
Polyoxometalate-supported metal carbonyl derivatives: from synthetic strategies to structural diversity and applications
Jingkun
Lu
 ,
Peipei
He
,
Jingyang
Niu
* and
Jingping
Wang
*
,
Peipei
He
,
Jingyang
Niu
* and
Jingping
Wang
*
Henan Key Laboratory of Polyoxometalate Chemistry, Institute of Molecular and Crystal Engineering, College of Chemistry and Chemical Engineering, Henan University, Kaifeng, Henan 475004, P. R. China. E-mail: jyniu@henu.edu.cn; jpwang@henu.edu.cn; Fax: (+86)371-23886876
First published on 27th August 2019
Abstract
Polyoxometalates (POMs) have demonstrated strong potential in various fields, such as catalysis, magnetism, medicine, photochemistry and materials science, mainly because of their remarkable physical and chemical properties. In the broad area of POM chemistry, the study of metal carbonyl-based POMs, as an important type of organometallic POM, has been a rather attractive direction in recent years. POM-supported metal carbonyl derivatives (PMCDs), which utilize the advantages of both POMs and metal carbonyls, have emerged as a promising class of molecules, and much effort has been devoted to their preparation and relevant applications in the last decade. Thus far, substantial development of the synthetic chemistry of PMCDs by using one-pot synthesis has been reported mainly by us. This review focuses on different structural complexes, accompanied by Lindqvist-type, Keggin-type, Dawson-type, and nonclassical-type PMCDs, and is meant to provide fodder and guidance for further exploration and discovery of more intriguing PMCDs with innovative architectures and remarkable functionality. Herein, we highlight and discuss the structural features of PMCDs based on various structural types. Furthermore, synthetic strategies and relevant applications, especially in terms of photochemical properties and catalysis, are reviewed. Ideally, these concepts and strategies can be extended to other organometallic POMs.
 Jingkun Lu | Jingkun Lu was born in Henan, China in 1995 and received her B.S. and M.S. degrees in chemistry from Henan University. In 2014, she began to conduct scientific research in the group of Prof. Jingyang Niu in Henan Key Laboratory of Polyoxometalate Chemistry. Her research interests focus on the preparation, characterization and relevant catalytic properties of polyoxometalate-based metal carbonyl complexes. |
 Peipei He | Peipei He received her B.S. degree in applied chemistry at Henan University Minsheng College University in 2017. And in 2018, she was admitted to Henan University to pursue a master's degree. In the first year of her graduate school, she conducted a series of experiments using polyoxometalates as catalysts at the Key Laboratory of Polyoxometalates of Henan University. Then she went to Dalian Institute of Chemical Physics to study the synthesis of boron-doped carbon material catalysts and their role in aldehyde amidation. |
 Jingyang Niu | Jingyang Niu is the Special Professor of Chemistry of Henan Province at Henan University. He obtained his BA, MA and PhD degrees in 1986, 1989 and 1996 from Henan University, Northeastern Normal University and Nanjing University, respectively. His research interests focus on the synthesis, crystal structure and properties of novel polyoxometalates. He has received several awards, including the Distinguished Young Scholars of Henan Province, National government special allowance recipients, and Thousands of intellectual plans of Central Plain. He is the leader of the Henan Province Key Laboratory of Polyoxometalate at Henan University. |
 Jingping Wang | Jingping Wang is the Special Professor of Chemistry of Henan University. She received her BA and MA from Northeastern Normal University at 1986 and 1989, respectively. From 1989 to 2002 she was an assistant professor, a lecturer and an associate professor and then she became a professor at Henan University. She has received several awards, including the May 1st Labour Medal of Henan Province and Excellent University Key Teacher of Henan Province. Her research expertise is the synthesis, crystal structure and properties of novel polyoxometalates and their potential application in different fields. |
1. Introduction
Polyoxometalates (POMs), a diverse family of anionic metal oxides based on early transition metals (e.g., Nb, Ta, V, Mo, W), have come to the fore due to their unique physical and chemical properties.1–7 These molecules are built from corner-, edge- and face-shared metal–oxygen {MOx} (x = 5, 6) polyhedra and span a very large diversity of morphologies (size and shape), resulting in a rich variety of structures involved in classical fields such as catalysis, photochemistry, electrochemistry, materials science, and medicine.8–17 The first synthesized POM, (NH4)3[PMo12O40]·nH2O, was reported by Berzelius in 1826.18 One century later, Keggin reported the crystallographic study of “H3[PMo12O40]·29H2O” based on powder X-ray diffraction,19 which was one of the most significant milestones in the history of POM research. The characterization of POMs and the systematic study of their properties began in the early 20th century with Rosenheim.20 In recent decades, POM research has seen tremendous growth, and a number of reviews have appeared that address different aspects of POM science.21–27 One of the most interesting aspects of POM chemistry is the underlying deceptively simple synthetic procedures, which require a small number of steps or even just one step (one-pot syntheses).28–33 Traditional “one-pot” synthesis with varying pH, temperature, concentration/type of metal oxide anion, heteroatom type/concentration, etc., has always been the main method to search for new POM clusters. Accompanying the development of new synthetic technologies, such as hydrothermal, microwave, refluxing and ionic liquid synthetic methods, applied to POM chemistry, the POM family has been significantly expanded and enriched.34–38The structural analogy between POMs and extended oxides was first noted by Baker and later expanded by Pope, Klemperer, Finke and Müller.39–48 In view of the extensive literature on POM chemistry and the rapid developments in the field of POM coordination chemistry, the development of functional POM-based materials has been rather slow because POMs are usually crystalline solids that are hard to prepare. Recently, the most common route to the integration of POMs into functional architectures and devices rests on inorganic/organic hybrids. The functionalization of POM species by organic or organic groups has been heavily investigated, resulting in a new class of molecular complexes, but also extended POM compounds. The organic–inorganic hybrids are classified into two types according to the nature of the interaction between the organic and inorganic components. The first class (class I) involving noncovalent interactions has been reviewed by Müller and Zubieta, such as host–guest systems, and intermolecular complexes between POMs and organic substrates.47,48 In the second class (class II), the organic and inorganic moieties are linked via strong covalent or non-covalent bonds. A special thematic issue in Chemical Reviews organized by Dolbecq divided class II into two parts.49 The first section gave an exhaustive list of hybrid POMs where organic groups are covalently linked to POM units via p-block elements. The second part described hybrid POMs where the organic ligand is bound to d- and f-block elements grafted to the surface or encapsulated in the vacancy of a POM. The incorporation of d-block elements into vacant POM matrices is one of the oldest and most studied reactions in POM chemistry. In recent years, numerous studies have been devoted to the directed grafting of organic substrates onto the nucleophilic oxygen atoms of the POM core and to the introduction of the organometallic fragments into vacant POM complexes.49,50 There are a limited number of POMs containing sufficient charge density at their surface oxygen atoms to covalently bind organometallics. And surface activation can be achieved by replacing MoVI or WVI centers by one or more lower valent metals. In addition, the chemistry of organometallic derivatives of POMs is an area of growing interest. The organometallic derivatives of POMs provide a novel strategy to synthesize POM-based organic–inorganic hybrid materials because they may serve as models of solid oxide-supported organometallic compounds and the combination of the hard oxo ligand and soft ligands may result in novel properties. Since the pioneering work published by the groups of Ho and Klemperer, Zonnevijlle and Pope and Knoth and Harlow, organic-functionalized POMs, i.e., species in which one or some oxo or {MoOx}n+ groups have been replaced with organic functional groups, have been studied extensively and now form the largest subclass of POM derivatives. In addition, Finke has emphasized that the organometallic POMs have two types, and the difference between organometallics supported on POMs and those incorporated into a POM is whether the organic species are firmly attached to a k3-O site of the POM surface oxygens.51
The research of POM-based organometallic derivatives is of huge significance in POM chemistry not only providing new dimensionalities in structures but also important owing to their materialistic advantages.52–55 In this context, immobilization of metal carbonyl units onto the POM surface is expected to obtain functional compounds and emerges as an important category. POM-supported metal carbonyl derivatives (PMCDs) possess the dual advantages of metal carbonyl groups and POMs. In particular, PMCDs have found immerse interest as the chromophoric metal carbonyl forms fascinating supramolecular arrays, structure peculiarities and various size modifications which expands them in various thrust areas of research like single molecular magnets, photoluminescence, and catalysis.56–59 However, among the reports of POM-supported organometallic compounds, very few papers describe the functionalization of POMs by metal carbonyl complexes. Despite their anionic charges, complete POMs have a rather low charge surface density. The nucleophilicity of the oxo ligands can be increased by substituting metals (Mo, W, V, Nb, Ta), and the resulting mixed addenda polyanions show enhanced reactivity towards organometallic fragments, which is exemplified by the grafting of various d6-fac-{ML3} units ({M(CO)3+}) Herein, we will focus our emphasis on the POM-supported metal carbonyl derivatives, and the purpose of this review is to give an extended survey of these compounds and their applications.
2. Synthetic strategies
In 1998, a comprehensive review by Gouzerh and Proust covered the sizeable literature on the organic and organometallic derivatives of POMs.50 Furthermore, as an important subclass of POM-based organometallic derivatives, PMCDs have been synthesized and characterized for nearly forty years. PMCDs possess the dual advantages of metal carbonyl groups and POMs. Since the report of the first metal carbonyl adducts, [Nb2W4O19M(CO)3]3− (M = Mn and Re), in 1980,60 the synthesis and characterization of POM-supported organometallic derivatives have received much attention, while tricarbonyl derivatives of the hexaniobate and hexatantalate anions have been recently reported by Pope.61 As a consequence, syntheses are very often relatively difficult (mainly because polyanions do not have sufficient charge density to combine with metal carbonyl groups, along with their poor solubility in the same solvent, their radiation- or thermoinstability and the high costs of metal carbonyl compounds), whereas those of POM-supported carbonyl metal derivatives are relatively less difficult. Moreover, the manganese/rhenium carbonyl derivatives of POMs, with rich behaviours displayed by the d6 metal carbonyl compounds and the POMs, are especially important. Synthetically, the routes to produce new PMCDs are often very simple synthetic manipulations, and most of these hybrid compounds have been obtained by one-pot aqueous/hydrothermal methods. The rational synthesis of PMCDs is still a great challenge because one or more of the reaction conditions, such as temperature, pH, stoichiometry, reaction time, medium, and template identity, can have a considerable influence on the reaction outcome. In most cases, the synthetic strategy has been used to react organic-soluble forms of the POMs, generally via tetrabutylammonium counterions, with metal carbonyl complexes in organic solvents such as CH3CN, CH2Cl2 and CH3COOH. To date, discrete integrated metal carbonyls, which are compounds where the metal carbonyl component is incorporated into the polyoxometal framework, have been obtained in several ways, including the incorporation of a metal carbonyl into a lacunary POM, the oxidation of carbonyl dimers [{Cp*M(CO)2}2](M = W or Mo) and various aggregation processes triggered by protons and/or Lewis acids in aqueous or non-aqueous media.62,63 Since 2008, our group has been dedicated to synthesizing novel PMCD structures, and we have obtained many new structures in H2O–CH3OH or H2O–CH3CN mixed solvents using one-pot aqueous synthesis. In this new review, we highlight the structural variety of reported PMCDs, and our aim is to survey the recent significant achievements and provide key information for designing novel structures with improved properties directed by their structure–property relationships.3. Representative structure types of PMCDs
3.1 Lindqvist-type PMCDs
As early as 1980, Klemperer et al. reported the synthesis and characterization of the first example of PMCDs, [(OC)3M(Nb2W4O19)]3− (M = Mn, Re) (Fig. 1), containing metal tricarbonyl units bonded to a triangle of oxygen atoms on the surface of the [Nb2W4O19]4− anion.60 The complexes were prepared by the reaction of [(n-C4H9)4N]4(Nb2W4O19), prepared from aqueous Na2[(CH3)4N]2(Nb2W4O19) by using a cation-exchange resin, with an equimolar amount of [(OC3)M(NCCH3)3](PF6) (M = Re/Mn) in acetonitrile and characterized by infrared (IR) and nuclear magnetic resonance (NMR) spectroscopy. It was not 1985 that Klemperer investigated these complexes in more detail by using single-crystal X-ray diffraction analysis.64 Specifically, the X-ray crystallographic study of [(OC)3M(Nb2W4O19)]3− (M = Mn, Re) indicated that both metal sites were assumed to be 1/3 Nb and 2/3 W and that M(CO)3+ (M = Re/Mn) was bound to three bridging oxygens of [Nb2W4O19]4− as expected from the 18-electron rule. Subsequently, Klemperer et al. reported the first polyoxoanion-supported rhodium and iridium carbonyl complexes,65 [{(OC)2Rh}5(Nb2W4O19)2][(n-C4H9)4N]3, [{(OC)2Rh}3(Nb2W4O19)2][(n-C4H9)4N]5, [{(OC)2Ir}2(Nb2W4O19)2][(n-C4H9)4N]5, and [(OC)2Ir(P3O9)][{(C6H5)3P}2N]2. In addition, [{(OC)2Rh}5(Nb2W4O19)2][(n-C4H9)4N]3 was prepared by the reaction of either [{(C7H8)Rh}5(Nb2W4O19)2][(n-C4H9)4N]3 with CO in a nitromethane solution or Nb2W4O19[(n-C4H9)4N]4 with [(OC)2RhCl]2 in a 1,2-dichloroethane solution. [{(OC)2Rh}3(Nb2W4O19)2][(n-C4H9)4N]5 was prepared by the reaction of Nb2W4O19[(n-C4H9)4N]4 with [(OC)2RhCl]2 in chloroform. The structure of the [{(OC)2Rh}3(Nb2W4O19)2]5− anion was obtained by removing two (OC)2Rh+ units from the structure of the [{(OC)2Rh}5(Nb2W4O19)2]3− anion. [{(OC)2Ir}2(Nb2W4O19)2][(n-C4H9)4N]5 was synthesized by bubbling CO into a CH3CN solution of [{(C8H12)Ir}2H(Nb2W4O19)2][(n-C4H9)4N]5, and in this structure, two octahedral Nb2W4O194− anions are linked together edge-to-edge by a proton and two (OC)2Ir+ cations.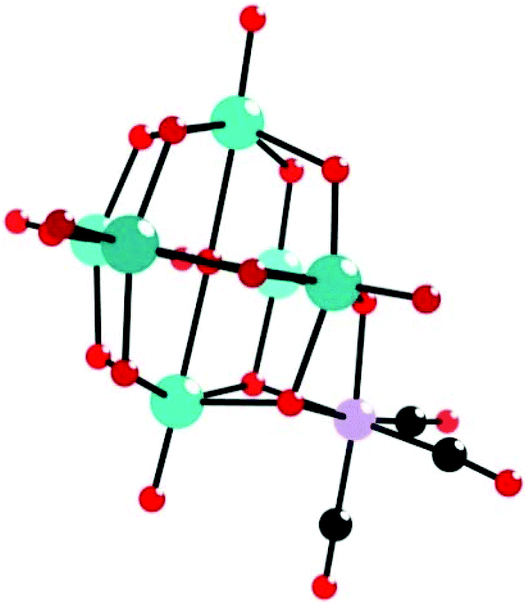 | ||
| Fig. 1 Ball-and-stick model of the [(OC)3M(Nb2W4O19)]3− (M = Mn, Re). Colour code: O, red; C, black; Nb/W, green; Mn/Re, purple. | ||
Similarly, [(OC)2Ir(P3O9)][{(C6H5)3P}2N]2 was prepared by substitution of cyclooctadiene ligands in [(C8H12)Ir(P3O9)][{(C6H5)3P}2N]2 with CO ligands.
In 1981, Day and co-workers reported preliminary results of the first X-ray crystallographic structure determinations of polyoxoanion-supported organometallic anions,66 [(η5-C5H5)Ti(Mo5O18)]3− and [(η5-C5H5)Ti(Mo5O18)MoO2Cl]2− (Fig. 2a and b), and the synthesis and structure of the first bifunctional polyoxoanion-supported organometallic, [(η5-C5H5)Ti(Mo5O18)Mn(CO)3]2−. [(η5-C5H5)Ti(Mo5O18)MoO2Cl]2− was prepared by the reaction of [(η5-C5H5)Ti(Mo5O18)]3− and 2 equiv. of aqueous HCl in a CH3CN solution. The X-ray diffraction results of the [(η5-C5H5)Ti(Mo5O18)MoO2Cl]2− polyoxoanion showed that a MoO2Cl+ unit is bonded to a triangle of three bridging oxygens in this structure. However, [(η5-C5H5)Ti(Mo5O18)MoO2Cl]2− is unstable towards water and heat, decomposing to [Mo6O19]2− in CH3CN. By replacement of MoO2Cl+ with a Mn(CO)3+ unit, [(η5-C5H5)Ti(Mo5O18)Mn(CO)3]2− was prepared from [(η5-C5H5)Ti(Mo5O18)]3− and [(OC)3Mn(NCCH3)3][PF6] in CH3CN and was relatively stable towards both water and heat in CH3CN. The existence of [(η5-C5H5)Ti(Mo5O18)Mn(CO)3]2− was confirmed by the carbonyl region in the IR spectrum and the NMR spectrum.
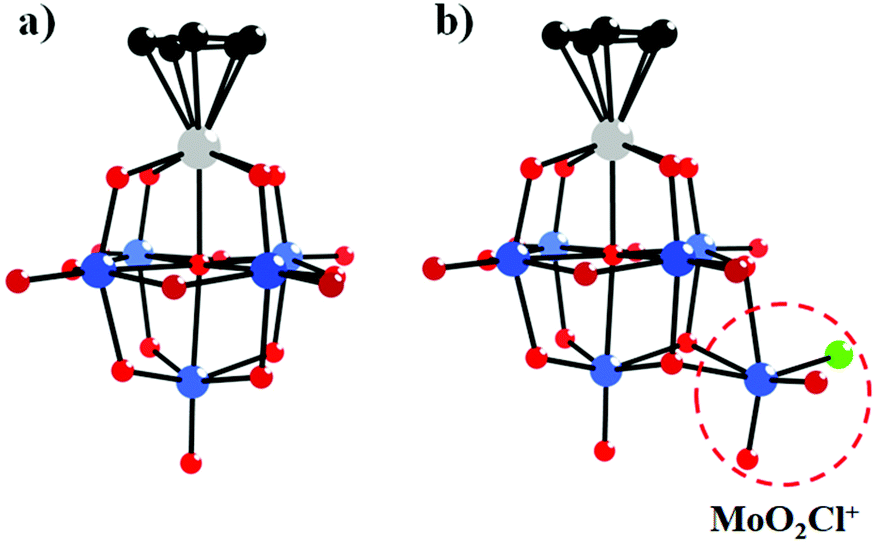 | ||
| Fig. 2 Ball-and-stick model of the [(η5-C5H5)Ti(Mo5O18)]3− (a) and [(η5-C5H5)Ti(Mo5O18)MoO2Cl]2− (b) polyoxoanions. Colour code: O, red; C, black; Ti, grey; Cl, light green. | ||
In 1993, Klemperer et al. reported five new RuI–RuI tetracarbonyl complexes: [(P3O9)2Ru2(CO)4]4−, [{CpTi(W5O18)}2Ru2(CO)4]4−, [(CH3CN)6Ru2(CO)4]2+, [(PPh3)2(CH3CN)4Ru2(CO)4]2+, and [(C5H5N)6Ru2(CO)4]2+.67 Notably, only two of these complexes were structurally characterized using single-crystal diffraction techniques: [(P3O9)2Ru2(CO)4][(n-C4H9)4N]4·2CH3CN and [(PPh3)2(CH3CN)4Ru2(CO)4](PF6)2 (Fig. 3a and b). [{CpTi(W5O18)}2Ru2(CO)4]4− was obtained by the reaction of [(CH3CN)6Ru2(CO)4](PF6)2 with 2 equiv. of TBA3[CpTiW5O18] in a CH2Cl2 solution.
 | ||
| Fig. 3 Ball-and-stick model of [(P3O9)2Ru2(CO)4]4− (a) and [(PPh3)2(CH3CN)4Ru2(CO)4]2+ (b). Colour code: O, red; Ru, orange; P, yellow; C, black; N, dark green. | ||
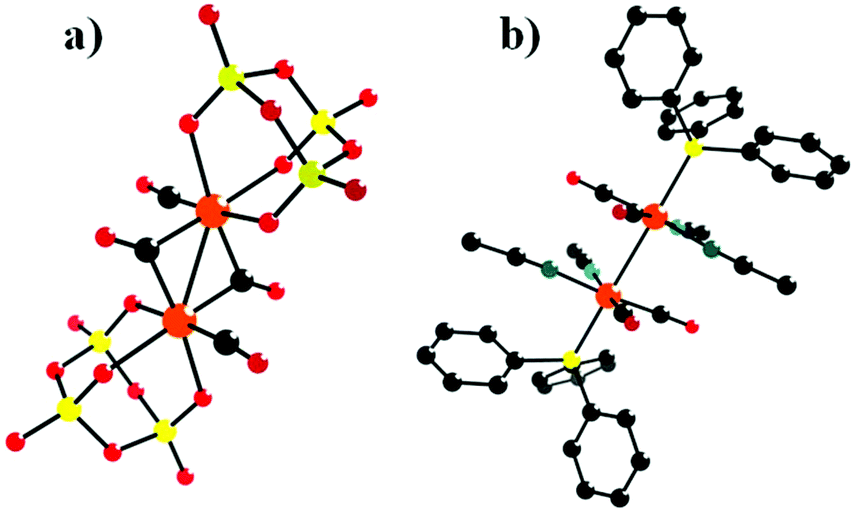
In 2001, Pope and co-workers reported ten 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 complexes of [Mn(CO)3]+ and [Re(CO)3]+ with [Nb6O19]8− and [Nb6O19]8− as potassium salts.68 The complexes contain M(CO)3 groups attached to the surface bridging oxygen atoms of the hexametalate anions to yield structures with nominal C3V (1:1), D3d (trans 2:1), and C2V (cis 2:1) symmetry (Fig. 4a, b, and c). The formation of 2:1 complexes and the existence of both cis and trans isomers were first deduced from the NMR spectra. K7[Re(CO)3Nb6O19] was first synthesized by using an aqueous hydrothermal reaction technique, while the other complexes were obtained conventionally in aqueous solution. The discovery of K7[Re(CO)3Nb6O19] not only enriches the diversity of the structural chemistry of PMCDs but also provides inspiring directions to design and prepare many more intriguing structures. The Re compounds are stable up to 400–450 °C in the solid state and can be formed under hydrothermal conditions at 130 °C and pH > 10, while the Mn compounds lose CO at temperatures above 200 °C. The high stability of Re compounds may be assumed to be associated with the loss of Re2O7 formed by oxidation or disproportionation.
:1 and 2:1 complexes of [Mn(CO)3]+ and [Re(CO)3]+ with [Nb6O19]8− and [Nb6O19]8− as potassium salts.68 The complexes contain M(CO)3 groups attached to the surface bridging oxygen atoms of the hexametalate anions to yield structures with nominal C3V (1:1), D3d (trans 2:1), and C2V (cis 2:1) symmetry (Fig. 4a, b, and c). The formation of 2:1 complexes and the existence of both cis and trans isomers were first deduced from the NMR spectra. K7[Re(CO)3Nb6O19] was first synthesized by using an aqueous hydrothermal reaction technique, while the other complexes were obtained conventionally in aqueous solution. The discovery of K7[Re(CO)3Nb6O19] not only enriches the diversity of the structural chemistry of PMCDs but also provides inspiring directions to design and prepare many more intriguing structures. The Re compounds are stable up to 400–450 °C in the solid state and can be formed under hydrothermal conditions at 130 °C and pH > 10, while the Mn compounds lose CO at temperatures above 200 °C. The high stability of Re compounds may be assumed to be associated with the loss of Re2O7 formed by oxidation or disproportionation.
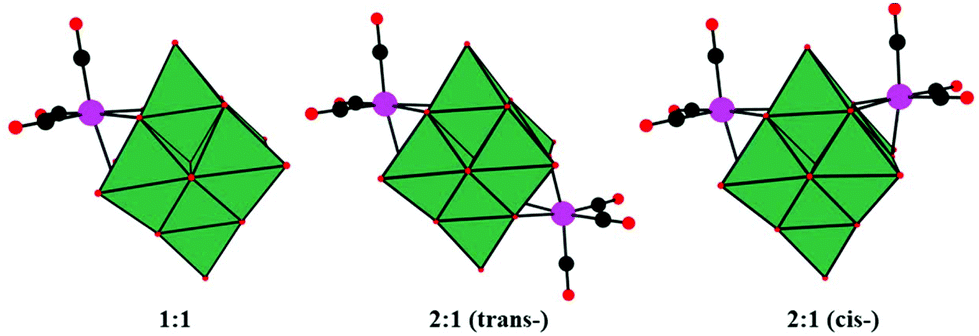 | ||
| Fig. 4 Polyhedral/ball-and-stick representations of [M6O19{M′(CO)3}n](8−n)− (M = Nb, Ta; M′ = Mn, Re; n = 1, 2). Colour code: O, red; C, black; Re/Mn, pink; MO6, dark green. | ||
In 2003, Gouzerh et al. reported three crystal structures of (nBu4N)2[Re(CO)3(H2O){Mo5O13(OMe)4(NO)}], (nBu4N)3[Na{Mo5O13(OMe)4(NO)}2{Mn(CO)3}2], and (nBu4N)4[Mn(H2O)2{Mo5O16(OMe)2}2{Mn(CO)3}2], which were obtained by the reaction of neutral or cationic manganese carbonyl species with the oxo-nitrosyl complex [Na(MeOH){Mo5O13(OCH3)4(NO)}]2− and characterized by single-crystal X-ray diffraction.69 Stirring of a 1:1 mixture of [Na(MeOH){Mo5O13(OCH3)4(NO)}]2− and {Mn(CO)3}+ in CH3OH at room temperature can lead to the isolation of (nBu4N)2[Re(CO)3(H2O){Mo5O13(OMe)4(NO)}]. In this paper, although (nBu4N)2[Re(CO)3(H2O){Mo5O13(OMe)4(NO)}] and (nBu4N)2[Mn(CO)3(H2O){Mo5O13(OMe)4(NO)}] are isomorphous, the crystal structure of only the Re-containing compound was obtained and fully analysed. As shown in Fig. 5a and b, the organometallic {Re(CO)3}+ fragment was bonded to two adjacent axial oxygen atoms of the {Mo5} ligand and achieved an 18-electron configuration by coordination to a molecule of water. (nBu4N)3[Na{Mo5O13(OMe)4(NO)}2{Mn(CO)3}2] was prepared by refluxing a mixture of [Na(MeOH){Mo5O13(OCH3)4(NO)}]2− with either {Mn(CO)3}+ or [MnBr(CO)5] in CH3OH. Each {Mn(CO)3}+ fragment was linked to a distinct {Mo5} unit through the oxygen atoms of two adjacent methoxo ligands and one bridging oxo ligand (Fig. 5c and d). Unlike the usual rule for PMCDs, where the {Mn(CO)3}+ fragment (M = Mn or Re) binds to a triangle of bridging oxygen atoms similar to d6-fac-ML3 fragments in general, such a coordination mode was unknown for the {Mo5} ligand until the characterization of (nBu4N)3[Na{Mo5O13(OMe)4(NO)}2{Mn(CO)3}2]. In detail, each [{Mo5O13(OMe)4(NO)}{Mn(CO)3}]2− unit interacts with Na+ through its four axial oxo ligands in such a way that Na+ displays distorted square-antiprismatic coordination. (nBu4N)4[Mn(H2O)2{Mo5O16(OMe)2}2{Mn(CO)3}2] can be obtained by refluxing a mixture of (nBu4N)2[Mo2O7] and [MnBr(CO)5] in non-deaerated methanol or by adding (nBu4N)2[Mo2O7] to a mixture of Mn(NO3)2·4H2O and [MnBr(CO)5] in boiling methanol. The structure of this complex can be viewed as consisting of two independent [Mo5O16(OMe)2}{Mn(CO)3}]3− units connected by a {Mn(H2O)2}2+ linker. Each {Mn(CO)3}+ fragment is linked to a distinct [Mo5O16(OMe)2]4− through the oxygen atoms of the methoxo ligands and one bridging oxo ligand (Fig. 5e and f). In addition, the [Mo5O16(OMe)2]4− ion is a new member of the family of monovacant Lindqvist-type POMs and has not been previously characterized because this complex has limited stability in an uncomplexed form. In addition to side-on coordination to a {Mn(CO)3}+ cation, octahedral coordination is achieved when each [Mo5O16(OMe)2]4− acts as a bidentate ligand towards the MnII centre and two mutually trans molecules of water are also present.
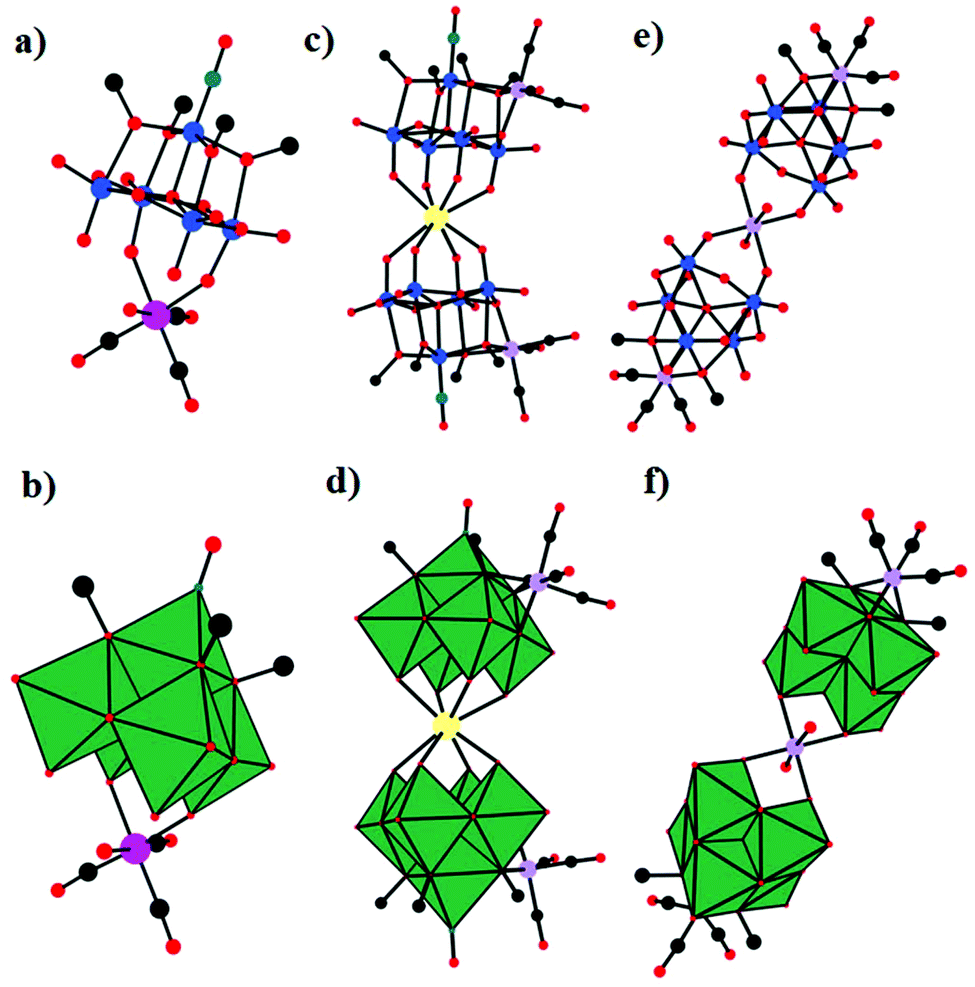 | ||
| Fig. 5 Ball-and-stick representations and polyhedral representations of [Re(CO)3(H2O){Mo5O13(OMe)4(NO)}]2− (a) and (b), [Na{Mo5O13(OMe)4(NO)}2{Mn(CO)3}2]3− (c) and (d), and [Mn(H2O)2{Mo5O16(OMe)2}2{Mn(CO)3}2]4− (e) and (f). Colour code: O, red; C, black; Mo, blue; Re, pink; Mn, lilac; Na, yellow; MoO6, dark green. | ||
With continuous research on PMCDs, our group has reported an isopentatungstate-supported rhenium carbonyl derivative, KH[(CH3)4N]3{[Re(CO)3]4[(μ2-OH)(μ3-O)(W5O18)]}·6H2O in 2016.56 As shown in Fig. 6, the [{Re(CO)3}4(μ2-OH)(μ3-O)(W5O18)]5− polyoxoanion can be regarded as a four-metal carbonyl unit attached to a [W5O18]6− subunit, which is closely related to the Lindqvist hexatungstate structure with one {WO6} octahedron removed from {W6O19}. More intriguingly, the trimetric metal model cluster {Re3} is capped on the [W5O18]6− subunit through four Re–O–W bonds, and the structure possesses the largest number of metal carbonyl groups among PMCDs. Furthermore, the compound demonstrated good capability of catalysing alkenes and was found to efficiently catalyse the epoxidation of cyclooctene with high conversion (98.9%) and excellent selectivity (99%). The high catalytic efficiency in epoxidation of alkenes may be attributed to the pentatungstate framework.
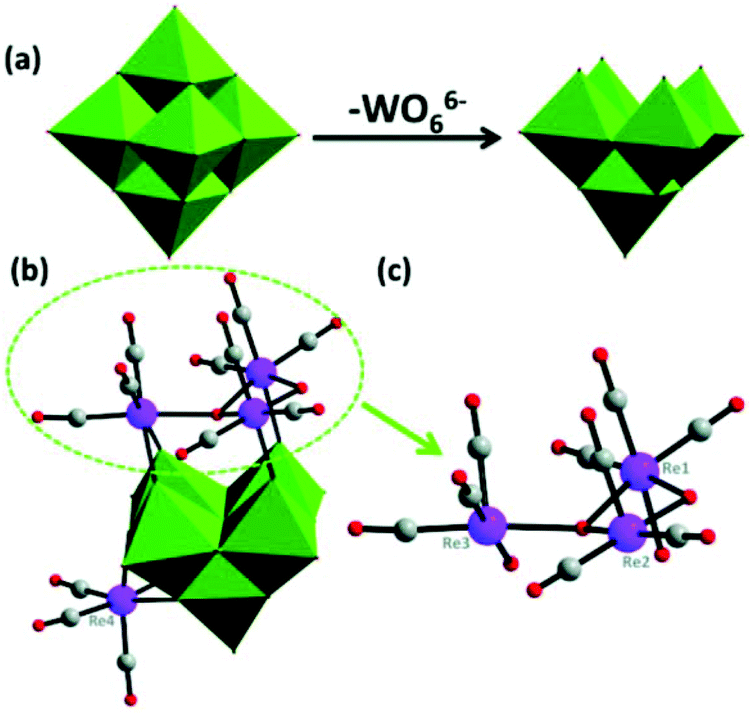 | ||
| Fig. 6 (a) Polyhedral representation of [W6O19]2− and [W5O18]6−; WO6 octahedral, green; (b) ball-and-stick representation of [{Re(CO)3}4(μ2-OH)(μ3-O)(W5O18)]5−; (c) ball-and-stick representation of {[Re(CO)3]4[(μ2-OH)(μ3-O)]}+. Colour code: Re, purple; O, red; C, gray; WO6, green. | ||
3.2 Keggin-type PMCDs
In 1979, Knoth et al. reported the first heteropolyanions that contain metal–metal bonds,70 including [CpFe(CO)2SnW11PO39]4− (Cp = π-C5H5), [(OC)3Co(SnW11SiO39)2]11−, [π-C3H5Pd(SnW11PO39)2]11−, and a high-molecular-weight completely inorganic polymer, [(OC)3CoGeW11SiO405−]n. These organic derivatives of Keggin-structure heteropolyanions are prepared by reacting the “unsaturated” Keggin fragments [SiW11O39]8−, [SiMo11O39]8−, and [PW11O39]7−, with transition metal complexes that contain trichlorostannane or trichlorogermane ligands. These polyoxoanions were isolated as trimethylammonium, tetramethylammonium, and trimethylsulfonium salts and further characterized by elemental analysis and IR and NMR spectroscopy. Four years later, Knoth et al. obtained a new disubstituted Keggin-type PMSC, [{CpFe(CO)2Sn}2W10PO38]5−, by reacting CpFe(CO)2SnCl3 with an aqueous solution of sodium dihydrogen phosphate at pH = 8.6. The structure of this complex was demonstrated by W NMR and P NMR spectra, but no crystallographic data were obtained.In 1987, Siedle et al. isolated several novel Keggin ion derivatives, [(Ph3P)2Rh(CO)(CH3CN)]nXM12O40 (X = P/Si; M = W/Mo; n = 3, 4), that contain coordinatively unsaturated [(Ph3P)2Rh(CO)]+ units.71 These complexes were structurally characterized by IR, NMR, and X-ray absorption spectroscopy three years later, and their catalytic activity and selectivity for olefin isomerization and hydroformation were also carefully described for the first time.
In 2008, Sadakane et al. reported a novel carbonyl–ruthenium-substituted undecatungstosilicate, [α-SiW11O39RuII(CO)]6−, which has been fully characterized in solution and in the solid state.72 More importantly, it was the first example of a metal carbonyl moiety being fully incorporated into the POM framework by means of a hydrothermal method. The incorporated ruthenium(II) shows a well-defined reversible redox couple, and the one-electron-oxidized ruthenium(III) derivative [α-SiW11O39RuIII(H2O)]5− is unexpectedly stable in aqueous solution compared to carbonyl–ruthenium coordination complexes. A CV study also indicated that the lacunary α-Keggin-type polyoxotungstate was able to stabilize the uncommon RuIII(CO) moiety, which usually releases its CO molecule in RuIII(CO)–organic derivatives too quickly to be detected.
In 2012, our group reported a series of compounds based on polyoxoanions and manganese carbonyl groups: [Mn(CO)3(CH3CN)3]n[α-XM12O40] [n = 3, X = PV, M = MoVI, WVI], [Mn(CO)3(CH3CN)3]n[α-XM12O40]·1.5H2O [n = 4, X = SiIV, M = MoVI], and [Mn(CO)3(CH3CN)3]nH[α-XM12O40] [n = 3, X = GeIV, M = MoVI].73 Owing to the relatively high activity of POM precursors in aqueous solution and the stability of [Mn(CO)5]Br in non-aqueous solvents, we chose the CH3CN/H2O mixed solvent to explore the reactions of POM precursors with [Mn(CO)5]Br. The molecular structural unit of these compounds consists of [Mn(CO)3(CH3CN)n]+ cations and a well-known α-Keggin-type [α-PMo12O40]3− anion. Considering the stationary fac-configuration of the [Mn(CO)3(CH3CN)n]+ cation, we attempted to obtain the isomer containing mer-[Mn(CO)3(CH3CN)n]+ cations by changing the experimental conditions; however, our attempts failed.
In the same year, our group reported two novel trivacant Keggin-type POM-based manganese carbonyl derivatives: K8[(OC)3Mn(A-α-H2GeW9O34)]·10H2O and K8[(OC)3Mn(A-α-H2SiW9O34)]·11H2O.57 The two compounds were synthesized by degradation of the metastable [γ-XW10O36]8− (X = GeIV, SiIV) in CH3CN–H2O solvent (1:2, vol.). X-ray diffraction analysis indicates that the two complexes are isomorphic and consist of a [(OC)3Mn]+ group and a trivacant [A-α-H2XW9O34]8− (X = SiIV, GeIV) fragment, representing the first examples of trivacant Keggin-type metal carbonyl derivatives (Fig. 7). In this case, the [γ-XW10] (X = SiIV, GeIV) unit as a starting material was transformed to the [A-α-XW9] unit in a CH3CN-H2O mixed solvent at pH = 7.04 or 7.16 at 80 °C. Moreover, the [γ-XW10] unit plays a vital role in the formation of the two compounds, and good crystals of both compounds can be obtained in the range of CH3CN–H2O = 1:2–1:5, where the optimum ratio is 1:2. It should be noted that the [Mn(CO)3] unit prefers to coordinate at the less nucleophilic bridging oxygens on the cap of the Keggin unit rather than at the lacunary sites. The reasons are as follows: firstly, the [(OC)3Mn(A-α-H2GeW9O34)]8− polyoxoanion has twelve terminal oxygen atoms at the lacunary sites, and all of which have W–O double bands in the range of 1.750–1.767 Å.56,59 From the viewpoint of the electronic structure, it is difficult for these terminal oxygen atoms to coordinate with the [Mn(CO)3] unit because of their poor bonding ability. Secondly, some documents have confirmed that several different two-electron donor ligands L can react with carbonyl metal units to afford kinetically stable d6 low-spin octahedral 18-electron moieties fac-[(OC)3ML3]+ (M = Mn, Re).66–69 So it is most likely that trigonal adjacent surface oxygens in Keggin POMs as two-electron L ligands coordinate with [(OC)3M]+ featuring the fac-[(OC)3MO3]+ moiety. In the trivacant [A-α-H2GeW9O34]9− polyoxoanion, there is only an intact W3O9 triad on the cap of the Keggin unit. From the viewpoint of coordination chemistry and steric configuration, in order to favour an 18-electron configuration, the [Mn(CO)3] unit prefers to coordinate at the (notionally) less nucleophilic bridging oxygens on the cap of the Keggin unit.
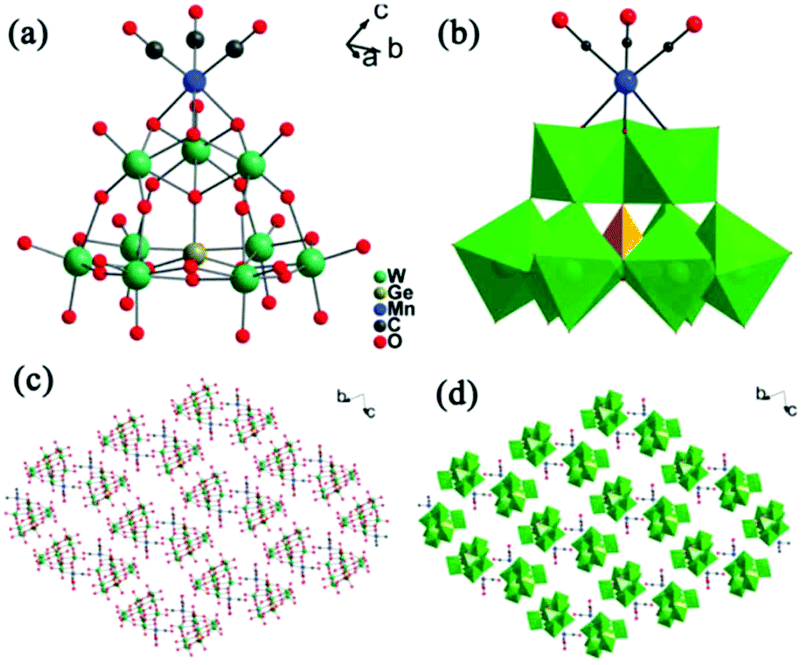 | ||
| Fig. 7 (a) Ball-and-stick representation of K8[(OC)3Mn(A-α-H2GeW9O34)]·10H2O. (b) Polyhedral/ball-and-stick representation of K8[(OC)3Mn(A-α-H2GeW9O34)]·10H2O. (c) and (d) The packing arrangements of K8[(OC)3Mn(A-α-H2GeW9O34)]·10H2O along the a-axis. The hydrogen atoms, K cations and lattice water molecules are omitted for clarity. | ||
In 2013, Hill et al. prepared a new series of complexes that contain two electron-donating groups, {M(CO)3}+ ions (M = Re/Mn), on one polytungstate electron-accepting group (Fig. 8).58 By utilizing [X2W22O74(OH)2]12− (X = Sb, Bi) as synthetic precursors, these authors used these POM ligands to coordinate fac-{M(CO)3}+ (M = Re/Mn) fragments and obtained four discrete Krebs-type “slipped-sandwich” structures: Na11H[Sb2W20O70{Re(CO)3}2]·34H2O, Na11H[Bi2W20O70{Re(CO)3}2]·33H2O, K9Na3[Sb2W20O70{Mn(CO)3}2]·32H2O, and K9Na3[Bi2W20O70{Mn(CO)3}2]·32H2O. The four compounds have been structurally characterized by single-crystal X-ray diffraction, and the convenient preparation of these structures was conducted in a weakly acidic aqueous solution (pH = 5–6). All four metal-donor-POM-acceptor compounds have similar structures and contain two identical β-B-[XW9O33]9− (X = Sb or Bi) units joined by two WO6 octahedra and two fac-{M(CO)3}+ moieties. The charge transfer (CT) dynamics, investigated by femtosecond transient absorption (TA) spectroscopy of Na11H[Sb2W20O70{Re(CO)3}2]·34H2O and Na11H[Bi2W20O70{Re(CO)3}2]·33H2O combined with the density functional theory (DFT) calculations, indicates that both complexes exhibit metal-to-POM charge transfer (MPCT) transitions from the Re centres to the POM ligands. The CT transition from the Mn centres to the POM ligands in K9Na3[Sb2W20O70{Mn(CO)3}2]·32H2O and K9Na3[Bi2W20O70{Mn(CO)3}2]·32H2O leads to decomposition of the starting compounds.
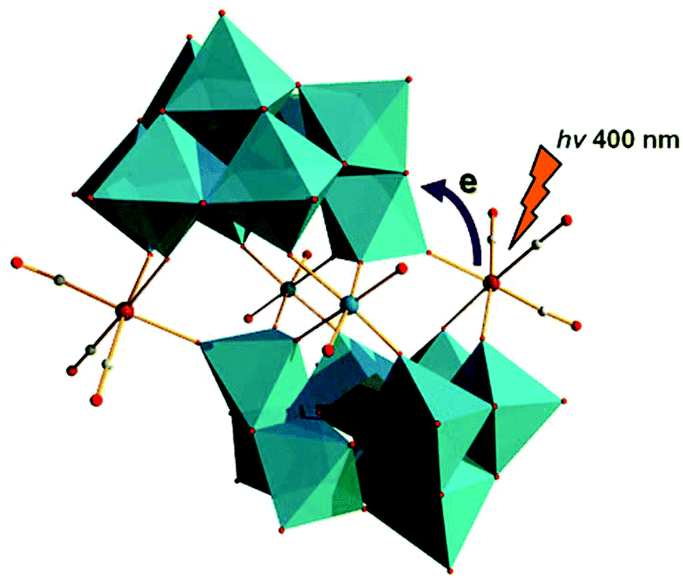 | ||
| Fig. 8 Polyhedral/ball-and-stick representation of [X2W20O70{M(CO)3}2]12− (X = Sb/Bi; M = Mn/Re). Colour code: Re, pink; O, red; C, grey; W, jasper; MO6, jasper. | ||
In the following year, Hill et al. reported several “twisted-sandwich” PMCDs,74 K7Na3P2W23O80{Re(CO)3}2·38H2O, (C3H10N)8Na2P2W23O80{Re(CO)3}2·10H2O, and (C3H10N)6KNa3P2W23O80{Mn(CO)3}2·7H2O, which were obtained by the reaction of [α-PW11O39]7− with solvated [M(CO)3]+ (M = Re, Mn) in acidic aqueous solutions (pH < 4). Upon decreasing the pH value, the solution colours become darker, indicating an ongoing dimerization process. During the self-assembly processes, the resulting di-coordinated intermediates are chiral building blocks, but they exist as a racemic enantiomeric pair in the unit cells. This study provides us with more convenient syntheses and a novel POM scaffold, named the “twisted-sandwich” (Fig. 9).
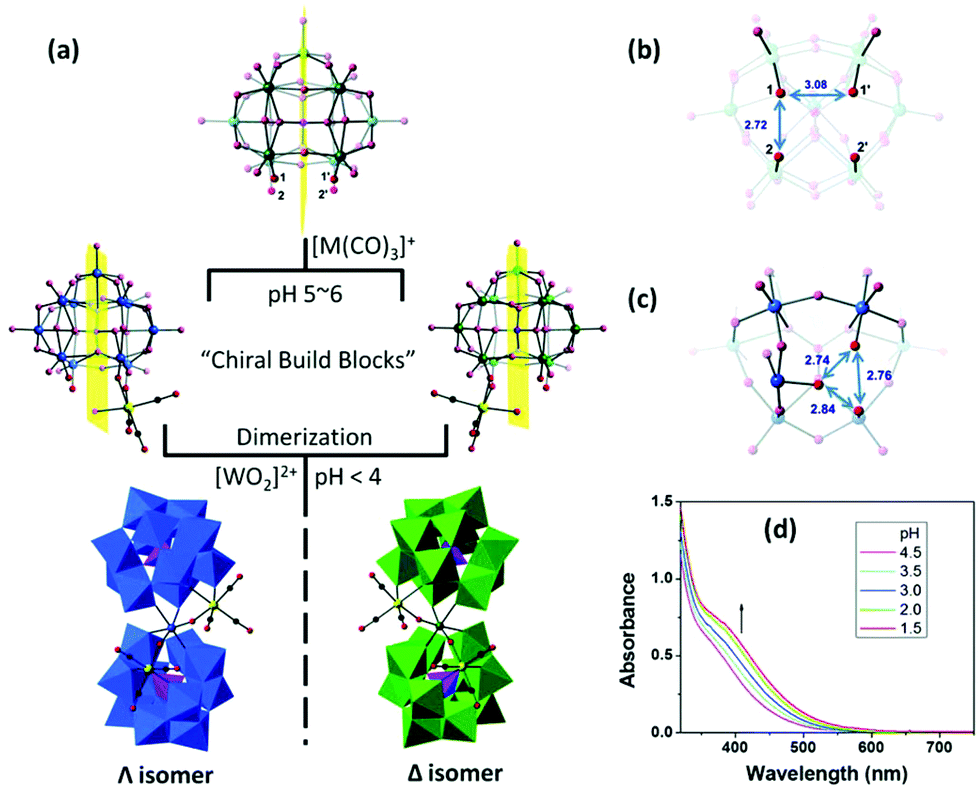 | ||
| Fig. 9 (a) Scheme of proposed synthetic pathways of K7Na3P2W23O80{Re(CO)3}2·38H2O and (C3H10N)6KNa3P2W23O80{Mn(CO)3}2·7H2O. Color code: O, red; C, black; P and PO4, violet; Re or Mn, yellow; W and WO6, blue or green. The yellow rectangles represent the mirror planes in [α-PW11O39]7−. (b) Representative distances (unit: Å) of the four terminal oxygens in [α-PW11O39]7−. (c) The trigonal coordination sites (Å). (d) UV-vis absorption spectra of K7Na3P2W23O80{Re(CO)3}2·38H2O. The first spectrum was recorded after 0.5 h of reaction. Spectra thereafter were recorded upon titration. | ||
In 2014, our group reported an undecatungstoarsenate-supported carbonyl rhenium derivative,75 [(AsW11O39){Re(CO)3}3(μ3-OH)(μ2-OH)]6−, which consists of one monovacant Keggin structure unit and a typical trimeric carbonyl rhenium cluster. [(CH3)4N]13[H11(AsW11O39)4{(Re(CO)3)3(μ3-OH)(μ2-OH)}4]·23H2O was synthesized by the reaction of Re(CO)5Cl and [(CH3)4N]8[HAsW9O34]·11H2O in the CH3CN-H2O mixed solvent under mild conditions. The trivacant metastable polyanion [HAsW9O34]8− could easily transform into the [AsW11O39]7− configuration in a weakly acidic environment (pH > 5 nearly). To the best of our knowledge, this complex is the only compound with a multinuclear {M(CO)3}+ unit incorporated into the Keggin-type POM (Fig. 10a). Additionally, this complex shows excellent catalytic activity for cycloaddition of epoxy chloropropane, and its potential applications are considered (Fig. 10b).
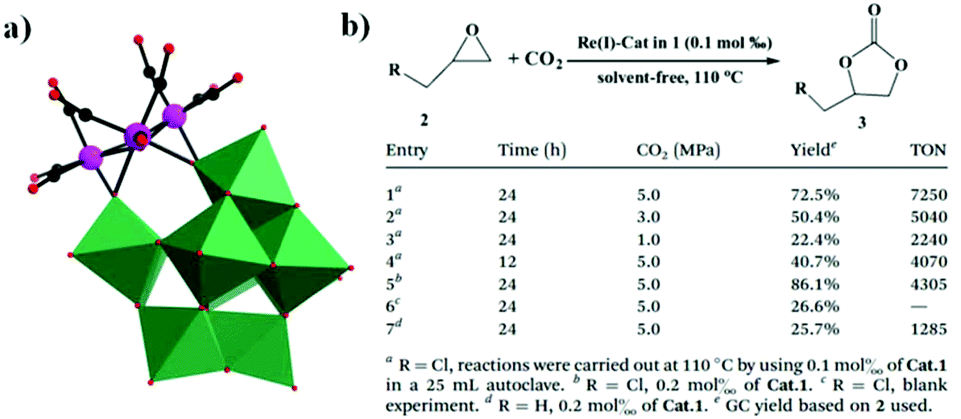 | ||
| Fig. 10 (a) Polyhedral/ball-and-stick representation of the [(AsW11O39){Re(CO)3}3(μ3-OH)(μ2-OH)]6− polyoxoanion. Colour code: O, red; C, black; Re, pink; WO6, green. (b) Cyclic carbonate synthesis from epoxides and CO2. | ||
In the same year, a monovacant Keggin-type POM-supported trirhenium carbonyl derivative, [(CH3)4N]5H23[(PW11O39){Re(CO)3}3(μ3-O)(μ2-OH)]4·24H2O, was synthesized by our group.59 Each lattice unit consisted of 4 [(PW11O39){Re(CO)3}3(μ3-O)(μ2-OH)]7− units, and the crystal structure of the polyoxoanion revealed a “cap” model of the trirhenium carbonyl cluster that was combined with the [PW11O39]8− fragment (Fig. 11a). The POM moiety can be considered an extremely active tetradentate ligand providing excellent coordination to the Re centres of the trirhenium carbonyl cluster. The ability of this compound to catalyse cyclic carbonate synthesis from CO2 and epoxides with an ionic liquid (1-ethyl-1-methylpyrrolidinium bromide) as a cocatalyst was also studied. DFT calculations demonstrated that the Re3–C7 bond is highly activated and more prone to breakage, thus providing proof that the Re3 centre becomes a strong Lewis acid mediator for activating the epoxide. As proposed, the epoxide is first activated by coordination to the Re3 centre, followed by epoxide ring opening upon nucleophilic attack by Br−, and subsequent interaction of the nucleophilic alkoxide intermediate with the electrophilic CO2 to form the cyclic carbonate. Both the Lewis acidic centre (Re3) and the nucleophile (Br−) have the same importance in this mechanism, and the excellent synergistic effects greatly promote the reactions. The proposed mechanism is supported by both theoretical and experimental results and provides new insights into the design of more powerful catalyst systems for the cycloaddition reaction (Fig. 11b and c).
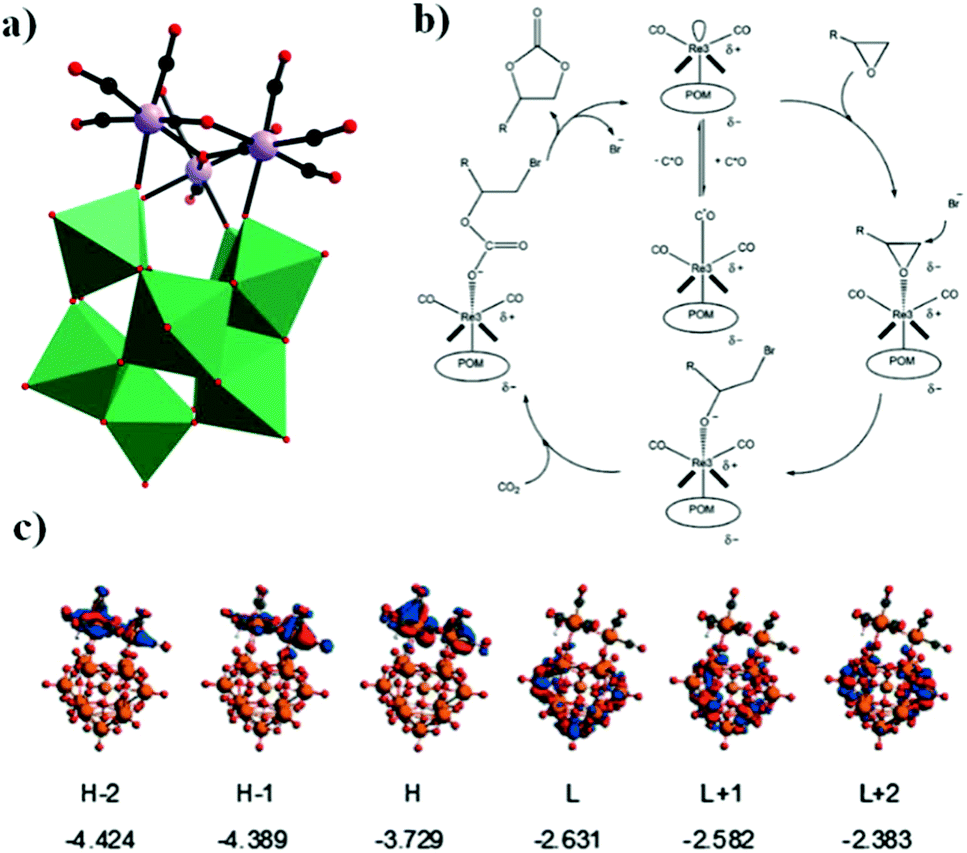 | ||
| Fig. 11 (a) Polyhedral/ball-and-stick representation of the [(PW11O39){Re(CO)3}3(μ3-O)(μ2-OH)]7− polyanion. Colour code: O, red; C, black; Re, purple; WO6, green. (b) Proposed reaction mechanism for the cycloaddition of CO2 to epoxides. (c) Several highest occupied (H = HOMO) and lowest unoccupied (L = LUMO) molecular orbitals of polyoxoanion, with the orbital energies given in eV. | ||
In 2015, our group prepared a new trivacant POM-based manganese carbonyl derivative,76 (NH4)3H3[{Mn(CO)3}(Mn(H2O)2)(Mn(H2O)3)(TeW9O33)]2·31H2O, which was isolated in the mixed solvent of acetonitrile and water at room temperature. Notably, the polyanion [{Mn(CO)3}(Mn(H2O)2)(Mn(H2O)3)(TeW9O33)]6− is composed of two [{Mn(CO)3}(β-B-TeW9O33)]7− building blocks linked via a [Mn4(H2O)10]8+ cluster, resulting in a stable “hamburger” structure (Fig. 12). This compound was synthesized by using a conventional method, and the raw material MnCl2 plays an important role in the synthetic process; the complex cannot be produced in the absence of MnCl2. In particular, the ratio of CH3CN to H2O also affects the crystal growth, and the optimal ratio is 1:4 to 1:5 because the solubilities of metal carbonyls and POMs are very different; hence, adjusting the mixing ratio of the solvent to optimize dissolution of both complexes is necessary.
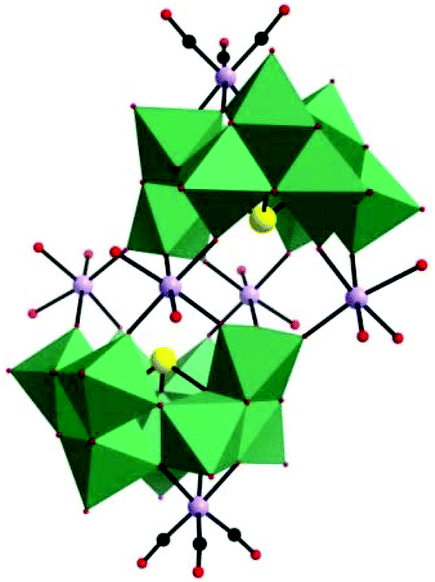 | ||
| Fig. 12 Polyhedral/ball-and-stick representation of [{Mn(CO)3}(Mn(H2O)2)(Mn(H2O)3)(TeW9O33)]6−. Colour code: O, red; C, black; Mn, light purple; Te, yellow; WO6, green. | ||
In the same year, our group reported a nonavacant Keggin-type rhenium tricarbonyl derivative, [(NH4)5]{[PMo3O16][Re(CO)3]4}·1.5H2O, which showed good catalytic activity for the cycloaddition reaction with pyrrolidinium bromide as a cocatalyst.77 The DFT calculations prove that the HOMOs are localized on the tricarbonyl rhenium fragment of [PMo3O16{Re(CO)3}4]5−; in contrast, the LUMOs localized entirely within the [PMo3O16]9− moiety are principally W–O antibonding orbitals (Fig. 13a and b). In this case, the Re2 atom is proven to be the Lewis acid centre when it is in the excited state, and the halide ions in ionic liquids act as Lewis base centres that could substantially lower the activation energy barrier of the cycloaddition reaction.
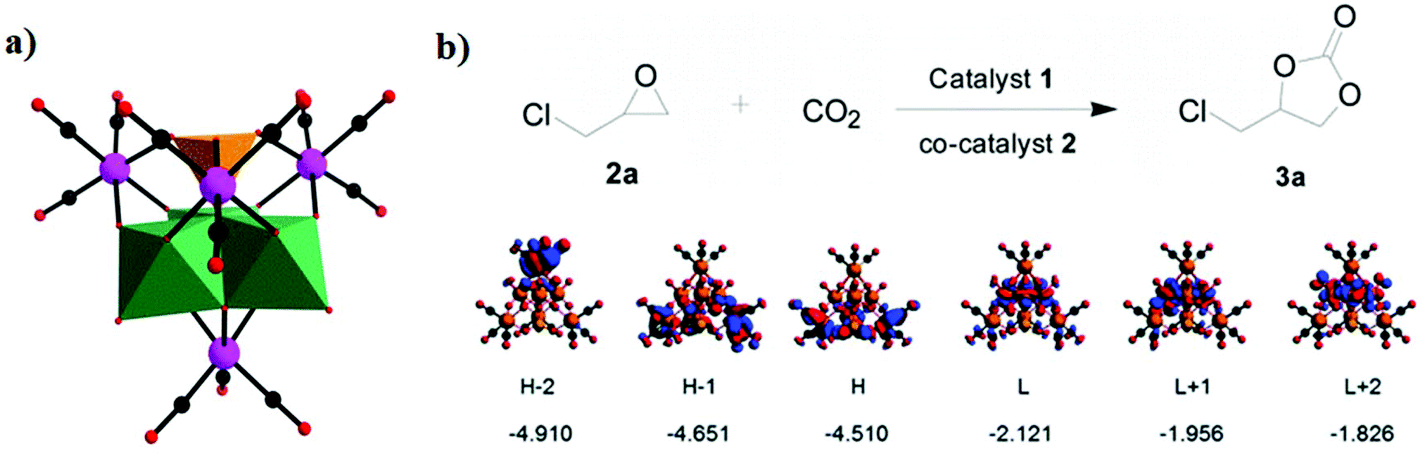 | ||
| Fig. 13 (a) Polyhedral/ball-and-stick representation of [PMo3O16{Re(CO)3}4]5−. Colour code: O, red; C, black; Re, pink; PO4, orange; WO6, green. (b) The cycloaddition of CO2 with epoxides with pyrrolidinium bromide as a cocatalyst and several highest occupied (H = HOMO) and lowest unoccupied (L = LUMO) orbitals of 1a and their orbital energies in eV. | ||
In 2016, our group reported four novel organic–inorganic hybrid Keggin-type PMCDs, [(M4(H2O)10)(XW9O33)2{Mn(CO)3}2]n− (X = Sb/Bi; M = Mn/Mn3.5W0.5), which were prepared by the reaction between [XW9O33]9− (X = Sb/Bi), Mn(CO)5Br and Mn2+ or the reaction between [{Mn(H2O)}3(SbW9O33)2]12−/[(Mn(H2O)3)2(WO2)2(BiW9O33)2]10− and Mn(CO)5Br.78 Single-crystal X-ray diffraction crystallography indicated that these complexes are the first examples of structurally characterized transition metal-substituted sandwich-type tungsto-antimonates/bismutates incorporated with carbonyl manganese groups. The organic–inorganic hybrids are composed of two {Mn(CO)3} groups attached to a dimeric heteropolytungstate {M4(B-β-XW9O33)2} unit via six MnI–O–W bonds. Furthermore, the {M4(B-β-XW9O33)2} unit consists of a central symmetric parallelogram-like tetra-MnII cluster sealed into two identical [B-β-XW9O33]9− fragments through MnII–O–W bonds. The [(Mn4(H2O)10)(XW9O33)2{Mn(CO)3}2]8− (X = Sb/Bi) proved to be efficient for the electrocatalytic reduction of NO2−.
In 2019, our group prepared a monomeric tellurotungstate(IV)-supported rhenium carbonyl derivative, Na2H2[(CH3)4N]6[Te2W20O70{Re(CO)3}2]·20H2O,79 by the one-pot reaction of Re(CO)5Cl, Na2WO4·2H2O and Na2TeO3 (Fig. 14). Single-crystal X-ray diffraction analysis revealed that this polyoxoanion was similar to the [X2W20O70{M(CO)3}2]12− (X = Sb, Bi and M = Re, Mn) polyoxoanions reported by Hill et al. The crystal structure of this compound comprises two [TeW10O35{Re(CO)3}]5− fragments, and each rhenium carbonyl group fac-{Re(CO)3}+ was stabilized by a [TeW10O35{Re(CO)3}]5− ligand in the “out-of-pocket” structural motif. In addition, this complex can be used as an excellent catalyst for the epoxidation of different alkenes using H2O2 as an oxidant in acetonitrile.
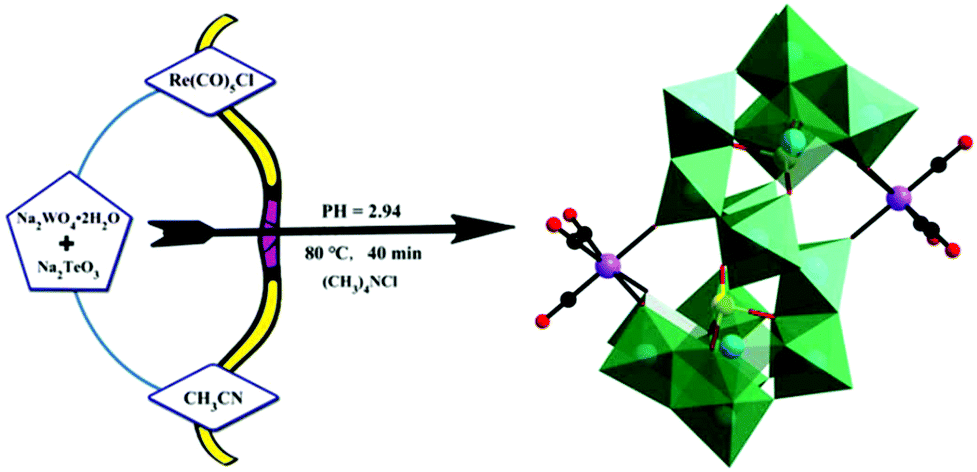 | ||
| Fig. 14 The preparation process of the [Te2W20O70{Re(CO)3}2]10− polyoxoanion. Colour code: O, red; C, black; Re, purple; Te, yellow; WO6, green. | ||
3.3 Dawson-type PMCDs
The first pioneering studies based on Dawson-type PMCDs were reported in 1997.80 Finke et al. reported two derivatives, [{Re(CO)3}P2W15Nb3O62]8− and [{Ir(CO)3}P2W15Nb3O62]8−, but the structures of these complexes have not been verified by single-crystal X-ray diffraction analysis. The [{Ir(CO)3}P2W15Nb3O62]8− complex is stable in the absence of water but decomposes quickly in the presence of even 1 equiv. of water. These authors also attempted to synthesize the analogous [P2W15Nb3O62]9−-supported Rh(CO)2+ complex; however, 31P NMR revealed that this compound was unstable in solution at room temperature. Importantly, in this study, the authors also provided the first direct evidence that the overall symmetry of organometallic cations supported on soluble basic oxides can be altered by the presence of cations such as Na+ cations.There were no reports of Dawson-type PMCDs from 1997–2010, but in 2011, Hill et al. reported a new complex comprising a [Re(CO)3]+ unit supported on the defect Wells–Dawson-type POM [α2-P2W17O61]10−, which exhibits good visible-light-induced photoredox activity.81 The resulting complex, [P4W35O124{Re(CO)3}2]16−, was prepared by mixing equivalent amounts of K10[α2-P2W17O61]·20H2O and Re(CO)3(CH3CN)3(BF4) in an acidic aqueous solution. In this structure, each [Re(CO)3]+ moiety is stabilized by an [α2-P2W17O61]10− ligand in an “out-of-pocket” structural motif (Fig. 15a). Comprehensive computational and spectroscopic studies have shown that the intense visible absorption of this complex can be attributed to an MPCT transition involving CT from the Re(I) centre to the POM. In addition, the orbitals and transition in this case are distinct from those in the well-documented heterobimetallic systems because the acceptor orbitals are delocalized and multimetallic.
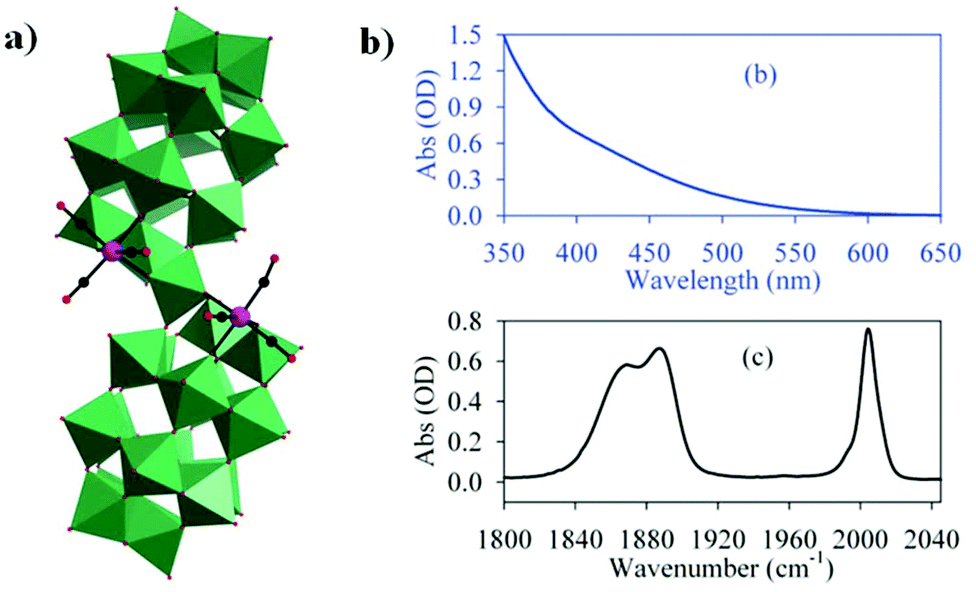 | ||
| Fig. 15 (a) Polyhedral and ball-and-stick representation of the [P4W35O124{Re(CO)3}2]16− polyoxoanion. Colour code: Re, pink; O, red; C, black; WO6, green. (b) UV-vis and (c) Fourier transform infrared (FTIR) spectra in CH2Cl2. | ||
In 2012, we showed that the introduction of Dawson-type precursors can result in several compounds that consist of [Mn(CO)3(CH3CN)3]+ cations and polyoxoanions.82 [Mn(CO)3(CH3CN)3]6[α-X2M18O62]H2O [X = PV, M = MoVI, WVI; X = AsV, M = MoVI, WVI] complexes were isostructural and were obtained in CH3CN/H2O solvents by exploring different solvent systems, such as CH3COCH3/H2O, C4H8O/H2O, CH3CH2OH/H2O, and CH3OH/H2O.
In 2013, a POM-supported trirhenium carbonyl cluster, [P2W17O61{Re(CO)3}3{ORb(H2O)}(μ3-OH)]9−, was synthesized and characterized.83 This complex was prepared by the reaction between a POM precursor [P4W35O124{Re(CO)3}2]16− and [Re(CO)3]+ complexes in a slightly acidic (PH = 5–6) aqueous solution. The crystal structure of this complex reveals an “out-of-pocket” motif with a trirhenium carbonyl “cap” grafted on the defect site of [α2-P2W17O61]10− (the “support”), and the three Re(I) centres reside in different coordination environments. Interestingly, this complex is the first example of an oxocentered multimetal electron-donor substructure, mimicking a metal oxide-supported interfacial dyadic structure.
A comprehensive investigation of the photophysical properties of this complex using computational and two different time-resolved spectroscopic methods clearly reveals the presence of MPCT. TA spectroscopy was used to characterize the CT dynamics occurring at the interfaces of this “double cluster”, and time-resolved infrared spectroscopy was applied to provide detailed information on the excited states of [Re(CO)3]+ complexes (Fig. 16). Additionally, it was found that the lifetime of this charge-separated excited state is short, and thus capturing it to drive a chemical reaction is challenging.
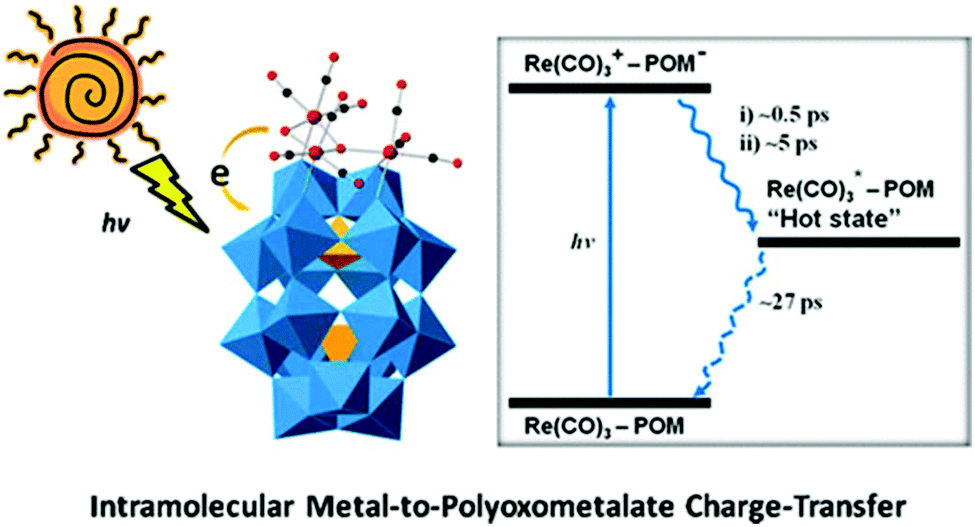 | ||
| Fig. 16 The scheme of intramolecular MPCT. Colour code: O, red; C, black; Re, pink; PO4, orange; WO6, blue. | ||
In 2015, Sadakane et al. reported an α2-isomer of a mono-Ru-substituted Dawson-type heteropolytungstate with a carbonyl (CO) ligand,84 [α2-P2W17O61RuII(CO)]8−, which was obtained by the reaction of [α2-P2W17O61]10− and Ru(acac)3 at 170 °C for 4 days. Note that this is the first single-crystal structure analysis of mono-Ru(CO)-substituted heteropolytungstate (Fig. 17).
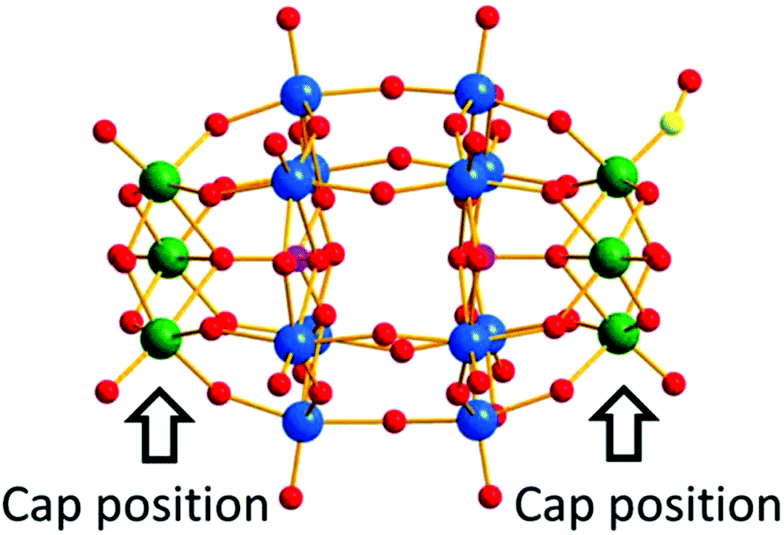 | ||
| Fig. 17 Ball-and-stick representation of [α2-P2W17O61Ru(CO)]8−. Green, blue, pink, yellow, and red balls represent belt tungsten atoms, cap tungsten atoms, and ruthenium, phosphorus, carbon, and oxygen, respectively. | ||
In 2017, our group obtained a POM-supported [Mn(CO)3]+ complex containing the mixed-metal cubane [Na(H2O)5](NH4)7[P2W15O56Co3(H2O)3(OH)3Mn(CO)3]·19H2O by the one-pot reaction of [α-P2W15O56]12− with Co(OAc)2·4H2O and Mn(CO)5Br in a slightly acidic (pH = 6.0) solution.85 Notably, this compound is the first example of a monomeric tricobalt(II)-substituted Dawson-type PMCD. This new polyanion can be described as grafting the metal carbonyl group {Mn(CO)3} onto the tricobalt-substituted POM framework {P2W15Co3O62}, which is similar to that of the previously reported Dawson-type [P2W15Nb3O62]9− polyoxoanion-supported Re(CO)3+ complex, [P2W15Nb3O62Re(CO)3]8−. The {P2W15Co3O62} subclass consists of a {Co3O6} cap embedded in the defect site of the [α-P2W15O56]12−, forming a P2W18-like saturated Well–Dawson structure (Fig. 18a). As shown in Fig. 18b, this complex shows weaker ferromagnetic interactions at low temperature. Furthermore, it also shows high catalytic activity in the cycloaddition of CO2 with epoxides under mild reaction conditions with pyrrolidinium bromide as a cocatalyst (Fig. 18c).
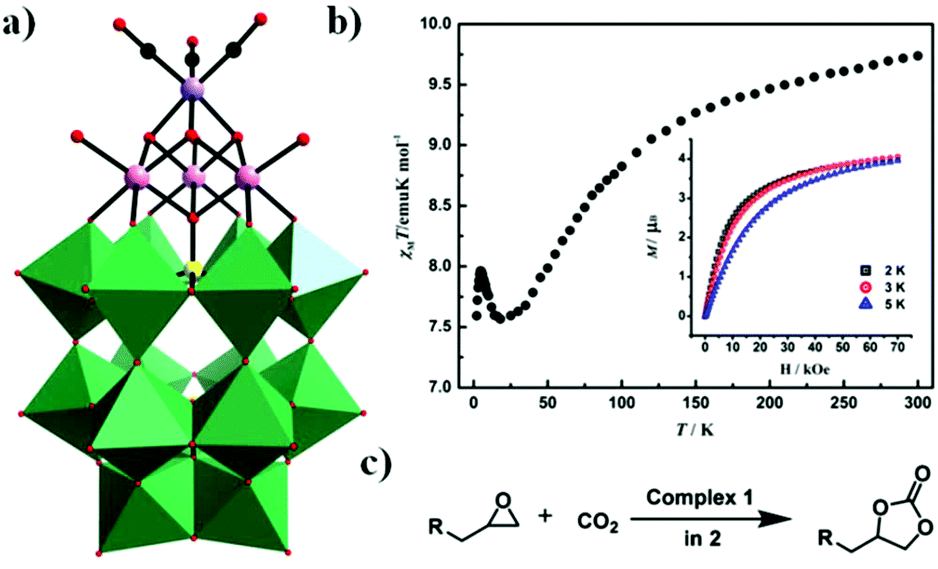 | ||
| Fig. 18 Polyhedral and ball-and-stick representation of [P2W15O56Co3(H2O)3(OH)3Mn(CO)3]8− Colour code: O, red; C, black; Mn, light purple; Co, light pink; P, yellow; WO6, green. (b) Plots of χMT versus T for the compound. Inset: Field dependence of magnetization at different temperatures. (c) The cycloaddition of CO2 to epoxides with pyrrolidinium bromide. | ||
In 2018, our group prepared a monomeric trinickel(II)-substituted Dawson-type PMCD,86 Na(NH4)5[P2W15O56Ni3(H2O)3(μ3-OH)3(Re(CO)3)3]·18H2O, which was isolated in the one-pot reaction of [α-P2W15O56]12− with Ni(OAc)2·4H2O and [Re(CO)5]+. The polyanion can be described as a hetero-metallic cluster {Ni3O6Re3(CO)9} stabilized by an [α-P2W15O56]12− ligand in the defect structural motif through six μ3-O bridges from six WO6 octahedra and one μ3-O bridge from the central PO4 tetrahedron (Fig. 19a). Notably, this polyanion is unprecedented and is the first example of a monomeric hexasubstituted Dawson structure decorated with low-valent transition metal Ni ions and metal carbonyl groups. A possible formation process of this polyanion is as follows: first, three scattered Ni(II) ions are captured by the [P2W15O56]12− polyanion to form a monomeric trisubstituted Dawson unit, β-Ni3P2W15. As depicted in Fig. 19b, three [Re(CO)3]+ fragments are attached to the β-Ni3P2W15 unit to form a {Ni3Re3} cluster-substituted Dawson POM.
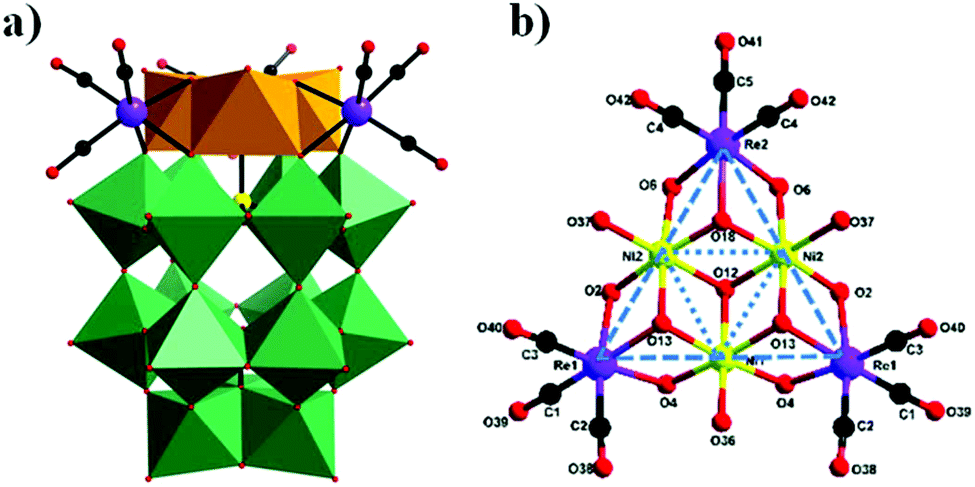 | ||
| Fig. 19 (a) Polyhedral /ball-and-stick representation of [P2W15O56Ni3(H2O)3(μ3-OH)3{Re(CO)3}3]6−. (b) Representations of the {Ni3Re3} cluster. Colour code: O, red; C, black; Re, purple; P, yellow; NiO6, orange; WO6, green. | ||
3.4 Nonclassical-type PMCDs
In parallel with most classical-type PMCDs possessing representative structural features, nonclassical-type compounds have also experienced rapid development in recent years. In 2000, Gouzerh et al. reported a series of POM-incorporated metal carbonyl complexes,87 such as (nBu4N)[Mo2O5(OMe)5{M(CO)3}2] (M = Mn/Re) (Fig. 20a), [Mo2O4(OMe)6{Mn(CO)3}2] (Fig. 20b), (nBu4N)2[Mo2O6(OMe)4{Re(CO)3}2] (Fig. 20c), (nBu4N)[Mo2O4{MeC(CH2O)3}2{Mn(CO)3}] (Fig. 20d), [Mo2O4{MeC(CH2O)3}2{Mn(CO)3}2] (Fig. 20e), and (nBu4N)2[Mo6O16(OMe)2{MeC(CH2O)3}2{Mn(CO)3}2] (Fig. 20f), characterized by X-ray crystal diffraction analysis. These structures were obtained by the reaction of [MBr(CO)5] or solvated {M(CO)3}+ ions (M = Mn/Br) with (nBu4N)2[Mo2O7] in methanol, sometimes in the presence of triols of the type RC(CH2OH)3 (R = Me or CH2OH). It should be noted that these clusters are the first examples of POMs incorporating {Mn(CO)3}+ and {Re(CO)3}+ units and constitute a family of organometallic complexes whose structures led to the recognition of topologically equivalent organometallic and oxo(alkoxo)metal units. Moreover, (nBu4N)[Mo2O5(OMe)5{M(CO)3}2] (M = Mn/Re) represent the first observation of discrete mono-cubane-type POM derivatives, as the frameworks of other complexes are based on tetranuclear units that display the common rhomb-like structure. Furthermore, these molecular structures have been determined by single-crystal diffraction analysis, and the structural relationships reveal some electronic connections between low-valent and high-valent complex fragments.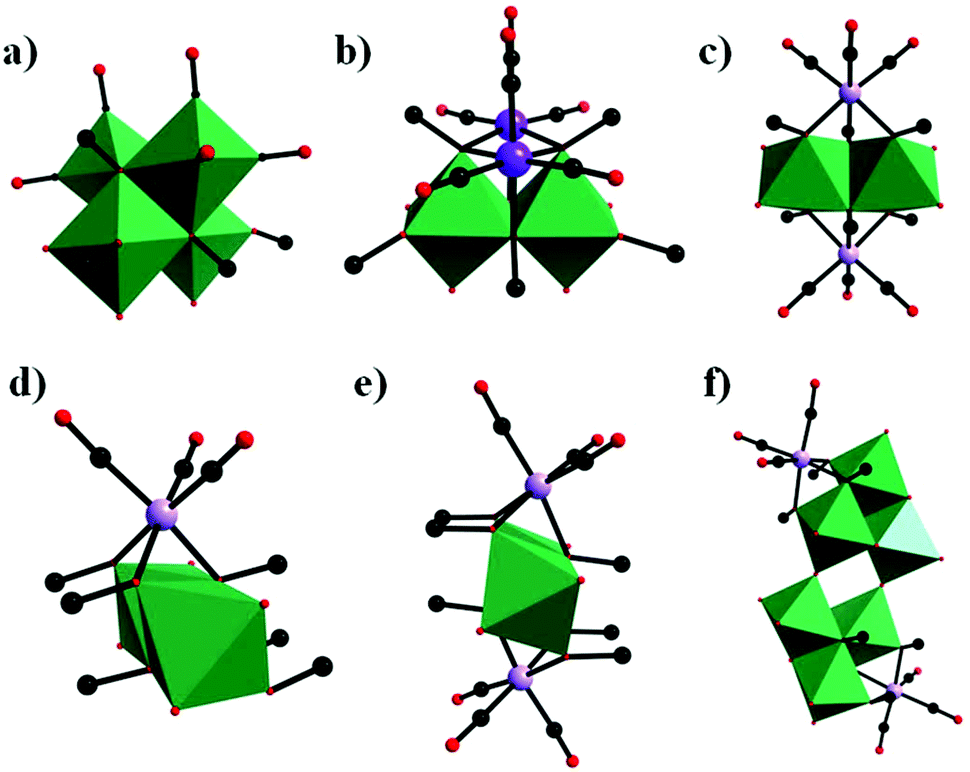 | ||
| Fig. 20 Polyhedral and ball-and-stick representations of [Mo2O5(OMe)5{M(CO)3}2]− (M = Mn/Re) (a), [Mo2O4(OMe)6{Mn(CO)3}2] (b), [Mo2O6(OMe)4{Re(CO)3}2]2− (c), [Mo2O4{MeC(CH2O)3}2{Mn(CO)3}]− (d), [Mo2O4{MeC(CH2O)3}2{Mn(CO)3}2] (e), and [Mo6O16(OMe)2{MeC(CH2O)3}2{Mn(CO)3}2]2− (f). Colour code: O, red; C, black; Re/Mn, purple; MoO6, green. | ||
Then, our group made a breakthrough in the field of nonclassical-type PMCDs. In 2008, we prepared a polyoxoanion-incorporated {Mn(CO)3+} complex,88 (n-Bu4N)2[Mo6O16(OCH3)2{HOCH2C(CH2O)3}2{Mn(CO)3}2], by the reaction of (n-Bu4N)4[Mo8O26] with Mn(CO)5Br in methanol in the presence of C(CH2OH)4. Furthermore, single-crystal structure analysis revealed that the polyoxoanion is located at a crystallographic inversion centre and consists of two tetranuclear [Mo3O8(OCH3){HOCH2C(OCH2)3}{Mn(CO)3}]− moieties (Fig. 21).
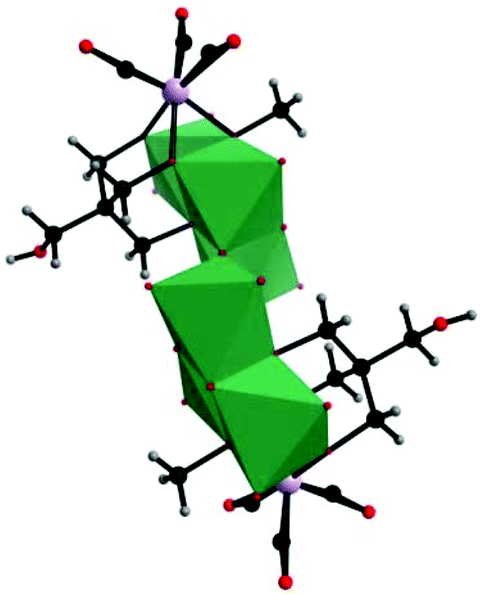 | ||
| Fig. 21 Polyhedral and ball-and-stick representation of [Mo6O16(OCH3)2{HOCH2C(CH2O)3}2{Mn(CO)3}2]2−. Colour code: O, red; C, black; H, grey; Mn, purple; MnO6, green. | ||
In the same year, we also reported three novel octatungstate-supported tricarbonyl metal derivatives,89 H6[Na(H2O)5]2{[H2W8O30][Mn(CO)3]2}·13H2O, H2[Na(H2O)5]2[Na(H2O)4]2[Na(H2O)2]2{[H2W8O30][Re(CO)3]2}·13H2O, and [Na(H2O)5]2 [Na2(μ2-H2O)2(H2O)4]2[Mn(H2O)2]{[H2W8O30][Mn(CO)3]2}, which were prepared by the reaction between Na2WO4·2H2O and M(CO)5Br (M = MnI, ReI). These structures not only are the first examples of isopolyoxotungstate-supported metal carbonyl compounds and enrich the structural diversity of POM-based organometallic compounds but also realize the merging of metal carbonyl chemistry and isopolyoxotungstate chemistry. X-ray diffraction analyses reveal that the first two structures are almost isomorphic and exhibit isolated topologies, whereas the third structure displays a 1D chain architecture. This grafting of a [Mn(CO)3]+ pendant cap on a POM unit via three bridging oxygen atoms is similar to the reported compounds (Fig. 22).
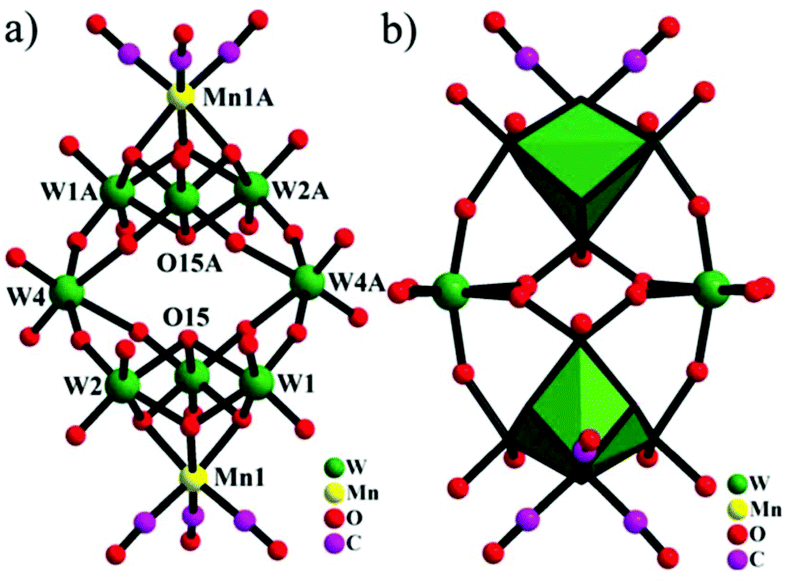 | ||
| Fig. 22 (a) Ball-and-stick representation of [H2W8O30{Mn(CO)3}2]8− with the selected labelling scheme. (b) Combined polyhedral/ball-and-stick representation of polyoxoanion with the selected labelling scheme. Cyan octahedra: {W3MnO4} cubanes. | ||
Five years later, we successfully obtained the first two examples of octamolybdate-supported tricarbonyl metal derivatives,90 (NH4)4[H4{[H2Mo8O30][Mn(CO)3]2}]·12H2O, and (NH4)4[{H6Mo8O30}{Re(CO)3}2]·14H2O, which were prepared by the reaction of (NH4)6[Mo7O24]·4H2O with Mn(CO)5Br and Re(CO)5Cl, respectively. Single-crystal X-ray diffraction analysis revealed that both polyanions are isostructural and contain a [H2Mo8O30]10− framework that is grafted by two tricarbonyl metal fragments. Notably, the [{H2Mo8O30}{M(CO)3}2]8− clusters have not been reported until now in the POM-based organometallic family. As a part of our continuous work, we also obtained another octatungstate-supported tricarbonyl metal polyanion, Na8(H2O)18(CH3COOH)2[{H2W8O30}{Mn(CO)3}2]·4H2O, with four crystallographically independent Na+ ions, by reacting Na2WO4·2H2O, ErCl3·6H2O, CH3COOH and Mn(CO)5Br. The preparations of these structures provide us with an effective and feasible method of designing novel isopolyoxomolybdate and isopolyoxotungstate metal carbonyl derivatives.
In 2018, our group reported two monomeric tellurium-containing heteropolymolybdate-supported metal carbonyl derivatives,91 (NH4)5H3{[Te2Mo12(OH)O44][Mn(CO)3]}·18H2O and (NH4)8{[Te2Mo12(OH)O44][Re(CO)3]}·13H2O, which were obtained by the conventional mixed-solvent solution method. To the best of our knowledge, these two compounds are the first examples of metal carbonyl groups supported on a telluromolybdate framework. A detailed crystallographic analysis reveals that the two compounds are isostructural and that the polyoxoanion [Te2Mo12(OH)O44]9− represents a nonclassical anion with considerably different bonding modes (Fig. 23). The irregular [Te2Mo12(OH)O44]9− framework comprises one upper [TeMo3(OH)O12]3− moiety and one lower [TeMo7O29]12− moiety, which are aggregated together by two corner-sharing {MoO6} octahedra, forming a like-Well–Dawson-type structure. Moreover, the complexes exhibit good electrocatalytic activity for the reduction of nitrite.
 | ||
| Fig. 23 (a) Formation of (NH4)5H3{[Te2Mo12(OH)O44][Mn(CO)3]}·18H2O and (NH4)8{[Te2Mo12(OH)O44][Re(CO)3]}·13H2O. (b) Polyhedral and ball-and-stick representations of [Te2Mo12(OH)O44]9−. Polyhedral representations of (c) P2W18; (d) Mo7O29 moiety in [Te2Mo12(OH)O44][Mn(CO)3]8−; (e) Mo7O24 unit in (NH4)6[Mo7O24]·4H2O. Colour code: Te, yellow; Mo/MoO6 octahedra, azure; Mn, blue; O, red; C, grey; WO6, light blue; PO4, pink. | ||
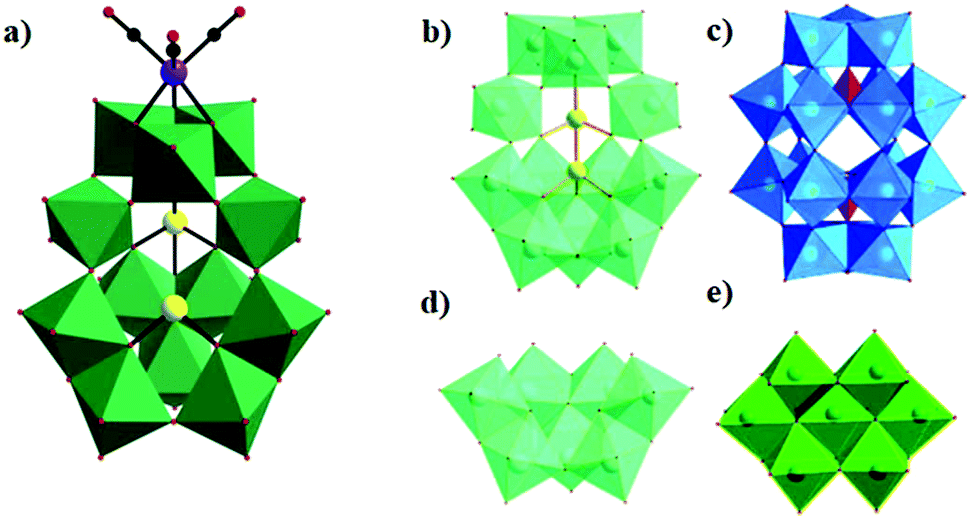
Then, we prepared a novel isotetramolybdate-supported rhenium carbonyl derivative, [(CH3)4N]4[{Re(CO)3}4(Mo4O16)]·H2O, by the reaction of Na2MoO4·2H2O with tetramethylammonium chloride and Re(CO)5Cl in a mixed solution (CH3CN:H2O = 3:10) at 80 °C. Single-crystal X-ray diffraction analysis reveals that the polyoxoanion can be described as grafting the four {Re(CO)3} units onto the {Mo4O4} cubane (Fig. 24).92 Moreover, the compound can be used as an efficient catalyst for the oxidation of thioanisole into the corresponding sulfoxide in the presence of hydrogen peroxide.
 | ||
| Fig. 24 (a) Ball-and-stick representation of [H2W8O30{Mn(CO)3}2]8− with the selected labelling scheme. (b) Combined polyhedral/ball-and-stick representation of polyoxoanion with the selected labelling scheme. Cyan octahedra: {W3MnO4} cubanes. | ||
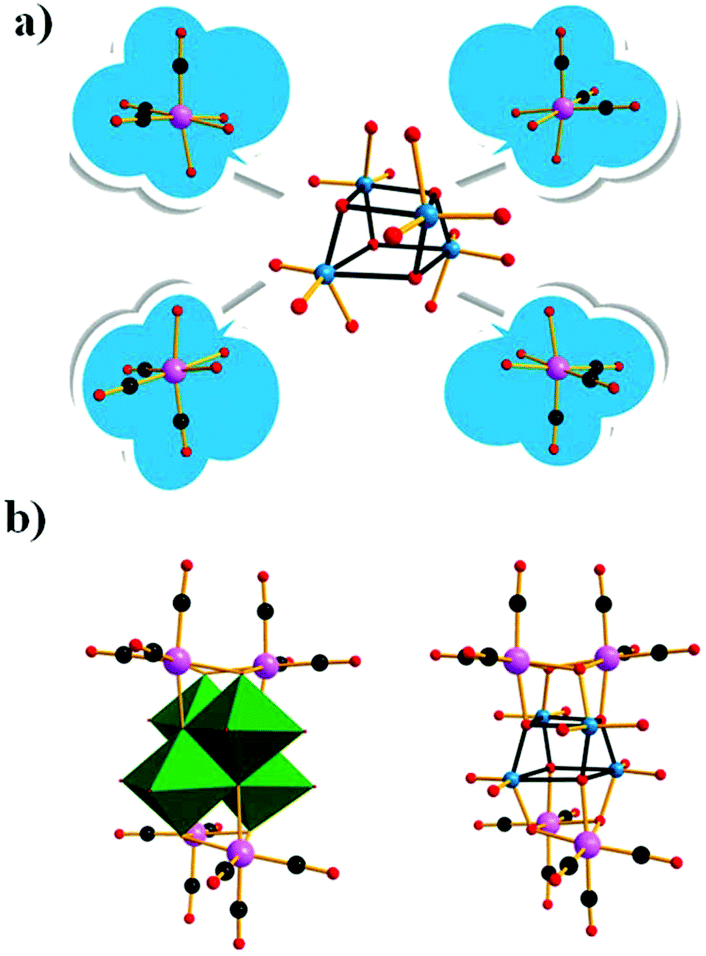
In the same year, we reported a novel tetracarbonyl metal selenotungstate derivative,93 Na1.5H4.5[(CH3)4N]2{[Mn(CO)3]4(Se2W11O43)}·9H2O, which was synthesized via one-pot synthesis by the reaction of Na2WO4, SeO2, and Mn(CO)5Br with a pH-controlled assembly (pH 4.8–5.6) in a mixed CH3CN/H2O solvent. In addition, the {Se2W11} fragment is composed of the novel {SeW3} moiety and {SeW8} unit via two μ2-O atoms and is completely different from the reported {Se2W11} fragment composed of the unreported {SeW4} and {SeW7} species, which are linked in a Wells–Dawson-like assembly (Fig. 25). The successful preparation of this compound provides us with an enlightening synthetic method for covalent grafting of metal carbonyl groups onto POM clusters and further guides the design of high-nuclear PMCDs, though it is quite difficult to obtain these complexes due to the steric hindrance of the organic groups. Moreover, this complex showed good activity in catalysing cyclic carbonate synthesis from epoxides and CO2 under mild reaction conditions in conjunction with 1-ethyl-1-methylpyrrolidinium bromide.
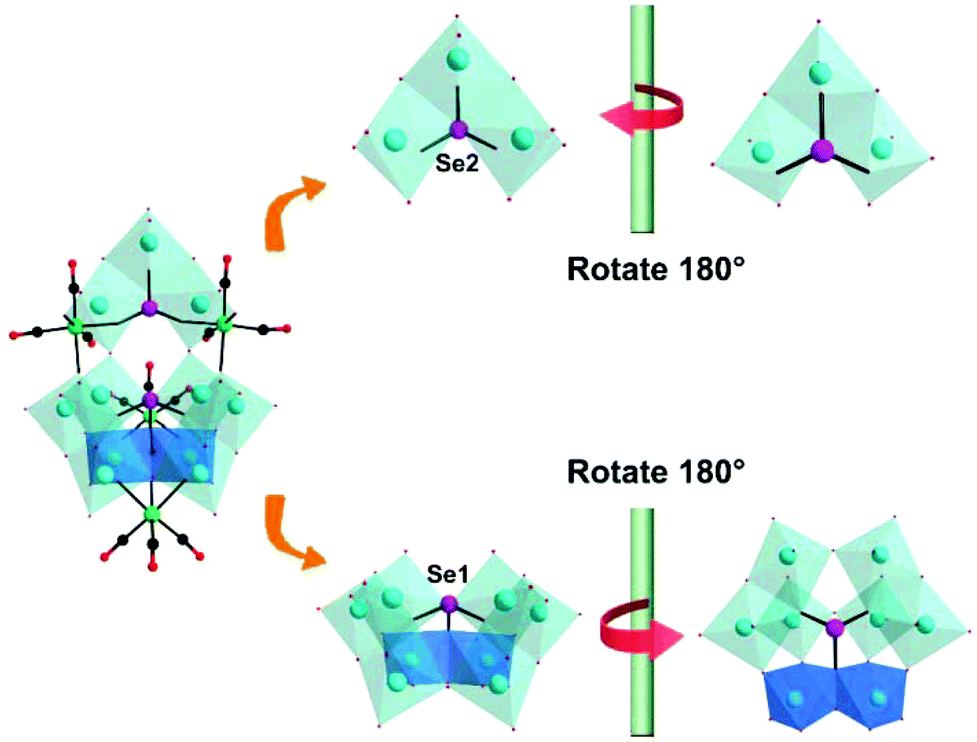 | ||
| Fig. 25 Polyhedral and ball-and-stick representations of the {Se2W11} fragment and its basic building blocks. Colour code: O, red; C, black; Re, green; W, cyan; Se, rose; O, red. | ||
4. Relative applications
4.1 Catalytic properties
As we know, POMs are a type of intriguing catalyst that can be applied for a wide range of applications owing to their reasonably high thermal stability, and reversible electron transfer ability under mild conditions. Moreover, metal carbonyl complexes can also exhibit interesting catalytic properties in many reactions. Consequently, relevant catalytic studies on PMCDs, possessing advantages of both POMs and metal carbonyl complexes, have been conducted. In 1987, Siedle et al. reported that [(Ph3P)2Rh(CO)(CH3CN)]8−nXM12O40 and [(Ph3P)2Rh(CO)]8−nXM12O40 (X = P/Si; M = W/Mo; n = 3, 4) were bifunctional catalysts in hydrogenation, which are capable of activating carbon monoxide and hydrogen at the rhodium centers as well as oxygen at tungsten oxide surfaces.71 Recently, in 2014, our group reported a monovacant Keggin-type PMCD, [(AsW11O39){Re(CO)3}3(μ3-OH)(μ2-OH)]6−, and it can be used as a catalyst for the synthesis of cyclic carbonates from carbon dioxide and epoxides under mild reactions with co-catalyst pyrrolidinium bromide.75 Using 0.1 mol% (relative to epoxide) of the catalyst, the yield achieved was 84.3% and the outstanding turnover frequency (TOF) of 843 h−1 was superior to that of the tricarbonyl rhenium(I) complex reported by Wong et al. (≈122) h−1.94 The mechanism proposed is supported by both theoretical and experimental results which provides new insights into the design of more powerful catalyst systems for the cycloaddition reaction. Then, [(AsW11O39){Re(CO)3}3(μ3-OH)(μ2-OH)]6−, [{PMo3O16}{Re(CO)3}4]5− and [{Mn(CO)3}4(Se2W11O43)]8−, reported by us in 2014, 2015, and 2018, respectively, could act as Lewis acid catalysts and promote the conversion of CO2 to cyclic carbonate under mild reaction conditions.75,77,93 In 2016, the use of [{Re(CO)3}4(μ2-OH)(μ3-O)(W5O18)]5− in the epoxidation of olefin with aqueous H2O2 provides the corresponding epoxides with a 98.9% conversion and 99% selectivity.564.2 Electrochemistry and electrocatalysis
As we all know, POMs can deliver up to 24 electrons to another species, making them powerful electron reservoirs for multielectron reduction processes. In 2008, Sadakane et al. addressed a new polyoxoanion [α-SiW11O39RuII(CO)]6−, the CV of which showed a well-defined reversible one-electron redox pair and two two-electron redox pairs.72 The controlled potential electrolysis confirmed that this well-defined reversible redox pair indeed corresponds to a single electron transfer. When the pH of the solution was raised, the RuIII/II redox potential did not change, whereas the two-electron redox potential shifted in a more negative direction. The redox potential of RuIII/II is independent of pH, indicating that no proton is involved in the redox reaction. In 2012, our group reported two novel trivacant Keggin-type PMCDs, and CV measurements of both were carried out in the pH 6.7 mixed solvent CH3CN–Na2SO4 (0.4 mol L−1).57 The cathodic peak currents are proportional to the scan rate, demonstrating that the redox processes of both compounds are surface-controlled. The electrocatalytic properties of the two complexes for NO2− oxidation were investigated, and the catalytic currents increase dramatically with the oxidation peak of NO2− with stepwise addition of nitrite. In 2016, two novel Keggin-type PMCDs [{Mn4(H2O)10(β-MW9O33)2{Mn(CO)3}2}]8− (M = Bi/Sb) proved to be efficient for the electrocatalytic reduction of NO2−. With the addition of modest amounts of sodium nitrite, an irreversible oxidation peak appears in the positive range at Epc = 0.85–0.90 V (this id expected for NO2−), while the reduction peak current of the WVI-based wave increase progressively.78 In 2018, two monomeric non-classical tellurium-containing PMCDs, [{Te2Mo12(OH)O44}{M(CO)3}]8− (M = Re/Mn), also exhibit well electrocatalytic activities toward the reduction of nitrite.915. Conclusion and outlook
In recent decades, a large number of PMCDs have been prepared, and there are strong motivations to study the excellent photoelectric and catalytic properties of these complexes. In this review, synthetic strategies, structural diversities and relevant applications of PMCDs are presented. Conventional solution synthesis has already been proved to be a powerful and efficient method for synthesizing PMCDs, while the hydrothermal synthesis approach is rarely used. Various structural PMCDs have been divided into four general families: Lindqvist-type, Keggin-type, and Dawson-type complexes and other nonclassical compounds. This article shows that a large variety of nonclassical-type PMCDs have been isolated by one-pot synthesis, but this probably represents only a small amount of what is possible, in view of the very large number of POM precursors available and the ability to covalently attach metal carbonyl groups to the POM. This approach can be extended to organic cations, and the solvent system can be extended to an aqueous/organic solvent mixture, commonly water/CH3CN. Moreover, the synthetic variables of greatest importance in synthesizing such clusters are the species and stoichiometric ratios of the raw materials, solution pH, ionic strength, type of carbonyl metals and reaction temperature, and all of these factors play an indispensable role in the formation and crystallization of the product phases. The self-assembly process makes the preparation of novel PMCDs a challenging task in synthetic chemistry because of the ambiguous reaction mechanism. Moreover, the poor solubility of metal carbonyl and the harsh reaction conditions make the preparation of new PMCDs a challenging task. To obtain new PMCDs with desired features and properties, the use of precursors or raw materials is another priority in the preparation process. It should be noted that the preparation of PMCDs is almost in the acid aqueous solution, and the dark environment during the preparation process is also necessary. In particular, among the organometallic POMs, the [M(CO)n]+ in PMCDs can act as face-directed cations to mediate the crystallization process, resulting in novel PMCDs with intriguing properties. And PMCDs can also be catalytically active and exhibit interesting visible-light-induced photo-redox activity. Thus, the development of PMCDs is a growing area of technical interest. Of course, there are challenges in the scientific community related to the further exploration of novel PMCDs and the improvement of many techniques. Therefore, we hope that a careful reading of this review will help to explore more intriguing PMCDs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21571050, 21573056, 21771053, and 21771054).Notes and references
- A. Müller, F. Peters, M. T. Pope and D. Gatteschi, Chem. Rev., 1998, 98, 239 CrossRef
.
- A. Blazevic and A. Rompel, Coord. Chem. Rev., 2016, 307, 42 CrossRef CAS
- M. T. Pope and A. Muller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34 CrossRef
- J.-D. Compain, P. Mialane, A. Dolbecq, I. M. Mbomekalle, J. Marrot, F. Sécheresse, E. Rivière, G. Rogez and W. Wernsdorfer, Angew. Chem., Int. Ed., 2009, 48, 3077 CrossRef CAS PubMed
- P. Ma, F. Hu, J. Wang and J. Niu, Coord. Chem. Rev., 2019, 378, 281 CrossRef CAS
- D. Li, P. Ma, J. Niu and J. Wang, Coord. Chem. Rev., 2019, 392, 49 CrossRef CAS
- J. Lu, Y. Wang, X. Ma, Y. Niu, V. Singh, P. Ma, C. Zhang, J. Niu and J. Wang, Dalton Trans., 2018, 47, 8070 RSC
- S. Omwoma, W. Chen, R. Tsunashima and Y.-F. Song, Coord. Chem. Rev., 2014, 258, 58 CrossRef
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893 CrossRef CAS PubMed
- D.-Y. Du, L.-K. Yan, Z.-M. Su, S.-L. Li, Y.-Q. Lan and E.-B. Wang, Coord. Chem. Rev., 2013, 257, 702 CrossRef CAS
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572 RSC
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC
- L. Cronin and A. Müller, Chem. Soc. Rev., 2012, 41, 7333 RSC
- H. N. Miras, J. Yan, D.-L. Long and L. Cronin, Chem. Soc. Rev., 2012, 41, 7403 RSC
- K. Kamata, K. Yonehara, Y. Nakagawa, K. Uehara and N. Mizuno, Nat. Chem., 2010, 2, 478 CrossRef CAS PubMed
- N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944 CrossRef CAS
- Z. M. Zhang, T. Zhang, C. Wang, Z. K. Lin, L. S. Long and W. B. Lin, J. Am. Chem. Soc., 2015, 137, 3197 CrossRef CAS PubMed
- J. J. Berzelius, Ann. Phys., 1826, 82, 369 CrossRef
- J. F. Keggin, Proc. R. Soc. London, Ser. A, 1934, 144, 75 CrossRef CAS
- A. Rosenheim and R. Bilecki, Chem. Ber., 1913, 4, 543 Search PubMed
- A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153 RSC
- J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Chem. Soc. Rev., 2012, 41, 7464 RSC
- M. Lechner, R. Güttel and C. Streb, Dalton Trans., 2016, 45, 16716 RSC
- J. M. Cameron, D. J. Wales and G. N. Newton, Dalton Trans., 2018, 47, 5120 RSC
- N. Narkhede, S. Singh and A. Patel, Green Chem., 2015, 17, 89 RSC
- J. Zhang, F. Xiao, J. Hao and Y. Wei, Dalton Trans., 2012, 41, 3599 RSC
- F. Pu, E. Wang, H. Jiang and J. Ren, Mol. BioSyst., 2013, 9, 113 RSC
- L.-H. Bi, U. Kortz, S. Nellutla, A. C. Stowe, J. Tol, N. S. Dalal, B. Keita and L. Nadjo, Inorg. Chem., 2005, 44, 896 CrossRef CAS PubMed
- W.-C. Chen, C. Qin, X.-L. Wang, Y.-G. Li, H.-Y. Zang, Y.-Q. Jiao, P. Huang, K.-Z. Shao, Z.-M. Su and E.-B. Wang, Chem. Commun., 2014, 50, 13265 RSC
- B. Botar, Y. V. Geletii, P. Kögerler, D. G. Musaev, K. Morokuma, I. A. Weinstock and C. L. Hill, J. Am. Chem. Soc., 2006, 128, 11268 CrossRef CAS PubMed
- B. S. Bassil, M. H. Dickman and U. Kortz, Inorg. Chem., 2006, 45, 2394 CrossRef CAS PubMed
- C. Pichon, A. Dolbecq, P. Mialane, J. Marrot, E. Riviere and F. Sécheresse, Dalton Trans., 2008, 71 RSC
- P. I. Molina, H. N. Miras, D.-L. Long and L. Cronin, Dalton Trans., 2014, 43, 5190 RSC
- J. P. Wang, J. Du and J. Y. Niu, CrystEngComm, 2008, 10, 972 RSC
- J. P. Wang, P. T. Ma, Y. Shen and J. Y. Niu, Cryst. Growth Des., 2008, 8, 3130 CrossRef CAS
- J. W. Zhao, D. Y. Shi, L. J. Chen, P. T. Ma, J. P. Wang and J. Y. Niu, CrystEngComm, 2011, 13, 3462 RSC
- J.-W. Zhao, J. Zhang, S.-T. Zheng and G.-Y. Yang, Inorg. Chem., 2007, 46, 10944 CrossRef CAS PubMed
- E. Ahmed and M. Ruck, Angew. Chem., Int. Ed., 2012, 51, 308 CrossRef CAS PubMed
- T. L. Jorris, M. Kozik and L. C. W. Baker, Inorg. Chem., 1990, 29, 4584 CrossRef CAS
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34 CrossRef
- K. Wassermann, M. H. Dickman and M. T. Pope, Angew. Chem., Int. Ed. Engl., 1997, 36, 1445 CrossRef CAS
- V. W. Day, T. A. Eberspacher, W. G. Klemperer and C. W. Park, J. Am. Chem. Soc., 1993, 115, 8469 CrossRef CAS
- Y. Lin, T. J. R. Weakley, B. Rapko and R. G. Finke, Inorg. Chem., 1993, 32, 5095 CrossRef CAS
- A. Müller, S. Sarkar, S. Q. N. Shah, H. Bögge, M. Schmidtmann, S. Sarkar, P. Kögerler, B. Hauptfleisch, A. X. Trautwein and V. Shnemann, Angew. Chem., Int. Ed., 1999, 38, 3238 CrossRef
- A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837 RSC
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed
- A. Müller, H. Reuter and S. Dillinger, Angew. Chem., Int. Ed. Engl., 1995, 34, 2328–2361 CrossRef
- J. Zubieta, Comments Inorg. Chem., 1994, 16, 153 CrossRef CAS
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed
- P. Gouzerh and A. Proust, Chem. Rev., 1998, 98, 77 CrossRef CAS PubMed
- L. S. Ott and R. G. Finke, Coord. Chem. Rev., 2007, 251, 1075 CrossRef CAS
- M. Tountas, Y. Topal, A. Verykios, A. Soultati, A. Kaltzoglou, T. A. Papadopoulos, F. Auras, K. Seintis, M. Fakis, L. C. Palilis, D. Tsikritzis, S. Kennou, A. Fakharuddin, L. Schmidt-Mende, S. Gardelis, M. Kus, P. Falaras, D. Davazoglou, P. Argitis and M. Vasilopoulou, J. Mater. Chem. C, 2018, 6, 1459 RSC
- Q. Li and Y. Wei, Chem. Commun., 2018, 54, 1375 RSC
- Y. Luo, M. Wächtler, K. Barthelmes, A. Winter, U. S. Schubert and B. Dietzek, Chem. Commun., 2018, 54, 2970 RSC
- J. Luo, K. Chen, P. C. Yin, T. Li, G. Wan, J. Zhang, S. T. Ye, X. M. Bi, Y. P. Yang, G. Wei and T. B. Liu, Angew. Chem., Int. Ed., 2018, 57, 4067 CrossRef CAS PubMed
- J. Li, J. Guo, J. Jia, P. Ma, D. Zhang, J. Wang and J. Niu, Dalton Trans., 2016, 45, 6726 RSC
- J. Zhao, J. Wang, J. Zhao, P. Ma, J. Wang and J. Niu, Dalton Trans., 2012, 41, 5832 RSC
- C. Zhao, W. R. Cordoba, A. L. Kaledin, Y. Yang, Y. V. Geletii, T. Lian, D. G. Musaev and C. L. Hill, Inorg. Chem., 2013, 52, 13490 CrossRef CAS PubMed
- Z. Huo, J. Zhao, Z. Bu, P. Ma, Q. Liu, J. Niu and J. Wang, ChemCatChem, 2014, 6, 3096 CrossRef CAS
- C. J. Besecker and W. G. Klemperer, J. Am. Chem. Soc., 1980, 102, 7600 CrossRef
- A. V. Besserguenev, M. H. Dickman and M. T. Pope, Inorg. Chem., 2001, 40, 2582 CrossRef CAS PubMed
- J. R. Harper and A. L. Rheingold, J. Am. Chem. Soc., 1990, 112, 4037 CrossRef CAS
- F. Bottomley and J. Chen, Organometallics, 1992, 11, 1707 CAS
- C. J. Besecker, V. W. Day, W. G. Klemperer and M. R. Thompson, Inorg. Chem., 1985, 24, 44 CrossRef CAS
- W. G. Klemperer and D. J. Main, Inorg. Chem., 1990, 29, 2355 CrossRef CAS
- V. W. Day, M. F. Fredrich, M. R. Thompson, W. G. Klemperer, R. S. Liu and W. Shum, J. Am. Chem. Soc., 1981, 103, 3597 CrossRef CAS
- W. G. Klemperer’ and B. Zhong, Inorg. Chem., 1993, 32, 5821 CrossRef
- A. V. Besserguenev, M. H. Dickman and M. T. Pope, Inorg. Chem., 2001, 40, 2582 CrossRef CAS PubMed
- R. Villanneau, A. Proust, F. Robert and P. Gouzerh, Chem. – Eur. J., 2003, 9, 1982 CrossRef CAS PubMed
- W. H. Knoth, J. Am. Chem. Soc., 1979, 101, 759 CrossRef CAS
- A. R. Siedle, C. G. Markell, P. A. Lyon, K. O. Hodgson and A. L. Roe, Inorg. Chem., 1987, 26, 219 CrossRef CAS
- M. Sadakane, Y. Iimuro, D. Tsukuma, B. S. Bassil, M. H. Dickman, U. Kortz, Y. Zhang, S. Ye and W. Ueda, Dalton Trans., 2008, 6692 RSC
- J. Zhao, J. Zhao, P. Ma, J. Wang, J. Niu and J. Wang, J. Mol. Struct., 2012, 1019, 61 CrossRef CAS
- C. Zhao, E. N. Glass, J. M. Sumliner, J. Bacsa, D. T. Kim, W. Guo and C. L. Hill, Dalton Trans., 2014, 43, 4040 RSC
- Y. Zhang, D. Zhang, Z. Huo, P. Ma, J. Niu and J. Wang, RSC Adv., 2014, 4, 28848 RSC
- Y. Liu, Y. Zhang, P. Ma, Y. Dong, J. Niu and J. Wang, Inorg. Chem. Commun., 2015, 56, 45 CrossRef CAS
- Z. Huo, J. Guo, J. Lu, Q. Xu, P. Ma, J. Zhao, D. Zhang, J. Niu and J. Wang, RSC Adv., 2015, 5, 69006 RSC
- J. Jia, Y. Zhang, P. Zhang, P. Ma, D. Zhang, J. Wang and J. Niu, RSC Adv., 2016, 6, 108335 RSC
- J. Lu, X. Ma, P. Wang, J. Feng, P. Ma, J. Niu and J. Wang, Dalton Trans., 2019, 48, 628 RSC
- T. Nagata, M. Pohl, H. Weiner and R. G. Finke, Inorg. Chem., 1997, 36, 1366 CrossRef CAS PubMed
- C. Zhao, Z. Huang, W. Rodríguez-Córdoba, C. S. Kambara, K. P. O'Halloran, K. I. Hardcastle, D. G. Musaev, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 20134 CrossRef CAS PubMed
- J. Zhao, J. Zhao, P. Ma, J. Wang, J. Niu and J. Wang, J. Mol. Struct., 2012, 1019, 61 CrossRef CAS
- C. Zhao, C. S. Kambara, Y. Yang, A. L. Kaledin, D. G. Musaev, T. Lian and C. L. Hill, Inorg. Chem., 2013, 52, 671 CrossRef CAS PubMed
- K. Nishiki, H. Ota, S. Ogo, T. Sano and M. Sadakane, Eur. J. Inorg. Chem., 2015, 2714 CrossRef CAS
- V. Singh, Y. Zhang, L. Yang, P. Ma, D. Zhang, C. Zhang, L. Yu, J. Niu and J. Wang, Molecules, 2017, 22, 1351 CrossRef PubMed
- J. Jia, P. Ma, P. Zhang, D. Zhang, C. Zhang, J. Niu and J. Wang, Dalton Trans., 2018, 47, 6288 RSC
- R. Villanneau, R. Delmont, A. Proust and P. Gouzerh, Chem. – Eur. J., 2000, 6(7), 1184–1192 CrossRef CAS
- J. Wang, J. Li and J. Niu, Chem. Res. Chin. Univ., 2008, 24, 675 CAS
- J. Niu, L. Yang, J. Zhao, P. Ma and J. Wang, Dalton Trans., 2011, 40, 8298 RSC
- D. Zhang, J. Zhao, Y. Zhang, X. Hu, L. Li, P. Ma, J. Wang and J. Niu, Dalton Trans., 2013, 42, 2696 RSC
- P. Zhang, V. Singh, J. Jia, D. Zhang, P. Ma, J. Wang and J. Niu, Dalton Trans., 2018, 47, 9317 RSC
- J. Lu, X. Ma, V. Singh, Y. Zhang, P. Ma, C. Zhang, J. Niu and J. Wang, Dalton Trans., 2018, 47, 5279 RSC
- J. Lu, X. Ma, V. Singh, Y. Zhang, P. Wang, J. Feng, P. Ma, J. Niu and J. Wang, Inorg. Chem., 2018, 57, 14632 CrossRef CAS PubMed
- W. L. Wong, K. C. Cheung, P. H. Chan, Z. Y. Zhou, K. H. Lee and K. Y. Wong, Chem. Commun., 2007, 2175 RSC
| This journal is © the Partner Organisations 2019 |