
Raman spectroscopy for the evaluation of the radiobiological sensitivity of normal human breast cells at different time points after irradiation by a clinical proton beam†
M.
Lasalvia
 ab,
G.
Perna
*ab,
P.
Pisciotta
cd,
F. P.
Cammarata
d,
L.
Manti
ef and
V.
Capozzi
ab
ab,
G.
Perna
*ab,
P.
Pisciotta
cd,
F. P.
Cammarata
d,
L.
Manti
ef and
V.
Capozzi
ab
aDipartimento di Medicina Clinica e Sperimentale, Università di Foggia, 71122 Foggia, Italy. E-mail: giuseppe.perna@unifg.it
bIstituto Nazionale di Fisica Nucleare – Sezione di Bari, 70126 Bari, Italy
cIstituto Nazionale di Fisica Nucleare, Laboratori Nazionali del Sud, INFN-LNS, Catania, Italy
dInstitute of Molecular Bioimaging and Physiology, National Research Council, 90015 Cefalù, Pa, Italy
eDipartimento di Fisica, Università di Napoli “Federico II”, 80126 Napoli, Italy
fIstituto Nazionale di Fisica Nucleare – Sezione di Napoli, 80126 Napoli, Italy
First published on 24th January 2019
Abstract
Among different radiotherapy techniques, proton irradiation is an established and effective method for treatment of several types of cancer, because less healthy tissue is exposed with respect to conventional radiotherapy by photons/electrons. Recently, proton therapy has been proposed for the treatment of breast cancer. In vitro studies of proton irradiated normal human breast cells can provide information about cellular radioresponse, particularly as far as healthy tissue is concerned. In this paper, a study of the effects at different time points, following proton irradiation at different doses, of human normal MCF10A breast cells is performed by Raman spectroscopy. The aim of this investigation is to detect the unwanted effects of proton treatment and to investigate the possibility of monitoring them and of making an assessment of the cellular sensitivity by means of such a technique. The obtained results seem to indicate a rather significant sensitivity of MCF10A cells to proton irradiation. In fact, even at doses as low as 0.5 Gy, biological effects are clearly detectable in Raman spectra. In particular, ratiometric analysis of the Raman spectra measured from the nucleoplasm compartment showed that DNA/RNA damage increases with time, suggesting that most cells are unable to repair DNA/RNA broken bonds. The results obtained by the Raman spectroscopy analysis exhibit a similar trend with regard to dose to those obtained by commonly used radiobiological assays (i.e. MTT, clonogenic assay, senescence, apoptosis and necrosis). The results of this study strongly suggest the possibility that the Raman technique can be used to identify molecular markers predicting radiation response.
Introduction
Among different radiotherapy modalities for cancer treatment, proton beam therapy offers the advantage of delivering the ionizing radiation dose in a more localized volume with respect to photon and electron beam therapy.1 Such a possibility is due to the presence of the Bragg peak in the absorbed dose vs. penetration depth curve. In fact, the absorbed dose increases very gradually with increasing penetration depth, suddenly rising to a peak when the protons are ultimately stopped. This allows the deposition of most of the dose into the tumor, sparing the healthy tissues to a much larger extent than that allowed by conventional radiotherapy.2 The particle range is energy-dependent and makes proton beam therapy suitable for treating deep seated cancers, especially if close to organs at risk. Due to the need to conform the high-dose gradient in the monochromatic beam Bragg peak to the physiological tumor region, a passive scattering technique is routinely used to broaden such a narrow proton beam into one that can achieve a biologically effective uniform dose of the target at all depths, delivering so-called spread-out Bragg peaks (SOBP).3 Therefore, there has been a rapid growth of proton therapy medical centers worldwide.4The motivations for using radiotherapy for cancer treatment are that ionizing radiation can cause lethal damage to cells both directly, causing the breakage of DNA bonds, and indirectly, by forming highly reactive radicals in the intracellular material that can chemically break bonds within the DNA macromolecule, causing a cell to lose its proliferative ability.5 The most deleterious types of lesion are DNA single- and double-strand breaks, i.e. the breakage of bonds in one or both DNA helices, respectively.6 Such damage is experienced both by malignant cells and cells in the exposed healthy tissue. With a sublethally damaged normal tissue increase, the probability of developing secondary malignant neoplasms might significantly increase.7 From a clinical point of view, this corresponds to unexpected side effects on normal tissue at the entrance position of the beam. Among the aims of a cancer treatment, achieving the right balance between the highest rate of local tumor cure and the lowest normal tissue complication probability is arguably a fundamental one.
Cells respond to radiation-induced DNA damage within a few hours by activating a complex set of biochemical signals aiming to restore DNA integrity.8 In current radiotherapy treatments, the total dose scheduled for the patient, typically about 50 Gy, depending on the tumor type, is fractioned in ∼2 Gy daily doses delivered over several weeks.9 The rationale for fractionation between two consecutive tumour tissue exposures is to allow the recovery of irradiated healthy cells so that they can repair sub-lethal damage minimizing the risk of early and late normal-tissue reactions.
Normal-tissue toxicity can vary in its severity and is patient-dependent.10 Effectiveness of proton-therapy could be improved if molecular markers predicting radiation response are identified and known before the start of treatment: in fact, the response of such markers could be used to tailor the radiation treatment as a good compromise between tumor control and normal tissue complications.
Proton irradiation of tumour tissue has been established as an effective tool for the radiotherapy treatment of ocular melanoma, brain and lung tumours.11 Recently, it has been suggested that proton therapy can be extended to the treatment of breast cancer.12,13 In order to propose proton therapy for the treatment of breast cancer, the response of healthy cells to the proton beam should be evaluated, especially at low doses, considering that the healthy tissues located at the entrance position of the SOBP are unavoidably irradiated. In vitro measurements of cell lines modelling human healthy breast tissue are a preliminary step to address this issue. In particular, the MCF10A human mammary epithelial cell line is a widely used in vitro model for studying normal breast cell functions.14
Based on these premises, the aim of our work is twofold. The first one is to evaluate, by means of biological assays (as viability, premature senescence, apoptosis and necrosis assays), the MCF10A cells’ response at different time points after irradiation. This will provide information on whether cellular physiological mechanisms are able to restore the balance among the cellular components existing in unexposed cells, particularly at low proton doses, and the time they need to eventually restore such a balance. This is important for the preservation of healthy breast tissue during proton-therapy treatments, in order to eventually review the time needed for the healthy cells to repair. Of course, the possibility of recovery also depends on cellular sensitivity to radiation. The second aim of this work is to investigate the capability of Raman spectroscopy (RS) to provide information about cellular sensitivity at low doses (on the order of 1 Gy) of proton radiation, in order to propose such a non-invasive and reagent-free technique as a complementary method to detect cell sensitivity, without neither affecting the cell sample with non-physiological chemicals nor waiting for a long time as is necessary for conventional radiobiological assays for measurement of cell survival and DNA damage (clonogenic assay, senescence induction, chromosome aberration analysis, micronuclei assay, etc.). RS is a vibrational technique based on the spectral analysis of a laser beam focused onto a sample: such a monochromatic beam induces molecular vibrations in the investigated sample, resulting in inelastically scattered photons whose frequencies and intensities are characteristic of the functional groups inside the molecules of the sample. Thus, RS is able to provide a molecular fingerprint of the analysed sample. In addition, RS is able to yield information at the single-cell level, unlike the abovementioned biological assays: therefore, it can be a complementary technique with respect to molecular biology assays traditionally employed in clinical radiobiology to detect the cellular response to radiotherapy.
In fact, numerous papers have been published in the last few years about the biological effects of cell lines exposed to ionizing radiation. Most of them concern tumour cell lines exposed to X- and γ-ray radiation15,16 as well as proton radiation.16–19 The irradiated cells have been frequently analyzed by means of conventional radiobiological assays for the measurement of cell survival and DNA damage (clonogenic assay, viability assays, immunofluorescence, flow cytometry analysis, etc.).16–18,20 Some authors have also used the RS technique to investigate the effects of the cells’ exposure under X- and γ-ray radiation both at high doses as for tumour cell lines21–24 and at low doses as for non-tumorigenic cell lines.25 In contrast, very few studies reported the RS analysis of proton irradiated cells, although such a technique has been proposed by Devpura et al.26 for application in radiation therapy response assessment.
In this study, we observed a significant presence of proton-induced cytogenetic effects in MCF10A cells, even at the lowest investigated dose (0.5 Gy), in terms of (i) cell death, assessed by MTT viability, clonogenic, apoptosis and necrosis assays, (ii) induction of premature cellular senescence and (iii) induction of DNA damage, estimated by the RS technique. In a recent study,27 we showed that the RS technique applied to single MCF10A cells, fixed immediately after proton-irradiation at low doses, can detect radiation-induced changes in the relative content of the functional groups characteristic of cellular components. In this work, we extend this method to the same cell type, but fixing cells at different time points after irradiation, in order to investigate either the capability of cellular mechanisms to repair the proton-induced damage or to propose the RS technique as an effective tool for the detection of cellular radiosensitivity. We observed that the radiation-induced changes to the DNA/RNA content increased after 24 and 48 h from irradiation, so that the exposed MCF10A cells were not able to recover the original proportions about the different components inside the cell nucleus. A quite anomalous behavior observed in the Raman spectra of cells fixed 72 h after irradiation can be explained by considering the breaking of chemical bonds in other cellular components (such as proteins) different from DNA/RNA. Since the above changes characterize also the spectra of cells exposed to a low dose of 0.5 Gy, we conclude that RS analysis performed at the single-cell level may serve as a useful tool to detect proton radiation cellular sensitivity.
Materials and methods
Cell culture
MCF10A cells (American Type Culture Collection, Manassas, VA) were cultured in DMEM/Ham's F-12 (Sigma-Aldrich, Milano, Italy) supplemented with 100 ng ml−1 cholera toxin, 20 ng ml−1 epidermal growth factor (EGF), 0.01 mg ml−1 insulin, 500 ng ml−1 hydrocortisone, and 5% horse serum (Life Technologies, Monza, Italy). Cells were grown in tissue-culture flasks and cultures were maintained at 37 °C, 5% CO2. One day before exposure, the samples to be analysed by RS were seeded in polylysine coated coverslips, located inside six-well plates, at densities of 5 × 104 cells for each well and grown for further 24 h, whereas the cells to be analysed by viability and senescence tests were seeded at a density of 5 × 103 in 24 well plates and grown for 24 h.Cell irradiation and fixation
Cell irradiation was carried out at the CATANA facility at the South National Laboratory – Istituto Nazionale di Fisica Nucleare (LNS-INFN) in Catania (Italy), where a 62 MeV proton beam was accelerated by a super-conducting cyclotron, yielding a pristine Bragg peak with a width of about 2 mm. In particular, glass coverslips containing adherent MCF10A cells were located in identical flasks, which were placed at the entrance position of a SOBP formed by the optimal stacking of multiple depth dose curves of pristine peaks of different energies using a polymethyl-methacrylate (PMMA) modulator wheel. The SOBP range was about 30 mm in water and the cells were located at a depth of 2.79 mm water equivalent (LET ∼ 2.91 keV μm−1), simulated by using PMMA beam degraders. The relative dose profile was measured with a Markus™ ionization chamber (PTW, Freiburg). A detailed description of beam line and dosimetry is presented elsewhere.28 Single fractions of 0, 0.5, 2 and 4 Gy were delivered to the plates. The uncertainty in dose measurements was within 3%.The plates containing the MCF10A exposed cells for RS measurements were incubated and three coverslips for each dose were fixed at 24, 48 and 72 h time points after the end of the exposure process, by means of 3.7% PFA in PBS solution.
Clonogenic survival assay
Twenty-four hours after irradiation, clonogenic survival assay was performed according to the protocol of Puck and Marcus.29 Colonies were allowed to grow under normal cell culture conditions for two weeks and then they were fixed and stained for 30 min with 6% glutaraldehyde and 0.5% crystal violet (both from Sigma-Aldrich, St Louis, MO, USA). Colonies with more than 50 cells were counted manually under a Zeiss Axiovert phase-contrast microscope (Carl Zeiss, Gottingen, Germany). As the control, untreated cells were seeded under the same conditions.MTT assay
Cell viability was assessed for each dose by using a thiazolyl blue tetrazolium blue (MTT) assay, which evaluates cellular survival on the basis of mitochondrial activity. This colorimetric test detects the reduction of yellow MTT [3-(4,5-dimethylthiazolyl)-2,5-diphenyl-tetrazolium bromide] into purple formazan crystals by mitochondrial dehydrogenases, which reflects the normal function of mitochondria. Results are provided in terms of the measurements of the absorbance value at a wavelength of 595 nm by using a multi-mode-microplate reader (Beckmann Coulter).Apoptosis and necrosis assay
The Annexin V-FITC apoptosis kit (Abnova Co., Taiwan) was used to detect apoptosis and necrosis in MCF10A cells. To distinguish necrotic and apoptotic cells, propidium iodide (PI) and Annexin-V were employed. The use of two fluorescent dyes allows imaging and discriminating living cells from dead ones. Furthermore, among the latter ones, the two dyes are able to distinguish those that underwent apoptosis from those that died by necrosis, according to two different colors: the apoptotic cells appear to be green and the necrotic cells appear to have a red nucleus. In contrast, live cells do not stain with either of the two dyes. In particular, necrotic cells with damaged cell membranes are permeable to PI, where it binds to the DNA: this causes necrotic cells to have a red fluorescence. Instead, Annexin V binds to the phosphatidylserine of the cell membranes. Since apoptotic cells expose phosphatidylserine residues on the extracellular side of the plasma membrane, they fluoresce with a green color. The staining procedure was performed by adding 5 μl of Annexin V-FITC and 5 μl of PI (50 μg ml−1) to the cells. After staining, adherent cells were incubated for 10 minutes, at room temperature and in the dark. Then, the cells were washed with 1× binding buffer. Finally, the cells were visualized with an inverted microscope (IX71, Olympus) by using a 20× objective.Senescence assay
For the senescence evaluation of MCF10A cells, a senescence cells histochemical staining kit (Sigma Aldrich) was used according to the manufacturer's protocol, based on a histochemical stain for β-galactosidase activity. The kit detects β-galactosidase, which is overexpressed in senescent cells.30 Specifically, cells were fixed in a fixation buffer containing formaldehyde and glutaraldehyde for 7 min followed by 3 washes in PBS solution. Next, 1 ml of staining mixture per well was added and the cells were incubated at pH 6.0 without CO2 for 5 hours at 37 °C, until they stained blue. After incubation, cells were washed in PBS and visualized using an Olympus IX71 microscope (20× objective magnification). The blue stained cells and the total number of cells were counted, in order to calculate the percentage of cells expressing β-galactosidase (senescent cells). The fraction of senescent cells was measured by examining a total of about 350 cells.Raman spectroscopy
Raman spectra were obtained at room temperature by means of a Raman confocal micro-spectrometer apparatus (Labram from Jobin–Yvon Horiba), as described elsewhere.31 The exciting laser beam, consisting of the 514.5 nm line of an Ar ion laser, was focused on the sample by using an Olympus optical microscope with a 100× oil-immersion objective (1.4 numerical aperture), resulting in a diffraction-limited spot about 0.5 μm in diameter. The laser power on the sample was properly fixed at 10 mW to obtain a good signal/noise ratio while preventing the thermal damage of the sample. The reproducibility of the Raman spectra, using such an excitation intensity, was verified by measuring three spectra in sequence at the same sample point. Each Raman spectrum was measured in the 700–1800 cm−1 spectral range, where information about the functional groups of the main cellular components (proteins, nucleic acids and lipids) is included. Raman spectra were obtained from about 30 randomly chosen single cells for each type of cellular sample (unexposed and exposed to different radiation doses). The acquisition time was set at 10 s for each single measured spectrum and the signal was averaged over three acquisitions. The light scattered from the sample was collected using the same 100× oil-immersion objective (in backscattering geometry) and passed through an edge filter (Omega Optical, Inc.) to suppress the laser line. The Raman scattered light was passed through a square confocal hole (300 μm diagonal) and a 200 μm entrance slit of a spectrometer equipped with a 600 grooves per mm grating. The signal was detected by means of a charge-coupled device (CCD) cooled at 223 K. A separate CCD camera was used to record white-light microscopy images of the cells to be probed. The spectral resolution was ∼5 cm−1 per pixel. Raman spectra were recorded from different single cells, randomly chosen on each glass coverslip containing the control cells and the cells exposed to the proton beam radiation. Raman spectra were measured by focusing the laser spot inside the nucleolus and the nucleoplasm region. Raman spectra of the background signal (the coated glass coverslip and the PBS solution) were acquired after the acquisition of each cell spectrum by moving the objective, without varying the focus position, to a nearby region where there were no cells.Spectral processing and data analysis
Each collected Raman spectrum was preprocessed firstly by subtracting the corresponding background signal; then, a subtraction of the cell fluorescence and stray light signal, described by a fifth-order polynomial function (based on the least squares fitting method of such polynomial function through some non-Raman points properly chosen on the measured spectrum) was performed; eventually, the intensity of each wavenumber channel was divided by the total spectral intensity to minimize the effect of fluctuation either in the sampling of the cellular volume or in the laser power on the spectral intensity of each single cell. The latter procedure makes each Raman spectrum independent of the total amount of biological material in the sampling volume. After this pretreatment, such normalized spectra of each cell sample were independently averaged to obtain an average spectrum for both the unexposed and exposed cells.Ratiometric analysis of several characteristic Raman peaks was performed by considering, for each of the about 30 normalized single spectra (corresponding to a specific fixation time and exposure dose), the intensity of these characteristic peaks, in order to calculate the intensity ratios between a couple of them. Therefore, a set of about 30 values for specific intensity ratios was obtained for each fixation time and dose. Then, for each set the mean value and standard error related to each specific intensity ratio were calculated.
Data compared in ratiometric analysis were expressed as mean ± standard error. The statistical difference between the exposed groups and control group was assessed by the Holm–Sidak method. P values <0.05 were considered to be statistically significant. All statistical analyses were performed by SigmaPlot software (version 12.5, Systat Software).
Results and discussion
Biological assays
The results of the cell growth curve between the 24 and 72 h time points and the normalized MTT absorbance obtained 24, 48 and 72 h after irradiation at different proton doses are shown in Fig. 1a and b, respectively. It is clearly evident in Fig. 1a that the cell growth trend is strongly influenced by the irradiation dose. In fact, unexposed cells considerably grow during the investigated time, although the growth rate decreases after the 48 h time point, because the cells tend to a confluence status on the plate. In contrast, the growth rate of exposed cells is lower than that of the control ones and it decreases with increasing exposure dose.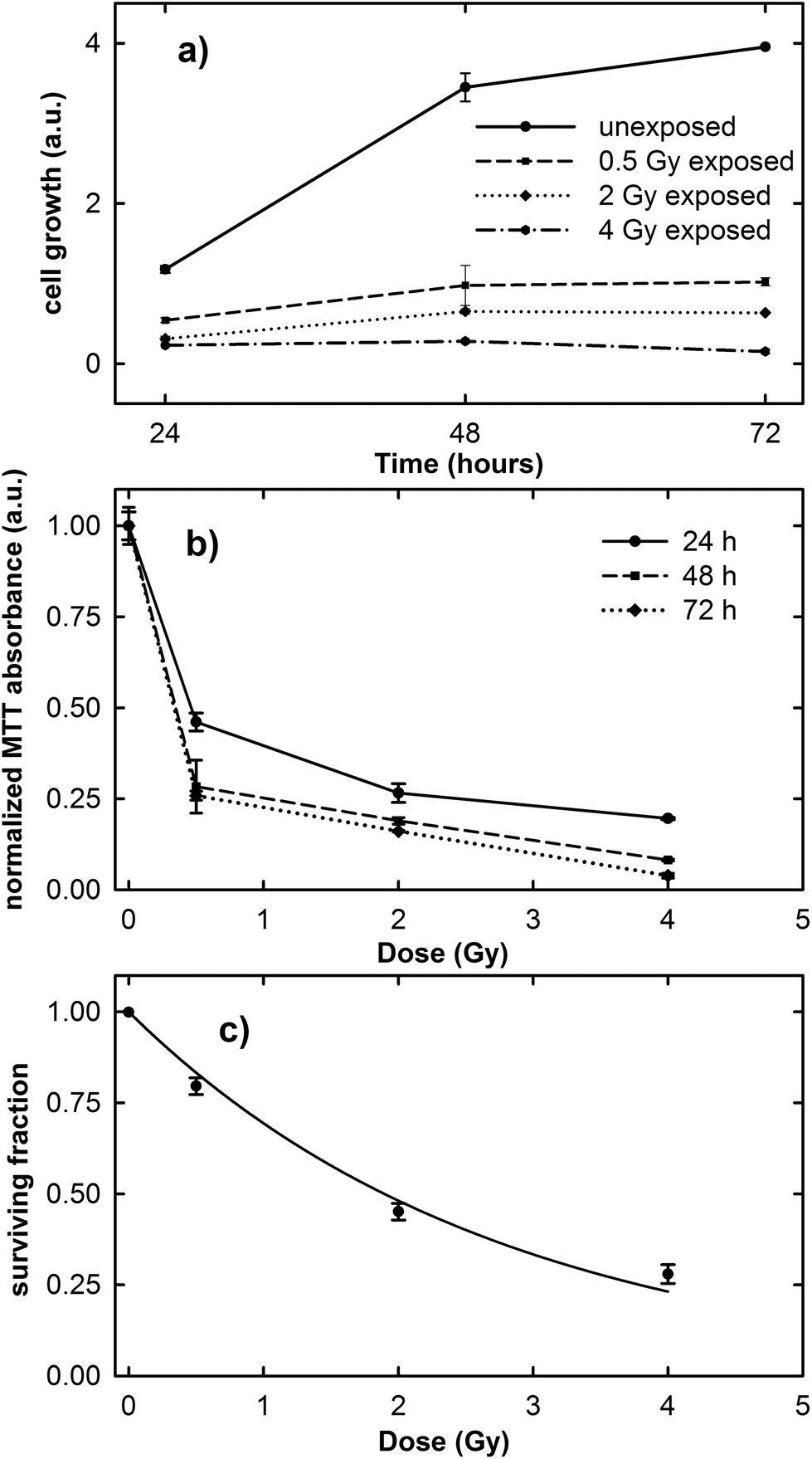 | ||
| Fig. 1 Time variation of cell growth (a), dependence of normalized MTT optical absorbance on proton irradiation dose (b) and the surviving fraction from the clonogenic assay (c) for MCF10A cells. Error bars depict the standard errors on the mean value. The lines in (a) and (b) are guides for the eyes, whereas the line in (c) is a data fitting (r2 = 0.986) to the linear-quadratic model S = exp(−αD − βD2). The value obtained for α is 0.366 ± 0.086, whereas the parameter β has a very low value. | ||
As the normalized MTT absorbance is proportional to the percentage of living cells, the data in Fig. 1b can be considered as a viability assay based on the inactivation of mitochondrial functions. In particular, Fig. 1b suggests that the viability of the exposed MCF10A cells at the three different investigated time points is characterized by a dose-dependent behavior. However, for cells measured 24 h after irradiation, the decreasing rate as a function of proton dose seems to be slower than for those measured 48 and 72 h after exposure. Overall, the surviving fraction after 2 Gy irradiation, known as the SF2 value, is less than 0.50.
The results obtained from the clonogenic assay performed two weeks after proton irradiation are shown in Fig. 1c. Survival data (points) are well fitted (continuous line) to a linear-quadratic model: S = exp(−αD − βD2), where S is the surviving fraction, D is the proton dose, and α and β are linear and quadratic coefficients describing the variation in cell survival at low and high dose, respectively.25 The value of the cell surviving fraction after 2 Gy irradiation, as obtained from the fitting procedure, corresponds to 0.48, in good agreement with that obtained by means of the MTT assay and also with previously published data about the MCF10A cell line subjected to proton irradiation.32 Therefore, the MCF10A cells can be considered as a radiosensitive cell line.16,22
These results confirm the risk to the health tissue as a result of the exposure to the clinical proton beam, not only at higher doses (e.g. 2 and 4 Gy), but also at doses as low as those to which the tissue is unintentionally exposed during the treatment (0.5 Gy).
The results of the Annexin V and PI staining assay on radiation-induced apoptosis and necrosis indicate a strong dose-dependence of such cellular events. Typical fluorescence images are shown in Fig. 2 for MCF10A cells exposed to different proton doses and fixed 24 h after exposure. Similar images were obtained for cells fixed 48 and 72 h after irradiation (ESI Fig. 1 and 2†). In particular, neither apoptotic nor necrotic cells are visible for unexposed (D = 0 Gy) MCF10A cells. In contrast, the heterogeneous cell population including apoptotic (green stained) and necrotic (red stained) cells, as well as living (non-stained) cells are present after 0.5 Gy exposure. Moreover, most of the cells exposed to 2 and 4 Gy of proton radiation appear to be in the necrotic state, regardless of the time point at which the staining was performed.
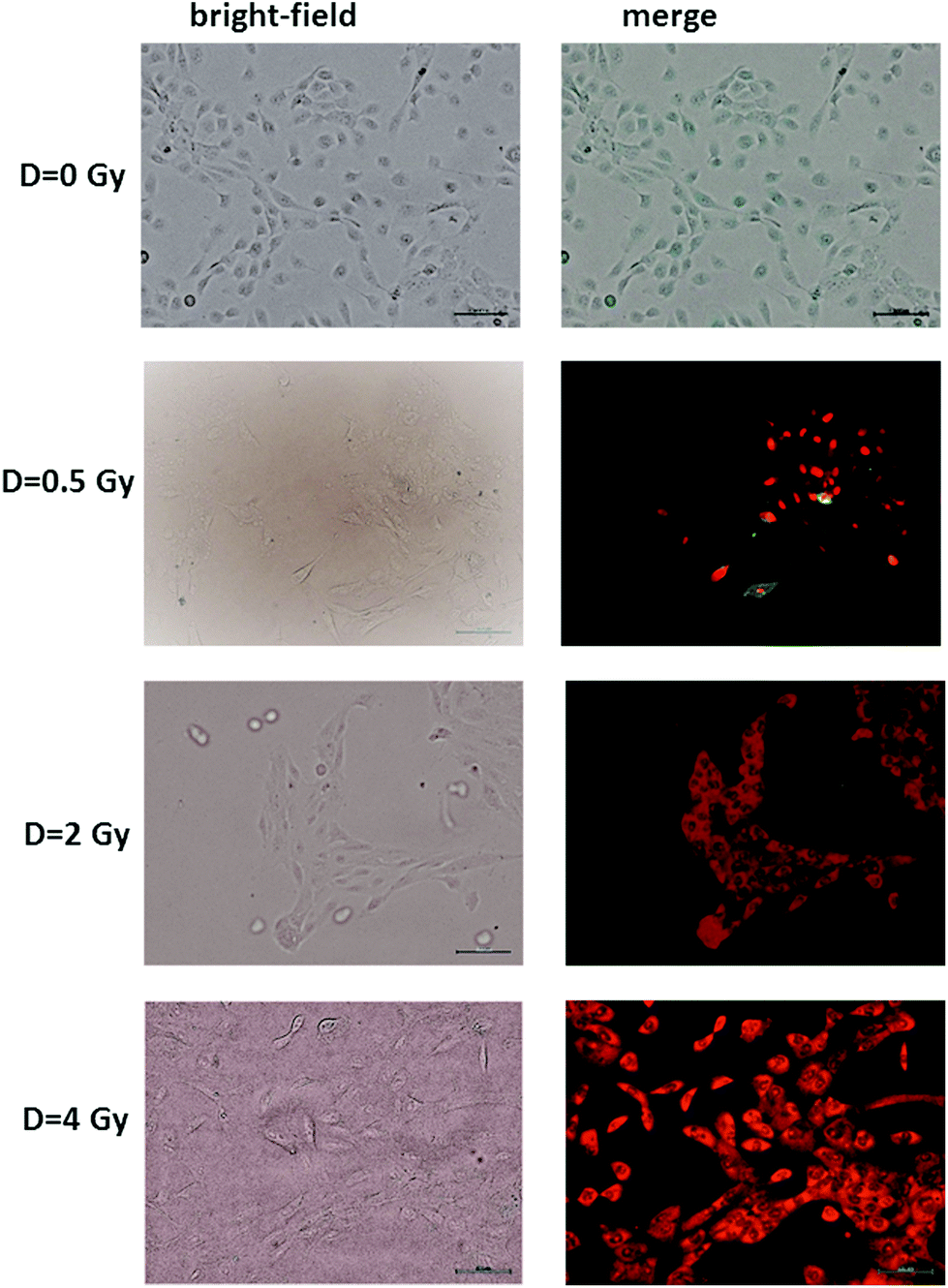 | ||
| Fig. 2 Representative bright-field (left side) and brightfield and fluorescence merged (right side) images of apoptotic and necrotic cells labeled with an Annexin V-FITC apoptosis kit, fixed 24 h after irradiation at different doses of a proton beam. Cells were visualized with an inverted fluorescence microscope (Olympus IX71) using a ×10 objective. Scale bar is 100 μm. Apoptotic cells (Annexin V conjugated) appear green, whereas necrotic cells (propidium iodide conjugated) appear red. Merged stands for overlay of propidium iodide and Annexin V conjugated. | ||
The induction of cellular death by means of apoptosis and necrosis processes is not the only damage caused by proton irradiation. In fact, the exposed cells are also subjected to premature senescence, as can be deduced from Fig. 3, where the percentage of β-galactosidase positive cells is reported at different time points and for different proton doses. In particular, the percentage of senescent cells among the unexposed samples slightly increases with time (1.39% after 24 h, 2.97% after 48 h and 5.01% after 72 h), as expected for cultured cells, because cellular senescence increases with aging.33 In contrast, the proton irradiation causes a strong time- and dose-dependent increase of cellular senescence, particularly after 48 and 72 h from the end of the exposure process. Instead, 24 h after exposure the proportion of senescent cells among irradiated cells is significantly larger than that of unexposed cells, but the results are almost dose-independent. Such results were to be expected, because ionising radiation is known to induce premature senescence at high and low doses.34 Moreover, the optical microscopy images show a fair number of detached death cells after 24 h for 2 Gy and 4 Gy exposure: it explains the decreasing percentage at higher doses after 24 h (Fig. 3a).
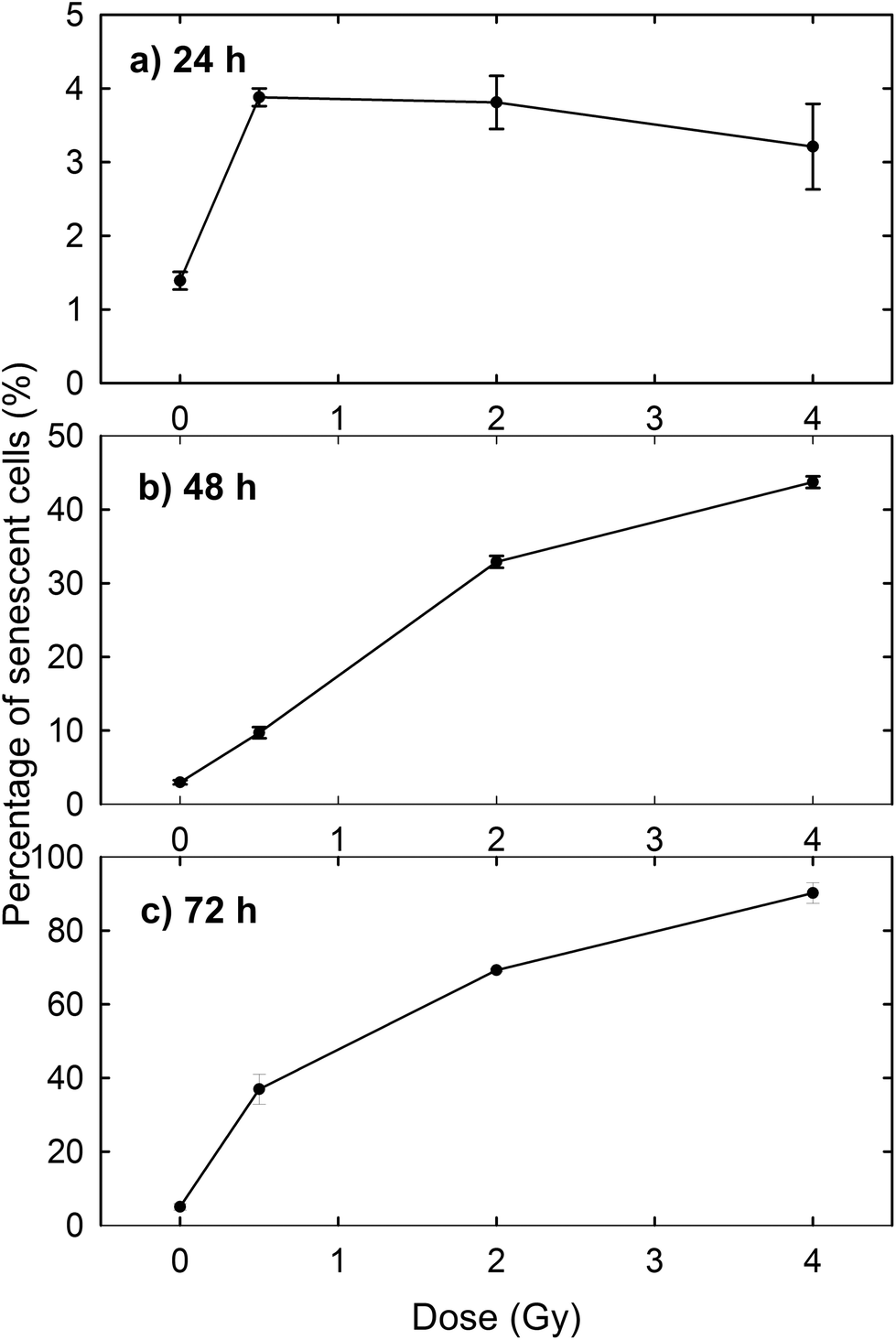 | ||
| Fig. 3 Premature senescence induced by proton irradiation of MCF10A cells vs. irradiation dose. Staining of β-galactosidase was performed after 24 h (a), 48 h (b) and 72 h (c) from the end of proton beam irradiation. | ||
Overall, fluorescence images highlight that proton radiation causes physiological modifications in most cells, including the induction of premature senescence and apoptosis and necrosis events, not only at high doses (2 and 4 Gy) but also at the lowest investigated one (0.5 Gy). Such events involve a modification of the morphology and distribution of biochemical components inside cells (cell shrinkage, nuclear condensation, DNA fragmentation, etc.). Therefore, they can be investigated by Raman microspectroscopy, which allows the biochemical analysis at the single cell level.
Raman spectroscopy
Raman spectra were obtained in two distinct subcellular compartments of the cells, i.e. by focusing the exciting laser beam within the nucleolus and within the nucleoplasm (i.e. inside the nucleus but outside of the nucleolus), so that the cellular components inside one or the other of the two compartments were mainly sampled in the corresponding measurements. Fig. 4 shows the average normalized spectra from each one of the two cellular regions for unexposed MCF10A cells. The 95% confidence intervals, reported as grey lines for both spectra, suggest that the single normalized spectra are quite similar, with the largest variability detected in the 870–950 cm−1 spectral range measured for the nucleoplasm region. In Fig. 4, it can also be seen that the spectral positions of the main peaks are very similar, although the relative intensities of some peaks of the average spectra from the nucleus and nucleolus regions are somewhat different, because of different concentrations of biomolecular components inside the two subcellular compartments. In fact, the biomolecular composition of the nucleolus mostly includes RNA and proteins involved in the RNA synthesis, processing, ribosomes formation and other cellular functions, whereas the nucleus mostly contains DNA, RNA, proteins and lipids.35 Moreover, in the nuclei of proliferating cells, the proteins are distributed nearly uniformly, with local accumulations in several nuclear structures, as the nucleolus, whereas during the different stages of the apoptosis process in the nucleoplasm, as well as in the nucleolus, a redistribution of proteins to a less uniform pattern is observed.36 In addition, the biomolecular material is denser in the nucleolus than in the nucleus, as commonly visible in optical microscopy images. Therefore, as the cytogenetic material is the main target of protons, for both the above nuclear regions it is worth analyzing by RS: the nucleolus because of the large density of nuclear material and, consequently, the large signal-to-noise ratio, and the nucleoplasm as a confirmation of the results obtained from the nucleolus. In addition, the investigation of both regions could clarify if one of them has a better diagnostic capability and/or sensitivity to external stress.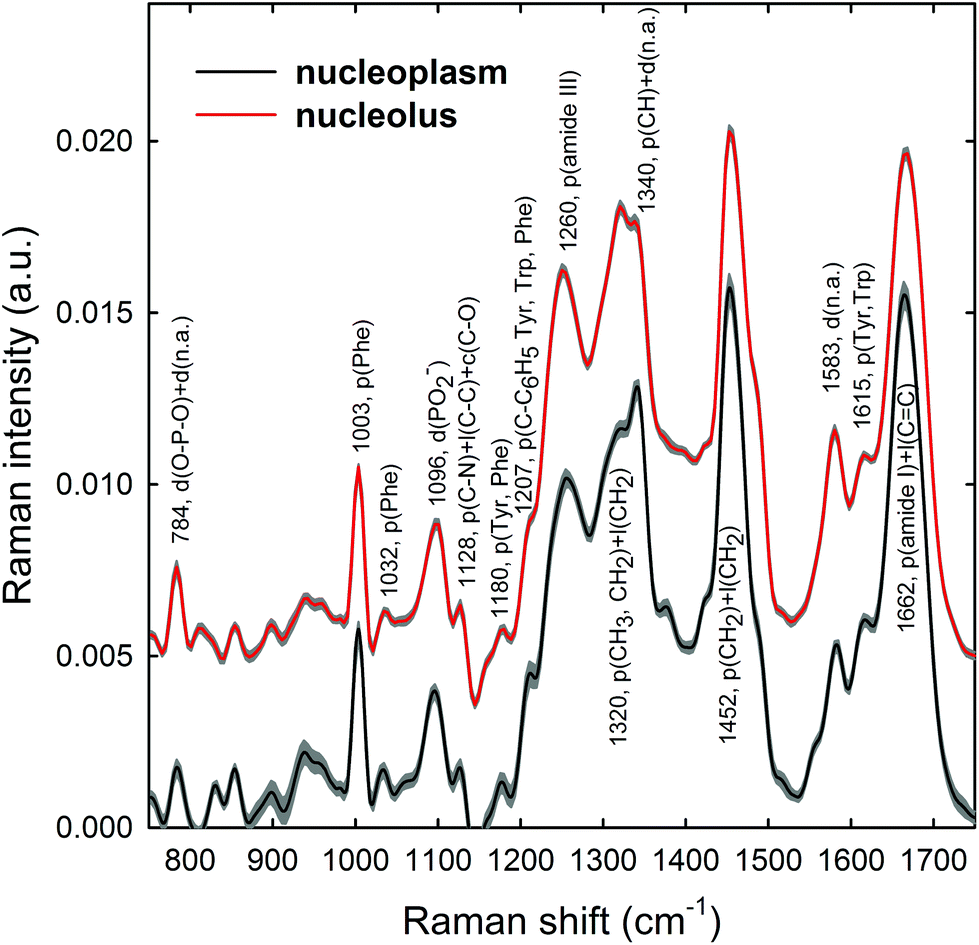 | ||
| Fig. 4 Average Raman spectra of (about 30) single control MCF10A cells. The spectra have been measured by focusing the exciting laser spot above the nucleolus (red line) and nucleoplasm (dark line) domains. Spectral 95% confidence intervals correspond to shaded grey areas for both spectra. The labels refer to the wavenumber value and the attribution of the most important spectral features. Abbreviations: d: DNA backbone; n.a: DNA bases; p: proteins; l: lipids; Tyr: Tyrosine; Phe: phenylalanine; Trp: tryptophan. The zero line of the average spectrum of the nucleolus has been intensity-shifted for clarity. | ||
The most intense bands visible in the Raman spectra in Fig. 4 are due to the contribution of the amide I (∼1662 cm−1), CH2 deformation (∼1452 cm−1), amide III (∼1260 cm−1) and phenylalanine ring breathing vibrations (1003 cm−1) of proteins. Other protein-related bands include aromatic ring vibrations associated with phenylalanine, tryptophan and tyrosine (e.g. 1032, 1128, 1180, 1207, 1340, and 1615 cm−1). The contribution of DNA and RNA components is mostly related to the peaks at 784 cm−1 (O–P–O stretching mode of the phosphodiester bond of the phosphate group), 1096 cm−1 (PO2− phosphodioxy bond of the phosphate group) and 1583 cm−1 (ring breathing vibrational modes characteristic of adenine and guanine).37 In contrast, lipid-related peaks scarcely contribute to the Raman spectra in Fig. 4, where they are mainly overlapped with the protein ones, as occurs at 1065, 1128, 1300 and 1440 cm−1.
Normalized and averaged Raman spectra measured from the nucleolus and nucleoplasm compartments of MCF10A cells exposed to different proton doses and fixed 24 h after irradiation are shown in Fig. 5a and b, respectively. For each average spectrum, the 95% confidence intervals are similar to those reported in Fig. 4 (data not shown). Among the many spectral features characterizing the spectra in Fig. 5, we investigated the dose dependent behavior of (i) the peaks at 784 and 1096 cm−1 as a pointer of radiation-induced modification of the phosphate groups in DNA and RNA, (ii) the peak at 1583 cm−1 as an index of the radiation-induced changes to DNA and RNA bases, (iii) the peak at 1003 cm−1 as related to the protein component modification induced by radiation and (iv) the peak at 1452 cm−1 as a marker of protein and lipid component changes following radiation exposure. This choice is due to the fact that such spectral features correspond to well-resolved peaks related to single cellular components. At first glance, it seems that the intensity of DNA and RNA related peaks (at 784, 1096 and 1583 cm−1) in Fig. 5 decreases with increasing exposure dose, particularly for the nucleoplasm spectra. However, it is worth remarking that, because of the normalization procedure, the relative intensity of specific Raman peaks, related to different vibrational bonds, should be considered as a proper marker of relative radiation-induced damage to the respective cellular components.
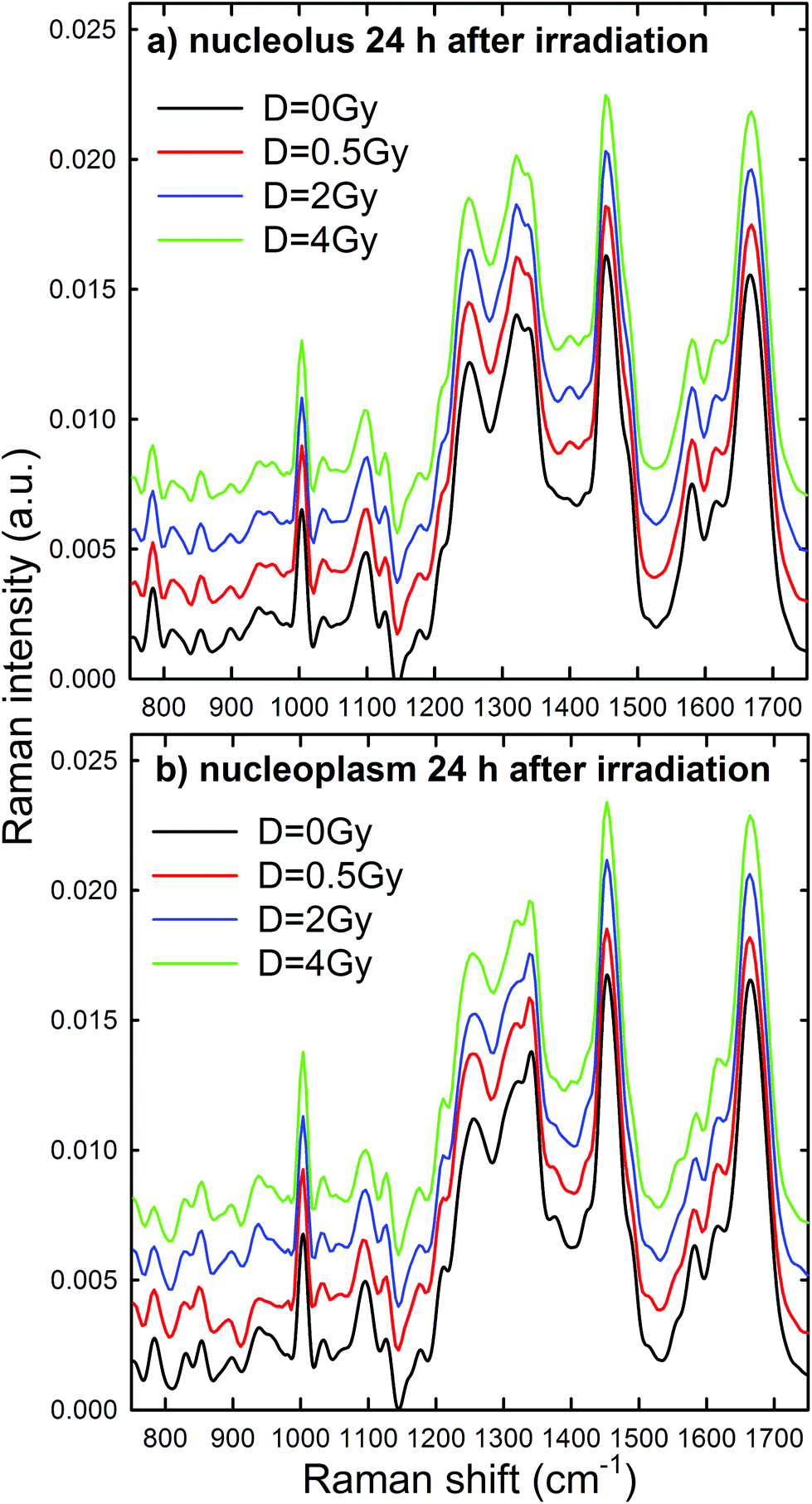 | ||
| Fig. 5 Average Raman spectra of single control (black line), 0.5 Gy (red line), 2 Gy (blue line) and 4 Gy (green line) exposed MCF10A cells (about 30 for each dose), fixed 24 h after irradiation. The spectra have been measured by focusing the exciting laser spot above the nucleolus (a) and nucleoplasm (b) domains. The zero line of the average spectra at 0.5 Gy, 2 Gy and 4 Gy has been intensity-shifted for clarity. | ||
Ratiometric analysis of Raman spectra
In order to gain a more detailed understanding of proton-induced effects on the different cellular components, we performed a ratiometric analysis by considering the dose dependence of the ratio of the intensity of the 784 cm−1 peak with respect to that of the 1003 cm−1 peak (I784/I1003) as a marker of the DNA-related modification with respect to that of proteins, similarly to the ratio of the intensity of the 1096 cm−1 peak with respect to that of the 1003 cm−1 peak (I1096/I1003). In addition, we also analyzed the ratio of the intensity of the 1583 cm−1 peak with respect to that of the 1452 cm−1 peak (I1583/I1452) as an indicator of the modifications of nucleic acids with respect to the other cellular components. These intensity ratios are shown in Fig. 6a and b for the Raman signal collected from the nucleolus and the nucleoplasm region, respectively. Each point in Fig. 6 represents the mean value and standard error of the corresponding ratio estimated for each cell of the whole investigated cell set. It is clearly evident in Fig. 6 that the ratio values for the control cells are quite larger than those for the exposed ones. Multiple comparisons of the exposed groups versus the control group revealed a statistically significant difference (p < 0.05) except for the comparison of the I1583/I1452 ratio at 0.5 Gy for spectra related to the nucleolus region and of the I784/I1003 ratio at 0.5 Gy for spectra related to the nucleoplasm region. Instead, it seems there is a lack of a strong decreasing trend of such ratios with increasing exposure dose above 0.5 Gy.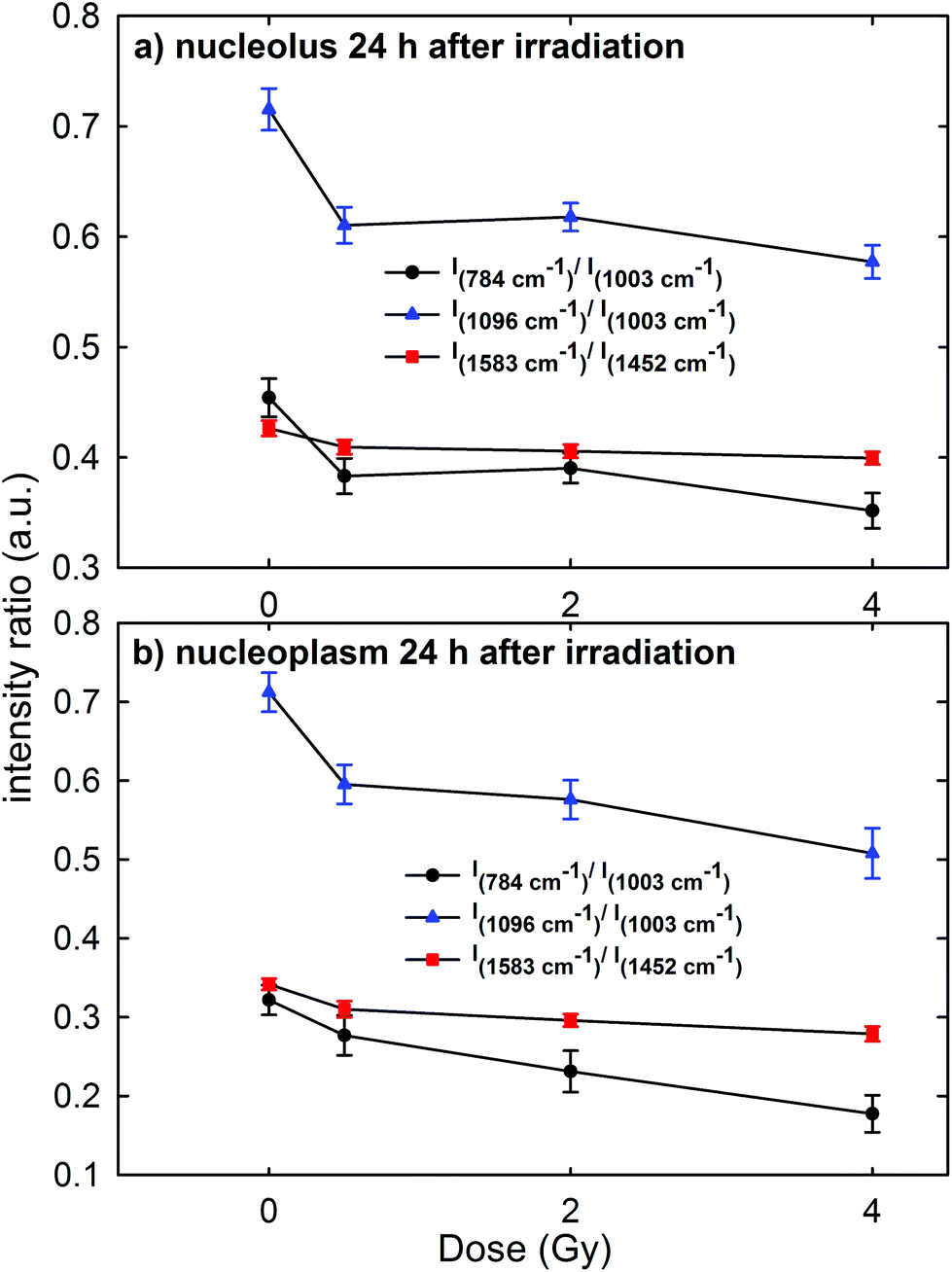 | ||
| Fig. 6 Dose dependence of the mean values of intensity ratios of: the Raman peak at 784 cm−1, due to the DNA vibration mode, with respect to the peak at 1003 cm−1, due to the protein vibration mode (I784/I1003, black circle); the Raman peak at 1096 cm−1, due to the DNA vibration mode, with respect to the peak at 1003 cm−1, due to the protein vibration mode (I1096/I1003, blue triangle); the Raman peak at 1583 cm−1, due to the DNA vibration mode, and with respect to the peak at 1452 cm−1, due to the protein and lipid vibration modes (I1583/I1452, red square). The intensity ratios were calculated by considering MCF10A cells fixed 24 h after irradiation and by focusing the exciting laser spot above the nucleolus (a) and the nucleoplasm (b) domains. Standard error bars are shown for each mean value. | ||
Overall, such results indicate that 24 h after irradiation a stronger damaging effect of irradiation involves the nucleic acid components with respect to the protein and lipid ones. Such damage mainly consists of the breaking of the O–P–O and PO2− bonds of the phosphate groups: in fact, the intensity ratios I784/I1003 and I1096/I1003 decrease by about 15% for the MCF10A cells exposed to a 0.5 Gy proton beam with respect to unexposed cells, both for nucleolus and nucleoplasm sampling. A minor damaging action involves the modification of chemical linkages inside single bases, because the intensity ratio I1583/I1452 decreases by about 4% and 8% in 0.5 Gy exposed cells with respect to the unexposed ones for nucleolus and nucleoplasm compartments, respectively. Similar results, consisting of decreasing Raman peak intensities from specific nucleic acids, were recently found also by S. Rangan et al. for CHO cultured cells undergoing chemically and physically inducted apoptosis and necrosis processes.38 In this work, the Raman signal of exposed cells is sampled from a heterogeneous population, comprising apoptotic, necrotic and living cells. Synytsya et al.39 also observed by means of RS a significant intensity decrease of the peaks related to the phosphodiester bond (at 784 cm−1) and DNA bases ring modes (at 1574 cm−1) in proton irradiated calf thymus DNA. Such results were observed only at high doses of radiation (50 Gy), whereas no significant intensity decrease of the spectral features after 0.5 Gy proton irradiation was reported. In addition, Lipiec et al. detected the intensity change and spectral shift of O–P–O stretching bands in the FTIR spectra of adenocarcinoma PC-3 cells irradiated by different doses of protons and fixed 24 h after exposure:19 they attributed such spectral modifications to DNA repair processes.
The cells sense DNA damage and begin to repair it through the formation of foci around the site of damage within 1 h of irradiation.39 Therefore, 24 h after irradiation, the repair of DNA and RNA simple lesions is supposed to be completed, whereas more complicated residual lesions can be present.40 In particular, the decreasing trend of DNA and RNA peak intensities as a function of the dose suggests that the repair process did not occur completely. Therefore, by considering also the results shown in Fig. 1–3, a strong sensitivity of MCF10A cells to proton radiation, even at the lowest investigated dose (0.5 Gy), can be deduced.
Raman measurements were also performed for the nucleolus and nucleoplasm compartments of MCF10A single cells exposed to different proton doses and fixed 48 h after irradiation (Fig. 3 of the ESI† shows the average normalized spectra). The above intensity ratios were estimated for each single spectrum and the obtained mean values vs. doses are shown in Fig. 7 for the Raman spectra collected from the nucleolus (Fig. 7a) and the nucleoplasm (Fig. 7b) region. The observed trend of such intensity ratios, similar to that reported in Fig. 6, suggests that the repair of DNA damage did not even occur 48 h after irradiation. Also for this time point multiple comparisons of exposed groups versus the control group revealed a statistically significant difference (p < 0.05), except for the comparison of the I1583/I1452 ratio at 4 Gy for spectra related to the nucleolus region. Moreover, it can be stated that 48 h after irradiation the amount of unrepaired damage to nucleic acid components increases with respect to what is reported after 24 h irradiation, particularly for the nucleoplasm region. In particular, I784/I1003 and I1096/I1003 decreased by about 42% and 20% for the MCF10A cells exposed to a 0.5 Gy proton beam with respect to unexposed cells. In contrast, such ratios for the nucleolus region are almost unchanged (they decreased by about 15% and 9%, respectively) if compared to the results obtained after 24 h irradiation. Instead, the intensity ratio I1583/I1452 decreased by about 10% and 13% in the 0.5 Gy exposed cells with respect to the unexposed ones for the nucleolus and nucleoplasm compartments, respectively.
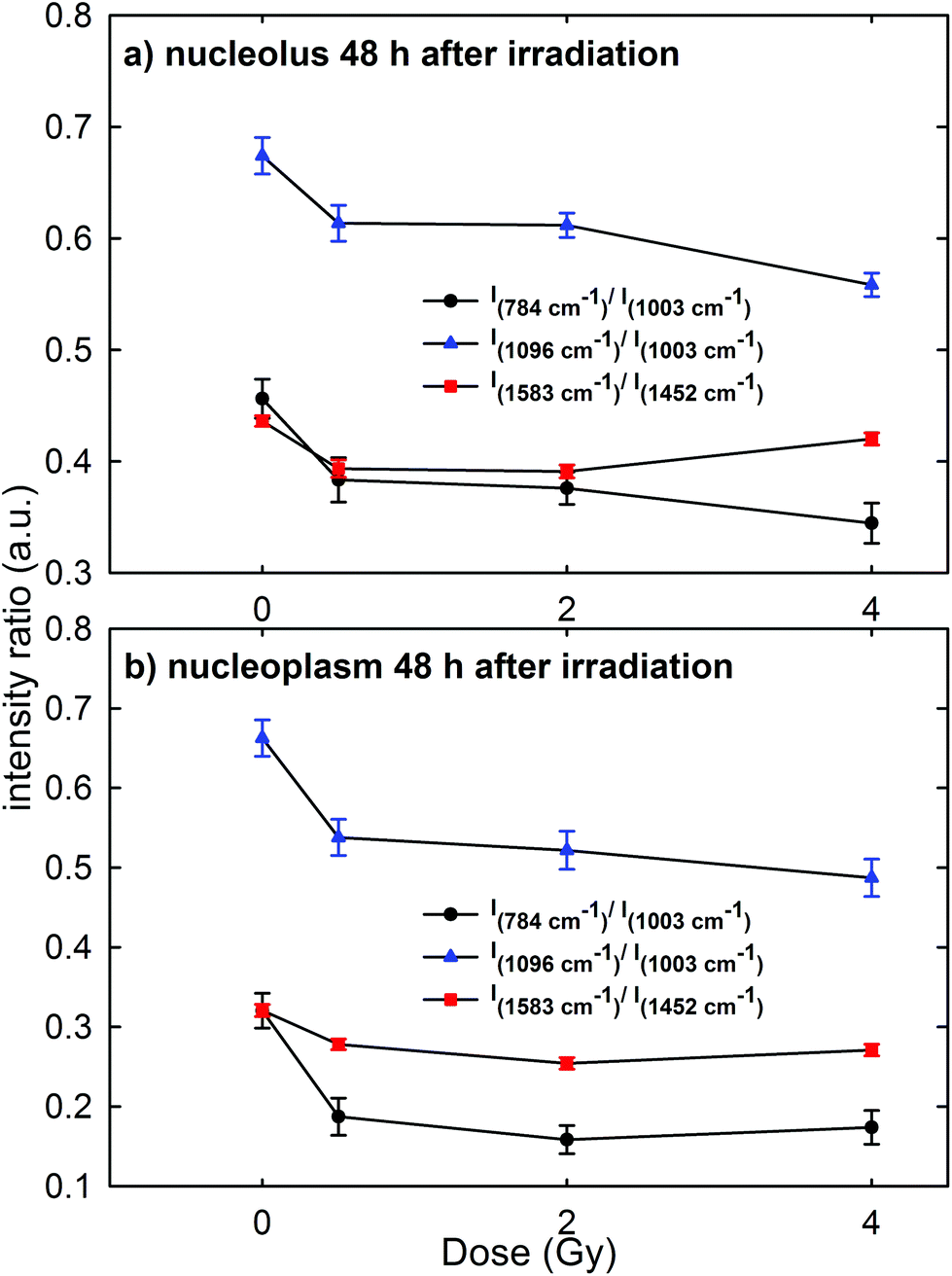 | ||
| Fig. 7 Dose dependence of the mean values of the intensity ratios of: the Raman peak at 784 cm−1, due to the DNA vibration mode, with respect to the peak at 1003 cm−1, due to the protein vibration mode (I784/I1003, black circle); the Raman peak at 1096 cm−1, due to the DNA vibration mode, with respect to the peak at 1003 cm−1, due to the protein vibration mode (I1096/I1003, blue triangle); the Raman peak at 1583 cm−1, due to the DNA vibration mode, with respect to the peak at 1452 cm−1, due to the protein and lipid vibration modes (I1583/I1452, red square). The intensity ratios were calculated by considering MCF10A cells fixed 48 h after irradiation and by focusing the exciting laser spot above the nucleolus (a) and nucleoplasm (b) domains. Standard error bars are shown for each mean value. | ||
Therefore, RS data suggest that proton radiation action mainly caused the breaking of chemical bonds inside phosphate groups and that such damage increases with time, i.e. physiological cellular mechanisms are unable to recover and repair the radiation induced chemical changes. These results are in agreement with those reported by Lipiec et al. about nuclei isolated from glioblastoma cells exposed to protons at 1 Gy and 10 Gy and were measured by means of the FTIR technique 48 h after exposure.41 In particular, they observed a decrease of the peak intensity due to the phosphodiester bond and an increase of the amide II protein peak in the exposed cells with respect to the unexposed ones. Such spectral changes were attributed to the DNA repair process, which involves a stopping of cells in the G1 phase and an increase of the amount of proteins.41 Probably, in our case an effective DNA repair process is inactive because of the larger radiation sensitivity of MCF10A cells with respect to the glioblastoma cells.
A contradictory behavior appears in the Raman spectra and the corresponding intensity ratios of MCF10A cells were fixed 72 h after irradiation. In particular, the mean values of the intensity ratio obtained from the Raman spectra of the cells (average normalized spectra shown in Fig. 4 of the ESI†) are shown in Fig. 8a and b for the spectra measured on the nucleolus and nucleoplasm compartment, respectively. In fact, there is neither a statistically significant difference for the I784/I1003, I1096/I1003 and I1583/I1452 intensity ratios of the exposed groups versus the control group, nor an evident decreasing trend of such ratios with increasing exposure dose, particularly for the nucleolus region. This behavior is in disagreement with the results obtained by Meade et al. about the Raman spectra of HaCaT cells γ-ray irradiated at different doses and measured 96 h after exposure.25 Indeed, an intensity decrease of DNA and RNA related peaks was observed in the Raman spectra of exposed cells with respect to those of the control ones.25
 | ||
| Fig. 8 Dose dependence of the mean values of the intensity ratios of: the Raman peak at 784 cm−1, due to the DNA vibration mode, with respect to the peak at 1003 cm−1, due to the protein vibration mode (I784/I1003, black circle); the Raman peak at 1096 cm−1, due to the DNA vibration mode, with respect to the peak at 1003 cm−1, due to the protein vibration mode (I1096/I1003, blue triangle); the Raman peak at 1583 cm−1, due to the DNA vibration mode, with respect to the peak at 1452 cm−1, due to the protein and lipid vibration modes (I1583/I1452, red square). The intensity ratios have been calculated by considering MCF10A cells fixed 72 h after irradiation and by focusing the exciting laser spot above the nucleolus (a) and nucleoplasm (b) domains. Standard error bars are shown for each mean value. | ||
In our opinion, such apparently anomalous behavior, characterized by an almost constant value of intensity ratios independent of the irradiation dose, cannot be explained by a repairing mechanism of DNA/RNA due to the cell response to the irradiation damage. In fact, both MTT and Annexin-V measurements suggest that at 72 h cell necrosis occurred, similar to that at 24 h and 48 h time points. In contrast, by remarking that the investigated intensity ratios concern the intensity of a nucleic acid-related Raman peak with respect to that of a protein one, this could be explained by considering that the damage elicited by proton radiation to DNA alters or stops the normal evolution of the biochemical content, specifically the synthesis of the protein component, in MCF10A cells. In fact, we reckon that most cells evolving towards an apoptotic or necrotic stage, but still living after 72 h irradiation, originate from parent cells that have not repaired the DNA bonds broken by a proton beam and, consequently, these are cells where the protein synthesis mechanism is strongly altered. Such unrepaired cells are also present in the samples fixed 24 and 48 h after irradiation, but the number of daughter cells is probably lower in the latter samples with respect to those fixed after 72 h, so that the direct DNA/RNA damage is predominant with respect to the indirect protein synthesis damage. Therefore, the intensity decrease of DNA and RNA peaks observed in HaCaT cells 96 h after photon irradiation25 could be related to the increased radioresistance of HaCaT cells compared to MCF10A and to a larger relative biological effectiveness (RBE) of proton radiation with respect to photon radiation.
Although previous studies by Matthews et al.21–23 have reported the use of RS to detect, at different time points, tumour cells’ response to clinical and high doses of ionizing radiation, it is very hard to compare such data with those reported by us, because of the marked differences about the investigated cell lines (tumour vs. normal line), ionizing radiation quality (photon vs. proton) and measured cell volume (several μm vs. less than 1 μm laser spot). However, Matthews et al. have demonstrated the ability of RS to identify radiation-induced cellular response by means of spectral changes occurring in the Raman signals of exposed cells with respect to the unexposed ones. On the other hand, they reported that the spectral changes are specific for a cell line and very subtle for the most radioresistant cell lines. Therefore, our results corroborate the utility and effectiveness of RS at the single cell level to identify the sensitivity of the investigated normal breast cellular model to proton beam exposure at low doses, as those unavoidably occurring during proton therapy treatments. A comparison with the proposed viability, apoptosis and necrosis assays reveals that RS is able to provide sensitivity information within a few hours after exposure.
Conclusions
The present study investigates the response of MCF10A cells to proton beam irradiation at different doses (from 0 Gy to 4 Gy) and at different time points (24 h, 48 h and 72 h following exposure). It was shown that the exposure to protons, even at doses as low as 0.5 Gy, can have strong radiobiological effects, clearly detectable in biological assays as well as in the Raman spectra. In particular, the Raman spectra showed that DNA/RNA related damage increases when the time allowed for the cells to repair increases from 24 to 48 h. We point out that most cells are unable to repair DNA/RNA broken bonds and initiate, according to the results of biological assays, a necrotic death pathway. These results are noteworthy for a better risk assessment in the field of proton radiotherapy because of the unavoidable proton exposure of healthy tissue close to the cancer region. In fact, the biochemical responses from healthy tissue are important because they may be a marker of gradual tissue alteration, which could lead to the late effects of radiation exposure.Raman spectra from both nucleolus and nucleoplasm compartments have been investigated by RS since the sampled spot size is less than the single cell area. A comparison of the results obtained by means of biological assays and Raman spectra indicates that the spectral response from the nucleoplasm region is somewhat more sensitive to the cytogenetic damage with respect to that from the nucleolus compartment. In particular, the intensity ratio of several Raman peaks, as I784/I1003, I1096/I1003 and I1583/I1452 discussed above, are sensitive to the DNA/RNA injury caused by the exposure to the proton beam, even at the lowest investigated dose: so, they could be considered as spectral markers of cytogenetic damage. A limitation about the use of RS for the detection of cellular sensitivity to proton radiation has emerged for measurements carried out 72 h after irradiation. Indeed, the corresponding results are not able to effectively discriminate the cytogenetic damage occurred. In contrast, the obtained results suggest that proper indications could be provided by RS analysis performed 24 h or 48 h after irradiation.
In conclusion, our investigation confirms the utility of RS for radiobiological studies, supporting the possibility of using such a technique as a complimentary method for non-invasive monitoring of radiation sensitivity in clinical practice.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The financial support of this research was provided by INFN (Istituto Nazionale di Fisica Nucleare), through the research project ETHICS (“Pre-clinical experimental and theoretical studies to improve treatment and protection by charged particles”).References
- J. Y. Chang, X. Zhang, X. Wang, Y. Kang, B. Riley, S. Bilton, R. Mohan, R. Komaki and J. D. Cox, Int. J. Radiat. Oncol., Biol., Phys., 2006, 65, 1087–1096 CrossRef PubMed.
- H. Paganetti. Proton Therapy Physics, CRC Press, Taylor and Francis Group, Boca Raton, FL, 2012 Search PubMed.
- M. Goitein, Radiation Oncology: A Physicist's Eye View, Springer-Verlag, New York, 2008 Search PubMed.
- PTCOG (Particle Therapy Coo-Operative Group). http://www.ptcog.ch/index.php/facilities-in-operation (last update: April 2018).
- D. Alloni, A. Campa, W. Friedland, L. Mariotti and A. Ottolenghi, Int. J. Radiat. Biol., 2012, 88, 77–86 CrossRef CAS PubMed.
- P. L. Olive, Radiat. Res., 1998, 150, S42–S51 CrossRef CAS PubMed.
- L. M. Burt, J. Ying, M. M. Poppe, G. Suneja and D. K. Gaffney, Breast, 2017, 35, 122–129 CrossRef PubMed.
- J. N. Kavanagh, K. M. Redmond, G. Schettino and K. M. Prise, Antioxid. Redox Signaling, 2013, 18, 2458–2472 CrossRef CAS PubMed.
- T. A. Koulis, T. Phan and I. A. Olivotto, Breast Cancer, 2015, 7, 363–370 Search PubMed.
- B. Emami, J. Lyman, A. Brown, L. Coia, M. Goitein, J. E. Munzenrider, B. Shank, L. J. Solin and M. Wesson, Int. J. Radiat. Oncol., Biol., Phys., 1991, 21, 109–122 CrossRef CAS.
- R. L. Foote, S. L. Stafford, I. A. Petersen, J. S. Pulido, M. J. Clarke, S. E. Schild, Y. I. Garces, K. R. Olivier, R. C. Miller, M. G. Haddock, E. Yan, N. N. Laack, C. A. Arndt, S. J. Buskirk, V. L. Miller, C. R. Brent, J. J. Kruse, G. A. Ezzell, M. G. Herman, L. L. Gunderson, C. Erlichman and R. B. Diasio, Radiat. Oncol., 2012, 7, 174 CrossRef PubMed.
- J. J. Cuaron, S. M. MacDonald and O. Cahlon, Chin. J. Clin. Oncol., 2016, 5, 52 CrossRef PubMed.
- J. A. Bradley, R. Dagan, M. W. Ho, M. Rutenberg, C. G. Morris, Z. Li and N. P. Mendenhall, Int. J. Radiat. Oncol., Biol., Phys., 2016, 95, 411–421 CrossRef PubMed.
- W. B. Kiosses, K. M. Hahn, G. Giannelli and V. Quaranta, Cell Commun. Adhes., 2001, 8, 29–44 CrossRef CAS PubMed.
- P. Voos, S. Fuck, F. Weipert, L. Babel, D. Tandl, T. Meckel, S. Hehlgans, C. Fournier, A. Moroni, F. Rödel and G. Thiel, Front. Immunol., 2018, 9, 922 CrossRef PubMed.
- O. Keta, D. Todorovic, N. Popovic, L. Koricanac, G. Cuttone, I. Petrovic and A. Ristic-Fira, Arch. Med. Sci., 2014, 3, 578–586 CrossRef PubMed.
- Y. Matsumoto, T. Matsuura, M. Wada, Y. Egashira, T. Nishio and Y. Furusawa, J. Radiat. Res., 2014, 55, 816–822 CrossRef PubMed.
- H. Hojo, T. Dohmae, K. Hotta, R. Kohno, A. Motegi, A. Yagishita, H. Makinoshima, K. Tsuchihara and T. Akimoto, Radiat. Oncol., 2017, 12, 111 CrossRef PubMed.
- E. Lipiec, K. R. Bambery, P. Heraud, C. Hirschmugl, J. Lekki, W. M. Kwiatek, M. J. Tobin, C. Vogel, D. Whelan and B. R. Wood, J. Mol. Struct., 2014, 1073, 134–141 CrossRef CAS.
- D. Juerß, M. Zwar, U. Giesen, R. Nolte, S. Kriesen, G. Baiocco, M. Puchalska, M. J. van Goethem, K. Manda and G. Hildebrandt, Radiat. Oncol., 2017, 12, 159 CrossRef PubMed.
- Q. Matthews, A. G. Brolo, J. J. Lum, X. Duam and A. Jirasek, Phys. Med. Biol., 2011, 56, 19–38 CrossRef CAS PubMed.
- Q. Matthews, A. Jirasek, J. J. Lum and A. G. Brolo, Phys. Med. Biol., 2011, 56, 6839–6855 CrossRef CAS PubMed.
- S. J. Harder, Q. Matthews, M. Isabelle, A. G. Brolo, J. J. Lum and A. Jirasek, Appl. Spectrosc., 2015, 69, 193–204 CrossRef CAS PubMed.
- J. Qi, B. Liu, Y. Li, D. Wu and W. Tang, Chin. Opt. Lett., 2009, 7, 080734 Search PubMed.
- A. D. Meade, O. Howe, V. Unterreiner, G. D. Sockalingum, H. J. Byrne and F. M. Lyng, Faraday Discuss., 2016, 187, 213–234 RSC.
- S. Devpura, K. N. Barton, S. L. Brown, O. Palyvoda, S. Kalkanis, V. M. Naik, F. Siddiqui, R. Naik and I. J. Chetty, Med. Phys., 2014, 41, 050901 CrossRef PubMed.
- M. Lasalvia, G. Perna, L. Manti, J. Rasero, S. Stramaglia and V. Capozzi, Int. J. Radiat. Biol., 2018 DOI:10.1080/09553002.2019.1547849.
- G. A. P. Cirrone, G. Cuttone, P. A. Lojacono, S. Lo Nigro, V. Mongelli, I. V. Patti, G. Privitera, L. Raffaele, D. Rifuggiato, M. G. Sabini, V. Salamone, C. Spatola and L. M. Valastro, IEEE Trans. Nucl. Sci., 2004, 51, 3568–3662 Search PubMed.
- T. T. Puck and P. I. Marcus, J. Exp. Med., 1956, 103, 653–666 CrossRef CAS PubMed.
- G. P. Dimri, X. Lee, G. Basile, M. Acosta, G. Scott, C. Roskelley, E. E. Medrano, M. Linskens, I. Rubelj and O. Pereira-Smith, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9363–9367 CrossRef CAS.
- I. Delfino, G. Perna, M. Lasalvia, V. Capozzi, L. Manti, C. Camerlingo and M. Lepore, J. Biomed. Opt., 2015, 20, 035003 CrossRef PubMed.
- R. Grasso, F. P. Cammarata, L. Minafra, V. Marchese, G. Russo, L. Manti, F. Musumeci and A. Scordino, Proc. SPIE, 2017, 10413, 104130L Search PubMed.
- L. Hayflick and P. S. Moorhead, Exp. Cell Res., 1961, 25, 585–621 CrossRef CAS PubMed.
- R. J. Sabin and R. M. Anderson, Genome Integr., 2011, 2, 7 CrossRef CAS PubMed.
- A. Pliss, A. N. Kuzmin, A. V. Kachynski and P. N. Prasad, Biophys. J., 2010, 99, 3483–3491 CrossRef CAS PubMed.
- A. Pliss, A. N. Kuzmin, A. V. Kachynski and P. N. Prasad, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12771–12776 CrossRef CAS PubMed.
- Z. Movasaghi, S. Rehman and I. U. Rehman, Appl. Spectrosc. Rev., 2007, 42, 493–541 CrossRef CAS.
- S. Rangan, S. Kamal, S. O. Konorov, H. G. Schulze, M. W. Blades, R. F. B. Turner and J. M. Piret, Biotechnol. Bioeng., 2018, 115, 401–412 CrossRef CAS PubMed.
- A. Synytsya, P. Alexa, J. de Boer, M. Loewe, M. Moosburger, M. Wurkner and K. Volka, J. Raman Spectrosc., 2007, 38, 1406–1415 CrossRef CAS.
- A. Noda, Y. Hirai, K. Hamasaki, H. Mitani, N. Nakamura and Y. Kodama, J. Cell Sci., 2012, 125, 5280–5287 CrossRef CAS PubMed.
- E. Lipiec, B. R. Wood, A. Kulik, W. M. Kwiatek and G. Dietler, Anal. Chem., 2018, 90, 7644–7650 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an02155d |
| This journal is © The Royal Society of Chemistry 2019 |