
Current progress of metallic and carbon-based nanostructure catalysts towards the electrochemical reduction of CO2
Liang
Hou†
a,
Jingze
Yan†
a,
Leta
Takele
b,
Yuanbin
Wang
a,
Xiaoqin
Yan
 *a and
Yan
Gao
*b
*a and
Yan
Gao
*b
aSchool of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, P. R. China. E-mail: xqyan@mater.ustb.edu.cn
bLaboratory of Nanomaterials, National Center for Nanoscience and Technology, Beijing, 100190, P. R China. E-mail: gaoyan@nanoctr.cn
First published on 15th July 2019
Abstract
The gradual increase in atmospheric carbon dioxide concentration has led to a series of environmental problems such as global warming; therefore, the large-scale conversion of carbon dioxide is urgent. In this regard, the electrochemical CO2 reduction (ECR) technology can reduce the greenhouse gas CO2 to low-carbon fuels and economically valuable chemicals, which is a promising method to achieve carbon cycle termination. Catalysts are critical for ECR due to high activation energy requirements and the endothermic nature of the reaction. This review describes the recent advances in the design of nanostructured inorganic catalysts for ECR, strategies to improve the catalytic performance of these catalysts, and particularly the structure–performance relationship of catalysts. After a brief introduction to the background and basic principles of ECR, we have summarized the crucial factors (size, morphology, crystal facets, defects, interface, surface, and oxide derivation) determining the performance of CO2 electroreduction. Finally, we have discussed the methods to improve the reaction efficiency and selectivity of catalysts and introduced the prospects for their future developments.
 Liang Hou | Liang Hou received his bachelor's degree in metal material and engineering from the Shandong University of Science and Technology in 2017. He is currently pursuing his master degree at the University of Science and Technology, Beijing, under the supervision of Professor Xiaoqin Yan. His research interest focuses on the design of advanced catalysts for the electrochemical reduction of CO2. |
 Jingze Yan | Jingze Yan obtained his bachelor's degree in metal material and engineering from Shandong University of Science and Technology in 2018. He is currently pursuing his master's degree at the University of Science and Technology Beijing under the supervision of Professor Xiaoqin Yan. His research focuses on catalysts for the electrochemical reduction of carbon dioxide and lithium-ion capacitors. |
 Leta Takele | Leta Takele received his BSc and MSc degrees in Applied Chemistry and Physical Chemistry from Ambo University and Haramaya University in 2010 and 2015 respectively. He is currently a Ph.D. candidate in Physical Chemistry under the supervision of Prof. Zhiyong Tang at the Chinese academy of sciences, institute of National Center for Nanosciences and Nanotechnology. His current research is focused on the development of nanostructured materials for electrochemical CO2 reduction, supercapacitors, and batteries. |
 Yuanbin Wang | Yuanbin Wang received her bachelor's degree in Materials Physics from Qingdao University in 2017. She is currently pursuing her master's degree at the University of Science and Technology Beijing under the supervision of Dr Xiaoqin Yan. Her research focuses on the design of a semiconductor photocathode that can generate photocurrent for water splitting at low bias. |
 Xiaoqin Yan | Xiaoqin Yan is a professor in the department of material physics and chemistry, University of Science and Technology, Beijing. She received her B.S. degree in Materials Science and Engineering from Taiyuan University of Technology in 1997. She received her Ph.D. degree from the Institute of Physics, Chinese Academy of Sciences in 2004. She held a postdoc position at the Institute for Materials Research, Tohoku University. She was promoted to a full professor in 2007 at the University of Science & Technology Beijing. Her research interests include fabrication and characterization of patterned nanomaterials and design and application of energy conversion and storage devices/system. |
 Yan Gao | Dr Yan Gao is a professor at the National Center for Nanoscience and Technology (NCNST). She received her Ph.D. degree from the Chinese Academy of Sciences in 2005. After this, she went to the Center of Advanced European Studies and Research in Bonn, Germany, as a post-doctor and Konstanz University as a Humboldt fellow. She joined NCNST in 2008, conducting the research work on controllable synthesis, photocatalytic and electrochemical performance of inorganic nanomaterials. |
1. Introduction
Since the industrial revolution, the progress of human society and the development of industry have increased people's demand for energy. The current sources of energy are mainly fossil fuels such as oil, coal, and natural gas; however, significant consumption of fossil fuels leads to the production of a large amount of CO2. The current concentration of CO2 in the atmosphere is 385 ppm, which is predicted to reach nearly 600 ppm in 2100.1–3 Carbon dioxide is the main cause of some adverse environmental changes such as the greenhouse effect, ocean acidification and desertification. Therefore, how to reduce the concentration of CO2 in the atmosphere has become an urgent problem in modern society.4,5To solve the current energy problem that is facing our world, renewable energy sources, such as solar energy and wind energy, have been developed. However, these energy sources are usually limited and currently occupy a very low total energy consumption ratio. Therefore, it is very important to develop a sustainable strategy to transform carbon dioxide into chemicals and fuels to form a sustainable recycling system for relieving environmental degradation. In the past few decades, various methods, including photochemical, electrochemical, thermochemical, and biochemical methods, have been proposed to conduct a comprehensive research on carbon dioxide reduction.5–8 Among these methods, the electrochemical reduction of carbon dioxide is particularly attractive because of its high reaction rate and efficiency, simple reaction units, controllable selectivity, and great potential for practical industrial applications.9,10
The electrochemical CO2 reduction (ECR) reaction is a multi-proton and electron transfer reaction that can be conducted in an aqueous solution using a suitable electrocatalyst; CO2 can be converted into one carbon and multi-carbon hydrocarbons such as CO, HCOOH, CH4, C2H4, EtOH and other hydrocarbons. Based on thermodynamic studies, Table 1 presents the various semi-reactions associated with standard hydrogen electrodes (SHE) in an aqueous solution and their corresponding electrode potentials (pH = 7, 25 °C).11–13 In fact, ECR does not easily occur, and the actual potential of the driving reaction is more negative than the standard electrode potential (i.e., large overpotential). The reason is that the energy required to form the intermediate substance CO2*− by transferring one electron to the CO2 molecule is as high as −1.9 V relative to SHE. Another complicating factor is the competitive proton reduction or the hydrogen evolution reaction, which is ubiquitous in aqueous media. Moreover, in terms of kinetics, the HER is more fast than the ECR. In addition, the thermodynamic reduction potentials of several products of ECR are very similar. Therefore, many products are generated in the reduction process; this particularly makes the generation of high-selectivity products difficult; therefore, it is very meaningful to develop efficient catalysts that can reduce the kinetic barriers and control the selectivity of ECR.
| Half-electrochemical thermodynamic reactions | Electrode potentials (V vs. SHE) under standard conditions |
|---|---|
| CO2(g) + e− = CO2*− | −1.90 |
| 2H+ + 2e− = H2 | −0.42 |
| CO2(g) + H2O(l) + 2e− = HCOO−(aq) + OH− | −0.43 |
| CO2(g) + H2O(l) + 2e− = CO(g) + 2OH− | −0.52 |
| CO2(g) + 3H2O(l) + 4e− = HCHO(l) + 4OH− | −0.89 |
| CO2(g) + 5H2O(l) + 6e− = CH3OH(l) + 6OH− | −0.81 |
| CO2(g) + 6H2O(l) + 8e− = CH4(g) + 8OH− | −0.25 |
| 2CO2(g) + 8H2O(l) + 12e− = CH2CH2(g) + 12OH− | −0.34 |
| 2CO2(g) + 9H2O(l) + 12e− = CH3CH2OH(l) + 12OH− | −0.33 |
Previous studies on the electrochemical reduction of carbon dioxide have focused on polycrystalline single metal catalysts. Based on the CO2 reduction of primary products, single-metal catalysts can be further divided into several subclasses: CO-selective metals (e.g., Au and Ag), formic acid-selective metals (e.g., Sn, In, and Pb), and hydrogen-selective metals (e.g., Fe, Ni and Pt). The Norskov team used DFT calculations to compare the binding energy and adsorption energy of intermediates in the electrochemical CO2 reduction processes on various metal electrodes and concluded that polycrystalline Cu was an excellent metal electrocatalyst for the production of hydrocarbons.14 The carbon dioxide reduction reaction (CO2RR) is sensitive to not only the type of electrocatalyst material, but also the structure of the electrocatalyst;15–17 structural sensitivities in other electrocatalytic applications, such as water decomposition and oxygen reduction reaction (ORR), are good examples of this viewpoint.18–21 The atomic arrangement, electronic structure, chemical composition and oxidation state of the catalyst significantly affect its performance. By constructing a surface nanostructure catalyst and changing the surface morphology and electronic structure, more active sites can be exposed; thereby, the performance of the catalyst can be improved.
The main challenges with respect to CO2 reduction electrocatalysts are how to increase the conversion efficiency of CO2, control the selectivity of specific products, and understand the relationship between catalyst structure and electrocatalytic performance; although several reviews22–24 have been reported on ECR, only few of them are focused on the optimization strategies to tune the materials for the active and selective generation of a specific product. In this review, we focused on the use of inorganic nanostructured catalysts for the electrochemical reduction of carbon dioxide. Specifically, we have summarized the latest strategies (Fig. 1) for the construction of advanced nanostructured electrocatalysts and presented representative cases of structure–performance relationships. Finally, we have introduced the prospects and future developments in this area, which will guide the design of advanced catalysts to further improve their ECR performance.
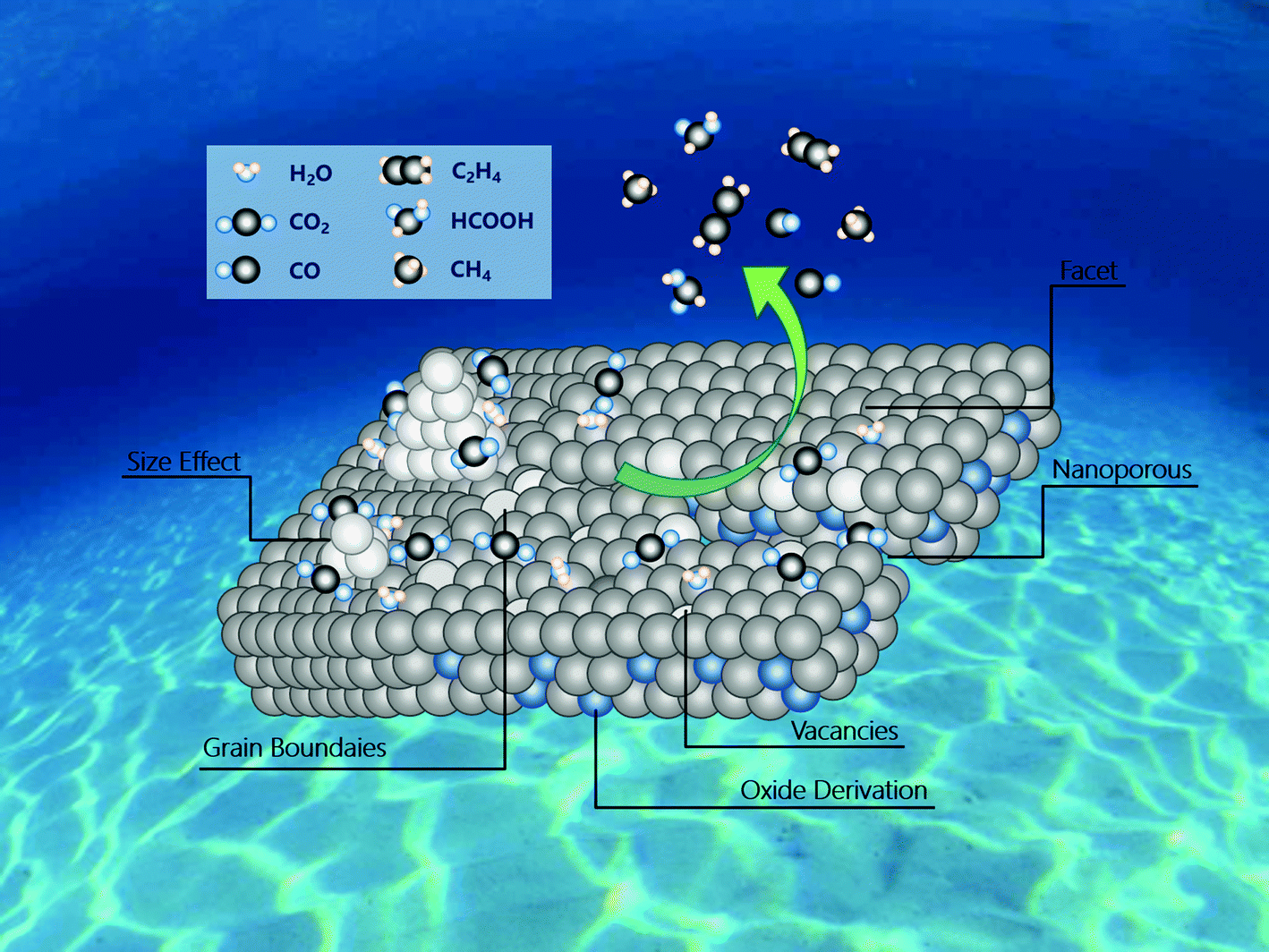 | ||
| Fig. 1 Overview of the strategies about activity and selectivity control in nanostructured electrocatalysts for ECR. | ||
2. Size and morphology effect in ECR
Carbon dioxide electroreduction is very sensitive to the structure of metal electrocatalysts. Nanosized metal catalysts generally exhibit significantly enhanced activity as compared to bulk metal catalysts due to the presence of low coordinating atoms, which may affect the free energy barrier in the reaction process and thus affect the reaction selectivity.Recently, Sun and co-workers25 reported a study on the tendency of ECR reactivity of Au nanoparticles (NPs) over a small size range of about 4–10 nm, in which 8 nm Au NPs had best ECR activity with the Faraday efficiency (FE) reaching 90% at −0.67 V (vs. RHE). Via DFT calculation of different models of Au nanoparticles based on different crystal facets of gold, they have found that the edge site favors CO evolution; however, the edge site is more active for the competitive HER (Fig. 2a). Therefore, the 8 nm Au NP can expose relatively high-density edge sites and minimize the number of corner portions, further enhancing the ECR performance. Cuenya et al.26 have also observed that when the NP size decreases, the current density increases sharply and the selectivity towards CO decreases (Fig. 2b). DFT calculations showed that the trend of this change was related to the number of low coordination sites on the NPs. The increase in the number of low coordination sites is conducive to the generation of H2 and inhibits the ECR process. Thus, they have proposed that by adjusting the size of the catalyst particles, the H2/CO ratio can be controlled for different industrial productions. Hwang27 and colleagues have synthesized a series of immobilized Ag nanoparticles of different sizes (3, 5 and 10 nm) and loaded them on carbon to study their CO2 reduction selectivity. The 5 nm Ag NPs exhibited highest FE for CO, up to 84.4% at −0.75 V vs. RHE in CO2-saturated 0.5 M KHCO3 (Fig. 2c). The authors used DFT calculations to account for this size-dependent effect of Ag NPs. The 5 nm Ag NP has best *COOH adsorption, showing maximum FE for CO production. Bao et al.28 reported the CO2 electroreduction performance of Pd nanoparticles in the range of 2.4–10.3 nm. As the size of the Pd NPs decreases, the FE for CO production increases (Fig. 2d). The FE of the 3.7 nm NPs for CO production was 91.2% at −0.89 VRHE, and the current density of CO was increased by 18.4 times. The DFT calculations show that the small-sized Pd has more corner and edge sites than the large Pd NPs and is beneficial to promote the adsorption of CO2 and reduce the evolution energy of *COOH in the ECR. Thus, the small size of Pd NPs is conducive to the production of CO.
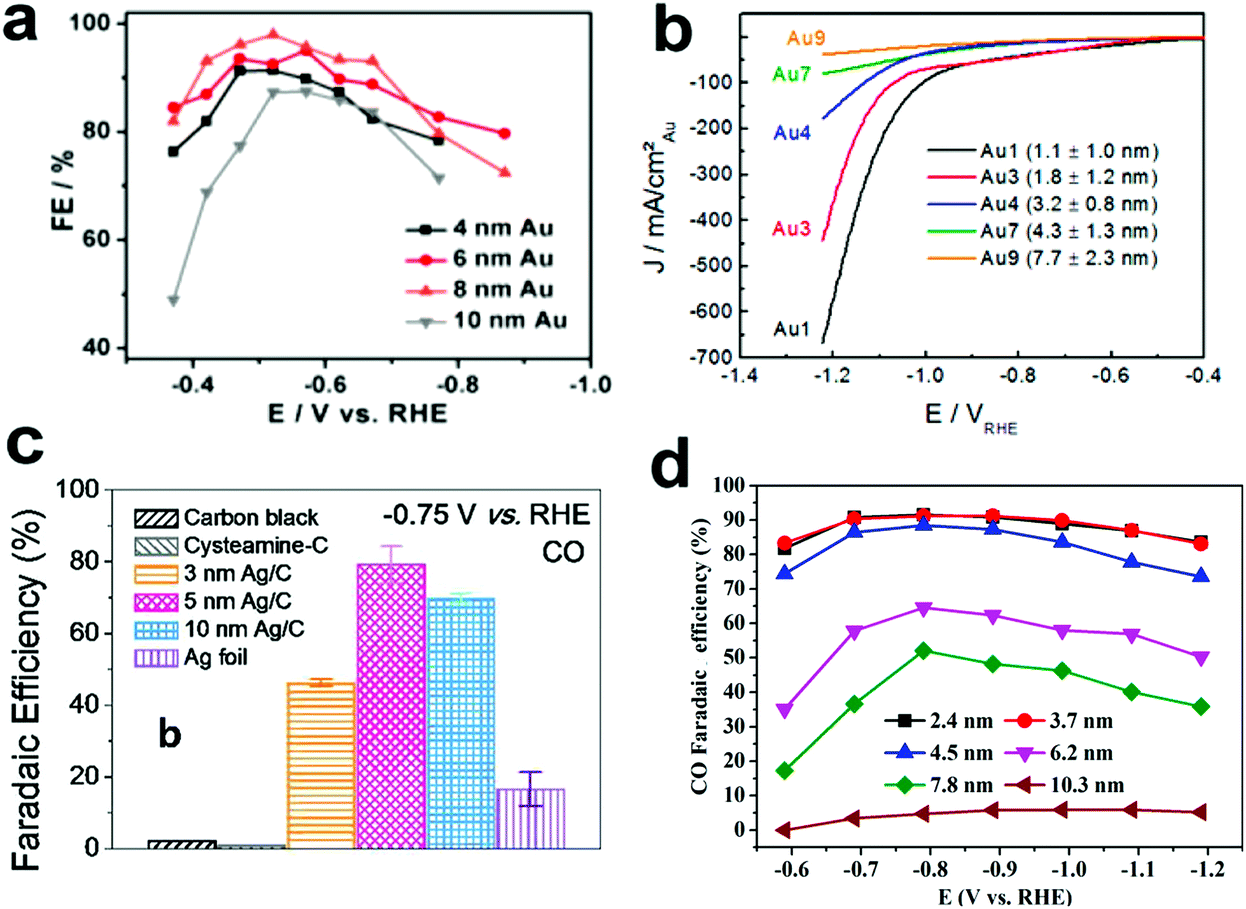 | ||
| Fig. 2 (a) CO FEs for different sizes of Au NPs loaded on carbon. Adapted with permission from ref. 25. Copyright 2013 American Chemical Society. (b) Current density of Au NPs of different sizes during ECR. Adapted with permission from ref. 26. Copyright 2014 American Chemical Society. (c) CO FEs for Ag NPs loaded on carbon at −0.75 V (vs. RHE). Adapted with permission from ref. 27. Copyright 2015 American Chemical Society. (d) CO FEs for Pd NPs with different sizes. Adapted with permission from ref. 28. Copyright 2015 American Chemical Society. | ||
As is well-known,29 copper generates many products, such as formate, CO and hydrocarbons (methane, ethylene), for ECR in an aqueous solution. Thus, how to enhance the catalytic selectivity performance of copper for ECR is a big challenge. In this regard, Strasser et al.30 have explored the size effect of Cu nanoparticles on ECR. As the particle size of Cu decreases, the catalytic activity for H2 and CO increases remarkably, whereas the selectivity of hydrocarbons (CH4 and C2H4) is suppressed. Especially, for particles below 5 nm, this enhancement effect is remarkable when compared with the case of the bulk Cu electrode. The authors established a spherical model of Cu nanoparticles to account for this difference in activity selectivity. It was found that the smaller the size of the copper NPs, the more the low coordination surface sites (CN < 8) on the surface. These binding sites increase the catalytic activity while accelerating the reduction of HER and CO2 to CO on the surface of Cu NPs, inhibiting hydrocarbon selectivity. In contrast, Manthiram et al. have prepared monodisperse 7 nm copper nanoparticles by reducing copper(I) in triethylamine with tetradecyl phosphonate as a blocking agent in trioctylamine.31 These Cu NPs exhibit high activity for CH4 while catalyzing the CO2RR; at −1.35 V, the FE is as high as 76%, which is significantly better than that achieved using a copper foil (FE = 44%); similar to the common problems of particulate or even monoatomic catalysts, these 7 nm copper nanoparticles are unstable in the reaction solution and condense into 25 nm nanoparticles after 10 minutes of electrochemical treatment at −1.25 V. Surprisingly, this morphological evolution does not appear to affect the evolution of methane, and its FE remains in the range of 71–90% over 60 minutes. In the case of monodisperse particulate catalysts, the difference in particle size results in different activities with significant selectivities.
Baturina et al.32 prepared carbon-supported Cu NPs ranging from 10 to 30 nm for the reduction of CO2. The results showed that the 12 nm Cu nanoparticles had higher proportion of C2H4/CH4, which was more active than the electrodeposited copper film for the formation of C2H4. These studies indicate that it is critical to tune the size of Cu NPs to achieve selectivity towards the reduction product, and the rational regulation of size can increase the selectivity towards hydrocarbons in ECR.
Exposing more edge sites for carbon dioxide reduction, one-dimensional nanowires have also been investigated as excellent catalysts. Sun et al. synthesized ultra-thin gold nanowires (NMs) of different lengths and explored their electrocatalytic properties for ECR.33 The 500 nm long gold nanowires had a lower initial potential (−0.2 VRHE) as compared to the 100 nm and 15 nm long nanowires, and the highest FE for the reduction of CO2 to CO was 94%. Further verification by DFT calculation reveals that the superior energy source on the Au nanowires is the edge sites, which facilitate the activation of CO2 to *COOH and promote the *CO release process. Li et al.34 synthesized Cu nanowires with a 20 nm width and 5 times twin structure by reducing CuCl2 and trimethylsilane in oleylamine at 160 °C. Due to the presence of high-density edge active sites along the twin boundaries, these NWs are more typical for the reduction of CO2 to form CH4 (55% FE at −1.25 V).
Recently, researchers have started to use three-dimensional porous electrodes as catalysts for carbon dioxide reduction.35–39 This structure exposes more catalytically active sites; this allows the reactants and intermediates to readily diffuse at these active sites, and thus, the electron and ion transport pathways are shortened. Three-dimensional porous electrodes regulate the selectivity and activity of ECR by adjusting the local pH and carbon dioxide concentration, thereby improving the electrocatalytic performance. Surendranath et al.40 reported a three-dimensional porous gold inverse opal (Au-IO) film that efficiently reduced CO2 to CO at moderate overpotentials. Among these Au-IO films, the 2.7 μm thick samples exhibited enhanced performance with the FE for CO reaching 75% at −0.4 VRHE. As the thickness of the porous gold film increases, the activity of HER decreases by a factor of 10, whereas the CO evolution activity remains substantially unchanged. This hydrogen suppression originates from the generation of a diffusion gradient in the holes of the mesoporous structure electrode rather than from a change in the facets or grain sizes. The study of Cheng et al.41 also supports a similar point of view. The porous gold film having the highest film thickness of 17.8 μm has the starting potential of CO at −0.25 VRHE, which is 100 mV lower than the initial potential of the polycrystalline gold electrode. The thickest porous gold film exhibited a higher FE (90.5% at −0.5 VRHE) for CO. As the reaction proceeds, a large amount of hydroxide ions accumulate on the surface of the catalyst. The thickest porous gold film can increase the local pH and reduce the proton concentration around the catalyst; this in turn inhibits the conversion of H2, resulting in an increase in CO selectivity.
Recently, our group42 has successfully synthesized nanoporous gold (NPGL) catalyst materials with high electrochemical specific surface area and a regional single crystal structure by a simple one-step de-alloying method. The nanoporous gold obtained by etching for 4 hours has good carbon dioxide reduction activity, and the max value of CO Faraday efficiency is 90% at −0.6 VRHE. Ding and colleagues43 have also developed nanoporous gold (NPG) with the max FE of 94% for CO, and this NPG performed excellently at all potentials as compared to Au octahedral nanoparticles (Fig. 3a). They elucidated the coordination–activity relationship for the reduction of CO2 to CO by establishing a nanoporous gold catalyst model. Different colors on the surface of the NPG correspond to atoms with different coordination numbers (Fig. 3b). The total fraction of under-coordinated surface atoms (GCN < 7.5) is as high as 37%, much larger than the octahedral nanoparticle fraction (Fig. 3c). Compared with the original (111) facet sites, the under coordinated surface atom has a lower free energy barrier, improving the evolution of COOH* intermediates and thus facilitating the process of CO production (Fig. 3d); this study not only proves the effect of under-coordinating atoms on CO2RR, but also provides an idea for understanding the mechanism of electrochemical CO2 reduction using NPG.
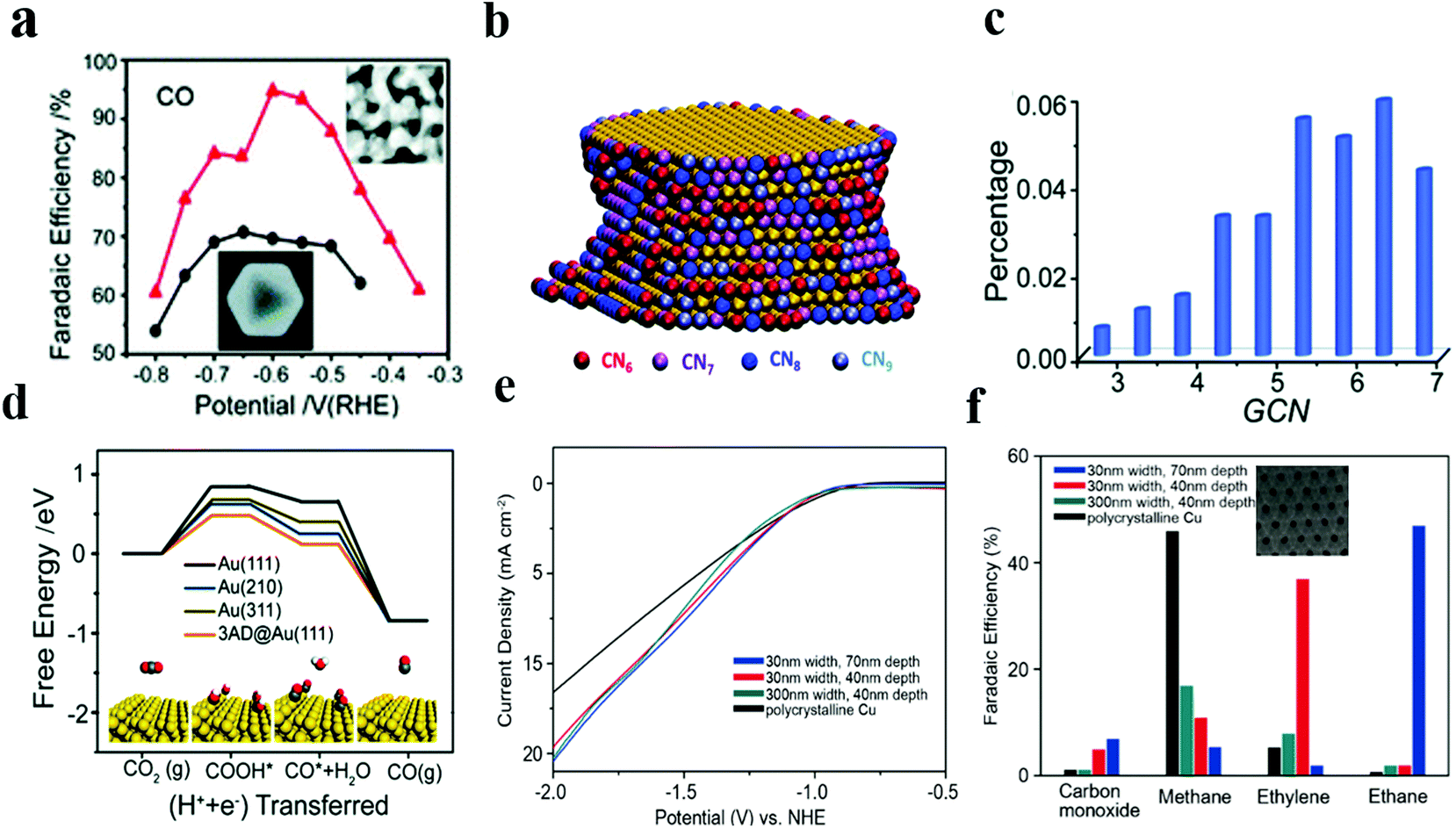 | ||
| Fig. 3 (a) CO FEs at various applied potentials on Au nanoporous (red) and Au octahedral nanoparticles (black). (b) 3D atomic configuration model of the NPG ligament. The color indicates the surface atoms with different coordination numbers. (c) Fractions of surface atoms with different generalized coordination numbers (GCN) out of the total surface atoms of NPG. (d) Free energy diagrams for electrochemical CO2RR to CO on various Au facets. Adapted with permission from ref. 43. Copyright 2018 the Royal Society of Chemistry. (e) Specific current density for ethylene production versus potential of 30 nm/40 nm (red), 30 nm/70 nm (blue), 300 nm/40 nm (cyan), and polycrystalline Cu (black). (f) Product distributions of porous Cu films and polycrystalline Cu foil at −1.7 V RHE. Adapted with permission from ref. 45. Copyright 2017 Wiley-VCH. | ||
Sen et al. used an electrochemical deposition method to construct a porous copper nanofoam with the pore size range of about 20–50 microns.44 While conducting electrocatalytic reduction performance test, this copper nanofoam helped to form HCOOH with a max FE (37%) at −1.5 V (vs. Ag/AgCl). The catalytic ability was significantly enhanced in the abovementioned case as compared to that in the case of a smooth Cu surface. However, large pore size distribution of this foam system makes it hard to determine the actual active sites for catalytic regulation and analyze the product selectivity. Faced with this problem, Yang et al.45 synthesized a nanoporous Cu structure with uniform pore size and depth. This ordered nanoporous Cu was prepared by magnetron sputtering copper on a nanoporous Al2O3 template prefabricated by an anodized aluminum foil. This method produces porous Cu structures of varying size widths and depths (Fig. 2e and f). When used as a catalyst for the CO2RR, the total FE of CO and CH4 produced by 300 × 40 nm porous Cu was about 24%, and the FE of the 30 × 40 nm sample for the production of C2H4 was about 38%. When the pore depth was increased to 70 nm, the reduced product was C2H6 (46% FE). The study strongly suggests that an increase in pore depth and a decrease in pore size promote the evolution of C2 products (C2H4 and C2H6). The increase in the C2 product is attributable to the specific structure of pores, resulting in an increase in local pH. As is well-known, alkaline solutions contribute to carbon dimerization and multi-carbon product formation.35,46 Since the diffusion control process has its own limitations in catalysis, it is necessary to further reduce the size of the pores of the catalyst to improve the activity of the catalyst.
Low local CO2 concentration around the electrode material is considered to be one of the problems that hinder the ECR in an aqueous solution. The electrode is made into a nanostructured needle-like morphology, which can effectively improve the slow dynamics of carbon dioxide reduction, and this has recently become a new method to solve the abovementioned problems; recently, Sargent47 and colleagues have reported that Au nanoneedles with a sharp tip enhancement effect can lead to high local electric fields at low overpotentials that further result in high local CO2 concentration around the gold electrode; moreover, this sharp tip enhancement effect may be explained by the activity of the tip towards the exposure of many active sites such as ridges and corners, which are regions of high local curvature.
3. Crystal facet control
A series of electrocatalytic studies have shown that crystal facets have a major impact on the reactivity and selectivity of the CO2 reduction product. The synthesis of nanoparticles with different shapes typically exposes a large number of crystal facets. Therefore, the shape of the custom NP can greatly improve the catalytic performance of the material. The effects of the crystal facets on metals, such as Au,48 Pd,49 Ag50,51 and Cu, have been studied for ECR, indicating the significance of crystal facets in the selectivity of the reduction product.Lee teams48 successfully synthesized concave rhombic dodecahedron Au nanoparticles. These gold nanoparticles exhibited outstanding activity in CO2 electrocatalytic reduction applications, the highest FE for the selective reduction of CO2 to CO was 93%, and the nanoparticles maintained good stability for the testing time of up to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s. Studies indicate that the surface of these Au NPs is rich in (331), (221), (553) and (775) high-index crystal facets (Fig. 4a–c). High-index crystal facets contain a large number of step and uncoordinated atoms. Higher chemical activity of these atoms makes them readily interact with reactant molecules; this leads to breaking of the chemical bonds between reactant molecules and formation of active sites. CO2 adsorption is more likely to occur in facets with low activation barriers and high adsorption energies.
000 s. Studies indicate that the surface of these Au NPs is rich in (331), (221), (553) and (775) high-index crystal facets (Fig. 4a–c). High-index crystal facets contain a large number of step and uncoordinated atoms. Higher chemical activity of these atoms makes them readily interact with reactant molecules; this leads to breaking of the chemical bonds between reactant molecules and formation of active sites. CO2 adsorption is more likely to occur in facets with low activation barriers and high adsorption energies.
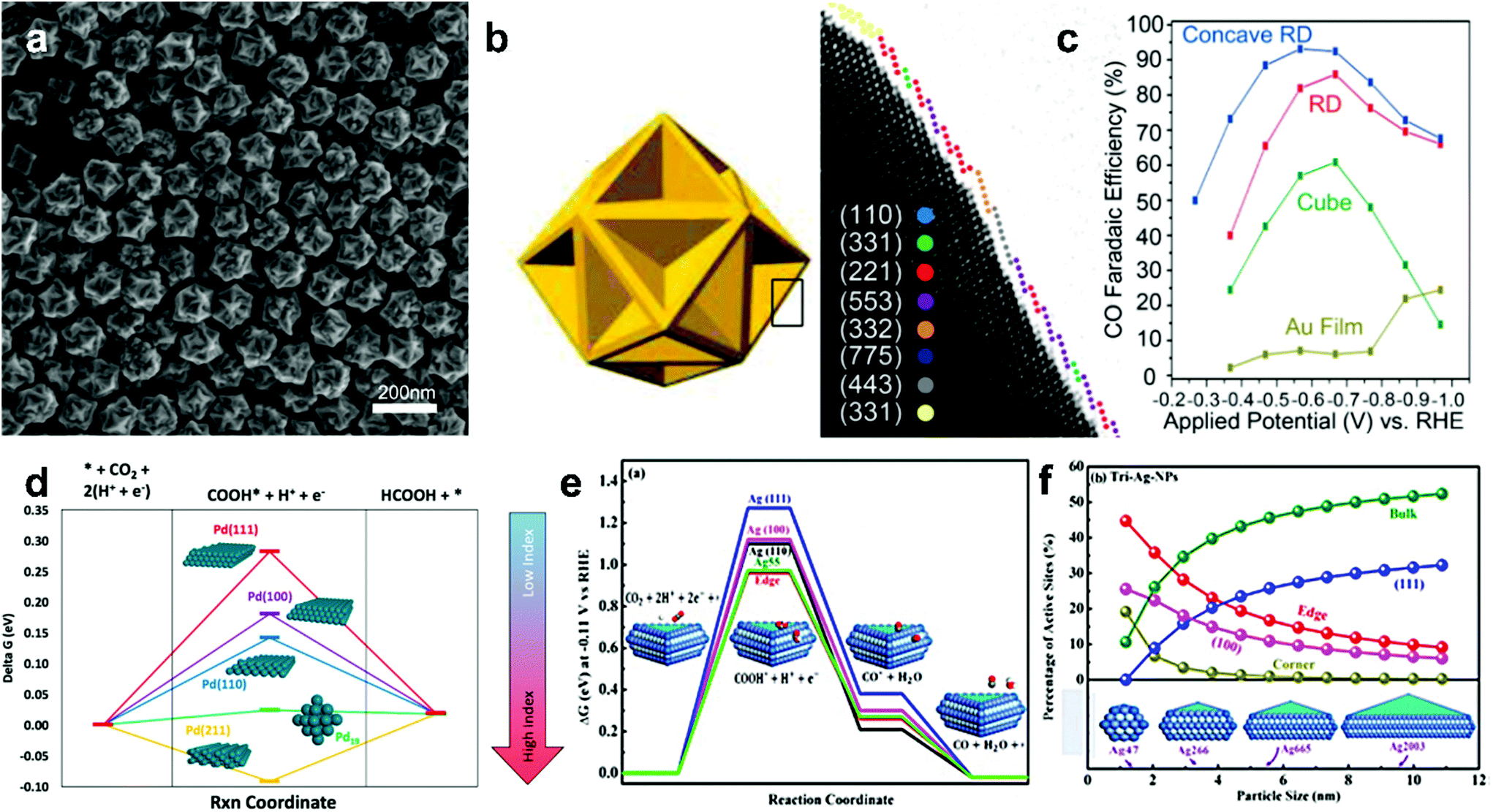 | ||
| Fig. 4 (a) SEM image of the concave RD Au. (b) The concave RD model and edge surface model. Different color spheres are assigned to each type of facets. (c) CO FE of the Au film and different Au NPs shapes (cubic, rhombic dodecahedron (RD) and concave RD) at −1.0 VRHE. Adapted with permission from ref. 48. Copyright 2015 American Chemical Society. (d) Thermodynamic barriers for the CO2RR to formic acid on different Pd single crystal facets and Pd19 cluster. Adapted with permission from ref. 49. Copyright 2016 American Chemical Society. (e) Free energy diagrams for CO2RR to CO and COOH* and CO*intermediates free energies on Ag nanosheets facets and Ag55 cluster. (f) Active adsorption site density on triangular Ag nanosheets as a function of particle size. Adapted with permission from ref. 51. Copyright 2017, American Chemical Society. | ||
There are similar rules for the selectivity of Pd metals. Sargent et al.49 developed a DFT calculation simulation on Pd nanoparticles with different facets and Pd19 clusters and revealed the relationship between crystal facets and CO2 reduction catalysis performance. The reaction energy barrier for the HCOOH reaction decreases as the index increases (Fig. 4d). This indicates that high index facets possess higher selectivity for HCOOH. Hori has studied different low-index crystal faces on a silver surface for CO2 electroreduction.50 The order of selectivity for CO formation among these facets is Ag(110) > Ag(111) > Ag(100). It can be explained by the fact that the Ag(110) roughest surface is rich in intrinsic active sites. Liu et al.51 have synthesized triangular silver nanoplates (tri-Ag-NPs), which are particularly selective for CO formation in the ECR. Compared with similar size Ag nanoparticles and Ag films, tri-Ag-NPs exhibit significantly increased current with the Faraday efficiency of up to 96.8% and a quite good durability (7 days). In addition, the initial potential of CO formation was 96 mV, which further confirmed the advantage of tri-Ag-NPs as a CO-forming catalyst for CO2 reduction. The DFT calculations suggest that the Ag(100) facet can require lower energy to initiate the rate-determination step of CO2 reduction (Fig. 4e–f) and is beneficial to lower the overpotential of the reaction.
Copper (Cu)-based catalysts can realize the production of various hydrocarbons and alcohols with higher economic value multiple carbons. The selectivity of CO2 reduction products is strongly dependent on the crystal facets of copper.50 The effect of Cu crystals on the CO2 reduction selectivity was studied through a series of experimental and DFT calculations. Hori et al. discussed the selectivity of the crystals of single crystal Cu for CO2 reduction products. Cu (100) favors the production of C2H4, whereas Cu (111) favors the production of CH4.52 In addition, the surface of Cu is exposed to much high-index surfaces such as (711) and (911), which effectively promotes the formation of C2H4 and inhibits the formation of CH4. High selectivity to the C2 product may be due to exposure of more active atoms on the surface of the high index facets. Koper and colleagues used online electrochemical mass spectrometry to confirm the selectivity of a copper facet. They also found that product selectivity is closely related to crystal orientation: Cu (111) favors the production of methane, whereas Cu (100) mainly favors the production of ethylene, and Cu (110) promotes the production of acetate and ethanol.53–55 They have suspected two different reaction mechanisms for the evolution of hydrocarbons from Cu (100) and Cu (111) by CO reduction: one path involves an intermediate formed on both facets; however, the formation of CH4 is easier on the Cu (111) facet. In another path, the intermediate *CO is dimerized by surface adsorption to form a C2 product on the Cu (100) facets.
Chen et al.56 synthesized a Cu mesocrystal rich in the (100) plane by the in situ reduction of a thin CuCl film, and they found the C2H4/CH4 FE ratio of 18 in a 0.1 M KHCO3 aqueous solution. A high selectivity for ethylene was achieved, and the study is in good agreement with the observation of Hori. Peterson et al.14 have suggested that the protonation of adsorbed CO to CHO* is a potential-determining step in the formation of methane on the Cu (211) and Cu (100) facets. To understand the difference between CO2 reduction products from different crystal facets reported in the experimental study, Luo et al.57 used DFT to explore the thermodynamic and kinetic pathways for CO2 reduction on different crystal facets of copper. The results indicate that Cu (111) facilitates the formation of COH*, which produces CH4. The Cu (100) crystal plane preferentially forms the CHO* species, which are coupled to ethylene. Moreover, on Cu (100), as the negative potential increases, the change in CO* coverage can trigger the conversion from ethylene to methane. Recently, Huang et al.58 have studied the selectivity to C2 products on the facets of Cu single crystals in the ECR. It has been found that the initial potential of C2H4 formation on the Cu electrode is 300–400 mV higher than the negative charge of CO evolution, and C2H4 is formed only after large amount of CO is produced. This indicates that the highest CO* coverage on the Cu(100) facet can further promote the formation of C2H4. Moreover, it was found that the cubic NP composed of the (100) plane showed increased C2H4 production while suppressing the CH4 selectivity.59 The Cu (751) facets are more selective for C2 production than the lower index surface at lower overpotentials.60 Therefore, the crystal facets of Cu play a significant role in catalytic activity and product selectivity during ECR. However, the role played by different crystal facets in CO2RR is not fully understood, and research is needed to use model surfaces for understanding the combined effects and other parameters, such as defects, to study the CO2 reduction.
4. Defect engineering
Electrochemical CO2 reduction requires higher overpotential to activate a stable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond due to its multiple reaction pathways and competition with the HER reaction. It is usually faced with the challenges of low Faraday efficiency and poor product selectivity. Researchers have improved the catalytic performance by making defects to regulate the distribution and structure of atoms on the catalyst surface.
O bond due to its multiple reaction pathways and competition with the HER reaction. It is usually faced with the challenges of low Faraday efficiency and poor product selectivity. Researchers have improved the catalytic performance by making defects to regulate the distribution and structure of atoms on the catalyst surface.
The catalytic selectivity and activity of nitrogen-doped carbon materials strongly depend on the level of nitrogen doping and the type of dopant in the ECR. Some studies have reported that the active site for the catalytic reduction of carbon dioxide is pyridinic N. For example, Ajayan et al.61 have prepared N-doped carbon nanotubes, which have the Faraday efficiency of 80% for the conversion of CO2 to CO and a better stability for at least 5 h. Later, the group62 has reported three-dimensional (3D) graphene foam combined with nitrogen defects; this foam has the lower initial overpotential of −0.19 V and exhibits activity superior to that of the noble metal Au for ECR. The maximum Faraday efficiency of CO production is 85% at −0.47 V, and the stability lasts for at least 5 hours. The authors have used DFT calculations to conclude that the active site for the ECR to CO is pyridinic N as it has lowest energy barrier for the *COOH intermediates as compared to other defect sites (Fig. 5a and b); subsequent studies63 have demonstrated that the pyridinic N defect contains a pair of lone pair electrons that assist in its bonding to carbon dioxide molecules. The presence of graphite and pyridinic N significantly reduces overpotentials and increases selectivity for CO formation. However, Zheng and coworkers101 have developed nitrogen-doped carbon nanotubes by steam etching, changed the type and proportion of nitrogen dopants, and obtained excellent CO2RR performance. The nitrogen-doped carbon nanotubes have the higher Faraday efficiency of −88% at −0.5 VRHE. The reason for the enhanced performance is that the etching process retains a higher proportion of pyrrolic N atoms (from 22.1% to 55.9%). This provides a new idea and method for tuning the activity sites of electrocatalysts.
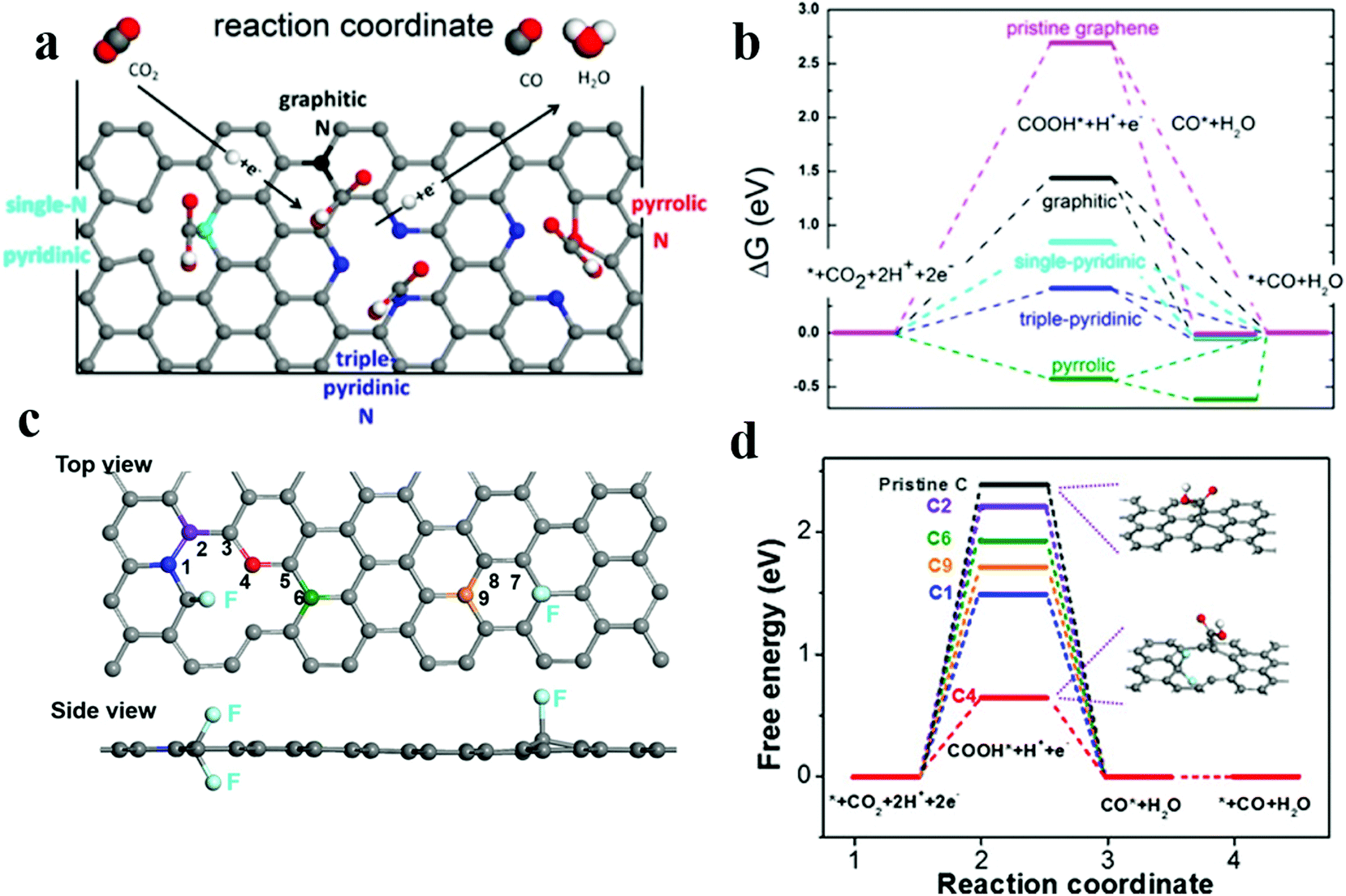 | ||
| Fig. 5 (a) Schematic of the CO2 reduction pathway at different types of nitrogen-doped active sites. (b) Free energy diagram of ECR on N-doped 3D graphene. Adapted with permission from ref. 62. Copyright 2015 American Chemical Society. (c) Image of the DFT model for the fluorine-doped carbon (FC) catalyst. (d)Free energy diagram of ECR on FC and pristine C. Adapted with permission from ref. 66. Copyright 2017 Wiley-VCH. | ||
In addition to N doping, other types of heteroatom doping/codoping have been investigated. Boron-doped graphene reduced CO2 to formic acid at 1.4 VSCE with the FE of 66%.64 DFT calculations indicate that the presence of boron introduces asymmetric spin density, making it suitable for the chemisorption of protonated CO2 (COOH*). Boron and nitrogen co-doped nanodiamonds65 achieved good ethanol selectivity with the high FE of 93.2% at −1.0 V RHE. Their superior performance is mainly attributed to the synergistic effect of N and B co-doping. Xie et al. have synthesized66 a fluorine-doped carbon (FC) catalyst to achieve high selectivity for CO with the max. Faraday efficiency of 90% at the low overpotential of 510 mV; the DFT calculations imply that fluorine doping enhances the intrinsic activity towards the electrochemical CO2 reduction such that the carbon atoms adjacent to fluorine enhance the adsorption of *COOH and weaken the force against *H (Fig. 5c and d). This is the first study on the application of halogen-doped carbon materials for CO2 reduction, which provides a new prospect for the improvement of catalytic performances.
The introduction of metal atom vacancy defects affects the electrocatalytic performance by adjusting the electronic structure of adjacent electrons and consequently the energy barrier of the rate-limiting reaction intermediate. Sargent102 used a solvothermal synthesis of Cu2S (V-Cu2S) nanoparticles with a core–shell structure in which the shell was rich in copper vacancies. Due to the presence of vacancies, the catalyst transferred selectivity from a competitive ethylene reaction to the formation of a liquid alcohol. The max. value of the Faraday efficiency obtained for multi-carbon alcohols was 32% (C2H5OH 25 ± 1% and C3H7OH 7 ± 0.5%). The DFT calculations have shown that the presence of vacancies on the copper shell with the Cu2S core increases the energy barrier in the ethylene pathway, whereas the ethanol pathway remains essentially unaffected. Zhang et al.103 tuned the surface electron transfer by O− vacancy engineering as an efficient strategy to develop enhanced catalysts for CO2 reduction. A series of distinct InOx NRs, namely, pristine (P-InOx), low vacancy (O-InOx) and high vacancy (H-InOx) NRs, with different numbers of O− vacancies have been prepared by simple thermal treatments. The H-InOx NRs show enhanced performance with the best formic acid (HCOOH) selectivity of up to 91.7% as well as high HCOOH partial current density over a wide range of potentials, largely outperforming those of the P-InOx and O-InOx NRs. Their catalysts show high stability with little decay over 20 hour electrolysis. Gao et al.104 studied the role of oxygen vacancies during the electroreduction of CO2. They constructed a model of oxygen vacancies confined in an atomic layer, taking the synthetic oxygen-deficient cobalt oxide single-unit-cell layers as an example. Density functional theory calculations demonstrate that the main defect is the oxygen(II) vacancy. Proton transfer has been theoretically/experimentally demonstrated to be the rate-limiting step, and via energy calculations, they have confirmed that the presence of oxygen(II) vacancies lowers the rate-limiting activation barrier from 0.51 to 0.40 eV by stabilizing the formate anion radical intermediate, as confirmed by the lowered onset potential from 0.81 to 0.78 V and decreased Tafel slope from 48 to 37 mV dec−1. Hence, the study reports that vacancy-rich cobalt oxide single-unit-cell layers exhibit the current densities of 2.7 mA cm−2 with an 85% formate selectivity during a 40 hour test.
Doping of metallic elements usually causes surface strain, which can help to improve the adsorption ability towards intermediates and tuning of the surface electronic structure in ECR. For instance, a small amount of (<3 at%) Ag atoms were doped into the Cu surface structure to obtain a surface alloy CuAg bimetal electrode. The Ag atom induces a compressive surface strain on the Cu lattice; this causes a change in the valence band density of the Cu state. Relative to the binding energy of *CO, the binding energy of copper to *H and *O is reduced; this results in the selective enhancement of the CO products.67 The study reported by Gewirth and Kenis et al. also demonstrates the effect of surface stress on the CO2 reduction selectivity.68 The nanoporous CuAg alloys were synthesized by electrodeposition, and a total of up to 60% of ethylene and 85% of C2 product were obtained. Cu and Ag can form an alloy on the surface. The study found that the surface Cu was concentrated in the local environment of the surface, and the surface strain was from the sublayer to the coverlayer. Zeng's group69 has developed octahedral and icosahedral Pd nanoparticles to study how tensile strain affects the CO2 reduction to CO. The nanoparticle model and DFT calculations show that the tensile strain causes the icosahedral Pd nanoparticles to move up the d-band center and strengthen the adsorption of the key intermediate COOH*; this explains that the FE (CO) is 1.7 times that of octahedral Pd nanoparticles. Sun et al. have reported that the trace element Te-doped Pd catalyst can convert CO2 to CO with high selectivity. The maximum CO Faraday efficiency is about 90% at −0.8 VRHE. The DFT calculations have shown that the Te atoms preferentially adsorb on the platform of Pd, inhibit the progress of HER and indirectly promote the adsorption and activation of CO2 at the high-index crystal plane of Pd.70
Defects play a significant role not only in catalysis but also in the synthesis of highly efficient catalysts. A defect can be used as a “trap” to capture a mononuclear metal precursor, and then, the single-atom catalyst can be stabilized by the charge-transfer effect between the single atom and the defect site. The single-atom catalysts (SAC) have excellent catalytic properties in CO2 reduction.71,72 For example, Li and his colleagues have designed a Ni single-atom catalyst. The Ni atoms were inserted into ZIF-derived carbons by ion exchange and subsequent carbonization. The Faraday efficiency of CO produced at the overpotential of 0.89 V exceeded 71.9%. The Ni single-atom doped carbon-based materials greatly improve the CO2 reduction performance as compared to Ni nanoparticles. Wang and coworkers73 have also found that the Ni single atoms dispersed into graphene nanosheets exhibit best selectivity to CO production. Electrochemical measurements show that Ni single atomic sites have 95% high CO selectivity at the overpotential of 620 mV. The valence state of the Ni atoms has good stability for over 20 hour continuous testing. The DFT calculation explains that the Ni atoms in a vacancy have lower COOH* free energy. These results provide evidence that Ni SAC has higher selectivity for ECR.
As multidimensional ‘defects’, grain boundaries (GB) may be the key to the significant enhancements in the selectivity and energy efficiency during the carbon dioxide reduction.74 Kanan et al. have reported that crystalline Cu prepared from Cu2O (oxide-derived Cu) produces multi-carbon oxygenates (ethanol and n-propanol) with the FE of 57%. They found a strong correlation between the density of grain boundaries and the performance of ECR. Subsequently, this group75 prepared polycrystalline gold rich in grain boundaries. The occurrence of local current across the grain boundary was detected by scanning electrochemical microscopy (SECCM), demonstrating that the grain boundary increased the CO2 electroreduction activity. The abrupt current jumps during line scan through GB under a CO2 atmosphere and an argon atmosphere together indicate that the activity of ECR originates from the grain boundary region.76 They envision the explanations that the grain boundary changes the binding energy of the intermediates, thereby reducing the overall energy barrier. Recently, Ajayan crystallized SnO2 NPs (<5 nm), which exhibited the high total FE of 97% (−0.95VRHE) for the formation of hydrocarbons. The grain boundary between crystallized SnO2 nanoparticles served as active sites for the adsorption/desorption of key intermediates. The “GB-enriched strategy” has significant implications for the improvement of carbon dioxide reduction performance.
5. Interface and surface modification
Low coordination number of atoms or the misalignment of atoms on the surface and at the interface will place them in a high-energy unstable state.22 These atoms are active sites in catalytic reactions with unique selectivity and activity.77 In addition, the interface can adsorb the intermediates produced in the catalytic process very well, thus promoting the occurrence of dimerization. Therefore, the interface and surface modification regulates the electronic structure of the atom, improving the adsorption capacity of the intermediate, further improving the performance of the ECR.78–81Metal–metal oxide interface is considered as the active region of catalysis with high activity and unique selectivity. Gao et al.82 have loaded Au NPs, CeOx NPs and Au-CeOx NPs on a carbon substrate. They demonstrated that Au NPs remained metallic in the composite catalyst, whereas the Ce element was in the form of Ce3+ coexisting with Ce4+. Moreover, the reduction of CeOx was promoted and the amount of Ce3+ was increased due to the strong interaction between Au and CeOx; thus, the activity of the catalyst was increased. The faradaic efficiency of the Au-CeOx/C catalyst for carbon monoxide could reach 89.1% at −0.9 V vs. RHE (Fig. 6a and b), which was much higher than that of CeOx/C and Au NPS/C. In addition, high catalytic current density indicated that the catalyst had better activities. Moreover, the Au-CeOx/C catalyst has very good stability. The interface was not inactivated during the catalytic process, and the catalyst was not deactivated for 10 hours.
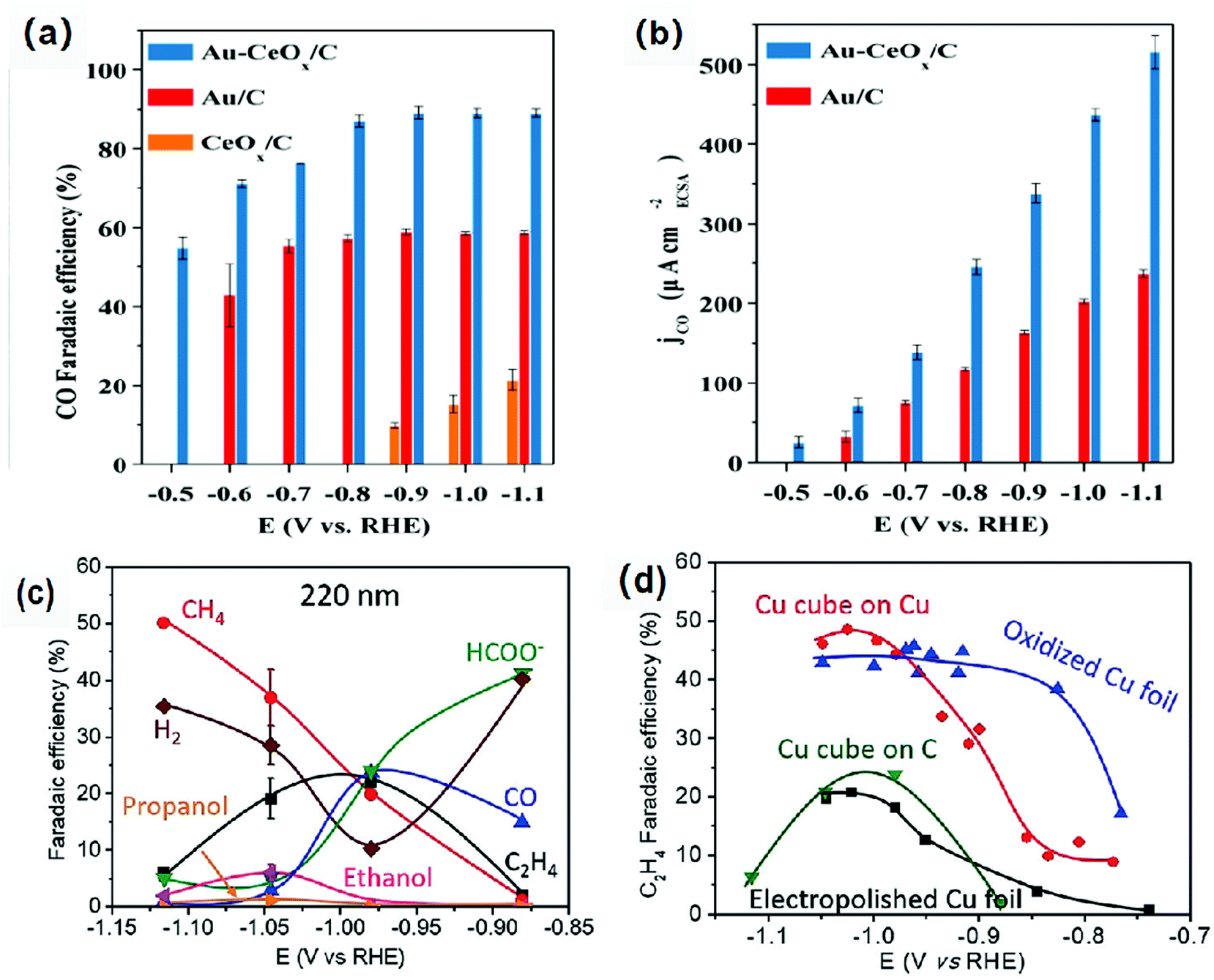 | ||
| Fig. 6 (a) CO faradaic efficiency, (b) Catalytic current density of the Au/C, CeOx/C and Au-CeOx/C catalysts for CO production at different overpotentials (a CO2-saturated 0.1 M KHCO3 solution). Reproduced with permission from ref. 82. Copyright 2019 Royal Society of Chemistry. (c) faradaic efficiency for each product of pristine 220 nm Cu cubes on C at different overpotentials after 1 h of ECR. (d) C2H4 faradaic efficiencies for the pristine 220 nm Cu cubes on C, 250 nm Cu cubes on a Cu foil and two references: electropolished Cu foils and O2-plasma treated Cu. Reproduced with permission from ref. 83. Copyright 2018 Wiley-VCH. | ||
The selectivity and activity of the same catalyst are greatly affected by the use of different carriers. Grosse et al.83 loaded copper cubes on a carbon paper and copper foil. There was a strong interaction between the copper cubes and the copper foil, which could stabilize the Cu+ species and increase the proportion of the Cu+ species. The dimerization reaction is more likely to occur at the interface of Cu/Cu+, which leads to high Faraday efficiency of C2 compounds. The Cu cube/C catalyst has weak adsorption capacity for the intermediate products because the interaction between carbon and copper is weak. For the Cu cube/C catalysts, the reduction of carbon dioxide to ethylene is inefficient because the dimerization of intermediates requires more energy (Fig. 6c and d). On the other hand, the morphology of copper cubes supported by carbon would change continuously in the catalytic process, the surface of Cu (100) would be roughened continuously, the cubic morphology of particles would change, and the number of particles would decrease; all these changes would seriously affect the selectivity of the catalysts and lead to poor catalytic stability. Baturina et al.84 loaded copper cubes on single-walled carbon nanotubes (SWNT), reduced graphene oxide (RGO) and onion-like carbon (OLC) (Fig. 7) and observed their catalytic performances and behaviors in the catalytic process. Copper cube/OLC had high selectivity for ethylene, and the faradaic efficiency for ethylene could reach 60% at −1.8 V vs. Ag/AgCl. The particle size of OLC was smaller than that of cubes; however, in general, the size of a support should be larger than that of the catalyst. The structure of the catalyst Cu-cubes surrounded by the OLC particles could increase the CO concentration on the surface of Cu-cubes, thus promoting CO dimerization on the surface of Cu (100) and improving the faradaic efficiency for ethylene. This is due to the fact that OLC can catalyze the production of CO; this will increase the pH around the catalyst and facilitate the occurrence of dimerization. Compared with copper cube/SWNT and copper cube/RGO, copper cube/OLC has good stability and long catalytic life. This is because the structure of copper cube/OLC can restrain the growth of copper cubes. After one hour of catalysis, the catalytic performance did not decrease, and the faradaic efficiency for ethylene was slightly improved.
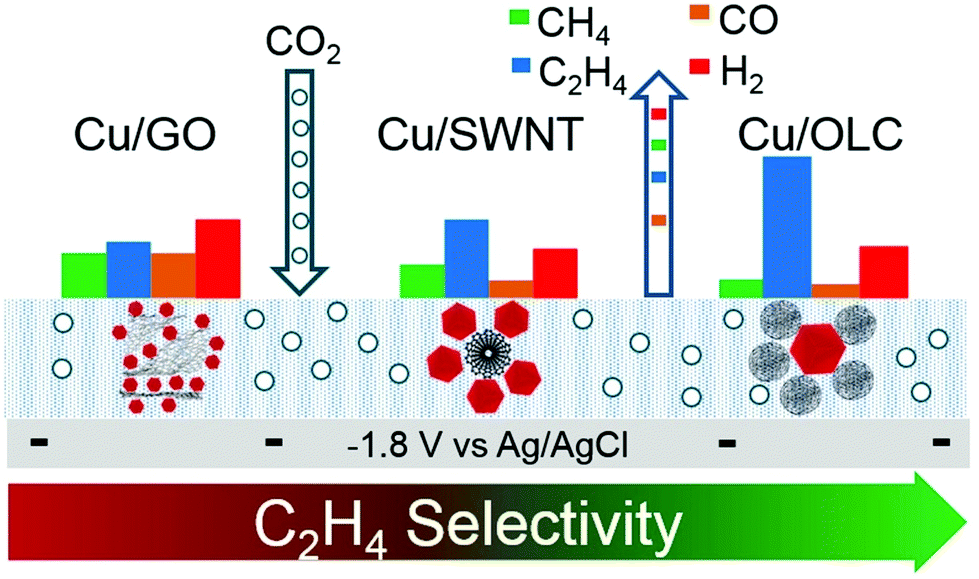 | ||
| Fig. 7 Different structures of Cu cube/SWNT, Cu cube/RGO and Cu cube/OLC. faradaic efficiency of each structure for each product. Reproduced with permission from ref. 84. Copyright 2017 Elsevier B.V. | ||
For the interface, the hybrid from different dimensionalities may bring distinct behavior. Gong et al.110 introduced metal Cu nanoparticles (NPs) on the Cu2O film to change the CO2 reduction product distribution in an aqueous solution. The Faraday efficiency of the Cu/Cu2O cathodes on CH3OH was as high as 53.6%. The specially designed Cu/Cu2O interface balances the binding strength of H* and CO* intermediates, which plays a key role in CH3OH production. Wang et al.111 prepared a nanoparticle-coated copper oxide nanowire (CuxO-Sn) catalyst for the efficient CO2 reduction. The CuxO nanowires (NWs) modified by the deposited Sn nanoparticles (NPs) showed a significantly improved CO Faraday efficiency from 6% to 82%. The high selectivity of CO in Cu–Sn hybrid nanowires is likely due to the synergistic interaction between the Sn NPs and the Cu NWs. The Sn atom at the interface can destroy multiple sites on the surface of pure Cu, which is not conducive to the adsorption of H, and the adsorption of CO is relatively undisturbed. Chen et al.112 constructed a method for the Cu atom pair site immobilized on the Pd10Te3 alloy nanowires, which stabilized the CO2 molecule chemisorption in the critical step of CO2 reduction. Due to this, the FE for CO exceeded 92%, which provided a novel and efficient method for the construction of functional interfaces at the atomic level.
Surface modification can change the surface structure and electronic distribution of the catalyst, which can improve its catalytic performance. The doping of electrode materials can increase the surface atom dislocation and lead them to a high energy state, which can improve the catalytic activity. Zhang et al.85 used a plasma treatment to treat CNT/GC (Glass carbon) and achieved NCNT (nitrogen-doped carbon nanotubes)/GC and OCNT (oxygen-doped carbon nanotubes)/GC. The specific surface area was measured, and the result showed that both dopings increased the specific surface area. Then, they modified NCNT/GC and OCNT/GC by PEI. The catalytic current densities of the doped NCNT and OCNT were higher than that of the single CNT. The faradaic efficiency of the doped 7% NCNT/GC for formic acid was 59% at −1.8 VSCE. The data showed that the optimum nitrogen doping concentration was 7%, and the performance of the catalyst began to decline after 7%; this was mainly because the increase in N concentration reduced the conductivity of the catalyst. The surface of PEI has strong adsorption capacity for CO2, which can improve the faradaic efficiency and reaction current density of formic acid. The interaction between carbon materials and PEI can reduce the overpotential required for catalysis. On the one hand, the Faraday efficiency of PEI-NCNT to formic acid is 85% at −1.8 V relative to SCE, and it has high catalytic current. On the other hand, the PEI-NCNT catalyst has good stability, and the catalytic current does not decrease after 24 hours of the catalysis.
Pure metal catalysts have the problems of selectivity and activity. Some studies have shown that metal catalysts modified by organic functional groups have good selectivity and activity. Xie et al.86 prepared glycine-modified copper nanowire thin film electrodes. XPS showed that the copper nanowire film electrode modified by glycine was attached with the *NH2 group and *COOH group, and the peak of the *NH2 group could still be detected after catalysis; this indicated that the *NH2 group could exist stably in the catalyst. The faradaic efficiency for hydrocarbons increased first and then decreased with an increase in the content of modified glycine. The author speculated that the reduction of catalytic active sites was a result of excessive coverage. The faradaic efficiency of the glycine-modified copper nanowire film electrode for the hydrocarbon catalytic products at −1.9 V vs. RHE was 34.1%, which was significantly higher than that of the unmodified electrode. This phenomenon is explained by the fact that the adsorbed group can combine with the catalytic reaction intermediate CHO* (Fig. 8), which promotes the intermediate dimerization to produce hydrocarbons.
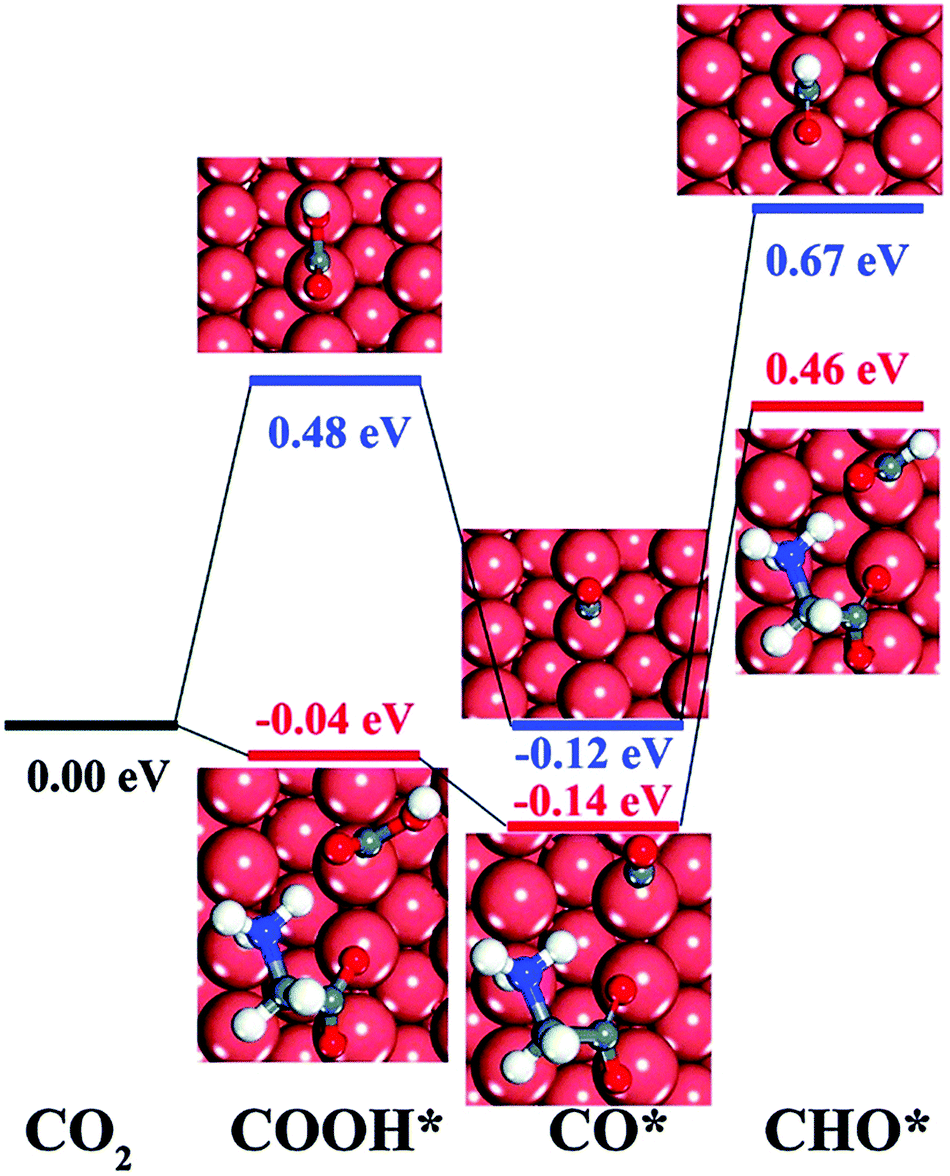 | ||
| Fig. 8 The DFT-calculated free energy change of CO2 and CO, each compound protonated without glycine (blue line) and with zwitterionic glycine (red line). Reproduced with permission from ref. 86. Copyright 2016 The Royal Society of Chemistry. | ||
6. Oxide derivation
Transition metal oxides are important catalysts and have been widely used in the catalytic field. Metal oxides have better catalytic performance after nanocrystallization. Metal cations in transition metal oxides easily gain or lose electrons and have strong redox properties. They also have semiconductor properties, which can enhance the adsorptive effect of the catalyst to reactants. Compared with metal catalysts, metal oxides have a certain catalytic performance as long as the metal has a variable valence state; thus, the range of materials available is wide. The stability of a metal oxide catalyst is better than that of a metal catalyst because of its high melting point and good heat resistance.Some metal oxides can be used as catalysts themselves, and many studies have used metal oxides as precursors to generate metal nanostructures that can be used for catalysis.87,88 Therefore, there are two main effects of oxide catalysts on ECR. The first is that metal oxides may have high catalytic activity and unique selectivity.89,90 The second is that metal oxides can be reduced at certain overpotential to form nanostructured metals.91
The DFT model calculation shows that Cu2O (200) also has a catalytic effect. Mandal et al.92 analyzed the catalysts using operando SIFT-MS, computational modeling and Raman spectroscopy technology. It was found that Cu2O decomposed before the catalytic reaction took place. Although their DFT model calculation found that Cu2O (200) had a catalytic effect, it decomposed and hence did not participate in the catalysis.
The structure of the catalyst often undergoes self-reconstruction during electrochemical reactions.105–109 Zheng and co-workers105 prepared an inverse opal structure cuprous oxide film electrode by the electrochemical reduction of copper lactate. In the process of CO2 reduction, the inverse opal oxide cuprous oxide film would be reduced to form a copper nanostructure film. The SEM images showed that it still retained the inverse opal structure. The diffraction peak of the copper metal in the XRD pattern indicated that cuprous oxide was reduced to form metallic copper. The maximum current density in the catalytic process could reach 1.2 mA cm−2, and the FE for CO was 45.3%, which was much higher than 14.6% of the Cu particle film. Sargent et al.109 have reported a facile surface reconstruction approach that enables tuning of CO2-reduction selectivity towards C2+ products on a copper-chloride (CuCl)-derived catalyst. Via a novel wet-oxidation process, both the oxidation state and the morphology of Cu surface were controlled to provide uniformity of the electrode morphology and abundant surface active sites. The Cu surface is partially oxidized to form an initial Cu(I) chloride layer, which is subsequently converted to a Cu(I) oxide surface. High C2+ selectivity on these catalysts has been demonstrated in an H-cell configuration, in which a 73% faradaic efficiency (FE) for C2+ products has been achieved with a 56% FE for ethylene (C2H4) and the overall current density of 17 mA cm−2. The record C2 + FE of ≈84% and the half-cell power conversion efficiency of 50% at the partial current density of 336 mA cm−2 obtained using the reconstructed Cu catalyst are also reported.
Kim et al.93 found that Cu2O partially reduced during catalysis; this resulted in a decrease of oxygen in the catalyst, leaving oxygen vacancies in the reduced copper nanostructures. Reduced copper nanostructures play an important role in the high faradaic efficiency for ethylene production. There would be residual Cu2O in the catalyst due to the partial reduction of Cu2O. Their research show that Cu2O remains on the surface of the catalyst during the catalytic process. At lower overpotentials, it could catalyze the decomposition of carbon dioxide efficiently and had good stability. The faradaic efficiency for ethylene production could still reach 20% at −1.9 V vs. AgCl under long-term conditions (Fig. 9a).
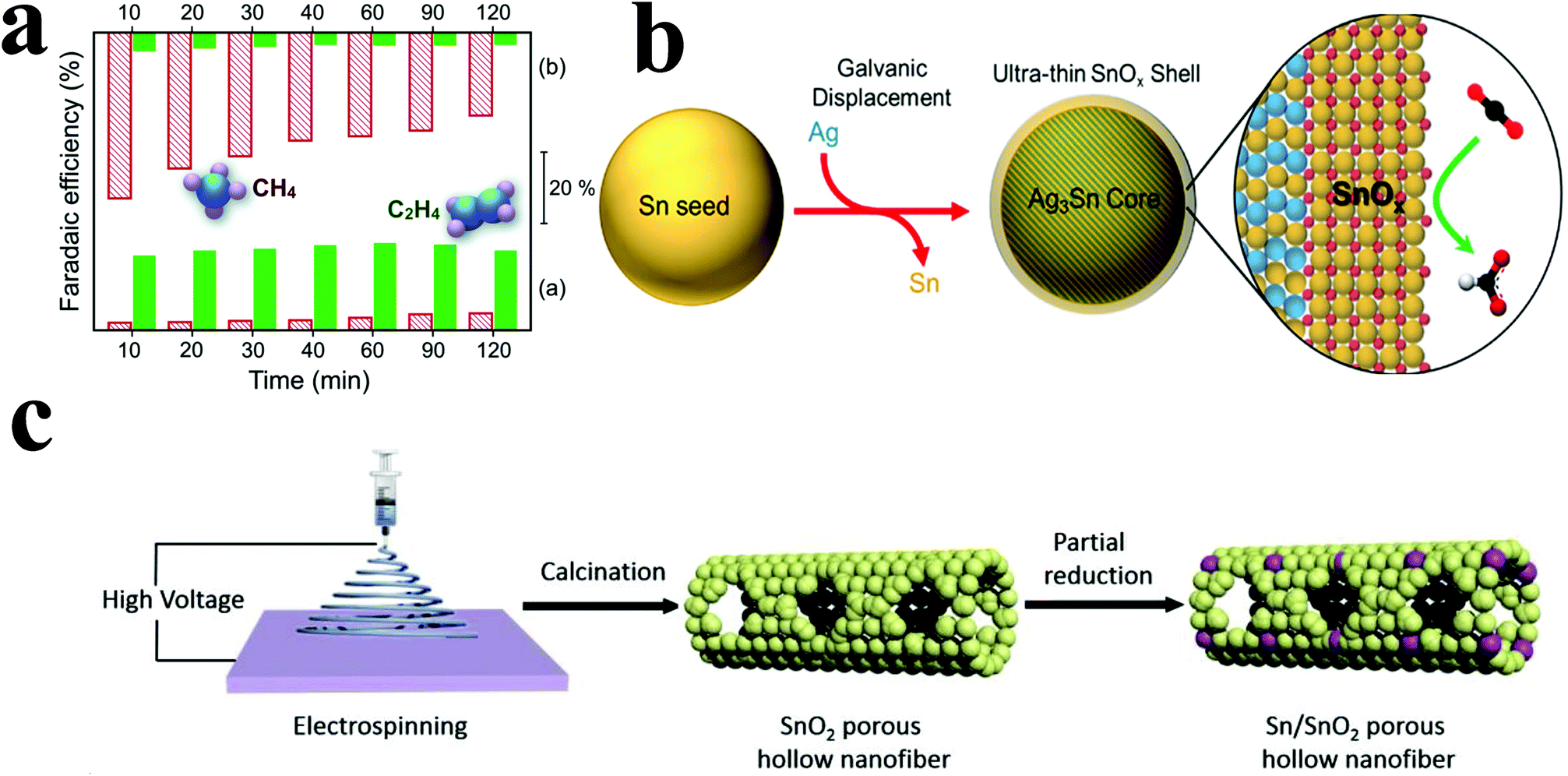 | ||
| Fig. 9 (a) Faradaic efficiencies for C2H4 and CH4 on the Cu2O (bottom) and Cu catalyst (top) at different times of catalysis. Both gas phase products were obtained and analyzed at each marked time. Reproduced with permission from ref. 93. Copyright 2015 Royal Society of Chemistry. (b) Structure diagram of the preparation of a core–shell structure tin oxide catalyst. Reproduced with permission from ref. 94. Copyright 2017 American Chemical Society. (c) Structure diagram of the preparation of the Sn/SnO2 PHF catalyst. Reproduced with permission from ref. 97. Copyright 2018 Elsevier B.V. | ||
At higher negative potentials, incompletely oxidized tin can be used as a catalyst for ECR. However, tin oxide has poor conductivity, which seriously affects its catalytic performance; thus, it needs to be loaded on a carrier with good conductivity. Luc et al.94 electrodeposited tin particles on Ag3Sn cores; this resulted in incompletely oxidized tin shells with nanometer thickness around the Ag3Sn cores (Fig. 9b). When the shell thickness was 1.7 nm, the faradaic efficiency for formic acid and carbon monoxide could reach 80% and 10% at −0.9 V vs. RHE, respectively. Catalysis for 24 hours could still maintain a relatively stable current density. Yiliguma et al.95 combined the hollow carbon structure with the hollow structure formed by the aggregation of tin oxide particles and made a nano-hollow carbon sphere structure modified by tin oxide. Sn–O–C bonding was formed in this structure, which could enhance the adsorption of carbon dioxide and increase the catalytic activity. The faradaic efficiency for formate reached 54.2% at −0.9 V vs. RHE, and its stability was better than that of the tin oxide particle-aggregated hollow structure catalyst.
In addition to tin oxide catalysts with core–shell structure, mesoporous tin oxide structure also has good catalytic performance to carbon dioxide. Ge et al.96 synthesized mesoporous tin oxide structure by hydrothermal method using cetyltrimethylammonium bromide. This structure had a high specific surface area and a mesoporous structure, conducive to adsorbing gas molecules. The faradaic efficiency for CO and formate can reach 80% at −0.8 V vs. RHE. Moreover, the highly ordered and uniform pore size of the catalyst had better catalytic performance. However, the life of the catalyst was short and the mesoporous structure would disappear during the catalysis process. Hu et al.97 prepared porous SnO2 hollow fibers by electrospinning, then partially reduced them to form porous Sn/SnO2 hollow fibers (Fig. 9c). The catalyst had a good adsorption effect to carbon dioxide, the faradaic efficiency for formate could reach more than 80% at −1.6 VSCE and the current density could reach 22.9 mA cm−2. The current density did not decrease significantly after 10 hours of catalytic reaction, which indicated that the catalyst had good stability.
The oxide-derived catalysts will decompose at certain overpotentials to form nanometer metal structures; in contrast, nanostructured materials can exhibit better catalytic activity and selectivity than a flat surface. Edges, defects and grain boundaries in nanostructures can directly boost the selectivity and activity of the catalysts; on the other hand, because of its high specific surface area and strong adsorption capacity for gases, a nanostructured material exhibits better selectivity and higher catalytic activity than the flat surface. In situ electrochemical reduction of metal oxides is the possible and effective way to prepare rough surfaces and high-defect density structures.
Lee et al.98 prepared an anodized copper (AN-Cu)-Cu(OH)2 catalyst by the electrochemical method. The catalyst had good selectivity for ethylene, and its faradaic efficiency for ethylene production could reach 38.1% at −1.08 V vs. RHE. Moreover, the catalyst exhibited long service life and no obvious deactivation after 30 hours. The SEM image of AN-Cu is shown (Fig. 10a). Its structure could be controlled by controlling the reaction time. They pointed out that Cu(OH)2 was reduced to Cu when the catalysis took place. Some copper was oxidized after the completion of catalysis, and then, a mixture of copper and oxidized copper was generated.
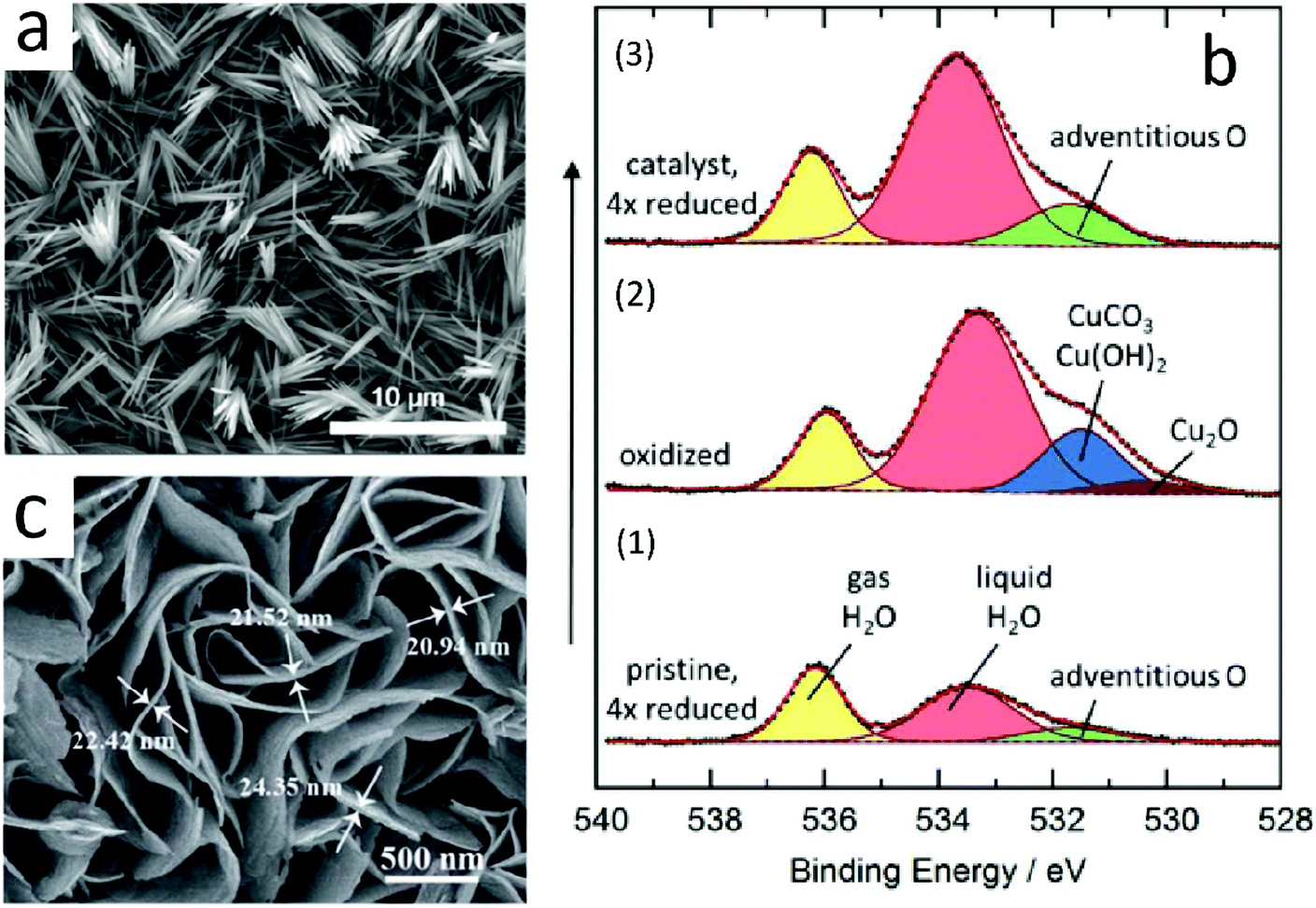 | ||
| Fig. 10 (a) SEM image of AN-Cu. Reproduced with permission from ref. 98. Copyright 2018 American Chemical Society. (b) In situ O 1s XPS spectra obtained with the incident photon energy of 3996 eV. (1) Pristine sample. (2) Oxidation of the pristine sample. (3) Initially oxidized and then reduced sample. Intensities not normalized. Reproduced with permission from ref. 99. Copyright 2016 American Chemical Society. (c) High-magnification SEM images of CuS@NF. Reproduced with permission from ref. 100. Copyright 2017 The Royal Society of Chemistry. | ||
Although metal oxides will be decomposed in the early stages of catalysis, there will be oxygen atoms remaining on the sub-surface of the catalyst, which would affect the selectivity and activity of the catalyst. Eilert et al.99 studied catalysis via in situ ambient pressure X-ray photoelectron spectroscopy and transmission electron microscopy. They found that copper oxide catalysts were reduced to nanostructured copper before ECR, and copper oxide disappeared completely during the reduction process (Fig. 10b); however, there was a large amount of residual oxygen that could enhance the selectivity of the catalyst for ethylene. They proposed that the presence of underground oxygen could reduce the binding energy of CO*. The reaction is more inclined to form a C–C bond because CO is adsorbed on the catalyst surface.
Zhao et al.100 loaded copper sulfide nanosheets on nickel foam to form nanostructured catalysts (Fig. 10c). This structure had high specific surface area and could adsorb the intermediate products of ECR well. It had good selectivity for methane, and the faradaic efficiency could reach 73.5% at −1.1 V vs. RHE. They speculated that Cu2+ was reduced to nanostructured copper, which played an important role in the catalytic process, and some S would remain in the catalyst, which could stabilize the intermediate products formed in the catalytic process. Thus, it had high faradaic efficiency for methane and catalytic activity.
7. Conclusion and outlook
This review focuses on the recent trends and strategies for the construction of nanostructure-based inorganic catalysts for ECR, especially the structure–performance relationship. Although many factors challenge the product selectivity and faradaic efficiency, size, morphology, crystal facets, defects, interface, surface and oxide derivation have a more direct impact on the enhancement of product selectivity and stability of catalyst during the electocatalysis of CO2. An appropriate modification and engineering of different catalysts allows binding of the catalysts to various intermediates to different extents, which leads to different products via control of the catalyst design.Although extensive experimental and computational studies have been repeatedly conducted over the past few years to accelerate the development of electrocatalysis for CO2 reduction and understand the structure–performance relationships, it is still very challenging to efficiently reduce CO2 to desirable products with maximum selectivity. This hinders the commercialization of electrocatalysis on an industrial scale; in this regard, the main challenges in this field include high overpotential, low catalytic activity, poor product selectivity, and unsatisfactory catalyst stability, which need further attention while designing a catalyst. To overcome these challenges, we have proposed the following future outlooks and research directions.
(1) Development of an innovative catalyst that enhances the catalytic activity and stability
Although many efforts have been made for achieving high catalytic activity, there exists a need for a breakthrough in the catalyst design in terms of product selectivity and stability for industrial applications; in this regard, the development of an innovative catalyst with optimal performance and good stability is an undeniable need. The catalyst surface plays a great role in performance enhancement. A more roughened surface has more active sites such as edges and defects. Besides, one of the problems in engineering the surface is the weak adsorption of the catalyst surface to the intermediate, which is evident in the case of C2 product formation, where dimerization of each CO molecule occurs at the surface of the catalyst. On the other hand, although oxide-derived catalysts have been widely employed as compared to Cu and Sn-derived catalysts, these catalysts have not gained popularity because of the difficulty in their complete decomposition; this leads to oxygen decrease; moreover, a definite mechanism for this action is not confirmed to date.(2) Explore more the structure–performance with the aid of DFT calculation
In the design of a catalyst with good activity, product selectivity and stability, it is vital to understand the fundamental principles that govern the catalyst both theoretically and experimentally. A better understanding of the catalyst mechanism for electroreduction is very necessary. For example, the definite mechanism of action of OD catalysts has not been explored to date; the fact that oxygen on the subsurface can influence the performance needs to be studied well with the aid of DFT and experimental observations; overall, a better understanding of catalyst performance needs to be achieved in depth. In addition to DFT, advanced characterization techniques can provide more insight into the catalyst nature and help more in the design of a new catalyst. An in situ technique can help in probing the morphology and the electronic state of the catalyst. Thus it gives a new verification method about the nature of catalyst for electrochemical CO2 reduction.(3) Optimization of reaction conditions and system design
One of the biggest problems in this area is rapid catalyst degradation and slow kinetics of CO2 reduction. This issue has more direct relation with the electrode/reactor design. Therefore, improvement of the catalyst working environment and optimization of the reaction conditions are very important to increase the working life of catalysts.In summary, to mitigate the current energy crises that our world faces, CO2 electroreduction to valuable fuels is an undeniable fact. Despite the great challenges that lie ahead in the commercialization of ECR, an extensive research in this field can lead to a highly efficient and robust catalyst for CO2 electroreduction in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support received from the National Natural Science Foundation of China (51772957, Y. G.; 51772024, X. Q. Y.).References
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- H. Yang, Z. Xu, M. Fan, R. Gupta, R. B. Slimane, A. E. Bland and I. Wright, J. Environ. Sci., 2008, 20, 14–27 CrossRef CAS.
- X. Xiaoding and J. A. Moulijn, Energy Fuels, 1996, 10, 305–325 CrossRef CAS.
- D. R. Feldman, W. D. Collins, P. J. Gero, M. S. Torn, E. J. M. Lawer and T. R. Shippert, Nature, 2015, 519, 339 CrossRef CAS PubMed.
- M. E. Mann, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4065–4066 CrossRef CAS PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille and P. J. Kenis, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- G. A. Ozin, Adv. Mater., 2015, 27, 1957–1963 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- J. P. Jones, G. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- D. T. Whipple and P. J. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- B. Innocent, D. Pasquier, F. Ropital, F. Hahn, J.-M. Léger and K. B. Kokoh, Appl. Catal., B, 2010, 94, 219–224 CrossRef CAS.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- H. Xie, T. Wang, J. Liang, Q. Li and S. Sun, Nano Today, 2018, 21, 41–54 CrossRef CAS.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, e1802066 CrossRef PubMed.
- Z.-L. Wang, C. Li and Y. Yamauchi, Nano Today, 2016, 11, 373–391 CrossRef CAS.
- W. Xia, A. Mahmood, Z. Liang, R. Zou and S. Guo, Angew. Chem., Int. Ed., 2016, 55, 2650–2676 CrossRef CAS PubMed.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- B. Braunchweig, D. Hibbitts, M. Neurock and A. Wieckowski, Catal. Today, 2013, 202, 197–209 CrossRef CAS.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem., 2017, 3, 560–587 CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- L. Zhang, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- W. Zhu, R. Michalsky, O. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- H. Mistry, R. Reske, Z. Zeng, Z. J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS PubMed.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS PubMed.
- O. A. Baturina, Q. Lu, M. A. Padilla, L. Xin, W. Li, A. Serov, K. Artyushkova, P. Atanassov, F. Xu, A. Epshteyn, T. Brintlinger, M. Schuette and G. E. Collins, ACS Catal., 2014, 4, 3682–3695 CrossRef CAS.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Nano Lett., 2017, 17, 1312–1317 CrossRef CAS PubMed.
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS.
- A. Dutta, M. Rahaman, M. Mohos, A. Zanetti and P. Broekmann, ACS Catal., 2017, 7, 5431–5437 CrossRef CAS.
- T. T. Hoang, S. Ma, J. I. Gold, P. J. Kenis and A. A. Gewirth, ACS Catal., 2017, 7, 3313–3321 CrossRef CAS.
- T. N. Huan, P. Simon, G. Rousse, I. Génois, V. Artero and M. Fontecave, Chem. Sci., 2017, 8, 742–747 RSC.
- H. Xie, T. Wang, J. Liang, Q. Li and S. Sun, Nano Today, 2018, 21, 41–54 CrossRef CAS.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed.
- C. Chen, B. Zhang, J. Zhong and Z. Cheng, J. Mater. Chem. A, 2017, 5, 21955–21964 RSC.
- X. Wen, L. Chang, Y. Gao, J. Han, Z. Bai, Y. Huan, M. Li, Z. Tang and X. Yan, Inorg. Chem. Front., 2018, 5, 1207–1212 RSC.
- W. Zhang, J. He, S. Liu, W. Niu, P. Liu, Y. Zhao, F. Pang, W. Xi, M. Chen, W. Zhang, S. S. Pang and Y. Ding, Nanoscale, 2018, 10, 8372–8376 RSC.
- S. Sen, D. Liu and G. T. R. Palmore, ACS Catal., 2014, 4, 3091–3095 CrossRef CAS.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796–800 CrossRef CAS PubMed.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., 2016, 128, 6792–6796 CrossRef.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382 CrossRef CAS PubMed.
- H.-E. Lee, K. D. Yang, S. M. Yoon, H.-Y. Ahn, Y. Y. Lee, H. Chang, D. H. Jeong, Y.-S. Lee, M. Y. Kim and K. T. Nam, ACS Nano, 2015, 9, 8384–8393 CrossRef CAS PubMed.
- A. Klinkova, P. De Luna, C.-T. Dinh, O. Voznyy, E. M. Larin, E. Kumacheva and E. H. Sargent, ACS Catal., 2016, 6, 8115–8120 CrossRef CAS.
- N. Hoshi, M. Kato and Y. Hori, J. Electroanal. Chem., 1997, 440, 283–286 CrossRef CAS.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- K. J. P. Schouten, Z. Qin, E. P. Gallent and M. T. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- K. J. P. Schouten, E. P. Gallent and M. T. Koper, ACS Catal., 2013, 3, 1292–1295 CrossRef CAS.
- K. J. P. Schouten, E. P. Gallent and M. T. Koper, J. Electroanal. Chem., 2014, 716, 53–57 CrossRef CAS.
- C. S. Chen, A. D. Handoko, J. H. Wan, L. Ma, D. Ren and B. S. Yeo, Catal. Sci. Technol., 2015, 5, 161–168 RSC.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2015, 6, 219–229 CrossRef.
- Y. Huang, A. Handoko, P. Hirunsit and B. Yeo, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS PubMed.
- C. Hahn, T. Hatsukade, Y.-G. Kim, A. Vailionis, J. H. Baricuatro, D. C. Higgins, S. A. Nitopi, M. P. Soriaga and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5918–5923 CrossRef CAS PubMed.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X.-D. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X.-D. Zhou, R. Vajtai, B. I. Yakobson, J. Lou and P. M. Ajayan, Nano Lett., 2015, 16, 466–470 CrossRef PubMed.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X. D. Zhou, Angew. Chem., Int. Ed., 2015, 54, 13701–13705 CrossRef CAS PubMed.
- N. Sreekanth, M. A. Nazrulla, T. V. Vineesh, K. Sailaja and K. L. Phani, Chem. Commun., 2015, 51, 16061–16064 RSC.
- Y. Liu, Y. Zhang, K. Cheng, X. Quan, X. Fan, Y. Su, S. Chen, H. Zhao, Y. Zhang, H. Yu and M. R. Hoffmann, Angew. Chem., Int. Ed., 2017, 56, 15607–15611 CrossRef CAS PubMed.
- J. Xie, X. Zhao, M. Wu, Q. Li, Y. Wang and J. Yao, Angew. Chem., Int. Ed., 2018, 57, 9640–9644 CrossRef CAS PubMed.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS PubMed.
- T. T. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- H. Huang, H. Jia, Z. Liu, P. Gao, J. Zhao, Z. Luo, J. Yang and J. Zeng, Angew. Chem., 2017, 129, 3648–3652 CrossRef.
- H. Tao, X. Sun, S. Back, Z. Han, Q. Zhu, A. W. Robertson, T. Ma, Q. Fan, B. Han, Y. Jung and Z. Sun, Chem. Sci., 2018, 9, 483–487 RSC.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504 CrossRef CAS PubMed.
- R. G. Mariano, K. McKelvey, H. S. White and M. W. Kanan, Science, 2017, 358, 1187–1192 CrossRef CAS PubMed.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819 CrossRef.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- O. A. Baturina, Q. Lu, M. A. Padilla, L. Xin, W. Li, A. Serov, K. Artyushkova, P. Atanassov, F. Xu, A. Epshteyn, T. Brintlinger, M. Schuette and G. E. Collins, ACS Catal., 2014, 4, 3682–3695 CrossRef CAS.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, J. Ma, Z. Zhang, G. Yang and B. Han, Green Chem., 2017, 19, 2086–2091 RSC.
- J. Yuan, M.-P. Yang, Q.-L. Hu, S.-M. Li, H. Wang and J.-X. Lu, J. CO2 Util., 2018, 24, 334–340 CrossRef CAS.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed.
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. Li, J. Zhu, P. Liu, F. Yang, G. Wang and X. Bao, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS PubMed.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2018, 57, 6192–6197 CrossRef CAS PubMed.
- O. Baturina, Q. Lu, F. Xu, A. Purdy, B. Dyatkin, X. Sang, R. Unocic, T. Brintlinger and Y. Gogotsi, Catal. Today, 2017, 288, 2–10 CrossRef CAS.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- F.-S. Ke, X.-C. Liu, J. Wu, P. P. Sharma, Z.-Y. Zhou, J. Qiao and X.-D. Zhou, Catal. Today, 2017, 288, 18–23 CrossRef CAS.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- R. Kas, R. Kortlever, A. Milbrat, M. T. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194–12201 RSC.
- L. Mandal, K. R. Yang, M. R. Motapothula, D. Ren, P. Lobaccaro, A. Patra, M. Sherburne, V. S. Batista, B. S. Yeo, J. W. Ager, J. Martin and T. Venkatesan, ACS Appl. Mater. Interfaces, 2018, 10, 8574–8584 CrossRef CAS PubMed.
- D. Kim, S. Lee, J. D. Ocon, B. Jeong, J. K. Lee and J. Lee, Phys. Chem. Chem. Phys., 2015, 17, 824–830 RSC.
- W. Luc, C. Collins, S. Wang, H. Xin, K. He, Y. Kang and F. Jiao, J. Am. Chem. Soc., 2017, 139, 1885–1893 CrossRef CAS PubMed.
- Y. Yiliguma, Z. Wang, C. Yang, A. Guan, L. Shang, A. M. Al-Enizi, L. Zhang and G. Zheng, J. Mater. Chem. A, 2018, 6, 20121–20127 RSC.
- H. Ge, Z. Gu, P. Han, H. Shen, A. M. Al-Enizi, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2018, 531, 564–569 CrossRef CAS PubMed.
- H. Hu, L. Gui, W. Zhou, J. Sun, J. Xu, Q. Wang, B. He and L. Zhao, Electrochim. Acta, 2018, 285, 70–77 CrossRef CAS.
- S. Y. Lee, H. Jung, N. K. Kim, H. S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2018, 140, 8681–8689 CrossRef CAS PubMed.
- A. Eilert, F. Cavalca, F. S. Roberts, J. Osterwalder, C. Liu, M. Favaro, E. J. Crumlin, H. Ogasawara, D. Friebel, L. G. Pettersson and A. Nilsson, J. Phys. Chem. Lett., 2017, 8, 285–290 CrossRef CAS PubMed.
- Z. Zhao, X. Peng, X. Liu, X. Sun, J. Shi, L. Han, G. Li and J. Luo, J. Mater. Chem. A, 2017, 5, 20239–20243 RSC.
- X. Cui, Z. Pan, L. Zhang, H. Peng and G. Zheng, Adv. Energy Mater., 2017, 7, 1701456 CrossRef.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- J. Zhang, R. Yin, Q. Shao, T. Zhu and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 5609–5613 CrossRef CAS PubMed.
- S. Gao, Z. Sun, W. Liu, X. Jiao, X. Zu, Q. Hu, Y. Sun, T. Yao, W. Zhang, S. Wei and Y. Xie, Nat. Commun., 2017, 8, 14503 CrossRef CAS PubMed.
- X. Zheng, J. Han, Y. Fu, Y. Deng, Y. Liu, Y. Yang, T. Wang and L. Zhang, Nano Energy, 2018, 48, 93–100 CrossRef CAS.
- A. S. Malkani, M. Dunwell and B. Xu, ACS Catal., 2018, 9, 474–478 CrossRef.
- R. Kas, R. Kortlever, A. Milbrat, M. T. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194–12201 RSC.
- C. S. Chen, J. H. Wan and B. S. Yeo, J. Phys. Chem. C, 2015, 119, 26875–26882 CrossRef CAS.
- M. G. Kibria, C. T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. García de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1804867 CrossRef PubMed.
- X. Chang, T. Wang, Z. J. Zhao, P. Yang, J. Greeley, R. Mu, G. Zhang, Z. Gong, Z. Luo, J. Chen, Y. Cui, G. A. Ozin and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 15415–15419 CrossRef CAS PubMed.
- Y. Zhao, C. Wang and G. G. Wallace, J. Mater. Chem. A, 2016, 4, 10710–10718 RSC.
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S.-F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |