
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-slaved protein motions accompany proton coupled electron transfer reactions catalysed by copper nitrite reductase†
Tobias M.
Hedison
,
Derren J.
Heyes
,
Muralidharan
Shanmugam
,
Andreea I.
Iorgu
and
Nigel S.
Scrutton
 *
*
The Manchester Institute of Biotechnology, University of Manchester, Manchester, M1 7DN, UK. E-mail: nigel.scrutton@manchester.ac.uk; Tel: +44 161 306 5152
First published on 3rd May 2019
Abstract
Through the use of time-resolved pH-jump spectroscopy, we demonstrate how proton transfer is coupled to inter-copper electron transfer in a copper nitrite reductase (CuNiR). Combined use of electron paramagnetic resonance spectroscopy with solvent viscosity- and pressure-dependence pH-jump stopped-flow spectroscopy is used to show that solvent-slaved protein motions are linked to this proton coupled electron transfer step in CuNiR.
Enzyme catalysed electron transfer reactions are ubiquitous in biology and are often coupled to protein conformational change or additional chemical steps.1,2 An example of a chemical event that is frequently linked to electron transfer reactions is proton transfer. The transfer of an electron and a proton together (either by a sequential or a concerted mechanism) is referred to as proton coupled electron transfer (PCET).3 PCET reactions are used by nature for processes essential to life, including respiration (i.e. by complex IV) and photosynthesis (i.e. by photosystem II).3 The highly conserved family of copper-containing nitrite reductases (CuNiRs) utilise a PCET reaction to catalyse the first committed step in denitrification, i.e. the reduction of soluble nitrite (NO2−) to gaseous nitric oxide (NO).4–6
CuNiRs are homotrimeric proteins.7 Within each monomer, there is an electron accepting T1Cu and a catalytic T2Cu centre that are located within discreet cupredoxin domains. From a mechanistic viewpoint, CuNiRs function by transferring electrons (originating from cytochrome c or azurin partner proteins), through a T1Cu centre to a catalytic T2Cu centre, where nitrite is converted to nitric oxide.4–6,8 The X-ray crystallographic structures of CuNiRs show that there is a 12.6 Å distance between the T1Cu and T2Cu sites in these enzymes (Fig. 1A).7 It has been hypothesised that electron transfer between these two copper sites is coupled to proton transfer. Nitrite reduction involves the consumption of two protons5 and active site residues D92 and H249 have been suggested to be the source of these protons (Fig. 1A).6 These protons are ultimately derived from bulk solvent and passed along a water channel (involving residue N90) that connects the T2Cu site to the surface of the protein.9,10 PCET in CuNiR has been shown to occur by a sequential mechanism with proton transfer occurring before electron transfer.10,11
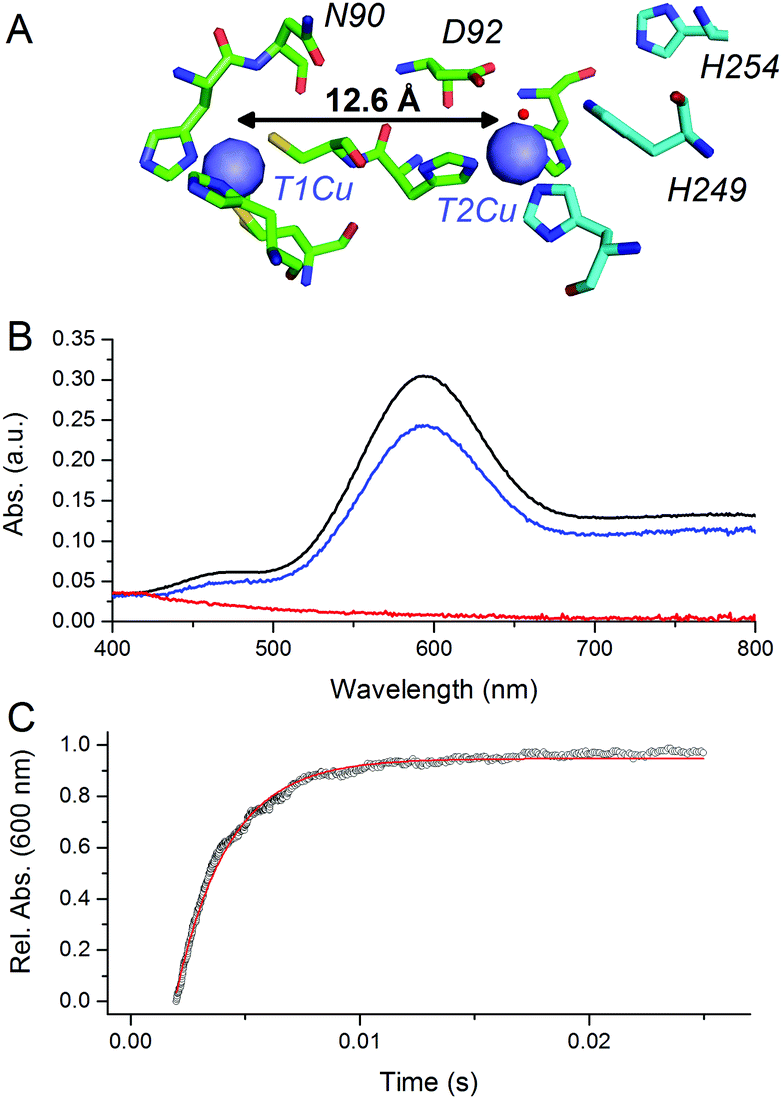 | ||
| Fig. 1 pH-jump method used to study PCET reactions of CuNiR. (A) Structure of T1 and T2Cu sites in AxNiR.7 (B) Absorbance spectra of ∼50 μM oxidised, nitrite-bound AxNiR present in MTEN buffer at pH 9 (black); T1Cu reduced, T2Cu oxidised nitrite-bound AxNiR present in MTEN buffer at pH 9 (red); and resulting absorbance spectra from mixing the species presented in red with an equal volume of MTEN buffer at pH 5.7 (blue; final pH 7). Partial recovery of the absorbance signal of the T1Cu is attributed to incomplete T2Cu incorporation.10 (C) An example of a stopped-flow transient and corresponding single-exponential fit for a pH-jump measurement used to track PCET in AxNiR (final pH 7). | ||
A number of studies have been used to probe PCET in CuNiRs. These include site-directed mutagenesis,6,8 solvent isotope effect measurements,5 proton inventory studies,5 and pH perturbation measurements.10,11 The importance or otherwise of protein dynamics in PCET reactions catalysed by CuNiRs has yet to be established experimentally. To date, protein motions have not been linked formally to discreet chemical steps in the catalytic cycle of these proteins.12 There is now growing evidence from a combination of biophysical and structural studies to suggest that protein motions are a feature of inter-copper (T1Cu to T2Cu) electron transfer. For example, in the absence of the nitrite substrate, electron transfer between T1Cu and T2Cu centres occurs in solution but not at cryogenic temperatures. This discrepancy is observed in room temperature solution-state laser flash spectroscopy and pulsed radiolysis studies compared to crystallographic analyses that are performed at cryogenic conditions (100 K).5,13,14 At cryogenic temperatures (i.e. below the glass transition temperature of ca. 200 K) the majority of large-scale collective motions of non-bonded atoms within proteins are effectively frozen out.15 Therefore, a potential explanation for the differences observed in solution and crystallographic studies is the involvement of protein motions in the CuNiR PCET reaction.
Here, we set out to examine the role of protein motions in PCET reactions catalysed by CuNiR. As an alternative to laser flash spectroscopy and pulsed-radiolysis techniques (which are limited by fractional reduction of T1Cu and non-discriminatory reduction of both copper centres in CuNiRs),5,13 we developed a new method to investigate PCET kinetics in CuNiRs. pH-jump spectroscopy is a kinetic relaxation method widely used to study protein (un)folding.16 Use of the pH-jump method has been limited, however, in studies of enzyme-catalysed electron transfer.17 Here, we explore the use of pH-jump spectroscopy in PCET, where proton transfer is coupled to an electron transfer reaction. We have chosen to investigate this reaction in Alcaligenes xylosoxidans CuNiR (AxNiR) as it is a well-characterised CuNiR.
At pH 9, the T1Cu (but not the T2Cu) of nitrite-bound AxNiR can be selectively reduced (Fig. 1B).10 Addition of acidic buffer (pH 5.7) leads to a lowering of the solution pH (pH 7 post mixing). This pH-jump leads to protonation of residues present in the major proton-channel (D92 and H249) and subsequent electron transfer from the reduced T1Cu to the catalytic T2Cu, which in turn leads to NO production.10,11 To determine if delivery of protons to the T2Cu site is kinetically coupled to inter-copper electron transfer, we monitored this PCET reaction by performing the pH perturbation (pH-jump) measurement in a stopped-flow instrument in both protonated and deuterated buffer.
Fig. 1C shows an example of an absorption transient recorded at 600 nm (λmax of oxidised T1Cu) and corresponding single exponential fit to the data for a reaction of T1Cu-reduced and nitrite-bound AxNiR (pH 9) mixed with an equal volume of buffered solution at pH 5.7. The final, post-mixing, pH value in the stopped-flow instrument was 7. The observed rate constant for inter-copper electron transfer in AxNiR determined using this approach was 377 ± 35 s−1. This observed rate constant is similar to that previously published using laser flash spectroscopy.5 The rate of inter-copper electron transfer measured in deuterated buffer was 502 ± 47 s−1. Therefore, the observed solvent isotope effect (SIE) recorded for inter-copper electron transfer of nitrite-bound AxNiR is 0.75 ± 0.08. Inverse SIEs in nitrite-bound AxNiR have been observed previously using the laser flash spectroscopy method.5 These findings demonstrate that inter-copper electron transfer is coupled to proton transfer in AxNiR. This inverse SIE has been argued previously to represent reaction routes that are differentially populated in H2O and D2O.5
We set out to clarify if electrons can transfer between the T1Cu and T2Cu sites in the absence of thermally activated protein motions by performing low temperature (80 K) spectroscopy studies with both nitrite-bound and nitrite-free enzyme.15 This was investigated using electron paramagnetic resonance (EPR) spectroscopy, which has multiple advantages over optical spectroscopies for use in low temperature investigations of CuNiRs. For example, EPR can be used to visualise the redox state of both the T1 and the T2Cu signals.18 It can also be used to determine if nitrite is bound to the T2Cu site. As transition metals (open-shell) have large anisotropic EPR signals with strong hyperfine couplings, enzymes such as CuNiRs are highly amenable to EPR studies. To ascertain if electron transfer from the T1Cu to the T2Cu in AxNiR is linked to protein motion, we used EPR spectroscopy to characterise cryolytically reduced wildtype and nitrite-bound AxNiR at 80 K. By holding samples at 80 K, the majority of motions within the enzyme should be frozen.
We utilised a 60Co-source to reduce the CuNIR enzyme. EPR spectra for oxidised and cryolytically reduced wildtype and nitrite-bound AxNiR are shown in Fig. 2. The EPR spectrum of wildtype AxNiR shows strong hyperfine couplings attributed to 63,65Cu nuclei of the T1 and T2Cu centres; A||(63,65Cu; T2); ∼330 MHz with g = 2.350 and A||(63,65Cu; T1); ∼208 MHz with g = 2.212, respectively. The spectrum shows no resolved hyperfine couplings at the perpendicular orientation. The EPR spectra show that the T2Cu centre is significantly altered (A||(63,65Cu; T2); ∼370 MHz with g = 2.290; Fig. S1 in the ESI†) when nitrite is present, while EPR changes at the T1Cu centre are either minimal or unaffected, as reported previously.18 This indicates nitrite binding to the catalytic pocket of AxNiR i.e., T2Cu.18 In Fig. 2, samples that have been subject to cryolytic reduction show that the signal of the T1Cu (but not T2Cu) is decreased, indicative of T1 reduction (ca. 30% reduced). There is essentially no reduction of the T2Cu centre (<5% reduction in T2Cu signal, which is significantly lower than would be expected based on the potentials of the copper redox centre).8 This is in agreement with previously published cryogenic crystallography studies showing selective reduction of the T1Cu at low temperatures.14 Moreover, as there appears to be no reduction of the T2Cu from the 60Co-source, or more specifically through T1 to T2Cu electron transfer at low temperature, we suggest that thermally driven protein motions are required to facilitate PCET reactions catalysed by AxNiR.
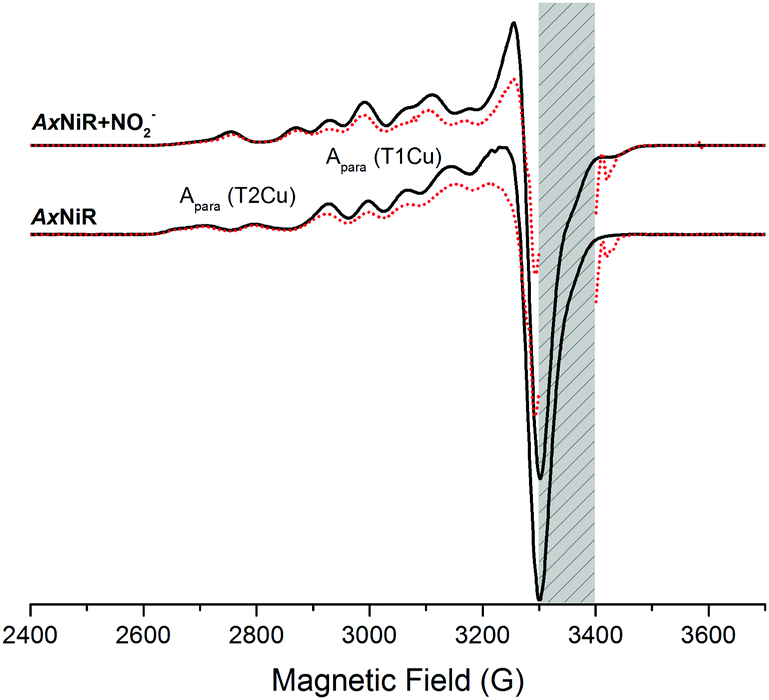 | ||
| Fig. 2 Comparisons of cw-EPR spectra of nitrite-bound and nitrite-free AxNiR before (black lines) and after cryolytic reduction (red, dotted lines) at 80 K. The weak parallel features arise from 63,65Cu nuclei of the T1 and T2Cu centres and are indicated on the EPR spectra of wildtype-AxNiR. The intense EPR signals observed between 3300–3400 G attributed to various paramagnetic species have been removed manually, as indicated by the grey shaded region. | ||
To investigate the potential importance of protein motions to inter-copper PCET in AxNiR, we have probed the effects of pressure on the rate of inter-copper electron transfer using the pH-jump stopped-flow method described above. Pressure can be used to perturb the underlying equilibrium found in dynamic protein systems, shifting the average ensemble conformation to a more compact state (as governed by the Le Chatelier's Principle).19–21 Pressure-dependent kinetic studies have proven to be useful in studies of domain motion coupled to electron transfer reactions (e.g. in the diflavin oxidoreductases) and also hydride and proton transfer reactions in a number of enzyme systems.19–21 Studies of the effects of pressure on enzyme catalysed PCET reactions are currently lacking. We performed stopped-flow pressure measurements that ranged from atmospheric pressure (1 bar) to 1500 bar using the pH-jump method to investigate the influence of perturbing the conformational equilibrium (through the use of pressure) on PCET reactions catalysed by AxNiR.
Fig. 3A shows example transients and the pressure dependence of observed rate constants for T1Cu to T2Cu electron transfer in AxNiR. To quantitatively draw conclusions from the pressure-dependence of the observed rate constant kobs, we fitted the pressure data in Fig. 3A to eqn (1):22
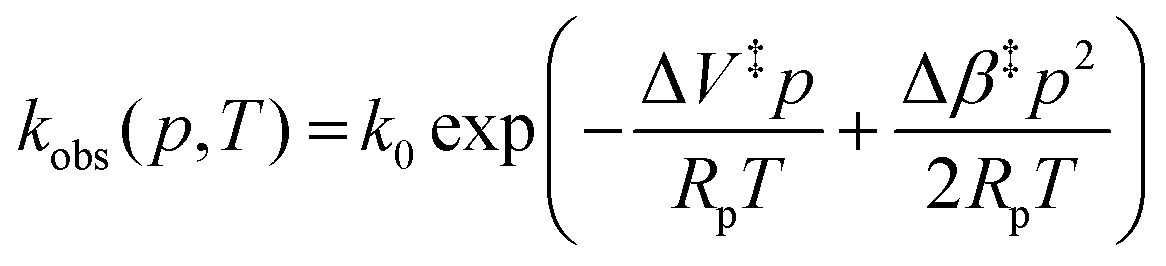 | (1) |
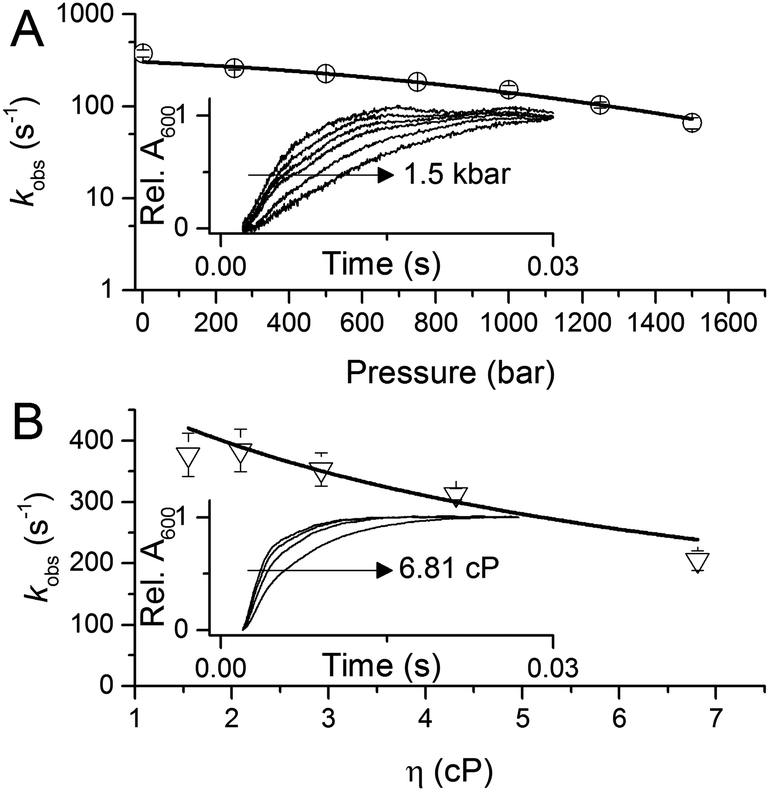 | ||
| Fig. 3 The influence of (A) hydrostatic pressure and (B) solvent viscosity on PCET reactions catalysed by AxNiR. The inserts in (A and B) show example transients for the pH-jump measurements used here to study PCET in AxNiR. Data in (A and B) are fit to eqn (1) and (2), respectively. | ||
We note that there is a significant effect of pressure on the observed rate of the PCET reaction catalysed by AxNiR (Tables S1 and S2, ESI†). Explicitly, with increasing pressure, the rate of T1Cu to T2Cu PCET in AxNiR is significantly impaired (over 80% change in observed rate constant between 1 and 1500 bar). While it is challenging to explain the direct origins of this pressure-dependence in terms of dynamic structural changes within AxNiR, our data point to the involvement of motions (protein and/or solvent) in facilitating PCET catalysed by AxNiR.
We have also investigated the effects of solvent viscosity on observed rates of PCET reactions catalysed by AxNiR. As protein motions are intimately linked to the solvent environment, the use of solvent viscosity stopped-flow measurements can provide insights into the type of protein motions related to catalytic steps.23–25 Motions that are directly related to the overall dielectric fluctuations of the bulk solvent are often termed ‘solvent-slaved’ motions. ‘Hydration’ and ‘non-slaved’ motions on the other hand are largely independent of the bulk solvent and are linked to motions of the hydration layers and thermally activated internal vibrations, respectively. To examine the properties of the motions related to PCET reactions in AxNiR, we performed solvent viscosity measurements using the pH-jump method over a range of glycerol concentrations (0–40%). Fig. 3B shows stopped-flow transients and the glycerol dependence of the PCET reaction catalysed by AxNiR. The glycerol dependence in Fig. 3B has been fitted to the Kramer model, used to describe the relationship between solvent viscosity and observed reaction rates through the means of a friction coefficient. This model is described in eqn (2):26
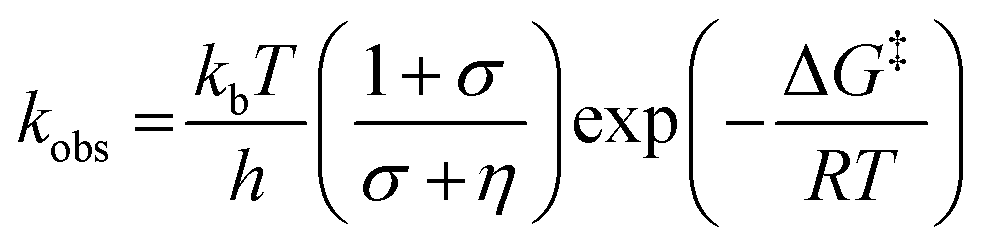 | (2) |
Using a pH-jump method, we have demonstrated the kinetic coupling of proton and electron transfer in a sequential PCET reaction (proton transfer followed by electron transfer) catalysed by AxNiR. Using this method in combination with pressure and viscosity dependence measurements, and low temperature EPR spectroscopy, we have shown how solvent-slaved protein motions accompany this PCET. To our knowledge, this is the first conclusive evidence for the involvement of protein motions in the PCET reaction catalysed by a CuNiR. While the origins of the catalytically relevant dynamic changes remain elusive, we hypothesise that they are linked to the catalytic aspartate and histidine residues (D92 and H249; Fig. 1A), which are seen in two different conformations in high resolution crystal structures of blue and green CuNiRs.7 Since all CuNiRs possess very similar structures and mechanisms,19,27 it is likely that similar solvent-slaved protein motions accompany PCET in other members of the CuNiR family.
Research conducted here was funded by the UK Biotechnology and Biological Sciences Research Council. We acknowledge colleagues from the Liverpool's Molecular Biophysics group (Samar Hasnain, Svetlana Antonyuk and Robert Eady) for discussions. The work on denitrifying enzymes in Manchester and Liverpool is supported by BBSRC (BB/L006960/1, BB/N013972/1) (BB/N013980/1 and BB/N013972/1). MS acknowledges Ruth Edge and DCF-Cumbria for the use of National Nuclear Facility, where gamma irradiations studies were carried out.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. M. Hedison and N. S. Scrutton, FEBS J., 2019 DOI:10.1111/febs.14757.
- V. L. Davidson, Acc. Chem. Res., 2008, 41, 730–738 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- W. G. W. Zumft, Microbiol. Mol. Biol. Rev., 1997, 61, 533–616 CAS.
- S. Brenner, D. J. Heyes, S. Hay, M. A. Hough, R. R. Eady, S. S. Hasnain and N. S. Scrutton, J. Biol. Chem., 2009, 284, 25973–25983 CrossRef CAS PubMed.
- S. Suzuki, K. Kataoka and K. Yamaguchi, Acc. Chem. Res., 2000, 33, 728–735 CrossRef CAS PubMed.
- M. J. Ellis, F. E. Dodd, G. Sawers, R. R. Eady and S. S. Hasnain, J. Mol. Biol., 2003, 328, 429–438 CrossRef CAS PubMed.
- N. G. H. Leferink, C. Han, S. V. Antonyuk, D. J. Heyes, S. E. J. Rigby, M. A. Hough, R. R. Eady, N. S. Scrutton and S. S. Hasnain, Biochemistry, 2011, 50, 4121–4131 CrossRef CAS PubMed.
- Y. Zhao, D. A. Lukoyanov, Y. V. Toropov, K. Wu, J. P. Shapleigh and C. P. Scholes, Biochemistry, 2002, 41, 7464–7474 CrossRef CAS PubMed.
- N. G. H. Leferink, R. R. Eady, S. S. Hasnain and N. S. Scrutton, FEBS J., 2012, 279, 2174–2181 CrossRef CAS PubMed.
- S. Ghosh, A. Dey, Y. Sun, C. P. Scholes and E. I. Solomon, J. Am. Chem. Soc., 2009, 131, 277–288 CrossRef CAS PubMed.
- H. B. Gray, B. G. Malmström and R. J. P. Williams, J. Biol. Inorg. Chem., 2000, 5, 551–559 CrossRef CAS PubMed.
- S. Suzuki, Deligeer, K. Yamaguchi, K. Kataoka, K. Kobayashi, S. Tagawa, T. Kohzuma, S. Shidara and H. Iwasaki, J. Biol. Inorg. Chem., 1997, 2, 265–274 CrossRef CAS.
- M. A. Hough, S. V. Antonyuk, R. W. Strange, R. R. Eady and S. S. Hasnain, J. Mol. Biol., 2008, 378, 353–361 CrossRef CAS PubMed.
- D. Ringe and G. A. Petsko, Biophys. Chem., 2006, 105, 667–680 CrossRef.
- T. P. Causgrove and R. B. Dyer, Chem. Phys., 2006, 323, 2–10 CrossRef CAS.
- R. J. Rohlfs, L. Huang and R. Hille, J. Biol. Chem., 1995, 270, 22196–22207 CrossRef CAS PubMed.
- B. D. Howes, Z. H. L. Abraham, D. J. Lowe, T. Brüser, R. R. Eady and B. E. Smith, Biochemistry, 1994, 33, 3171–3177 CrossRef CAS PubMed.
- C. R. Pudney, D. J. Heyes, B. Khara, S. Hay, S. E. J. Rigby and N. S. Scrutton, FEBS J., 2012, 279, 1534–1544 CrossRef CAS PubMed.
- A. Sobolewska-Stawiarz, N. G. H. Leferink, K. Fisher, D. J. Heyes, S. Hay, S. E. J. Rigby and N. S. Scrutton, J. Biol. Chem., 2014, 289, 11725–11738 CrossRef CAS PubMed.
- S. Hay, M. J. Sutcliffe and N. S. Scrutton, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 507–512 CrossRef CAS PubMed.
- D. B. Northrop, J. Am. Chem. Soc., 1999, 121, 3521–3524 CrossRef CAS.
- D. Beece, L. Eisenstein, H. Frauenfelder, D. Good, M. C. Marden, L. Reinisch, K. T. Yue, A. H. Reynolds and L. B. Sorensen, Biochemistry, 1980, 19, 5147–5157 CrossRef CAS PubMed.
- A. Ansari, C. M. Jones, E. R. Henry and J. Hofrichter, Science, 1992, 256, 1796–1798 CrossRef CAS PubMed.
- P. W. Fenimore, H. Frauenfelder, B. H. McMahon and R. D. Young, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14408–14413 CrossRef CAS PubMed.
- M. M. Ivković-Jensen and N. M. Kostić, Biochemistry, 1997, 36, 8135–8144 CrossRef PubMed.
- S. Horrell, D. Kekilli, R. W. Strange and M. A. Hough, Metallomics, 2017, 9, 1470–1482 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c9cc01026b |
| This journal is © The Royal Society of Chemistry 2019 |