
Novel fluorescent amphiphilic copolymer probes containing azo-tetraphenylethylene bridges for azoreductase-triggered release†
Xiaojie
Yuan
a,
Zhe
Wang
a,
Lishan
Li
a,
Jiawei
Yu
a,
Yuqing
Wang
a,
Hongkun
Li
 a,
Jiandong
Zhang
a,
Zhengbiao
Zhang
*a,
Nianchen
Zhou
*a and
Xiulin
Zhu
ab
a,
Jiandong
Zhang
a,
Zhengbiao
Zhang
*a,
Nianchen
Zhou
*a and
Xiulin
Zhu
ab
aSuzhou Key Laboratory of Macromolecular Design and Precision Synthesis, Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, State and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou Industrial Park, Suzhou 215123, China. E-mail: nczhou@suda.edu.cn; zhangzhengbiao@suda.edu.cn
bGlobal Institute of Software Technology, No 5. Qingshan Road, Suzhou National Hi-Tech District, Suzhou 215163, China
First published on 10th April 2019
Abstract
Enzyme-sensitive amphiphilic polymers play an important role as smart materials in drug delivery and biological detection. Azobenzene, an azoreductase active site, has received significant attention in recent years. Herein, we synthesized a novel aggregation-induced emission (AIE) fluorescent probe of amphiphilic block copolymer PCL-TPE-Azo-PEG based on an azoreductase response, in which tetraphenylethylene (TPE) conjugated to an azo group provides the junction between the hydrophilic and hydrophobic (PEG and PCL) chain segments. In phosphate buffer solution, the resulting amphiphilic polymers self-assembled into rod-like micelles that are non-fluorescent owing to the quenching effect of the azo moieties on the AIE activity of TPE. In the presence of a reducing agent (such as azoreductase or Na2S2O4), the azo bond was broken and the assemblies fragmented into PEG and PCL chain segments. Following disassembly of the micelles, the AIE effect of TPE was activated because the TPE moiety is encapsulated in the resulting PCL aggregation. Furthermore, the amphiphilic polymer could encapsulate drugs, such as DOX, to form drug-loaded micelles in PB solution. And the micelles indicated an increasing fluorescence with drug release under the action of reducing agent. This amphiphilic block copolymer could potentially be applied in biosensing and controlled drug delivery for colonic conditions.
Introduction
Stimuli-responsive materials can undergo changes in their structures or states in response to external stimuli, such as light, temperature, pH, ionic strength, and enzymes, amongst others. In the last few decades, stimuli-responsive polymeric materials have attracted significant attention in sensing, drug delivery, and biotechnology owing to their versatile and dynamic material properties.1–8 Developing controlled assembly/disassembly polymeric micellar nanostructures is becoming increasingly important in order to fulfill the requirements of a wide range of smart materials.1Enzymes play an important role in living organisms. The concentrations of various enzymes in diseased cells and tissues can be higher than normal, leading to high activity. As a result, the enzyme response at a specific site could be used to trigger drug delivery and controlled release under physiological conditions.9,10 Currently a popular approach is to encapsulate the drug in polymeric nano-assemblies and then disassemble the delivery carriers to afford release.
Azobenzene compounds not only have a well-known light response due to reversible cis–trans isomerization but are also redox-responsive because the azo double bond can be disrupted under reducing conditions (such as in the presence of azoreductase, Na2S2O4, or hydrazine). Azoreductase is primarily found in the human intestine and plays an important role in a number of processes. Azobenzene is an artificial enzyme active site that is attracting increasing research interest.11–25
In recent years, azoreductase-responsive amphiphilic block copolymers have attracted significant attention owing to their facile self-assembly in selected solvents to form nanoparticles with various morphologies.11–19 Khan et al. successfully prepared a series of nanoparticles composed of diblock copolymers with azobenzene side chains or as a bridge.16,17,21 Gao et al. prepared polymeric vesicles with good stability through the self-assembly of poly(glycidyl methacrylate) with grafted hydrophobic azobenzene and hydrophilic cyclodextrins. The azo bond was broken by Na2S2O4 in a simulated colon chemistry environment, leading to the release of bovine serum protein from the cargo-loaded vesicles.14
Fluorescent probes have shown to have an important role in the fields of bioimaging and biosensing owing to their efficiency and sensitivity.5,19,20,22–27 The emergence of enzyme-responsive fluorescent probes for drug delivery, the identification of enzymes, and biological imaging, has excited interest for various applications.20,22–28 In recent years, fluorescence switches derived from azobenzene have attracted significant attention for their interesting stimuli-responsive properties. A useful design strategy is the use of pro-fluorophores containing an azo moiety as a fluorophore quenching unit, based on the concept of fluorescence resonance energy transfer (FRET) or the ultrafast azo-bond rotation mechanism. Fluorescence activation of the pro-fluorophores takes place through reductive cleavage of the azo moieties under stimulated redox conditions.22–24,28–33 Some azo-based small molecule fluorescence probes have been studied and have shown promise for application in the fields of biosensing and bioimaging. Renard et al. modified a bio-reductive probe suitable for the detection of azoreductases by diazo coupling to connect azobenzene to rhodamine 110 dye (Rh110). Hanaoka et al.22,24,30 designed and synthesized a library of fluorescent probes based on azo rhodamine derivatives.
Over the last decade, fluorescent probes based on the aggregation-induced emission (AIE) effect have been successfully applied to in vivo cell imaging and enzyme activity determination.34,35 Tetraphenylethylene (TPE), a typical AIE chromophore, has been widely investigated owing to its high quantum yield and facile synthesis. Recently, He et al. prepared self-assembling aggregates containing azobenzene and TPE moieties through azo coupling reaction-induced macromolecular self-assembly, with enzyme-triggered fluorescent switching behaviors based on the FRET concept.23
Compared with small molecules, the molecular size of polymers can effectively reduce the absorption of the probe molecules, giving the polymer a higher cargo loading. Therefore, macromolecular fluorescent probes based on stimulus response have a broad range of potential applications. Various polymeric assembly-based azo reductions have been documented. However, to our knowledge, reports are limited for azo polymeric nanoparticles, either as controlled delivery carriers or biosensors.
In this work, we designed and prepared a small molecule pro-fluorophore with a TPE-azo moiety based on azo reduction response and AIE, in which an azobenzene group is directly conjugated to the TPE. This small molecule was then used as a scaffold for the fabrication of a macromolecular fluorescence probe. The amphiphilic block copolymer PCL3k-TPE-Azo-PEG5k, which had a bridging TPE-azo moiety to connect the hydrophilic and hydrophobic chain segments. It was efficiently produced using a Cu-catalyzed azide/alkyne cycloaddition (CuAAC) “click” reaction by connecting hydrophilic segments (PEG) and hydrophobic segments (PCL) to the small molecule. The synthetic route to PCLn-TPE-Azo-PEGm is outlined in Scheme 1. The resulting amphiphilic block copolymer could self-assemble in PB solution to form rod-like micelles, which were non-fluorescent as the azo moieties quenched the TPE fluorescence. In the presence of the reducing agent, the azo bond was broken, resulting in disassembly of the micelles. The AIE effect of TPE was activated because the TPE moiety after the broken azo bond was encapsulated in the regenerating PCL aggregation. Furthermore, the amphiphilic polymer was able to encapsulate drugs such as DOX, to form drug-loaded micelles, resulting in an increased fluorescence with drug release. Azoreductase exists predominantly in the human colon, and therefore this amphiphilic block copolymer has shown potential for application in biosensing and controlled drug release in the colon.
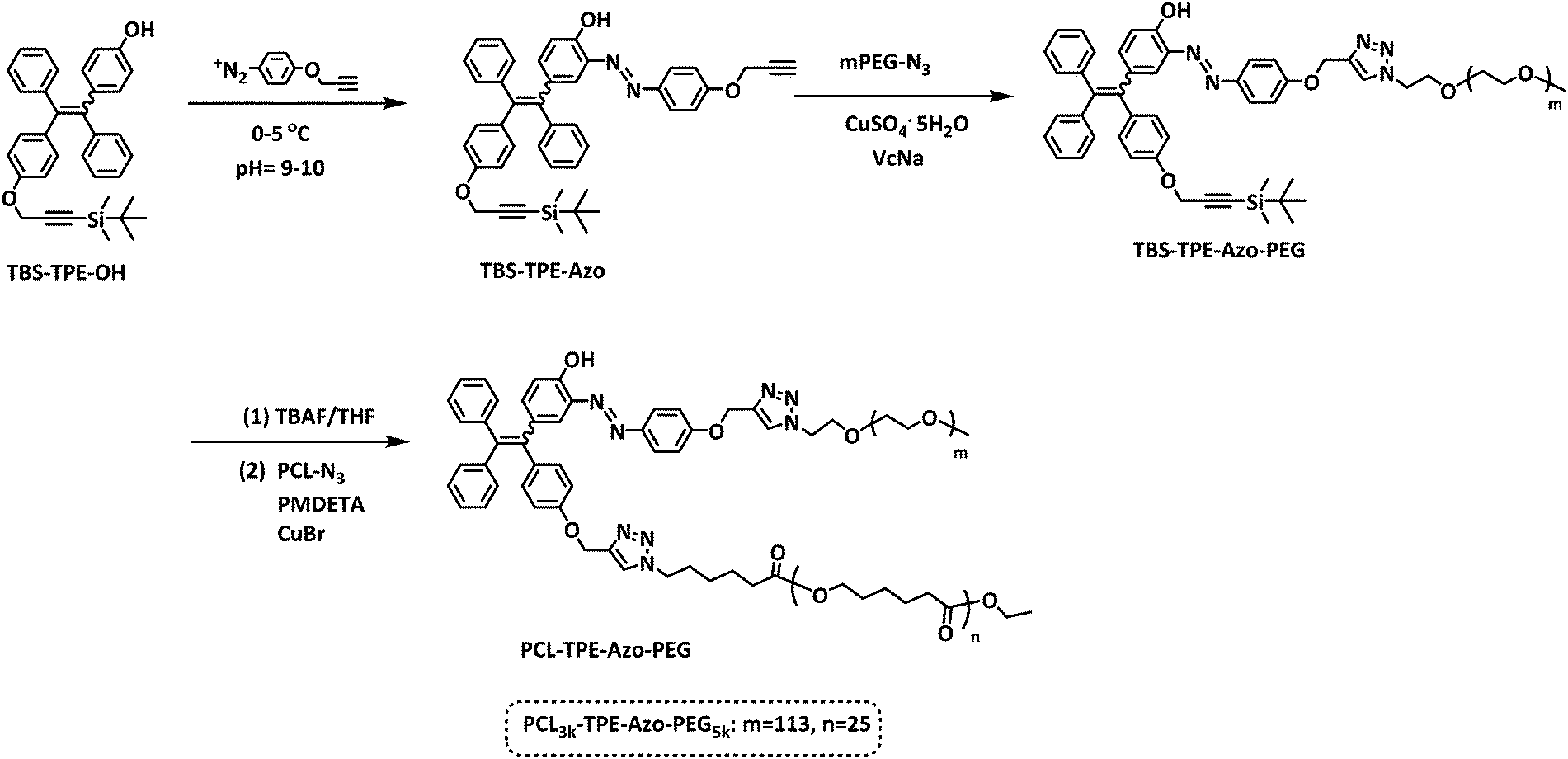 | ||
| Scheme 1 Synthetic routes of the block copolymer PCLn-TPE-Azo-PEGm. | ||
Materials
N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine (PMDETA; 98%, J&K) was dried with 4 Å molecular sieves and then distilled under vacuum. Copper(I) bromide (CuBr) was purified by washing with acetic acid, water, and ethanol, dried under vacuum. Zinc dust (99.99% trace metals basis, 600 mesh, Aladdin), 4-hydroxylbenzophenone (98%, Energy Chemical), titanium tetrachloride (TiCl4, AR, Enox), tetrabutylammonium fluoride (TBAF; 1.0 mol L−1 in tetrahydrofuran (THF), Energy Chemical), tetrahydrofuran (THF, AR, Enox), dimethylformamide (DMF; AR, Enox), polyethylene glycol monomethyl ether mesylate with Mn of 5000 g mol−1 Sigma-Aldrich), sodium azide (99.5%, Aldrich), 6-bromohexanoylchloride, and 3-bromo-1-(tert-butyldimethylsilyl)-1-propyne were used as received. ε-Caprolactone (ε-CL, AR, Aladdin) and stannous octoate (Sn(Oct)2, 95%, Aldrich) were purified using distillation from calcium hydride under reduced pressure prior to use. Ethanol, (95+%, Enox) was purified by distillation from anhydrous MgSO4 under reduced pressure prior to use. Nile Red was purchased from J&K. Doxorubicin hydrochloride (DOX·HCl, 99%) was purchased from Sigma. Azoreductase (DT-diaphorase human (NQO 1)) and coenzyme NADPH (β-nicotinamide adenine dinucleotide phosphate reduced tetra(cyclohexylammonium) salt) were purchased from Sigma-Aldrich. The synthetic route to the new copolymer is shown in Scheme 1. The syntheses of mPEG5k-N3 and PCL3k-N3 are given in the ESI.†Synthesis of TBS-protected-alkyne-azobenzene unimer TBS-TPE-Azo
The coupling reaction was as follows: TBS-TPE-OH (3.10 g, 6.00 mmol) and 10 mL tetramethylene oxide (THF) were added to a 25 mL beaker. A solution of the diazonium salt of 4-(prop-2-ynyloxy)aniline (al-N2, synthesis given in the ESI†) was then added to the beaker dropwise, slowly with stirring at 0–5 °C. The NaOH solution was then added to adjust the pH of the coupling solution to 9–10. The final mixture was stirred for another 4 h and then extracted with ethyl acetate. The organic layer was dried using anhydrous Na2SO4 and evaporated under vacuum. The crude product was purified by column chromatography on silica gel (ethyl acetate/petroleum ether (V/V = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20–1:25) to obtain TBS-TPE-Azo (0.67 g, 17%). This compound was characterized by 1H NMR. 1H NMR (300 MHz, CDCl3): δH (TMS, ppm) = 7.80–7.75 (dd, 2H), 7.57–7.54 (dd, 1H), 7.12–6.92 (m, 15H), 6.76–6.70 (m, 3H), 4.76 (d, 2H), 4.61 (d, 2H), 2.55 (t, 1H), 0.90–0.87 (d, 9H), 0.09–0.07 (d, 6H) (Fig. S2, ESI†).
:20–1:25) to obtain TBS-TPE-Azo (0.67 g, 17%). This compound was characterized by 1H NMR. 1H NMR (300 MHz, CDCl3): δH (TMS, ppm) = 7.80–7.75 (dd, 2H), 7.57–7.54 (dd, 1H), 7.12–6.92 (m, 15H), 6.76–6.70 (m, 3H), 4.76 (d, 2H), 4.61 (d, 2H), 2.55 (t, 1H), 0.90–0.87 (d, 9H), 0.09–0.07 (d, 6H) (Fig. S2, ESI†).
Synthesis of PCL3k-TPE-Azo-PEG5k
A typical synthesis of PCL3k-TPE-Azo-PEG5k was as follows: a mixture of 448 mg TPE-Azo-PEG5k and 120 mg PCL3k-N3 in 6 mL of dried anhydrous toluene was added to a 25 mL Schlenk flask. 42 mg of CuBr and 72 μL of PMDETA were then added to the flask. The mixture was degassed using three freeze/pump/thaw cycles and stirred for 24 h at 60 °C. Subsequently, the solution was stirred with ion exchange resin for 24 h to remove the copper salts, then filtered and evaporated under reduced pressure to obtain the concentrated product. The concentrated solution was then slowly dripped into cold anhydrous ether. An orange solid was obtained after drying under high vacuum. The crude product was further purified with Pre-SEC to obtain the amphiphilic copolymer (yield: 28%, Mn,NMR = 8900 Da, Mn,SEC = 12100 Da, Mw/Mn = 1.02). 1H NMR (300 MHz, CDCl3): δH (TMS, ppm) = 8.23 (s, 1H), 7.80–7.75 (dd, 2H), 7.57–7.54 (dd, 1H), 7.12–6.96 (m, 15H), 6.78–6.70 (dd, 3H), 5.43 (s, 2H), 5.27 (s, 2H), 4.67 (t, 2H), 4.57 (dd, 2H), 4.08–4.04 (dt, 50H), 3.90 (t, 2H), 3.70–3.60 (m, 452H), 3.38 (s, 3H), 2.33–2.28 (t, 50H), 1.70–1.60 (m, 96H), 1.43–1.33 (m, 50H), 1.28–1.23 (m, 3H) (Fig. S4, ESI†).
Self-assembly of PCL3k-TPE-Azo-PEG5k in PB (phosphate buffered) solution (pH = 7.4)
Self-assembly was typically conducted as follows: the PCL3k-TPE-Azo-PEG5k amphiphilic polymer was first dissolved in DMF at an initial concentration of 5 mg mL−1. PB solution (50 mM, pH 7.4) was then added to the solution at a rate of 0.2 mL h−1 with stirring steadily to obtain the stable micelles. After dialyzing the micelles against PB solution (50 mM, pH 7.4) using dialysis tubing (MWCO 3500) to remove the organic solvent, transmission electron microscopy (TEM) and dynamic light scattering (DLS) were used to characterize the micelle morphology.Determination of the critical aggregation concentration (CAC)
Nile red, a hydrophobic fluorescent dye, was selected as the fluorescent probe for determination of the critical aggregation concentration (CAC) of the amphiphilic block copolymer. By measuring the fluorescence intensity of Nile red in a series of different concentrations of micelle solution (1.0 × 10−3–1.0 × 10−1 mg mL−1), the CAC could be easily established. The final concentration of Nile red in each sample was maintained at 5 × 10−6 M. The fluorescence emission at 580–700 nm was monitored with excitation at 550 nm. The CAC was estimated at the intersection point by extrapolating the intensity I630 of the low and high concentration zones.Reduction response and fluorescence variation of micelles
The enzyme azoreductase (DT-diaphorase, human) was dispersed in PB (phosphate buffer) solution (1 mg mL−1, pH 7.2–7.4). Then the solution (0.2 mL) was added into freshly prepared self-assembled solution (1.0 mL) of PCL3k-TPE-Azo-PEG5k in PBS. After that, the reduction reaction was initiated by the addition of co-enzyme NADPH solution (1.0 mL) in PBS (1 mg mL−1). The reaction mixture was stirred at 37 °C for 24 h to 48 h under argon atmosphere and placed at room temperature for 3 days. Meanwhile, Na2S2O4 was also used to break the azo bonds. Under anaerobic conditions, 0.5 mg of Na2S2O4 was added to 3 mL of self-assembled micelles in PB solution (0.3 mg mL−1) at 37 °C.Fluorescence spectrophotometry was then used to monitor the change in fluorescence intensity of the micelle solution. The effective breaking of azo bonds in the assembly was demonstrated by TEM and UV-vis spectroscopy.
DOX loading of micelles
The following process was carried out in the dark. A mixture of 5.0 mg DOX·HCl and 3.6 μL of triethylamine (TEA) in 1.0 mL of dimethyl sulfoxide (DMSO) was added to a 2 mL ampoule. After stirring overnight, excess TEA was removed by rotary evaporation to obtain the hydrophobic DOX solution. 4 mL of PB solution (50 mM, pH 7.4) was added dropwise to the mixture of copolymer in DMF (1.0 mL, 5 mg mL−1) and DOX base solution (50 μL, 5.0 mg mL−1) with stirring at room temperature. Afterwards, in order to remove both unencapsulated DOX and the organic solvent, the mixture was dialyzed against PB solution (50 mM, pH 7.4) for 24 h.The amount of DOX was determined by fluorescence (FL4600) measurement (excitation at 480 nm). Firstly, a calibration curve was obtained by measuring the fluorescence intensity of DOX/DMSO solutions with different concentrations. Secondly, the fluorescence intensity of DOX-loaded micelles dissolved in DMSO was analyzed. The amount of DOX loaded in the micelles could be determined using the calibration curve.
The drug loading content (DLC) and drug loading efficiency (DLE) were calculated using the following formulas:
| DLC (wt%) = (weight of loaded drug/weight of (polymer + loaded drug)) × 100% |
| DLE (wt%) = (weight of loaded drug/weight of drug in feed) × 100% |
Cytotoxicity tests
The cytotoxicity of the blank micelles was evaluated using Caco2 human colon cancer cells and the MTT method. The (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT reagent) was added to the Caco2 human colon cancer cells. The cells were then incubated with blank micelles with a range of concentrations from 10 to 200 μg mL−1 for 72 h.Characterization
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker 300 MHz nuclear magnetic resonance (NMR) instrument using CDCl3 or DMSO-d6 as the solvent with tetramethylsilane (TMS) as the internal standard. The 1H NMR spectra were referenced to δ 7.26 ppm in CDCl3 or δ 2.50 ppm in DMSO-d6. The number average molecular weight (Mn) and molecular weight distribution (Mw/Mn) of the obtained polymers were determined using a size exclusion chromatograph (SEC) TOSOH HLC-8320 equipped with both refractive-index and UV detectors. Two TSKgel Super Mutipore HZ-N (3 μm bead size) columns with a molecular weight range of 500–190000 Daltons were used, calibrated with narrow-distribution PS standard samples. THF was used as the eluent at a flow rate of 0.35 mL min−1 operated at 40 °C. An Agilent PL-50 reparative SEC system equipped with a manual injector was used to purify the crude polymers. THF was used as the eluent at a flow rate of 3 mL min−1. Separations were obtained using a PLgel 10 μm MIXED-D, 300 × 25 mm preparative SEC column maintained at 40 °C. Each composition was determined using the TOSOH HLC-8320 SEC column as described above. Fourier transform-infrared (FT-IR) spectra were recorded on a Bruker TENSOR-27 FT-IR spectrometer using the KBr disc technique. Ultraviolet visible (UV-vis) absorption spectra of the polymer solution were measured on a Shimadzu UV-2600 (Kyoto, Japan). The fluorescence emission spectra were obtained on a HITACHI F-4600 fluorescence spectrophotometer at room temperature. The TEM images were recorded on a HITACHI HT7700 TEM operating at a voltage of 120 kV. Before measurement, the samples were prepared by dropping the freshly dialyzed assembly solution onto a carbon-covered copper grid. After drying in air at room temperature, the samples were negatively stained with 1.0 wt% phosphotungstic acid. Hydrodynamic diameter (Dh) was determined by DLS using a Brookhaven NanoBrook 90Plus PALS instrument at 25 °C with a scattering angle of 90°.
Results and discussion
Optimized configurations and UV-vis spectra of TBS-TPE-Azo
To better understand the novel azobenzene-containing TPE-azo, in which an azo group is directly conjugated to the TPE, we simulated and calculated the optimized structures of the cis/trans isomers of TBS-TPE-Azo and their dipole moments and energies. The details are described in the ESI† (Scheme S2, Fig. S1, Tables S1 and S2). The UV-vis spectrum of TBS-TPE-Azo in THF was contrasted with that of TPE-2OH (Fig. S3, ESI†). Following the conjugation of the azo moiety, the absorption peak in TBS-TPE-Azo, corresponding to TPE, underwent a red shift from 320 nm to 360 nm. This shift was not observed for TPE-2OH because of the extension of the conjugate structure.Fluorescence characterization of TBS-TPE-Azo
The fluorescence of TBS-TPE-Azo was investigated in detail by contrasting with TPE-2OH. As shown in Fig. 1(a), when the initial solutions of both TPE-2OH and TBS-TPE-Azo in THF were excited at 360 nm, no fluorescence was detected because TPE is non-fluorescent in good solvents. However, as water was added to the THF solution (the water content reached 90% in the THF/water mixture), the TPE-2OH showed strong fluorescence emission around 450–500 nm owing to the AIE activity of TPE in the aggregated state. On the contrary, the TBS-TPE-Azo still showed no fluorescence, indicating no AIE behavior.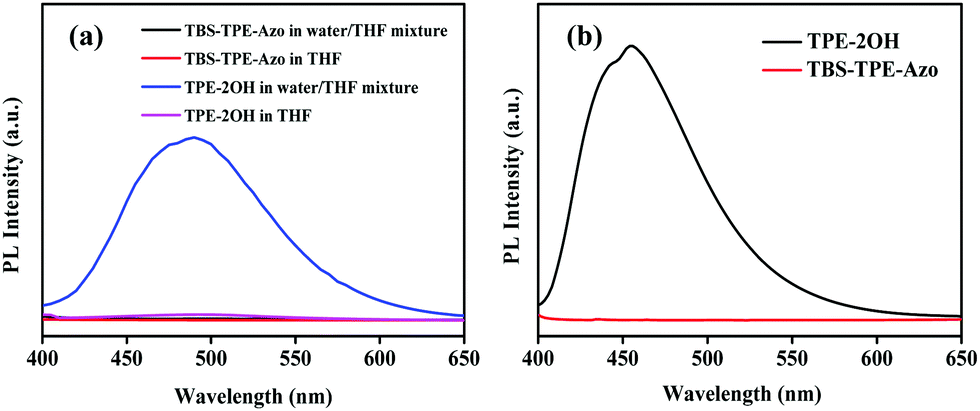 | ||
| Fig. 1 (a) Fluorescence spectra of TBS-TPE-Azo and TPE-2OH in THF and water/THF mixture with a water fraction of 90% (λex = 360 nm). All concentrations were 5 × 10−5 mol L−1. (b) Fluorescence emission spectra of TBS-TPE-Azo and TPE-2OH in the solid state (λex = 395 nm). | ||
It is well known that the azobenzene chromophore is non-fluorescent in solution owing to ultrafast conformational changes around the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond after photoexcitation.22,28,31 However, when azobenzene chromophores are conformationally strained, such as in aggregates or when ring-strained, hampering their main nonradiative relaxation, azobenzene compounds can show intense fluorescence emission.36–39 To better understand the mechanism for the azo group quenching to TPE fluorescence in the aggregated state, we studied the emissions of TBS-TPE-Azo and TPE-2OH in the solid state (Fig. 1(b)). No TBS-TPE-Azo fluorescence was observed. We speculate that no fluorescence emission of TBS-TPE-Azo in the solid state or aggregated state may be related to the mechanisms of intramolecular photo-induced electron transfer or intramolecular electronic energy transfer. More investigation is needed to determine the exact reason.40–44
N bond after photoexcitation.22,28,31 However, when azobenzene chromophores are conformationally strained, such as in aggregates or when ring-strained, hampering their main nonradiative relaxation, azobenzene compounds can show intense fluorescence emission.36–39 To better understand the mechanism for the azo group quenching to TPE fluorescence in the aggregated state, we studied the emissions of TBS-TPE-Azo and TPE-2OH in the solid state (Fig. 1(b)). No TBS-TPE-Azo fluorescence was observed. We speculate that no fluorescence emission of TBS-TPE-Azo in the solid state or aggregated state may be related to the mechanisms of intramolecular photo-induced electron transfer or intramolecular electronic energy transfer. More investigation is needed to determine the exact reason.40–44
Synthesis and characterization of amphiphilic block copolymer PCL3k-TPE-Azo-PEG5k
Based on the fluorescence characterization of TBS-TPE-Azo, we used TBS-TPE-Azo as a scaffold to fabricate a macromolecular fluorescent probe. This probe was based on AIE as the fluorescence would be “on” when the azo bond was broken by reduction in the aggregated state. As shown in Scheme 1, we first explored the synthesis of TBS-TPE-Azo using the traditional method of diazo coupling. TBS-TPE-Azo has two alkyne end groups to facilitate easy reaction with various amphiphilic block polymers that have different hydrophilic and hydrophobic segments. PCL3k-TPE-Azo-PEG5k was obtained via CuAAC reaction of mPEG5k-N3 and PCL3k-N3 with TBS-TPE-Azo in succession.1H NMR, FT-IR and SEC curves conformed the successful synthesis of the amphiphilic block copolymers (Fig. S4 and S5, ESI,†Fig. 2). Symmetric SEC elution curves and a significant shift of the SEC curves of both PCL3k-N3 and TPE-Azo-PEG5k to PCL3k-TPE-Azo-PEG5k were observed (Fig. 2). In addition, the disappearance of the azide peak at 2100 cm−1 and alkynyl peak at 3300 cm−1 was observed by FT-IR (Fig. S5, ESI†), which suggested that the click reaction had successfully completed. The molecular weight of PCL3k-TPE-Azo-PEG5k and the block ratio of the hydrophilic chain segment (m) and hydrophobic chain segment (n), were obtained by 1H NMR (Fig. S4, ESI†) and eqn (S1), (S2) and (S3) (ESI†), m/n = 113/25.
 | ||
| Fig. 2 SEC traces of PCL3k-TPE-Azo-PEG5k, PCL3k-N3 and TPE-Azo-PEG5k. | ||
Self-assembly of PCL3k-TPE-Azo-PEG5k in PB solution (pH = 7.4)
The self-assembly of PCL3k-TPE-Azo-PEG5k was initiated in a mixture of DMF and water by carefully tuning the experimental parameters. The critical aggregation concentration (CAC) in PB solution was determined using the hydrophobic dye Nile red as a fluorescent probe, based on a previously reported method,18 and was found to be about 32 mg L−1 for PCL3k-TPE-Azo-PEG5k (Fig. S6, ESI†). The size and morphology of the self-assembled aggregates formed by the amphiphilic block copolymer in PB solution, were characterized by TEM (Fig. 3(a) and Fig. S8(a), ESI†) and DLS (Fig. S7 and S9(a), ESI†). As shown in the TEM images, the amphiphilic block copolymer formed stable nano-rod micelles. | ||
| Fig. 3 TEM images of (a) PCL3k-TPE-Azo-PEG5k micelles before azoreductase treatment and (b) after azoreductase treatment for 48 h. | ||
Reduction response of micelles and reductant-activated AIE fluorescence
Based on the literatures,11,17 enzyme azoreductase can fully induce the cleavage of the azobenzene units in micelles solution of the amphiphilic diblock copolymer with an azobenzene linkage. In this work, azoreductase and dithionate sodium hydrosulfite (Na2S2O4) were used as the reducing agent to investigate the reduction responsiveness and activated-AIE fluorescence of self-assembled micelles of PCL3k-TPE-azo-PEG5k, respectively. The morphologies and sizes of the degradation products above were detected by TEM and DLS (Fig. 3(b) and Fig. S7, S8(b), S9(b), ESI†). AS shown in Fig. 3(b), the short rod-like micelles were dissociated and regenerated spherical aggregates of diameter about 40–60 nm and larger aggregates as a result of the interruption of the azo bond. The hydrophobic block PCL re-aggregated into spherical aggregates after the azo bond fracture. The aggregates further combined to form large aggregates due to the lack of hydrophilic block to stabilize the aggregates. The aggregates precipitated after being placed for a period of time. It can also be verified from the DLS data (Fig. S7, ESI†), which show obvious difference between the sizes of aggregates before and after the enzymatic hydrolysis.Fluorescence intensity was measured at different time intervals after the addition of the reducing agent (excitation wavelength, 360 nm). The intensity of the fluorescence emission of micelle solution around 500 nm increased significantly after the treatment of reducing agent (Fig. 4(a and c)). By tracking the UV-vis spectra of the enzyme degradation solution, the absorption peaks of the azobenzene of TPE-Azo at around 365 nm distinctly weakened after reduction for 48 h (Fig. 4(b)). The results make it clear that azoreductase is capable of efficiently cleaving the azo bonds of PCL-TPE-Azo-PEG in micelles solution, led to the significant increase in fluorescent emission. Nevertheless, the weaker absorption peak around 365 nm was still observed from the enzyme degradation product of micelles solution in UV-vis spectra. This should be attributed to small amounts of incomplete reduction of azo bonds from TPE-azo after the enzymolysis. Contrasting UV-vis spectra of the degradation product of micelles solution in the presence of Na2S2O4 (Fig. 4(d)), the absorption peaks of azobenzene of TPE-Azo completely disappeared after reduction reaction. The disappearance of azobenzene indicated that the reducing agent of small molecule such as Na2S2O4 was more effective for the azo bond reduction compared with macromolecular enzyme.
 | ||
| Fig. 4 (a) Fluorescence intensity variation for the micelles of PCL3k-TPE-Azo-PEG5k in PB solution before (black) and after (red) the enzyme treatment for 48 h in the presence of azoreductase (0.16 mg mL−1 of PCL3k-TPE-Azo-PEG5k in PB solution, at 37 °C), λex = 360 nm. (b) UV-vis spectra of the micelles of PCL3k-TPE-Azo-PEG5k in PB solution before (black) and after (red) the enzyme treatment. (c) Fluorescence intensity variation for the micelles of PCL3k-TPE-Azo-PEG5k in PB solution before and after the Na2S2O4 treatment for 24 h (0.1 mg mL−1 of PCL3k-TPE-Azo-PEG5k, at 37 °C). (d) UV-vis spectra of the micelles of PCL3k-TPE-Azo-PEG5k in PB solution (0.1 mg mL−1) before (black line) and after (red) the Na2S2O4 treatment for 24 h. | ||
In order to explore the efficiency of the enzymatic catalytic action on the self-assembling micelles of PCL3k-TPE-Azo-PEG5k, control experiments, i.e., the enzymolysis of aqueous solution of TPE-Azo-PEG5k containing PEG hydrophilic segments were conducted under equal conditions as the enzymolysis of micelles solution. The variation of fluorescence intensity and absorption change before and after the enzyme treatment were measured by fluorescence and UV-vis spectra, as shown in Fig. S10 (ESI†). The characteristic peaks of azobenzene in TPE-Azo after the enzyme treatment was also observed in UV-vis spectra, similar to the results of enzyme treatment of the PCL3k-TPE-Azo-PEG5k micellar solution. It was speculated that the direct connection of the propeller-like and bulky TPE moiety to azo bond could lead to a larger steric hindrance.
Reduction-triggered drug release and fluorescence changes
The reduction-triggered structural transformation gives rise to potential applications as encapsulation and release systems for colonic diseases.45 Here we focused on the “turn-on” fluorescence behavior for the fluorescent polymer probe during reduction-triggered drug delivery. The anticancer drug doxorubicin (DOX) was chosen as the model molecule for encapsulation. The self-assembly of PCL3k-TPE-Azo-PEG5k was conducted in PB solution in the presence of DOX. A typical encapsulation experiment is described in the Experimental section. DOX was loaded into micelles at a polymer concentration of 5.0 mg mL−1. The PCL3k-TPE-Azo-PEG5k micelles showed DOX loading efficiencies of 41%. The morphology of the micelles obtained in the presence of DOX was characterized by TEM, which revealed that stable rod-like micelles were produced (Fig. S13(a) and S14(a), ESI†).Reduction-triggered drug release studies in vitro were investigated at pH 7.4 (PB, 50 mM) and 37 °C by using Na2S2O4 and azoreductase that exists in the colon, respectively. A control experiment was carried out in the absence of reducing agent. After reduction for a specific time, the fluorescence emission of DOX was monitored at 480 nm excitation wavelength (Fig. S11, ESI†). The fluorescence intensity of DOX at ∼590 nm decreased gradually owing to the release of hydrophobic DOX from the micelle carriers. The percentage release was calculated based on the fluorescence changes at 590 nm. The percentage release profile was plotted as a function of time (Fig. 5(a and c)). As expected, the minimal DOX release was observed in the control experiment, which showed that the DOX-loaded polymer micelles were stable in the absence of a stimulus. DOX release from the micelles was markedly accelerated with time in the presence of Na2S2O4, and the percentage release reached a maximum value (about 70%) for 35 min. The percentage release increased slowly with time in the presence of reductases. After enzyme treatment for 48 h, the percentage release of DOX reached a maximum value (about 28%). The percentage release of DOX in the presence of reductases is obviously less than that in the presence of Na2S2O4, which can be attributed that the macromolecular enzyme catalyst has a steric hindrance to enter into the micelles for the interruption of the azo bond compared with that of Na2S2O4. The absorption and morphology of the DOX-loaded micelles during reduction were characterized by UV-vis spectroscopy and TEM. TEM images showed that the nanorod boundaries became blurred and gradually dissociated (Fig. S13 and S14, ESI†). In addition, UV-vis spectra showed that the characteristic peak of azobenzene at 450 nm dissipated owing to the breaking of the azo double bonds (Fig. S12, ESI†).
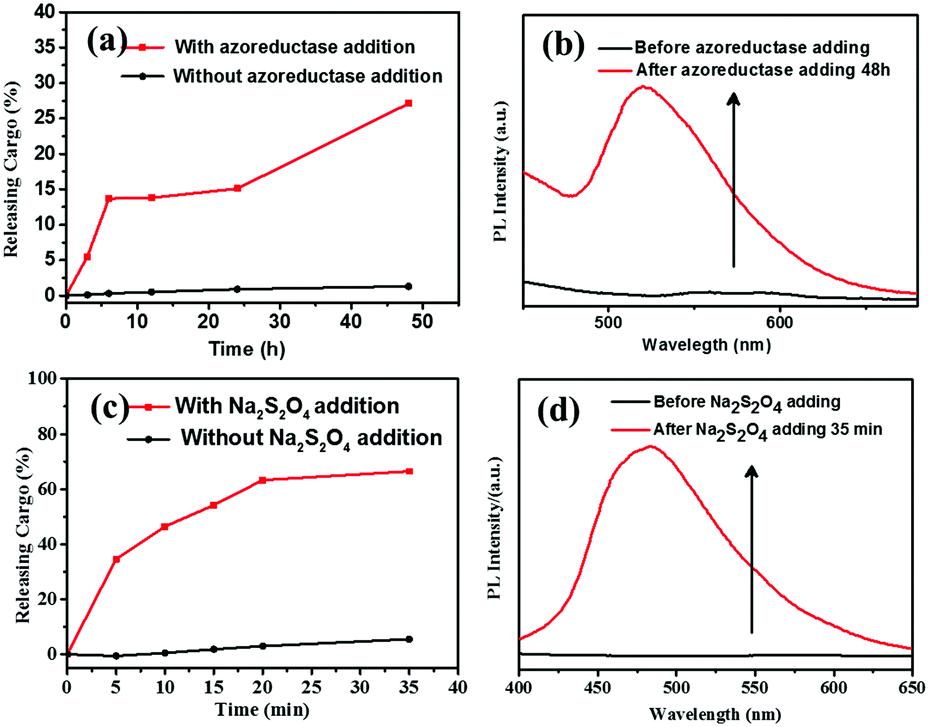 | ||
| Fig. 5 Drug release profiles (a) fluorescence changes (b) for the DOX-loaded micelles of PCL3k-TPE-Azo-PEG5k in PB solution during azoreductase treatment for different time periods (0.1 mg mL−1 PCL3k-TPE-Azo-PEG5k in PBS at 37 °C) (λex = 360 nm). Drug release profiles (c) fluorescence changes (d) for the DOX-loaded micelles of PCL3k-TPE-Azo-PEG5k in PB solution during Na2S2O4 treatment for different time periods (0.3 mg mL−1 PCL3k-TPE-Azo-PEG5k in PB at 37 °C) (λex = 360 nm). | ||
The fluorescence emission based on the TPE (λex = 360 nm) of the DOX-loaded micelles in PB solution was tested during the reduction release, as shown in Fig. 5(b and d). With increasing release time, the fluorescence intensity of the micelles solution distinctly enhanced after reduction reaction in the presence of Na2S2O4/azoreductase. Therefore, based on the “Latent fluorescence” concept, the Azo-TPE caged nanoparticles would be versatile in biosensing and bioimaging applications.
Cytotoxicity tests
The cytotoxicity of the blank micelles was evaluated using Caco2 human colon cancer cells and the MTT method. The results showed that the cell viability was ∼90%, indicating that this type of block copolymer has good biocompatibility (Fig. 6). | ||
| Fig. 6 Cytotoxicity of polymeric micelles against Caco2 human colon cancer cells at various concentrations. | ||
Conclusions
We successfully synthesized a fluorescent amphiphilic block copolymer probe, PCL3k-TPE-Azo-PEG5k, based on an azo reduction response and AIE mechanism, using the CuAAC click reaction. The amphiphilic block copolymer can be self-assembled in PB solution (pH 7.4) to form nanorod like micelles with a hydrophobic interface of TPE-Azo, which is nonfluorescent as the azo moieties quench the AIE activity of TPE. Furthermore, the micelles can encapsulate hydrophobic model drugs such as DOX. Under reducing conditions, such as in the presence of enzyme azoreductase, the micelles disassembled owing to the disruption of the NN double bond. Increasing fluorescence was observed with the micelles dissociation because the AIE effect was activated by the reducer stimulus. Reduction-triggered drug release showed turn-on behavior of fluorescence for the polymer micelles during drug release. Thus, the amphiphilic block copolymer has revealed potential for application in biosensing and controlled drug release for colonic conditions. We anticipate that the valuable data in this work will provide new insights into the AIE mechanism and the reduction-responsiveness of TPE-azo containing derivatives, broaden the variety of macromolecular fluorescent probes based on AIE. Ongoing work to modulate the morphology and size of the drug-loaded assemblies is underway in our laboratory, in order to achieve better drug loading and more controlled release in the presence of azoreductase.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Nature Science Foundation of China (21574090), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions, and the Program of Innovative Research Team of Soochow University. The authors are very grateful for the kind supports on enzymolysis experiments provided by Prof. Yaning He at Tsinghua University, China.Notes and references
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278 CrossRef CAS.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289 RSC.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866 CrossRef CAS PubMed.
- C. De Las Heras Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276 RSC.
- J. N. Liu, W. Bu and J. Shi, Chem. Rev., 2017, 117, 6160 CrossRef CAS PubMed.
- X. F. Ji, Y. Li, H. Wang, R. Zhao, G. P. Tangb and F. H. Huang, Polym. Chem., 2015, 6, 5021 RSC.
- G. C. Yu, M. M. Zhang, M. L. Saha, Z. W. Mao, J. Chen, Y. Yao, Z. J. Zhou, Y. J. Liu, C. Y. Gao, F. H. Huang, X. Y. Chen and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 15940 CrossRef CAS PubMed.
- G. C. Yu, R. Zhao, D. Wu, F. W. Zhang, L. Shao, J. Zhou, J. Yang, G. P. Tang, X. Y. Chen and F. H. Huang, Polym. Chem., 2016, 7, 6178 RSC.
- M. Zelzer, S. J. Todd, A. R. Hirst, T. O. McDonald and R. V. Ulijn, Biomater. Sci., 2013, 1, 11 RSC.
- R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217 RSC.
- J. L. Wang, S. Li, B. Wu and Y. N. He, Eur. Polym. J., 2016, 84, 236 CrossRef CAS.
- A. D. Wong, A. L. Prinzen and E. R. Gillies, Polym. Chem., 2016, 7, 1871 RSC.
- J. Wang, B. Wu, S. Li and Y. J. He, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2450 CrossRef CAS.
- W. X. Gu, Q. L. Li, H. Lu, L. Fang, Q. Chen, Y. W. Yang and H. Gao, Chem. Commun., 2015, 51, 4715 RSC.
- A. D. Wong, T. M. Güngör and E. R. Gillies, ACS Macro Lett., 2014, 3, 1191 CrossRef CAS.
- J. Rao, C. Hottinger and A. Khan, J. Am. Chem. Soc., 2014, 136, 5872 CrossRef CAS PubMed.
- J. Rao and A. Khan, J. Am. Chem. Soc., 2013, 135, 14056 CrossRef CAS PubMed.
- J. Lu, F. Zhou, L. Li, Z. Zhang, F. Meng, N. Zhou and X. Zhu, RSC Adv., 2016, 6, 58755 RSC.
- M. Bates, B. Huang, G. T. Dempsey and X. Zhuang, Science, 2007, 317, 1749 CrossRef CAS PubMed.
- S. M. Borisov and O. S. Wolfbeis, Chem. Rev., 2008, 108, 423 CrossRef CAS PubMed.
- J. Rao and A. Khan, Polym. Chem., 2015, 6, 686 RSC.
- W. Piao, S. Tsuda, Y. Tanaka, S. Maeda, F. Liu, S. Takahashi, Y. Kushida, T. Komatsu, T. Ueno, T. Terai, T. Nakazawa, M. Uchiyama, K. Morokuma, T. Nagano and K. Hanaoka, Angew. Chem., Int. Ed., 2013, 52, 13028 CrossRef CAS PubMed.
- S. Li, J. Wang, J. Shen, B. Wu and Y. He, ACS Macro Lett., 2018, 7, 437 CrossRef CAS.
- W. Piao, K. Hanaoka, T. Fujisawa, S. Takeuchi, T. Komatsu, T. Ueno, T. Terai, T. Tahara, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2017, 139, 13713 CrossRef CAS PubMed.
- L. L. Lock, A. G. Cheetham, P. Zhang and H. Cui, ACS Nano, 2013, 7, 4924 CrossRef CAS PubMed.
- Y. Fan, F. Li and D. Chen, Biomaterials, 2014, 35, 7870 CrossRef CAS PubMed.
- R. Zhan, A. J. H. Tan and B. Liu, Polym. Chem., 2011, 2, 417 RSC.
- A. Chevalier, P. Y. Renard and A. Romieu, Chem. – Asian J., 2017, 12, 2008 CrossRef CAS PubMed.
- N. Shin, K. Hanaoka, W. Piao, T. Miyakawa, T. Fujisawa, S. Takeuchi, S. Takahashi, T. Komatsu, T. Ueno, T. Terai, T. Tahara, M. Tanokura, T. Nagano and Y. Urano, ACS Chem. Biol., 2017, 12, 558 CrossRef CAS PubMed.
- S. Luo, Y. Liu, F. Wang, Q. Fei, B. Shi, J. An, C. Zhao and C. H. Tung, Analyst, 2016, 141, 2879 RSC.
- A. Chevalier, C. Mercier, L. Saurel, S. Orenga, P. Y. Renard and A. Romieu, Chem. Commun., 2013, 49, 8815 RSC.
- N. J. Rattray, W. A. Zalloum, D. Mansell, J. Latimer, C. H. Schwalbe, A. J. Blake, E. V. Bichenkova and S. Freeman, Chem. Commun., 2012, 48, 6393 RSC.
- K. Kiyose, K. Hanaoka, D. Oushiki, T. Nakamura, M. Kajimura, M. Suematsu, H. Nishimatsu, T. Yamane, T. Terai, Y. Hirata and T. Nagano, J. Am. Chem. Soc., 2010, 132, 15846 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- A. Qin, J. W. Y. Lam and B. Z. Tang, Prog. Polym. Sci., 2012, 37, 182 CrossRef CAS.
- M. Hammerich, C. Schutt, C. Stahler, P. Lentes, F. Rohricht, R. Hoppner and R. Herges, J. Am. Chem. Soc., 2016, 138, 13111 CrossRef CAS PubMed.
- M. Han and M. Hara, J. Am. Chem. Soc., 2005, 127, 10951 CrossRef CAS PubMed.
- Y. Li, N. Zhou, W. Zhang, F. Zhang, J. Zhu, Z. Zhang, Z. Cheng, Y. Tu and X. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4911 CrossRef CAS.
- Q. Bo and Y. Zhao, Langmuir, 2007, 23, 5746 CrossRef CAS PubMed.
- Y. Liu, Y. Yu, J. W. Lam, Y. Hong, M. Faisal, W. Z. Yuan and B. Z. Tang, Chem. – Eur. J., 2010, 16, 8433 CrossRef CAS PubMed.
- S. Speiser, Chem. Rev., 1996, 96, 1953–1976 CrossRef CAS PubMed.
- W. K. Feng, Y. Y. Feng, S. F. Wang, W. Feng, W. H. Yi and Q. H. Gong, Chin. Phys. B, 2010, 19, 113401 CrossRef.
- T. Ueno, Y. Urano, K. i. Setsukinai, H. Takakusa, H. Kojima, K. Kikuchi, K. Ohkubo, S. Fukuzumi and T. Nagano, J. Am. Chem. Soc., 2004, 126, 14079 CrossRef CAS PubMed.
- J. Huang, Y. Wu, H. Fu, X. Zhan, J. Yao, S. Barlow and S. R. Marder, J. Phys. Chem. A, 2009, 113, 5039 CrossRef CAS PubMed.
- S. Q. Gao, Z. R. Lu, B. Petri, P. Kopeckova and J. Kopecek, J. Controlled Release, 2006, 110, 323 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, calculation, characterization. See DOI: 10.1039/c8qm00672e |
| This journal is © the Partner Organisations 2019 |