
Hydrofunctionalization of alkenols triggered by the addition of diverse radicals to unactivated alkenes and subsequent remote hydrogen atom translocation†
Na
Wang‡
ab,
Liu
Ye‡
c,
Zhong-Liang
Li
c,
Lei
Li
b,
Zhuang
Li
b,
Hong-Xia
Zhang
a,
Zhen
Guo
 a,
Qiang-Shuai
Gu
*c and
Xin-Yuan
Liu
*b
a,
Qiang-Shuai
Gu
*c and
Xin-Yuan
Liu
*b
aCollege of Materials Science & Engineering, Key Laboratory of Interface Science and Engineering in Advanced Materials, Ministry of Education, Taiyuan University of Technology, Shanxi, 030024, China
bDepartment of Chemistry and Shenzhen Grubbs Institute, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: liuxy3@sustc.edu.cn
cAcademy for Advanced Interdisciplinary Studies, Southern University of Science and Technology, Shenzhen, 518055, China. E-mail: guqs@sustc.edu.cn
First published on 22nd August 2018
Abstract
Diverse anti-Markovnikov hydrofunctionalization of alkenols triggered by the addition of S-, P-, and C-centered radicals to alkenes followed by intramolecular 1,5(6)-hydrogen atom transfer (HAT) with remote α-C–H bonds of alcohols has been developed. The strategy simultaneously realized the hydrofunctionalization of alkenes and remote alcohol oxidation. This mild and versatile method allows for direct access to valuable sulfonyl-, phosphonyl-, and malonate-substituted ketones or aldehydes from a wide range of alkenols.
Introduction
The hydrofunctionalization of alkenes provides a facile access to a wide range of functionalized alkanes.1 Among intensive research in this area, the radical-mediated alkene hydrofunctionalization initiated by the addition of radicals to alkenes followed by trapping of the incipient radical with a hydrogen donor represents an efficient and powerful tool for alkene hydrofunctionalization in an anti-Markovnikov manner.2 The use of efficient hydrogen donors is critical for the realization of alkene hydrofunctionalization because of the high propensity of alkyl radical intermediates, particularly for linear alkene substrates, to undergo undesired competitive pathways such as 1,2-difunctionalization and β-hydrogen elimination.3 Compared with the well-established external hydrogen donors, intramolecular hydrogen delivery via 1,n-hydrogen atom transfer (HAT) for hydrofunctionalization of unactivated alkenes has recently attracted considerable attention due to its high regio-selectivity and concomitant remote C–H bond functionalization.4 In this respect, our group and others have independently reported the general and efficient hydrofluoroalkylation of alkenes with suitably positioned alcohols/ethers as internal hydrogen donors to afford a variety of fluoroalkyl-containing carbonyl products (Scheme 1a).5 However, the research on this chemistry was confined to fluoro-containing radical sources, which unnecessarily limited both the scope and the potential of this method for practical applications. Hence, the realization of hydrofunctionalization of alkenols via this strategy with diverse heteroatom- or carbon-centered radical sources is highly desirable.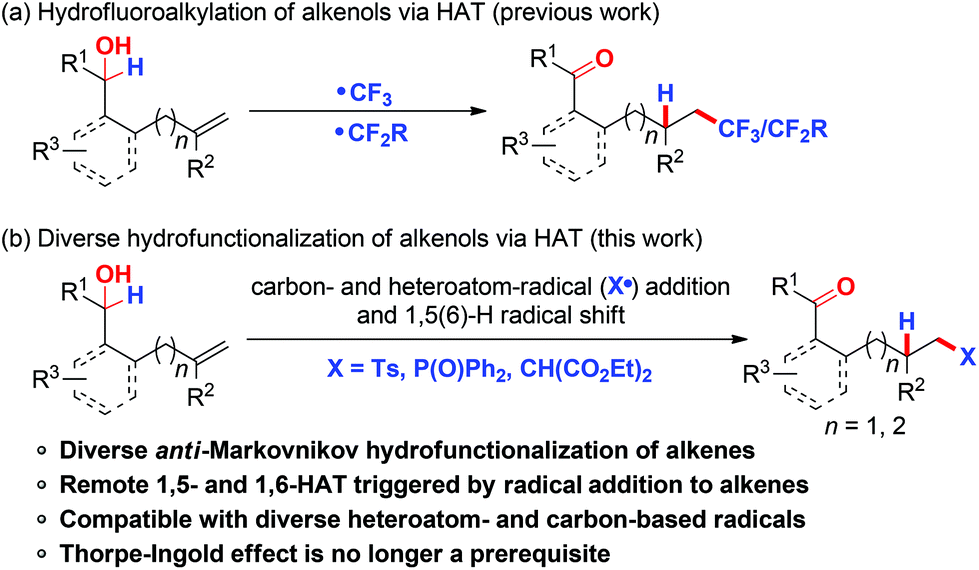 | ||
| Scheme 1 Hydrofunctionalization with diverse alkenols via 1,5(6)-HAT. | ||
Organosulfones and organophosphorus compounds are common structural motifs widely displayed in agrochemicals, pharmaceuticals, and catalysts.6 They have also been employed as highly useful building blocks in organic synthesis. Recently, the direct functionalization of alkenes triggered by S-7 or P-centered8 radical addition to alkenes has emerged as a promising tool for the synthesis of functionalized organosulfones and organophosphorus compounds. Thus, much progress has been made in the development of sulfonylation and phosphonylation of activated olefins. However, the application of similar transformations to unactivated alkenes is more challenging,9 which is probably due to the unfavorable reversible generation of unstable radical intermediates.8a,10 As our continuing interest in the functionalization of unactivated alkenes via the addition of radicals to unactivated alkenes followed by subsequent remote functional group translocation,11 we describe herein a general and efficient protocol for hydrosulfonylation, hydrophosphonylation, and hydroalkylation of alkenols in the presence or absence of a gem-disubstituent effect via intramolecular 1,5(6)-HAT in a highly controlled site-selective manner. It generally provides valuable sulfonyl-, phosphonyl-, and malonate-functionalized carbonyl compounds in good yields (Scheme 1b).
Results and discussion
At first, we began our investigation by exploring the hydrosulfonylation reaction of linear alkenol 1A with p-toluenesulfinic acid 2a. Various oxidants were investigated and only K2S2O8 gave the desired product 3Aa (entries 1–4, Table 1).7e An inert atmosphere was slightly beneficial for the reaction (entry 5). Three equiv. of 2a together with two equiv. of K2S2O8 were proven to be necessary for the full conversion of 1A and the good yield of 3Aa (entries 6–8). Switching the oxidant to (NH4)2S2O8 unexpectedly led to significantly diminished yield (entry 9). Co-solvent systems exhibited high reaction efficiency and resulted in a clear reaction mixture. Finally, we found that the reaction of 1A with p-toluenesulfinic acid (2a) proceeded smoothly by simply using the cheap K2S2O8 (2 equiv.) as the oxidant in CH3CN/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) at 80 °C for 16 h, providing sulfonylated ketone 3Aa in 82% isolated yield.
:2) at 80 °C for 16 h, providing sulfonylated ketone 3Aa in 82% isolated yield.
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant (x equiv.) | 2a (y equiv.) | Solvent (ratio) | Yieldb (%) |
| a Reaction conditions: 1A (0.2 mmol), 2a, solvent (2.0 mL) at 80 °C for 16 h under an argon atmosphere. b Determined by NMR spectroscopy using 1,3,5-trimethylbenzene as an internal standard. c p-Toluenesulfonylhydrazine was utilized. d Isolated yield. | ||||
| 1 | Fe(NO3)3·9H2O (0.10), air | 3.0 | MeCN | Trace |
| 2c | TBAI (0.10), TBHP (3.0) | 3.0 | Dioxane/H2O (4/1) | Trace |
| 3 | Pyridine (4.0), air | 5.0 | DCE | NR |
| 4 | K2S2O8 (1.0), air | 3.0 | MeCN/H2O (1/2) | 50% |
| 5 | K2S2O8 (1.0), argon | 3.0 | MeCN/H2O (1/2) | 55% |
| 6 | K2S2O8 (2.0), argon | 1.0 | MeCN/H2O (1/2) | 48% |
| 7 | K2S2O8 (2.0), argon | 2.0 | MeCN/H2O (1/2) | 70% |
| 8 | K 2 S 2 O 8 (2.0), argon | 3.0 | MeCN/H 2 O (1/2) | 82% |
| 9 | (NH4)2S2O8 (2.0), argon | 3.0 | MeCN/H2O (1/2) | 35% |
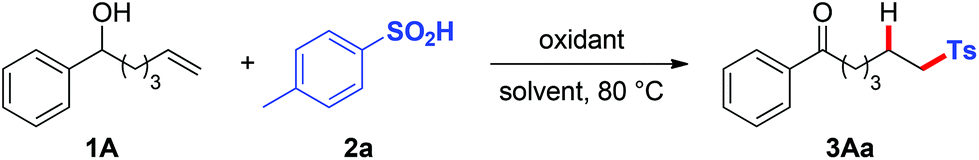
With the optimal reaction conditions in hand, we set out to explore the scope with respect to various alkenols (Table 2). A range of linear alkenols bearing diversely functionalized aryl groups (electron-rich one: Me for 1B; electron-deficient ones: Cl for 1C–1E and Br for 1F) were found to be suitable substrates to give the corresponding products 3Ba–3Fa selectively in 48–80% yields. It should be noted that substrate 1I, with an isopropyl group in place of a phenyl one, also afforded the expected product 3Ia albeit with lower yield, thus clearly demonstrating that the current process was not limited to benzylic alcohol. Comparable to linear alkenyl alcohols, the aryl-tethered substrates 1J–1N were also applicable to this process, affording highly substituted Ts-containing aryl aldehydes 3Ja–3La and aryl ketones 3Ma–3Na in moderate to good yields. Notably, substrate 1N with a gem-disubstituted alkenyl group reacted efficiently to afford aryl ketone 3Na in 70% yield.
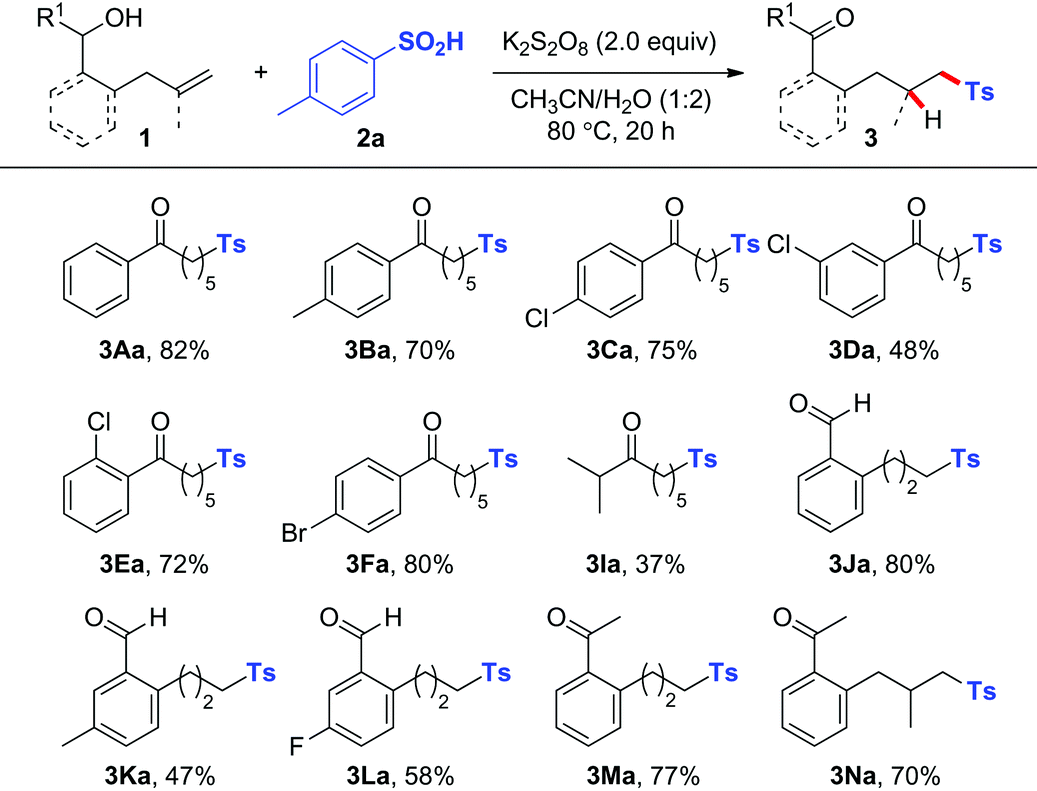
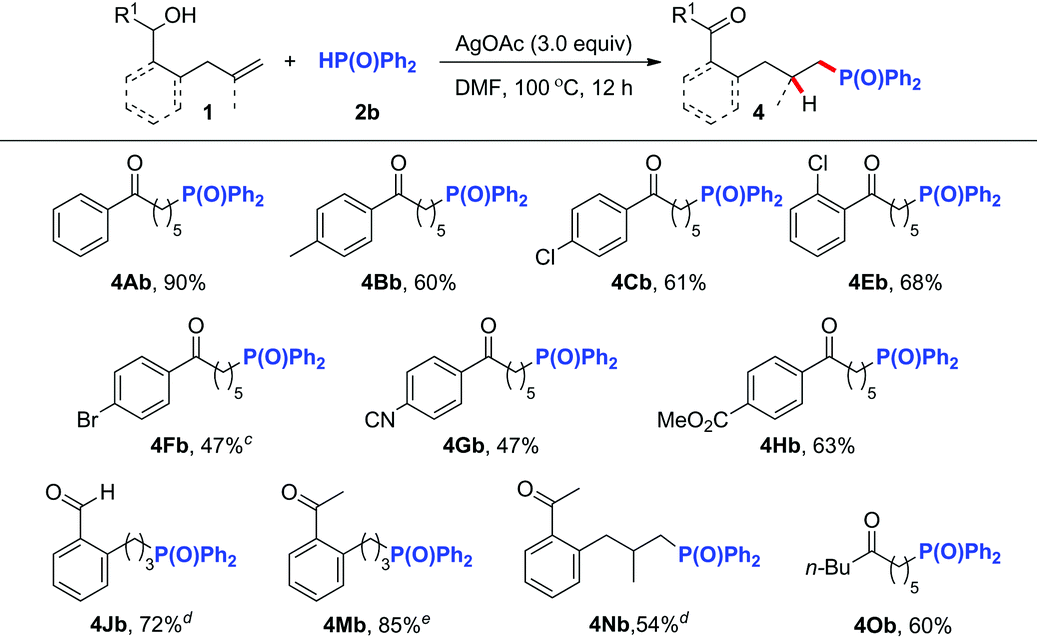
Inspired by the above successful results, we next focused our attention on other challenging heteroatom- and carbon-centered radicals. A phosphonyl radical was then selected as the heteroatom-centered radical to probe whether a similar radical process could be realized.8,9 After systematic optimization of different reaction parameters (Table S1†), we were delighted to find that in the presence of 3.0 equiv. of AgOAc8i or 50 mol% of AgNO3,8d the reaction of alkenol substrates with 2.0 equiv. of Ph2P(O)H (2b) in DMF at 100 °C afforded the corresponding phosphonylated ketones in good yields (Table 3). The reaction tolerated a variety of linear alkenols containing aryl groups with different substituents to yield products 4Ab–4Cb and 4Eb–4Hb in 47–90% yields. Similarly, the aryl-tethered substrates 1J and 1M exhibited high efficiency in this hydrophosphonylation reaction. Good reactivity was also observed for the gem-disubstituted alkene substrate 1N and aliphatic substrate 1O, thus illustrating the wide applicability of the current hydrophosphonylation reaction.
In order to further expand the synthetic utility of this methodology, we next investigated the hydroalkylation of alkenols with C-centered radical precursors via the HAT strategy. Malonate, which is prone to undergoing oxidation to produce C-centered radicals, was then used to test our hypothesis.12 After screening lots of reaction parameters (Table S2†), we optimized the reaction conditions of this hydroalkylation as follows: 3.0 equiv. of Mn(OAc)3 as the oxidant and 2,2,2-trifluoroethanol as the solvent at 110 °C for 36 h.12d The reaction of various substrates 1A, 1C, 1F, 1M, and 1O with malonate 2c as the radical precursor gave the desired malonate-substituted ketone products 5Ac, 5Cc, 5Fc, 5Mc, and 5Oc in moderate yields (Scheme 2).
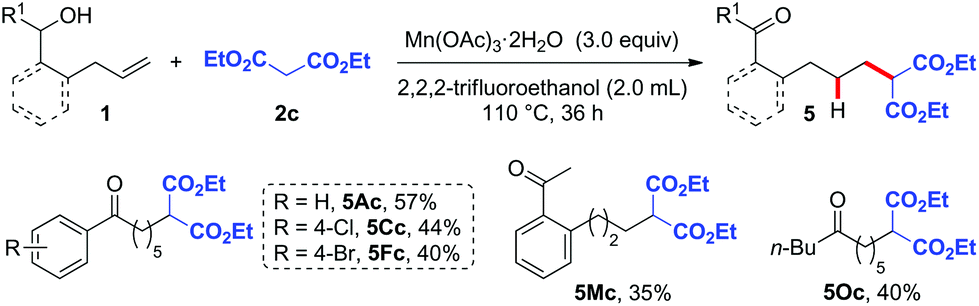 | ||
| Scheme 2 Substrate scope of hydroalkylation. Reaction conditions: 1 (0.2 mmol), 2c (0.3 mmol), Mn(OAc)3·2H2O (0.6 mmol), 2,2,2-trifluoroethanol (2.0 mL), 110 °C, 36 h. | ||
Inspired by the above success in the 1,5-HAT process, we subsequently switched our attention to the use of alkenols 6 with one-carbon-longer chains to probe whether the more challenging 1,6-HAT obtained via a higher energy seven-membered cyclic transition state13 could be realized under the current reaction system. Gratifyingly, under the reaction conditions identical to those of the 1,5-HAT process (Tables 2, 3 and Scheme 2), remotely sulfonyl- (7Aa, 7Ba), phosphonyl- (7Ab, 7Cb), and malonate-substituted (7Ac, 7Bc) aldehydes/ketones were obtained in moderate to good yields from alkenyl alcohols 6A–6C (Table 4).
| a Reaction conditions: 6 (0.2 mmol), 2a (0.6 mmol), K2S2O8 (0.4 mmol), CH3CN (0.7 mL), H2O (1.4 mL), 80 °C. b Reaction conditions: 6 (0.2 mmol), 2b (0.4 mmol), AgOAc (0.6 mmol), DMF (2.0 mL), 100 °C. c Reaction conditions: 6 (0.2 mmol), 2c (0.3 mmol), Mn(OAc)3·2H2O (0.6 mmol), 2,2,2-trifluoroethanol (2.0 mL), 110 °C. d Reaction conditions: 6 (0.2 mmol), 2b (0.4 mmol), AgNO3 (0.6 mmol), DMF (2.0 mL), 100 °C. |
|---|
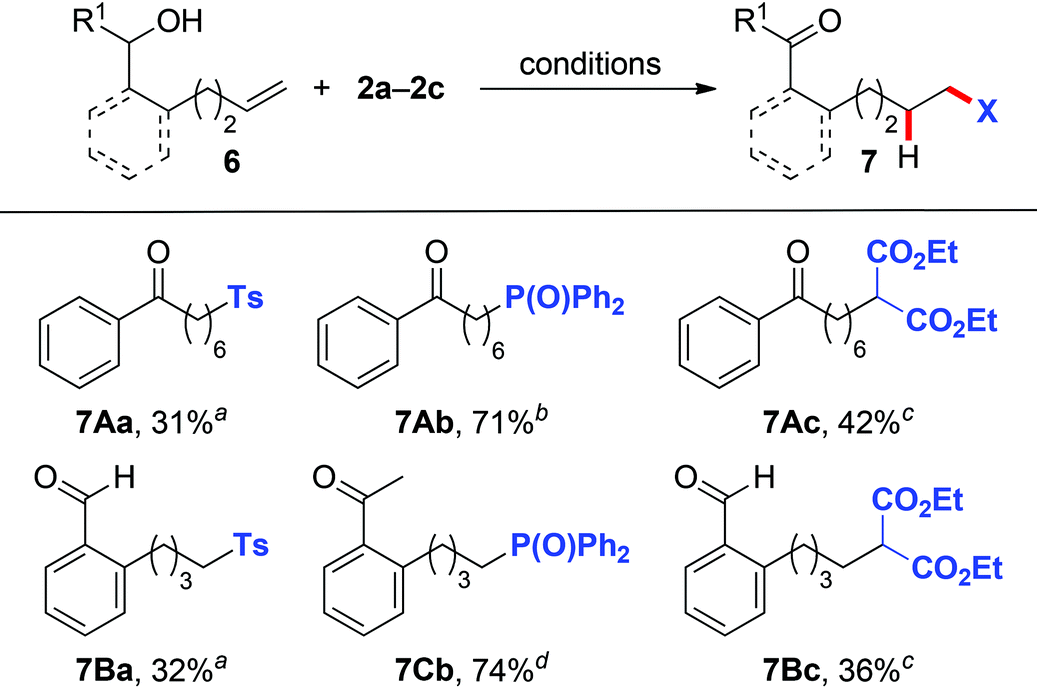
|
To gain insights into the reaction mechanism, several control experiments were performed. Firstly, the sulfonylation reaction was run under the standard conditions with the addition of radical scavengers such as benzoquinone (BQ) and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO). The reactions were significantly inhibited by these reagents (Scheme 3, eqn (1)), indicating radical intermediates in the reaction. Secondly, the use of [D1]-1A under the standard sulfonylation conditions led to the formation of [D1]-3Aa with complete transposition of the α-D atom (Scheme 3, eqn (2)). Thirdly, no crossover products 3Aa or [D1]-3Pa were detected when equal molar amounts of [D1]-1A and 1P were used under the standard conditions, which excludes an intermolecular HAT process (Scheme 3, eqn (3)).
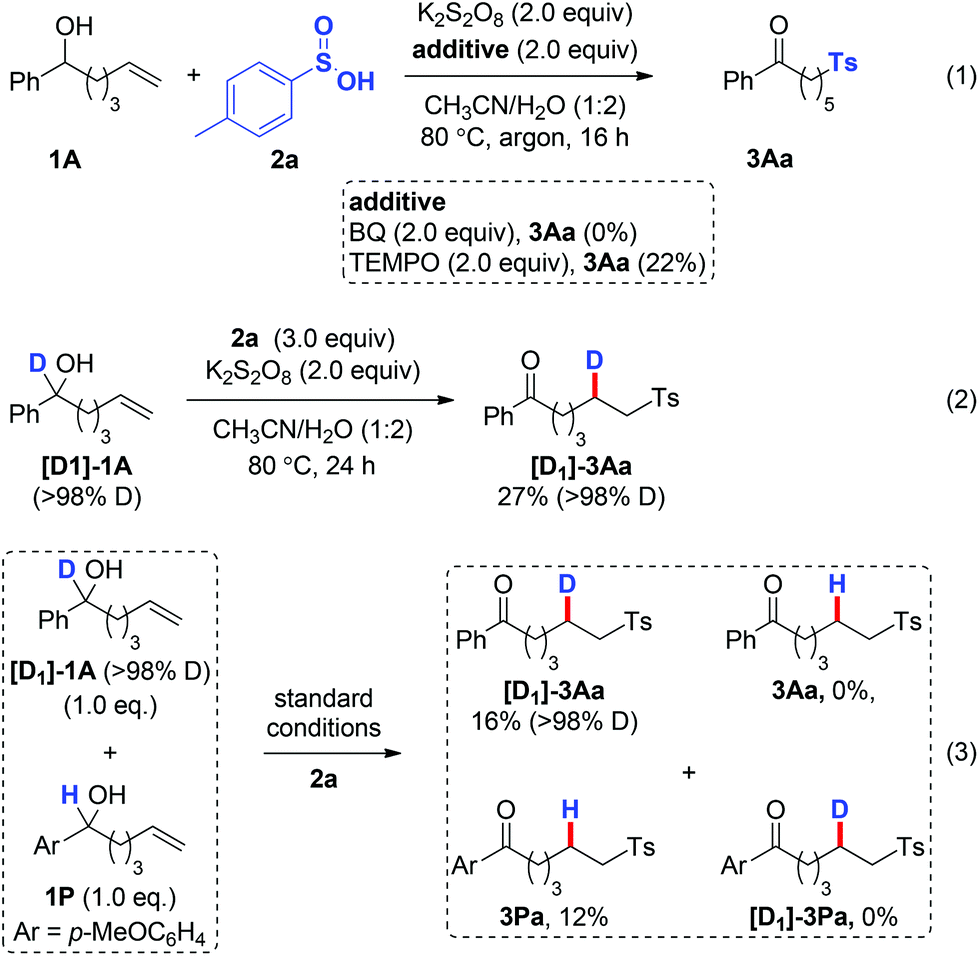 | ||
| Scheme 3 Control experiments. | ||
To illustrate the potential synthetic applicability of this protocol, 3Aa was prepared in large scale to indicate that no significant erosion in the reaction efficiency had occurred (Scheme 4, eqn (1)). The utility of our protocol was further demonstrated by cyclization of the obtained ketone 3Ma under basic conditions at room temperature to afford the α,β-unsaturated sulfone product 8 in 73% yield; 8 is a good Michael acceptor widely applied in biotransformation and organic synthesis (Scheme 4, eqn (2)).
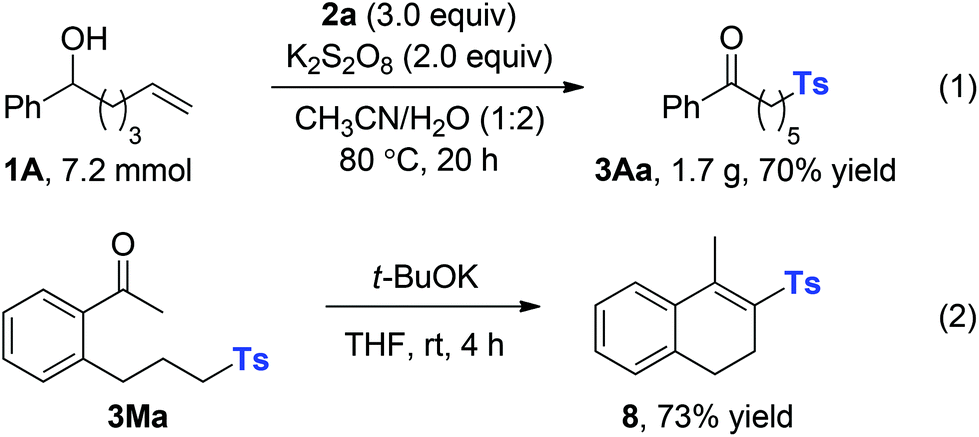 | ||
| Scheme 4 Large scale preparation and transformation. | ||
Conclusions
In summary, we have successfully developed a general and efficient radical protocol for the concomitant diverse hydrofunctionalization of alkenes and oxidation of remote alcohols. It provides valuable sulfonyl-, phosphonyl-, and malonate-substituted ketones or aldehydes through ketyl radical intermediates via 1,5(6)-HAT triggered by addition of the corresponding S-, P-, and C-centered radicals to alkenes in an anti-Markovnikov manner. This mild and versatile method exhibits a broad substrate scope in regard to alkenols with or without gem-disubstituted tethering groups and tolerates a series of carbon- and heteroatom-centered radical precursors.Experimental
General procedure for the radical hydrosulfonylation reaction system
To a flame-dried Schlenk tube equipped with a magnetic stir bar were added 1 (0.2 mmol), 2a (0.6 mmol), and K2S2O8 (0.4 mmol). The tube was evacuated and backfilled with argon three times, and then MeCN (0.7 mL) and H2O (1.4 mL) were added. The tube was stirred at 80 °C for 18 h and then H2O (5 mL) was added. EtOAc was used to extract the product from the aqueous layer (3 × 20 mL). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to afford the crude product, which was purified using flash column chromatography to afford the product 3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21722203, 21702093, and 21572096), the Shenzhen special funds for the development of biomedicine, Internet, new energy, and new material industries (No. JCYJ20170412152435366 and JCYJ20170307105638498), and the Shenzhen Nobel Prize Scientists Laboratory Project (No. C17213101) is greatly appreciated.Notes and references
- (a) Hydrofunctionalization, in Topics in Organometallic Chemistry, ed. V. P. Ananikov and M. Tanaka, Springer, 2013, vol. 43 Search PubMed; (b) M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef PubMed; (c) K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997 CrossRef PubMed.
- For representative anti-Markovnikov hydrotrifluoromethylation examples, see: (a) S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheelhouse, G. Rassias, M. Médebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505 CrossRef PubMed; (b) D. J. Wilger, N. J. Gesmundo and D. A. Nicewicz, Chem. Sci., 2013, 4, 3160 RSC; (c) X. Wu, L. Chu and F.-L. Qing, Angew. Chem., Int. Ed., 2013, 52, 2198 CrossRef PubMed; (d) S. Choi, Y. J. Kim, S. M. Kim, J. W. Yang, S. W. Kim and E. J. Cho, Nat. Commun., 2014, 5, 4881 CrossRef PubMed; (e) H. Egami, Y. Usui, S. Kawamura, S. Nagashima and M. Sodeoka, Chem. – Asian J., 2015, 10, 2190 CrossRef PubMed; (f) L. Zhu, L.-S. Wang, B. Li, B. Fu, C.-P. Zhang and W. Li, Chem. Commun., 2016, 52, 6371 RSC; (g) Y. Cheng and S. Yu, Org. Lett., 2016, 18, 2962 CrossRef PubMed. For a representative anti-Markovnikov hydroazidation example, see: (h) A. Kapat, A. Konig, F. Montermini and P. Renaud, J. Am. Chem. Soc., 2011, 133, 13890 CrossRef PubMed.
- (a) T. Nishikata, Y. Noda, R. Fujimoto and T. Sakashita, J. Am. Chem. Soc., 2013, 135, 16372 CrossRef PubMed; (b) Q. Liu, H. Yi, J. Liu, Y. Yang, X. Zhang, Z. Zeng and A. Lei, Chem. – Eur. J., 2013, 19, 5120 CrossRef PubMed.
- For selected reviews, see: (a) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef PubMed; (b) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (c) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef PubMed; (d) Y. Zeng, C. Ni and J. Hu, Chem. – Eur. J., 2016, 22, 3210 CrossRef PubMed; (e) Y. Tian, S. Chen, Q.-S. Gu, J.-S. Lin and X.-Y. Liu, Tetrahedron Lett., 2018, 59, 203 CrossRef; (f) W. Li, W. Xu, J. Xie, S. Yu and C. Zhu, Chem. Soc. Rev., 2018, 47, 654 RSC. For representative examples, see: (g) X. Nie, C. Cheng and G. Zhu, Angew. Chem., Int. Ed., 2017, 56, 1898 CrossRef PubMed; (h) W. Shu, E. Merino and C. Nevado, ACS Catal., 2018, 8, 6401 CrossRef.
- (a) L. Li, L. Ye, S.-F. Ni, Z.-L. Li, S. Chen, Y.-M. Du, X.-H. Li, L. Dang and X.-Y. Liu, Org. Chem. Front., 2017, 4, 2139 RSC; (b) G. H. Lonca, D. Y. Ong, T. M. H. Tran, C. Tejo, S. Chiba and F. Gagosz, Angew. Chem., Int. Ed., 2017, 56, 11440 CrossRef PubMed; (c) J. Zhang, W. Jin, C. Cheng and F. Luo, Org. Biomol. Chem., 2018, 16, 3876 RSC.
- (a) S. Patai, C. Z. Rappoport and J. M. Stirling, The Chemistry of Sulfones and Sulfoxides, Wiley, New York, 1988 Search PubMed; (b) N. S. Simpkins, Sulfones in Organic Synthesis, Pergamon, Oxford, 1993 Search PubMed; (c) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863 CrossRef; (d) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef PubMed.
- (a) Radicals in Organic Synthesis, ed. M. Sibi and P. Renaud, Wiley, Weinheim, Germany, 2001, vol. 1 and 2 Search PubMed; (b) Z. Yuan, H.-Y. Wang, X. Mu, P. Chen, Y.-L. Guo and G. Liu, J. Am. Chem. Soc., 2015, 137, 2468 CrossRef PubMed; (c) Q. Lu, J. Zhang, F. Wei, Y. Qi, H. Wang, Z. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7156 CrossRef PubMed; (d) T. Shen, Y. Yuan, S. Song and N. Jiao, Chem. Commun., 2014, 50, 4115 RSC; (e) W. Wei, J. Wen, D. Yang, J. Du, J. You and H. Wang, Green Chem., 2014, 16, 2988 RSC; (f) C. S. Gloor, F. Dénès and P. Renaud, Angew. Chem., Int. Ed., 2017, 56, 13329 CrossRef PubMed.
- For reviews on phosphonylation of alkenes, see: (a) D. Leca, L. Fensterbank, E. Lacôte and M. Malacria, Chem. Soc. Rev., 2005, 34, 858 RSC; (b) A. Dondoni and A. Marra, Org. Biomol. Chem., 2015, 13, 2212 RSC. For selected recent examples, see: (c) W. Wei and J.-X. Ji, Angew. Chem., Int. Ed., 2011, 50, 9097 CrossRef PubMed; (d) C. Zhang, Z. Li, L. Zhu, L. Yu, Z. Wang and C. Li, J. Am. Chem. Soc., 2013, 135, 14082 CrossRef PubMed; (e) Y.-M. Li, M. Sun, H.-L. Wang, Q.-P. Tian and S.-D. Yang, Angew. Chem., Int. Ed., 2013, 52, 3972 CrossRef PubMed; (f) W. Kong, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2014, 53, 5078 Search PubMed; (g) N. Fuentes, W. Kong, L. Fernández-Sánchez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2015, 137, 964 CrossRef PubMed; (h) W. Kong, N. Fuentes, A. García-Domínguez, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 2487 CrossRef PubMed; (i) X.-Q. Chu, Y. Zi, H. Meng, X.-P. Xu and S.-J. Ji, Chem. Commun., 2014, 50, 7642 RSC.
- For representative examples on sulfonylation and phosphonylation of unactivated alkenes, see: (a) F. Chen, N.-N. Zhou, J.-L. Zhan, B. Han and W. Yu, Org. Chem. Front., 2017, 4, 135 RSC; (b) D. Xia, Y. Li, T. Miao, P. Li and L. Wang, Chem. Commun., 2016, 52, 11559 RSC; (c) L. Li, Q.-S. Gu, N. Wang, P. Song, Z.-L. Li, X.-H. Li, F.-L. Wang and X.-Y. Liu, Chem. Commun., 2017, 53, 4038 RSC; (d) L. Coudray and J. Montchamp, Eur. J. Org. Chem., 2008, 3601 CrossRef PubMed.
- (a) V. I. Timokhin, S. Gastaldi, M. P. Bertrand and C. Chatgilialoglu, J. Org. Chem., 2003, 68, 3532 CrossRef PubMed; (b) J. Pellon, J. Am. Chem. Soc., 1961, 83, 1915 CrossRef.
- (a) P. Yu, J.-S. Lin, L. Li, S.-C. Zheng, Y.-P. Xiong, L.-J. Zhao, B. Tan and X.-Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 11890 CrossRef PubMed; (b) P. Yu, S.-C. Zheng, N.-Y. Yang, B. Tan and X.-Y. Liu, Angew. Chem., Int. Ed., 2015, 54, 4041 CrossRef PubMed; (c) L. Huang, S.-C. Zheng, B. Tan and X.-Y. Liu, Org. Lett., 2015, 17, 1589 CrossRef PubMed; (d) L. Huang, S.-C. Zheng, B. Tan and X.-Y. Liu, Chem. – Eur. J., 2015, 21, 6718 CrossRef PubMed; (e) L. Huang, J.-S. Lin, B. Tan and X.-Y. Liu, ACS Catal., 2015, 5, 2826 CrossRef; (f) T.-T. Li, P. Yu, J.-S. Lin, Y. Zhi and X.-Y. Liu, Chin. J. Chem., 2016, 34, 490 CrossRef; (g) L. Li, Z.-L. Li, F.-L. Wang, Z. Guo, Y.-F. Cheng, N. Wang, X.-W. Dong, C. Fang, J. Liu, C. Hou, B. Tan and X.-Y. Liu, Nat. Commun., 2016, 7, 13852 CrossRef PubMed; (h) Z.-L. Li, X.-H. Li, N. Wang, N.-Y. Yang and X.-Y. Liu, Angew. Chem., Int. Ed., 2016, 55, 15100 CrossRef PubMed; (i) N. Wang, L. Li, Z.-L. Li, N.-Y. Yang, Z. Guo, H.-X. Zhang and X.-Y. Liu, Org. Lett., 2016, 18, 6026 CrossRef PubMed; (j) L. Li, Z.-L. Li, Q.-S. Gu, N. Wang and X.-Y. Liu, Sci. Adv., 2017, 3, e1701487 CrossRef PubMed.
- For selected reviews on the addition of 1,3-dicarbonyl C-centered radicals to alkenes, see: (a) B. B. Snider, Chem. Rev., 1996, 96, 339 CrossRef PubMed; (b) G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668 RSC. For selected recent examples, see: (c) L. Zhu, H. Chen, Z. Wang and C. Li, Org. Chem. Front., 2014, 1, 1299 RSC; (d) S. He, W. Yang, L. Zhu, G. Du and C.-S. Lee, Org. Lett., 2014, 16, 496 CrossRef PubMed; (e) H. Wang, L.-N. Guo and X.-H. Duan, Org. Lett., 2013, 15, 5254 CrossRef PubMed; (f) X. Wang, Z. Ma, J. Lu, X. Tan and C. Chen, J. Am. Chem. Soc., 2011, 133, 15350 CrossRef PubMed.
- M. Nechab, S. Mondal and M. P. Bertrand, Chem. – Eur. J., 2014, 20, 16034 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and characterization of compounds. See DOI: 10.1039/c8qo00734a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |