
Metal-free deoxygenative sulfonylation of quinoline N-oxides with sodium sulfinates via a dual radical coupling process†
Long-Yong
Xie
a,
Sha
Peng
a,
Fang
Liu
a,
Guan-Rong
Chen
a,
Wen
Xia
b,
Xianyong
Yu
b,
Wen-Feng
Li
c,
Zhong
Cao
c and
Wei-Min
He
 *a
*a
aHunan Provincial Engineering Research Center for Ginkgo biloba, Hunan University of Science and Engineering, Yongzhou 425100, China. E-mail: weiminhe2016@yeah.net
bKey Laboratory of Theoretical Organic Chemistry and Functional Molecule of Ministry of Education, Hunan University of Science and Technology, Xiangtan 411201, China
cHunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation, Changsha University of Science and Technology, Changsha, 410114, China
First published on 7th August 2018
Abstract
The first example of a metal- and reductant-free deoxygenative sulfonylation of quinoline N-oxides with sodium sulfinates via a dual radical coupling process is reported. In this reaction, sodium sulfinates play dual roles of a sulfonylation reagent and activating agent. This procedure is expected to complement the current methods for the radical reaction of quinoline N-oxides.
Introduction
Radical chemistry has played an increasingly important role in modern synthesis. The utilization of a controlled radical reaction to synthesize structurally diverse organic molecules has been of increasing interest in both academia and industry.1 In recent decades, a large number of radical reactions have been catalyzed by various transition metal complexes, which result in inevitable metal residuals in the terminal products and disposal of these metal salts often causes environmental pollution.2 Therefore, the development of metal-free radical reactions is of great importance and has been extensively studied over the past years.The C–H bond functionalization of quinoline N-oxides has received extensive attention in organic synthesis, as it has been demonstrated to be a powerful and versatile tool for direct incorporation of new functionalities at the C-23 and C-8 positions4 of quinoline skeletons with excellent atom economy. However, although the nucleophilic addition to quinoline N-oxides generating 2-substituted quinolines has been well established, direct construction of such motifs5via radical pathways remains a long-standing challenge. In fact, most radical reactions of quinoline N-oxides through C–H bond activation result in substituted quinoline N-oxides (Scheme 1a).6 These protocols require additional deoxygenative functionalization with superstoichiometric amounts of harmful reagents as reductants for obtainment of the expected 2-substituted quinolines. The one-pot synthesis of 2-sulfonylquinolines from quinoline N-oxides through C–H bond activation7 has attracted considerable attention due to their potential biological activities and valuable synthetic utilities as well as the availability of readily accessible quinoline N-oxides.8 However, to the best of our knowledge, the only example of direct synthesis of 2-sulfonylquinolines via deoxygenative radical sulfonylation9 of quinoline N-oxides in the presence of copper salts as the catalyst under an argon atmosphere was reported by Pan and Han (Scheme 1b).10 Considering the inconveniences and manufacturing costs in eliminating the trace metal contamination, especially for the late-stage functionalization of pharmacologically active compounds, a metal-free radical reaction for the synthesis of 2-sulfonylquinolines would be greatly desired. As part of our program devoted to the study of eco-friendly organic synthesis,11 herein, we report for the first time a facile metal- and reductant-free protocol for the direct construction of 2-sulfonylquinolines via a dual radical coupling process (Scheme 1c).
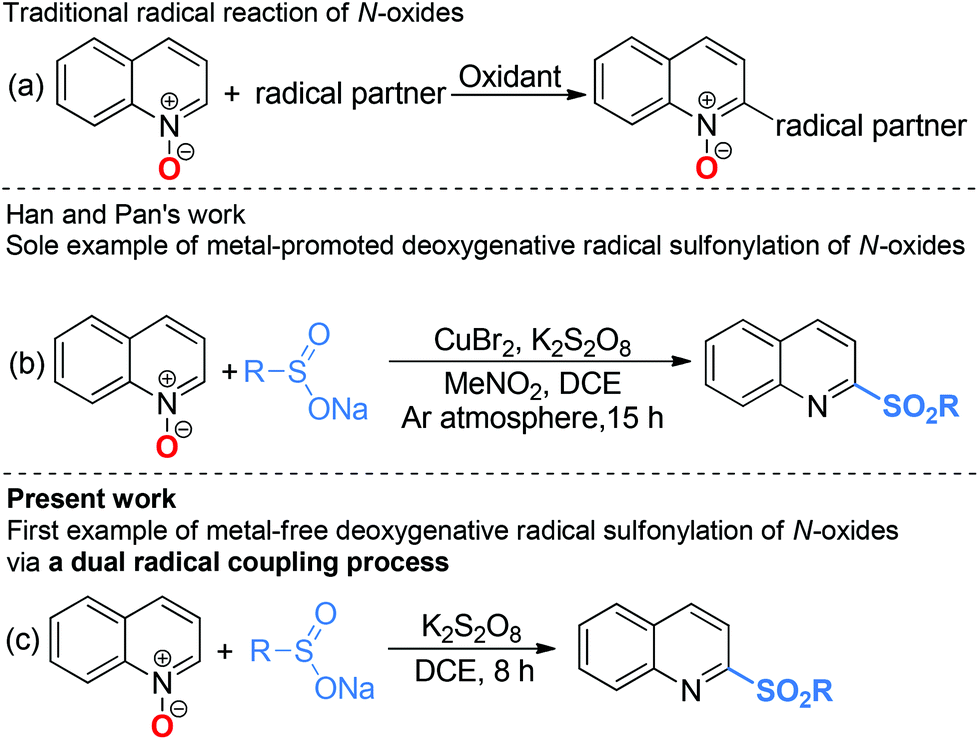 | ||
| Scheme 1 Radical coupling reactions of N-oxides. | ||
Results and discussion
Our investigation started with the cross coupling reaction of quinoline N-oxide (1a) and sodium p-toluensulfinate (2a, 2 equiv.) in the presence of K2S2O8 (1.2 equiv.) in 1,2-dichloroethane (DCE) at 100 °C for 8 h, and it led to the production of the desired 2-tosylquinoline 3aa in 55% yield based on 65% conversion of the starting material 1a (Table 1, entry 1). No 4-sulfonylquinoline was observed. Increasing the amount of oxidant to 2 equiv. resulted in the complete consumption of 1a and an 85% yield of 3aa (entry 2). However, further increasing the loading of K2S2O8 was not beneficial for the reaction (entry 3). Unexpectedly, inferior outcomes of the reaction were detected when employing related persulfate salt oxidants (entries 4–6), whereas a markedly lower yield of 3aa or no reaction was observed when organic oxidants were used (entries 7–12). The solvent also played a key role in the reaction outcome. Among the solvents examined, DCE proved to be the best for this reaction (entries 2, 13–17). Performing the transformation at a concentration of 0.1 M did not affect the reaction outcome; however, a higher concentration of 0.25 M was chosen that reduces solvent waste (entries 18 vs. 1). Further increasing the concentration of 1a resulted in a slightly decreased yield (entry 19). Increasing the reaction temperature from 100 °C to 110 °C did not improve the reaction outcome, whereas a distinct decrease in the yield of 3aa and the 1a conversion was detected when the temperature was decreased to 90 °C (entries 20 and 21). Furthermore, reducing the amount of 2a led to a decrease in the yield of 3aa (entry 22). No reaction occurred in the absence of K2S2O8, and the raw material 1a was quantitatively recovered (entry 23).|
|
||||
|---|---|---|---|---|
| Entry | Oxidant (equiv.) | Solvent (mL) | Temp. | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), oxidant, solvent, 100 °C, 8 h. b Estimated by 1H NMR using diethyl phthalate as the internal reference. c 1.5 equiv. of 2a were used. N.R: no reaction. | ||||
| 1 | K2S2O8 (1.2) | DCE (1) | 100 °C | 55 |
| 2 | K2S2O8 (2) | DCE (1) | 100 °C | 85 |
| 3 | K2S2O8 (2.5) | DCE (1) | 100 °C | 85 |
| 4 | Na2S2O8 (2) | DCE (1) | 100 °C | 68 |
| 5 | (NH4)2S2O8 (2) | DCE (1) | 100 °C | 61 |
| 6 | Oxone (2) | DCE (1) | 100 °C | 36 |
| 7 | PhI(OAc)2 (2) | DCE (1) | 100 °C | 21 |
| 8 | PCC (2) | DCE (1) | 100 °C | N.R. |
| 9 | m-CPBA (2) | DCE (1) | 100 °C | N.R. |
| 10 | TBHP (2) | DCE (1) | 100 °C | N.R. |
| 11 | H2O2 (2) | DCE (1) | 100 °C | N.R. |
| 12 | O2 balloon | DCE (1) | 100 °C | N.R. |
| 13 | K2S2O8 (2) | MeCN (1) | 100 °C | 52 |
| 14 | K2S2O8 (2) | MeNO2 (1) | 100 °C | 56 |
| 15 | K2S2O8 (2) | DMSO (1) | 100 °C | 32 |
| 16 | K2S2O8 (2) | DMF (1) | 100 °C | 41 |
| 17 | K2S2O8 (2) | THF (1) | 100 °C | N.R. |
| 18 | K 2 S 2 O 8 (2) | DCE (0.4) | 100 °C | 85 |
| 19 | K2S2O8 (2) | DCE (0.3) | 100 °C | 70 |
| 20 | K2S2O8 (2) | DCE (0.4) | 110 °C | 83 |
| 21 | K2S2O8 (2) | DCE (0.4) | 90 °C | 45 |
| 22c | K2S2O8 (2) | DCE (0.4) | 100 °C | 73 |
| 23 | — | DCE (1) | 100 °C | N.R. |
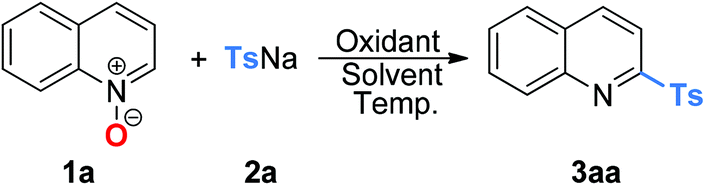
The optimal reaction conditions (Table 1, entry 18) were applicable for the oxidative coupling reaction of quinoline N-oxides and sodium sulfinates. As shown in Table 2, quinoline N-oxides bearing sterically hindered, electron-rich or electron-poor substituents in the quinoline ring all reacted well to yield the desired sulfonated products in moderate to good yields (3aa–3pa). Notably, a variety of functional groups are tolerated, including methyl (3aa–3ga), isopropyl (3ha), methoxy (3ia), phenyl (3ja), fluoride (3ka), chloride (3la and 3ma) and bromide (3na–3pa). When isoquinoline N-oxide 1q was employed as the substrate, a total 78% yield of the isomer products 3qa-1 and 3qa-2 was obtained in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. However, when pyridine N-oxide or quinoxaline N-oxide was used as the substrate, only a trace amount of the tosylation product could be detected.
:1 ratio. However, when pyridine N-oxide or quinoxaline N-oxide was used as the substrate, only a trace amount of the tosylation product could be detected.
| a All reactions were carried out in a sealed tube in the presence of 1 (0.3 mmol), 2 (0.6 mmol), K2S2O8 (0.6 mmol) and DCE (1.2 ml); isolated yields are reported. |
|---|
|
Sodium arenesulfinates with various synthetically useful functional groups present on the phenyl ring such as alkyl (Me and t-Bu), methoxyl, halo (F, Cl and Br), trifluoromethyl, acetyl, nitrile and phenyl were well compatible (3ab–3am). Regardless of their electronic characteristics, quinoline N-oxides coupled smoothly with sodium arenesulfinates bearing both electron-donating and electron-deficient substituents, to produce the expected products in moderate to excellent yields. Furthermore, the yield was slightly lower in the case of ortho-substituted sodium arenesulfinate (3am) than those obtained with the para-substituted ones, which might be due to steric factors. Moreover, di-substituted aryl sulfinates and 2-naphthalenesulfinates also afforded the corresponding compounds in good yields (3an and 3ao). When sodium thiophene-2-sulfinate or sodium pyridine-3-sulfinate was used as the substrate, no sulfonylation product was formed and the quinoline N-oxide starting material was quantitatively recovered. No sulfonylation reaction occurred when an aliphatic sodium sulfinate was employed as the substrate, which might be ascribed to the unstable aliphatic sulfonyl intermediate.
Finally, we investigated the scalability of this new sulfonylation reaction. More than 1 g of quinoline N-oxide 1a and sodium p-toluensulfinate 2a was subjected to the standard reaction conditions (Scheme 2). As anticipated, the reaction proceeded well by delivering 3aa in 78% yield.
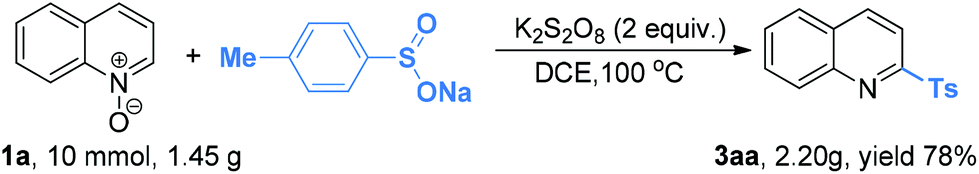 | ||
| Scheme 2 Gram-scale synthesis. | ||
To understand the reaction mechanism of the present oxidative coupling reaction, a series of control experiments were conducted. Firstly, no sulfonylation reaction occurred between quinoline 4a and 2a under optimal reaction conditions, which indicated that the N–O group played a key role in the transformation (Scheme 3a). Secondly, when 1a was treated under standard reaction conditions in the presence of 2 equiv. of radical scavenger (TEMPO or BTH) (Scheme 3b), the reaction was completely inhibited. To further verify whether the radical species is involved in the overall chemical process, electron paramagnetic resonance (EPR) experiments were carried out to gain insight into the radical coupling reaction. When K2S2O8 with 2a in DCE at room temperature was tested, no radical signal (g = 2.003, AN = 1.43 mT, AHβ = 1.36 mT, AHγ = 0.21 mT) was observed (Fig. 1a). An oxygen-centered sulfate radical could also be detected through treatment of K2S2O8 in DCE at 80 °C for 5 min (Fig. 1b). After the reaction of 2a and K2S2O8 in DCE at 80 °C, the signal of the sulfur-centered 4-methylbenzenesulfonyl radical (g = 2.002, AN = 1.54 mT, AH = 2.206 mT) was clearly observed (Fig. 1c).12 These observations suggested that both an oxygen-centered sulfate radical and a sulfur-centered 4-methylbenzenesulfonyl radical were generated in this reaction. 1H NMR analysis can provide valuable information about the reaction process. Pleasingly, a characteristic spectrum was obtained when the reaction mixture was investigated by 1H NMR. Both the S-p-tolyl 4-methylbenzenesulfonothioate (5a) and TsOH were detected by 1H NMR and MS (Scheme 3c). Further treatment of 1a with 5a and 6a with 2a under standard reaction conditions could not produce the desired 3aa (Schemes 3d and 3e), which indicated that 5a and 6a might not be the possible reaction intermediates. A clear kinetic isotope effect value (1.18) of the competitive intermolecular experiment involving a 1:1 mixture of 1a/[D1]-1a was observed (Scheme 3f), which revealed that the scission of the quinoline N-oxide C(sp2)–H bond may not be involved in the rate-limiting step. Treatment of 2-methylquinoline N-oxide 1r under standard reaction conditions could not deliver the sulfonylation product and the starting material 2-methylquinoline N-oxide was quantitatively recovered.
 | ||
| Scheme 3 Control experiments. | ||
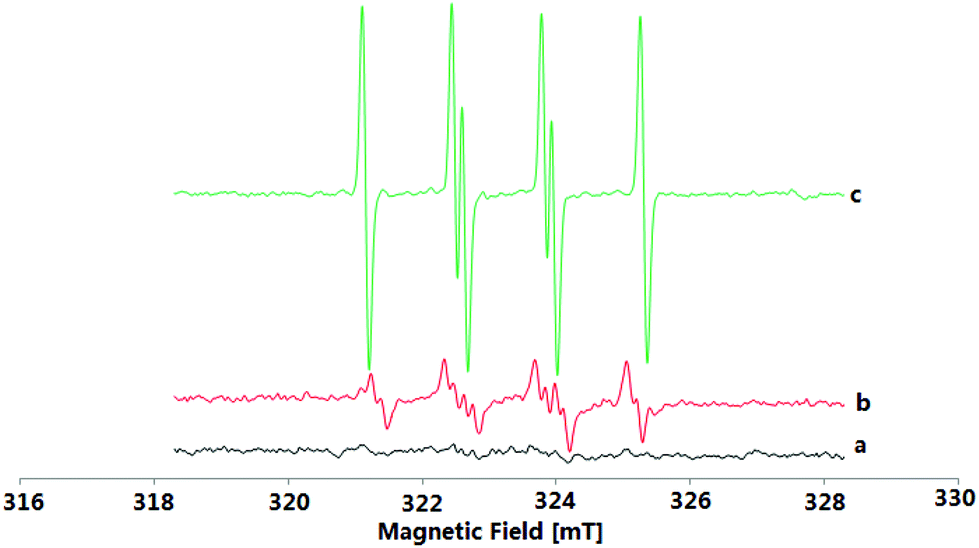 | ||
| Fig. 1 Electron paramagnetic resonance experiments (a) K2S2O8 + TsNa-DCE-RT-5 min; (b) K2S2O8-DCE-80 °C-5 min, g = 2.003, AN = 1.43 mT, AHβ = 1.36 mT, AHγ = 0.21 mT, an oxygen-centered sulfate radical; (c) K2S2O8 + TsNa-DCE-80 °C-5 min, g = 20056, AN = 1.39 mT, AH = 1.48 mT, a sulfur-centered 4-methylbenzenesulfonyl radical. | ||
According to the mechanism research in previous literature10 and the above experimental observation, a plausible reaction mechanism is proposed as shown in Scheme 4. First, in the presence of K2S2O8, sodium arenesulfinate 2 was easily transformed into an oxygen-centered radical A resonating with the sulfonyl radical Bvia single electron transfer (SET).9a,13 Subsequently, the radical B reacted with quinoline N-oxide 1 through a Minisci-like radical reaction to produce an intermediate C. Then, the intermediate C coupled with the oxygen-centered sulfate radical B to form intermediate D, which underwent an aromatization reaction to account for the formation of 2-sulfonylquinoline 3 with concomitant release of sulfonic acid.
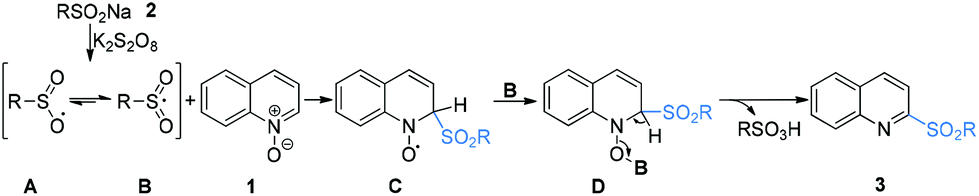 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In conclusion, we have reported the first example of a metal- and reductant-free deoxygenative sulfonylation of quinoline N-oxides with sodium sulfinates via a dual radical coupling process. Various functional groups in both quinoline N-oxides and arenesulfinates are accepted to afford a broad range of 2-sulfonylquinoline derivatives. In this reaction, sodium sulfinates play dual roles of an oxidant and activating agent. A possible mechanism is proposed on the basis of detailed mechanistic studies and suggests that in situ generation of sulfonyl radicals might be a key factor for obtaining deoxygenative sulfonated products. This procedure is expected to complement the current methods for Minisci-type radical-coupling reaction.Experimental
General procedure for the synthesis of compound 3
In a pressure tube were consecutively placed quinoline N-oxides 1 (0.3 mmol), DCE (1.2 mL), sodium sulfinates 2 (0.6 mmol) and K2S2O8 (0.6 mmol), and then the mixtures were heated to 100 °C. The progress of the reaction was monitored by TLC. The reaction typically took 8 h–12 h. Upon completion, the reaction was cooled to room temperature, and then water (5 mL) was added to the reaction mixture; it was extracted with CH2Cl2 (5 mL × 3) and the organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (eluent: petroleum ether/EtOAc = 10:1–4:1) to obtain 2-sulfonylquinolines 3.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the post-funded projects of Hunan University of Science and Engineering, the National Natural Science Foundation of China (No. 21273068 and 21545010), the Scientific Research Hunan Provincial Education Department (No. 17A081) and the 9th Science and Technology Innovation Program (2017) of China Hunan Provincial Science & Technology Department.Notes and references
-
(a) A. Studer and P. Curran Dennis, Angew. Chem., Int. Ed., 2015, 55, 58 CrossRef PubMed
; (b) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef PubMed
-
(a) L. J. Sebren, J. J. Devery and C. R. J. Stephenson, ACS Catal., 2014, 4, 703 CrossRef PubMed
-
(a) J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888 CrossRef PubMed
-
(a) J. Jeong, P. Patel, H. Hwang and S. Chang, Org. Lett., 2014, 16, 4598 CrossRef PubMed
-
(a) G. Li, C. Jia and K. Sun, Org. Lett., 2013, 15, 5198 CrossRef PubMed
-
(a) Z. Wu, C. Pi, X. Cui, J. Bai and Y. Wu, Adv. Synth. Catal., 2013, 355, 1971 CrossRef
-
(a) Z. Wu, H. Song, X. Cui, C. Pi, W. Du and Y. Wu, Org. Lett., 2013, 15, 1270 CrossRef PubMed
-
(a) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642 CrossRef PubMed
- Selected examples of radical sulfonylation reaction:
(a) D. Xia, Y. Li, T. Miao, P. Li and L. Wang, Chem. Commun., 2016, 52, 11559 RSC
- B. Du, P. Qian, Y. Wang, H. Mei, J. Han and Y. Pan, Org. Lett., 2016, 18, 4144 CrossRef PubMed
-
(a) Y. Pan, G.-W. Chen, C.-H. Shen, W. He and L.-W. Ye, Org. Chem. Front., 2016, 3, 491 RSC
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259 CrossRef PubMed
-
(a) D. Xia, T. Miao, P. Li and L. Wang, Chem. – Asian J., 2015, 10, 1919 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qo00661j |
| This journal is © the Partner Organisations 2018 |