plasma having been reported over the past two decades.5–8 Trends appear to be correlated with geographic and temporal exposure. Although the correlation between the industrial output and biological levels of these contaminants is likely, their fate and persistence in the environment lacks definitive research, including the mechanisms behind biological uptake.8 Toxicological research into the reported quantities of PFASs in mammals is also still in its infancy, but studies have suggested that they are particularly related to developmental toxicity, immunotoxicity, and hepatotoxicity.5
As the toxicological impacts of PFASs have become evident, treatment methods for PFAS polluted groundwater and wastewater have been explored. Unfortunately both traditional wastewater remediation techniques and even many advanced oxidation processes (AOPs) have not been successful in treatment of PFAAs due to their extreme chemical stability.9 Development of certain techniques has reported some success in treating PFAAs such as photochemical decomposition,10–12 ultrasonic irradiation,13 and heat induced persulfate oxidation,14 but the application of these technologies is limited because of their high energy input and harsh operating conditions.
Recent research into electrochemical oxidation has emerged at the forefront of PFAS treatment because of its robustness and ability to operate under benign conditions. Electrochemical degradation of PFAAs has been successful in recent studies using boron doped diamond (BDD) and Sb and Pb doped titanium based anodes under ambient conditions for spiked samples,15–18 and BDD has demonstrated success using real world wastewater effluents.19 Although efficient degradation and mineralization has been achieved in some studies, drawbacks exist.20 These drawbacks regarding the high cost of BDD based anodes and generation of disinfection by-products and potentially toxic by-products released by Sb and Pb doping agents have been major limitations in the implementation of these technologies.16
We have recently investigated the use of Magnéli phase titanium suboxide (TSO) anodes for PFAS degradation because of their potentially low cost commercial production, high conductivity, and robustness.21 These materials consist of a series of sub-stoichiometric titanium oxides with the formula TinO2n−1, where n is an integer between 3 and 10; Ti4O7 is the most desired variant because it has the highest electrical conductivity.21 Recent studies have shown the viability of Ti4O7 based anodes for electrochemical remediation of aqueous organic pollutants such as phenols and tetracycline.22–24 Early work done on the remediation of PFOA and PFOS using TSO anodes yielded promising results,25–27 removing more than 95% of PFOA and PFOS in a batch system at 10 mA cm−2 after 3 hours25 and more than 99% of PFOA and PFOS in a reactive electrochemical membrane (REM) system at an anodic potential of 2.9 V vs. SHE with a constant permeate flux of 36 LMH (liters per square meter per hour).27 However, the impact of different compositions of PFASs is still unknown, both in terms of their respective degradation rates and possible competition behaviors, and requires further exploration.25 Knowledge on the behavior of PFAS mixtures is much needed, because multiple PFASs are often used and present together.3 This study assessed eight PFAAs (PFBA, PFPeA, PFHxA, PFHpA, PFOA, PFBS, PFHxS and PFOS), either individually or in a mixture, by TSO-based electrooxidation across a range of anodic potentials in solutions of different compositions. The treatment performance was explored in terms of key operation conditions as well as the molecular features of the chemicals.
2. Experimental section
2.1 Chemicals and standards
Mass-labelled perfluoro-n-[13C8]octanoic acid (M8PFOA) and sodium perfluoro-n-[13C8]octanesulfonate (M8PFOS) were obtained from Wellington Laboratories Inc. (Guelph, Ontario, Canada,). Perfluorooctanoic acid (PFOA, >97%) and perfluorohexanesulfonic acid (PFHxS, 98%) were purchased from Sigma Aldrich Chemical Co., Ltd (St. Louis, Missouri). Perfluorobutanoic acid (PFBA, 98%), perfluorobutanesulfonic acid (PFBS, 97%), perfluoropentanoic acid (PFPeA, 97%), and perfluoroheptanoic acid (PFHpA, 97%) were also from Sigma Aldrich. Perfluorooctanesulfonic acid (PFOS, 98%) was purchased from Indofine Chemical Company, Inc. (Hillsborough, NJ). Perfluorohexanoic acid (PFHxA, 98%) was purchased from Tokyo Chemical Industries (Tokyo, Japan). Sodium sulfate (>99.0%), sodium nitrate (>99.1%), and sodium hydroxide (>98%) were purchased from J.T. Baker (Phillipsburg, New Jersey). Monobasic sodium phosphate (H2NaPO4·2H2O) (99%) was purchased from Alfa Aesar (Ward Hill, Massachusetts). Dibasic sodium phosphate (Na2HPO4·7H2O) (>98%) was from EM Science (Gardena, CA).2.2 Electrochemical oxidation experiment
The electrochemical oxidation experiments were conducted in a rectangular reactor (10 cm × 5 cm × 2.5 cm) containing 200 mL target solution, with one TSO plate (10 cm × 5 cm) placed in the middle as an anode and two stainless steel plates of the same size on either side in parallel as cathodes. The reactor was made of acrylic materials, and the TSO electrodes were custom-made according to a method used in our previous study.26,28 The distance between the electrodes is 2.5 cm (see Fig. S1† for a picture). A DC power supply unit (303 DM supplied by Electro Industries Inc.) was used to supply electricity at constant current, and the solution was stirred using a magnetic stirrer at a constant speed throughout each experiment. The electric current density was calculated using the submerged surface area on both sides of the anode (the total geometric surface area is 78 cm2). The anodic potential was monitored using a CHI 660E electrochemical workstation (CH Instruments, Inc., Austin, TX) with an Ag/AgCl reference electrode placed close to the anode, and all potentials are reported against a standard hydrogen electrode (SHE) with internal resistance compensation. The reaction solution was prepared with each PFAA at an initial concentration of 2.0 μM, either individually or in a mixture, with 100 mM Na2SO4 as a supporting electrolyte, unless otherwise specified. The electric current was applied after 30 minutes of solution stirring in the reactor. Aliquots of samples, 400 μL each, were collected at prescribed time intervals, and the samples were taken after pausing the current for 90 seconds while maintaining stirring to ensure solution homogeneity. The pH was also measured during this period using an ion selective electrode (Oakton pH 300 series). A washing procedure was used on electrodes between experiments, including submersion in deionized water while sonicating for one hour, in methanol for one hour, in 0.1 M HCL for one hour, and finally in deionized water again for one hour. The PFAA concentration used in this study was greater than those typically present in contaminated groundwater for ease of sample handling and analysis.2.3 Chemical analysis
Each sample was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with 0.10 μM M8PFOA and M8PFOS in MeOH and filtered through a nylon-based syringe filter. The samples were processed through a UPLC-MS/MS system (Waters I-class UPLC; Water Xevo TQD triple quadrupole mass spectrometer) in negative electrospray ionization mode using multiple reaction monitoring (MRM). The UPLC was operated with methanol (A) and water (B) (5 mM ammonium acetate in each) as the mobile phases at a flow rate of 0.3 mL min−1 using gradient conditions listed in Table S1.† Electrospray ionization (ESI) was operated in a negative mode with a capillary voltage of 1.14 kV, a cone voltage of 60 V, a source temperature of 400 °C and a desolvation temperature of 550 °C. The mass transitions and spectrometry conditions for MRM are specified in Table S2.† Mass labeled PFOS and PFOA were used as internal standards, with M8PFOS for perfluoroalkyl sulfonates (PFSAs) and M8PFOA for perfluoroalkyl carboxylic acids (PFCAs). Quantification was achieved by the ratio of the MRM signal of the chemical to that of the internal standard in reference to a five-point calibration curve.
:1 with 0.10 μM M8PFOA and M8PFOS in MeOH and filtered through a nylon-based syringe filter. The samples were processed through a UPLC-MS/MS system (Waters I-class UPLC; Water Xevo TQD triple quadrupole mass spectrometer) in negative electrospray ionization mode using multiple reaction monitoring (MRM). The UPLC was operated with methanol (A) and water (B) (5 mM ammonium acetate in each) as the mobile phases at a flow rate of 0.3 mL min−1 using gradient conditions listed in Table S1.† Electrospray ionization (ESI) was operated in a negative mode with a capillary voltage of 1.14 kV, a cone voltage of 60 V, a source temperature of 400 °C and a desolvation temperature of 550 °C. The mass transitions and spectrometry conditions for MRM are specified in Table S2.† Mass labeled PFOS and PFOA were used as internal standards, with M8PFOS for perfluoroalkyl sulfonates (PFSAs) and M8PFOA for perfluoroalkyl carboxylic acids (PFCAs). Quantification was achieved by the ratio of the MRM signal of the chemical to that of the internal standard in reference to a five-point calibration curve.
Fluoride ion concentrations were analyzed in selected samples. The analysis was performed using a F− ion selective electrode (Thermo Scientific™ Orion™) with a reported detection limit of 0.01 ppm by a standard-addition method,29 with details described in Text S3.†
2.4 Electrode characterization
The characterization of TSO anodes used in this study has been reported in our earlier study.25 They are primarily composed of Ti4O7 as indicated by X-ray diffraction analysis (Fig. S2A and B†) and have interconnected micropores (Fig. S2C†) with sizes ranging roughly from 0.8 to 9 μm as shown by mercury intrusion analysis (Fig. S2D†). Linear sweep voltammetry (LSV) was performed at a scan rate of 100 mV s−1 using a CHI 660E electrochemical workstation (Austin, TX) with a three-electrode setup, with Ti4O7 as the working electrode (1 cm × 1 cm), a 304 stainless steel rod (10 cm × 0.3 mm) as the counter electrode, and a single-junction saturated silver chloride electrode (SCE) as the reference electrode (Pine Research Instruments, Grove City, PA, USA).2.5 Molecular simulation
Quantum mechanical computation was performed using Gaussian 09 based on the density functional theory (DFT) at the B3LYP/6-311G** level. The molecular geometry was optimized and the vibrational frequencies were computed for each PFAA, based on which a suite of molecular descriptors were calculated, including the frontier electron densities (FEDs) of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), the energy levels of the HOMO (EHOMO) and LUMO (ELUMO) and their gap (ELUMO − EHOMO), and molecular polarizabilities.3. Results and discussion
3.1 PFAA degradation
Experiments were performed to examine electrooxidation in solutions having each of the eight PFAAs spiked individually using a TSO anode at 5 mA cm−2 current density. Fig. 1 shows the results for PFOA and PFOS. It is evident that the concentration of PFOA or PFOS decreased rapidly over time upon electrolysis, and the time-course data are well fitted by the pseudo-first order rate equation (Text S1†). Both linear and branched PFOS (L-PFOS and B-PFOS) were contained in the PFOS sample tested, and the ratio between them was approximately 16.5 (L-PFOS/B-PFOS) based on their responses in MRM. Both L-PFOS and B-PFOS were monitored separately during the experiment, and the data of each are shown in Fig. S3.† The degradation behaviors of L-PFOS and B-PFOS are close to each other (Fig. S3†), and thus only L-PFOS data were reported in the ensuing discussion. Control experiments were performed in the same system with no current applied (Fig. S4†). The result indicates that adsorption of PFOS and other PFAAs on the TSO anode is minimal.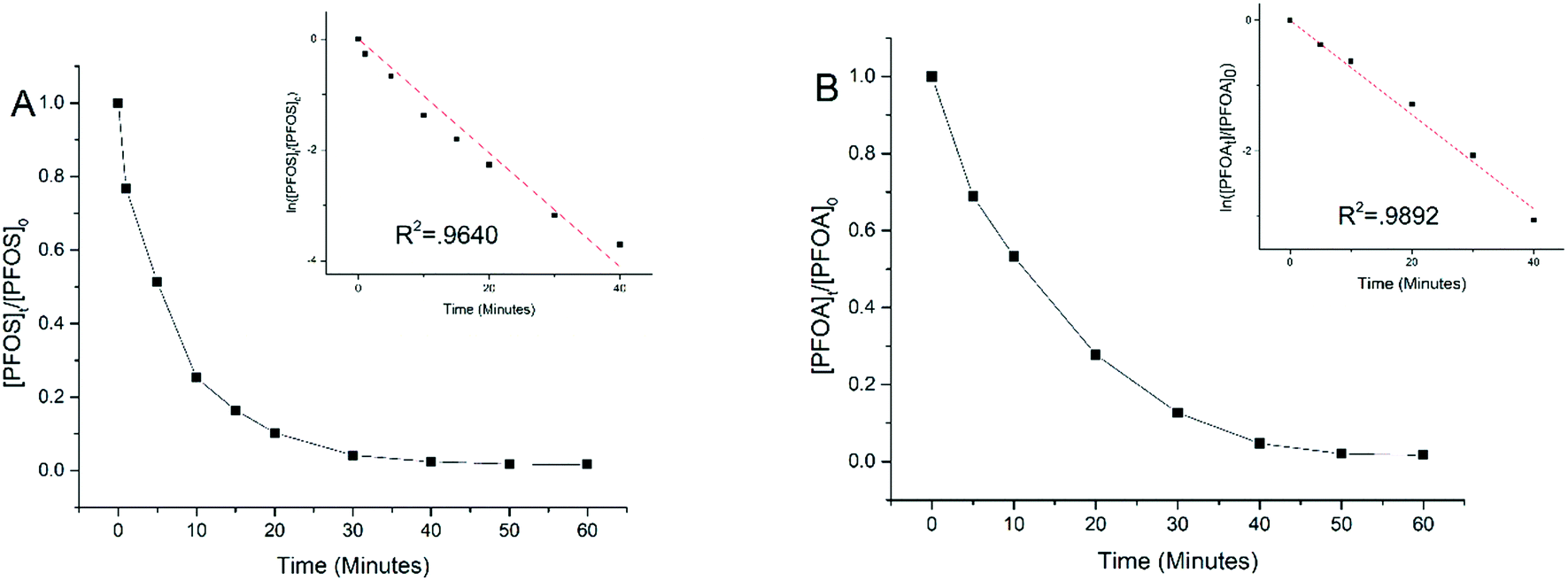 | ||
| Fig. 1 The concentration of PFOS (A) and PFOA (B) over time during electrooxidation with the TSO anode at 5.0 mA cm−2. The test solutions contained each PFAA individually at the initial concentration of 2.0 μM in 100 mM Na2SO4 as a supporting electrolyte. Insert shows fit to the pseudo-first rate model. | ||
It is possible that shorter chain PFAAs may be formed from the degradation of longer chain PFAAs. Therefore, when the electrooxidation of PFOA was tested individually, all PFAAs tested in this study were quantified. Results showed minimal potential by-product formation (<1%) for PFOA (Fig. S5†), and this result is consistent with our earlier findings.26 This indicated that the negatively charged substrates may be held on the anode and undergo further degradation and mineralization.
The time-course data of all eight PFAAs were fitted to the pseudo-first order rate model to calculate the rate constant which was then normalized to the effective electroactive surface area of the anode (Text S1†) to obtain the surface area normalized rate constant (kSA). The effective electroactive surface area (EESA) of the anode is 468.05 cm2, and the method for calculating the effective electroactive surface area is presented in Text S2.† Based on previous work,30 the potential drop in the pores of TSO may have some effect on the EESA, albeit not significant (Text S2†). However, since the pore size is not uniform in the TSO anode used in this study, the potential drop cannot be accurately calculated and thus it is not addressed in this study.
Table 1 lists the kSA values for all eight PFAAs for comparison. In general, the degradation of longer chain PFAAs appeared to be faster, and the degradation of PFSAs was faster than that of PFCAs under the experimental conditions. A discussion based on a quantitative structure–activity relationship study is presented later in the study. The kSA values obtained in this study for PFOA (5.74 × 10−6 m s−1) and PFOS (8.81 × 10−6 m s−1) are comparable to those in our previous study using a similar batch reactor setup,26 but are significantly lower than those of a recent study using a reactive electrochemical membrane (REM) reactor, which are 4.40 × 10−5 m s−1 for PFOA and 1.30 × 10−4 m s−1 for PFOS.27 This is not surprising because a REM operation can significantly enhance mass transfer and thus electrochemical reactions on the anode surface.
| Chemicals | Individual kSA (m s−1) | Mixture kSA (m s−1) | Literature (m s−1) |
|---|---|---|---|
| a The initial concentration of PFOS was 0.1 mM, the electrolyte was 20 mM NaClO4. b The initial concentration of PFOA/PFOS was 10 μM, the electrolyte was 100 mM K2HPO4. c The initial concentration of PFOA was 0.5 mM, the electrolyte was 20 mM NaClO4. | |||
| L-PFOS | 8.81 × 10−6 ± 1.02 × 10−6 | 1.11 × 10−5 ± 3.87 × 10−7 | 4.30 × 10−6 in batch system26a |
| 4.40 × 10−5 in REM system27b | |||
| PFOA | 5.74 × 10−6 ± 5.49 × 10−8 | 4.09 × 10−6 ± 4.56 × 10−8 | 1.13 × 10−5 in batch system26c |
| 1.30 × 10−4 in REM system27b | |||
| PFHpA | 1.26 × 10−6 ± 7.04 × 10−9 | 1.62 × 10−6 ± 2.04 × 10−8 | |
| PFHxS | 2.57 × 10−6 ± 1.64 × 10−7 | 2.59 × 10−6 ± 2.02 × 10−7 | |
| PFHxA | 5.02 × 10−7 ± 5.87 × 10−9 | 3.42 × 10−7 ± 4.01 × 10−9 | |
| PFPeA | 5.99 × 10−8 ± 2.45 × 10−9 | 1.19 × 10−7 ± 5.12 × 10−9 | |
| PFBS | 1.75 × 10−7 ± 7.88 × 10−9 | 1.59 × 10−7 ± 4.44 × 10−9 | |
| PFBA | 3.49 × 10−8 ± 4.17 × 10−9 | 7.94 × 10−8 ± 7.76 × 10−9 | |
3.2 Effect of pH and the electrolyte
The impact of pH on PFOS degradation during electrooxidation was evaluated with the TSO anode at a current density of 5.0 mA cm−2 in solutions of 50 mM H2SO4, 100 mM NaOH and 100 mM phosphate buffer, respectively. The surface area normalized rate constants (kSA) of L-PFOS degradation obtained in these solutions are shown in Table 2. The kSA value was slightly smaller in the acidic solution, probably because of the lower anodic potential in this solution than those in the other two solutions. The influence of the bulk solution pH is expected to be minimal because PFOS degradation occurred on the anode surface where the pH was controlled more by the anodic reactions rather than the bulk solution.| Electrolyte | pH | Anodic potential (V vs. SHE) | k SA (m s−1) |
|---|---|---|---|
| 50 mM H2SO4 | 1.95–2.20 | 3.070 | 5.72 × 10−6 ± 5.46 × 10−7 |
| 100 mM NaOH | 12.40–12.55 | 3.104 | 7.89 × 10−6 ± 1.94 × 10−7 |
| 100 mM phosphate buffer | 7.03–7.29 | 3.109 | 7.89 × 10−6 ± 6.02 × 10−7 |
A set of experiments were also performed to evaluate the electrooxidation of PFOS with TSO in solutions containing different supporting electrolytes, including 10 mM, 25 mM and 100 mM Na2SO4, 100 mM NaNO3 and 100 mM NaClO4. The kSA values of L-PFOS degradation obtained in different electrolytes are summarized in Table 3. At the same current density, the anodic potential was usually higher in lower concentration electrolytes as seen in Table 3. Therefore, kSA values decreased with increasing electrolyte concentration. The kSA did not vary much for the three different electrolyte solutions at 100 mM.
| Electrolyte | Anodic potential (V vs. SHE) | k SA (m s−1) |
|---|---|---|
| 10 mM Na2SO4 | 3.830 | 1.30 × 10−5 ± 2.34 × 10−7 |
| 25 mM Na2SO4 | 3.622 | 1.26 × 10−5 ± 1.94 × 10−7 |
| 100 mM Na2SO4 | 3.322 | 8.81 × 10−6 ± 2.42 × 10−7 |
| 100 mM NaNO3 | 3.441 | 9.26 × 10−6 ± 4.14 × 10−7 |
| 100 mM NaClO4 | 3.506 | 9.91 × 10−6 ± 2.90 × 10−7 |
Taken together, the change in the electrolyte type, concentration and pH of the reaction solution appeared to have minimal, if any, impact on PFOS degradation by electrooxidation on TSO anodes. This may indicate that the bulk solution conditions have very little effect on the anode surface conditions that are mainly controlled by the water oxidation reactions on the anode under our experimental conditions. This may be one advantage in applications to waters of varying conditions. It should be noted though that the conditions having been tested here may differ from those of many PFAS-contaminated wastewaters that have lower electrolyte concentrations and high concentrations of co-existing contaminants. More experiments are needed to draw more conclusive guidelines regarding applications.
3.3 Mixture of PFAAs
Experiments were conducted to evaluate the performance of electrooxidation with the TSO anode in a solution containing a mixture of PFAAs including PFBA, PFPeA, PFHxA, PFHpA, PFOA, PFBS, PFHxS and PFOS at varying current densities. Fig. 2 shows the concentration profiles of each PFAA in the mixture solution during electrooxidation at current densities of 5 mA cm−2 (Fig. 2A) and 15 mA cm−2 (Fig. 2B) as examples to show the degradation behavior. The degradation of all 8 PFAAs was evident, with the rates being greater at the higher current density, while the degradation rates of PFAAs followed roughly the similar general order for both high and low current densities: L-PFOS > PFOA > L-PFHxS > PFHpA > PFHxA > L-PFBS > PFPeA > PFBA. Over 90% PFOA and PFOS were removed within 30 minutes at 5 mA cm−2 and within 10 minutes at 15 mA cm−2. At 15 mA cm−2, all PFAAs were over 90% degraded except for PFBA, which was about 50% degraded after 8 hours of electrooxidation treatment. The concentration of fluoride in the solution was also quantified at the end of the treatment, and the defluorination ratio was calculated via dividing the released F− concentration by the total fluorine in the PFAA that has been removed from the system (Text S3†). The results are listed in Table S3,† and the defluorination ratio was above 90% when the current density was above 10 mA cm−2.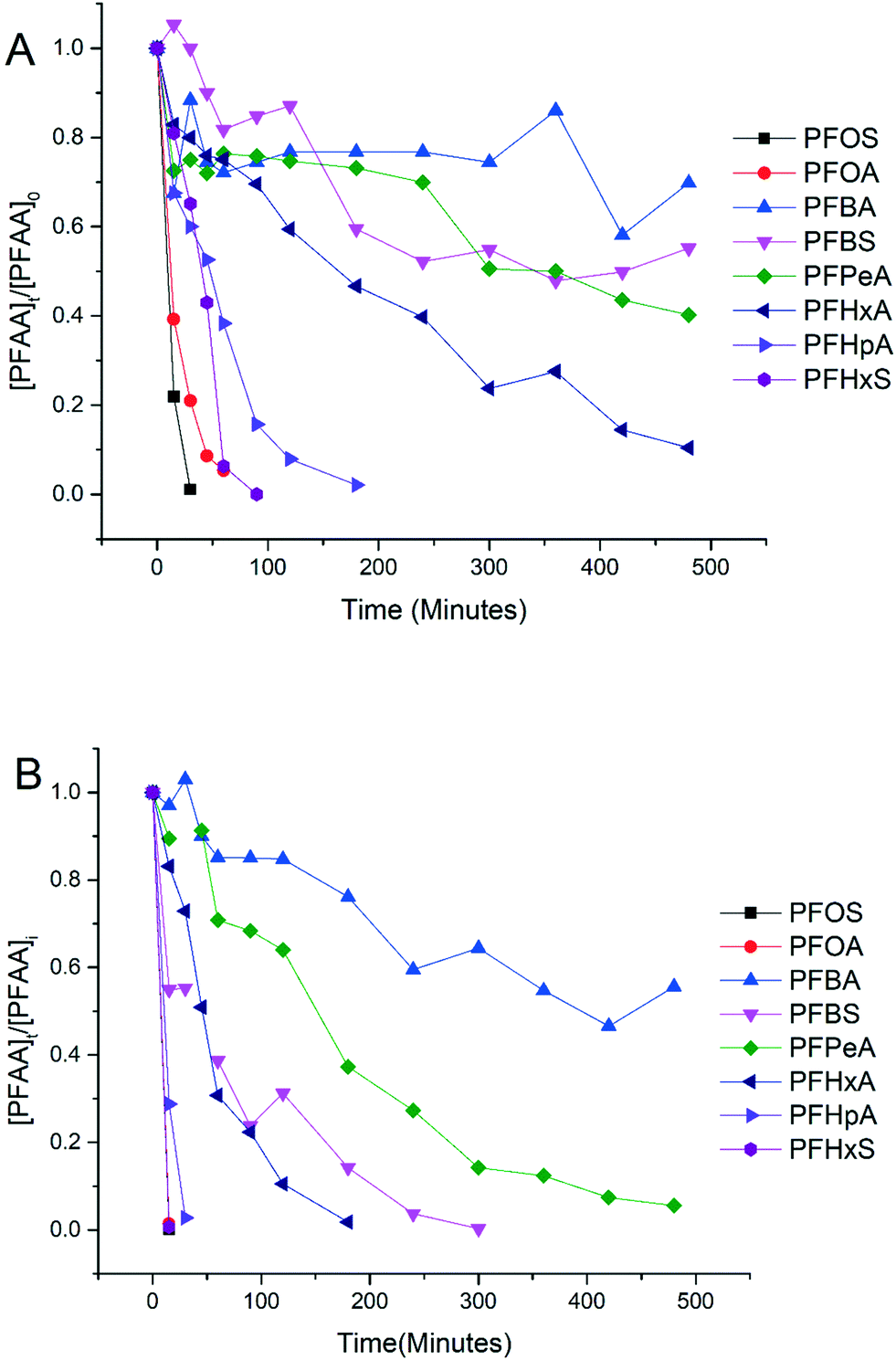 | ||
| Fig. 2 The concentration of each PFAA over time during electrooxidation with the TSO anode at 5 mA cm−2 (A) and 15 mA cm−2 (B). The test solutions contained all PFAAs in the mixture with each at the initial concentration of 2.0 μM in 100 mM Na2SO4 as a supporting electrolyte. | ||
The surface area normalized pseudo-first order rate constants for PFAA degradation obtained from the electrooxidation treatment of the PFAA mixture at 5 mA cm−2 are also listed in Table 1 to compare with those obtained from the experiment with each PFAA tested individually at the same initial concentration (2.0 μM). It appears that the kSA values in the solutions with PFAAs spiked individually are not significantly different from those in the mixture, suggesting a minimal competition effect, if any, among PFAAs at the tested concentration (2.0 μM). The competition effect may however become evident as the concentrations of PFAAs become higher, such as at the level tested in our previous studies (0.5 mM).25 Notably, although the relative order in the PFAA degradation rate constants remains the same between solutions with PFAAs spiked individually and in a mixture, the kSA values of certain PFAAs increase in the mixture compared to those in the individual solution (e.g. PFOS) while others decrease (e.g. PFOA). Such variation may be attributed to the interference or interactions between species during electrooxidation (EO) reactions, which warrants further investigation.
The degradation of PFAAs was evaluated with all PFAAs spiked in a mixture under different current densities, and the obtained kSA values are plotted in Fig. 3 in relation to the chain lengths and the functional head groups. For each PFAA, the kSA increased with increasing current density. For the PFAAs having the same functional group (PFSAs vs. PFCAs), the increase in carbon chain length led to greater reactivity. This is in agreement with other studies showing shorter chain PFAAs being more recalcitrant to electrooxidation.17,31 For the PFAAs of the same carbon chain length, the ones with the sulfonate head group tend to degrade faster than those with the carboxylic group.
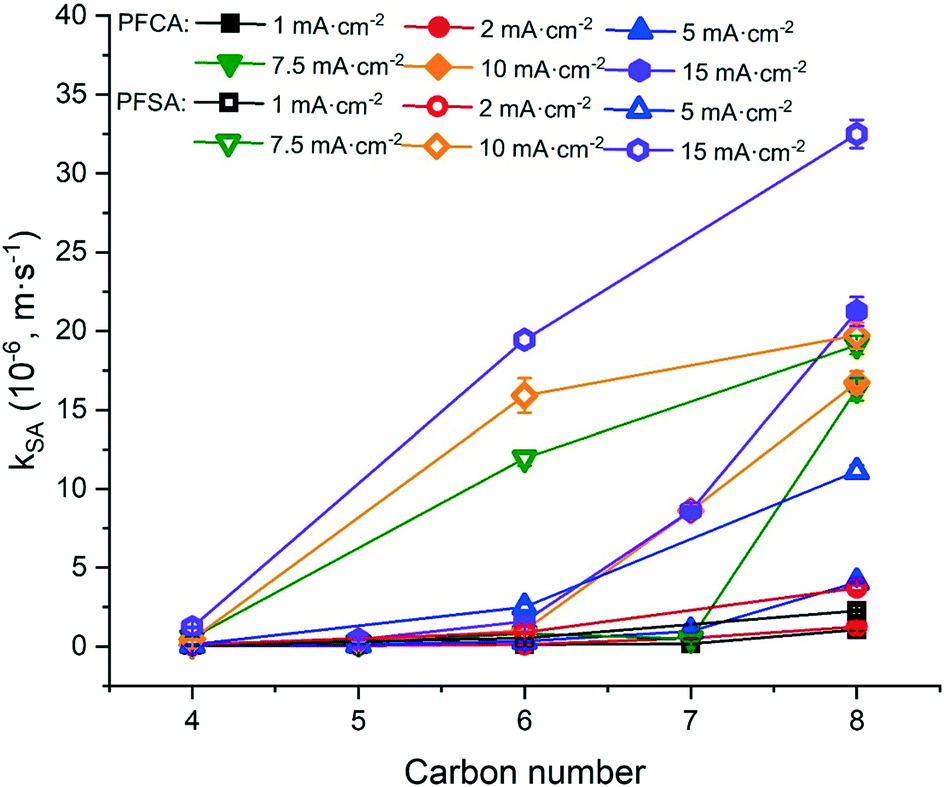 | ||
| Fig. 3 The kSA values of PFAAs of different carbon chain lengths and functional head groups, obtained in electrooxidation experiment with TSO anodes at different current densities. The test solutions contained all PFAAs in the mixture with each at the initial concentration of 2.0 μM in 100 mM Na2SO4 as a supporting electrolyte. Error bar represents standard deviation. | ||
The increased PFAA removal under higher current densities may be ascribed to the higher anodic potentials as the current density increases before reaching the point at which mass transfer becomes limiting. The limiting current method was used to measure the mass transfer rate of each PFAA on the TSO anode (Text S4†). The mass transfer rates of all the PFAAs are summarized in Table S5.† The comparison of the data in Tables 3 and S5† indicates that all PFAA degradation was controlled by the EO process under the experimental conditions. The surface area normalized degradation rate constants for each PFAA obtained from the experiments with mixture solutions are plotted against the anodic potential as shown in Fig. 4. It is again evident that the PFAAs with relatively longer carbon chains tend to have greater degradation rates across the tested anodic potential range than the shorter ones, and the PFSAs have higher rates than the PFCAs (Fig. 4). Regardless of the length of carbon chains, all PFAA degradation rates increased along with the anodic potential. The PFAAs started to exhibit degradation behavior when the anodic potential was above about 2.87 V vs. SHE (standard hydrogen electrode). It is where water oxidation occurs according to linear sweep voltammetry (LSV) as shown in Fig. S6,† which corroborates that the hydroxyl free radicals generated via water oxidation play an essential role in PFAA degradation during electrooxidation on the TSO anode.26 It is regarded that direct electron transfer (DET) and hydroxyl radical attack take place in a concerted manner that lead to PFAA degradation during electrochemical oxidation.17,32
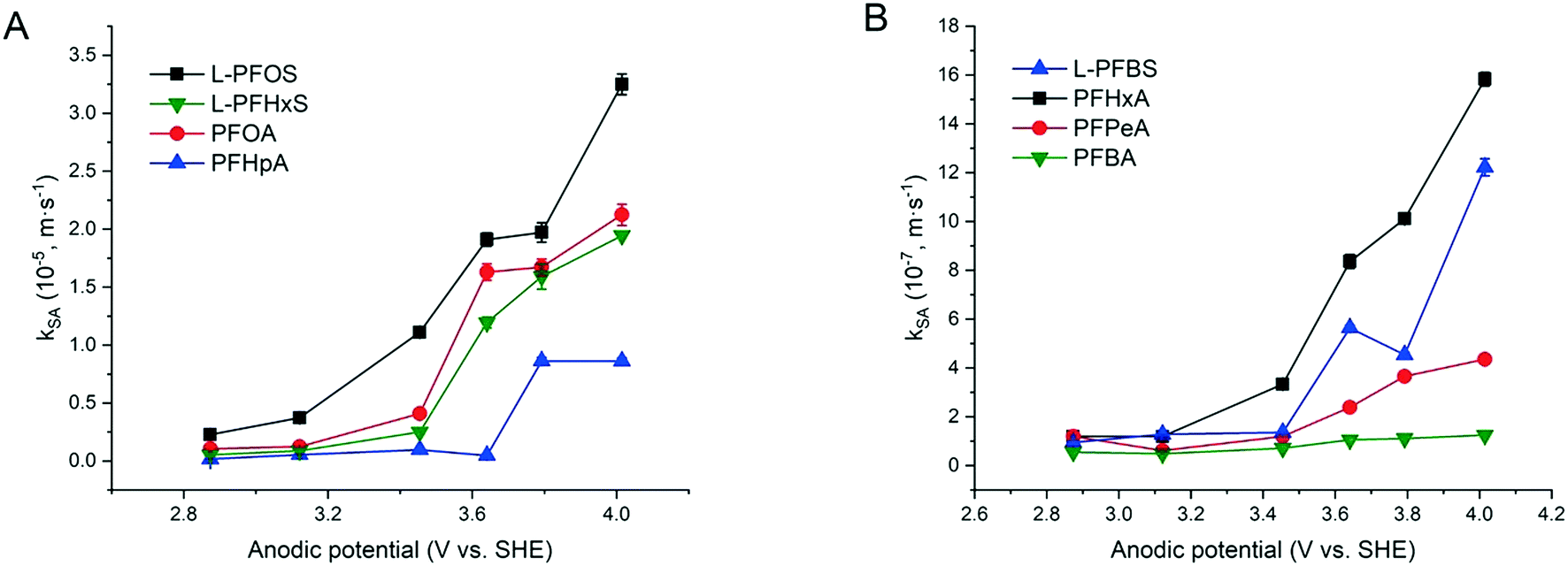 | ||
| Fig. 4 The kSA values of 8 PFAAs plotted against the anodic potential vs. SHE (standard hydrogen electrode) for electrooxidation. The test solutions contained all PFAAs in the mixture with each at the initial concentration of 2.0 μM in 100 mM Na2SO4 as a supporting electrolyte. Error bar represents standard deviation. The data are shown in two figures according to orders of magnitude for better observation. | ||
3.4 Influence of molecular structures
In an effort to explain the trends in different degradation rates across tested PFAAs found in this study, the relationships were explored between certain molecular descriptors and the surface area normalized rate constant of PFAA degradation measured in the experiments with solutions each containing an individual PFAA at the current density of 5 mA cm−2. It has been proposed that the combination of direct electron transfer and hydroxyl radical attack plays a major role in the electrooxidative degradation of PFAAs.26 Therefore, certain molecular and electrical distribution properties of the PFAAs related to their reactivity with hydroxyl radicals and electron transfer have been examined.Frontier molecular orbital energies often play an important role in determining the reactivity of organic chemicals.33 The energies of the highest occupied molecular orbital (EHOMO) and the lowest unoccupied molecular orbital (ELUMO) represent the ability of a molecule to donate or gain an electron, respectively, and the gap between them implies the stability of the molecule in terms of redox reactivity.34,35 In this study, greater ELUMO − EHOMO gaps were seen for shorter chained PFAAs ranging from 0.244 hartrees for PFBA to 0.155 hartrees for PFOA. It thus makes sense that we have seen a negative correlation between the ELUMO − EHOMO gap and the logkSA (Fig. 5A). Indeed, these values form a cohesive negative linear trend through each of the functional groups (PFSAs vs. PFCAs) when plotting logkSA against the ELUMO − EHOMO gap. The energy gap however does not capture the rate constant trend between the functional groups, although this is not surprising given the large difference the functional groups make to molecular properties.
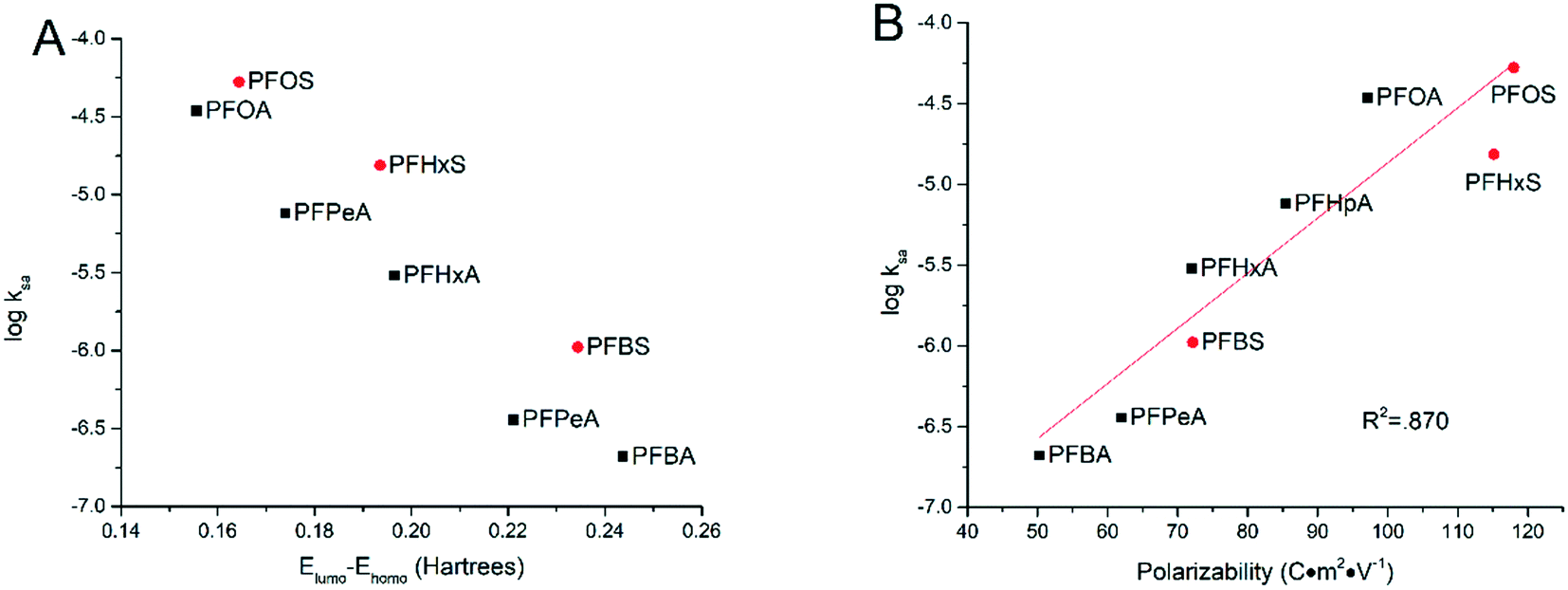 | ||
| Fig. 5 Relationship between the logarithm of the surface area normalized rate constants and ELUMO − EHOMO gap (A) and molecular polarizability (B). | ||
Another molecular descriptor of interest, molecular polarizability, indicates the electronic flexibility of each molecule, thus, in connection to the tendency of oxidative reactivity as well as the direct electron transfer on the anode.34,36 When plotted against the experimental degradation rate constants found in this study, the molecular polarizability has a nice positive correlation across both functional groups, serving as a good indicator for the reactivity of PFAAs. This suggests that the direct electron transfer may be the rate-limiting step in PFAA degradation in TSO-based electrooxidation systems, although the hydroxyl radicals generated via water oxidation may also play a role in concert.26
4. Conclusions
Novel TSO anodes have been shown to be effective in removing a range of PFCAs and PFSAs in solutions of varying compositions and under a range of operation conditions in this study. The change in the electrolyte type, concentration and pH of the reaction solution appeared to have minimal, if any, impact on PFAA degradation. This is probably because the bulk solution conditions have very little effect on the anode surface conditions that are controlled by the water oxidation reactions on the anode under our experimental conditions. The kSA for PFAAs in the mixture was not much different from that obtained when each PFAA was spiked in the solution individually, indicating the absence of a strong competitive effect among the PFAAs in the mixture under the experimental conditions. For the PFAAs having the same head acid group (PFSAs vs. PFCAs), the increase in carbon chain length led to greater reactivity. For the PFAAs of the same carbon chain length, the ones with the sulfonate head group tend to degrade faster than those with the carboxylic group. The PFAAs started to exhibit degradation behavior when the anodic potential was above about 2.83 V vs. SHE where water oxidation occurs, suggesting that the hydroxyl free radicals generated via water oxidation play a role in PFAA degradation.26 PFAA degradation rates increased along with the anodic potential. Molecular polarizability, a molecular descriptor of electron flexibility and transfer tendency, appears to be a good indicator for PFAA degradation, suggesting that the direct electron transfer may be the rate-limiting step for PFAA degradation in TSO-based electrooxidation systems. Our research indicates the effectiveness of TSO-based electrooxidation and provides a basis for further studies leading to process design and optimization for remediation of PFAS contaminated waters. It should be noted that the experiments in this study were performed with selected PFAAs in simple electrolyte solutions. More studies are warranted to examine a wider range of PFAS compounds in solutions reflecting actual conditions of PFAS-contaminated water/wastewater.Conflicts of interest
A patent application is pending on “Methods and Systems for Electrochemical Oxidation of Polyfluoroalkyl and Perfluoroalkyl Contaminants” (62/377,120) with QH as one of the inventors.Acknowledgements
This study was supported in part by the U.S. Department of Defense SERDP ER-2717 and ER-1320. Dr. Shangtao Liang, Dr. Lei Li and Ms. Lu Wang are acknowledged for their helpful discussion. Dr. Sayed Hassan is acknowledged for help with the fluoride analysis.References
- S. Fujii, C. Polprasert, S. Tanaka, N. P. Hong Lien and Y. Qiu, New POPs in the water environment: distribution, bioaccumulation and treatment of perfluorinated compounds – a review paper, J. Water Supply: Res. Technol.--AQUA, 2007, 56, 313–326 CrossRef CAS.
- J. P. Giesy and K. Kannan, Global Distribution of Perfluorooctane Sulfonate in Wildlife, Environ. Sci. Technol., 2001, 35, 1339–1342 CrossRef CAS PubMed.
- I. Ross, J. McDonough, J. Miles, P. Storch, P. T. Kochunarayanan, E. Kalve, J. Hurst, S. S. Dasgupta and J. Burdick, A review of emerging technologies for remediation of PFASs, Remediation, 2018, 28, 101–126 CrossRef.
- S. Mejia-Avendano, S. V. Duy, S. Sauve and J. X. Liu, Generation of Perfluoroalkyl Acids from Aerobic Biotransformation of Quaternary Ammonium Polyfluoroalkyl Surfactants, Environ. Sci. Technol., 2016, 50, 9923–9932 CrossRef CAS PubMed.
- C. Lau, K. Anitole, C. Hodes, D. Lai, A. Pfahles-Hutchens and J. Seed, Perfluoroalkyl acids: a review of monitoring and toxicological findings, Toxicol. Sci., 2007, 99, 366–394 CrossRef CAS PubMed.
- C. M. Butt, U. Berger, R. Bossi and G. T. Tomy, Levels and trends of poly- and perfluorinated compounds in the arctic environment, Sci. Total Environ., 2010, 408, 2936–2965 CrossRef CAS PubMed.
- X. Wang, C. Halsall, G. Codling, Z. Xie, B. Xu, Z. Zhao, Y. Xue, R. Ebinghaus and K. Jones, Accumulation of perfluoroalkyl compounds in tibetan mountain snow: temporal patterns from 1980 to 2010, Environ. Sci. Technol., 2014, 48, 173–181 CrossRef CAS PubMed.
- G. W. Olsen, D. C. Mair, C. C. Lange, L. M. Harrington, T. R. Church, C. L. Goldberg, R. M. Herron, H. Hanna, J. B. Nobiletti, J. A. Rios, W. K. Reagen and C. A. Ley, Per- and polyfluoroalkyl substances (PFAS) in American Red Cross adult blood donors, 2000-2015, Environ. Res., 2017, 157, 87–95 CrossRef CAS PubMed.
- M. F. Rahman, S. Peldszus and W. B. Anderson, Behaviour and fate of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in drinking water treatment: A review, Water Res., 2014, 50, 318–340 CrossRef CAS PubMed.
- Y. R. Gu, T. Z. Liu, H. J. Wang, H. L. Han and W. Y. Dong, Hydrated electron based decomposition of perfluorooctane sulfonate (PFOS) in the VUV/sulfite system, Sci. Total Environ., 2017, 607, 541–548 CrossRef PubMed.
- L. Jin and P. Y. Zhang, Photochemical decomposition of perfluorooctane sulfonate (PFOS) in an anoxic alkaline solution by 185 nm vacuum ultraviolet, Chem. Eng. J., 2015, 280, 241–247 CrossRef CAS.
- L. Jin, P. Y. Zhang, T. Shao and S. L. Zhao, Ferric ion mediated photodecomposition of aqueous perfluorooctane sulfonate (PFOS) under UV irradiation and its mechanism, J. Hazard. Mater., 2014, 271, 9–15 CrossRef CAS PubMed.
- J. Cheng, C. D. Vecitis, H. Park, B. T. Mader and M. R. Hoffmann, Sonochemical Degradation of Perfluorooctane Sulfonate (PFOS) and Perfluorooctanoate (PFOA) in Groundwater: Kinetic Effects of Matrix Inorganics, Environ. Sci. Technol., 2010, 44, 445–450 CrossRef CAS PubMed.
- S. Park, L. S. Lee, V. F. Medina, A. Zull and S. Waisner, Heat-activated persulfate oxidation of PFOA, 6:2 fluorotelomer sulfonate, and PFOS under conditions suitable for in-situ groundwater remediation, Chemosphere, 2016, 145, 376–383 CrossRef CAS PubMed.
- K. E. Carter and J. Farrell, Oxidative destruction of perfluorooctane sulfonate using boron-doped diamond film electrodes, Environ. Sci. Technol., 2008, 42, 6111–6115 CrossRef CAS PubMed.
- H. Lin, J. F. Niu, S. Y. Ding and L. L. Zhang, Electrochemical degradation of perfluorooctanoic acid (PFOA) by Ti/SnO2-Sb, Ti/SnO2-Sb/PbO2 and Ti/SnO2-Sb/MnO2 anodes, Water Res., 2012, 46, 2281–2289 CrossRef CAS PubMed.
- Q. F. Zhuo, S. B. Deng, B. Yang, J. Huang, B. Wang, T. T. Zhang and G. Yu, Degradation of perfluorinated compounds on a boron-doped diamond electrode, Electrochim. Acta, 2012, 77, 17–22 CrossRef CAS.
- Q. C. Ma, L. Liu, W. Cui, R. F. Li, T. T. Song and Z. J. Cui, Electrochemical degradation of perfluorooctanoic acid (PFOA) by Yb-doped Ti/SnO2-Sb/PbO2 anodes and determination of the optimal conditions, RSC Adv., 2015, 5, 84856–84864 RSC.
- B. G. Ruiz, S. Gomez-Lavin, N. Diban, V. Boiteux, A. Colin, X. Dauchy and A. Urtiaga, Efficient electrochemical degradation of poly- and perfluoroalkyl substances (PFASs) from the effluents of an industrial wastewater treatment plant, Chem. Eng. J., 2017, 322, 196–204 CrossRef.
- J. Radjenovic and D. L. Sedlak, Challenges and Opportunities for Electrochemical Processes as Next-Generation Technologies for the Treatment of Contaminated Water, Environ. Sci. Technol., 2015, 49, 11292–11302 CrossRef CAS PubMed.
- F. C. Walsh and R. G. A. Wills, The continuing development of Magneli phase titanium sub-oxides and Ebonex (R) electrodes, Electrochim. Acta, 2010, 55, 6342–6351 CrossRef CAS.
- A. M. Zaky and B. P. Chaplin, Mechanism of p-Substituted Phenol Oxidation at a Ti4O7 Reactive Electrochemical Membrane, Environ. Sci. Technol., 2014, 48, 5857–5867 CrossRef CAS PubMed.
- P. Geng, J. Y. Su, C. Miles, C. Comninellis and G. H. Chen, Highly-Ordered Magneli Ti4O7 Nanotube Arrays as Effective Anodic Material for Electro-oxidation, Electrochim. Acta, 2015, 153, 316–324 CrossRef CAS.
- S. T. Liang, H. Lin, X. F. Yan and Q. G. Huang, Electro-oxidation of tetracycline by a Magneli phase Ti4O7 porous anode: Kinetics, products, and toxicity, Chem. Eng. J., 2018, 332, 628–636 CrossRef CAS.
- S. T. Liang, R. Pierce, H. Lin, S. Y. Chiang and Q. Huang, Electrochemical oxidation of PFOA and PFOS in concentrated waste streams, Remediation, 2018, 28, 127–134 CrossRef.
- H. Lin, J. F. Niu, S. T. Liang, C. Wang, Y. J. Wang, F. Y. Jin, Q. Luo and Q. G. Huang, Development of macroporous Magneli phase Ti4O7 ceramic materials: As an efficient anode for mineralization of poly- and perfluoroalkyl substances, Chem. Eng. J., 2018, 354, 1058–1067 CrossRef CAS.
- T. X. H. Le, H. Haflich, A. D. Shah and B. P. Chaplin, Energy-Efficient Electrochemical Oxidation of Perfluoroalkyl Substances Using a Ti4O7 Reactive Electrochemical Membrane Anode, Environ. Sci. Technol. Lett., 2019, 6, 504–510 CrossRef CAS.
- S. Liang, R. D. Pierce, H. Lin, S.-Y. Chiang and Q. Huang, Electrochemical oxidation of PFOA and PFOS in concentrated waste streams, Remed. J., 2018, 28, 127–134 CrossRef.
- D. A. Skoog, F. J. Holler and T. A. Nieman, Principles of instrumental analysis, Brooks/Cole, Belmont (Calif.), 5th edn, 1998 Search PubMed.
- A. Lasia, Porous electrodes in the presence of a concentration gradient, J. Electroanal. Chem., 1997, 428, 155–164 CrossRef CAS.
- C. E. Schaefer, C. Andaya, A. Urtiaga, E. R. McKenzie and C. P. Higgins, Electrochemical treatment of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS) in groundwater impacted by aqueous film forming foams (AFFFs), J. Hazard. Mater., 2015, 295, 170–175 CrossRef CAS PubMed.
- B. P. Chaplin, Critical review of electrochemical advanced oxidation processes for water treatment applications, Environ. Sci.: Processes Impacts, 2014, 16, 1182–1203 RSC.
- Z. W. Cheng, B. W. Yang, Q. C. Chen, W. C. Ji and Z. M. Shen, Characteristics and difference of oxidation and coagulation mechanisms for the removal of organic compounds by quantum parameter analysis, Chem. Eng. J., 2018, 332, 351–360 CrossRef CAS.
- H. Kusic, B. Rasulev, D. Leszczynska, J. Leszczynski and N. Koprivanac, Prediction of rate constants for radical degradation of aromatic pollutants in water matrix: A QSAR study, Chemosphere, 2009, 75, 1128–1134 CrossRef CAS PubMed.
- C. G. Zhan, J. A. Nichols and D. A. Dixon, Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: Molecular properties from density functional theory orbital energies, J. Phys. Chem. A, 2003, 107, 4184–4195 CrossRef CAS.
- X. H. Jin, S. Peldszus and P. M. Huck, Predicting the reaction rate constants of micropollutants with hydroxyl radicals in water using QSPR modeling, Chemosphere, 2015, 138, 1–9 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ew00759h |
| ‡ These authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2020 |