
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comparative perspective of electrochemical and photochemical approaches for catalytic H2O2 production
Yanyan
Sun
a,
Lei
Han
*b and
Peter
Strasser
 *a
*a
aDepartment of Chemistry, Technical University of Berlin, 10623 Berlin, Germany. E-mail: pstrasser@tu-berlin.de
bCollege of Materials Science and Engineering, Hunan University, 410082, Changsha, Hunan, China. E-mail: hanlei@hnu.edu.cn
First published on 6th August 2020
Abstract
Hydrogen peroxide (H2O2) has a wide range of important applications in various fields including chemical industry, environmental remediation, and sustainable energy conversion/storage. Nevertheless, the stark disconnect between today's huge market demand and the historical unsustainability of the currently-used industrial anthraquinone-based production process is promoting extensive research on the development of efficient, energy-saving and sustainable methods for H2O2 production. Among several sustainable strategies, H2O2 production via electrochemical and photochemical routes has shown particular appeal, because only water, O2, and solar energy/electricity are involved during the whole process. In the past few years, considerable efforts have been devoted to the development of advanced electrocatalysts and photocatalysts for efficient and scalable H2O2 production with high efficiency and stability. In this review, we compare and contrast the two distinct yet inherently closely linked catalytic processes, before we detail recent advances in the design, preparation, and applications of different H2O2 catalyst systems from the viewpoint of electrochemical and photochemical approaches. We close with a balanced perspective on remaining future scientific and technical challenges and opportunities.
 Yanyan Sun | Yanyan Sun received her PhD degree in Physical Chemistry and Electrochemistry from Technical University of Berlin (Germany) under the supervision of Prof. Peter Strasser in 2018. Currently, she is continuing her postdoctoral research with Prof. Peter Strasser. Her research focuses on the design and fabrication of carbon-based functional materials for the oxygen reduction reaction in fuel-cells and hydrogen peroxide production. |
 Lei Han | Lei Han is a professor at the School of Materials Science and Engineering, Hunan University, P. R. China. He received his PhD degree from Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, under the supervision of Prof. Shaojun Dong in 2015. After that, he did postdoctoral research at the Nanyang Technological University, University of Waterloo, and Free University of Berlin before joining Hunan University in 2018. His research interests include the design and application of nanostructured materials for solar energy conversion and storage systems including photocatalysis, photoelectrochemical devices and oxygen electrochemistry. |
 Peter Strasser | Peter Strasser is the chaired professor of “Electrochemistry and Electrocatalysis” in the Department of Chemistry at the Technical University of Berlin. Prior to his appointment, he was Assistant Professor at the Department of Chemical and Biomolecular Engineering at the University of Houston, after he served as Postdoctoral Scientist and later Senior Member of staff at Symyx Technologies, Inc., Santa Clara, USA. He earned his PhD in Physical Chemistry and Electrochemistry from the Fritz-Haber-Institute of the Max-Planck-Society in Berlin under the direction of Gerhard Ertl. He studied chemistry at Stanford University, the University of Tübingen, Germany, and the University of Pisa, Italy. He was awarded the ISE “Brian Conway Prize” in Physical Electrochemistry, the IAHE “Sir William Grove” award, the “Otto-Roelen” medal by the German Catalysis Society, the “Ertl Prize”, as well as the “Otto-Hahn Research Medal” by the Max-Planck Society. |
1 Introduction
Hydrogen peroxide (H2O2) has been widely used as an eco-friendly oxidant in chemical industry and environmental treatment such as organic/inorganic chemical synthesis, pulp and paper bleaching, medical disinfection and wastewater treatment.1–5 Moreover, H2O2 could also be utilized as an ideal oxidant and sustainable energy carrier alternative to oxygen and hydrogen in fuel cells since it possesses the advantages of easy storage and safe operation, high oxidation potential, and only water as a by-product.6,7 Currently, the well-established industrial anthraquinone-based method for H2O2 production involves the hydrogenation and oxidation of the anthraquinone molecule, and extraction, purification and concentration of H2O2 solution.4 This method is very beneficial for the large-scale production of highly concentrated H2O2, but there are still some serious sustainability challenges to be solved. First, this method involves a sequential multi-step procedure including many purification steps, thus consuming a significant amount of energy and resources. Moreover, the use of a large amount of H2 during the hydrogenation step leads to difficulty in handling and storage. The by-products of anthraquinone/anthrahydroquinone require the use of a large amount of organic solvents during the process, which will become waste at the end. Furthermore, highly concentrated H2O2 is a hazardous chemical and requires great caution in transport, handling and storage, leading to an increase in cost and some safety issues. Alternatively, considerable efforts have been dedicated to developing the direct production of H2O2 from a mixture of H2 and O2 using chemical catalysts.8–11 However, to catalyze this reaction, noble-metal-based catalysts such as Pd, Au and their alloys are needed, which is a potential barrier in view of scale-up practical applications owing to their high cost and low abundance. Moreover, the activity and selectivity toward H2O2 production are also quite low, because water production is thermodynamically more favorable than H2O2 production. Last but not least, the incoming feed of H2 and O2 constitutes an explosive and potentially hazardous mixture. To avoid this last issue, hydrogen-permeable membranes possessing catalytic activity have also been developed,12 which are generally made of thin Pd and Pd–Ag alloy membranes, Pd-coated V, Nb, or Mo metal membranes, and porous inorganic membranes. These bi-functional membranes enable the direct production of H2O2 without mixing H2 and O2. However, they lack sufficient stability and reproducibility, and have thus remained unattractive for industrial-scale production of H2O2 at the moment. Another important aspect that needs to be considered in the development of a future, sustainable synthetic H2O2 process is down-scalability of the process and product, that is, the direct production of dilute H2O2 from O2 or H2O locally and on smaller scales. Dilute H2O2 is more suitable for a wide range of applications, and also addresses the safety issue and largely decreases the cost mentioned above.The electricity- and light-driven oxygen reduction reaction (ORR) and/or water oxidation reaction (WOR) through proton-coupled electron transfer (PCET) have been considered to be appealing processes for direct efficient and economic H2O2 production, chiefly because they merely require water, O2, and solar/electric energy,13–22 and enable scalable H2O2 production without H2/O2 gas explosion risks, thus dramatically decreasing the cost of transport, storage and handling of highly concentrated H2O2. The ORR process plays critical roles in various sustainable energy conversion/storage devices such as rechargeable fuel cells and rechargeable metal–air batteries.23–26 Generally, there are three kinds of reaction pathway during the ORR process (eqn (1)–(3)): (1) the four-electron ORR for water production; (2) the two-electron ORR for H2O2 production; and (3) the one-electron ORR to ˙OOH. It should be mentioned that all the reduction potentials in the present work are relative to the standard hydrogen electrode (SHE) without a special statement. Nevertheless, massive efforts have been made to develop highly advanced four-electron ORR catalysts for boosting this reaction and thus maximizing the energy conversion efficiency of devices, probably because H2O2 production could result in low energy conversion efficiency and cause instability issues such as catalyst corrosion and chemical degradation of the polymer electrolyte membrane in a proton-exchange membrane fuel cell (PEMFC) resulting from the self-decomposition of the produced H2O2.27 Moreover, the four-electron ORR process (+1.23 VSHE) is thermodynamically favored compared to the two-electron ORR process (+0.68 VSHE) due to its more positive equilibrium potential. Recently, the direct electrochemical two-electron ORR for H2O2 production began to receive increasing attention from the perspective of chemical synthesis purposes. H2O2 production through the electrochemical two-electron ORR process can be performed in several types of electrochemical devices such as electrolytic cells,15 fuel cells,28,29 and microbial fuel cells.30,31 In an aqueous electrolytic device, H2O2 production can be achieved through the electrolysis of water and O2, where the anodic half-cell reaction proceeds by water oxidation and the cathodic half-cell reaction proceeds by the ORR. It should be here highlighted that on-site production of H2O2 has been achieved at the point of use through electrochemical generation devices by the HPNow company.32 Moreover, electrolyte-free H2O2 could also be obtained in the case of using a solid polymer electrolyte.1,15 Besides, in combination with intermittent renewable power sources like solar or wind, H2O2 could be produced in an eco-friendly fashion without additional energy input. What's more, electrochemical devices also enable in situ, on-demand H2O2 production for some practical applications, thus avoiding the cost of storage and transportation. In particular, if the two-electron ORR process is carried out in fuel cells and microbial fuel cells, additional chemical electricity could be recovered along with H2O2 production. Meanwhile, in the fuel cell system, the potential for explosion could be avoided due to the separation of O2 and H2 by an ion-exchange membrane. Additionally, in the microbial fuel cell system, some organic compounds in wastewater could be directly oxidized by electrochemically active bacteria on the anode, thus achieving simultaneously wastewater treatment. In contrast, differently from the two-electron ORR process, the direct two-electron WOR has recently also attracted growing interest for electrochemical on-site production of H2O2 recently since it requires only the use of water as a raw material and enables the simultaneous production of valuable H2 at the cathode by a single water electrolysis device. Similarly, there are generally three reaction pathways during the WOR process (eqn (4)–(6), e.g. in acid electrolyte),33 including the one-electron pathway to form ˙OH, the two-electron pathway for H2O2 production and the four-electron pathway for O2 evolution. Among these three water oxidation routes, currently, most research has mainly focused on the development of oxygen electrocatalysts for promoting the four-electron WOR (also called the oxygen evolution reaction, OER) since the OER is an important half-reaction in electrochemical water splitting and rechargeable metal–air batteries.34 So, improvement of the catalyst is necessary to achieve efficient energy conversion and storage. In contrast, two-electron water oxidation to H2O2 production has been long overlooked. This may be because the equilibrium potential of the two-electron WOR (+1.77 VSHE) is thermodynamically higher than that of the four-electron WOR (+1.23 VSHE). Therefore, the critical challenge for this route lies in the development of effective WOR catalysts capable of significantly promoting the two-electron pathway and suppressing the thermodynamically favorable four-electron pathway owing to its more negative potential.
| O2 + 4H+ + 4e− → 2H2O E0 = +1.23 VSHE | (1) |
| O2 + 2H+ + 2e− → H2O2 E0 = +0.68 VSHE | (2) |
| O2 + H+ + e− → ˙OOH E0 = −0.13 VSHE | (3) |
| H2O → ˙OH + H+ + e− E0 = +2.73 VSHE | (4) |
| 2H2O → H2O2 + 2H+ + 2e− E0 = +1.77 VSHE | (5) |
| 2H2O → O2 + 4H+ + 4e− E0 = +1.23 VSHE | (6) |
Since the ORR is an inherently sluggish process involving proton-coupled multi-electron transfer and multiple reaction paths,1,18,24 a highly efficient and selective ORR for H2O2 production via the electrochemical and photochemical methods remains a significant challenge and faces many fundamental scientific and technical hurdles.1,13 The first study on electrochemical production of H2O2 came from Traube in 1887, who produced H2O2 from O2 at a Hg–Au electrode.41 Subsequently, Berl et al. reported the electrochemical performances of a carbon electrode for H2O2 production in 1939.42 After that, there was a long time in which most research mainly focused on the investigation of experimental influencing parameters on the H2O2 production in electrochemical devices, such as high internal resistance and a low concentration of oxygen dissolved in the electrolyte.43,44 Some methods have also been developed to solve these issues above, including the usage of gas diffusion electrodes,43 an applied external voltage,30 and buffered electrolytes, and reducing the distance between the anode and the cathode.45 In contrast, little attention was paid to the development of cost-effective advanced two-electron ORR catalysts with high catalytic activity, selectivity, and stability, which is actually also very important for H2O2 production. Recently, various ORR catalysts toward H2O2 production have sprung up including noble-metal-based materials, transition-metal-based materials, and metal-free carbon-based materials. Besides, some metal oxides such as ZnO, WO3, SnO2, BiVO4, and TiO2 have been both theoretically estimated and experimentally investigated as potential catalyst candidates for H2O2 production from two-electron water oxidation. On the other hand, the history of photocatalytic H2O2 production can be traced back to the 20th century. Baur and Neuweiler first observed the photo-driven ORR for H2O2 production over ZnO in the presence of glycerine and glucose under light illumination in 1927.46 In subsequent years, various photocatalysts were developed to catalyze H2O2 production through the two-electron ORR, including metal oxides, metal–organic frameworks or coordination polymers, and metal-free graphitic carbon nitride (g-C3N4). Meanwhile, some photoelectrochemical systems based on electrocatalysts and photocatalysts have been designed and developed for H2O2 production. In the past few years, considerable research efforts were reported with a focus on the electrocatalytic and photocatalytic production of H2O2, and many exciting results have been achieved that merit a careful review to establish the conceptual links between the recent advances in materials and methodologies.
This review begins with a brief introduction of the fundamentals principles and some important parameters for evaluating the catalytic performances of electricity/photo-driven ORR and WOR catalysts designed for H2O2 production. Thereafter, we summarize recent progress in the design, preparation, and applications of different catalysts for H2O2 production from the perspective of electrochemical and photochemical routes. Finally, the critical challenges and opportunities for future development in this field are also presented. We hope that this review could provide guidance for the design and preparation of novel electricity/photo-driven ORR and WOR catalysts with high performance, and promote their practical applications in energy conversion/storage systems.
2 Fundamentals of electrocatalytic and photocatalytic H2O2 production
Electrocatalytic and photocatalytic H2O2 production through the ORR and WOR processes are both thermodynamically uphill reactions. What complicates the H2O2 production chemistry further is the fact that there are several competing reduction products (e.g. H2O, H2O2, and ˙O2−) formed via distinct reaction pathways of the ORR and WOR processes with one-, two- and four-electron transfer. The dominant reaction pathway is mainly determined by the nature of the electrocatalysts and photocatalysts as well as the experimental conditions.47–49 As a result of these competing reaction pathways, electrocatalytic and photocatalytic H2O2 production face severe challenges in terms of low energy efficiency and limited Faradaic efficiency (chemical selectivity).2.1 Fundamentals of electrocatalytic H2O2 production
The mechanism of the ORR process has been widely investigated. Following a popular 4-step proton-coupled electron transfer mechanism from the early 2000s, the process involves three reaction intermediates (HOO*, O*, and HO*) and proceeds as follows in acid medium:50| O2 + H+ + e− + * → HOO* | (7) |
| HOO* + H+ + e− → O* + H2O | (8) |
| O* + H+ + e− → HO* + H2O | (9) |
| HO* + H+ + e− → * + H2O | (10) |
| O2 + H+ + e− + * → HOO* | (11) |
| HOO* + H+ + e− → * + H2O2 | (12) |
The catalytic activity and selectivity toward H2O2 production can be evaluated using the Rotating Ring-Disk Electrode (RRDE) technique. For the RRDE technique, linear sweep voltammetry (LSV) was generally performed on the disk electrode with a constant applied potential on the ring electrode (e.g. +1.2 VRHE) for detecting the H2O2 produced on the disk electrode. So, the current assigned to the production of H2O2 (IH2O2, in units of mA) can be calculated from the ring current (Iring, in units of mA) and the electrode geometry-dependent collection efficiency (N) of the ring electrode as follows:
 | (13) |
| Idisk = IH2O2 + IH2O | (14) |
 | (15) |
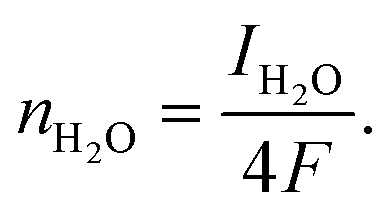 | (16) |
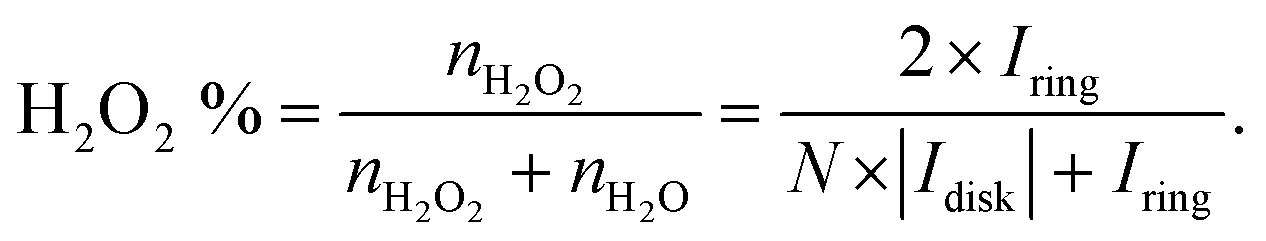 | (17) |
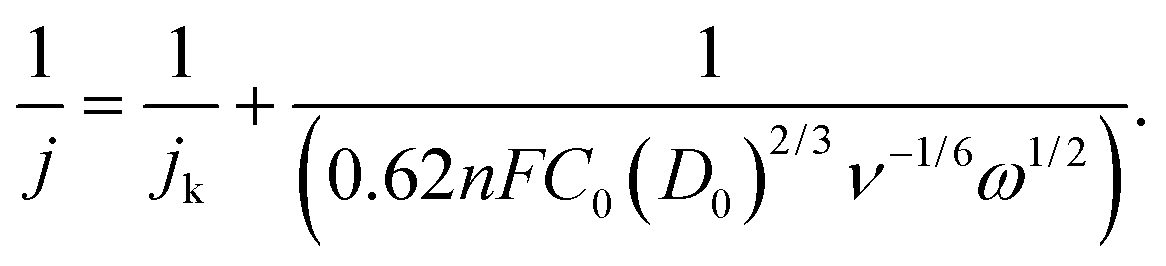 | (18) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), D0 is the diffusion coefficient of oxygen in the electrolyte (cm2 s−1), C0 is the bulk concentration of oxygen in the electrolyte (mol cm−3), ν is the kinetic viscosity of the electrolyte (cm2 s−1), and ω is the angular velocity of the disk (ω = 2πN, N is the linear rotation speed (rpm)).
485 C mol−1), D0 is the diffusion coefficient of oxygen in the electrolyte (cm2 s−1), C0 is the bulk concentration of oxygen in the electrolyte (mol cm−3), ν is the kinetic viscosity of the electrolyte (cm2 s−1), and ω is the angular velocity of the disk (ω = 2πN, N is the linear rotation speed (rpm)).
The produced H2O2 may be further catalytically reduced to H2O by the ORR catalyst, which is detrimental for overall H2O2 production. Starting from this point, the catalytic activity toward the H2O2 reduction reaction (H2O2RR) also needs to be investigated for accurately evaluating the actual H2O2 production capacity. Typically, the evaluation of the H2O2RR was performed under the same conditions as the ORR measurement except the addition of H2O2 in the nitrogen-saturated electrolyte.18
The evaluation of the H2O2 productivity in various reaction media has been typically performed in membrane-separated two-compartment H-cells, H2/O2 fuel cells or electrolysis cells by measuring the concentration of the produced H2O2 at the cathode within defined time intervals. During this process, one of the biggest problems facing ORR routes to H2O2 production is the low solubility of O2 in various electrolytes, such as the bulk concentration of O2 up to 1.2 mM in 0.1 M KOH and 1.1 mM in 0.5 M H2SO4.54 Moreover, the solubility of O2 decreases with increasing the temperature and the concentration of the electrolyte but increases as the pressure increases. In order to solve this issue efficiently, gas diffusion layers (GDLs) modified with ORR catalysts (then referred to as gas diffusion electrodes, GDEs) have been recently deployed to promote the mass transport of O2. Enhanced mass transport and larger local O2 concentrations at the catalyst surface prolonged the contact time of O2 with the catalyst surface at a 3-phase interface (solid catalyst, liquid solvent/electrolyte, and gaseous oxygen), further supported by unique porous and hydrophobic structures, ultimately resulting in enhanced space time yields of H2O2 by the ORR.18,43 To date, most of the investigated ORR catalysts as cathodes in these systems were commercial carbon-based materials (e.g. active carbon and vapor-grown-carbon-fiber)55 and metal complexes (e.g. iron and cobalt-phthalocyanine).56,57 Moreover, some researchers demonstrated that electrolyte-free neutral H2O2 solution can also be obtained through the use of a solid polymer electrolyte (SPE), which is more useful and flexible for practical applications.29,58 Besides, continuous electrolytic flow reactors, also called flow-through electrolyzer cells, overcame the above issues of mass transport by continuously circulating the reactants and products to and away from the electrodes, and inhibit the subsequent decomposition of H2O2 due to the accumulation of H2O2, thus resulting in an increased amount of produced H2O2.18,43,59
The H2O2 concentration within certain time intervals can be determined by the UV-vis spectrophotometric method, and chemical titration of potassium permanganate or iodometry solution,60,61 as well as by the chemiluminescence method based on the catalytic oxidation of luminol by hydrogen peroxide.14 Among these methods, the UV-vis spectrophotometric method has been well developed and only requires the use of a spectrophotometer with easy operation and low-cost. Moreover, several indicators have been proposed for the measurement of the H2O2 concentration including ammonium molybdate/KI solution,62 potassium titanium(IV) oxalate,30 TiOSO4,18 and a commercial hydrogen peroxide test.63 Nevertheless, this method is only suitable for the determination of low-concentration hydrogen peroxide, so in order to ensure accuracy, further dilution is generally necessary prior to measurement. Besides, standard solutions of H2O2 also need to be prepared and measured to obtain the calibration curve. In contrast, the operation of chemical titration is relatively simple without the need for a calibration curve and suitable for high-concentration hydrogen peroxide, whereas the accuracy may be inferior to the spectrophotometric method. Differently, additional flow-injection is needed during the measurement for the chemiluminescence method, which makes the overall operation complex.
The H2O2 production Faradaic efficiency (H2O2 FE) can then be calculated by dividing the charge transferred to the produced H2O2 by the total charge passed through the circuit as the following equation:63
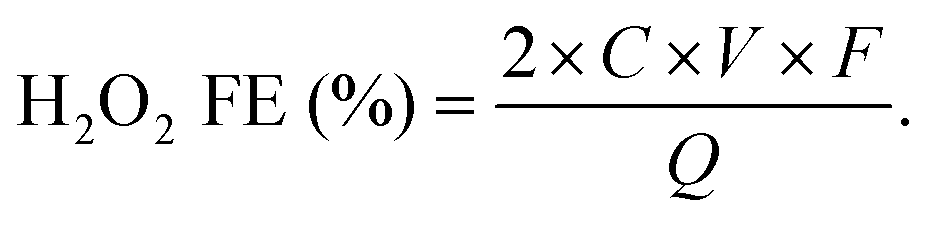 | (19) |
485 C mol−1), and Q is the amount of charge passed (C).
In addition to the catalytic activity and selectivity above, long-term stability is also of importance for any H2O2 production catalysts in practical applications. In this regard, there are two reported methods for the evaluation of the catalytic stability of catalysts including chronoamperometry (CA) or chronopotentiometry (CP), and accelerated durability tests (ADTs).
Similar to the ORR process, the WOR process also involves three oxygenated reaction intermediates on the surface of catalysts including O*, HO*, and HOO*.33,64 The activity and selectivity of the WOR process are related to the adsorption free energies of these relevant intermediates (ΔGO*, ΔGHO*, and ΔGHOO*), and theoretical results demonstrated that the proper range of ΔGHO* from 1.6 eV to 2.4 eV is beneficial for H2O2 production, especially with zero theoretical over-potential under the condition of ΔGHO* ≈ 1.76 eV. Generally, there are also some parameters for evaluating the performances of WOR catalysts toward H2O2 production, including the onset potential, selectivity or Faradaic efficiency, and stability. For the onset potential, there are two kinds of different definition, that is, the experimental potential at a current density of 0.2 mA cm−2 and the experimental potential for the H2O2 concentration to go up to 1 ppm after testing for 10 min, respectively.64 The Faradaic efficiency for H2O2 production via the WOR process could be calculated in a similar manner to the ORR process, that is, the ratio between the amount of experimentally produced H2O2 and the theoretically calculated H2O2 according to the current density.65 Also, the stability could be evaluated by the CA or CP method.
2.2 Fundamentals of photocatalytic H2O2 production
Typically, there are five continuous steps over a semiconductor photocatalyst during the photocatalytic H2O2 production process, including light harvesting, photo-generated carrier separation, O2 adsorption, surface redox reactions and H2O2 desorption. Electrons and holes are firstly generated from the valence band and conduction band within the semiconductor photocatalyst under illumination with incident photons with energy identical to its band gap. In order to better utilize solar energy, it is thus very necessary to develop visible-light-driven semiconductor photocatalysts since about 40% of the solar spectrum is located in the visible light region. Besides, the use of UV light also induces the decomposition of the produced H2O2 on the surface of the semiconductor photocatalyst.66 Prior to participation in the subsequent redox reactions, photo-generated electrons and holes first need to be separated with the minority carrier being transferred to the surface of the semiconductor photocatalyst. Charge recombination is a competing detrimental process, which is influenced by several factors such as the crystallinity, morphology, and surface properties of the photocatalyst.36,39 The proper modification of material structures is the most effective way to improve the overall photocatalytic efficiency by means of regulating the separation and recombination processes of photo-generated carriers. After transferring to the surface, photo-generated electrons and holes can drive the ORR to either H2O2 or the superoxide radical (˙OOH), while catalyzing water oxidation to molecular oxygen, respectively.37,67 Conduction band edge positions located between the redox potential of oxygen reduction to H2O2 (+0.68 VSHE) and ˙OOH (−0.13 VSHE) are most favorable for H2O2 production.37,68 At the same time, regardless of H2O2 and ˙OOH production, the valence band edge position should be more positive than the redox potential of water oxidation (+1.23 VSHE), which is energetically favorable for water oxidation. In practice, kinetic over-potentials are inevitable during these electrochemical processes. In this regard, considerable efforts have been devoted to engineering the band structure of semiconductor photocatalysts by means of heteroatom-doping, surface modification, and the formation of hetero-junctions, aiming at meeting the ideal requirements. Especially, the incorporation of two-electron ORR or water oxidation catalysts as co-catalysts could significantly accelerate the kinetic process of the interfacial redox reaction, and thus facilitate charge transfer and separation, clearly demonstrating the close connection between electrocatalysis and photocatalysis.Photocatalytic H2O2 production is usually performed with the photocatalyst suspended in a mixture of water and an electron donor such as ethanol, methanol, formic acid, and 2-propanol under illumination. It should be noted that the use of electron donors leads to difficulties in the subsequent separation of H2O2, which is why it is highly desirable to develop a photocatalytic system without the need for an electron donor. In addition, semiconductor photocatalysts have also been used to fabricate photoelectrodes for photoelectrochemical production of H2O2via the two-electron ORR or two-electron WOR. The catalytic performance of the photocatalyst is usually expressed using the H2O2 production rate in terms of μmol h−1 or μmol L−1 h−1 under given conditions including temperature and light intensity. The concentration of the produced H2O2 can be determined by potassium permanganate (KMnO4) and iodometric titration or HPLC in conjunction with an electrochemical analyzer.37 The H2O2 selectivity over semiconductor photocatalysts can also be evaluated by the RDE technique according to Koutecky–Levich plots.37 In addition, from the viewpoint of practical application, the behavior of H2O2 decomposition over the semiconductor photocatalyst also needs to be investigated by dispersing the semiconductor photocatalyst into nitrogen-saturated H2O2 solution under illumination.37 Besides, there are two additional important parameters for evaluating the photocatalytic performances of photocatalysts including the apparent quantum yield (AQY) and solar-to-chemical conversion (SCC) efficiency. In the reported literature, the AQY is defined as the ratio of the number of electrons transferred toward H2O2 production relative to the incident photons at a given wavelength, and thus could be calculated according to the following equations:37,69
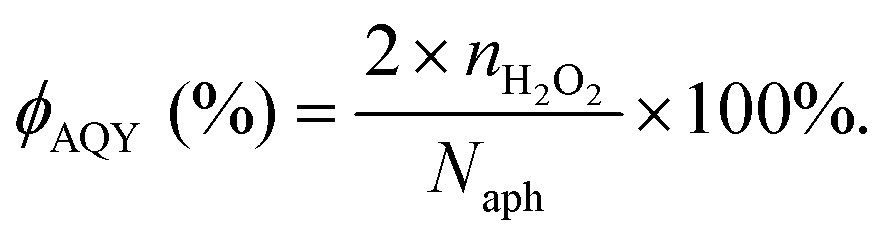 | (20) |
 | (21) |
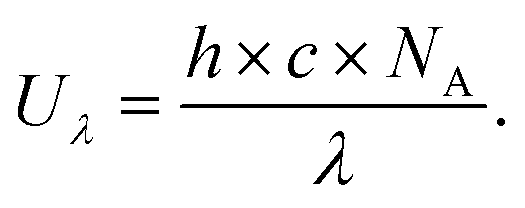 | (22) |
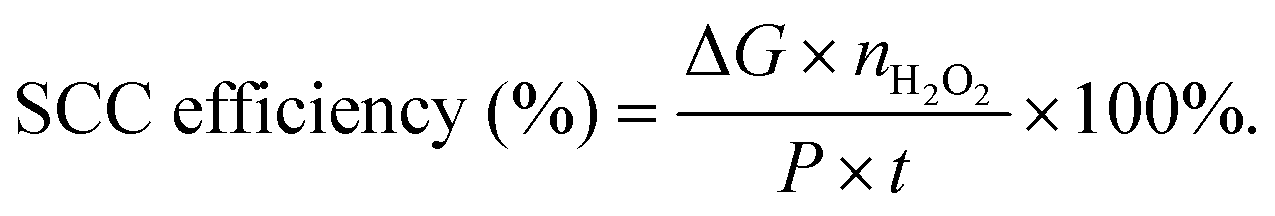 | (23) |
3 Electrocatalytic materials for H2O2 production
ORR electrocatalysts are considered to be one of the most important components in electrochemical devices for H2O2 production. To date, the developed ORR electrocatalysts can be categorized into noble-metal-based materials, transition-metal-based materials, and metal-free carbon-based materials. All these developed ORR electrocatalysts will be discussed in detail below.3.1 Noble-metal-based materials
Platinum-based catalysts are considered to be the state-of-the-art ORR electrocatalysts, and have also been widely used in commercialized PEMFCs due to their high catalytic activity, selectivity, and durability in acidic environments.24 Generally, the ORR process over platinum-based catalysts mainly proceeds by a four-electron reaction pathway for selective production of water, which is also highly desirable from the perspective of the achievement of highly efficient energy conversion. Besides, during the ORR process, the reaction pathway is usually considered to be strongly dependent on the ability of the catalyst for the dissociation of the O–O bond, whereas the cleavage of the O–O bond is undesirable for H2O2 production. In this regard, two configurations have been proposed for O2 adsorption on the surface of these platinum-based catalysts, including a “side-on” adsorption configuration and an “end-on” adsorption configuration (Fig. 1a and b).71 For the “side-on” adsorption configuration, the four-electron ORR reaction pathway is dominant because the adjacent noble metal atoms favor the dissociative adsorption of O2 and the continual reduction of H2O2 through providing separate binding sites for the two oxygen atoms in O2 and H2O2, which was also called ensemble effects. By contrast, the elimination of accessible ensembles of noble metal active sites (site isolation strategy) could induce the “end-on” adsorption configuration of O2 on the surface of the catalyst, thus making the ORR process occur through both four-electron and two-electron reaction pathways, which was beneficial for the selective production of H2O2. On the basis of these considerations above, Choi's group coated amorphous carbon layers on the surface of a commercial Pt/C catalyst (Johnson–Matthey, 60 wt% Pt) through chemical vapor deposition (CVD) in order to eliminate accessible Pt ensemble sites, thus resulting in the increased selective production of H2O2 during the ORR process. Furthermore, the amorphous carbon layers also efficiently suppressed the consecutive chemical decomposition of H2O2 by a disproportionation reaction or electrochemical reduction reaction, and also made the catalyst possess excellent stability. | ||
| Fig. 1 (a and b) ORR pathways on Pt surfaces: (a) on the pristine Pt/C, molecular O2 prefers to adsorb on the Pt surface as a side-on configuration and then reduces to H2O through (1) dissociative, (2) associative, and (3) non-dissociative mechanisms; (b) on the carbon-coated Pt, molecular O2 absorbs on the Pt surface as an end-on configuration and then produces H2O and H2O2via competitive dissociation and desorption steps, respectively. The produced H2O2 is not further reduced on the Pt surface due to hindrance by the carbon layer. (c–f) Trends in activity and selectivity for H2O2 production: (c) Schematic representation of oxygen reduction to H2O2 on a model Pd2Hg5(001) surface. Palladium atoms are represented in green, mercury in blue, oxygen in red, and hydrogen in yellow. (d) Partial kinetic current density to H2O2 as a function of the applied potential, corrected for mass transport losses. (e) Potential required to reach 1 mA cm−2 kinetic current density to H2O2 on polycrystalline catalysts as a function of the calculated HOO* binding energy. The solid lines represent the theoretical Sabatier volcano. The dotted line represents the thermodynamic potential for oxygen reduction to H2O2. (f) H2O2 selectivity for different catalysts at 2.5 mA cm−2 total current density. All electrochemical experiments were performed at 50 mV s−1 and 1600 rpm in O2-saturated 0.1 M HClO4 at room temperature with corrections for ohmic drop. The surface area was normalized to the geometrical value. (a and b) Reprinted with permission.71 Copyright 2014, American Chemical Society. (c–f) Reprinted with permission.51 Copyright 2014, American Chemical Society. | ||
Theoretically, according to Sabatier's principle, the binding of the reaction intermediates on the surface of the catalyst should be neither too strong nor too weak for the ideal catalyst, that is, a moderate interaction between the catalyst surface and the reaction intermediates.2,51 Therefore, as the sole reaction intermediate during the H2O2 production process, HOO* should be bound on the surface of the catalyst within a moderate range, which is thus conducive to the preservation of the O–O bond. Nevertheless, for conventional Pt-based catalysts, the HOO* dissociation to O* and HO* (the four-electron ORR intermediates) is generally preferred over the HOO* hydrogenation to H2O2 due to their strong binding of HOO* on their surface. Rossmeisl and Stephens recently reported the tunable binding of the reaction intermediates through regulating the composition of the catalysts, and screened a series of oxygen electrocatalysts based on noble metal alloys for H2O2 production using density functional theory (DFT) calculations (Fig. 1c–f).51 In their work, the binding of HOO* could be optimized by the modification of reactive noble metals including Pt, Pd, and Ag with inactive metals such as Au and Hg, in order to regulate the catalytic activity and selectivity toward H2O2 production. Electrochemical results demonstrated that the resultant Pt–Hg, Ag–Hg, and Pd–Hg exhibited high selectivity toward H2O2 production owing to the elimination of the accessible active metal ensemble sites through the isolation of active sites using Hg. Besides, starting from the viewpoint of both composition and morphology optimization, Mahata et al. also studied three cuboctahedral core–shell nanoclusters of Au19@Pt60, Co19@Pt60 and Au10Co9@Pt60 by performing DFT calculations to get better understanding of the influence of the core metal (Co and Au) on improving the selectivity and efficiency of H2O2 production.72 The optimal binding of HOO* can be achieved by the formation of a mixed alloy consisting of Co and Au due to the synergetic effect of Co and Au with different oxygen binding energies. Among them, the Au10Co9@Pt60 NCs showed the lowest over-potential and highest selectivity for H2O2 production.
The particle size of nanostructured materials has been reported to show significant influences on the catalytic performances including the catalytic activity and reaction pathway. For instance, Anderson's group prepared different size Ptn clusters supported on indium tin oxide (ITO) films in an ultrahigh vacuum, and found size-dependent catalytic activity and selectivity toward H2O2 production,73 where the smallest Ptn clusters were most beneficial for H2O2 production. Other typical examples of the size effect are single-atom catalysts (SACs) with the advantages of minimal noble metal usage and unique catalytic properties. Extensive efforts have been devoted to the preparation of Pt SACs dispersed on various substrates such as MgO, FeOx, zeolites, TiO2, and MoS2, showing high catalytic activity for different catalytic applications such as propane combustion,74 CO oxidation,75 nitroarene hydrogenation,76 and photocatalytic hydrogen production.77,78 Besides, the elimination of Pt ensemble sites in these Pt SACs also induced the pathway change of some chemical reactions such as formic acid oxidation.79 Generally, formic acid oxidation proceeds by the indirect dehydration pathway over conventional Pt nanoparticles. However, Pt SACs can catalyze formic acid oxidation through the direct dehydration pathway. Recently, Pt SACs have also been employed as ORR electrocatalysts for selective H2O2 production. For instance, Lee's group demonstrated high mass activity and selectivity toward H2O2 production over single-atom Pt supported on TiN nanoparticles because of the absence of Pt ensemble active sites,79 and also observed the support effect on the catalytic performances of Pt SACs toward the electrochemical ORR, indicating that the substrate was not only used as anchoring sites to stabilize the single metal atoms, but also may participate in the surface catalytic reaction.80 Meanwhile, the composition and structure of the substrate have significant effects on the loading of atomically dispersed Pt. A high loading of atomically dispersed Pt up to 5 wt% can be achieved using sulfur-doped zeolite-templated carbon as a support with a high sulfur content of 17 wt% and highly curved three dimensional networks of graphene nanoribbons, which was attributed to the abundant S-functionalities and unique carbon structure.81 Very recently, the loading of single atom Pt sites was further improved to 24.8% by atomically dispersing platinum on the surface of an amorphous CuSx hollow nanosphere support due to the strong Pt–S interaction (Fig. 2),82 which exhibited a high H2O2 selectivity of 92–96% over a wide potential range as well as excellent stability even after 10000 cyclic voltammetry cycles within a potential range from +0.1 to +0.8 VRHE in HClO4 electrolyte. In addition, the loading amount of the Pt-based catalyst on the electrode showed a significant influence on the H2O2 production during the ORR process, and showed an inverse correlation with the H2O2 selectivity.49 A similar phenomenon was also observed for some non-Pt-based ORR electrocatalysts like Fe–N–C and Se/Ru/C catalysts.83,84
 | ||
| Fig. 2 (a) Schematic illustration of the structure evolution of h-Pt1–CuSx (blue, purple and white balls represent Cu, Pt, and S atoms, respectively); and (b–g) characterization of h-Pt1–CuSx nanoparticles: (b) TEM image of h-Pt1–CuSx (the inset is the photograph of the nanoparticles dispersed in cyclohexane); (c) high-magnification TEM image (the inset is the SAED pattern); (d) XRD pattern of the Cu1.94S nanoparticle seeds and the h-Pt1–CuSx nanoparticles; (e) EDS elemental mapping of Pt, S, and Cu; and (f and g) AC-HAADF-STEM images of h-Pt1–CuSx. Reprinted with permission.61 Copyright 2019, Elsevier Inc. | ||
Au has been demonstrated to be a promising ORR electrocatalyst for H2O2 production, and its selectivity is strongly influenced by the crystallographic orientation and experimental conditions.82 However, the weak binding of HOO* on its surface resulted in a low coverage of O2 and thus limited the catalytic activity.82 Recently, rational design of alloying with other metals, such as Pd, Pt, and Rh, has been proposed to optimize the catalytic properties of Au. The typical example reported is the Au–Pd alloy system,17 which has been investigated as a heterogeneous catalyst for direct catalytic production of H2O2 from H2 and O2.85 For instance, the Au–Pd alloy with variable Pd content has also been investigated as an ORR electrocatalyst for H2O2 production, and increased H2O2 selectivity was observed at the optimal content of Pd owing to the ensemble effect arising from the presence of finely dispersed Pd within Au. In contrast, excess content of Pd resulted in a decrease of H2O2 selectivity due to the strong binding of HOO* on the surface of Pd. In addition to Pd, some DFT calculation results demonstrated that the combination of Ni with Au resulted in increased coverage of O2, maintaining simultaneously weak dissociation of the O–O bond. Inspired by this point, Amal's group reported the designed synthesis of Au–Ni core–shell nanorods though the epitaxial growth method,82 which exhibited improved catalytic activity and selectivity toward H2O2 production. The enhanced results could be attributed to the following aspects: (1) the decreased tendency for the dissociation of the O–O bond resulting from Au–Ni; and (2) the complex electron interaction and the lattice strain effect resulting from the epitaxial growth process. What's more, a Pt layer was also further introduced to improve the catalytic activity and selectivity toward H2O2 production by tuning the electrical properties of the Ni shell and the degree of lattice strain caused by the high work function of Pt. Similarly, the substrate has a significant influence on selective H2O2 production over Au-based ORR electrocatalysts. Camargo's group reported the synthesis of hybrid materials consisting of Au and TiO2 with different morphologies including spheres and wires.86 Interestingly, the TiO2 spheres had a positive effect on the selectivity for H2O2 production whereas the H2O2 selectivity decreased for the TiO2 wires.
3.2 Transition-metal-based materials
Transition-metal-based materials, mainly including transition-metal complexes, metal–N–C materials, metal/C materials, and metal oxides, showed good catalytic activity and selectivity toward the ORR to H2O2via the two-electron reaction pathway. The most popular examples of transition-metal complex ORR electrocatalysts for H2O2 production are cobalt-based porphyrins and iron-based phthalocyanines.56,57 Their unique geometric configuration, consisting of reactive metal atoms surrounded by supporting and coordinating atoms such as N and C, could efficiently restrain the dissociation of the O–O bond, thus realizing high selectivity toward two-electron H2O2 production. Besides, DFT calculation results revealed that the neither too strong nor too weak binding of the reaction intermediate HOO* on the surface of these catalysts proved most beneficial for H2O2 production.87 However, the high cost and unsatisfactory durability of these catalysts largely prohibited their practical applications. Nevertheless, some research studies also reported the significant improvement of their catalytic activity and durability through heat treatment. Inspired by this, extensive efforts have been made for the synthesis of Co–N–C catalysts from various nitrogen-containing precursors, carbon supports, and Co salts. Research demonstrated that the catalytic activity and selectivity of these Co–N–C catalysts were largely related to the nature of the carbon supports and nitrogen-containing precursors. For instance, bidentate N-ligands were found to be optimal nitrogen-containing carbon precursors because of the favorable formation of such structures with Co-coordinated Co–N2–C sites during the heat treatment process, which were found to be active sites for the selective ORR for H2O2 production.88 In addition to the formation of Co–N2–C sites, metal Co nanoparticles coated with Co oxides were also generally produced during the heat treatment process, where the produced H2O2 was further electrochemically reduced to H2O. Besides, there have also been conflicting opinions on the roles of Co–Nx–C sites (x represents the coordination number), that is, Co–N4–C sites are considered to be active sites for H2O2 production through the two-electron ORR, whereas Co–N2–C sites promote the four-electron ORR for H2O production.90 Recently, our group observed the activity–selectivity trends for electrochemical H2O2 production over single-site metal–N–C (metal = Mn, Fe, Co, Ni, and Cu) by the combination of computational calculations and experimental results. The as-prepared Co–N–C catalyst was found to possess the most optimal binding energy of the HO* intermediate near the top of the volcano of the two-electron ORR, which well explained its outstanding H2O2 productivity with high ORR activity, highest H2O2 selectivity, and lowest H2O2 reduction reaction activity (Fig. 3a–e).18 Moreover, industrial H2O2 productivity over the prepared Co–N–C catalyst in a micro flow cell was achieved with a production rate of more than 4 mol peroxide gcatalyst−1 h−1 at a current density of 50 mA cm−2. Later, the introduction of oxygen functional groups (OFGs) into carbon-based materials possessing Co–Nx–C active sites could further improve the catalytic activity and selectivity toward electrochemical H2O2 production, where Co–Nx–C sites mainly contributed to the catalytic ORR reactivity, whereas the H2O2 selectivity was attributed to OFGs (Fig. 3f and g).89 Very recently, Hyeon's group also observed nearly the same phenomenon, that is, Co–N4 moieties surrounded by oxygen species (C–O–C epoxides) as an ideal system for highly active H2O2 production from both electrochemical results and DFT calculations.91 Besides, Wang's group also reported Fe–O–C as an efficient ORR catalyst for H2O2 production with a relative positive onset potential of +0.822 VRHE and high selectivity of more than 95% in both alkaline and neutral medium,92 which was completely different from the previously reported Fe–N–C ORR catalysts. Therefore, the fundamental understanding of the nature of the active sites of these M–N–C catalysts toward the ORR remains important yet challenging, and more work is still needed. | ||
| Fig. 3 (a) Linear sweep voltammetry (LSV) in a rotating ring-disk electrode (RRDE) setup with the Pt ring held at +1.2 VRHE. (b) H2O2 selectivity (H2O2%) and the number of electrons (n) at +0.1 VRHE derived from RRDE data. (c) Background-corrected H2O2RR performance in N2-saturated 0.5 M H2SO4 electrolyte containing 1 mM H2O2. (d) Thermodynamic relations (volcano) lines for the two- (green solid line) and four-electron ORR (black solid line). The DFT calculated ORR onset potential values (circles) are on the left y-axis, while the experimental current densities (crosses and triangles), reported as ln(|j|), are on the right y-axis. Both are shown as functions of the chosen reaction descriptor, the DFT calculated HO* binding free energy (GHO). (e) Scheme of the micro-flow cell setup. (f) Schematic of the synergistic strategy of atomic Co–Nx–C sites and OFGs for H2O2 electrosynthesis on noble-metal-free electrocatalysts. (g) Performance comparison in regard to reactivity and selectivity for H2O2 electrosynthesis on Co–POC–O, Co–POC–R, and POC–O electrocatalysts. The inset in (g) shows the mechanism scheme for synergistic H2O2 electrosynthesis. The carbon, nitrogen, oxygen, and cobalt atoms are marked with black, blue, yellow, and red, respectively. (a–e) Reprinted with permission.18 Copyright 2019, American Chemical Society. (f and g) Reprinted with permission.89 Copyright 2019, Wiley-VCH. | ||
3.3 Metal-free carbon-based materials
In the past few decades, metal-free carbon-based materials have been widely used as supports in many energy-related applications due to their advantages of earth-abundance, low-cost, facile preparation, large specific surface area, excellent electrical conductivity, chemical resistance, and mechanical stability.23,25,93 Recently, metal-free carbon-based materials, such as activated carbon fibers and graphite, have also shown great promise as alternative catalysts to noble metal materials for electrochemical production of H2O2 through the two-electron ORR process in alkaline solution.14–16,42,94,95 It has been well accepted that the electrochemical performances of metal-free carbon-based materials are strongly determined by their geometric and electronic structures. Starting from these two aspects, considerable efforts have been made for the synthesis of advanced metal-free carbon-based materials with high activity, selectivity, and stability for electrochemical H2O2 production.One of the most promising geometric structures for H2O2 production is reported to be porous structures since such structures with large surface area and high pore volume are beneficial for mass transfer and also help in exposing more catalytically active sites. Metal–organic frameworks (MOFs) as a typical porous multi-functional material have received increasing attention due to their high surface area and uniform porosity, and have been proposed as promising precursors for the fabrication of porous carbon materials. For instance, hierarchically porous carbon (HPC) was fabricated under an atmosphere of H2 through the direct pyrolysis of MOF-5, which was synthesized using zinc nitrate and terephthalic acid.14 The resultant porous carbon exhibited high catalytic activity and selectivity for electrochemical reduction of O2 to H2O2 production in a wide range of pH from 1 to 7. These outstanding performances can be attributed to the high amount of sp3-C and defects, high surface area, and favorable mass transfer. In addition, the pore size also showed a significant influence on the catalytic properties of porous carbon materials. Two kinds of porous carbon materials with mesopore-dominant porous carbon (Meso-C) and micropore-dominant porous carbon (Micro-C) have been investigated for electrochemical production of H2O2, where Meso-C exhibited superior ORR performance for H2O2 production compared to Micro-C.94,96 This result may be explained by the fact that the favorable mass transport within the mesoporous structure resulted in the fast release of the produced H2O2 from the surface of the catalyst, thus avoiding the subsequent reduction of H2O2.
Heteroatom doping, such as with oxygen, nitrogen, sulphur, and fluorine, has been reported as another promising strategy to regulate the catalytic activity and selectivity of metal-free carbon materials through tailoring their electronic structures.16,94,97 Santos's group compared the electrochemical performance of Vulcan XC-72R and Printex carbon supports for H2O2 production.98 They found that there are more oxygenated functional groups on the surface of Printex L6 compared to Vulcan XC-72R, resulting in improved H2O2 selectivity. Moreover, the same group also further investigated the influence of different surface modification on the selectivity of these two carbon supports toward H2O2 production through pre-treatment with nitric acid and ammonia.99 A similar phenomenon was also observed that acid-treated Printex L6 exhibited the highest selectivity toward H2O2 production due to the largest concentration of oxygenated functional groups. Very recently, Cui and co-workers also reported the increased activity and selectivity of carbon materials toward H2O2 production through surface oxidation treatment.3 Moreover, a linear correlation of the catalytic activity and selectivity with the oxygen content was observed, and DFT results demonstrated that the existence of the –COOH functional group in the armchair edge and the C–O–C functional group in the basal plane of graphene resulted in the high activity and selectivity toward H2O2 production. These results implied that the introduction of oxygenated functional groups plays an important role in regulating the chemical selectivity.
In addition to oxygen functionalization, nitrogen doping has also been reported to be an efficient means for improving the ORR performances of metal-free carbon materials since the incorporation of nitrogen with higher electronegativity could induce the charge redistribution of the π conjugated system of the carbon frameworks and thus tailor the adsorption properties of carbon materials for ORR reactive intermediates.100 Nevertheless, most of the reported nitrogen-doped carbon materials led to the four-electron ORR to H2O, and only a few samples were reported to exhibit high selectivity toward H2O2 production.63,97,101–104 For example, Anderson's group investigated the ORR performance of nitrogen-doped Ketjenblack for H2O2 production from both experimental and theoretical calculation viewpoints.105 Experimental results demonstrated that the nitrogen-doped Ketjenblack showed a lower onset potential and mainly followed the two-electron ORR process for H2O2 production. Meanwhile, DFT results also indicated that the formed carbon radical sites neighboring substitutional N in graphite were active for the electrochemical ORR to H2O2. In addition, Iglesias et al. also reported the synthesis of nitrogen-doped graphitized single wall carbon nanohorns (CNHs) by coating polydopamine (PDA) followed by annealing,106 which also exhibited high catalytic activity with a very positive onset potential, high Faradaic efficiency, and excellent stability in a wide pH range from 1.0 to 13.0. In their work, the outstanding performances were assigned to their specific Npyridinic/Npyrrolic ratios and microporosity. Besides, our group also reported improved ORR activity and H2O2 selectivity induced by nitrogen doping and observed the different roles of nitrogen doping species during the ORR process by means of ex situ XPS, where pyridinic-N contributed to the catalytic ORR process in acid medium while graphitic-N groups appeared to be catalytically active moieties in neutral and alkaline conditions.63,107 Further, our group investigated the effect of graphene precursors with different in-plane carbon lattice defect density on the subsequent nitrogen doping and the catalytic performances of their derived nitrogen-doped graphene. The resultant nitrogen-doped graphene derived from oxo-G with lower carbon lattice defect density exhibited the highest H2O2 selectivity. Similar to nitrogen, fluorine doping can also create active sites through inducing the polarization of adjacent carbon, which is beneficial for improving the ORR performances. Moreover, the fluorine doping content and different fluorine species were also found to influence the catalytic activity and selectivity toward H2O2 production. For instance, Zhao et al. reported the synthesis of fluorine-doped hierarchically porous carbon (FPC) from an aluminum-based MOF.108 Electrochemical measurements and DFT calculations demonstrated that the incorporation of CF2,3 atoms into carbon frameworks could facilitate the adsorption of O2 and desorption of reactive intermediate HOO*, thus resulting in significantly increased activity and selectivity toward H2O2 production.
In addition to the two-electron ORR process, the two-electron WOR process has also been widely investigated theoretically and experimentally for H2O2 production recently.111 For instance, Nørskov's group presented a thermodynamic picture of the adsorption free energies of the intermediates (HO*) on the surface of different catalysts for the three different reaction pathways using thermodynamic analysis based on DFT calculations, and SnO2 and TiO2 were identified as promising candidate materials with good selectivity for H2O2 production.33,112 Subsequently, they further adopted DFT calculations to investigate the trends in activity of four different oxides including WO3, SnO2, TiO2 and BiVO4 for water oxidation toward H2O2 production.109 BiVO4 was identified theoretically and experimentally as the best catalyst for H2O2 production with a high FE of 70% in the dark and 98% under 1 sun illumination under the condition of the optimal bias range (Fig. 4a). Nevertheless, there also exist some limitations of the BiVO4 catalyst, including high over-potential and poor stability from VO43− dissolution. Recently, Zheng's group developed an efficient method to improve the activity and stability of BiVO4 for H2O2 production by doping rare earth element gadolinium (Gd).65 Theoretical calculation results demonstrated that Gd doping not only makes several facets of BiVO4 more active for H2O2 production, but also results in the increase of the energy barrier of VO43− dissolution. Experimentally, the Gd-doped BiVO4 exhibited a significant decrease of the onset potential for H2O2 production by around 110 mV with a high FE of around 99.5% and a substantially prolonged catalytic lifetime under illumination. Meanwhile, the same group also employed DFT calculations to identify another two efficient and selective WOR catalysts for H2O2 production, including CaSnO3 and ZnO, which were also verified experimentally (Fig. 4b–d).64,110
 | ||
Fig. 4 (a) Activity volcano plots based on calculated limiting potentials as a function of calculated adsorption energies of HO* (ΔGHO*) for two-electron oxidation of water to hydrogen peroxide evolution (black) and four-electron oxidation to oxygen evolution (blue). The corresponding equilibrium potentials for each reaction are shown as dashed lines. (b) Volcano plots showing the calculated limiting potential UL for four-electron H2O oxidation to O2 (blue dashed line) and two-electron H2O oxidation to H2O2 (black solid line) as a function of ΔGHO*. The convention is such that a low value of UL (a high value of −UL) corresponds to a low overpotential and thus a high rate. The full lines correspond to the trend model described in the text, and DFT calculated values for different catalyst materials are included. The 0.5 and 1 ML H ter. of (0001) ZnO indicate two different terminations at the bottom of the slabs. (c) Overall current density J–V curves for (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) ZnO, (0001) ZnO, and several reported metal oxides. (d) Corresponding JH2O2–V curves for the current density toward H2O2 formation, for which JH2O2 was calculated by multiplying the overall current Joverall by the Faraday efficiency at each potential. (a) Reprinted with permission.109 Copyright 2017, Nature publishing group. (b–d) Reprinted with permission.110 Copyright 2019, American Chemical Society. 0) ZnO, (0001) ZnO, and several reported metal oxides. (d) Corresponding JH2O2–V curves for the current density toward H2O2 formation, for which JH2O2 was calculated by multiplying the overall current Joverall by the Faraday efficiency at each potential. (a) Reprinted with permission.109 Copyright 2017, Nature publishing group. (b–d) Reprinted with permission.110 Copyright 2019, American Chemical Society. | ||
4 Photocatalytic materials for H2O2 production
Semiconductor photocatalysts have been widely applied in the field of various solar energy storage and conversion systems including dye-sensitized solar cells (DSSCs), photocatalytic water splitting and CO2 reduction since Fujishima and Honda first demonstrated the photocatalytic application of TiO2 in 1972.113–115 Recent studies also began to focus on the design and development of advanced semiconductor photocatalysts with novel compositions and structures for photocatalytic H2O2 production. To date, the reported semiconductor photocatalysts mainly include metal oxides, metal–organic complexes, and metal-free graphitic carbon nitride (Table 1). Subsequently, recent progress on these photocatalysts will be discussed in detail below.| Photocatalyst | Electron donor | Light wavelength/nm | H2O2/μM h−1 | Φ AQY/%@420 nm | Ref. |
|---|---|---|---|---|---|
| Note: APTMS is aminopropyltrimethoxysilane. HTNT–CD is proton-form titania nanotubes with carbon dots. IO g-C3N4 means inverse opal g-C3N4. “/” means without the use of an electron donor. | |||||
| TiO2 | Benzyl alcohol | >280 | 3300 | 29.1@334 nm | 116 |
| Au/TiO2 | Ethanol | >300 | 291.7 | — | 60 |
| Au/TiO2–CO32− | Formic acid | >430 | 640 | 5.4@530 nm | 117 |
| Au–Ag alloy/TiO2 | Ethanol | >280 | 283.3 | — | 66 |
| Pd/APTMS/TiO2 | / | — | 150 | — | 120 |
| SN-GQD/TiO2 | 2-Propanol | ≥420 | 451 | — | 118 |
| rGO/TiO2 | 2-Propanol | ≥320 | 1512 | — | 48 |
| HTNT–CD | / | >420 | 2118 | 0.7 | 121 |
| Co@TiO2 | Methanol | =400 | 1650 | — | 122 |
| Au/BiVO4 | Ethanol | >420 | 40.2 | 0.24 | 37 |
| Ni/MIL-125-NH2 | Benzyl alcohol | >420 | 986.8 | — | 123 |
| Alkylated MIL-125-R | Benzyl alcohol | >420 | 766.7 | — | 124 |
| Cd3(C3N3S3)2 | Methanol | ≥420 | 2200 | — | 125 |
| Cd3(C3N3S3)2/rGO | Methanol | ≥420 | 297.6 | 3.5@450 nm | 126 |
| g-C3N4 | Ethanol | >420 | 1641 | — | 127 |
| Mesoporous g-C3N4 | Ethanol | >420 | 742.8 | — | 67 |
| g-C3N4 with C vacancies | / | >420 | 91.3 | — | 128 |
| IO g-C3N4 with C vacancies | Ethanol | ≥420 | 162.9 | — | 36 |
| C3N4–carbon | 2-Propanol | — | 317.8 | — | 129 |
| g-C3N4 with N vacancies | Ethanol | >400 | 288.9 | — | 130 |
| Holey defective g-C3N4 | 2-Propanol | >420 | 80 | 10.7 | 131 |
| Reduced g-C3N4 | / | >420 | 170 | 4.3 | 132 |
| O-Enriched g-C3N4 | 2-Propanol | ≥420 | 1200 | 10.2 | 21 |
| K, P, O co-doped g-C3N4 | Ethanol | ≥420 | 241.3 | 8.0 | 38 |
| KPF6-modified g-C3N4 | Ethanol | >420 | 312.5 | 24.3 | 133 |
| K, P co-doped g-C3N4 | Ethanol | ≥420 | 507 | — | 134 |
| g-C3N4/PDI | / | >420 | 20.8 | — | 135 |
| g-C3N4/PDI/graphene | 2-Propanol | >420 | 40.3 | 5.68 | 20 |
| g-C3N4/PDI/BN-rGO | 2-Propanol | >420 | 3095 | 7.3 | 136 |
| g-C3N4/MTI | / | >420 | 27.8 | 6.3 | 39 |
| g-C3N4/BDI | 2-Propanol | >420 | 29.2 | 4.5 | 70 |
| Perylene imide/g-C3N4 | / | ≥420 | 1200 | 3.2 | 137 |
| g-C3N4–SiW11 | Methanol | — | 152 | 6.7 | 138 |
| 3DOM g-C3N4–PW11 | / | ≥320 | 350 | — | 139 |
| g-C3N4–PWO | / | ≥420 | 1007 | — | 140 |
| g-C3N4–CoWO | / | ≥420 | 97 | 14.6 | 141 |
| Metal oxide/g-C3N4 | / | — | 42 | — | 142 |
| g-C3N4–CNT | Formic acid | ≥400 | 487 | — | 143 |
| Au/g-C3N4 | 2-Propanol | — | 330 | 3.63@400 nm | 144 |
| Au/g-C3N4 | Ethanol | >420 | 63.8 | — | 145 |
| CoP/g-C3N4 | Ethanol | ≥420 | 70 | — | 146 |
4.1 Metal oxides
Metal oxide semiconductors have been proposed as promising catalysts for photocatalytic H2O2 production because of their general superior stability to liquid electrolytes and facile preparation.60,116 Among the various metal oxide semiconductors, TiO2 is the most widely investigated but usually showed unsatisfactory efficiency for H2O2 production within the micro molar range. This is largely a consequence of the intrinsic properties of TiO2 related to its band gap width and band gap structure, coupled with the sluggish intrinsic surface reaction kinetics of water oxidation, low selectivity toward the two-electron ORR for H2O2 production, and the decomposition of the produced H2O2 by the absorbed UV light and photo-generated electrons/holes, thus significantly hampering the overall photocatalytic performances.37 In order to mitigate the problems above, various strategies have been explored to improve the photocatalytic performances for H2O2 production over TiO2-based photocatalysts, including the incorporation of noble metal60,66,117 and carbon materials,48,118 and reaction medium optimization.47,119The introduction of noble metals is one powerful route for the design and preparation of advanced TiO2-based photocatalysts for H2O2 production. The most commonly used noble metals include Au,60 the Au–Ag bimetallic alloy,66 and Pd.120 For instance, Tada's group demonstrated a significant improvement in H2O2 production through loading Au nanoparticles on the surface of TiO2.60 On one hand, the formation of a junction between Au and TiO2 was beneficial for efficient photo-induced interfacial electron transfer from TiO2 to Au. On the other hand, these transferred electrons were favorable to selectively follow the two-electron ORR process toward H2O2 production over the Au nanoparticles. These two effects both resulted in efficient H2O2 production. Nevertheless, Au particles were also found to promote the decomposition of H2O2 by reduction with the transferred electrons due to their strong adsorption for H2O2 molecules.66 In this regard, the introduction of Au–Ag bimetallic alloy particles as a substitute for Au was reported to address this dilemma and showed improved efficiency for H2O2 production by means of the decreased adsorption of H2O2 onto the Au atoms (Fig. 5a and b).66 Recently, Kim's group reported the synthesis of electronically tuned Pd nanoparticles loaded on a TiO2 substrate by coordinating organic ligands on the surface of Pd.120 The increased negative charge on the surface of Pd nanoparticles induced by electron donation from amine groups of the coordinated ligands was found to facilitate enhanced catalytic activity and selectivity for the two-electron ORR for H2O2 production (Fig. 5c–e). In short, the introduction of noble metals plays several different vital roles in enhancing the photocatalytic performances: (1) promoting the separation of photo-generated electron–hole pairs through the formation of a hetero-junction between the metal and metal oxide semiconductor; (2) reducing the decomposition of the produced H2O2 through suppressing the adsorption of H2O2 on the surface of photocatalysts; and (3) improving the H2O2 selectivity by promoting the desired two-electron ORR. Nevertheless, despite improved H2O2 efficiencies, there still exist other challenges that need viable solutions, such as the low quantum efficiency resulting from UV illumination and wide band gap semiconductors, and the continuous need for a suitable electron donor molecule in the liquid phase.
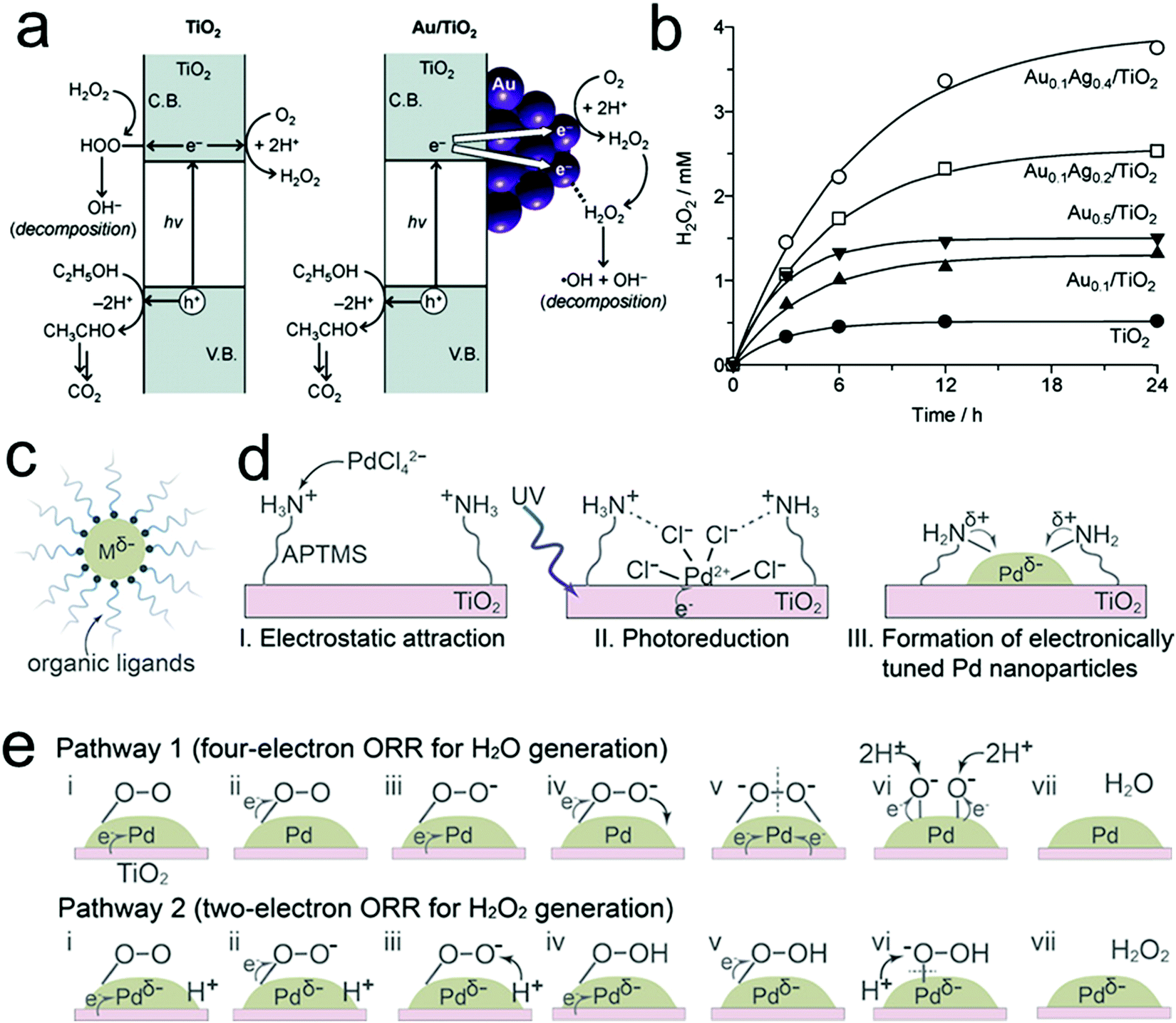 | ||
| Fig. 5 (a) Photocatalytic formation and decomposition of H2O2 on TiO2 and Au/TiO2 catalysts; (b) time-dependent change in H2O2 concentration during the photoreaction with the respective catalysts; (c) solvent-dispersed metal nanoparticles that are electronically tuned by coordinating with dissolved organic ligands; (d) preparation of electronically tuned Pd nanoparticles on TiO2 by coordinating with immobilized organic ligands; and (e) mechanisms of oxygen reduction on the surface of Pd nanoparticles. (a and b) Reprinted with permission.66 Copyright 2012, American Chemical Society. (c–e) Reprinted with permission.120 Copyright 2018, American Chemical Society. | ||
In addition to noble metals, carbon materials were considered as promising candidates for the fabrication of high performance metal-oxide-based photocatalysts due to low cost, large surface area, and high conductivity.48,118 In particular, carbon quantum dots (CQDs), a kind of amorphous carbon along with sp2 hybridized graphitic carbon, have received considerable attention in the fields of bio-imaging, biosensors, and other opto-electrical devices due to their unique features such as fluorescence properties with excitation wavelength-dependent multi-color emission.147 This fascinating feature made CQDs a promising concept in order to expand the absorption capability of photocatalysts into smaller band gaps, thereby harvesting photons in the range of the UV-visible spectral region. Recently, Xiao's group reported a hybrid photocatalyst of protonated TiO2 nanotubes and CQDs (PTNT–CQD) with an H2O2 productivity of 3.42 mmol gcat−1 h−1, a solar-to-H2O2 efficiency of 5.2%, and good continuous cyclic stability under visible-light illumination.121 The protons on PTNT–CQD were found to play critical roles in boosting the two-electron ORR for H2O2 production and suppressing the decomposition of the produced H2O2. Long's group also utilized highly luminescent sulfur and nitrogen co-doped graphene quantum dots (SN-GQDs) that were loaded on the surface of TiO2 in order to extend the light absorption into the visible light region and promote the migration of photo-generated electrons.118 Moreover, the introduction of SN-GQDs resulted in a promoted two-electron ORR accompanied by the suppressed decomposition of H2O2, thus achieving photocatalytic performances with an H2O2 productivity of 451 μmol L−1. Besides, a similar role of graphene was also observed in the photocatalytic systems of WO3 and TiO2/WO3 composites for enhanced performances for photocatalytic H2O2 production.148,149
The photocatalytic performance of a semiconductor photocatalyst is usually dependent on the bulk (e.g. light absorption, separation and recombination of photo-generated electron and hole pairs, and band edge position) and surface (e.g. structural defects and reconstruction, and surface charge) properties.119 However, the optimization of the entire catalytic solid–liquid interface is of the essence to arrive at a viable photocatalytic reaction system. For instance, a recent study examined the role of the reaction medium in the regulation of the surface properties of semiconductor photocatalysts for H2O2 production. More specifically, the study focused on the electrolyte's pH, the addition of inorganic anions and cations, and the nature of the sacrificial electron donor. The authors highlighted how surface passivation through complexation with metal cations or non-metal anions leads to blocking of the trapping sites (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ti–OH) on the surface of TiO2 for photo-generated electrons and holes, and thereby suppresses the formation of Ti–OOH complexes for the decreased decomposition of H2O2. For instance, the introduction of Cu ions and Zn ions into the photocatalytic system resulted in an increased H2O2 yield over the TiO2 photocatalyst and the surface coverage of Zn ions was also observed to be strongly dependent on the electrolyte's pH.119,150 In addition, the introduction of F− also achieved enhanced efficiency for photocatalytic H2O2 production through surface fluorination to prohibit the surface complexation of superoxide/peroxide species and the decomposition of the produced H2O2.47 These results demonstrated the important roles of surface speciation during the photocatalytic process and also provided insights on improving the photocatalytic H2O2 production efficiency by the combined effect of pH and surface passivation. On the other hand, ethanol is usually used as an electron and proton donor in photocatalytic systems for H2O2 production in order to promote the separation of photo-generated electrons and holes by replacing water oxidation with sluggish kinetics. Recently, the use of benzylic alcohols as a substitute for ethanol was found to further improve the H2O2 yield up to ca. 40 mM, which was attributed to the efficient formation of side-on coordinated peroxo species on the surface of the TiO2 photocatalyst through the reaction between benzylic alcohol and O2.116 The formed peroxo species can be easily converted into H2O2, thus facilitating highly efficient H2O2 production.
Ti–OH) on the surface of TiO2 for photo-generated electrons and holes, and thereby suppresses the formation of Ti–OOH complexes for the decreased decomposition of H2O2. For instance, the introduction of Cu ions and Zn ions into the photocatalytic system resulted in an increased H2O2 yield over the TiO2 photocatalyst and the surface coverage of Zn ions was also observed to be strongly dependent on the electrolyte's pH.119,150 In addition, the introduction of F− also achieved enhanced efficiency for photocatalytic H2O2 production through surface fluorination to prohibit the surface complexation of superoxide/peroxide species and the decomposition of the produced H2O2.47 These results demonstrated the important roles of surface speciation during the photocatalytic process and also provided insights on improving the photocatalytic H2O2 production efficiency by the combined effect of pH and surface passivation. On the other hand, ethanol is usually used as an electron and proton donor in photocatalytic systems for H2O2 production in order to promote the separation of photo-generated electrons and holes by replacing water oxidation with sluggish kinetics. Recently, the use of benzylic alcohols as a substitute for ethanol was found to further improve the H2O2 yield up to ca. 40 mM, which was attributed to the efficient formation of side-on coordinated peroxo species on the surface of the TiO2 photocatalyst through the reaction between benzylic alcohol and O2.116 The formed peroxo species can be easily converted into H2O2, thus facilitating highly efficient H2O2 production.
Direct production of H2O2 without the need for sacrificial organic electron donors in the photocatalytic system is desirable from the viewpoint of green chemistry and sustainability. To that effect, Choi's group reported enhanced H2O2 production up to a milli molar level over reduced graphene oxide (rGO)/TiO2 composites in the presence of phosphate and cobalt ions in the absence of organic electron donors.48 On the one hand, the improvements were attributed to the synergistic effect of RGO, phosphate and cobalt ions. The introduced RGO can be used as an electron mediator to facilitate the separation of photo-generated electron–hole pairs thanks to the lower Fermi level compared to the conduction band edge of TiO2. Furthermore, RGO can also be used as a co-catalyst to promote the two-electron ORR for H2O2 production. On the other hand, two different roles of phosphate were observed in their work: (1) phosphate can be used as a surface passivation agent to retard the adsorption and decomposition of the produced H2O2 within a wide range of pH; and (2) phosphate reacts with cobalt ions for in situ formation of cobalt phosphate complexes on the surface of the RGO/TiO2 composite as a water oxidation co-catalyst to promote water oxidation.
Given that visible light is dominant in the solar spectrum and UV light induces the decomposition of the produced H2O2, the development of visible-light-driven photocatalysts is highly desirable for efficient and sustainable H2O2 production. In this regard, Shiraishi's group reported the design and synthesis of an advanced inorganic photocatalyst consisting of Au nanoparticles loaded on BiVO4 for highly efficient H2O2 production from water and O2 under visible light illumination (Fig. 6).37 In their work, BiVO4 possesses not only a narrow band gap for a visible-light response, but also a proper position of the CB edge (+0.02 VSHE) between the one-electron ORR potential (−0.13 VSHE) and the two-electron ORR potential (+0.68 VSHE), thus facilitating selective H2O2 production. Besides, the formation of a junction between Au and BiVO4 was also favorable for the separation of photo-generated electron–hole pairs and the two-electron ORR for H2O2 production.
 | ||
| Fig. 6 (a) Energy diagrams for Au/TiO2 and Au/BiVO4 and the reduction potential of O2; (b) time-dependent changes in the H2O2 concentrations during the photoreaction on Au0.2/TiO2 (λ > 300 nm) and Au0.2/BiVO4 prepared by calcination at 673 K (λ > 420 nm); (c) linear-sweep voltammograms of Au0.2/TiO2 and Au0.2/BiVO4 prepared by calcination at 673 K, measured on a rotating-disk electrode at different rotating speeds and (d) Koutecky–Levich plots of the data obtained at a constant potential (−0.3 V). Reprinted with permission.37 Copyright 2016 American Chemical Society. | ||
4.2 Metal–organic-frameworks or metal-coordination polymers
Metal–organic-frameworks (MOFs), a family of coordination materials consisting of secondary building units interconnected by organic linkers, have been widely investigated in various fields of gas storage/separation, drug delivery, sensors, and catalysis due to their significant advantages of facile preparation, high surface area, and adjustable composition and structure.151–155 Recently, MOFs with amine-functionalization of terephthalic acid as an organic linker have been demonstrated to catalyze various photocatalytic reactions such as water splitting, CO2 reduction and oxidation of organic compounds.156–159 For instance, Yamashita's group reported photocatalytic H2O2 production over a MOF photocatalyst consisting of Ti8O8(OH)4 clusters and 2-aminoterephthalic acid linkers (MIL-125-NH2) in the presence of triethanolamine (TEOA) and benzyl alcohol under visible-light illumination.123 In this case, Ti(IV) within the Ti8O8(OH)4 clusters was first reduced to Ti(III) by ligand-to-cluster electron transfer (LCET), which can be used to further reduce O2 to produce the ˙O2− intermediate with subsequent fast disproportionation for H2O2 production, whereas benzyl alcohol was oxidized to benzaldehyde by photo-generated holes. Nevertheless, the resultant mixture of H2O2 and benzaldehyde dissolved in acetonitrile brought difficulty in the separation process and thus resulted in high energy consumption. Later, they applied a two-phase system consisting of benzyl alcohol and water to achieve efficient separation between H2O2 and benzaldehyde accompanied by enhanced photocatalytic H2O2 production, which mainly benefited from the hydrophobization of the MOF by the modification of alkyl chains (Fig. 7).124 Moreover, the concentration of the obtained H2O2 solution can be easily tuned by the volume of the aqueous phase.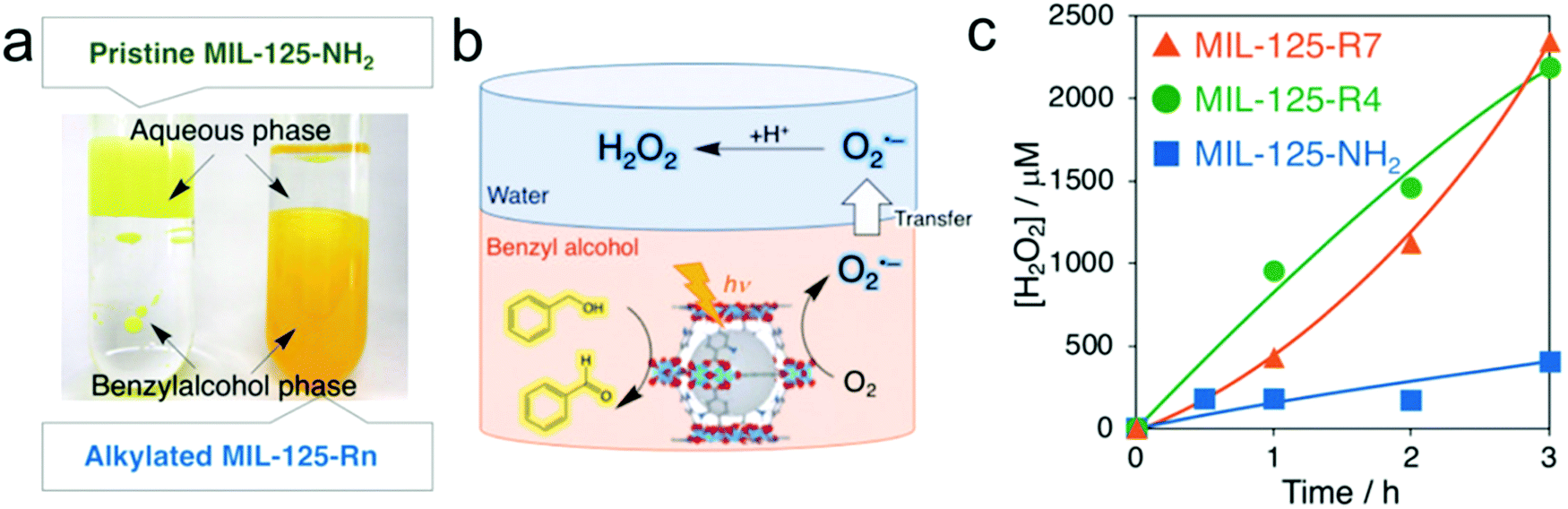 | ||
| Fig. 7 (a) Digital photographs of two-phase systems composed of an aqueous phase and a benzyl alcohol phase containing MIL-125-NH2 (left) and MIL-125-Rn (right); (b) photocatalytic H2O2 production utilizing the two-phase system; and (c) time courses of H2O2 production under photoirradiation (λ > 420 nm) of the two-phase system composed of benzyl alcohol (5.0 mL) and water (2.0 mL) catalyzed by 5.0 mg of MIL-125-NH2 (blue), MIL-125-R4 (green), and MIL-125-R7 (orange). Reprinted with permission.124 Copyright 2019 Wiley-VCH. | ||
In addition, metal coordination polymers have also been investigated as robust photocatalysts for H2O2 production, which was mainly inspired by the structure of superoxide dismutases (SODs) in nature systems consisting essentially of late transition metal ions as central atoms and proteins as organic ligands.160 Long's group developed an octahedral Cd3(C3N3S3)2 coordination polymer as a kind of novel noble-metal-free photocatalyst for photocatalytic H2O2 production in a mixture of methanol and water solution, where high H2O2 productivity of ca. 110.0 mmol L−1 g−1 at pH = 2.8 could be achieved under visible-light illumination.125 In order to further improve the H2O2 production efficiency, they introduced RGO as a 2D support to modulate the growth and formation of Cd3(C3N3S3)2, leading to accelerated photo-generated charge transfer and thus achieving 2.5-fold enhancement.126
4.3 Metal-free graphitic carbon nitride
Graphitic carbon nitride (g-C3N4) possesses a graphitic stacking structure of C3N4 layers comprising tri-s-triazine units connected through planar amino groups.67,127,135,161 To date, g-C3N4-based materials as metal-free organic polymeric semiconductor photocatalysts have received wide attention in the fields of water splitting,162–164 the ORR,165 CO2 reduction,166,167 organic photosynthesis,168 and organic pollutant degradation,169 due to their facile preparation, environmentally friendly nature, low-cost, reliable thermal and chemical stability and, more importantly, appropriate band gap structure for a visible light response and target reactions,165 since the first report about the excellent performances in photocatalytic hydrogen production by Antonietti's group in 2009.161 Recently, Shiraishi's group extended the application of the g-C3N4 photocatalyst into photocatalytic H2O2 production.127 They pointed out that the efficient formation of 1,4-endoperoxide species on the surface of g-C3N4 could suppress the one-electron ORR to produce ˙OOH and promote the selective two-electron ORR to produce H2O2, thus resulting in high H2O2 selectivity. Nevertheless, the overall photo-conversion efficiency is still low because of the limited absorption and utilization in the visible-light region, the poor separation efficiency of photo-generated electrons and holes, and the sluggish kinetics for water oxidation, and thus needs to be further improved prior to practical applications. In this regard, various strategies have been actively developed in order to improve the photocatalytic performances of g-C3N4 for H2O2 production such as self-modification, chemical modification, and hybridization with other materials including noble metals and metal phosphides.In recent years, self-modification of g-C3N4 could be achieved by the introduction of carbon vacancies, nitrogen vacancies, and heteroatom doping. Wang's group demonstrated improved photocatalytic H2O2 production efficiency over g-C3N4 through the incorporation of carbon vacancies.128 Two important roles of carbon vacancies were proposed: (1) reducing the symmetry and band gap of C3N4 facilitated the extension of the visible-light absorption and the increase of photo-generated electrons; and (2) increasing the catalytic sites enhanced the adsorption and activation of O2 molecules and induced the transformation of the H2O2 production pathway from the two-step one-electron ORR to the one-step two-electron ORR. An inverse opal (IO) structure was also introduced into g-C3N4 with carbon vacancies to further improve the efficiency for H2O2 production through the enhanced absorption and utilization of visible light, and increased surface area, which benefited from the unique structural advantages of the IO structure such as the slow photon effect, Bragg diffraction and scattering.36 Besides, a series of g-C3N4 with tunable doping of foreign carbon was also prepared by a facile hydrothermal reaction of glucose with subsequent thermal treatment.129 The energy levels of the resultant carbon-doped g-C3N4 were found to vary with the doping content of carbon, accompanied by the positive shift of the conduction and valence band, which was favorable for a selective two-electron ORR and achieved two-fold enhancement of H2O2 production.
In addition to carbon vacancies, nitrogen vacancies have also been developed to modify the intrinsic electronic structure to narrow the band gap and create more catalytically active sites for photocatalytic reactions. For instance, Zhang's group developed the thermal reduction treatment of g-C3N4 in the presence of NaBH4 to introduce nitrogen vacancies through the formation of CN functional groups.132 This structural change resulted in the narrowing of the band gap and the positive shift of the band edge, thus extending the light absorption within the visible-light region, and facilitating water oxidation and a selective two-electron ORR for H2O2 production. The optimal reduced g-C3N4 photocatalyst exhibited an H2O2 productivity of 170 μmol L−1 h−1 with a solar-to-H2O2 chemical conversion efficiency of 0.26% and AQY of 4.3% under visible-light illumination without organic electron donors. Besides, nitrogen vacancies have been successfully in situ embedded into g-C3N4 moieties through dielectric barrier discharge (DBD) plasma under an H2 atmosphere.130 Compared with the direct annealing treatment under an H2 atmosphere, H2 plasma treatment can achieve more nitrogen vacancies embedded in g-C3N4 moieties, which facilitated the adsorption of O2 molecules and the transfer of photo-generated electrons for the subsequent two-electron ORR. Nevertheless, these methods have inherent shortcomings such as high energy consumption and explosive potential. In this case, Ye's group reported a novel photo-assisted thermal reduction route for the preparation of holey defective g-C3N4 photocatalysts (Fig. 8).131 The developed method could achieve the introduction of abundant nitrogen vacancies and holey structure within the g-C3N4 photocatalyst simultaneously, thus facilitating the increase of the visible-light absorption range, the separation of photo-generated electrons and holes, and the accessibility of reactants to the surface active sites. The optimal holey defective g-C3N4 (DCN) photocatalysts show ten-fold enhanced photocatalytic activity for H2O2 production compared to pristine bulk g-C3N4 (BCN).
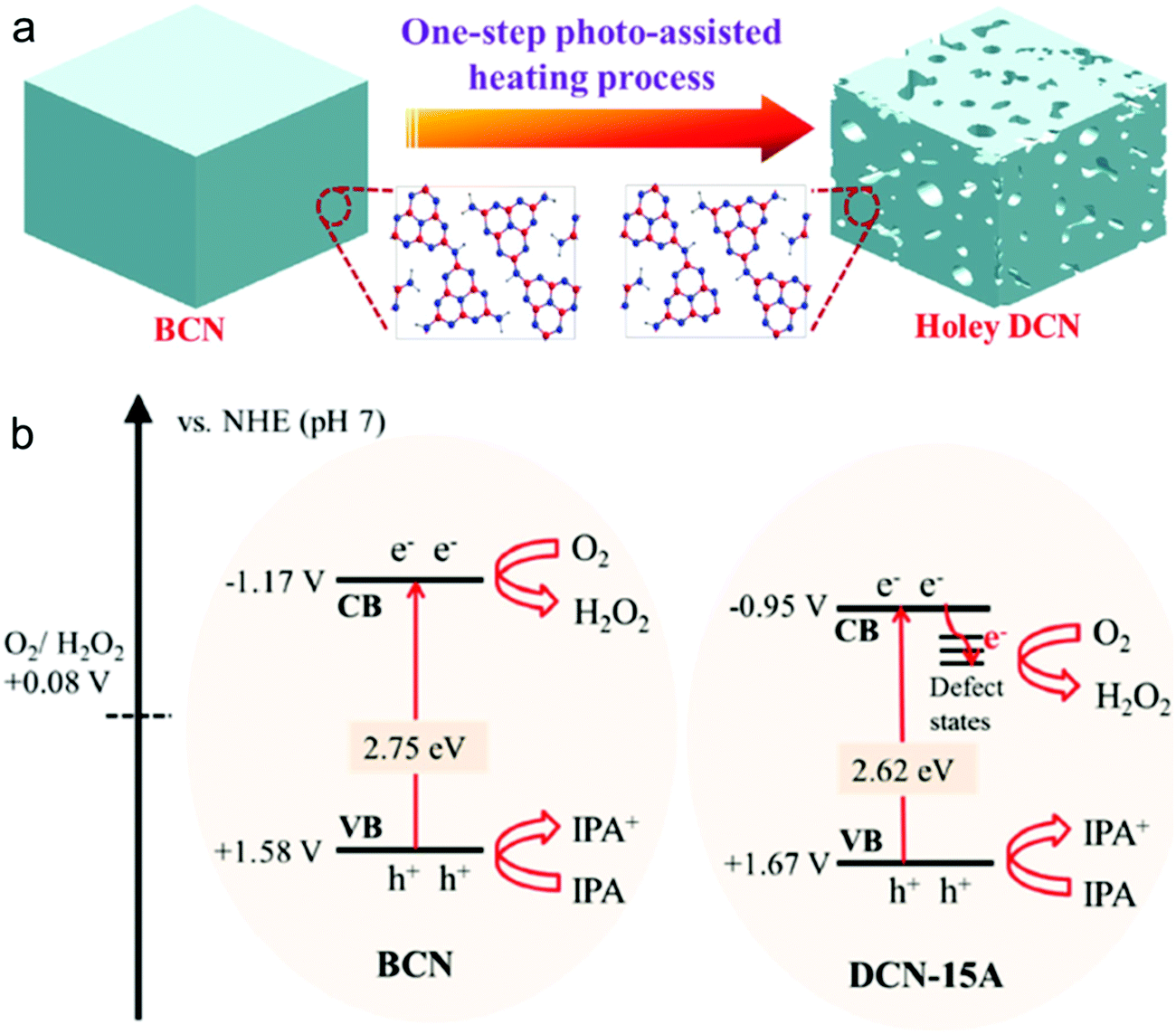 | ||
| Fig. 8 (a) Illustration of the preparation process of holey DCN, and the insets are the simulated structure (H, C, and N atoms are represented by the gray, red, and blue balls) and (b) schematic of mechanisms underlying the photoexcited dynamics involved in photocatalytic H2O2 evolution over BCN and DCN-15A. Reprinted with permission.131 Copyright 2018 Wiley-VCH. | ||
Recent research demonstrated that the introduction of oxygen function groups (–COOH and C–O–C) and other heteroatom doping into carbon frameworks can significantly improve the selective electrochemical two-electron ORR for H2O2 production.3 Similarly, the doping of earth-abundant heteroatoms (potassium, phosphorus, oxygen, and fluorine) into the C3N4 framework was reported to improve the efficiency of photocatalytic H2O2 production through the formation of photo-generated charge trapping sites on the surface of the g-C3N4 photocatalyst. Zhu's group prepared oxygen-enriched g-C3N4 (OCN) using ammonium para-tungstate and dicyandiamide,21 which exhibited a 3.5-fold higher AQY for H2O2 production of 10.2% at 420 nm compared to pristine bulk g-C3N4 due to the relatively easy formation of 1,4-endoperoxide species and the highly selective ORR to H2O2. Potassium-, phosphorus-, and oxygen-doped g-C3N4 can also be successfully prepared through the one-pot thermal polymerization of urea or melamine in the presence of phosphates.38 The incorporation of these heteroatoms resulted in the narrowing of the band gap, the increase of the generation, transfer, and lifetime of photo-generated electrons and holes, and the inhibition of the decomposition of the produced H2O2, thus achieving high AQY for H2O2 production.
Chemical modification with electron-deficient or π-conjugated organic monomers was another efficient way to improve the photocatalytic performances of g-C3N4 for H2O2 production. For instance, the incorporation of pyromellitic diimide (PDI) with high electron affinity into the lattice network of g-C3N4, which was achieved by a facile thermal condensation of melem and pyromellitic dianhydride (PMDA), resulted in the simultaneous positive shift of the valence and conduction band edge, thus promoting water oxidation and maintaining a highly selective two-electron ORR for H2O2 production.135 Subsequently, graphene with high charge carrier mobility was also introduced into the g-C3N4/PDI moiety by a hydrothermal-calcination process (Fig. 9a–c).20 The introduction of graphene facilitated the transfer of photo-generated electrons from g-C3N4/PDI to graphene and then promoted the selective two-electron ORR for H2O2 production on the graphene moiety. In order to further improve the photocatalytic activity for H2O2 production, layered boron nitride (BN) was also introduced into the above hybrid catalyst of g-C3N4/PDI/graphene to promote the transfer of photo-generated electrons and holes to graphene and BN, thus promoting the two-electron ORR for H2O2 production on the surface of graphene and water oxidation on the surface of BN, respectively.136 Meanwhile, they also utilized three-directional mellitic triimide (MTI) and biphenyl diimide (BDI) units as a substitute for PDI to incorporate into the g-C3N4 moiety to promote H2O2 production from the viewpoint of enhanced electrical conductivity and charge separation.39,70
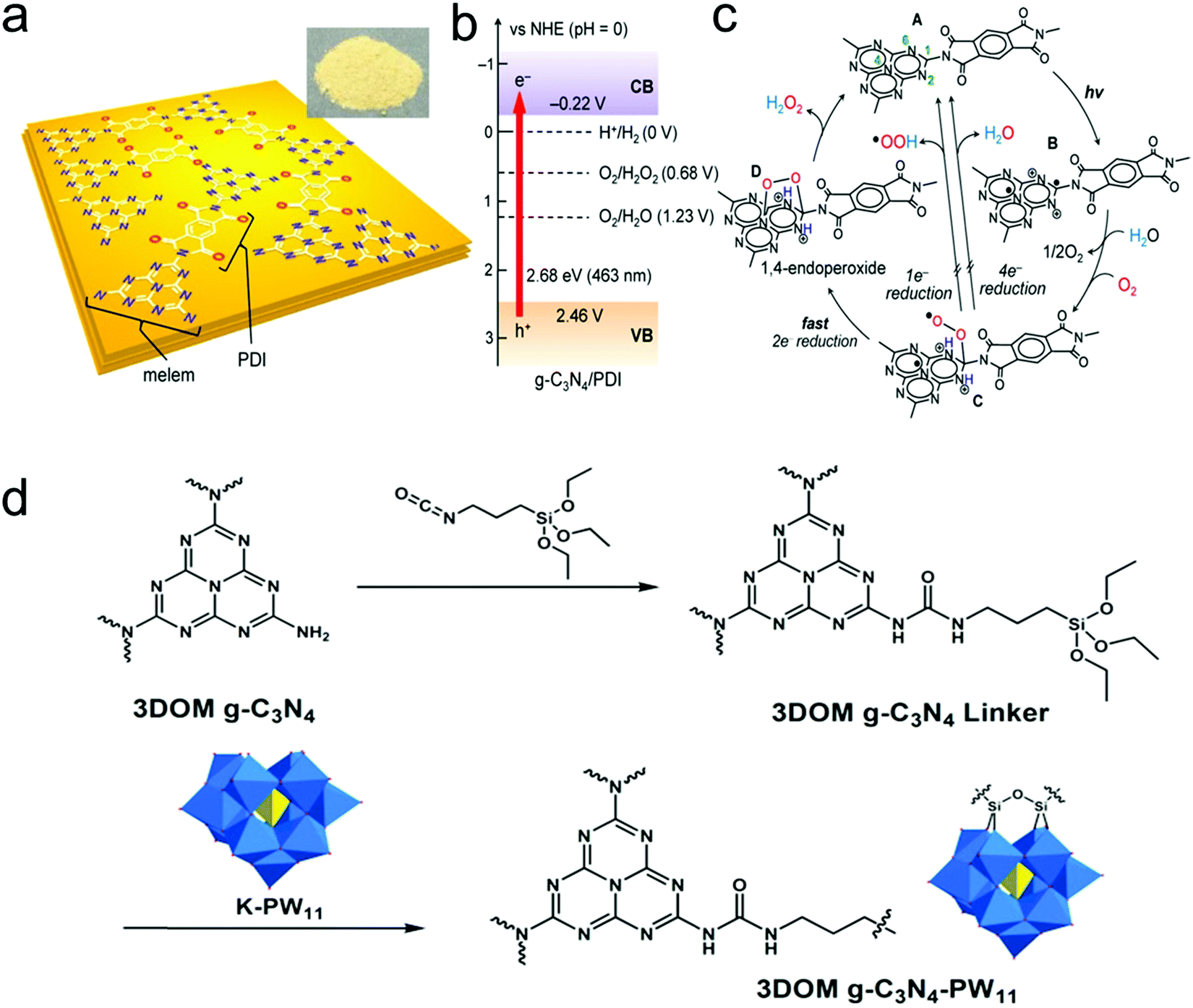 | ||
| Fig. 9 (a–c) g-C3N4/PDI structures and mechanism for photocatalytic H2O2 production: (a) three-dimensional structure, (b) electronic band structure of g-C3N4/PDI (containing 51% PDI units), and (c) mechanism of photocatalytic H2O2 production. (d) Preparation process of 3DOM g-C3N4–PW11. (a–c) Reprinted with permission.20 Copyright 2016 American Chemical Society. (d) Reprinted with permission.139 Copyright 2017 Elsevier Ltd. | ||
Polyoxometalates (POMs), consisting of cations and polyanion clusters, were reported to produce a hole center (O−) and trapped electron center (M(n−1)+) pair as electron acceptors and donors through the charge transfer between O2− and Mn+ (n = 5, 6) under light illumination, making them promising guest molecules for chemical modification of the g-C3N4 host. Based on this point, Zhu's group demonstrated the covalent combination of a POM cluster of [PW11O39]7− (PW11) with three dimensionally ordered macroporous (3DOM) g-C3N4 through the reaction between the amine groups of the g-C3N4 framework and (triethoxysilyl)-propyl isocyanate (Fig. 9d).139 The resultant 3DOM g-C3N4/PW11 hybrid photocatalyst exhibited efficient photocatalytic H2O2 production without the use of organic electron donors, which was attributed to the suppressed one-electron ORR to ˙OOH and the promoted charge separation and two-electron ORR to H2O2. Later, they applied a similar organic linker strategy to fabricate the hybrid catalyst of a POM cluster of [SiW11O39]8− (SiW11) possessing a more negative CB edge and g-C3N4 containing more amine groups obtained by thermal decomposition of urea.138 Recently, Zhao's group reported the incorporation of POM-derived metal oxides into the g-C3N4 framework by the thermal treatment of the g-C3N4 precursor and the POM precursor.140,141 The incorporation of POM-derived metal oxides resulted in the negative shift of the CB edge of g-C3N4 and thus promoted the one-electron ORR to ˙OOH, whereas it is also thermodynamically favored to oxidize ˙OOH by photo-generated holes to singlet oxygen (1O2), both of which can promote photocatalytic H2O2 production. A similar effect was also achieved through the covalent combination of carbon nanotubes (CNTs) with g-C3N4 because of their unique π-conjugated structure capable of accepting, transporting and storing electrons.143
The hybridization of g-C3N4 with catalytically active metal nanoparticles (NPs) as a co-catalyst has also been used to improve the photocatalytic performances for H2O2 production by promoting the separation efficiency of photo-generated electrons and holes. For instance, loading Au nanoparticles on the surface of g-C3N4 resulted in highly efficient and stable H2O2 production because of the favorable two-electron ORR and the inert nature for catalyzing the decomposition of the produced H2O2 over the hybrid catalyst.144,145 As a noble-metal-free co-catalyst, CoP has also been loaded on the surface of g-C3N4 to improve the photocatalytic H2O2 production efficiency under visible-light illumination by extending the light absorption range and promoting charge separation and electron transfer.146
Last but not least, a new type of photoelectrochemical (PEC) system without an external bias has also been developed to simultaneously achieve the generation of H2O2 and electricity in recent years.174,175 Generally, the PEC configuration for H2O2 production can be divided into three categories based on where H2O2 was produced, at the cathode, anode, or both. Early research demonstrated the indirect ORR for H2O2 production through the photoelectrochemical reduction of anthraquinone derivative molecules followed by reaction with O2 (Fig. 10a).170,176,177 Nevertheless, for this kind of configuration, the use of organic solvents is unavoidable and the energy conversion efficiency is low, arising from sluggish electrode kinetics, which hinders wide application from the viewpoint of sustainability. Recently, the direct ORR to H2O2 has also been achieved with high Faradaic efficiency and decent photocurrent on a dye-sensitized NiO photocathode in aqueous electrolyte,171,178 where photo-generated electron–hole pairs from the excited dye upon illumination performed fast hole injection into NiO and one-electron transfer to O2 for the formation of ˙OOH, followed by reaction with a proton and disproportionation into hydrogen peroxide in protic electrolytes (Fig. 10b). It should be highlighted here that the design of dye molecules has a significant effect on the energy conversion efficiency and electrode stability. Currently, the investigated dye molecules mainly include porphyrin, coumarin, and ruthenium dyes, and BH4 dye (a kind of hydrophobic donor–double-acceptor dye). In addition, some organic polymeric semiconductors, like polyterthiophene (pTTh),179 polymeric metal salen-type complexes,180 poly-tetrakis-5,10,15,20-(4-aminophenyl)porphyrin (pTAPP) and its cobalt derivative (pCoTAPP),173 have been directly explored as efficient photocathodes without dye sensitization for H2O2 production via the two-electron ORR (Fig. 10c and d). In order to further optimize the performances, an organic hetero-junction photocathode comprising phthalocyanine and tetracarboxylic perylenediimide has been developed, which exhibited continuous generation of high concentrations of peroxide with the Faradaic efficiency remaining at around 70%.174 Besides, cathodic H2O2 production has also been performed in such a PEC configuration, where semiconductor photocatalysts were used as photoanodes to drive water oxidation, and electrocatalysts as cathodes to promote the selective two-electron ORR for H2O2 production.5,172,181,182 For instance, Fukuzumi's group systematically investigated WO3 and BiVO4 as a durable photoanode with a cobalt chlorine complex supported on a glassy carbon substrate as a cathode to construct a two-compartment PEC cell separated by a Nafion membrane for efficient H2O2 production from water and O2 under solar illumination.5,172 Especially, when iron(III) oxide(hydroxide) (FeO(OH)) was modified as a water oxidation catalyst on the surface of BiVO4, the produced H2O2 with a high concentration up to 61 mM can be used directly as a fuel to generate electricity in an H2O2 fuel cell.172
 | ||
| Fig. 10 (a) Illustration of the working principle of dye sensitized photo-electrochemical cells (DSPECs) for H2O2 production by using AQ redox mediators (a, top), and the corresponding schematic representation (a, bottom). (b) Schematic representation of the DSPECs for H2O2 production in protic electrolyte and nucleophilic substitution in aprotic electrolyte. (c) Schematic reaction diagram of a photoelectrochemical cell for hydrogen peroxide production. (d) Band gap diagram showing photoresponse behaviors in the photosynthetic reduction of oxygen by pTAPP. (a) Reprinted with permission.170 Copyright 2020 Wiley-VCH. (b) Reprinted from Chemical Science,171 published by The Royal Society of Chemistry. (c) Reprinted with permission.172 Copyright 2016 American Chemical Society. (d) Reprinted with permission.173 Copyright 2017 American Chemical Society. | ||
On the other hand, H2O2 production has also been reported through two-electron water oxidation over semiconductor photoanodes.40,185–189 For instance, Sayama's group reported the production and accumulation of H2O2 through two-electron water oxidation using a WO3/BiVO4 photoanode with simultaneous H2 production on a Pt cathode in an aqueous solution of KHCO3 at an applied voltage far lower than the theoretical electrolysis voltage under simulated solar light.40 Particularly, H2O2 formation was proposed through the hydrolysis of percarbonate intermediates (HCO4− and C2O62−) from the oxidation of HCO3− by the photo-generated holes within BiVO4. Moreover, highly concentrated HCO3− could effectively suppress the oxidation degradation of the produced H2O2. In order to further improve the selective H2O2 production by two-electron water oxidation, the surface modification of the WO3/BiVO4 photoanode with an Al2O3 layer has also been developed by the metal–organic decomposition method and chemical vapor deposition method to suppress the oxidative decomposition of the produced H2O2, thus leading to high Faradaic efficiency for H2O2 production.68,185 Further, the introduction of Au-supported fluorine-doped tin oxide (FTO) glass or ordered mesoporous carbon as a substitute for the Pt cathode into the above system successfully achieved the simultaneous production of H2O2 and electricity without the need for an external bias from both sides of the PEC cell through the two-electron ORR and two-electron water oxidation (Fig. 11), respectively.183,184
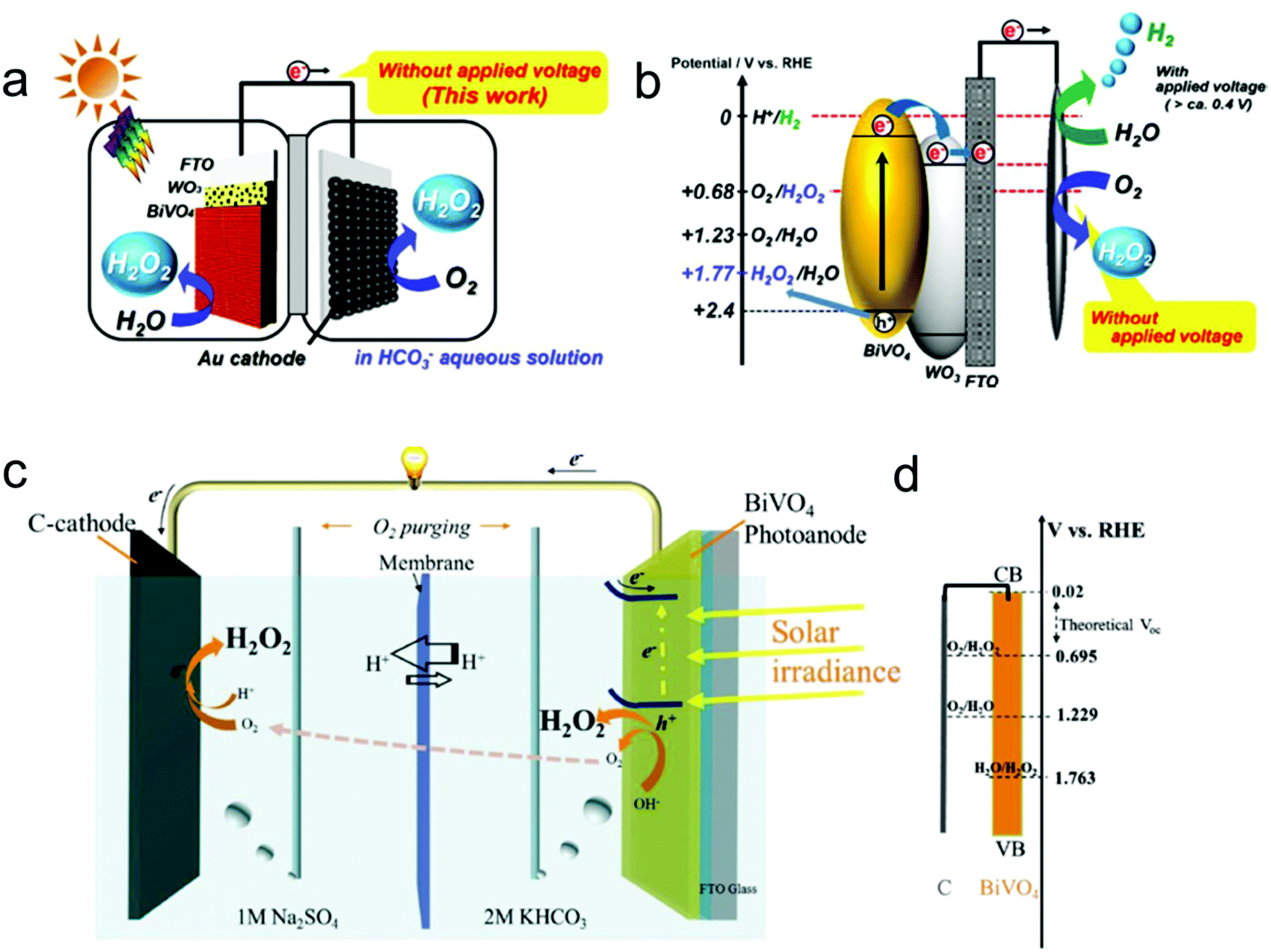 | ||
| Fig. 11 (a) Diagram of the photoelectrode system for producing only H2O2 by using a two-electron oxidation system of H2O on a WO3/BiVO4 photoanode under solar light irradiation; (b) energy diagram of the photoelectrode systems; (c) schematic illustration of the design of a light-driven fuel cell with spontaneous H2O2 generation; and (d) the band diagram of the system. The conduction band (CB) and valence band (VB) edge positions of BiVO4 straddle the redox potentials of O2/H2O2 and H2O/H2O2, suggesting the possibility of unassisted H2O2 production. The theoretical Voc for the light-driven fuel cell is 0.693 V, estimated from the CB of BiVO4 and the O2/H2O2 redox potential. (a and b) Reprinted with permission.183 Copyright 2017 Wiley-VCH. (c and d) Reprinted with permission.184 Copyright 2018 Wiley-VCH. | ||
5 Conclusions and perspectives
H2O2 plays an essential role in the fields of chemical industry, environmental treatment, and sustainable energy conversion/storage. Therefore, the development of efficient, energy-saving and sustainable methods for H2O2 production is of great significance and urgency to address the contradiction between the growing H2O2 demand and market, on one side, and the severe unsustainability of today's industrial production methods, on the other. Future H2O2 production pathways involving either electrochemical or photochemical approaches are currently considered most promising and sustainable, because only water, O2, solar energy or electricity from renewable power sources are involved during the entire process. The key challenge lies in the development of new scalable catalysts with low-cost, high efficiency and excellent electrochemical stability. In the past few years, various catalysts have been widely studied from the viewpoint of both electrochemical and photochemical H2O2 production, including ORR electrocatalysts such as noble-metal-based materials, transition-metal-based materials, and metal-free carbon-based materials, and semiconductor photocatalysts such as metal oxides, metal–organic complexes, and metal-free graphitic carbon nitride. Recent advances have been summarized herein. Importantly, upon comparing various ORR electrocatalysts and semiconductor photocatalysts, it becomes obvious that most strategies that have been explored to improve the catalyst activity and selectivity for H2O2 production largely follow two aspects, that is, geometric structural engineering and electronic structure engineering. Geometric structure engineering of catalysts can be achieved through accurately controlling the morphology of both ORR electrocatalysts and semiconductor photocatalysts. Electronic structure engineering of ORR electrocatalysts, on the other hand, can be achieved through surface passivation or alloying with inactive elements, single-atom dispersion, and surface heteroatom doping, whereas for semiconductor photocatalysts, electronic structure engineering can be achieved through self-modification by the introduction of carbon or nitrogen vacancies and heteroatom doping, chemical modification with electron-deficient or π-conjugated organic monomers and POMs, hybridization with other materials such as noble metal and carbon materials, and surface passivation by anion and cations. Besides, the simultaneous generation of H2O2 and electricity can also be achieved by the combination of semiconductor photocatalysts as photoanodes with ORR electrocatalysts as cathodes in photoelectrochemical systems.Generally, there are three different reaction intermediates during the ORR and WOR processes for H2O2 production, including HOO*, O*, and HO*. According to Sabatier's principle, the optimal catalyst for H2O2 production should possess balanced binding of HOO* on the surface of the catalyst during the ORR process, whereas balanced binding of HO* during the WOR process. In reported studies, most of them mainly focused on tuning the binding of the reaction intermediates through coating a carbon layer on the surface of catalysts, forming single metal atoms dispersed on the support, or regulating the composition of the catalyst like the formation of alloys comprising reactive metals with inactive metals, making the overall reaction pathway favorable for the two-electron pathway. In addition, optimizing the morphological structure of catalysts has been reported as an efficient method to promote H2O2 production. For instance, the mass transport within a mesoporous structure favored the fast release of the produced H2O2 from the surface of the catalyst, thus avoiding the subsequent reduction of H2O2. Besides, for photocatalytic systems, the experimental conditions have a certain effect on the reaction intermediates during the H2O2 production process. For instance, using benzylic alcohols as electron donors was reported to result in the formation of new reaction intermediates, coordinated peroxo species, which facilitated H2O2 production.
Even though there are some significant advances in the development of both electrocatalytic and photocatalytic H2O2 production over various nanostructured heterogeneous catalysts, there remains a grand challenge to further improve their performances prior to commercialization becoming industrially viable and economical. Future work in this area should focus on the following aspects.
5.1 Exploiting further improved catalytic materials
Seeking materials with new compositions and structures is still at the heart of research work on electrocatalytic and photocatalytic H2O2 production. To date, the developed noble-metal-based materials are considered to be most efficient ORR electrocatalysts for H2O2 production, but their inherent disadvantages of high cost and scarcity significantly hamper their practical industrial application. In view of cost reduction, it is thus urgently needed that ORR electrocatalysts with low-cost and high efficiency are developed. Recently, H2O2 production by two-electron water oxidation over metal oxides has also shown great potential application in the field of water electrolysis, capable of the simultaneous generation of two important valuable products of H2O2 and H2 in a single electrochemical system using water as the only raw material.109,110,190 Nevertheless, very few kinds of metal oxides were explored, such as ZnO, WO3, SnO2, BiVO4, and TiO2, and thus considerable efforts need to be made for the development of other metal oxides and beyond. For photocatalytic H2O2 production, considering that UV light only accounts for ca. 4% and also induces H2O2 decomposition, it is thus essential to design and develop visible-light-driven photocatalysts with highly selective promotion of the two-electron ORR to produce H2O2 and efficient suppression of subsequent decomposition of the produced H2O2, thus enhancing the SCC efficiency. Nevertheless, the reported visible-light-driven photocatalysts such as BiVO4 and g-C3N4 still have some critical issues including sluggish kinetics of water oxidation and unsatisfactory stability. In addition, careful observation demonstrates that the only difference between photocatalytic H2O2 production and photocatalytic water splitting lies in the surface redox reaction process. This point provides a possibility for the integration of the existing photocatalysts for water splitting with two-electron ORR and water oxidation electrocatalysts as co-catalysts to achieve highly efficient H2O2 production.5.2 Mechanistic investigations
The introduction of porous structure and heteroatom doping has been reported to play an important role in improving the capability of carbon-based materials for electrochemical H2O2 production through the two-electron ORR. However, the precise role of the pore size and heteroatom doping species is still unclear and even conflicting. Similarly, the introduction of metal catalytically active sites into nitrogen-doped carbon materials can further improve the catalytic performances through the formation of metal-coordinated metal–Nx–C moieties, but there have also been conflicting opinions on which coordination number is more favorable for the two-electron ORR for H2O2 production. For photocatalytic H2O2 production, most studies demonstrated the negative effect of the one-electron ORR on the efficiency of the ORR and H2O2 production, and thus various methods have been developed to suppress the one-electron ORR by the positive shift of the CB edge within semiconductor photocatalysts. Nevertheless, recent research also reported the negative shift of the CB edge of g-C3N4-based catalysts to enhance the one-electron ORR for ˙OOH formation and thus promote the sequential two-step one-electron ORR for H2O2 production. Besides, the type of electron donor used has been found to play distinct roles in influencing the ORR reaction pathways in photocatalytic systems, but the detailed reason is still unknown and thus requires further study. All in all, more studies are needed to understand the nature of active sites and the reaction pathways, and how they contribute to the high catalytic performances for H2O2 production, which provides guidance for the design of novel catalytic systems for electrochemical and photochemical H2O2 production.5.3 Scalable photo-/electro-chemical interfaces and devices
Most of the recently reported research work mainly evaluated the electrochemical performances of ORR electrocatalysts for H2O2 selectivity by the RRDE technique, whereas only a few studies about the practical H2O2 productivity in home-made two-compartment H-cells and flow cells were reported. Nevertheless, it should be highlighted that flow cells may be more relevant to a commercial scale H2O2 production system compared to H-cells, providing a key stepping stone for the translation of fundamental lab discoveries into practice. In addition, the electrochemical systems discussed above generally produce a mixture of H2O2 and solutes in traditional liquid electrolytes, thus requiring extra separation processes for the purification of the produced H2O2 solutions. Recently, Wang's group produced a new strategy to achieve the direct electrochemical production of pure H2O2 solutions by using a porous solid electrolyte, and various concentrations of pure H2O2 solutions could be easily achieved by the change of the water flow rate.1 Based on this point, we expect more widespread use of solid electrolytes in PEC systems for simultaneous generation of H2O2 and electricity. Anyway, the research based on such a thought is still in its infancy, and more work is needed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. S. and Y. S. acknowledge financial support from FCH Joint Undertaking (CRESCENDO project, Grant Agreement No. 779366) and the German Ministry of Economics and Energy (BMWi) through project “ChemEFlex” (FKN 0350013A). L. H. gratefully acknowledges financial support from Fundamental Research Funds for the Central Universities (No. 531118010232) and Huxiang High-Level Talent Gathering Project of Hunan Province (No. 2019RS1012).References
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef CAS.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS.
- L. Han, S. Guo, P. Wang and S. Dong, Adv. Energy Mater., 2015, 5, 1400424 CrossRef.
- S. A. Mousavi Shaegh, N.-T. Nguyen, S. M. Mousavi Ehteshami and S. H. Chan, Energy Environ. Sci., 2012, 5, 8225 RSC.
- J. K. Edwards, J. Pritchard, L. Lu, M. Piccinini, G. Shaw, A. F. Carley, D. J. Morgan, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2014, 53, 2381–2384 CrossRef CAS.
- S. Shibata, T. Suenobu and S. Fukuzumi, Angew. Chem., Int. Ed., 2013, 52, 12327–12331 CrossRef CAS.
- R. Arrigo, M. E. Schuster, S. Abate, G. Giorgianni, G. Centi, S. Perathoner, S. Wrabetz, V. Pfeifer, M. Antonietti and R. Schlögl, ACS Catal., 2016, 6, 6959–6966 CrossRef CAS.
- V. Paunovic, J. C. Schouten and T. A. Nijhuis, Appl. Catal., A, 2015, 505, 249–259 CrossRef CAS.
- I. Yamanaka, Y. Satake, P. Pantira, D. Hiraki and H. Ogihara, ChemistrySelect, 2017, 2, 464–468 CrossRef CAS.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- Y. Liu, X. Quan, X. Fan, H. Wang and S. Chen, Angew. Chem., Int. Ed., 2015, 54, 6837–6841 CrossRef CAS.
- I. Yamanaka and T. Murayama, Angew. Chem., Int. Ed., 2008, 47, 1900–1902 CrossRef CAS.
- T. P. Fellinger, F. Hasche, P. Strasser and M. Antonietti, J. Am. Chem. Soc., 2012, 134, 4072–4075 CrossRef CAS.
- J. S. Jirkovsky, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. Schiffrin, J. Am. Chem. Soc., 2011, 133, 19432–19441 CrossRef CAS.
- Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef.
- Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, J. Am. Chem. Soc., 2016, 138, 10019–10025 CrossRef CAS.
- Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan and Y. Zhu, Energy Environ. Sci., 2018, 11, 2581–2589 RSC.
- J. Song and S. Cho, APL Mater., 2020, 8, 050701 CrossRef.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- P. Strasser, M. Gliech, S. Kuehl and T. Moeller, Chem. Soc. Rev., 2018, 47, 715–735 RSC.
- D.-W. Wang and D. Su, Energy Environ. Sci., 2014, 7, 576 RSC.
- R. Chattot, O. Le Bacq, V. Beermann, S. Kuhl, J. Herranz, S. Henning, L. Kuhn, T. Asset, L. Guetaz, G. Renou, J. Drnec, P. Bordet, A. Pasturel, A. Eychmuller, T. J. Schmidt, P. Strasser, L. Dubau and F. Maillard, Nat. Mater., 2018, 17, 827–833 CrossRef CAS.
- T. Najam, S. S. A. Shah, W. Ding, J. Jiang, L. Jia, W. Yao, L. Li and Z. Wei, Angew. Chem., Int. Ed., 2018, 57, 15101–15106 CrossRef CAS.
- I. Yamanaka, T. Onizawa, S. Takenaka and K. Otsuka, Angew. Chem., Int. Ed., 2003, 42, 3653–3655 CrossRef CAS.
- I. Yamanaka, S. Tazawa, T. Murayama, R. Ichihashi and N. Hanaizumi, ChemSusChem, 2008, 1, 988–992 CrossRef CAS.
- J.-y. Chen, L. Zhao, N. Li and H. Liu, J. Power Sources, 2015, 287, 291–296 CrossRef CAS.
- D. Ki, S. C. Popat, B. E. Rittmann and C. I. Torres, Environ. Sci. Technol., 2017, 51, 6139–6145 CrossRef CAS.
- https://www.hpnow.eu/# .
- S. Siahrostami, G. L. Li, V. Viswanathan and J. K. Norskov, J. Phys. Chem. Lett., 2017, 8, 1157–1160 CrossRef CAS.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS.
- A. Torres-Pinto, M. J. Sampaio, C. G. Silva, J. L. Faria and A. M. T. Silva, Appl. Catal., B, 2019, 252, 128–137 CrossRef CAS.
- J. Lei, B. Chen, W. Lv, L. Zhou, L. Wang, Y. Liu and J. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 16467–16473 CrossRef CAS.
- H. Hirakawa, S. Shiota, Y. Shiraishi, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2016, 6, 4976–4982 CrossRef CAS.
- G.-h. Moon, M. Fujitsuka, S. Kim, T. Majima, X. Wang and W. Choi, ACS Catal., 2017, 7, 2886–2895 CrossRef CAS.
- Y. Kofuji, S. Ohkita, Y. Shiraishi, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, ACS Sustainable Chem. Eng., 2017, 5, 6478–6485 CrossRef CAS.
- K. Fuku and K. Sayama, Chem. Commun., 2016, 52, 5406–5409 RSC.
- M. Traube, Ber. Dtsch. Chem. Ges., 1887, 2, 1041–1050 Search PubMed.
- E. Berl, J. Electrochem., 1939, 76, 359 Search PubMed.
- R. M. Reis, A. A. G. F. Beati, R. S. Rocha, M. H. M. T. Assumpção, M. C. Santos, R. Bertazzoli and M. R. V. Lanza, Ind. Eng. Chem. Res., 2011, 51, 649–654 CrossRef.
- J. F. Pérez, A. Galia, M. A. Rodrigo, J. Llanos, S. Sabatino, C. Sáez, B. Schiavo and O. Scialdone, Electrochim. Acta, 2017, 248, 169–177 CrossRef.
- M. N. Young, M. J. Links, S. C. Popat, B. E. Rittmann and C. I. Torres, ChemSusChem, 2016, 9, 3345–3352 CrossRef CAS.
- E. Baur and C. Neuweiler, Helv. Chim. Acta, 1927, 10, 901–907 CrossRef CAS.
- V. Maurino, C. Minero, G. Mariella and E. Pelizzetti, Chem. Commun., 2005, 2627–2629 RSC.
- G.-h. Moon, W. Kim, A. D. Bokare, N.-e. Sung and W. Choi, Energy Environ. Sci., 2014, 7, 4023–4028 RSC.
- A. Bonakdarpour, T. R. Dahn, R. T. Atanasoski, M. K. Debe and J. R. Dahn, Electrochem. Solid-State Lett., 2008, 11, B208 CrossRef CAS.
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Norskov, J. Phys. Chem. Lett., 2012, 3, 2948–2951 CrossRef CAS.
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. Stephens, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134–145 CrossRef CAS.
- D. R. Lawson, J. Electrochem. Soc., 1988, 135, 2247 CrossRef CAS.
- G. Yang, W. Choi, X. Pu and C. Yu, Energy Environ. Sci., 2015, 8, 1799–1807 RSC.
- I. Yamanaka, T. Hashimoto, R. Ichihashi and K. Otsuka, Electrochim. Acta, 2008, 53, 4824–4832 CrossRef CAS.
- R. B. Valim, R. M. Reis, P. S. Castro, A. S. Lima, R. S. Rocha, M. Bertotti and M. R. V. Lanza, Carbon, 2013, 61, 236–244 CrossRef CAS.
- W. R. P. Barros, R. M. Reis, R. S. Rocha and M. R. V. Lanza, Electrochim. Acta, 2013, 104, 12–18 CrossRef CAS.
- J. Choi, S. H. Hwang, J. Jang and J. Yoon, Electrochem. Commun., 2013, 30, 95–98 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS.
- M. Teranishi, S. Naya and H. Tada, J. Am. Chem. Soc., 2010, 132, 7850–7851 CrossRef CAS.
- R. Shen, W. Chen, Q. Peng, S. Lu, L. Zheng, X. Cao, Y. Wang, W. Zhu, J. Zhang, Z. Zhuang, C. Chen, D. Wang and Y. Li, Chemistry, 2019, 5, 2099–2110 CrossRef CAS.
- Z. Chen, S. Chen, S. Siahrostami, P. Chakthranont, C. Hahn, D. Nordlund, S. Dimosthenis, J. K. Nørskov, Z. Bao and T. F. Jaramillo, React. Chem. Eng., 2017, 2, 239–245 RSC.
- Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. Dresp, S. Kühl, C. Spöri, H. Schmies, H. Wang, D. Bernsmeier, B. Paul, R. Schmack, R. Kraehnert, B. Roldan Cuenya and P. Strasser, ACS Catal., 2018, 8, 2844–2856 CrossRef CAS.
- S. Y. Park, H. Abroshan, X. Shi, H. S. Jung, S. Siahrostami and X. Zheng, ACS Energy Lett., 2018, 4, 352–357 CrossRef.
- J. H. Baek, T. M. Gill, H. Abroshan, S. Park, X. Shi, J. Nørskov, H. S. Jung, S. Siahrostami and X. Zheng, ACS Energy Lett., 2019, 4, 720–728 CrossRef CAS.
- D. Tsukamoto, A. Shiro, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, ACS Catal., 2012, 2, 599–603 CrossRef CAS.
- Y. Shiraishi, Y. Kofuji, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2015, 5, 3058–3066 CrossRef CAS.
- Y. Miyase, S. Takasugi, S. Iguchi, Y. Miseki, T. Gunji, K. Sasaki, E. Fujita and K. Sayama, Sustainable Energy Fuels, 2018, 2, 1621–1629 RSC.
- H. Joo, Korean J. Chem. Eng., 2006, 23, 931–934 CrossRef CAS.
- Y. Kofuji, S. Ohkita, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2016, 6, 7021–7029 CrossRef CAS.
- C. H. Choi, H. C. Kwon, S. Yook, H. Shin, H. Kim and M. Choi, J. Phys. Chem. C, 2014, 118, 30063–30070 CrossRef CAS.
- A. Mahata and B. Pathak, Nanoscale, 2017, 9, 9537–9547 RSC.
- A. von Weber, E. T. Baxter, H. S. White and S. L. Anderson, J. Phys. Chem. C, 2015, 119, 11160–11170 CrossRef CAS.
- K. Asakura, H. Nagahiro, N. Ichikuni and Y. Iwasawa, Appl. Catal., A, 1999, 188, 313–324 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS.
- J. Deng, H. Li, J. Xiao, Y. Tu, D. Deng, H. Yang, H. Tian, J. Li, P. Ren and X. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC.
- Y. H. Li, J. Xing, X. H. Yang and H. G. Yang, Chem. – Eur. J., 2014, 20, 12377–12380 CrossRef CAS.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS.
- S. Yang, Y. J. Tak, J. Kim, A. Soon and H. Lee, ACS Catal., 2017, 7, 1301–1307 CrossRef CAS.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H. T. Kim, K. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS.
- Z. Zheng, Y. H. Ng, D. W. Wang and R. Amal, Adv. Mater., 2016, 28, 9949–9955 CrossRef CAS.
- A. Bonakdarpour, M. Lefevre, R. Yang, F. Jaouen, T. Dahn, J.-P. Dodelet and J. R. Dahn, Electrochem. Solid-State Lett., 2008, 11, B105 CrossRef CAS.
- A. Bonakdarpour, C. Delacote, R. Yang, A. Wieckowski and J. R. Dahn, Electrochem. Commun., 2008, 10, 611–615 CrossRef CAS.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- F. V. E. dos Reis, V. S. Antonin, P. Hammer, M. C. Santos and P. H. C. Camargo, J. Catal., 2015, 326, 100–106 CrossRef CAS.
- S. Siahrostami, M. E. Bjorketun, P. Strasser, J. Greeley and J. Rossmeisl, Phys. Chem. Chem. Phys., 2013, 15, 9326–9334 RSC.
- I. Yamanaka, S. Tazawa, T. Murayama, T. Iwasaki and S. Takenaka, ChemSusChem, 2010, 3, 59–62 CrossRef CAS.
- B. Q. Li, C. X. Zhao, J. N. Liu and Q. Zhang, Adv. Mater., 2019, 31, e1808173 CrossRef.
- I. Yamanaka, R. Ichihashi, T. Iwasaki, N. Nishimura, T. Murayama, W. Ueda and S. Takenaka, Electrochim. Acta, 2013, 108, 321–329 CrossRef CAS.
- E. Jung, H. Shin, B. H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. Hooch Antink, S. Park, K. S. Lee, S. P. Cho, J. S. Yoo, Y. E. Sung and T. Hyeon, Nat. Mater., 2020, 19, 436–442 CrossRef CAS.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Norskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839 RSC.
- J. Park, Y. Nabae, T. Hayakawa and M.-A. Kakimoto, ACS Catal., 2014, 4, 3749–3754 CrossRef CAS.
- F. Yu, M. Zhou, L. Zhou and R. Peng, Environ. Sci. Technol. Lett., 2014, 1, 320–324 CrossRef CAS.
- S. Chen, Z. Chen, S. Siahrostami, T. R. Kim, D. Nordlund, D. Sokaras, S. Nowak, J. W. F. To, D. Higgins, R. Sinclair, J. K. Nørskov, T. F. Jaramillo and Z. Bao, ACS Sustainable Chem. Eng., 2017, 6, 311–317 CrossRef.
- Y.-H. Lee, F. Li, K.-H. Chang, C.-C. Hu and T. Ohsaka, Appl. Catal., B, 2012, 126, 208–214 CrossRef CAS.
- M. H. M. T. Assumpção, R. F. B. De Souza, D. C. Rascio, J. C. M. Silva, M. L. Calegaro, I. Gaubeur, T. R. L. C. Paixão, P. Hammer, M. R. V. Lanza and M. C. Santos, Carbon, 2011, 49, 2842–2851 CrossRef.
- A. Moraes, M. H. M. T. Assumpção, F. C. Simões, V. S. Antonin, M. R. V. Lanza, P. Hammer and M. C. Santos, Electrocatalysis, 2015, 7, 60–69 CrossRef.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS.
- Y. Yang, F. He, Y. Shen, X. Chen, H. Mei, S. Liu and Y. Zhang, Chem. Commun., 2017, 53, 9994–9997 RSC.
- T. Xing, Y. Zheng, L. H. Li, B. C. Cowie, D. Gunzelmann, S. Z. Qiao, S. Huang and Y. Chen, ACS Nano, 2014, 8, 6856–6862 CrossRef CAS.
- L. Han, Y. Sun, S. Li, C. Cheng, C. E. Halbig, P. Feicht, J. L. Hübner, P. Strasser and S. Eigler, ACS Catal., 2019, 9, 1283–1288 CrossRef CAS.
- R. A. Sidik, A. B. Anderson, N. P. Subramanian, S. P. Kumaraguru and B. N. Popov, J. Phys. Chem. B, 2006, 110, 1787–1793 CrossRef CAS.
- D. Iglesias, A. Giuliani, M. Melchionna, S. Marchesan, A. Criado, L. Nasi, M. Bevilacqua, C. Tavagnacco, F. Vizza, M. Prato and P. Fornasiero, Chemistry, 2018, 4, 106–123 CrossRef CAS.
- Y. Sun, S. Li, Z. Jovanov, D. Bernsmeier, H. Wang, B. Paul, X. Wang, S. Kuehl and P. Strasser, ChemSusChem, 2018, 11, 3388–3395 CrossRef CAS.
- K. Zhao, Y. Su, X. Quan, Y. Liu, S. Chen and H. Yu, J. Catal., 2018, 357, 118–126 CrossRef.
- X. Shi, S. Siahrostami, G. L. Li, Y. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. Zheng and J. K. Norskov, Nat. Commun., 2017, 8, 701 CrossRef.
- S. R. Kelly, X. Shi, S. Back, L. Vallez, S. Y. Park, S. Siahrostami, X. Zheng and J. K. Nørskov, ACS Catal., 2019, 9, 4593–4599 CrossRef CAS.
- A. Nadar, S. S. Gupta, Y. Kar, S. Shetty, A. P. van Bavel and D. Khushalani, J. Phys. Chem. C, 2020, 124, 4152–4161 CrossRef CAS.
- V. Viswanathan, H. A. Hansen and J. K. Norskov, J. Phys. Chem. Lett., 2015, 6, 4224–4228 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- Y. Shiraishi, S. Kanazawa, D. Tsukamoto, A. Shiro, Y. Sugano and T. Hirai, ACS Catal., 2013, 3, 2222–2227 CrossRef CAS.
- M. Teranishi, R. Hoshino, S. Naya and H. Tada, Angew. Chem., Int. Ed., 2016, 55, 12773–12777 CrossRef CAS.
- L. Zheng, H. Su, J. Zhang, L. S. Walekar, H. Vafaei Molamahmood, B. Zhou, M. Long and Y. H. Hu, Appl. Catal., B, 2018, 239, 475–484 CrossRef CAS.
- V. Maurino, C. Minero, E. Pelizzetti, G. Mariella, A. Arbezzano and F. Rubertelli, Res. Chem. Intermed., 2007, 33, 319–332 CrossRef CAS.
- C. Chu, D. Huang, Q. Zhu, E. Stavitski, J. A. Spies, Z. Pan, J. Mao, H. L. Xin, C. A. Schmuttenmaer, S. Hu and J.-H. Kim, ACS Catal., 2018, 9, 626–631 CrossRef.
- R. Ma, L. Wang, H. Wang, Z. Liu, M. Xing, L. Zhu, X. Meng and F.-S. Xiao, Appl. Catal., B, 2019, 244, 594–603 CrossRef CAS.
- T. Baran, S. Wojtyła, A. Minguzzi, S. Rondinini and A. Vertova, Appl. Catal., B, 2019, 244, 303–312 CrossRef CAS.
- Y. Isaka, Y. Kondo, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Chem. Commun., 2018, 54, 9270–9273 RSC.
- Y. Isaka, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 5402–5406 CrossRef CAS.
- H. Zhuang, L. Yang, J. Xu, F. Li, Z. Zhang, H. Lin, J. Long and X. Wang, Sci. Rep., 2015, 5, 16947 CrossRef CAS.
- J. Xu, Z. Chen, H. Zhang, G. Lin, H. Lin, X. Wang and J. Long, Sci. Bull., 2017, 62, 610–618 CrossRef CAS.
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS.
- S. Li, G. Dong, R. Hailili, L. Yang, Y. Li, F. Wang, Y. Zeng and C. Wang, Appl. Catal., B, 2016, 190, 26–35 CrossRef CAS.
- R. Wang, X. Zhang, F. Li, D. Cao, M. Pu, D. Han, J. Yang and X. Xiang, J. Energy Chem., 2018, 27, 343–350 CrossRef.
- X. Qu, S. Hu, P. Li, Z. Li, H. Wang, H. Ma and W. Li, Diamond Relat. Mater., 2018, 86, 159–166 CrossRef CAS.
- L. Shi, L. Yang, W. Zhou, Y. Liu, L. Yin, X. Hai, H. Song and J. Ye, Small, 2018, 14 Search PubMed.
- Z. Zhu, H. Pan, M. Murugananthan, J. Gong and Y. Zhang, Appl. Catal., B, 2018, 232, 19–25 CrossRef CAS.
- S. Kim, G.-H. Moon, H. Kim, Y. Mun, P. Zhang, J. Lee and W. Choi, J. Catal., 2018, 357, 51–58 CrossRef.
- J. Tian, T. Wu, D. Wang, Y. Pei, M. Qiao and B. Zong, Catal. Today, 2019, 330, 171–178 CrossRef CAS.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS.
- Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, ChemCatChem, 2018, 10, 2070–2077 CrossRef CAS.
- L. Yang, G. Dong, D. L. Jacobs, Y. Wang, L. Zang and C. Wang, J. Catal., 2017, 352, 274–281 CrossRef CAS.
- S. Zhao, X. Zhao, S. Ouyang and Y. Zhu, Catal. Sci. Technol., 2018, 8, 1686–1695 RSC.
- S. Zhao, X. Zhao, H. Zhang, J. Li and Y. Zhu, Nano Energy, 2017, 35, 405–414 CrossRef CAS.
- S. Zhao and X. Zhao, J. Catal., 2018, 366, 98–106 CrossRef CAS.
- S. Zhao and X. Zhao, Appl. Catal., B, 2019, 250, 408–418 CrossRef CAS.
- R. Wang, K. Pan, D. Han, J. Jiang, C. Xiang, Z. Huang, L. Zhang and X. Xiang, ChemSusChem, 2016, 9, 2470–2479 CrossRef CAS.
- S. Zhao, T. Guo, X. Li, T. Xu, B. Yang and X. Zhao, Appl. Catal., B, 2018, 224, 725–732 CrossRef CAS.
- X. Chang, J. Yang, D. Han, B. Zhang, X. Xiang and J. He, Catalysts, 2018, 8, 147 CrossRef.
- G. Zuo, S. Liu, L. Wang, H. Song, P. Zong, W. Hou, B. Li, Z. Guo, X. Meng, Y. Du, T. Wang and V. A. L. Roy, Catal. Commun., 2019, 123, 69–72 CrossRef CAS.
- Y. Peng, L. Wang, Y. Liu, H. Chen, J. Lei and J. Zhang, Eur. J. Inorg. Chem., 2017, 4797–4802 CrossRef CAS.
- S. Gogoi and N. Karak, Nano-Micro Lett., 2017, 9, 40 CrossRef.
- B. Weng, J. Wu, N. Zhang and Y. J. Xu, Langmuir, 2014, 30, 5574–5584 CrossRef CAS.
- C. Wang, M. Long, B. Tan, L. Zheng, J. Cai and J. Fu, Electrochim. Acta, 2017, 250, 108–116 CrossRef CAS.
- R. Cai, Y. Kubota and A. Fujishima, J. Catal., 2003, 219, 214–218 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- L. Han, X. Y. Yu and X. W. Lou, Adv. Mater., 2016, 28, 4601–4605 CrossRef CAS.
- M. Wen, K. Mori, Y. Kuwahara and H. Yamashita, ACS Energy Lett., 2016, 2, 1–7 Search PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS.
- M. Wen, K. Mori, T. Kamegawa and H. Yamashita, Chem. Commun., 2014, 50, 11645–11648 RSC.
- Y. Kuwahara, H. Kango and H. Yamashita, ACS Sustainable Chem. Eng., 2016, 5, 1141–1152 CrossRef.
- J. Collén, M. J. Del Rio, G. García-Reina and M. Pedersén, Planta, 1995, 196, 225–230 CrossRef.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS.
- D. Zheng, X. N. Cao and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 11512–11516 CrossRef CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef.
- Y. He, L. Zhang, B. Teng and M. Fan, Environ. Sci. Technol., 2015, 49, 649–656 CrossRef CAS.
- P. Niu, Y. Yang, J. C. Yu, G. Liu and H. M. Cheng, Chem. Commun., 2014, 50, 10837–10840 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS.
- H. Zhang, L. Zhao, F. Geng, L.-H. Guo, B. Wan and Y. Yang, Appl. Catal., B, 2016, 180, 656–662 CrossRef CAS.
- J. Sun and Y. Wu, Angew. Chem., Int. Ed., 2020, 59, 10904–10908 CrossRef CAS.
- J. Sun, Y. Yu, A. E. Curtze, X. Liang and Y. Wu, Chem. Sci., 2019, 10, 5519–5527 RSC.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, ACS Energy Lett., 2016, 1, 913–919 CrossRef CAS.
- N. U. Day and C. C. Wamser, J. Phys. Chem. C, 2017, 121, 11076–11082 CrossRef CAS.
- M. Gryszel, A. Markov, M. Vagin and E. D. Głowacki, J. Mater. Chem. A, 2018, 6, 24709–24716 RSC.
- X. Zong, H. Chen, B. Seger, T. Pedersen, M. S. Dargusch, E. W. McFarland, C. Li and L. Wang, Energy Environ. Sci., 2014, 7, 3347–3351 RSC.
- G. S. Calabrese and M. S. Wrighton, J. Electrochem. Soc., 1981, 128, 1014–1018 CrossRef CAS.
- G. S. Calabrese, R. M. Buchanan and M. S. Wrighton, J. Am. Chem. Soc., 1983, 105, 5594–5600 CrossRef CAS.
- O. Jung, M. L. Pegis, Z. Wang, G. Banerjee, C. T. Nemes, W. L. Hoffeditz, J. T. Hupp, C. A. Schmuttenmaer, G. W. Brudvig and J. M. Mayer, J. Am. Chem. Soc., 2018, 140, 4079–4084 CrossRef CAS.
- W. Fan, B. Zhang, X. Wang, W. Ma, D. Li, Z. Wang, M. Dupuis, J. Shi, S. Liao and C. Li, Energy Environ. Sci., 2020, 13, 238–245 RSC.
- A. S. Konev, M. Y. Kayumov, M. P. Karushev, Y. V. Novoselova, D. A. Lukyanov, E. V. Alekseeva and O. V. Levin, ChemElectroChem, 2018, 5, 3138–3142 CrossRef CAS.
- I. Papagiannis, E. Doukas, A. Kalarakis, G. Avgouropoulos and P. Lianos, Catalysts, 2019, 9, 243 CrossRef.
- F. Ye, T. Wang, X. Quan, H. Yu and S. Chen, Chem. Eng. J., 2020, 389, 123427 CrossRef CAS.
- K. Fuku, Y. Miyase, Y. Miseki, T. Funaki, T. Gunji and K. Sayama, Chem. – Asian J., 2017, 12, 1111–1119 CrossRef CAS.
- X. Shi, Y. Zhang, S. Siahrostami and X. Zheng, Adv. Energy Mater., 2018, 8, 1801158 CrossRef.
- K. Fuku, Y. Miyase, Y. Miseki, T. Gunji and K. Sayama, RSC Adv., 2017, 7, 47619–47623 RSC.
- J. Zhang, X. Chang, Z. Luo, T. Wang and J. Gong, Chem. Commun., 2018, 54, 7026–7029 RSC.
- Y. Miyase, S. Takasugi, S. Iguchi, Y. Miseki, T. Gunji, K. Sasaki, E. Fujita and K. Sayama, Sustainable Energy Fuels, 2018, 2, 1621–1629 RSC.
- T. Baran, S. Wojtyła, A. Vertova, A. Minguzzi and S. Rondinini, J. Electroanal. Chem., 2018, 808, 395–402 CrossRef CAS.
- T. H. Jeon, H. Kim, H.-I. Kim and W. Choi, Energy Environ. Sci., 2020, 13, 1730–1742 RSC.
- K. Fuku, Y. Miyase, Y. Miseki, T. Gunji and K. Sayama, ChemistrySelect, 2016, 1, 5721–5726 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |