
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom 3D to 2D zeolite catalytic materials
J.
Přech
ab,
P.
Pizarro
cd,
D. P.
Serrano
 cd and
J.
Čejka
*ae
cd and
J.
Čejka
*ae
aDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University, Hlavova 8, 128 43 Prague 2, Czech Republic
bNormandie Univ, ENSICAEN, UNICAEN, CNRS, Laboratoire Catalyse et Spectrochimie, 14000 Caen, France
cThermochemical Processes Unit, IMDEA Energy Institute, 28935, Móstoles, Madrid, Spain
dChemical and Environmental Engineering Group, ESCET, Rey Juan Carlos University, 28933, Móstoles, Madrid, Spain
eJ. Heyrovský Institute of Physical Chemistry, Czech Academy of Sciences, Dolejškova 3, 182 23, Prague 8, Czech Republic. E-mail: jiri.cejka@jh-inst.cas.cz
First published on 31st August 2018
Abstract
Research activities and recent developments in the area of three-dimensional zeolites and their two-dimensional analogues are reviewed. Zeolites are the most important industrial heterogeneous catalysts with numerous applications. However, they suffer from limited pore sizes not allowing penetration of sterically demanding molecules to their channel systems and to active sites. We briefly highlight here the synthesis, properties and catalytic potential of three-dimensional zeolites followed by a discussion of hierarchical zeolites combining micro- and mesoporosity. The final part is devoted to two-dimensional analogues developed recently. Novel bottom-up and top-down synthetic approaches for two-dimensional zeolites, their properties, and catalytic performances are thoroughly discussed in this review.
 J. Přech | Jan Přech obtained his bachelor's and master's (engineer's) degree from the University of Chemistry and Technology in Prague in 2012. For his PhD, he joined the group of Prof. Jiří Čejka at J. Heyrovský institute of physical chemistry of ASCR. He obtained PhD in physical chemistry in 2016 for a study on the synthesis and modifications of 2-dimensional titanosilicates in selective oxidation catalysis. Dr. Přech obtained the Jean-Marie Lehn prize in Chemistry 2016 for the best PhD thesis. Then Dr. Přech, joined the Laboratory of Catalysis and Spectrochemictry at Universite de Normandie in Caen (France) in the group of Dr. Valentin Valtchev working on the synthesis of metal–zeolite composite catalysts. Currently (2018) he is working on the CUCAM project at Charles University in Prague. His current research interests include synthesis, characterisation and application of Lewis acidic zeolite based catalysts, 2-dimensional zeolites and zeolite catalysed reactions in Fine Chemistry. |
 P. Pizarro | Patricia Pizarro is an Associate Professor of Chemical Engineering at Rey Juan Carlos University and a Senior Associate Researcher at IMDEA Energy Institute. She obtained her PhD from Rey Juan Carlos University (2005) and an Extraordinary Doctorate Award. She was a Visiting Researcher at Laboratoire d'application de la Chimie a L'environnement (France, 2003) and MPIK (Germany, 2006). Her research is mainly focused on the design of heterogeneous catalysts and materials for different chemical processes such as energy storage and biofuels generation. She has participated in 27 research projects, and is the co-author of 54 indexed publications (H-index = 23), 6 book chapters and of >90 congress communications as well as the supervisor of 2 PhD theses. |
 D. P. Serrano | D. P. Serrano is a Full Professor of Chemical Engineering at Rey Juan Carlos University (since 2002) and the Director of the IMDEA Energy Institute (since 2007). He obtained his PhD from Complutense University of Madrid (1990). He was a Visiting Researcher in CALTECH (1991) and UCSB (2006). He was the Vice-rector for Research and Technological Innovation (2001–2002) and the Head of the Chemical and Environmental Technology Department (2002–2007) at Rey Juan Carlos University. He has participated in 60 research projects, and he is the author of >200 indexed publications (= 47) and of >300 congress communications as well as the supervisor of 21 PhD theses. He was the Coordinator of the FP7 EU CASCATBEL project (2013–2017) and the President of the Spanish Zeolite Group (since 2015). |
 J. Čejka | Prof. Jiří Čejka is currently employed at the J. Heyrovský Institute of Physical Chemistry and holds the position at the CUCAM project at the Faculty of Science, Charles University in Prague. In 2005 he chaired the 3rd FEZA Conference on Zeolites in Prague and is an organizer of Workshops on zeolites and catalysis, e.g. the EFCATS School on Catalysis in 2018. His research interests include synthesis of zeolites and nanoporous materials, sorption, and catalysis. Jiří Čejka is the co-author of 310 papers with more than 9200 citations, co-editor of 6 books, and has H-index = 52. |
1. Introduction
Zeolites are widely used in petroleum and chemical industries due to their exceptional catalytic and selective adsorption properties in combination with thermal and chemical stability as well as environmental friendliness. At present, the dominant role of zeolites (as catalysts) is in crude oil upgrading. Most petrochemical processes employ zeolites, in particular for shape-selective reactions. Recently, a number of exciting examples of highly active zeolites have been shown in selective synthesis of fine chemicals and environmental protection.1–3The name zeolite, coined in 1756 and meaning ‘boiling stone’, refers to a diverse but uniform class of natural and synthetic crystalline aluminosilicates and related compositions with framework structures containing well-defined micropores, which are able to adsorb and discriminate molecules based on their size and shape. Zeolites crystallize under hydrothermal conditions from suitable synthesis mixtures. There is a continuous effort in numerous academic and industrial laboratories to discover new framework structures for particular processes and to gain a better fundamental understanding of synthetic mechanisms.
Despite a significant effort in the synthesis of zeolites, there are still limitations in increasing their pore sizes. While some zeolites have entrance windows consisting of up to 30 T-atoms (silicon or other atoms with tetraheral coordination in the framework),4 the largest industrially relevant windows are formed by 12 T-atoms in the case of zeolite FAU. This strongly limits possibilities of using zeolites for transformation of larger (bulkier) substrates. As a result, several important approaches have been recently developed to overcome this limitation. All of them target at an increased accessibility of active sites in zeolites but in different ways. In the case of nanozeolites, size limitation of zeolite crystals to dozens of nanometers increases the external surface of zeolite crystals and decreases diffusion paths inside the crystals. In contrast, hierarchical zeolites consist of both micropores and mesopores facilitating diffusion of reactants and products.5–7 Both bottom-up and top-down approaches have been described providing hierarchical zeolites with improved catalytic properties. While traditional zeolites have been conceptualized as 3D networks, a recent breakthrough revealed their formation as crystalline layered solids,8–11 which can lead e.g. to creation of expanded architectures. Fig. 1 summarizes the major advances achieved in tailoring the properties of zeolites.
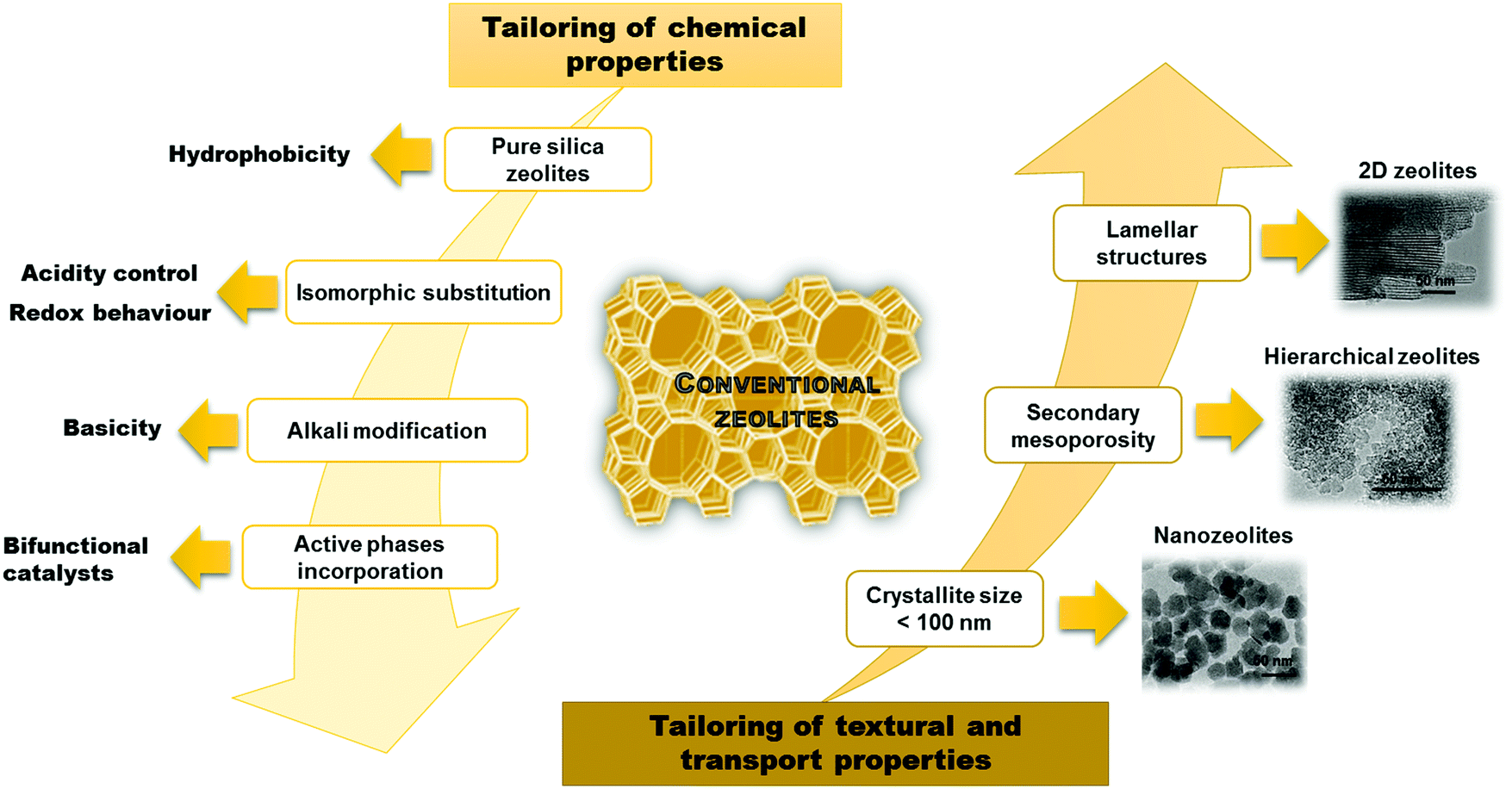 | ||
| Fig. 1 Main strategies applied for tailoring the zeolite properties. TEM pictures adapted with permission from ref. 12–14. | ||
The aim of this review is to discuss the relationship among synthesis – properties – catalytic performance of three-dimensional (3D) zeolites including nanozeolites and hierarchical ones in comparison to the variety of forms with two-dimensional (2D) structures (Fig. 1). Similarities and differences in chemical and textural properties of different zeolites are highlighted based on different examples of catalytic performance including cracking reactions, biomass transformations, shape selective aromatic chemistry and fine chemical synthesis.
2. Classical three-dimensional zeolites
2.1. Synthesis
Although the first known zeolites were naturally occurring minerals, those having currently the most relevant applications are synthetic zeolites, which have been discovered and developed in laboratory research.15 Synthetic zeolites are typically obtained by crystallization of gels containing water, silica and alumina sources (other metals) under hydrothermal conditions at basic pH, temperatures in the range 60–200 °C and autogenous pressure. The synthesis gel usually contains also basic agents and alkali cations, as well as organic compounds that may act as structure-directing agents.16The Si/Al ratio is a key parameter of the synthesis as it may determine the type of zeolite structure being formed, its crystallinity, as well as the kinetics of the crystallization process. In general, the Si/Al ratio of the zeolite is lower than that of the gel. A variety of silica and alumina sources can be employed including polymeric and monomeric reagents.17
Due to the low solubility of silica and alumina in water, mineralization agents, like NaOH or KOH, must be added to the gel in order to solubilize the precursors and promote the formation of Si–O–Si and Si–O–Al bonds through condensation reactions. The alkali concentration is also a key parameter of the synthesis that must be conveniently balanced. Similarly, severe crystallization conditions, such as high temperature and long time, favour the formation of more thermodynamically stable phases.
Stirring also has a strong influence on zeolite crystallization. It may be accomplished in different ways, such as using mechanical agitators placed internally or by rotating the crystallization vessel. Many zeolite syntheses carried out in the laboratory proceed under static conditions, which may lead to a non-uniform composition of the gel and contamination. In general, stirring causes the crystallization to take place with a more favourable kinetics, leading to the formation of smaller crystals with a narrower size distribution.17
The crystallization mechanism of zeolites is usually described by a classical combination of nucleation and crystal growth steps.18,19 An amorphous solid phase (hydrogel) is often formed at the beginning of the crystallization. In the solution-mediated mechanism, both nucleation and crystal growth are assumed to proceed just with the participation of soluble species, whereas any amorphous solid phase, that could be present, acts as a reservoir of nutrients. In contrast, in the hydrogel-mediated mechanism the zeolite crystallization is considered to occur by the reorganization of the amorphous solid phase formed during the early stages of the hydrothermal treatment. An intermediate scheme, which tries to reconcile the previous ones, proposes nucleation to occur within the hydrogel, whereas the crystal growth would take place by incorporation of soluble species. Taking into account the great diversity of gel compositions, synthesis conditions and zeolite structures, it is a majority opinion that a general zeolite crystallization mechanism does not exist.
Incorporation of organic compounds into the synthesis gel has been instrumental in the discovery of new zeolite structures (mainly with high silica compositions) over the past few decades revealing their role as structure-directing agents.2,17 They influence the crystallization process via modification of the gel chemistry, as pore filling agents or templates. In the last case, a very high specificity exists between the organic template and the zeolite structure. This is, for instance, the case of tetrapropylammonium cation and the MFI zeolitic structure due to the almost perfect fitting of the organic molecule and the zeolite microporosity.16
Seeding has been widely applied in zeolite synthesis to accelerate the crystallization process as it provides an external source of nuclei, thus reducing the induction period associated with the nucleation step. Seeding is also effective for the organotemplate-free synthesis of zeolites, which has important advantages in terms of reagent cost reduction and avoiding the zeolite calcination treatment for removal of organics.20,21
The presence of water is necessary for zeolite crystallization as it makes possible the solubilisation of the Si and Al precursors and is deeply involved in the gel chemistry. However, an excess of water may have also undesired effects, such as a decrease in the zeolite yield and generation of aqueous waste streams. Accordingly, a number of methods have been developed to minimize the water content during the zeolite crystallization. These are dry-gel conversion, vapour-phase transport and steam-assisted synthesis.22 Zeolite synthesis has also been conducted by hydrothermal treatment of wetness-impregnated xerogels.23,24 These non-classical approaches have been reviewed together with contributions related to green routes for the synthesis of zeolites in a recent work.25
An alternative to hydroxide based syntheses is the use of fluoride-containing media. It allows the crystallization of zeolites at neutral or even slightly acidic pH.26–28 Under these conditions, fluoride anions replace hydroxide anions as mineralizers and the solubility of silica increases significantly due to the formation of hexafluorosilicate species. Typically, zeolite synthesis in fluoride media leads to large crystals with a lower concentration of defects and highly hydrophobic properties.
Microwave heating has been employed for zeolite synthesis, dramatically reducing synthesis times compared to conventional heating.25 Moreover, microwave heating has been very effective for the preparation of zeolite membranes, enabling the control of a number of properties of the membrane i.e. morphology, orientation and permeation characteristics.29 Ultrasound application in zeolite synthesis has also resulted in shorter crystallization times, as it contributes to a reduction of the induction period. Moreover, ultrasonication influences the size and morphology of the crystals and improves the crystallinity of the zeolite.30
Recently, the ADOR (assembly–disassembly–organisation–reassembly) methodology has been developed for the preparation of novel zeolite structures that could not be obtained by conventional hydrothermal synthesis.31 This approach is discussed in Section 5.3.
2.2. Zeolite structures and properties
According to the classical definition, zeolites are crystalline microporous aluminosilicates with the general formula M2/nO·Al2O3·ySiO2, where “n” represents the valence of the cation M. The structure of the zeolites is formed by a 3D arrangement of TO4 tetrahedra (T corresponds to Si and Al atoms), connected by oxygen bridges, which generates an open framework with pores and cavities accessible to molecules having kinetic diameter small enough to penetrate into the zeolite channels.Taking into account all the possible arrangements of the TO4 units, a great number of zeolitic structures could be generated.32 However, in practice, just a relatively small number of structures have been synthesised. Nevertheless, the number of known zeolites is increasing continuously and by April 2018, the International Zeolite Association (http://www.iza-online.org/) has recognized 235 known structures, represented by three-letter framework codes.
The pore sizes existing within zeolite structures are mostly below 1 nm, so they are clearly within the micropore range (<2 nm). The size of these channels is directly related to the number of T atoms present in the pore rings. They are usually classified as small-pore zeolites (8-rings), medium-pore zeolites (10-rings), large-pore zeolites (12-rings) and extra-large pore zeolites (>12-rings). Accordingly, the pore size is typically specified by its minimum and maximum dimensions. In the case of zeolites with multidimensional pore systems, the occurrence of intersections between the channels has a very positive effect on their catalytic behaviour as this attenuates significantly their deactivation by pore blockage when coke is deposited.
Classical aluminosilicate zeolites are known by their acidic and cation-exchange properties, which derive from the extra negative charge of the framework associated with the Al tetrahedra that needs to be balanced by external counter-cations. The counter-cations can be exchanged by contacting the zeolite with a solution of another cation until reaching the equilibrium between the zeolite and the liquid phase. Typically, zeolites are synthesized in sodium or potassium form (related to the mineralisation agent) that can be transformed into the protonic form by ion-exchange with ammonium salts followed by calcination to remove ammonia. Acidic zeolites, prepared in this way, are used in a wide variety of acid-catalysed reactions.33,34 In principle, acidity in zeolites arises from the presence of Brønsted acid sites directly linked to Al atoms in tetrahedral framework positions (see Section 6.1.1); however, zeolites may also contain significant amounts of Lewis acid sites, formed by dehydration of the Brønsted sites or linked to extra-framework Al species which can be formed during the synthesis or calcination treatments. The ratio between Brønsted/Lewis acid sites typically decreases for low Si/Al ratios in the framework. Thus, whereas high silica zeolites exhibit mainly Brønsted type acidity, both Brønsted and Lewis acid sites are present in significant concentrations in zeolites with low Si/Al ratios. It is remarkable that, compared to amorphous aluminosilicates, the acid sites in zeolites are stronger and more uniform, which is a direct consequence of the crystalline nature of zeolites. Various theoretical methods and experimental techniques have been developed and applied for the characterization of the zeolite acid sites35,36 (see Section 6.1).
Although quite less developed than the acidic features, zeolites may also present basic properties.37,38 This is the case of zeolites ion-exchanged with alkali cations, as well as of zeolites containing occluded basic metals and metal oxides. Thus, reactions such as alkylation of toluene with methanol, cyclo-addition of carbon dioxide to ethylene oxide, isomerization of 1-butene, and alkylation of toluene with ethylene occur e.g. over basic X zeolites.
The size of the zeolite micropores is on the level of molecular dimensions of small organic molecules. As a result, they exhibit so-called shape selectivity and molecular sieving effects.39–41 A zeolite may discriminate between molecules having just a small variation in their size or shape, so the larger molecule cannot penetrate the zeolite micropores or it diffuses very slowly through channels while the smaller one can diffuse substantially faster. As a result, great differences have been observed in the uptake and reactivity of compounds such as xylenes or n- vs. iso-alkanes. The concept of shape selectivity was introduced in the 1960s, being initially classified according to three types: reactant, product and transition state shape selectivity depending on which of the species the most important steric restriction is related to.40,41 Subsequently, a number of terms have been coined closely related to shape selectivity effects, such as molecular traffic control, surface pocket catalysis, and pore-mouth selectivity.
2.3. Zeolite modification
While traditional zeolites were known as aluminosilicates, it was realized early on that there is a possibility of preparing some of the zeolitic structures in pure silica form. Resulting materials possess almost no acidity or ion-exchange capacity, but the objective was to use them as supports for other catalytically active phases or in separation and purification processes taking advantage of their high hydrophobicity and molecular sieving effects.42,43 However, the number of zeolitic structures, which have been prepared as pure silica zeolites, is relatively limited since the presence of Al species in the gel is essential in many cases for the synthesis of the desired zeolite with high crystallinity. Thus, for instance, the pure silica form of zeolite Beta was difficult to synthesize in alkaline media, which was achieved instead by the fluoride route.44Increasing the Si/Al ratio has been commonly employed as a method for enhancing the zeolite stability. For low silica zeolites this is achieved by post-synthesis treatments that cause the extraction and removal of Al species. This is the case of the USY (ultrastable Y) zeolite, prepared usually by steam treatment to extract a great part of the Al from the structure followed by washing the extra-framework species with dilute acid.45 USY is a remarkable catalyst used in the cracking of heavy oil fractions in the FCC process.46
A variety of metallosilicates with a zeolite structure have been prepared through direct synthesis by isomorphous substitution of the Al3+ atoms by other trivalent cations, such as Sb3+, B3+, Ga3+ and Fe3+. These zeolitic materials contain Brønsted acid sites with a diversity of strengths, which have a remarkable effect on their catalytic properties in many reactions. Thus, antimonosilicate, borosilicate, ferrisilicate, and gallosilicate with a MFl structure have been found to exhibit much higher para-selectivity for the alkylation of ethylbenzene with ethanol than the HZSM-5 zeolite, since the weaker acid sites of the metallosilicates catalyse to a lesser extent the undesired isomerization of p-diethylbenzene.47 Likewise, zeolites containing Ga3+ in the framework have shown excellent catalytic properties in alkane aromatization,48 whereas zeolites with Fe3+ incorporated into them have exhibited a remarkable catalytic activity for NOx decomposition.49
Replacement of Al by tetravalent cations has yielded materials with Lewis acidity, showing catalytic properties very different from those exhibited by the corresponding aluminosilicates. Thus, incorporation of Ti4+ into zeolitic frameworks opened the way for the development of materials, like titanium silicalite 1 (TS-1) zeolite, with redox properties and remarkable catalytic performance in partial oxidation reactions.50,51 Moreover, zeolitic catalysts containing Sn, Zr, Hf or Nb atoms in the structure have shown remarkable properties in a number of biomass conversion reactions due to the unique Lewis acid character of the corresponding tetrahedral metal sites, which promotes the activation and conversion of oxygen-containing molecules.52 Last but not least, isomorphous incorporation of Ge4+ opened a way towards a number of large and extra-large pore zeolites53 and in addition some of these zeolites can be transformed into others via the ADOR mechanism (see Section 5.3).
Zeolites have also been employed as supports for a wide variety of active phases in order to modify and complement their properties.54–59 These active phases include metals, metal oxides, metal salts and metal complexes, generating materials with multifunctional properties and applications in a great variety of reactions in various sectors such as oil refining, petrochemistry, fine chemicals, waste valorization, pollutant removal in gaseous and liquid streams, biofuel production, etc.
Finally, a huge research effort has been made aiming to tailor zeolite properties directly related to their porosity and accessibility of the active sites in order to improve their catalytic behaviour, mainly in reactions involving bulky substrates that are controlled by steric and diffusional limitations. Accordingly, a new type of zeolitic material showing enhanced accessibility and external/mesopore surface area has been developed, which includes nano-sized, hierarchical and 2-dimensional zeolites, as reviewed in the following sections.
3. Nanozeolites
The intrinsic microporosity of zeolites, with uniform pore shapes and narrow size distributions, provides excellent properties as molecular sieves and as shape-selective catalysts. However, it is their main weakness when zeolites need to process bulky compounds that cannot access the internal active sites. This limitation has motivated the worldwide intensive research attempting to develop zeolitic materials with enhanced accessibility.60Decreasing the crystal size to the nanometer scale is an effective way to enlarge the external surface area and the ratio of external/internal active sites, with the consequent improvement of their accessibility to bulky molecules. Besides, the diffusion path lengths are shortened enhancing the mass transport of reactants/products. According to these modifications, the catalytic applications of zeolites can be expanded to those reactions involving large molecules. Other benefits reported include an increased lifetime and a higher tolerance to coke than conventional zeolites.60,61
Among the different crystal sizes obtainable within the category of nanozeolites, there is a special interest on reaching values below 100 nm due to the unique properties they provide.60 Below this size, nanozeolites can be obtained in the form of stable colloidal suspensions with narrow particle size distributions, which are excellent precursors for the preparation of either membranes, films, composites or hierarchical structures, with potential applications in a variety of fields such as heterogeneous catalysis, molecular separation, ion exchange, chemical sensors and medicine.12 There are several reviews presenting the latest advances in the study of synthesis strategies, mechanisms of formation, properties, applications and types of zeolites prepared as nanocrystals.12,60–64 Traditional synthesis methods to obtain zeolite nanocrystals use clear solutions or gels containing zeolite precursors. Clear solutions are recommended when stable suspensions with smaller and narrow particle size distribution are desired. The synthesis conditions must be carefully controlled in order to promote nucleation over crystal growth. For instance, it is preferable to work at low synthesis temperatures, since the activation energy of nucleation is generally lower than that of crystal growth.12,65,66 In order to avoid aggregation of zeolite nanocrystals, large concentrations of organic structure-directing agents (OSDA) are employed, whereas the alkali cation content is reduced as such cations neutralize the negatively charged (alumino)silicate sub-colloidal particles. Moreover, if the objective is to attain a colloidal suspension of nanocrystals, the as-synthesized suspension must be subjected several times to the sequence of high-speed centrifugation–redispersion in a liquid (using an ultrasonic device).
It can be inferred that traditional synthesis strategies of nanozeolites present serious difficulties to reach the industrial viability due to the large amounts of OSDA employed, together with the long synthesis times and low mass zeolite yields. Likewise, removal of the organic templates from as-synthesized nanozeolites is done by calcination at relatively high temperatures, which may cause aggregation of nanocrystals with some loss of size homogeneity and external surface area, or even destruction of the zeolite structure.63 For these reasons, template-free strategies have been explored. The generation of small and homogeneous nanocrystals is more successful under mild synthesis conditions, vigorous stirring, and crystallizations in several steps. One of the first zeolites successfully prepared in the absence of an organic template was FAU-type, in particular Y and X zeolites.67–69 Control of both the amount of water in the initial gel and temperature of crystallization, carried out in 2 or 3 stages, allowed obtaining individual particles as small as 30 nm.67,68 Quite recently, the low-cost synthesis of NaX nanozeolites by the hydrothermal method without organic template addition and using agricultural wastes, in particular stem seep ash (SSA), as silica source has been reported.70,71 Aggregates of nanoparticles with a mean size below 70 nm were attained from a clear solution hydrothermally treated at 60 °C and containing sodium aluminate, sodium hydroxide, water and silica extracted from SSA. Rice husk ash (RHA) has been also used as a low-cost source of silica by Ng et al.72 to synthesize EMT-type nanocrystals by an organic-free hydrothermal route. Highly crystalline EMT-type nanocrystals with a mean size of 15 nm, narrow particle size distributions and relatively high yields based on silica (75 wt%) were obtained after 28 h of hydrothermal crystallization at 28 °C under strongly basic conditions.
The use of organic structure-directing agents can also be avoided by seeding the initial synthesis gel with small amounts of zeolite seeds, as is the case reported for Mordenite,73 Beta74 and ZSM-5 zeolites.75,76 Together with the synthesis conditions, the amount of seeds introduced influences the final crystallite sizes. For instance, when the seeding was applied to prepare nanosized ZSM-5, it was observed that lower synthesis temperatures and higher seed concentrations led to smaller nanoparticles, with values ranging from 140 and 230 nm depending on the conditions.76 Moreover, the solid yields were significantly higher (80%) than the typical values corresponding to the traditional synthesis routes. More examples regarding the use of seeds on the preparation of nanozeolites can be found in the review literature.61
Many other approaches have been proposed for the preparation of nanosized zeolites in the absence of organic templates.60,61 Some of them are modifications of the traditional synthesis procedures, such as replacing conventional heating with microwave-irradiation,29,77,78 fluid microreactors79,80 or inert solid matrixes as templates (confined-space synthesis).81–83 Top-down strategies have also been reported, as is the case of milling the primary zeolites with micro-meter sizes, usually followed by recrystallization.84–86 Nevertheless, top-down routes are characterized by broader particle size distributions.
After appropriate post-synthesis treatments (intensive washing and pH neutralization), nanosized zeolites are usually very stable in different solvents. This allows their further processing to final shapes or macroscopic structures depending on their application (Fig. 2). For sorption and catalytic applications, nanosized zeolites are usually moulded as extrudates with different shapes (spheres, cylinders) with the aid of binders (silica or alumina). For optical devices and chemical sensors, thin-to-thick layers of nanozeolites can be created by different routes, including screen-printing, sol–gel techniques, dip- or spin-coating and direct growth of the substrates. Zeolites are also usually employed to fabricate membranes for gas–vapour and liquid–liquid separation processes.64
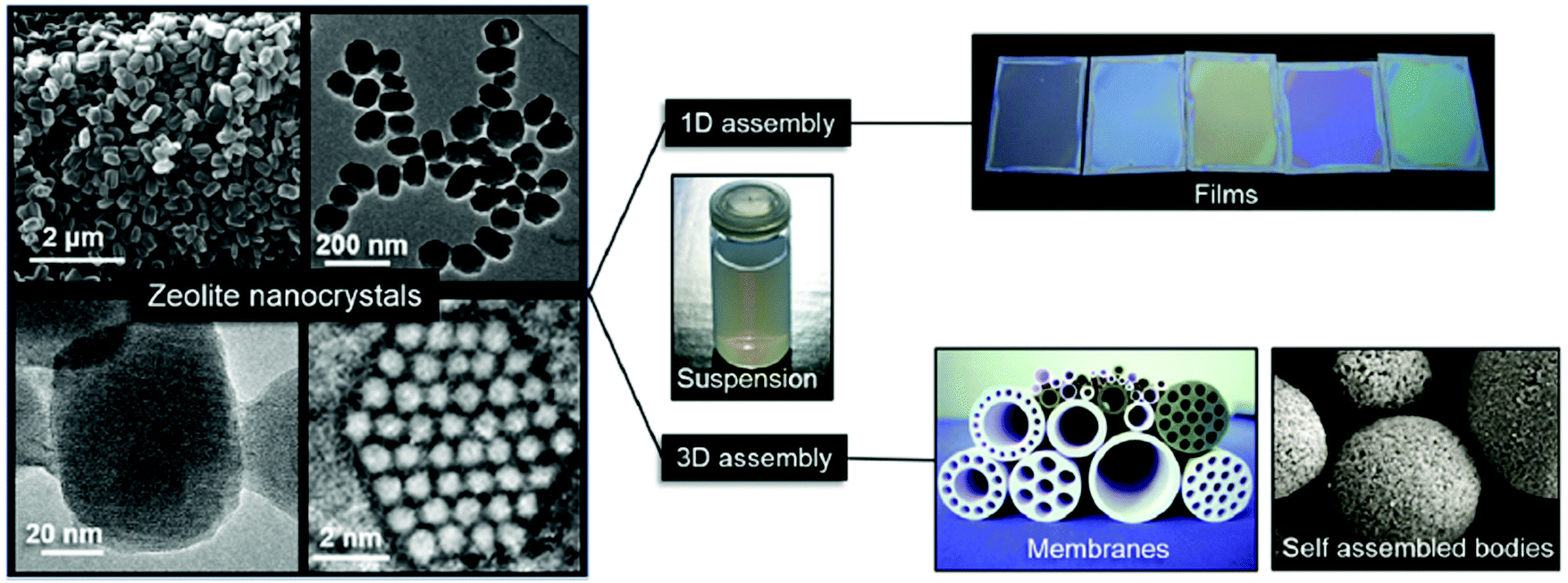 | ||
| Fig. 2 Zeolite nanocrystals with diverse morphologies and sizes in colloidal suspensions and examples of processed forms as self-supported shapes, porous membranes, and optical quality films. Reproduced from ref. 64 S. Mintova, J. Grand and V. Valtchev, C. R. Chim., 2016, 19, 183–191. Copyright (c) 2016, published by Elsevier Masson SAS. All rights reserved. | ||
At present, there exist around 20 types of zeolites synthesized at the nanoscale (including SOD, LTA, MTW, MEL, FAU, EMT, MFI, BEA, MOR),64 but it is presumable that this number will increase progressively. The challenge is to develop efficient strategies to stabilize the nanocrystals into small (<100 nm), discrete and homogeneous particles as well as to improve the control of their physico-chemical properties. The success on these targets will allow their expansion into new applications in different industrial fields, including food, cosmetics, medicine and nanotechnology.
4. Hierarchical zeolites
The term “hierarchical zeolite” was first applied at the beginning of the XXI century to zeolitic materials possessing a bimodal pore size distribution,87–89 consisting of zeolitic micropores and a secondary porosity (usually in the range of mesopores). This mesoporosity can be generated by a variety of top-down or bottom-up synthesis strategies.5 Some authors also call these materials as “mesoporous zeolite”,90,91 which can be somewhat misleading as it may suggest the presence of only mesopores, while in reality they still contain a great proportion of micropores. Accordingly, the term “hierarchical zeolites” seems to be more convenient.Hierarchical zeolites exhibit enhanced accessibility of the active sites, faster mass transport of molecules and they are usually more resistant against coke deactivation. Therefore, they often show a higher catalytic activity than conventional zeolites, in particular in those reactions suffering from steric and/or diffusional limitations. Moreover, the secondary porosity provides an ideal space for the incorporation and grafting of other components and phases, opening a great diversity of routes for the preparation of multifunctional materials.
In the past 15 years, a large number of original papers have been published on the synthesis, properties and catalytic applications of hierarchical zeolites, which are now briefly reviewed. It must be noted that several review articles have also been published on this emerging and relevant topic.60,92–98
4.1. Synthesis strategies for the preparation of hierarchical zeolites
The methods reported for the preparation of hierarchical zeolites were classified as bottom-up and top-down approaches, depending on whether the creation of the secondary porosity is induced during or after the zeolite crystallization.5 However, this classification is rather simplistic taking into account the great diversity of synthesis strategies that have been developed in the past few years, such as dealumination, desilication and degermanation treatments, fluoride etching, zeolitization of preformed solids, assembly of zeolite nanocrystals, hard-templating by carbon materials, use of organosilanes, templating by polymers and addition of surfactants to the synthesis gel. Those routes that have been more widely applied in the literature are briefly commented below.Dealumination is a classical treatment of low silica zeolites that very often provokes the development of mesoporosity, being carried out by steaming at elevated temperatures and acid leaching.99 This treatment has been traditionally applied to zeolite Y to remove a part of the aluminium in the zeolite framework, thus increasing its hydrothermal stability aimed to improve its performance when used as a catalyst in fluid catalytic cracking (FCC), which has also been noted to create some mesoporosity.
Degermanation is a method based on the liability of Ge–O bonds, which are usually located anisotropically in the framework of some microporous germanosilicates.100–102 This method is discussed in more detail in Section 5.3 dedicated to the ADOR mechanism.31
Desilication is based on the treatment of zeolites with a base (usually sodium hydroxide), which causes creation of mesopores due to preferential removal of silica from the framework.103 The alkali treatment can also be performed using organic bases (e.g. tetrapropylammonium hydroxide and tetrabutylammonium hydroxide) allowing mesopore volume to be optimized,104 since these organic cations act as pore growth moderators. Desilication is a quite versatile method as it has been successfully applied to a large number of zeolite structures,105 although a significant destruction/dissolution of the zeolite structure can happen.
Fluoride etching is somewhat similar to desilication; however, it is not selective to silicon or aluminium (or any other element). It starts from the boundaries of intergrown crystals or crystal domains, thus separating twins into single crystal parts.106,107 Under certain conditions, the formation of uniform rectangular mesopores was observed yielding a material with an Emmental cheese-like morphology, but being fully crystalline and with the same acidic properties as the parent one.108
Hard-templating by carbon materials is another route widely employed for the synthesis of hierarchical zeolites. In this approach, the zeolite crystallizes around and/or within particles of a carbon matrix, which is removed by combustion after the formation of the zeolite crystals. Thus, mesopores are generated between or inside the zeolitic entities. A diversity of carbon sources have been employed, including active carbons (both micro- and mesoporous), colloid imprinted carbons, carbon nanoparticles, multi-walled carbon nanotubes (MWNT), carbon aerogels and ordered mesoporous carbons (CMK-3).109–115 In the case of carbon nanoparticles, a substantial effect of microwave regime has been documented.116
Hierarchical zeolites have also been prepared starting from pre-formed solids, which are then subjected to crystallization into zeolites, preserving their initial shape and meso-/macro-porosity. The crystallization is performed by the Vapour Phase Transport (VPT) method, in which a mixture of water and structure directing agents is vaporized and brought into contact with the dry gel. Alternatively, Steam-Assisted Conversion (SAC) can be applied for promoting the zeolitization process. In the SAC route only water is vaporized and the structure directing agents (non-volatile compounds) are already included in the solid gel. These approaches have been used for the preparation of hierarchical zeolites from different types of solid precursors, such as dry gels,117 silica-based nanoparticles,118 mesostructured solids,119 or macroscopic hierarchical amorphous solids.120,121
Different types of polymers (cationic, amphiphilic and block polymers) have been added to the zeolite synthesis gel to act as porogens of the secondary porosity. Thus, mesoscale polydiallyldimethylammonium chloride has been used for the preparation of hierarchical Beta zeolites,122 whereas polystyrene spheres have been employed for obtaining hierarchical zeolites with a microporous/macroporous pore structure.123 Likewise, ZSM-5 single crystals with b-axis-aligned mesopores have been prepared using a designed cationic amphiphilic copolymer as a mesoscale template.124 In the same way, hierarchical zeolite Y has been directly synthesized using a block copolymer (Pluronic F127) as the template, which guides the aluminosilicate gel to assemble into a zeolite with significant mesoporosity.125
Organosilane-based methods have also been reported as very effective strategies to develop mesoporosity in zeolites.126 They are based on the addition of different types of organosilanes (simple organosilanes, silylated polymers or amphiphilic organosilanes) to the zeolite synthesis gel in order to perturb the crystallization process, causing the generation of secondary porosity.127–132 Their effect is noticeable even when incorporating relatively small amounts of the organosilane. Amphiphilic organosilanes become linked to the aluminosilicate species in the gel, promoting their arrangement around the formed amphiphile micelles. In the case of using simple organosilanes or silylated polymers, these reagents are anchored on the external surface of protozeolitic nano-units, partially hindering their aggregation and, therefore, the growth of standard zeolite crystals (Fig. 3). In all cases, the removal of the organosilane generates the mesoporosity finally present in the hierarchical zeolite.
 | ||
| Fig. 3 TEM images of hierarchical ZSM-5 prepared using organosilanes in the synthesis gel.133 | ||
Hierarchical zeolites have also been prepared by surfactant-assisted crystal rearrangement of the zeolite framework.134–137 Thus, dealuminated zeolite Y was treated with an NH4OH/surfactant (cetyltrimethylammonium bromide) solution, followed by heating at 150 °C under autogenous pressure.134 This treatment augmented the mesoporosity initially present in the sample, leading to a material showing a quite uniform mesopore size distribution. The authors proposed that a local rearrangement of the zeolite structure takes place around the micelles of the surfactant, which provokes the transformation of the initial mesoporosity into a more uniform one, so the final materials exhibit mesostructured features. Fig. 4 illustrates digital analyses of TEM micrographs of hierarchical USY samples obtained by this approach, as well as of the parent zeolite.
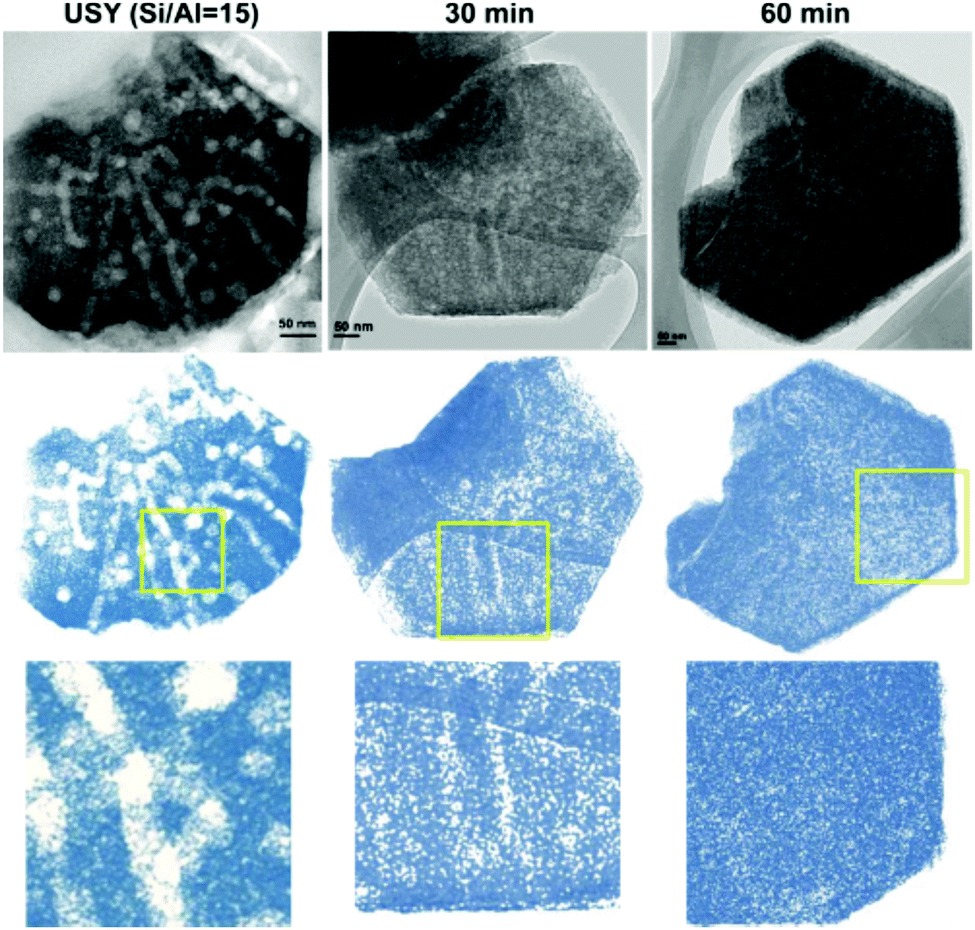 | ||
| Fig. 4 TEM micrographs and digitally treated TEM micrographs of the parent USY zeolite (left) and ex situ surfactant-templated USY zeolites treated at 373 K for 30 min (center) and 60 min (right). Reprinted with permission from ref. 138. | ||
Other methods for the preparation of hierarchical zeolites have been envisaged by combining several of the above mentioned strategies in order to achieve a better control of the properties of the final zeolite, and in particular of its mesoporosity. In this way, the sequential combination of desilication/dealumination treatments has been explored for the synthesis of a number of zeolites, such as ferrierite, clinoptilolite and L zeolites.139,140 The main challenge in the post-synthetic modification of these zeolites is their high Al content, requiring a tailored dealumination prior to the desilication step. On the other hand, a combination of seed silanization and surfactant-assisted crystal rearrangement strategies has been very effective for obtaining zeolites TS-1, MFI and *BEA with quite uniform mesopore size distribution.141–143
4.2. Properties of hierarchical zeolites
The presence of bimodal porosity in hierarchical zeolites causes sharp changes in many physicochemical properties of these materials compared to conventional zeolites. Thus, hierarchical zeolites typically exhibit enhanced accessibility, improved textural properties, faster intraparticle transport properties, changes in the acidity and better resistance against deactivation by coke formation compared with their conventional counterparts. In addition, as has been indicated earlier, hierarchical zeolites can be transformed into multifunctional materials by incorporation and/or grafting of other components taking advantage of their large share of mesopore volume and surface.The mesopores occurring in hierarchical zeolites, as well as their connectivity with the zeolitic micropores, have been visualized using transmission electron microscopy,96,145,146 as well as by other techniques, such as hyperpolarized 129Xe-NMR,147 positron annihilation148 and electron tomography.149 A variety of 2D and 3D microscopic techniques have been demonstrated to probe the pore structure of hierarchical zeolites.96 Thus, zeolite single crystals with intracrystalline mesopores incorporating metal oxide particles have been characterised by direct TEM stereo-imaging,145 whereas polarized-light microscopy has been used to unravel the pore geometry of ZSM-5 crystals containing mesopores.146 Likewise, one- and two-dimensional 129Xe-NMR spectroscopy has been employed to study the porosity of hierarchical zeolites under the continuous flow of laser-hyperpolarized xenon gas, demonstrating that these hierarchical pores have connected networks that facilitate xenon diffusion and exchange.147 On the other hand, the porosity of hierarchical zeolites has also been characterized by high-contrast electron tomography, the samples being supported with platinum.149 The resultant tomograms showed disordered and interconnected networks of Pt nanowires and nanosheets, which corresponded to the shape of the surfactant-directed mesopores. It was proposed that the micropores and mesopores could be connected through apertures, in which the neck of the surfactant molecule had been located.
Hierarchical zeolites usually present enhanced BET areas in comparison with conventional ones. This fact is a result of the lower restrictions that exist for the adsorption of gases over the mesopore surface, as it occurs also on the external surface of zeolite nanocrystals.150 Accordingly, a general trend is observed for hierarchical zeolites, showing an increase in the BET area due to the contribution of the secondary mesoporosity. In the case of hierarchical ZSM-5 zeolites, values of the BET area of up to 800 m2 g−1 have been reported for samples prepared using organosilanes, which represents almost twice that of a conventional zeolite sample.151 Regarding the assessment of the pore size distribution, the application of NLDFT models has made calculation of the individual contribution of the micro- and the mesoporosity to the overall zeolite textural properties possible, also providing a continuous pore size distribution curve extending from micro- to mesopores.151Fig. 5 shows the cumulative pore volume and pore size distribution curves obtained applying the NLDFT model to Ar adsorption isotherms (87 K) of a hierarchical Beta zeolite before and after being subjected to (above mentioned) mesopore narrowing treatment with a surfactant/ammonia solution.
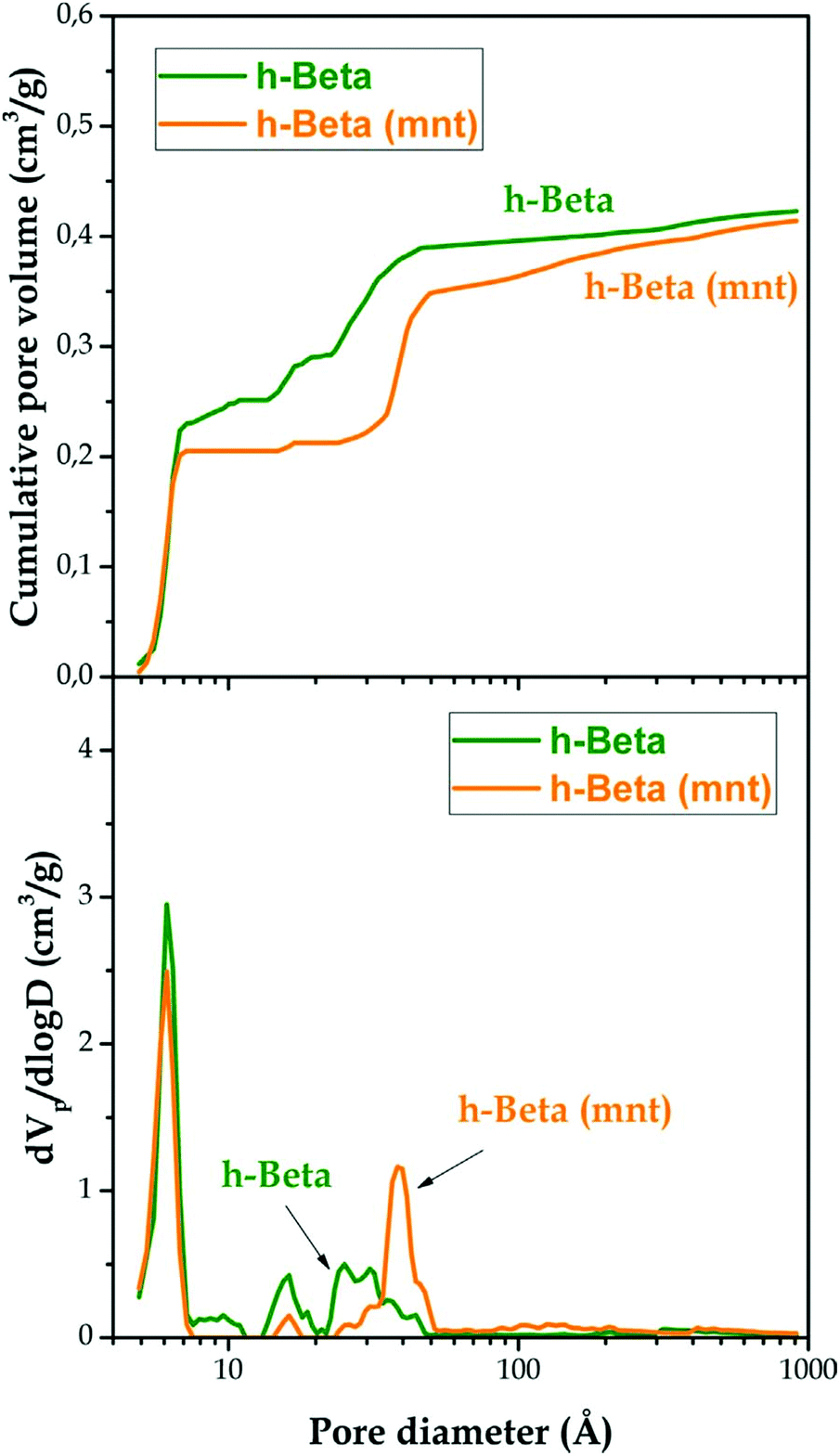 | ||
| Fig. 5 Cumulative pore volume curves (upper graph) and pore size distribution curves (lower graph) of the h-Beta and h-Beta (mnt) samples obtained using the NLDFT method to analyse Ar adsorption–desorption isotherms measured at 87 K. Reprinted with permission from ref. 143. | ||
Different literature studies investigated experimentally the expected enhancement of the mass transport rates in hierarchical zeolites. Two-order of magnitude improvement in the diffusion rate of neopentane within desilicated ZSM-5 has been observed, due to both shorter diffusion path length and the presence of an accessible network of mesopores.153 Similarly, water diffusion is favoured in Na+ zeolites in which, in addition to the micropores, the crystals are traversed by a network of mesopores.154 For instance, for zeolite *BEA, an increase in water diffusion rate by a factor of 3 was observed. The effect of mesopores on the adsorption and diffusion properties of MFI zeolites was studied for n-heptane and toluene using the zero length column (ZLC) method.155 The effective diffusion coefficients of these hydrocarbons increased greatly in the presence of mesopores, while the corresponding activation energy decreased. Expectably (having in mind size and shape of the two molecules), a higher enhancement was observed for toluene relative to that for n-heptane.
The increase in the mass transport rate is preserved even when the hierarchical zeolites are shaped into technical forms using binders. The influence of shaping on the adsorption and diffusion properties of hierarchical ZSM-5 has been assessed by studying gravimetric uptake of 2,2-dimethylbutane over powders and millimeter-sized bodies.7 Improved intracrystalline diffusivity (due to the presence of interconnected mesopores) was observed in powder as well as in the macroscopic bodies, independently of the shaping method or binder applied.
For hierarchical zeolites prepared by desilication, it has been concluded that the acid strength of the Brønsted sites of zeolites does not change significantly after the generation of mesopores.156 For mesoporous ZSM-5 prepared using amphiphilic organosilane, a lower concentration of Brønsted acid sites compared to conventional ZSM-5 was assessed by infrared-mass spectroscopy/temperature-programmed desorption (IRMS-TPD) of ammonia.157 Characterization by FTIR/pyridine of hierarchical ZSM-5 zeolites, prepared using organosilanes, has shown that they contain a higher proportion of Lewis acid sites compared to conventional samples, which arise probably from the larger concentration of extra-framework Al species in the former.158
The formation of Lewis acid sites in hierarchical ZSM-5 samples during a typical air calcination has been investigated,159 showing that it occurs to a higher extent than for conventional ZSM-5, due to lower stability of the Al atoms located on the mesopores. However, when a two-step (nitrogen/air) calcination treatment was applied, the so obtained hierarchical ZSM-5 preserved most of the Al atoms in tetrahedral coordination having the acid sites mainly of Brønsted type. This result noted the possibility of limiting the formation of extra-framework Al species, as well as of the associated Lewis acid sites, providing a convenient calcination treatment.
To describe in detail acid sites in hierarchical ZSM-5 materials, the so-called “accessibility index” has been determined from the FTIR spectra obtained after adsorbing substituted alkylpyridines with different molecular size: pyridine (0.57 nm), 2,6-lutidine (0.67 nm) and 2,4,6-collidine (0.74 nm).160 The accessibility index is defined as the ratio between the number of Brønsted acid sites probed by the employed alkylpyridine and the total amount of Brønsted acid sites determined from pyridine adsorption. While 2,6-lutidine has access to roughly 50% of the ZSM-5 acid sites, collidine only probes the acid sites present on the external/mesopore surface.
Accessibility studies of acid sites in hierarchical ZSM-5 zeolites have also been carried out by IR measurements after adsorption of pivalonitrile, determining the corresponding accessibility factor (AF) referenced to pyridine.161 The main advantage of this probe molecule is that it can interact and, therefore, assess both Brønsted and Lewis acid sites. This technique was applied to desilicated ZSM-5 zeolites, observing that the accessibility of Lewis acid sites in the desilicated samples is even more enhanced than that of Brønsted sites. The authors proposed that in desilicated zeolites the majority of Lewis sites originate from dehydroxylation of the Si–OH–Al groups, previously formed by the reinsertion of Al extracted from zeolite during alkaline treatment. Accordingly, newly formed Lewis sites are expected to be located mainly on the mesopore surfaces, which facilitates the accessibility of bulky molecules like pivalonitrile.
Retardation of deactivation has been observed in a number of reactions over ZSM-5 zeolites,162 such as isomerisation of 1,2,4-trimethylbenzene, cumene cracking and esterification of benzyl alcohol with hexanoic acid. In all cases, the hierarchical ZSM-5 showed a lower deactivation rates than conventional ZSM-5. In the methanol-to-hydrocarbon reaction,163 the catalyst lifetime was more than three times prolonged using the hierarchical ZSM-5 in comparison with a conventional ZSM-5. The impact of the textural properties of both hierarchical and nanocrystalline ZSM-5 zeolites has been investigated in conversion of ethanol to hydrocarbons.164 The lifetime of the catalysts is not correlated with the coking rate but with the number of pore mouths. Thus, the longest catalyst lifetime (>100 h) was observed with a hierarchical nanosized zeolite even though most of its acid sites were poisoned.
Extension of the catalyst lifetime has also been reported for other types of hierarchical zeolites. Thus, isomerization and cracking of n-hexadecane have been investigated over ZSM-12 zeolites containing mesopores, showing significantly improved resistance to poisoning by coke formation.165 Similarly, hierarchical zeolite Y has been found to exhibit remarkably higher resistance to deactivation in the aldol condensation of benzaldehyde with n-butyl alcohol than a commercial zeolite Y, which is attributed to its highly mesoporous structure minimizing the diffusion length of coke precursors out of the zeolite matrix.166
Different metals, such as Pt, Pd, Ni, Co and Cu, have been added to hierarchical zeolites by impregnation of the precursors.167–171 These metals are present usually in the form of nanoparticles located mainly in the mesopores in close contact with the support and with an improved dispersion compared with the use of conventional zeolites. These findings have been related to the existence of a significant mesoporosity in hierarchical zeolites, as well as to the occurrence of a high concentration of structural defect sites, which act as preferred deposition points for dispersion of metal phases.172 Such materials exhibit bifunctional properties, being applied in hydrogenation, hydroisomerization, Fischer–Tropsch synthesis, and hydrocarbon abatement.
Metal oxides, sulphides and phosphides have also been supported over hierarchical zeolites in order to expand their catalytic applications in reactions such as aromatization, alkylation, desulphurization and hydrodeoxygenation.173–176 Thus, ZnO-containing MFI zeolite catalysts with bimodal and trimodal hierarchical pore structures have been studied for the conversion of methanol to aromatics. The presence of highly dispersed ZnO clusters enhanced the selectivity for aromatics.173 Interestingly, a catalyst with basic properties has been synthesized by supporting of MgO over hierarchical silicalite-1, showing an improved catalytic activity for the side chain alkylation of toluene with methanol compared to bulk MgO.174 On the other hand, mesoporous ZSM-5 modified with metal sulfides (NiMoS/ZSM-5 and CoMoS/ZSM-5) was active in deep hydrogenation of phenanthrene.175 Ni2P has been supported over hierarchical ZSM-5 zeolites, showing an enhanced activity in hydrodeoxygenation reactions of bio-oil model compounds.176
In addition to metals, metal oxides and salts, hierarchical zeolites have been used as supports for other catalysts: encapsulation of Fe–Schiff bases and Ti–salen complexes, incorporation of heteropolyacids and enzymes, and functionalization by grafting of sulfonic groups.177–181
5. Zeolites with 2-dimensional forms
A zeolite is considered 2-dimensional when one of the dimensions of its crystals is less than several nanometres, corresponding to about one or two unit cells. In such a case, the well accessible external surface per mass/volume unit is highly increased (in comparison with 3D (conventional) zeolites) and a number of post-synthesis modifications (e.g. swelling, pillaring, exfoliation etc.), characteristic of layered materials, become possible.9,182From the synthesis point of view, 2D zeolites have been reviewed recently. Reviews by Roth and coworkers9,183 and Eliášová and coworkers31 are recommended for more details.
In principle, there exist 3 general approaches to the synthesis of 2D zeolites. (I) Some zeolites (FER, MWW, NSI, SOD, and several others) can be directly prepared as lamellar precursors (which can be processed or modified in a way that their 2D character is preserved). (II) 2D zeolites can be obtained via a restricted crystal growth mechanism using a specially designed (surfactant) structure directing agent which blocks crystal growth in one of the crystallographic directions forming sheet-like single crystals. (III) Top-down post-synthesis modification of zeolites with anisotropic structures (namely germanosilicates).
5.1. Hydrothermal synthesis
3D zeolites traditionally crystallize under hydrothermal conditions from appropriate synthesis gels or from clear solutions. Some of them, however, form lamellar precursors, which are intercalated with the structure directing agent (SDA). The molecules of the SDA, inter alia, keep the layers organised in a way that opposite silanol groups on the surface of two neighbouring layers can condense into oxygen bridges thus forming a fully 4-connected zeolite upon calcination. In some cases the condensation does not produce the 3D framework (EU-19, RUB-20, RUB-40).184The first zeolite, for which the formation of a layered precursor was observed (and so far the most studied of the 2D zeolites), is the MWW. In its 3D form (reported independently as PSH-3,185,186 SSZ-25,187 MCM-22,188 ERB-1189), it is a medium pore zeolite with two independent 2-dimensional channel systems of 10-ring pores. One of the pore systems contains 7.1 × 18.1 Å supercages.190 The layered character of the as-synthesised material was disclosed by the Mobil researchers in 1990s and confirmed by others.189,191 Later on, synthesis routes yielding directly 3D MWW (MCM-49192), delaminated MWW (MCM-56; material which does not condense into a 3D zeolite upon calcination even without any further modification193) and, recently, in situ swollen MWW (ECNU-7)194 were also disclosed.
Ferrierite195 (IZA code FER) is another important zeolite found to form a lamellar precursor (denoted as PREFER).196 In fact, the PREFER layers do not condense only to form ferrierite but when prepared using a different procedure, they are shifted and condensed to form a CDO structure (CDS-1 zeolite), which is composed of the same layers but stacked in a different symmetry.197,198 Similarly, RUB-36 (CDO lamellar precursor) can be either directly calcined to form the CDO zeolite, or swollen with cetyltrimethylammonium cations (see Section 5.4) and de-swollen with EtOH/HCl which results in a layer shift yielding PREFER and after calcination 3D FER.199 In fact it is possible to switch between the CDO and FER arrangements by intercalation and pH adjustment.200 Such pairing on 3D zeolites composed of same layers, where in one of them the layers are propagated by translation while in the other by mirror plane, is observed also for NSI/CAS, RRO/HEU and FAU/EMT structures9 (for FAU and EMT, lamellar precursors were not disclosed yet). Rich chemistry of the lamellar zeolite precursors has been recently reviewed by Roth et al.9 and Díaz et al.201
5.2. Surfactant templated synthesis
Until 2009, the only known lamellar zeolitic materials were those forming lamellar precursors during crystallization. These lamellar precursors can then condense into fully four-connected 3-dimensional zeolites upon calcination (e.g.MWW, FER; see previous chapter). The main role of the SDA in crystallization of these zeolites is the same as for those forming 3-dimensional network directly; that is to fill and stabilize void volumes in the structure of zeolites against dissolution or transformation into more stable (denser) phases.202In 2009, the Ryoo group introduced the concept of surfactant structure directing agents enabling to prepare lamellar (or nanosponge; vide infra) forms of zeolites such as MFI (Fig. 6) or MTW, which were not yet known to form lamellar precursors directly.203 Quaternary ammonium surfactants (e.g. cetyltrimethylammonium (CTMA)) were used before in the synthesis of mesoporous molecular sieves;204,205 however, their templating effect was not strong enough to promote zeolite crystallization. Only very recently was a synthesis of hierarchical MFI using CTMA in combination with KOH reported.206 Choi et al. augmented the structure directing effect designing a di-quaternary ammonium surfactant, where the ammonium groups in the hydrophilic part of the molecule are separated by an organic linker of an appropriate structure and size (n-C6 for MFI).203 In the pioneering work,203 a surfactant C22H45–N+(–CH3)2–C6H12–N+(–CH3)2–C6H13 (denoted as C22–6–6) was used to form MFI in a form of nanosheets of about 2.5 nm thickness. The surfactant nature of the SDA is a key to form the layered structure. The hydrophilic end with two quaternary ammonium groups templates the zeolite crystallization while the long hydrophobic chain prevents the crystal growth in one of the crystallographic directions (b-axis for MFI). In addition, the hydrophobic chains support (under certain conditions) regular stacking of the formed (nanosheet) crystals. As a result, bigger aggregates of the nanosheets (not a colloidal suspension) are formed and these are easy to process. Note an important difference in comparison with zeolites forming lamellar precursors: the MFI nanosheets (and generally all materials prepared using surfactant SDAs) do not condense into a 3-dimensional zeolite upon calcination but the layers stack randomly one to another forming a material with slit-shaped mesopores because the nanosheets do not fit regularly one on another (Fig. 6). Condensation of surfactant templated nanosheets into a 3D structure is still a challenge and particularly in the case of MFI nanosheets it may lead to a new zeolite.
 | ||
| Fig. 6 TEM images of nanosheet MFI viewed along the b-axis (left), pillared nanosheet MFI viewed along the a–c plane (middle) and self-pillared-pentasil zeolite (right). Reprinted with permission from ref. 207 (left, middle) and ref. 208 (right). | ||
Deeper investigations of the surfactant templated syntheses209 revealed that shorter hydrophobic chains (C18, C16) also provide the lamellar MFI phase and in some cases it is beneficial to use them to shorten the crystallization time (e.g. for layered titanium silicalite-1210). The minimum hydrophobic chain length to form lamellar MFI is about C10209 and the hydrophobic chain length enables to control the interlayer d-spacing.211 Tailoring of the hydrophilic part of the SDA controls the thickness of the MFI nanosheet. Addition of third and fourth quaternary ammonium groups results in the formation of nanosheets composed of 5 resp. 7 pentasil layers instead of 3 for C22–6–6.209 On the other hand, the nanosheet thickness can be as low as 2 pentasil layers (MFI unit cell) when a surfactant with two hydrophobic tails and 3 quaternary ammonium groups is used [C18H35–N+(–CH3)2–C6H12–N+(–CH3)2–C6H12–N+(–CH3)2–C18H35] (in short C18–N3–C18).212 Under certain conditions the C18–N3–C18 surfactant can also promote the formation of a hexagonally ordered mesoporous material with walls of crystalline MFI instead of lamellar (nanosheet) MFI213 bridging the boundary between zeolites and mesoporous molecular sieves. This shows that the surfactant SDA is not the only driving parameter determining the properties of the resulting material but the gel composition, alkalinity and other parameters are also important. Choi et al. demonstrated that concentration of Na+ ions influences the stacking of the formed nanosheets (forming a material denoted as multilamellar (ordered stacking) or unilamellar (disordered stacking))203 and studies on the synthesis gel composition were conducted by Machoke et al.214 and Wei et al.215
The concept of surfactant templating was applied also to other zeolites but the results were not so straightforward. Nanosheet MTW was reported together with MFI203 using a –CH2–(p-phenylene)–CH2– spacer in addition to n-C6. The same mechanism as for MFI restricted crystal growth has been proposed (cf. TEM images in the supplementary information of ref. 203); however, further studies on this material have not yet been reported.
Interestingly, the surfactants with phenylene spacers among ammonium groups can also be used for disordered mesoporous molecular sieves with walls of zeolite beta213 and similar materials with *MRE and MTW structures.216,217 Based on the TEM images,213 it appears that the crystal growth is restricted by the template in all 3 crystallographic directions in this case. Nevertheless, these materials (later denoted as nanosponge zeolites) have a hierarchical sponge-like structure composed of intergrown zeolite nanocrystals with an intercrystalline system of mesopores. In contrast to the above (*BEA, *MRE and MTW), nanosponge MFI is obtained when the synthesis mixture for nanosheet MFI is seeded with bulk MFI crystals218,219 or a polymer (polystyrene) randomly grafted with –CH2–N+(–CH3)2–C6H12–N+(–CH3)2–C6H12–N+(–CH3)2–C6H13 groups is used.220 The nanosponge MFI keeps the lamellar morphology according to the TEM images although it is a 3-dimensionally intergrown material and post-synthesis layer manipulations applicable to 2-dimensional materials have not been reported (and most probably are not possible).
A similar sponge-like morphology of intergrown lamellar MFI and MEL crystals was prepared also by Zhang et al.208 (referred to as self-pillared pentasil zeolite (SPP); Fig. 6 left) and Chen et al.221 Interestingly, these materials were obtained using tetrabutylphosphonium and tetrabutyl ammonium hydroxide SDAs208 and no surfactant was used to support the formation of lamellar or hierarchical architecture.
The nanosheet MFI, the SPP, and nanosponge MFI zeolites typically exhibit increased BET area (510–610 m2 g−1) in comparison with conventional MFI (300–400 m2 g−1) and almost an order of magnitude increased external surface area (270, 420, 360 m2 g−1 respectively vs. 20–60 m2 g−1), which is a direct consequence of their lamellar character.207 Similarly, their total adsorption capacity (total pore volume) increases depending on their layer arrangement as follows: nanosheet MFI 0.36–0.65 cm3 < nanosponge MFI 0.56–0.62 cm3 < SPP 0.73–1.0 cm3vs. conventional MFI 0.16–0.25 cm3 g−1.207,208,215,222 The improvement of transport properties of the above MFI catalysts was nicely documented in etherification of benzyl alcohol in the presence of di-tert-butylpyridine (DTBP) over SPP, pillared nanosheet MFI (vide infra) and several conventional MFI catalysts.208 DTBP was used to deactivate external acid sites thus allowing observation of the transport phenomena via the etherification occurring exclusively in micropores. Observed data are in perfect agreement with the plot of effectiveness factor vs. Thiele modulus (Fig. 7) documenting that commonly observed improvements of apparent reaction rates in 2D catalysts truly account for suppressing the diffusion limitations.
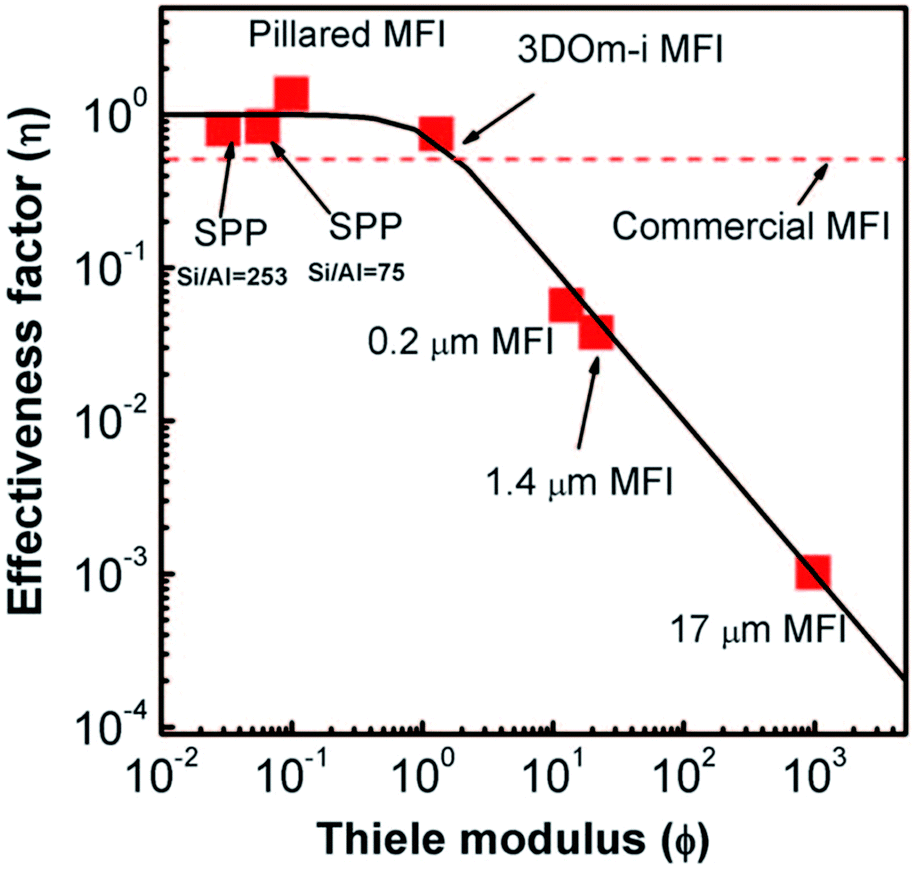 | ||
| Fig. 7 Effectiveness factor vs. Thiele modulus plot and experimental data for etherification of benzyl alcohol over different MFI catalysts. Reprinted with permission from ref. 208. | ||
Summarising the above discussed findings, it is clear that the surfactant templating is a useful and versatile tool for preparation of layered forms of zeolites, for which layered precursors have not yet been synthesized by hydrothermal synthesis; however, in some cases the nanocrystalline form rather than the layered one (nanosheet) can be obtained.
5.3. ADOR
Synthesis routes to 2D zeolites described vide supra discussed bottom-up approaches either with conventional or specially designed templates.8,182 A top-down synthetic protocol is an opposite way, which is applicable for zeolites with anisotropic structures affording the possibility of chemically selective removal of some structural units. This is nicely documented for a series of germanosilicates, originally started with a UTL zeolite followed by UOV and SAZ-1.31,223–225Introduction of Ge allowed synthesizing a number of new zeolites with large or extra-large pores. Simultaneously, it resulted in the formation of relatively labile Ge–O bonds. Location of germanium in double-four-rings (d4r) is a characteristic feature of germanosilicate zeolites (sometimes even d3rs).226,227 The d4r units form connecting pillars between individual layers (porous or non-porous depending on the zeolite structure) in the framework. It was observed that Ge–O bonds can be easily hydrolysed in aqueous solutions not depending on the pH.31,228 This hydrolysis utilizes the chemical weakness of Ge–O bonds and leads to the removal of species from the destroyed d4r units leaving the rest of the zeolite structure (mostly siliceous zeolitic layers) unaffected. In the case of the UTL zeolite, a layered material called IPC-1P is formed.100,229 Obtaining the layers of IPC-1P from the UTL zeolite opened new possibilities to manipulate with the layers in different ways. The most important is the use of IPC-1P and related layered zeolites prepared by the top-down approach for the preparation of new zeolites. Hydrolysis of the UTL zeolite results in the formation of 4 silanol groups at each layer instead of 4 connections to each original d4r. Thus, layers possess very high concentration of available silanol groups, which can be used either for condensation or for another interlayer chemistry.230
Roth et al. described the formation of IPC-1P layers from zeolite UTL when it was hydrolysed at ambient or increased temperatures.100 Removal of the debris formed from d4r units from the interlayer space depends on the pH of the solution. While in diluted solutions practically all such species are removed to the aqueous phase, at higher concentrations these species remain partly in the interlayer space. As a result, calcination of layered materials formed under different pH leads to the different outcome. In the case of IPC-1P formed under diluted conditions, calcination provided a new zeolite PCR having the individual layers connected just by oxygen bridges (10–8-rings).101 When part of original d4r remained in the interlayer space, calcination led to the OKO zeolite having single-4-rings (s4r) connecting layers.231
The mechanism leading to PCR or OKO zeolites has been named ADOR (A – assembly, D – disassembly, O – organization, R – reassembly), see Fig. 8.31 This mechanism offers a variety of possible outcomes in particular due to the top-down preparation of layered precursors not available by direct synthesis. At present, zeolites PCR, OKO, IPC-6, and IPC-7 have been prepared by different condensation of IPC-1P layers. OKO has also been prepared using high pressure (about 1 GPa) at 200 °C, which means at a much lower temperature than that usually used for calcination (500–550 °C).232 General applicability of the ADOR mechanism has been recently confirmed starting from the UOV zeolite and obtaining a new zeolite IPC-12224 by substituting d4r units for s4r ones and recently from SAZ-1 creating IPC-15 and IPC-16.225
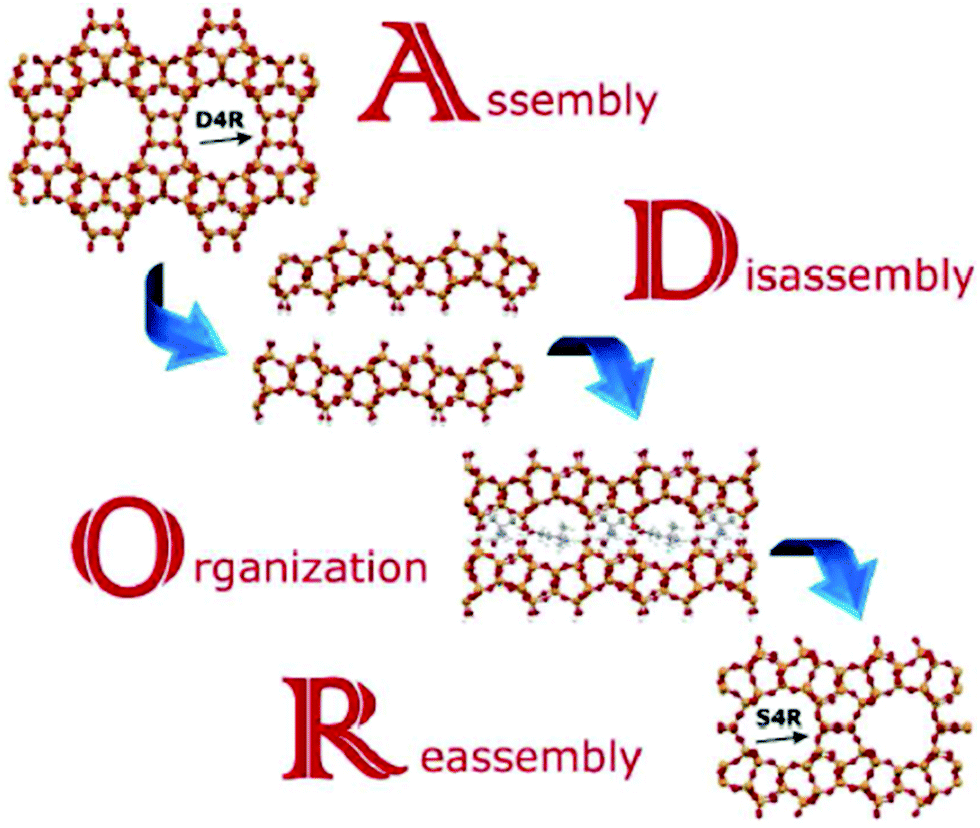 | ||
| Fig. 8 Scheme of the ADOR process for transformation of the germanosilicate parent zeolites, Reproduced from ref. 232 with permission from the Royal Society of Chemistry, copyright 2017. | ||
In addition, the individual IPC-1P layers can be connected with different organic or inorganic pillars making stable systems with larger layer distance. This achievement opens new possibilities in preparing porous hybrid materials being amenable to different post-synthesis modifications involving addition of functional groups with catalytic activity.233
Next and very interesting target for the ADOR mechanism was shifting of the individual layers prior to calcination, which could lead to new frameworks. In such a case, zeolites with odd number of channels and having relatively high energy on the energy/density plot proposed by Deem should be achieved.32,234 Those zeolites were considered as “unfeasible”. Mazur and coworkers succeeded in this by intercalation of choline chloride at a specific pH into the interlayer space, which induced shift of the layers.228 The condensation resulted in a new zeolite IPC-9 having a 10–7-ring zeolite while addition of silica to the interlayer space followed by condensation produced zeolite IPC-10 (12–9-rings).194 Structural features of novel zeolites are summarized in Table 1. These zeolites have not yet been synthesized by direct solvothermal synthesis.
| Parent zeolite | New zeolite | Channel structure |
|---|---|---|
| UTL | IPC-2 (OKO) | 12 × 10 |
| IPC-4 (PCR) | 10 × 8 | |
| IPC-6 | 10 × 8 + 12 × 10 | |
| IPC-7 | 12 × 10 + 14 × 12 | |
| IPC-8 | 10 × 8 + 14 × 12 | |
| IPC-9 | 10 × 7 | |
| IPC-10 | 12 × 9 | |
| UOV | IPC-12 | 12–8 (1D) |
| CIT-13 | IPC-13 | 12 × 8 |
| SAZ-1 | IPC-15 | 10 |
| IPC-16 | 12 × 8 | |
| IWR | IPC-17 | 12 × 8 |
| IWW | IPC-18 | 12–8 × 8 |
To increase the stability of these zeolites the substitution of Ge for more stable Si was carried out. Tuel, Eliášová or Wu and their teams replaced substantial part of Ge with Si, which increased dramatically the stability of these zeolites. In addition, Si can also be replaced by Al introducing acid sites to the structure sites.235 Opanasenko et al. evidenced for zeolites ITH and IWW that aluminium can be incorporated either directly into the synthesis mixture or via post-synthesis substitution of Ge with Al, thus introducing activity for acid-catalysed reactions.236,237
There is no doubt that the ADOR protocol represents a substantial breakthrough in the synthesis of new zeolites complementing the traditional synthesis approaches. Until now ADOR has been limited to germanosilicate zeolites, posing an important challenge for synthesis, namely how to extend ADOR applicability to zeolites without germanium.
5.4. Post-synthesis modifications
The great advantage of 2D zeolites lies in their structural flexibility and rich post-synthesis modification chemistry,183 especially layer manipulations with increasing, decreasing or preserving the interlayer distance (Fig. 9). The present figure is based on the modifications of the MWW zeolite (a zeolite with hydrothermally synthesised lamellar precursor); however, it can be considered universal for all of the 2D zeolites. The variations for surfactant templated synthesis and ADOR mechanism are discussed below.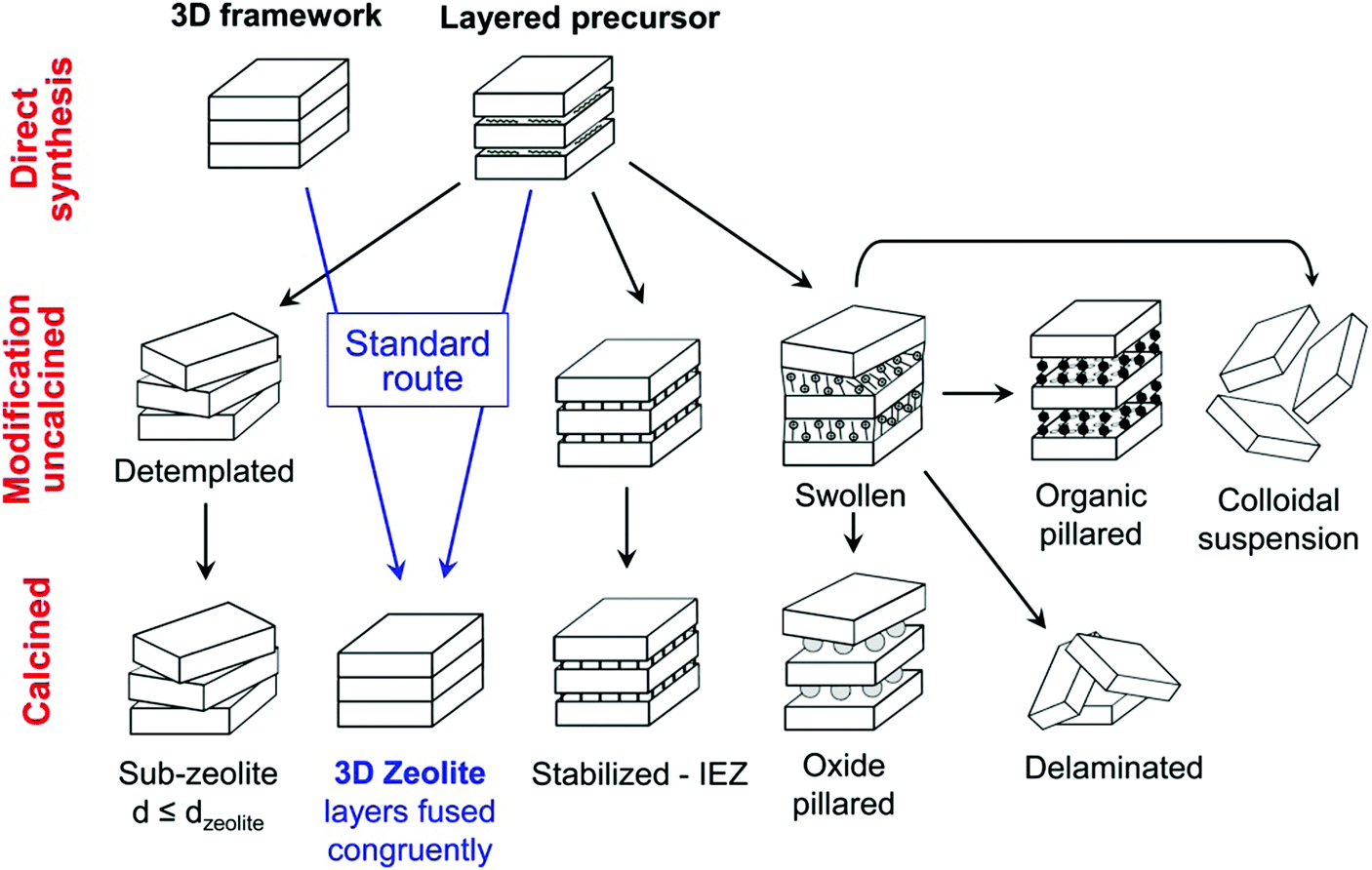 | ||
| Fig. 9 Post-synthesis modifications of layered zeolites. Reprinted with permission from ref. 182. | ||
As shown in the scheme in Fig. 9, a 2D zeolite precursor consists of alternating crystalline siliceous layers and the SDA molecules in between the layers. In contrast to the 3D zeolites (where the structure is fully covalently bonded directly after the synthesis), there are no covalent bonds between the individual layers and the whole structure (crystalline layers and SDA molecules) is kept together only by interactions with SDA molecules and hydrogen bonds.9
In the case of ADOR transformation, the layered precursor is formed without the SDA (e.g. IPC-1(P)) but subsequent intercalation with an appropriate molecule (the organisation step in the ADOR mechanism) is the key to the layer manipulation and even lateral shift of the layers yielding “unfeasible” zeolites IPC-9 and IPC-1031,100,101,228 (vide supra). The third approach – surfactant template synthesis yields a swollen 2D zeolite intrinsically.
The SDA molecules prevent full 3-dimensional covalent connection (during the hydrothermal synthesis) but keep the layers in regular orientation one to another (or intercalated molecules helps to orient the layers in the ADOR mechanism). Upon calcination, the SDA molecules are removed and subsequently a dehydration of two corresponding silanol groups occurs forming a covalent oxygen bridge connecting the discrete layers into a fully connected 3D framework (Fig. 9, “standard route”).
In some cases, the SDA can be removed from the lamellar precursor not only by calcination, but also at low temperature (below 80 °C; Fig. 9 “detemplated”), under conditions when the silanol condensation into oxygen bridges does not occur. For instance, the template from MCM-22(P) can be removed by diluted HNO3 solution (<2 M).238 In the resulting material, called a MCM-56 analogue, the layers are randomly oriented one on another and the detemplated MWW material exhibits increased external surface area (150 vs. 117 m2 g−1) and decreased micropore volume (0.13 vs. 0.17 cm3 g−1) with respect to the 3D-MWW. Similarly, textural properties of IPC-1 (a partially disordered material formed by disassembly of UTL) exhibit lower micropore volume in comparison with corresponding reassembled PCR (0.095 cm3 g−1vs. 0.106 cm3 g−1).100,101 These collapsed materials were called sub-zeolites.239
Besides simple calcination, forming a fully connected 3D framework, also an additional bridging group (either additional silicon atom or e.g. organic bridge240) can be inserted in between the layers and connected to the silanol groups. As a result so-called interlayer expanded zeolites (IEZ) are obtained.241 The process of interlayer expansion is sometimes referred to as stabilisation. The IEZ zeolites are characterised by larger interlayer pore openings in comparison with their parent structures. For instance IEZ-MWW has 12-ring pores and 2.7 nm basal d-spacing while conventional MWW has 10-ring pores and a basal d-spacing of 2.5 nm. Except for IPC-2 and IPC-10 materials,101,228 the IEZs are not fully connected 3D-frameworks and free silanol groups are present on the bridging atoms.
A critical step in the 2D zeolite modification is breaking of the interlayer hydrogen bonds and expansion of the interlayer space. This process is called swelling (Fig. 9) and it is well described for lamellar materials such as clays, phyllosilicates and layered metal oxides.242 In the case of 2D zeolites, the swelling was first performed using a concentrated (25 wt%) quaternary ammonium surfactant (e.g. cetyltrimethylammonium hydroxide) in a high pH medium.191 The swollen material exhibits a highly increased interlamellar distance (e.g.MWW: d-spacing 5.2 nm vs. 2.6 nm in MWW(P)243). It should be noted that the materials prepared using a surfactant203 are swollen intrinsically but in contrast the surfactant tail is not easily removed from interlayer space as it is part of the SDA.
The swelling is not a final step of the post-synthesis modification and once layers are separated, a number of other materials can be prepared. The layers are kept apart from each other by organic surfactants and when these are removed e.g. by calcination, the layers collapse. However, if an additional support, which can withstand calcination, is inserted, the interlayer distance can be preserved. It was found that the tetraethyl orthosilicate (TEOS) easily penetrates among the surfactant chains in the interlayer space, where it is converted into mesoporous amorphous silica pillars242 (Fig. 6 middle). The first pillared zeolite material is MWW denoted as MCM-36.191 Pillaring is nowadays one of the standard modifications of 2D zeolitic materials (as well as of e.g. clays244); however, the true structure of the pillars remains uncertain because of lack of regularity.9 Besides pure TEOS, also its mixtures with other metal-alkoxides (e.g. titanium(IV) butoxide, tin(IV) iso-propoxide) can be used for pillaring, which results in the formation of additional catalytically active sites.222,245,246
Besides amorphous silica-based pillaring, also other groups, such as silsesquioxanes or organic linkers may be inserted.233,240,247 The latter provide hybrid organic–inorganic materials with interesting adsorption properties. Recently, also covalent insertion of iron, titanium, tin, zinc and europium has been reported into RUB-36 (CDO lamellar precursor).248 Besides that, metallic species, which are turned into metallic nanoparticles upon calcination, can be inserted using the advantage of enlarged space between the zeolite layers.249,250
A delaminated material is composed of randomly oriented lamellae (ultimately a house-of-cards morphology). A highly open structure is the main feature of this material and ideally the whole external surface of the lamellae is accessible from the interparticle space or wide mesopores. Corma et al. established this group of materials by ultrasonic treatment of swollen MWW layers, forming a material denoted as ITQ-2.251 However, the MWW delamination procedure is not generally applicable and examples of other delaminated zeolites are sparse (only NSI, FER, RWR precursors).9 Several attempts to delaminate nanosheet MFI using conditions similar to the ITQ-2 have been made; however, TEM analysis revealed that the product (although having BET area up to 800 m2 g−1 and total adsorption capacity 1.50 cm3 g−1) is only a physical mixture of parent nanosheet MFI and amorphous silica coming out from partial dissolution of the zeolite in the basic medium.252
Last but not least, the swollen material can be dispersed into a single lamellae colloidal solution.253,254 The so-called exfoliation into single layers has been reported for zeolites MWW255 and MFI.256
6. Catalytic investigations of 2D vs. 3D materials
6.1. Active sites in 2D zeolites
The main advantage of 2D zeolites in comparison with their 3D analogues lies in an improved accessibility of the active sites (resulting from their location on the external surface of the lamellae) while their other characteristics (such as geometry, coordination, strength of acidity etc.) are desired to remain the same as for conventional zeolites. Here we review ways of analysis and the literature data confirming or challenging the above statement.In both 2D and 3D zeolites, active sites in zeolite-based catalysts are usually represented by isomorphously substituted silicon atoms for three- or four-valent elements and/or defects in the crystalline silica structure. To introduce catalytic activity to the structure of zeolites, appropriate heteroatoms (Al, Ti, Sn, Zr, Fe, Ga, Ge, B etc.) need to replace silicon atoms in the zeolite frameworks. Exceptionally, various atoms in extra-framework positions or vacancies (defects) as silanol groups (e.g. for Beckmann rearrangement) can also be catalytically active. In addition, the concentration of silanol groups defines other important properties such as hydrophilicity.257
All mentioned heteroatoms isomorphously incorporated into the framework represent acid sites of Brønsted or Lewis nature. Strong Brønsted acid sites (comparable to mineral acids) are formed by bridging [Si(OH)Al] sites and other trivalent metal ions can also form this type of sites. The other tetravalent (divalent Zn) heteroelements form mostly Lewis acid sites of a different strength. Extra-framework heteroatoms are in a number of cases considered undesirable but there are examples when they significantly contribute to the catalytic activity.222,245,258
Brønsted acid sites can be observed directly using IR spectroscopy as the acidic [Si–(OH)–Al] group exhibits a vibration band clearly distinguishable from other silanol signals. Its precise position alternates with the zeolite structure (e.g.FER: ν(OH) = 3605 cm−1vs.FAUν(OH) = 3645 cm−1 (ref. 264)) and it is also dependent on the zeolite dimensionality (e.g.MWW 3-dimensional MCM-22: ν(OH) = 3625 cm−1 (ref. 264) vs. 2-dimensional ITQ-2: ν(OH) = 3620 cm−1 (ref. 265)). Molecular modelling showed that the precise position of the band even reflects the crystallographic position of the corresponding aluminium atom;264 however, distinguishing between several bands corresponding to different positions (which are close to each other) as well as their proper assignment is rarely possible. Laforge et al. distinguished acidic OH groups in supercages (ν(OH) = 3621 cm−1) and in the sinusoidal channel (ν(OH) = 3608 cm−1) of the MWW zeolite and provided a comparison of its 3-dimensional (MCM-22) and 2-dimensional (MCM-36) forms.266 The signal belonging to Brønsted sites in supercages was about 7 times weaker for MCM-36 in comparison with MCM-22 reflecting that the supercages are not formed when the zeolite does not condense into a 3D connected structure. In contrast, Lewis sites cannot be observed directly.
To analyze the character (Lewis or Brønsted), location and (relative) strength of the (not only) aluminum acid sites, techniques using basic probe molecules need to be employed. In general there are three approaches for identification and quantification of the adsorbed probe molecules: (i) in situ IR analysis,183,264–268 (ii) 31P-NMR spectroscopy261,269,270 and (iii) temperature programmed desorption (TPD) experiment.271,272 The in situ IR analysis is a widespread method, used in combination with probe molecules including CO, amines, nitriles, pyridine and its derivatives and others, which exhibit bands characteristic for the corresponding species interacting with Brønsted or Lewis acid sites.27331P-NMR can be used for observation of trialkylphosphine oxides, which adsorb on the acid sites in a one-to-one manner. The TPD experiments give information about the strength of acid sites (the higher the desorption temperature, the stronger the site) but the adsorbed species are not observed directly.274 The in situ IR and TPD techniques can also be combined into so called IRMS-TPD providing a deep understanding of the surface processes as well as quantitative information.275,276
The choice of the probe molecule determines the information possible to obtain similarly to Hammett indicators used to estimate acid strength in non-aqueous media (i.e. in homogeneous catalysis).277,278 In fact, it was demonstrated that a carefully chosen set of Hammett indicators can be used to probe even zeolite acidity (namely large-pore zeolite Y); however, steric limitations restrict the application for porous materials.279 For 2D materials, the enhanced accessibility of the acid sites is the key feature. Therefore, a combination of a small probe molecule, which can access all the acid sites, and a bulky analogue, which cannot enter the system of micropores, provides information about the sites located on the external surface of the crystals or lamellae. For instance, a commonly used pair of such molecules, in connection with IR spectroscopy, is pyridine (kinetic diameter 0.54 nm) together with lutidine (2,6-dimethylpyridine, 0.67), collidine (1,3,5-trimethylpyridine; 0.74 nm)267 or 2,6-di-tert-butylpyridine (DTBpy, 1.05 nm). The pyridine molecule is small enough to enter medium-pore zeolites such as MFI or MWW while DTBpy in particular is too bulky to enter even the channel systems of extra-large zeolites and its nitrogen atom is strongly hindered so it can interact only with Brønsted acid sites (not Lewis).265 All the (substituted) pyridines exhibit characteristic vibrations of the aromatic ring attached to different acid sites (and silanol groups) in the region 1400–1800 cm−1, where they do not overlap with e.g. framework vibrations of the zeolite.
An accurate quantification of the acid sites requires reliable determination of the extinction coefficients (either for a single wavenumber or integrated molar extinction coefficient for the whole band), which is a complex problem reviewed e.g. in ref. 273.
Phosphine oxides are also bases and therefore they can be used similarly to substituted pyridines and other amines. Their size (determining their ability to enter the porous system of the catalyst) can by adjusted by choosing an appropriate length of the alkyl substituents. Trimethylphosphine oxide (TMPO) has a kinetic diameter of 0.55 nm and therefore it can enter even 10-ring pores. On the other hand, e.g. tributylphosphine (TBPO, kinetic diameter 0.82 nm) is suitable to assess external surface acidity. The acidic proton interacts with the oxygen atom of the phosphine oxide, resulting in a change in the electron density and thus chemical shift of the phosphorus atom. Hence, the phosphine oxide species can be observed and distinguished using solid state 31P-NMR. Pure solid TMPO exhibits a chemical shift of 30 ppm, TMPO interacting with silanol has a chemical shift of 42 ppm and the signal of TMPO bound to Brønsted acid sites is observed between 66 and 86 ppm depending on the acid site strength.261 The stronger the acid site, the higher the phosphorus chemical shift. Considerable drawbacks of this method are high demands on experimental equipment (glovebox, MAS-NMR spectrometer) in comparison with FT-IR techniques. In addition, quantification of Lewis sites is complicated and uncertain because the corresponding TMPO signal (37 ppm)269,270 is very close to the signal of TMPO interacting with silanol (42 ppm), which can be very intensive especially in layered or hierarchical zeolites.
In titanosilicate zeolites, the titanium atoms can basically adopt 3 different states. Generally, the desired species is an isolated titanium atom (not connected to another titanium atom via an oxygen bridge) with tetrahedral coordination, isomorphously incorporated into the framework. Titanium can also be present as isolated 5- or 6-coordinated species in the extra-framework position or it can form clusters of more titanium atoms connected directly with oxygen bridges (referred to as anatase or anatase-like phase). Catalytic activity of isolated extra-framework titanium atoms is a subject of debates. Previously, they were considered inactive; recently multiple pieces of evidence have been found proving their catalytic activity.222,282–284 On the other hand, the anatase phase is inactive in selective oxidation but it has its irreplaceable role in photocatalysis.
The titanium species can be qualitatively distinguished using UV/Vis spectroscopy in diffuse-reflectance mode (DR-UV/Vis). Tetrahedrally coordinated titanium atoms exhibit absorption centred at about 205–210 nm (corresponding to ligand-to-metal charge transfer from oxygen to titanium atom), 6-coordinated titanium species absorbs in the range 260–290 nm and anatase phase exhibits a band centred at 330 nm. In fact, the position of the absorption band red-shifts with increasing coordination number and a change of the geometry from tetrahedral to octahedral.281 The isomorphous incorporation of titanium is evidenced by an IR vibration band at 960 cm−1. The true origin of this band was a subject of debates but the most reliable assignment appears to be to antisymmetric stretching mode of a TiO4 unit.285
Isolated titanium atoms act as weak Lewis acids, which are able to coordinate molecules such as H2O, NH3, H2O2 and organic peroxides. In particular, in the case of peroxides, Ti withdraws electrons from adsorbed species making the O–O bond more susceptible towards a nucleophilic attack e.g. of an alkene, which can be then oxidized to the corresponding epoxide.
It is very important to note that the initial step, a titanosilicate catalysed reaction, is the weakening or even hydrolysis of one of the Ti–O–Si bridges, forming a Ti(OSi)3(OH) defective species (which is then able to extend its coordination).286 This is proof for the catalytic activity of extra-framework isolated titanium species, which are, most probably, attached to the framework by 2 or 3 oxygen bridges (especially in the case of post-synthesis titanium incorporation).222,245,258,287 Note however, that leaching of titanium from a titanosilicate zeolite is rarely observed.
Just as with titanium, tin can be isomorphously incorporated into the zeolite framework or it can be present in the extra-framework positions or to form the bulk SnO2 phase. Isomorphously incorporated tin atoms are the desired active sites. The tin species can be observed and to some extent distinguished using DR-UV/Vis spectroscopy. Similarly to titanium, 4-coordinated (framework) tin atoms exhibit an absorption band centred at 205 nm and bulk SnO2 absorbs at 280 nm.294,297–299 In contrast to titanium, a Sn active site can be easily hydrated extending its coordination number to 5 or 6 resulting in a red-shift of the absorption band to 220 nm resp. 255 nm. This may cause some confusion, because extra-framework Sn species (or small SnO2 domains) also absorb in this wavelength range (around 240 nm). The band shifts accompanying dehydration/rehydration can be easily observed and therefere can be used to distinguish qualitatively the framework Sn sites from SnO2 domains.294
The framework Sn site has a Lewis acid character and can be investigated by probes e.g. by adsorbed CD3CN, which exhibits a characteristic vibration band at 2311 cm−1. Physisorbed CD3CN is observed at 2273 cm−1.293 Boronat et al.300 and later in more detail Harris et al.294 showed that in fact, two distinct bands of CD3CN on Sn sites can be observed around 2311 cm−1 characterising two different types of sites. One of them (2308 cm−1) characterises adsorption on a closed site (Sn atom is bound by four oxygen bridges) while the other (2316 cm−1) characterises an open site, where two hydroxyl groups (one on Sn, one on neighbour Si) are present instead of one of the oxygen bridges. The latter sites are considered more active in sugar isomerisation.294 Note the similarity to opening of the titanium site (vide supra). However, in most publications these two bands are not distinguised. In some reports, a vibration band at about 970 cm−1 (ref. 297 and 301) is reported. A similar band is observed for titanosilicates (being considered as proof of titanium isomorphous incorporation)285 and vanadium silicates;302 however, for tin silicates its origin, as well as whether it proves Sn incorporation, is unclear.
An important feature of Sn-silicates, and, in fact, the origin of their unique catalytic activity, is their ability to bind and activate a carbonyl group. This is evidenced e.g. by a shift of the cyclohexanone ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibration band (48 cm−1 for Sn-Beta) towards lower wavenumbers.289,290,303
O) vibration band (48 cm−1 for Sn-Beta) towards lower wavenumbers.289,290,303
Sn active sites can also be studied using 119Sn-MAS-NMR spectroscopy. The SnO2 phase exhibits a sharp signal at −604 ppm.291,295,298,301 In contrast, 4-coordinated Sn sites are observed at −420 to −450 ppm in dehydrated samples. The signal of 4-coordinated Sn disappears upon hydration and a new signal appears in the range −690 to −740 ppm (characteristic of the hydrated 5- or 6-coordinated Sn).290,293,295,301 This observation is in accordance with the DR-UV/Vis observations (vide supra). It was mentioned above that there exist two types of framework Sn-sites (open and close) and these have been distinguished, besides IR analysis of adsorbed CD3CN, also in the 119Sn-MAS-NMR spectra: open site −423 ppm; closed site −443 ppm.304
6.2. Industrial processes
Zeolites dominate the world catalyst global market due to their enormous use in oil refining and petrochemistry with increasing applications in fine chemical synthesis and environmental catalysis. Fluid catalytic cracking (FCC) and hydrocracking are the most important processes using zeolites, in particular USY (ultra stable zeolite Y, FAU topology). FCC catalysts have been developed using hierarchical USY zeolites, stabilized by ion exchange with Rare Earth elements, showing improved hydrothermal stability, increased bottom conversion capacity and better product selectivity in comparison with a conventional commercial USY based catalyst.305 Additions of MFI zeolites are important to increase the “sharpness” of the catalyst to produce more propylene. Regardless of the knowledge gained on cracking of oil-based feedstocks, catalytic cracking of other bulky feedstocks remains widely investigated particularly over hierarchical zeolites.233 Thus, it has been applied to many diverse raw materials such as vacuum gas–oil,306 vegetable oils307 and polyolefins, as main components of plastic wastes.308Zeolites were firstly used in large-scale applications in the 1960s for cracking, which was followed by the breakthrough discovery of high-silica zeolites including the most important ZSM-5 (MFI topology). This enabled relating the catalytic behaviour of different structural types of zeolites to different topologies and understanding in detail the structure–activity–selectivity relationship. At present there are about 130 industrial processes using zeolite catalysts from about 850 of total processes.309 The combination of acid functionalities, uniform micropores providing shape selectivity and easy regeneration allowed zeolites to revolutionise the chemical industry through replacement of hazardous catalysts like H2SO4, HF, and AlCl3 in the early 1960s. Table 2 provides a list of the most important processes over zeolites indicating the type of zeolite catalyst employed in each process. An in-depth discussion of industrial applications of zeolites is outside the scope of this review, but for an excellent survey the readers are referred to the chapter by Abdo and Wilson.310
| Structural type | Catalytic process |
|---|---|
| a Zeotype – silicoaluminophosphate. | |
| FAU (zeolite Y) | Catalytic cracking, hydrocracking, NOx reduction, acylations |
| MOR (mordenite) | Alkane hydroisomerisations, hydrocracking, dewaxing, aromatic alkylations, olefin oligomerisation |
| MFI (ZSM-5, TS-1) | MTO, MTG, olefin cracking, olefin oligomerisation, aromatic alkylations, isomerisations, disproportionations, aromatisation, NOx reduction, selective oxidations, hydration, amination, Beckmann rearrangement, cyclodimerisation |
| *BEA (Beta) | Benzene alkylations, aliphatic alkylations, acetylation, Baeyer–Villiger reaction, etherification |
| LTL (zeolite L) | Alkane aromatisation |
| MWW (MCM-22) | Benzene alkylations |
| CHA (SAPO-34a) | MTO |
| AEL (SAPO-11) | Long-chain alkane hydroisomerisation, Beckmann rearrangement |
| ERI (Erionite) | Selectoforming |
| RHO (Rho) | Amination |
| TON (Theta-1) | Long-chain alkane hydroisomerisation |
It should be noted that further expansion of zeolites as industrial catalysts has been connected with successful isomorphous substitution of Si for Ti by Eni researchers.280 They succeeded in synthesising a TS-1 zeolite (MFI morphology) without the presence of aluminium. The lowest Si/Ti molar ratio is close to 30 and this zeolite is currently used in large scale applications for phenol hydroxylation.312 In addition to titanium, a Sn-containing zeolite (Sn-Beta, BEA) is also currently used for Baeyer–Villiger oxidation of some cyclic ketones to lactones using hydrogen peroxide as the oxidation agent. Last but not least, SAPO-34 (CHA) with an 8-ring channel system is used for the methanol-to-olefin (MTO) reaction, providing substantially higher selectivities to ethylene and particularly propylene when compared with MFI zeolites.
Hierarchical zeolites have been investigated in the following industrially relevant reactions:
• Aromatization of light alkanes and alcohols to produce aromatic hydrocarbons with applications in the formulation of fuels or as raw chemicals. Aromatization of hexane and propane has been investigated over Pt promoted mesoporous gallium-containing ZSM-11 zeolites, improved conversion and selectivity being obtained owing to the enhanced accessibility to the active extra-framework Ga species due to the presence of mesopores.313 ZnO-containing ZSM-5 zeolite catalysts with bimodal and trimodal hierarchical pore structures have been studied for the conversion of methanol to aromatics.173 The authors propose that the presence of highly dispersed ZnO clusters enhances the selectivity towards aromatics by catalysing the dehydrogenation pathway, whereas the hierarchical pore structure facilitates the transfer of reaction intermediates and thus accelerates the formation of aromatics.
• Isomerization and rearrangement reactions of different substrates, such as xylenes, epoxides and oximes.314–316 Hierarchical Beta zeolites have been investigated as catalysts for epoxide rearrangement reactions, being compared with hybrid zeolitic-mesoporous materials. When tested in the liquid phase rearrangement of cyclic, branched, and linear epoxides, hierarchical Beta zeolites exhibited the best combination of catalytic activity and selectivity due to enhanced accessibility and appropriate balance of Brønsted and Lewis acid sites.315 Likewise, hierarchical Beta zeolites with different Si/Al molar ratios showed significant improvements in the catalytic behaviour in liquid-phase Beckmann rearrangement of cyclohexanone and cyclododecanone oximes compared to conventional Beta samples due to a number of factors: faster intracrystalline diffusion, availability of non-sterically hindered mesopore/surface area and lower deactivation through product inhibition.316
• Incorporation of noble metals into hierarchical zeolites has allowed preparing catalysts for hydrotreating processes, such as alkane hydroisomerization and hydrogenation of aromatic compounds. Hydroisomerization and hydrocracking of n-decane, n-nonadecane and pristane (2,6,10,14-tetramethylpentadecane) over a hierarchical Pt/ZSM-22 zeolite noted the contributions of both the acid sites located in the pore mouths and within the micropores to the skeletal rearrangement and cracking reactions.317 Pd supported on a hierarchical Beta zeolite evaluated in both absence and presence of 4,6-dimethyldibenzothiophene showed a better sulphur tolerance than a Pd/Al-MCM-41 catalyst in hydrogenation of naphthalene and pyrene.318 Likewise, hierarchical Ni/Beta zeolites have been studied in the hydroreforming of the oil obtained from the thermal cracking of low-density polyethylene, showing to be an adequate catalyst for obtaining gasoline and diesel fractions that could be employed in the formulation of transportation fuels.169
• Transesterification reactions of fatty feedstock. Transesterification of fatty acids to produce bio-diesel is traditionally a domain of homogeneous base or acid catalysts (KOH, NaOH, H2SO4);319 however, heterogeneous catalysts are also a subject of investigation due to their obvious advantages. Among these, hierarchical Beta zeolites exhibited a remarkable activity in biodiesel production using microalgae lipids as feedstock. Hierarchical character of the catalyst is essential in this case.320 Likewise, hierarchical zeolite Beta has exhibited significant activity in the transesterification of triolein to afford methyl oleate.321
• Hydrodesulfurization of oil fractions has also been studied over hierarchical zeolites. Thus, zeolite L containing mesopores322 has been incorporated as a component of a catalyst for gasoline hydrodesulfurization, showing excellent performances in terms of desulfurization, aromatization, isomerization and preservation of the RON value compared with the catalyst into which ordinary microporous zeolite L was introduced.323 CoMo catalysts supported on ZSM-5 zeolite–alumina composites have been tested in the hydrodesulfurization of 4,6-dimethyldibenzothiophene.324
• Finally, an emerging and relevant field of application of hierarchical zeolites is their use in catalytic treatments for the removal of pollutants in gaseous and liquid streams. Hierarchical Cu/H-ZSM-5 zeolite-based systems, with improved Cu dispersion, have been employed with remarkable performance as a catalytic trap for the retention of propene and toluene, enabling their further conversion to carbon dioxide and water instead of being emitted in the exhaust.171 Cu-Modified ZSM-5 and ZSM-11 type zeolite catalysts, both containing mesopores, have been used as catalysts for direct NO decomposition, concluding that the presence of mesoporosity leads to a significant improvement of the catalytic activity regarding conventional zeolites.325 Fe-Containing ZSM-5 and ZSM-12 catalysts, possessing mesoporosity, have been tested in the selective catalytic reduction (SCR) of NOx with NH3, exhibiting a significantly higher activity than conventional samples.326 This difference has been assigned to both faster diffusion of reactants and products in the mesopores and better dispersion of the iron particles. Pd-Containing hierarchical ZSM-5 zeolites have been explored in hydrodechlorination of trichloroethylene in the aqueous phase, showing enhanced catalytic performance compared to the use of a pure microporous zeolite sample.327 Hierarchical ZSM-5 zeolites, containing iron, have been tested in H2O2 decomposition reactions in the absence and presence of an iron-complexing agent, as well as in oxidation of low and high molecular weight organic compounds by H2O2, noting that the presence of secondary porosity in the zeolite resulted in a significant improvement of the catalyst properties.328
6.3. Biomass transformations
The versatility of the catalytic properties of zeolites has allowed their expansion to new applications demanded by worldwide socioeconomic progress. This is the case of the search for new and sustainable routes to deliver for the needs of humanity driven by the climate change. Among these routes, there are all those processes based on the use of biomass as a raw material, such as the production of bio-based chemicals and biofuels within the “biorefinery” concept.329Biomass is defined as any kind of organic material of either animal or plant origin.330 Its renewable character, abundance and worldwide distribution have prompted its use as an energy source. This encouraging scenario is even more attractive when the biomass feedstocks comprise any material not competing with the food sector, such as forestry and agricultural residues.
The routes to convert biomass into both bio-based chemicals and biofuels can be grouped in terms of the chemical nature of the biomass feedstock (sugars, lipids or lignocelluloses). Some general aspects regarding the most relevant conversion routes are outlined here:
Sugars or starch as feedstocks are used to produce biofuels, in particular bio-ethanol. The process comprises saccharification or hydrolysis into simple sugars and fermentation. Recently, the use of sugars has been expanded to produce platform molecules by catalytic routes, such as isomerization (e.g. from glucose to fructose), dehydration (e.g. production of 5-(hydroxymethyl)furfural (5-HMF), furfural and levulinic acid) and hydrogenation (e.g. conversion of glucose and xylose into sorbitol and xylitol, respectively).
Lipids or oleaginous materials can be transformed into biodiesel by the transesterification reaction with alcohols. Together with bioethanol they are currently commercially available as biofuels and blended with conventional fossil-derived fuels in the transportation sector. Both of them represent the so-called “first generation biofuels” as they are produced from raw materials competing with the food industry. However, lipidic feedstocks may be achieved from non-edible plants, called energy crops, solving the sustainability concerns associated with the first generation biofuels. In addition, alternative technologies have been proposed, such as hydroprocessing and catalytic cracking, to yield hydrocarbons with similar properties to those of conventional fossil-fuels.
Lignocellulose, according to its more complex composition, has been evaluated as feedstock for different thermochemical and biochemical routes to obtain biofuels or bio-based chemicals. Syngas is the main product of gasification for Fischer–Tropsch synthesis. Sugars can be extracted from lignocellulose by means of hydrolysis. Such sugars are further processed into bioethanol (via fermentation) or into chemicals and/or biofuels by other catalytic processes. Residual lignin from the hydrolysis treatment can be pyrolyzed or gasified. Finally, pyrolysis of lignocellulose yields bio-oils which, after catalytic upgrading, may produce valuable chemicals and biofuels. This catalytic upgrading can be classified into two main groups: (a) hydroprocessing of the liquid bio-oil (hydrodeoxygenation; HDO) and (b) bringing the pyrolysis vapours into contact with a catalyst (catalytic pyrolysis) promoting dehydration and decarboxylation as preferred deoxygenation routes.
A detailed discussion of each technology is outside the scope of this review and it can be found in the literature.331–334 All these processes incorporate one or several catalytic steps. Zeolites may be especially relevant catalysts because of their acidic and porous properties.335–337 The main reactions over zeolites are summarized in Fig. 10. However, their application in the field of biomass transformation is quite challenging as they face several difficulties intrinsic to the features of the raw material: (a) complex composition, with bulky compounds and multiple functionalities; (b) presence of heteroatoms affecting the quality of the final product (e.g. oxygen in biofuels), interfering the catalytic processes (e.g. Na+, K+) or poisoning the catalyst (e.g. sulphur); (c) high water content, which may deteriorate the catalyst structure and activity, and (d) strong trend to form carbon deposits (coke) over the catalysts, causing their deactivation.
 | ||
| Fig. 10 Main catalytic routes of biomass conversion involving zeolites. | ||
This section focuses on the role of zeolite properties in the performance of the most relevant biomass transformation processes and, in particular, on the impact of moving from 3D to 2D topologies in terms of their activity, selectivity and stability to deactivation.
6.3.1.1. Catalytic pyrolysis of solid biomass. Zeolites exhibited excellent performance in catalytic pyrolysis of solid biomass, demonstrating their ability to increase the production of aromatics and, therefore, yielding final oils with lower oxygen content and higher quality as biofuels.332,333,338–343 Acid sites promote the dehydration of levoglucosan into furans, and their further conversion into aromatics by Diels–Alder reactions with light olefins. Brønsted acid sites are particularly active because of their higher acid strength. Li et al.344 tested zeolites with different acidity in the pyrolysis (500 °C) and subsequent catalytic upgrading (550 °C) of corncob hydrolysis residue using an Auger-type reactor coupled to a downstream fixed-bed reactor. Specifically, FAU and MFI zeolites were used as catalysts. In the case of the FAU zeolite, the strength of acid sites is rather low, whereas MFI presents both mild and strong acid sites. Accordingly, USY zeolites exhibited very low activity yielding oxygenate amounts (50%) very close to the non-catalytic bio-oil (51.3 area%). In contrast, ZSM-5 was able to catalyse the bio-oil transformation reducing the amount of oxygenated compounds below 10 area% and increasing the content of phenols and aromatics (above 80%).
Increasing the density of zeolite Brønsted sites is an effective route to upgrade pyrolysis-oils via deoxygenation and aromatization.333 The concentration of Brønsted acid sites in MFI zeolites from 17 to 212 has been explored.345–347 However, together with a higher yield of aromatics, the acidity of MFI at low Si/Al ratios has been found to be detrimental leading to the formation of gases and coke.
Alternatively, cation-exchange and impregnation with metals or metal oxides have been explored to modulate the acidity of zeolites to slow down the coke formation while keeping or even maximizing the deoxygenation and aromatization activity.329 The most successful results have been attained when metallic species, exchanged or impregnated on the zeolite, possess some intrinsic catalytic activity for any deoxygenating reactions. For instance, when Ga, Ni or Zn are either cation exchanged348 or impregnated338,349 in MFI, the selectivity to aromatics in the upgraded bio-oil is increased. Similar effects have been found with Pt dispersed on MFI in the catalytic pyrolysis of Miscanthus to promote cracking, hydrogenolysis, hydrocracking, and dehydrogenation reactions.350 It has been reported that impregnation of MFI with NiO–MgO also leads to a higher selectivity to alkylbenzenes and less coke formation.351 In particular, the authors found that using MFI (Si/Al ratio = 50) having 6% Ni + 2% Mg, the upgraded bio-oil contained the highest amount of hydrocarbon compounds, with a yield of 35.52 chromatogram area% (12.94% for aliphatic hydrocarbons and 22.58% aromatic hydrocarbons), versus 10.22% in the case of the non-catalytic run. Moreover, coke formation was half (1.65 wt%) of the value attained with the parent zeolite (3.75 wt%). Nevertheless, there is an optimum amount of metallic or metal oxide species to be incorporated into zeolites as excessive loadings may lead to a dramatic loss of acidity, especially Brønsted acid sites, also necessary to drive the formation of aromatics via cyclization and oligomerization of light components.352,353
In addition to acidity, porosity is also a key feature of zeolites in terms of activity, selectivity and deactivation resistance by coke deposition in catalytic pyrolysis.332,333,338 It is quite difficult to correlate its direct impact on the product yield and distribution as changes in porosity are usually accompanied by variations in acidity (strength and accessibility). However, there is consistent evidence that zeolites with too small micropores (e.g. SAPO-34, with 6 and 8-rings) are hardly active in upgrading pyrolysis vapours, which are mostly composed of bulky molecules that cannot access the internal zeolite space.354 On the other hand, zeolites with large mesopore volumes39 or relatively large micropores (e.g.*BEA and FAU, having 12-membered rings)355,356 are prone to generate more coke as polymerization of large molecules is less restricted. The combination of acidic properties and a micropore size of 10-rings makes ZSM-5 one of the most promising zeolites for catalytic pyrolysis.
6.3.1.2. Hierarchical zeolites in catalytic pyrolysis. Pyrolysis vapours contain many large molecules, including phenolic oligomers coming from lignin decomposition, whose dimensions are too large to enter into any zeolite micropores. For that reason, zeolites with larger external surface areas and hierarchical structures have also been recently explored. Typically, good results have been obtained over hierarchical ZSM-5 due to a strong aromatization activity of this zeolite, which leads to the production of a highly deoxygenated bio-oil.357–359 In the case of hierarchical ZSM-5 prepared by desilication,359 it has been proposed that the mesopores can be seen as “highways” where big molecules (such as levoglucosan) can diffuse to the micropore mouths, where the Brønsted acid sites present are active for their conversion into small fragments. Moreover, mesopores also promote the transport of reaction products, thus enhancing the selectivity of mono-aromatic hydrocarbons.
It seems that the key is a right balance between high mesopore accessibility and external acid density. By this way, conversion of bulky char precursors and oxygenates could be promoted on the external surface, hindering their polymerization to coke within the micropores, and increasing the aromatic yields. This was evidenced by Gamliel et al.,358 when comparing different synthetic routes to introduce mesoporosity in MFI zeolites: (a) desilication with organic hydroxides; (b) desilication with NaOH at different concentrations (mild and strong desilication) and (c) surfactant-assisted desilication. The best performance was achieved with mildly-desilicated zeolite as it was able to increase the production of monoaromatics from 11.3% (measured as carbon yield) for the parent zeolite to 19%. However, a harsher desilication, despite generating a larger mesopore volume, reduced the monoaromatics to 15.9% and increased the coke yield (from 43.4 to 46.5%, measured as coke + char).
Hydrodeoxygenation of bio-oils (from biomass pyrolysis) and fatty compounds has also been investigated using hierarchical zeolites as supports of different active phases. Thus, hydrodeoxygenation of bio-oil model compounds has been probed using catalysts based on Pt and Ni2P, respectively, supported on hierarchical ZSM-5 zeolites.176,360 Likewise, Ni supported on hierarchical USY zeolites has exhibited a high efficiency in the hydrodeoxygenation of fatty acids, esters, and palm oil.361
6.3.1.3. 2D vs. 3D zeolites in catalytic pyrolysis. Very recently, MCM-22 zeolite samples (MWW framework) with different Si/Al ratios have been studied in the catalytic pyrolysis of acid-washed wheat straw at 400–450 °C.362 The materials were characterized by a significant contribution of external surface area (160–190 m2 g−1) and a higher concentration of Brønsted acid sites than Lewis ones. In terms of overall deoxygenation degree and aromatics production, both zeolites exhibited similar behaviour being worse than the MFI reference catalyst. In addition, a higher amount of coke was deposited over the MWW zeolites. This coke was lighter and presented significant oxygen content, denoting that its formation was mostly mediated by oxygenated compounds.
2D zeolites have been applied in catalytic pyrolysis of solid biomass to take advantage of their higher external surface areas and pore dimensions, diminishing the effect of diffusion, for converting bulky compounds in primary pyrolysis vapours. Lee et al.363 employed unilamellar mesoporous MFI nanosheets (UMNs) to upgrade the pyrolysis vapours generated from the individual fractions of lignocellulose: cellulose, hemicellulose and lignin. For comparison purposes, the same reactions were carried out without a catalyst (thermal pyrolysis) using Al-SBA-15 as the representative of an acid mesoporous catalyst. The UMN catalyst was composed of randomly assembled zeolite nanosheets with a thickness of 2.5 nm (see Section 5.2), BET area of 600 m2 g−1 and a mean mesopore size of 6.3 nm. BET area and mean pore size of the Al-SBA-15 sample was 500 m2 g−1 and 7.8 nm, respectively. Regarding acidity, unilamellar MFI exhibited both weak and strong acid sites whereas the ordered mesoporous Al-SBA-15 contained only weak acid sites. Due to the superior acidity, the UMN catalyst exhibited higher activity for cracking and deoxygenation and it produced a bio-oil with lower oxygen content and with a better overall quality. While monoaromatics were hardly detected in bio-oils from thermal pyrolysis of each lignocellulose fraction, their production was promoted using the catalysts. This effect was especially important over the zeolite which almost tripled the monoaromatics selectivity compared to the Al-SBA-15 catalyst (e.g. 16% of total distribution of compounds vs. around 5%, respectively, in catalytic pyrolysis of lignin).
MCM-22 and its delaminated derivative, ITQ-2, have been tested in the catalytic pyrolysis of rice husk by Naqvi et al.364–367 at 450 °C in a lab scale fixed-bed pyrolyzer and compared to conventional zeolites with different micropore sizes (8, 10 and 12-rings). Despite scarce information that is given regarding the formation of coke, some conclusions were provided. As expected, small pore zeolites (8-rings: SAPO-34) did not produce aromatics and exhibited the lowest deoxygenation capacity. Large pore size zeolites (12-rings: MOR) exhibited high solid yields (char + coke) and low aromatics production. Medium pore sizes with 10 membered rings, and specially ZSM-5 and ITQ-2, resulted in the best performance with similar results. The combination of medium pore size and high acid strength of ZSM-5 was considered to be mainly responsible for achieving the highest deoxygenation degree and increased aromatics production when compared with thermal pyrolysis. In the case of ITQ-2, its higher external surface area than the parent MCM-22 (442 m2 g−1vs. 100 m2 g−1 of MCM-22) effectively reduced the steric hindrance allowing a higher conversion of sugar compounds, which resulted in higher aromatics production and the second position in the deoxygenation capacity.
Lamellar and pillared MFI zeolites, and their modifications by MgO or ZnO impregnation, have also been studied for the catalytic pyrolysis of eucalyptus woodchips in a lab-scale fixed-bed reactor operating at 500 °C.368 The use of zeolites promoted decarboxylation and decarbonylation as deoxygenation routes for pyrolysis vapours. The parent layered ZSM-5 and pillared ZSM-5 produced the monocyclic aromatic hydrocarbons most effectively. Their share in the total bio-oil increased from 1.4 (thermal pyrolysis) to 11.9 and 7.5%, respectively. The modification with both metal oxides remarkably reduced the porosity and acidity of the zeolitic supports. As a result, the formation of aromatic hydrocarbons was reduced in favour of oxygenated aromatics (3,4,5-trimethoxytoluene, 1,2,4-trimethoxybenzene and 2,6-dimethoxyphenol being the most abundant, Fig. 11). On the other hand, formation of coke and undesired polyaromatic hydrocarbons was also reduced.
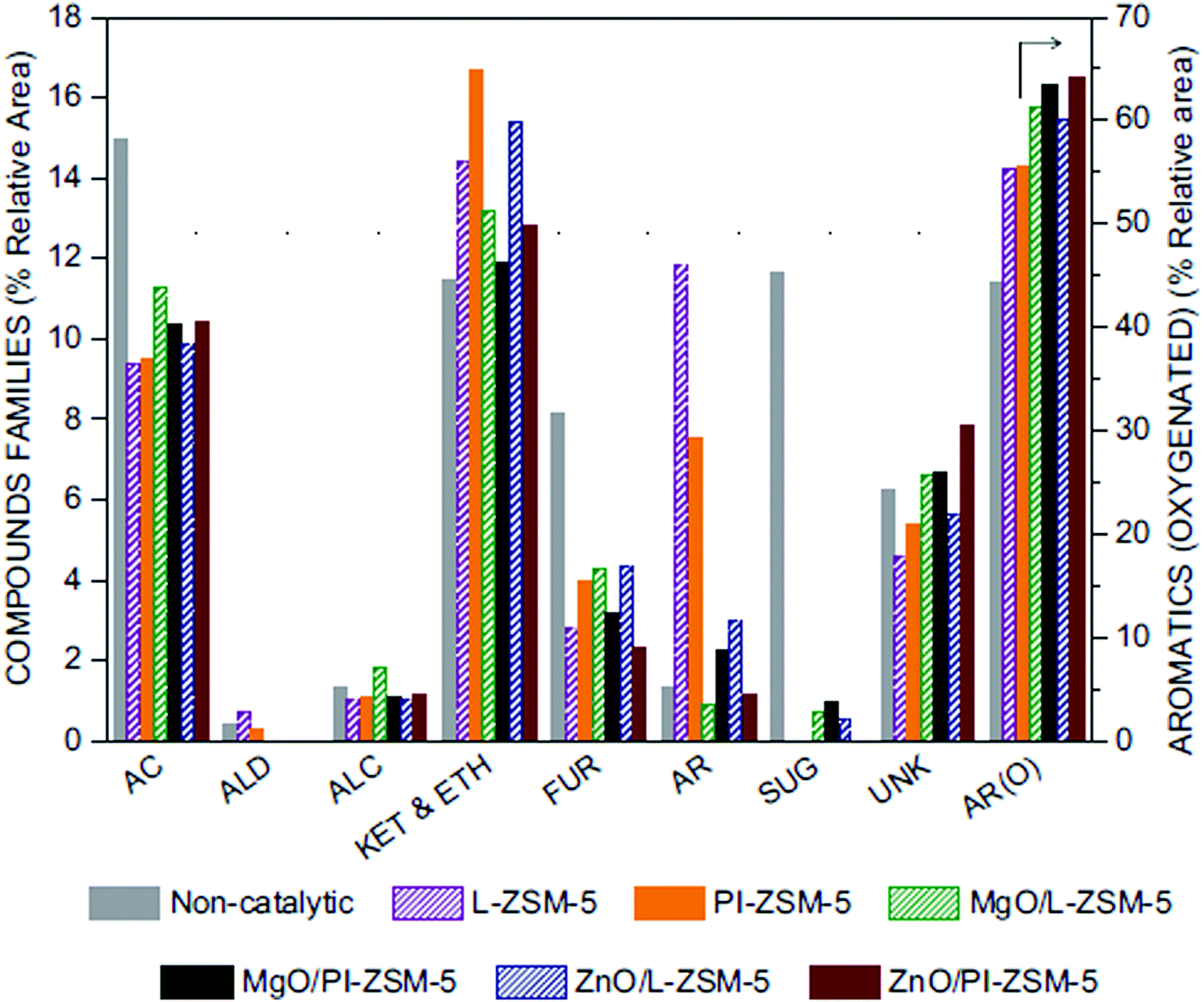 | ||
| Fig. 11 Impact of catalyst type on the deoxygenation degree of the bio-oil obtained by catalytic pyrolysis of rice husk. Reprinted with permission from ref. 368. | ||
Numerous condensation reactions have been traditionally applied in organic chemistry where homogeneous acid or base catalysts are mostly used. Homogeneous catalysts cannot be recovered, and there are economic and environmental concerns to initiate searching for solid catalysts as substitutes. One example is aldol condensation of aldehydes or ketones in the presence of homogeneous base catalysts, such as sodium hydroxide.372–374 As aldehydes and ketones are present in pyrolysis bio-oils in significant amounts, this reaction has been considered as a very reasonable way to upgrade them. Aldol condensation can be performed in both liquid and vapour phase. Particularly the vapour phase option allows its direct coupling to the exhaust of the pyrolysis reactor without necessity to condensate the primary vapours. Although most of the solid catalysts under investigation possess basic properties, some acidic materials (in particular zeolites) have also been tested showing promising results.375–378 Large pore zeolites, especially *BEA zeolites, were found to be good catalysts of aldol condensation. However, the existence of Brønsted acid sites in this kind of zeolite promotes the catalyst deactivation by coke formation. Therefore, efforts in the development of catalysts with enhanced resistance to deactivation have been made. Puértolas et al.379 attempted to modulate the USY zeolite acidity by impregnation with a second active phase and used the resulting catalysts in aldol condensation of oak wood pyrolysis vapours at 400 °C. The authors modified the zeolite by K-exchange or supporting MgO. The exchanged catalyst exhibited better performance than MgO/USY (24.7% oxygen reduction vs. 15.2%, respectively), but at the expense of producing more coke (26.1 vs. 16. 5 wt%, respectively).
Similarly, hierarchical ZSM-5 zeolites have shown much higher catalytic activity than their conventional counterparts for aldol condensations involving large molecules, especially in the synthesis of vesidryl.370 Impregnation of hierarchically structured ZSM-5 zeolites with metal cations (Sn, Cu, Ni, or Mg) has led to catalysts that promote selectively bio-oil oxygen removal reactions. Ketonization reactions of acids with aldehydes to produce ketones seem to be favoured over the Lewis acid sites created after incorporation of Mg cations by ion exchange.371
6.3.2.1. 2D vs. 3D zeolites in aldol condensation. Owing to their enhanced external surface areas compared to conventional zeolites, two-dimensional zeolites are considered more appropriate catalysts for aldol condensation reactions involving bulky compounds, as they occur in primary pyrolysis vapours from biomass. Kikhtyanin et al.380 explored a variety of layered zeolites belonging to the MWW family in aldol condensation of furfural and acetone. A batch reactor operating at 100 °C under autogenous pressure was employed and their activity was compared with that of Beta zeolites. All the MWW-type catalysts (MCM-22 and MCM-49, characterized by a 3D layered structure, as well as their delaminated and pillared derivatives, MCM-56 and MCM-36) were more active than the *BEA zeolite and other conventional zeolites previously tested by the same authors.378 Moreover, selectivity to FAc (4-(2-furyl)-3-buten-on) was always above 80%. Among the four MWW-type zeolites, MCM-22 gave the best performance, which was attributed to the participation of the active sites located within the supercages, in addition to those located on the external surface. On the other hand, the pillared MCM-36 showed the lowest conversion of its family, denoting that introducing the silicate pillars deteriorated its activity. Moreover, MCM-36 was deactivated much faster by coke due to polymerization reactions on the easily accessible external active sites created by delamination and pillaring.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :225 Sn:glucose molar ratio) as the catalyst for 60 min at 110 °C. The reaction occurred without any decrease in catalyst activity even at pH = 2. This is beneficial either for combination of the isomerisation with acid hydrolysis of starch to produce glucose for isomerisation, or to combine the isomerisation with further acid catalysed dehydration leading e.g. to 5-HMF.
:225 Sn:glucose molar ratio) as the catalyst for 60 min at 110 °C. The reaction occurred without any decrease in catalyst activity even at pH = 2. This is beneficial either for combination of the isomerisation with acid hydrolysis of starch to produce glucose for isomerisation, or to combine the isomerisation with further acid catalysed dehydration leading e.g. to 5-HMF.
Another interesting isomerization reaction using acidic zeolites is the conversion of xylose into xylulose.385,388–390 M. Paniagua et al.389 compared zeolites FAU (Y), *BEA (Beta) and MFI (ZSM-5), observing the first one as the most active leading to xylulose yields up to 47%. Li et al.,391 used periodic DFT calculations to determine the mechanism of glucose isomerization to fructose over a variety of Sn-zeolites (MOR, *BEA, MFI and MWW). Despite a higher glucose adsorption energy caused by the narrower 10-ring micropores of MWW, it was predicted that Sn-MWW could provide high conversions due to the presence of a small fraction, but highly reactive, of accessible sites.
The presence of mesopores to overcome limited accessibility of the active sites was also found to be beneficial in isomerisation reactions. Hierarchical Sn-ZSM-5 materials have been able to isomerize selectively glucose, xylose, and lactose, which suffer from access or diffusion limitations in purely microporous Sn-ZSM-5.392 Likewise, Sn-Beta facilitated a selective conversion of sugars into important intermediates for producing alkyl lactate.393 Ethanolysis of renewable furfuryl alcohol has been performed over hierarchical ZSM-5 with high yield into ethyl levulinate.394 In the same way, hierarchical niobium-containing zeolites have been applied for isomerization of dihydroxyacetone to alkyl lactate and lactic acid with a yield over 95%.395
6.3.3.1. 2D vs. 3D zeolites in isomerization reactions. The application of layered zeolites in the isomerization reaction of biomass-derived compounds has been scarcely explored so far. Sn-self-pillared-pentasil (Sn-SPP)293 and pillared Sn-MWW(SP)-SSE396 (analogue of aluminosilicate MCM-36) were demonstrated to be very effective catalysts in glucose to fructose isomerisation using an alternative approach, when glucose is turned into ethylfructoside (which helps in shifting the isomerisation equilibrium) and fructose is obtained after hydrolysis of the fructoside with water at the end of the reaction. Fructose yield up to 60% has been reported. These self-pillared resp. pillared catalysts isomerize even disaccharides (lactose to lactulose resp. maltose to maltulose), where catalysts such as Sn-BEA fail because the substrates are too sterically demanding.293,396
Isomerization of β-pinene, as a representative of terpenes present in the essential oil of pine trees, has been attempted over MCM-22 and its hierarchical microporous-mesoporous derivatives obtained by acid post-treatment.397 The purpose of such a reaction is to produce a mixture of limonene and camphene, compounds of particular interest in the field of cosmetics and medicine. An excellent performance was obtained over the hierarchical MCM-22, with full conversion and 93.7% selectivity to the mixture of limonene and camphene (76.2% limonene and 17.5% camphene) under the reaction conditions employed. Such good catalytic behaviour was attributed to the right balance between acidic and textural properties. Moreover, the catalyst could be efficiently recycled by mild washing treatments, denoting good structural stability and removal of organic compounds facilitated by the existence of mesopores.
The dehydration of hexoses (glucose and fructose) to 5-HMF revealed that the yield of 5-HMF is highly dependent on the reaction conditions, type of sugar used and zeolite properties. For instance, fructose leads to higher 5-HMF yields than glucose. On the other hand, Brønsted acid sites are poorly active in the conversion of glucose; thus most efforts are made in the development of zeolitic catalysts with well-balanced Lewis and Brönsted acidity. This can be addressed by preparing mixtures of catalysts, such as Ti-Beta + Sn-Beta, as Lewis center carriers, and HCl as Brönsted acid,398 or Sn-Beta (as Lewis center carrier) + Amberlyst (as Brønsted center carrier).399 However, it would be more practical to apply bifunctional zeolite catalysts containing both types of acidity.
The use of zeolites to produce furfural by dehydration of biomass derived xylose is also a field of interest as it would allow replacing the hazardous and non-recoverable sulphuric acid used industrially as a catalyst. However, it is a tricky issue because dehydration of C5 sugars to furfural, which is promoted by Brønsted acid sites, can interfere with isomerization reactions to -xylulose, which are catalysed by Lewis acidity.332 A variety of 3D zeolites have been used for xylose dehydration, including MFI (ZSM-5), MOR (Mordenite), FAU (Y), *BEA (Beta) and silicoaluminophosphates (SAPO-5, SAPO-11, SAPO-40).400–404 Excellent results have been reported while combining the use of Mordenite with a solvent composed of γ-valerolactone (GVL) and 10 wt% of water.403 Under these conditions 80% yield of furfural was achieved, which was attributed to hindering of side condensation reactions between furfural and pentose intermediates. More recently, Iglesias et al.404 reported that zeolites with smaller pores (ZSM-5) are poorly active in xylose dehydration. They also found that the nature of the acid sites was the key parameter driving the main reaction pathways for transforming xylose into furfural. Thus, Lewis acid sites promoted the direct dehydration of xylose, whereas Brønsted-type catalysts produce alkyl xylosides as intermediates. In addition, Beta-zeolite with a proper balance between Brønsted and Lewis acidity was postulated as the most promising catalyst to maximize furfural production.
6.3.4.1. 2D vs. 3D zeolites in dehydration reactions. Lima et al.400 applied a delaminated zeolite (Si/Al = 29) as a catalyst for the liquid phase dehydration of xylose to furfural at 170 °C, using a water–toluene biphasic reactor system. The delaminated zeolite was prepared by swelling and ultrasonication of a laminar precursor of Nu-6(2), resulting in a BET surface area close to seven times higher (151 m2 g−1) than that of the parent material (25 m2 g−1). Such an increase in the textural properties and its consequent accessibility enhancement, led to a significant improvement with a furfural yield of 47%, higher than that (34%) obtained over H-Mordenite used as a reference catalyst. Moreover, the authors evidenced no Al-leaching and quite stable activity in recycling.
Very recently, Abdelrahman et al.405 proposed an alternative thermochemical pathway to produce butadiene from biomass based on the dehydration of sugars to furfural, followed by decarbonylation and hydrogenation to THF and a final dehydration and decyclization step. Regarding the last catalytic step, dehydration and ring opening of THF, they compared a phosphorous-containing siliceous self-pillared pentasil sample (P-SPP) with a wide variety of catalysts, including ZSM-5, HY, Sn-Beta, ZrO2 and silica–alumina. Among them, P-SPP demonstrated very promising performance with very high selectivity to butadiene (87–92%) and conversions up to 83%.
Another interesting application of zeolites based on dehydration reactions of biomass-derived molecules is the conversion of glycerol to acrolein. Acrolein is an intermediate for the production of acrylic acid, the platform molecule used in the manufacture of paints, plastics, adhesives and super-adsorbents. Recently, two similar studies employing MWW-type catalysts in this reaction have been reported.406,407 In both cases, MCM-22, pillared MCM-36 and delaminated ITQ-2 zeolites were compared under gas phase conditions. ITQ-2 stood out as the best catalyst in terms of conversion, selectivity (Fig. 12) and regeneration capability. The observed features were explained by the higher accessibility of their active sites (ITQ-2 possesses a house-of-cards morphology) together with an easier removal of coke from mesopores.
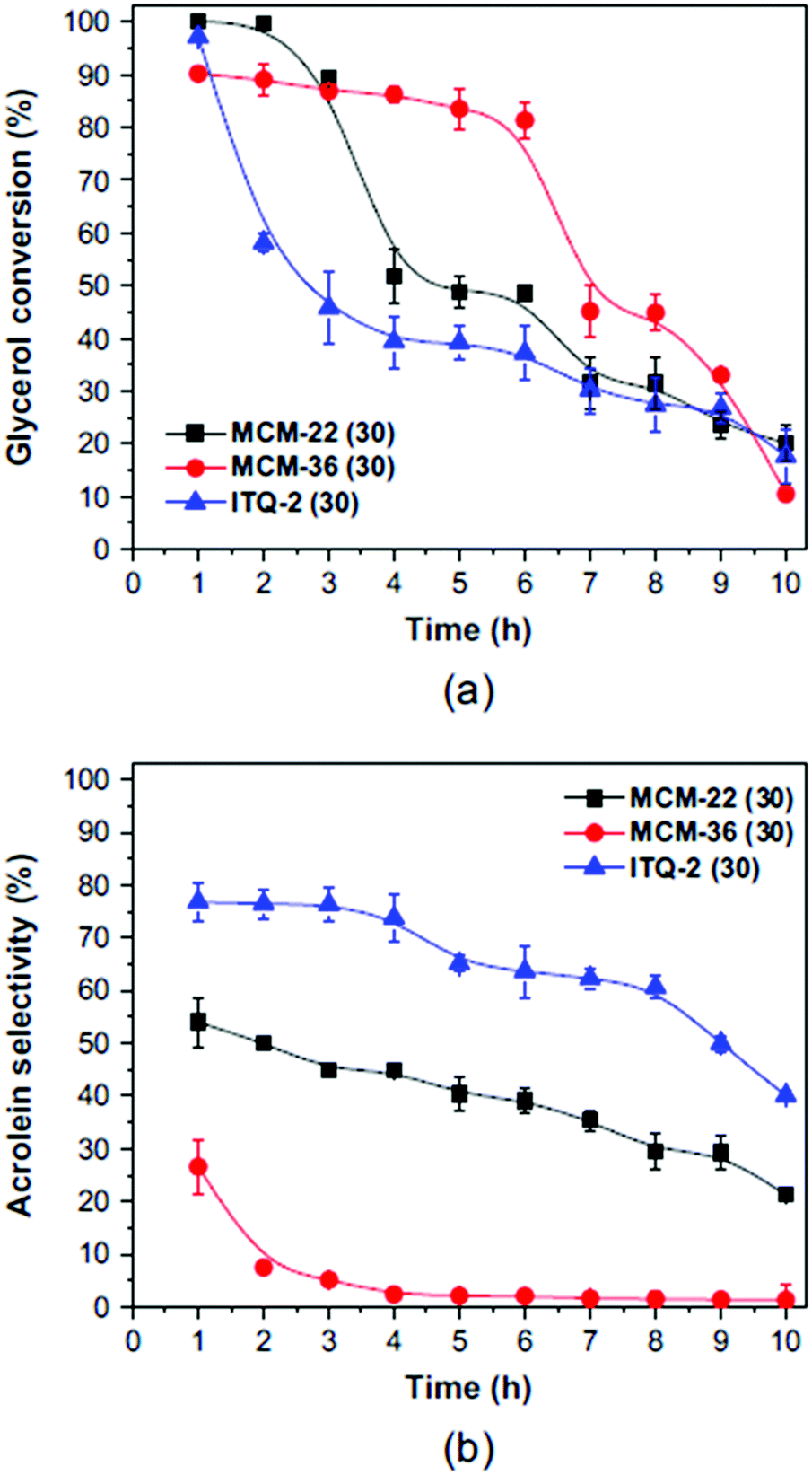 | ||
| Fig. 12 Glycerol conversion and acrolein selectivity over MWW catalysts. Reprinted with permission from ref. 407. | ||
One example is the study reported by Yoon et al.,408 in which they demonstrated an improved activity of Rh supported on swollen MCM-22 and its pillared derivative MCM-36 compared with other acidic supports (MCM-41 and silica–alumina aerogel), when they were applied in the hydrodeoxygenation (HDO) of guaiacol and 1,3,5-trimethoxybenzeze (1,3,5-TMB). Among the zeolites tested, MCM-36 was particularly the most efficient catalyst, due to a higher dispersion of Rh particles. Despite Rh dispersion onto MCM-41 and silica–alumina aerogel being even higher, the activity over Rh/MCM-36 was the largest due to the higher accessibility of the acid sites located on the external surface, thus, minimizing mass transfer limitations.
Alkylation using conventional zeolites, layered 2D and 3D acidic zeolites has been explored as a useful reaction for either improving the H/C ratio in bio-oils,409 producing biofuels410 or for the production of high value chemicals, such as xylenes, ethylbenzene and ethyltoluene.411 Large pore zeolites are more convenient than small and medium pore size ones due to lower mass transfer restrictions. Among 2D zeolites, delaminated ITQ-2 is specially active and selective to alkylation products thanks to the enhancement of accessible surface and to the shorter diffusion path lengths.
Encapsulation of cobalt clusters in a hierarchical ZSM-5 zeolite has afforded a catalyst for the direct production of middle isoparaffins412 by means of the hydrogenation of carbon monoxide into hydrocarbons (the Fischer–Tropsch (FT) synthesis). In the same way, hierarchical zeolite Y supported Co catalysts has been tested in FT synthesis.413 Middle hydrocarbons, with a remarkable selectivity to isoparaffins, were the main products due to the optimized hydrocracking and isomerization function brought by the hierarchical zeolite. Also for this reaction, addition of Mn to Co/Na-hierarchical zeolite Y caused a significant improvement in the diesel fuel selectivity by suppressing the formation of CH4 and lighter hydrocarbons.414
Delaminated and pillared MCM-22 zeolites have also been applied as a support of cobalt for the FT reaction.415 It has been found that using either delaminated or pillared derivatives allows narrowing the product distribution of the FT reaction as the selectivity to C4–C12 molecules is increased. Nevertheless, compared to the parent MCM-22, the delaminated support leads to a higher proportion of C21+ hydrocarbons, which was attributed to the loss of acidity caused during the swelling and delaminating process. Last but not least, a Co impregnated nanosponge ZSM-5 catalyst exhibited high selectivity for branched hydrocarbons and olefins in the gasoline range (C5–C11).416 The above observed selectivity towards lighter and branched hydrocarbons results from close intimacy between the metal and zeolite active phase resulting in an in situ processing of the primary (linear) FT hydrocarbons.
6.4. Selective oxidation/reduction reactions
Selective oxidation is an important part of heterogeneous catalysis. For instance the world ethylene oxide production capacity was 34.5 million tons per year in 2016417 and all the production was perfomed by direct oxygen or air oxidation of ethylene over a silver oxide-based catalyst. Here we discuss 3D and 2D zeolite based oxidation catalysts including epoxidation as one of the important reactions.Zeolites with Lewis acid sites (namely titanosilicates, tin-silicates, and zirconium silicates) catalyse a number of selective oxidation/reduction reactions, of which the epoxidation, hydroxylation of aromatics, ammoxidation, Baeyer–Villiger oxidation and Meervein–Ponndorf–Verley reduction/Oppenauer oxidation are the most important; the titanosilicate catalysed propylene epoxidation, cyclohexanone ammoxidation and phenol hydroxylation are carried out even at the industrial scale.312,418,419 One of the strong points of oxidation reactions catalysed by zeolites is the use of a “green“ and simple oxidant: hydrogen peroxide. Unfortunatelly, to our best knowledge, there are no data (except for phenol hydroxylation over IEZ-Ti-CDO (denoted as Ti-COE-3, Ti-COE-4420)) on phenol hydroxylation and ammoxidation using 2D titanosilicates.
C double bond is an important selective oxidation reaction and epoxides are important reactive intermediates. Except for ethylene oxide and propylene oxide, it is traditionally performed by the so-called chlorhydrine route in which a hypochloric acid is added to the CC bond and the epoxide is obtained by subsequent dehydrochloration with a strong base. Alternatively, it can be performed directly using hydrogen peroxide or organic hydroperoxides (e.g. tert-butyl hydroperoxide (TBHP) or cumeme hydroperoxide). However, peroxides require activation, which can be performed effectively with titanosilicate zeolites. Titanium-zeolite catalysed epoxidation occurs under mild conditions in a liquid phase and is advantageous for suppression of consecutive reactions, safety, and particularly for a considerable side product/waste reduction when hydrogen peroxide is used. Data from a continuous setup experiments are rare (Deng et al.421 is one of the few examples); typically the epoxidations are carried out batchwise using acetonitrile or methanol as solvent at temperatures between 30 and 60 °C (depending on substrate).
Titanium silicalite-1 (TS-1)280 and Ti-MWW422,423 based materials are the most studied. Both of them exist in 3D and 2D forms; see recent review.424
The substrates subjected to the epoxidation can be divided into two groups. Small substrates (linear alkenes up to about C8, allylchloride, allylalcohol, etc.) and bulky substrates (cyclic alkenes, terpenes, branched alkenes etc.). For the first group, the highest yields and selectivity (when using H2O2 as oxidant) are obtained using conventional 3D catalysts while their two dimensional analogues provide significantly lower conversions and yields, although e.g. the titanium content and characteristics of the titanim species are the same. Such behaviour is nicely documented in epoxidation of 1-hexene e.g. in the case of conventional TS-1 (conversion 5.5% after 2 h) vs. nanosheet TS-1 (conversion 3.8 resp. 3.8% after 2 h under the same conditions).257,258 Similarly, IEZ-Ti-MWW (Si/Ti = 94)425 fails in comparison with Ti-MWW (Si/Ti = 44)426 (conversion 12.1% vs. 54.2% after 2 h, under the same conditions, exhibiting similar selectivity (95%)). Note that the difference in titanium content is about 2 times while the difference in conversion is almost 5 times. Qualitatively the same results were also observed when comparing data for 1-octene.424,427,428 Reasons for such behaviour are not fully clear, but different hydrophobicity of the 3D frameworks, confinement effects or slight differences in Ti sites geometry are assumed to be responsible.
A direct relationship has been identified also between the catalytic activity and the mesopore surface area of hierarchical TS-1 in 1-hexene and cyclohexene epoxidation (Fig. 13).429,430 In addition, the connectivity between micro- and mesopores may have also a relevant effect on the catalytic properties.
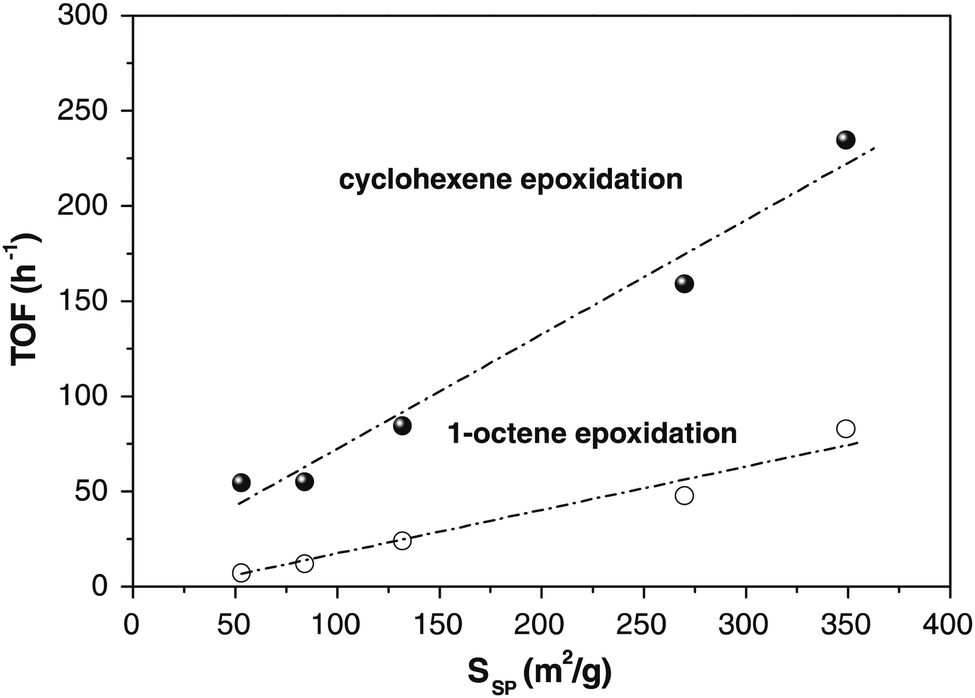 | ||
| Fig. 13 Variation of the catalytic activity (TOF) in olefin epoxidation with the surface area related to the secondary porosity of TS-1 samples. Reprinted with permission from ref. 430. | ||
For bulky substrates, the data show the expected advantages of the open structures of hierarchical and 2D catalysts. Cyclohexene is the most frequently reported bulky substrate, although it is the smallest, and it is strongly prone to side reactions such as allylic oxidation (using the H2O2). Nevertheless, it is epoxidised with higher yield and selectivity over IEZ-Ti-MWW catalysts426,431,432 (e.g. yield 33.3% after 5 h) in comparison with Ti-MWW (yield 4.7% after 5 h) and TS-1.
Cyclooctene and cyclodecene oxides are more stable than cyclohexene oxide under the reaction conditions and less prone to oxidation of allylic carbon and in contrast their diffusion even in large pore titanosilicates such as Ti-Beta is more limited. The difference between cyclooctene conversion over TS-1 (0.2% after 2 h) and layered TS-1 (2.8% after 2 h) reaches an order of magnitude.258 In different experimental setups, silica–titania pillared TS-1 (Si/Ti = 20) provided 21% conversion222 and silica–titania pillared Ti-IPC-1-PITi (Si/Ti = 69) provided even 35% conversion245 of cyclooctene both exhibiting 80% selectivity while Ti-Beta (Si/Ti = 119) provided only 14% conversion with 32% selectivity and TS-1 8% conversion with 42% selectivity. Note that the silica–titania pillared catalysts bear additional titanium sites on their external surface. In the case of cyclodecene the difference between conventional and layered TS-1 is expectably higher (TS-1: 0.2% conversion vs. layered TS-1 5.1% conversion258).
Epoxidation over 3D medium pore titanosilicates (TS-1, Ti-MWW) using TBHP as an oxidant is not possible (or at least not convenient) because the TBHP is too bulky to penetrate the microporous system and therefore only the active sites on the external surface participate in the reaction (e.g. TS-1: conversion 1.2% after 2 h,221 Ti-MWW: 4.2% after 2 h210 in epoxidation of cyclohexene). In contrast, hierarchical catalysts (e.g. TS-1 prepared using silanized zeolite seeds433), the 2D catalysts, have well accessible external surface of the layers with titanium sites434 and therefore, they are much more effective (layered TS-1: conversion 17% after 2 h under the same coditions210). Note, that in contrast to the epoxidation with hydrogen peroxide, epoxide selectivity of 90–100% is observed even at cyclohexene conversion above 40%.424 In addition to 2D zeolites, also mesoporous molecular sieves (e.g. Ti-MCM-41) are effective catalysts, when TBHP is used instead of H2O2.210,435 Recently, a layered silicate (not a zeolite) HUS-7 intercalated with Ti(IV) acetylacetonate was reported to be very active in epoxidation of cyclohexene with TBHP (TOF up to 266 h−1). In this case, the catalyst is not calcined and the mentioned TOF is achieved in the 3rd recycling of the catalyst. Changes in the catalyst structure during the reaction were observed (changing of the interlamellar spacing) but titanium species leaching was not observed. Thus the results can be interpreted as successful immobilization/stabilization of a titanium organometallic complex, which provides the catalytic activity.436
The results observed for the cyclic alkenes are illustrative also for substrates like norbonene. The readers are invited to check the review424 for a detailed comparison.
BV oxidation of 2-adamantanone is the most used model reaction when investigating novel BV catalysts. The kinetic diameter of 2-adamantanone is about 7 Å which makes it too bulky not to enter the 10-ring pores but small enough to enter large pores of Sn-Beta. Its polycyclic structure also brings high stability and rigidity to the molecule and therefore high selectivity of the BV oxidation is usually observed (typically >98%).301 Similar behaviour can be observed e.g. in BV oxidation of norcamphor.303 On the other hand cyclopentanone and cyclohexanone are not so easy to oxidise and observed selectivities are usually lower (<80%).246,442 In most cases the above 2D catalysts (particularly the MFI and MWW families) provide only similar or lower conversions and lactone yields in BV oxidation of 2-adamantanone with hydrogen peroxide (vide infra). In this sense, a strong difference in comparison with titanosilicate catalysed epoxidation with hydrogen peroxide was observed, where the 2D catalysts brought strong enhancement of the catalytic performance. Also within a group of germanosilicate zeolites tested in BV oxidation of 2-adamantanone, ITQ-17 (BEC, Beta polymorph C) was also the most active catalyst, followed by extra-large pore zeolite IM-12 (UTL)441 but in comparison with Sn-UTL and Sn-Beta, germanosilicate UTL provided significantly lower conversion in BV oxidation of 2-adamantanone and cyclohexanone.446
Luo et al.296 tested Sn containing MFI nanosheets, which were considerably more active than 3D Sn-MFI (initial turn-over-frequency TOF = 38 h−1 resp. 5 h−1) but Sn-Beta provided initial TOF = 210 h−1. The authors suggest that lower TOF of Sn-MFI nanosheets (and Sn-MCM-41) in comparison with Sn-Beta can be caused by the more hydrophilic character of the 2D (resp. amorphous silica) catalyst which results in stronger concurrent adsorption of water (introduced with the H2O2). Decreasing of the H2O2 dose (and therefore also water concentration in the reaction mixture) resulted in an increase of TOF (Sn-MFI nanosheets 86 h−1). On the other hand, silylation (hydrophobisation) of the surface resulted in blocking of the access to the Sn sites rather than improvement of the catalytic activity. Liu et al.295 investigated BV oxidation of 2-adamantanone with Sn-MWW based catalysts in comparison with Sn-MFI and Sn-Beta. An advantage of the partially delaminated structure of Sn-MCM-56 over 3D Sn-MWW was observed, but still Sn-Beta provided higher conversion (Sn-Beta 57% > Sn-MCM-56 42.3% > Sn-MWW 29.5% > blank 3.5% after 2 h at 90 °C). Recently Ren et al.396 reported pillared Sn-MWW(SP)-SSE (MCM-36 analogue) to provide 90.5% conversion of 2-adamantanone while Sn-Beta with the same Sn content gave 82.3%.396 In addition, much stronger difference was observed when the catalysts were tested in sugar isomerisation (see Section 6.5). The observation evidences that 2-adamantanone is still too small to assess the advantages of 2D stannosilicates in BV oxidation.
Regarding the pillared catalysts, Přech et al.246 recently extended his silica–titania pillaring concept,222,245 which was successfully used to prepare highly active epoxidation catalysts, also to tin-silica pillaring. The layered MFI based and (UTL based) ICP-1-SnPI catalysts showed remarkable activity in BV oxidation of 2-adamantanone, norcamphor and cyclopentanone. Although reference Sn-Beta exhibited higher turn-over-numbers (Sn-Beta TON = 69 vs. e.g. IPC-1-SnPI TON = 28 in BV oxidation of norcamphor) than the tin-silica pillared catalysts, the overall conversions (e.g. norcamphor conversion: Sn-Beta: 13% vs. IPC-1-SnPI: 36% after 8 h) and yields were higher for the pillared catalysts because the tin-silica pillaring creates a large amount of well accessible Sn sites located preferentially on the external surface of the crystalline layers. A similar feature (Sn sites close to the external surface of crystals) is probably responsible also for the high performance of the Sn-Y (Sn-FAU) catalyst prepared by dealumination of zeolite Y and subsequent tin incorporation by SnCl4 vapour.301
In the same way, organic sulphones and particularly sulphoxides (intermediate products in the S-oxidation) can be prepared by oxidation of corresponding sulphides with hydrogen peroxide over titanosilicate catalysts. When 2D TS-1 and Ti-IPC-1-PI catalysts were tested in this reaction, it was found that the use of these 2D catalysts with a highly open structure is beneficial for both small and bulky substrates.461 In the case of small substrates (represented by methylphenyl sulphide), selectivity of the reaction is diffusion driven enabling the attainment of a sulphoxide:sulphone ratio of 94:6 at 40% conversion (3D TS-1 exhibited only 60–74% sulphoxide selectivity depending on crystal size under the same conditions). In the case of bulky substrates (represented by diphenyl sulphide, dibenzothiophene and dioctyl sulphide) the enhanced accessibility of the active sites resulted in a significantly increased conversion in comparison with 3D counterparts.
6.5. Chemical specialities
Zeolites are traditionally considered as catalysts for large-scale industrial processes and much less they represent a toolkit for synthetic organic chemists. However, researchers and manufacturers, dealing with pharmaceuticals, fragrances or food additives, can benefit from the same advantages known from large-scale processes. In most research papers from the area of molecular sieves, thorough characterisations of novel catalysts are reported and catalytic testing serves in the first instance as a characterisation tool. Properly chosen standard catalytic tests may provide ultimate information on the abilities of the catalyst under investigation. However, beyond standard reactions such as toluene alkylation and disproportionation,1 methanol-to-olefins breakthrough curves,203,206 cyclohexene epoxidation and Baeyer–Villiger oxidation of adamantanone (vide supra), there exist many other interesting and useful transformations (e.g. acylation of methoxynaphthalene, vitamin E synthesis), which can be performed over zeolites383,462 (the referenced book462 contributes to bridging the gap between materials science and organic chemistry) and many of them are still waiting for disclosure or more detailed investigation.463 In this section, there are highlighted examples of transformations, that are performed over 2D catalysts and which either represent non-classical zeolite-catalysed reactions or extend a classical approach to unusual substrates. Note also the overlap of fine chemical area with biomass processing (Section 6.3).The use of 2D catalysts opens possibilities to effectively couple two relatively bulky molecules. In this sense, alkylation of benzene or more bulky mesitylene with benzyl alcohol was demonstrated in the liquid phase. Kim et al.469 performed the benzylation of benzene over nanosponge and conventional zeolites (*BEA, MTW, MRE, MFI). Both reactants can enter the zeolite micropores but the bulky product diphenylmethane is, most probably, unable to leave the micropores, particularly of MTW, MRE and MFI resulting in almost zero conversion per Al site (a turn-over-number, TON). The active sites on the external surface, strongly augmented in the nanosponge form, catalyse the reaction effectively. Selective poisoning of the external acid sites then switches the catalytic behaviour of nanosponge Beta into commercial Beta. The nanosponge Beta provided 15% conversion after 1 h and the conversion increased steadily up to about 80% conversion (after 50 h) while reference commercial Beta and poisoned nanosponge Beta provided 10% resp. 7% conversion after 1 h and the reaction died out after about 10 h, reaching conversion below 20%. High conversion (93% vs. 30% over 3D MFI) and diphenyl methane selectivity (94%) have also been reported using single-pore nanosheet MFI.212
Mesitylene has a kinetic diameter of 0.87 nm not entering the micropores even of large-pore zeolites. Therefore, its alkylation is restricted to the external surface of the catalyst. Selective poisoning of external acid sites switches the selectivity from 1-benzyl-2,4,6-trimethylbenzene to dibenzylether because the concurrent etherification of benzyl alcohol can occur in the micropore system of catalysts,470 but the main reaction cannot (Scheme 1). Almost complete suppression of the diffusion limitations is observed for pillared MFI and self-pillared-pentasil as their observed effectiveness factors are close to 1 (Fig. 7) in the etherification of benzylalcohol while external acid sites are switched-off (poisoned) with di-tert-butylpyridine208 not to influence the observation of transport phenomena.
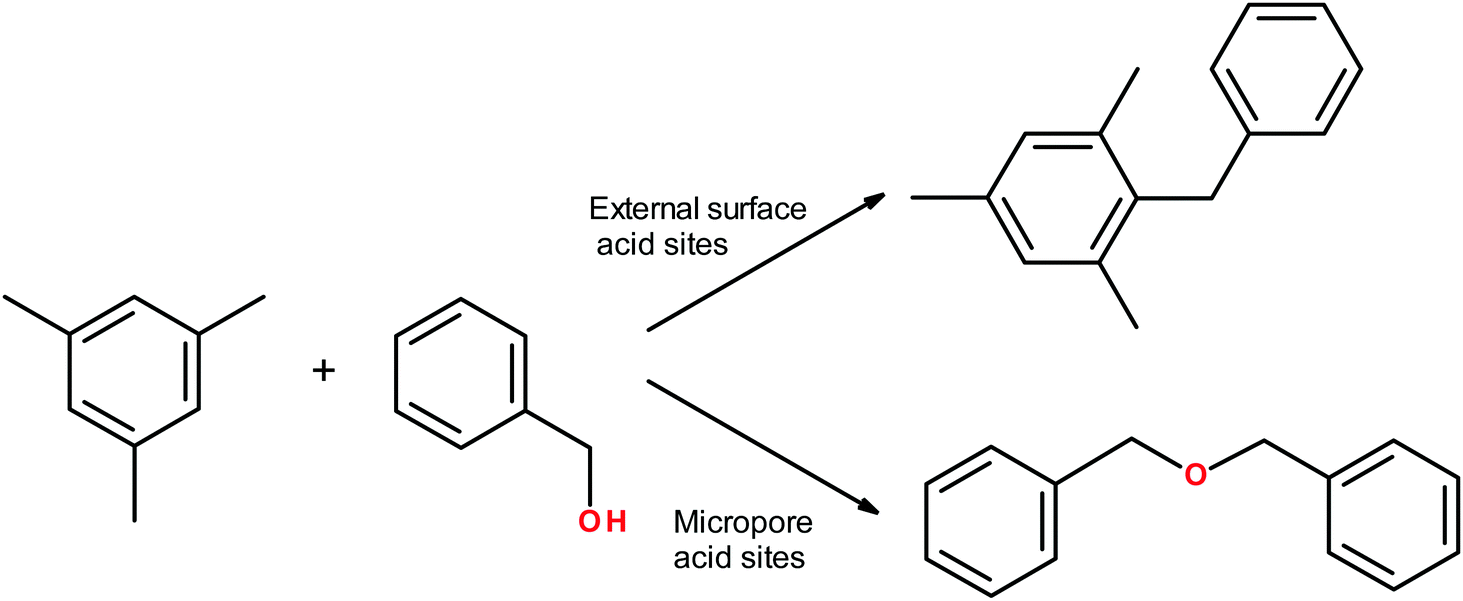 | ||
| Scheme 1 Mesitylene alkylation with benzylalcohol and parallel self-etherification of benzyl alcohol. | ||
Mesostructured molecular sieves with zeolite-like walls (materials combining features of nanocrystalline and nanosheet MFI and *BEA, prepared by surfactant templating) were demonstrated to catalyse even alkylation of pyrene with 9-phenyl-9-fluorenol (Scheme 2).213 The reaction was performed in an autoclave at 130 °C and the substrate/catalyst mass ratio was 2; however, in 2 h, the hexagonally mesostructured sieve with MFI walls provided 82% conversion, while the conventional Beta zeolite and mesoporous MCM-41 were almost inactive (conversion <5%). The mesostructured catalysts were composed of crystalline MFI phase with a similar Si/Al ratio to reference catalysts. Thus the observed catalytic activity can be ascribed to the improvement of the active site accessibility even for very bulky molecules such as the mentioned polyaromatic hydrocarbons. Similarly, these catalysts were active in Friedel–Crafts acylation of 1-methoxynaphthalene with benzoic anhydride to form 4-benzoyl-1-methoxynaphthalene (with more than 95% selectivity), and annulation of trimethylhydroquinone with isophytol (1-ethylen-1-methylheptadekan-1-ol) to form α-tocopherol (vitamin E, Scheme 3). Opanasenko et al.471 reported annulation of phenol, 1- and 2-naphthol with 2-methyl-3-buten-2-ol. This reaction combines alkylation of the corresponding phenol in the first step with a subsequent intramolecular ring closing reaction of isoprenylphenol to form 3,4-dihydro-2H-1-benzopyran (chromane). The reaction was performed over unilamellar MFI and several mesostructured MFI zeolites prepared by surfactant templating. These catalysts provided the highest conversion while the selectivity of the reaction was, particularly for 1-naphthol and 2-naphthol, independent of the used catalyst.
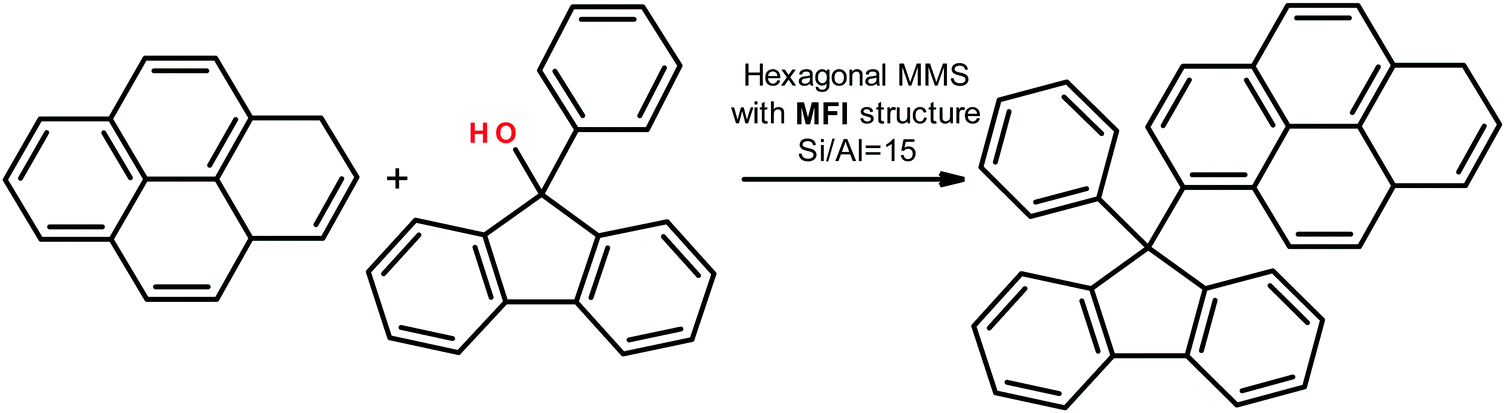 | ||
| Scheme 2 Alkylation of pyrene with 9-phenyl-9-fluorenol. | ||
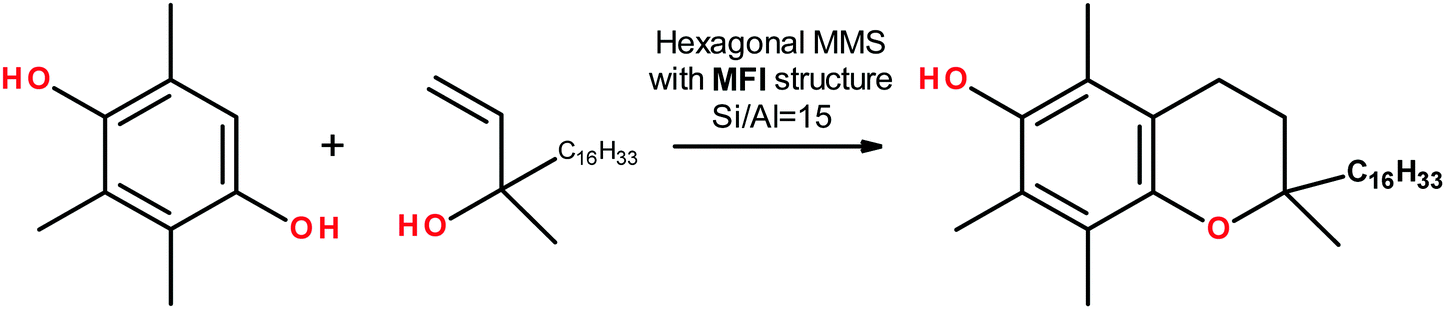 | ||
| Scheme 3 Annulation of trimethylhydroquinone with isophitol yielding vitamin E. | ||
Similarly, acylation of anisole with acetanhydride to p-acetophenone occurs considerably faster over lamellar and pillared ECNU-7 (MWW layers), and delaminated MCM-56194 in comparison with 3D MFI and *BEA. Interlamellar expanded IEZ-PLS-3 (FER layers) having 12 × 10 ring channels instead of a 10 × 8 ring, characteristic of FER, provided conversion comparable with *BEA.472 Partially delaminated ERB-1-del-135 (MWW layers) catalysed acylation of 2-methoxynaphthalene with acetic anhydride giving 1-acetyl-2-methoxynaphthalene,473 which is kinetically favoured but more sterically demanding than 2-acetyl-6-methoxynaphthalene. Position 2 was favoured when large-pore zeolites (FAU, MOR, *BEA) were used to catalyse naphthalene acylation.474 However, in the above case, the partial delamination brought no significant improvement in comparison with 3D MWW. Note that the 2-methoxynaphthalene acylation is a key reaction in one of the syntheses of a pain-reliever drug naproxen (Scheme 4).475
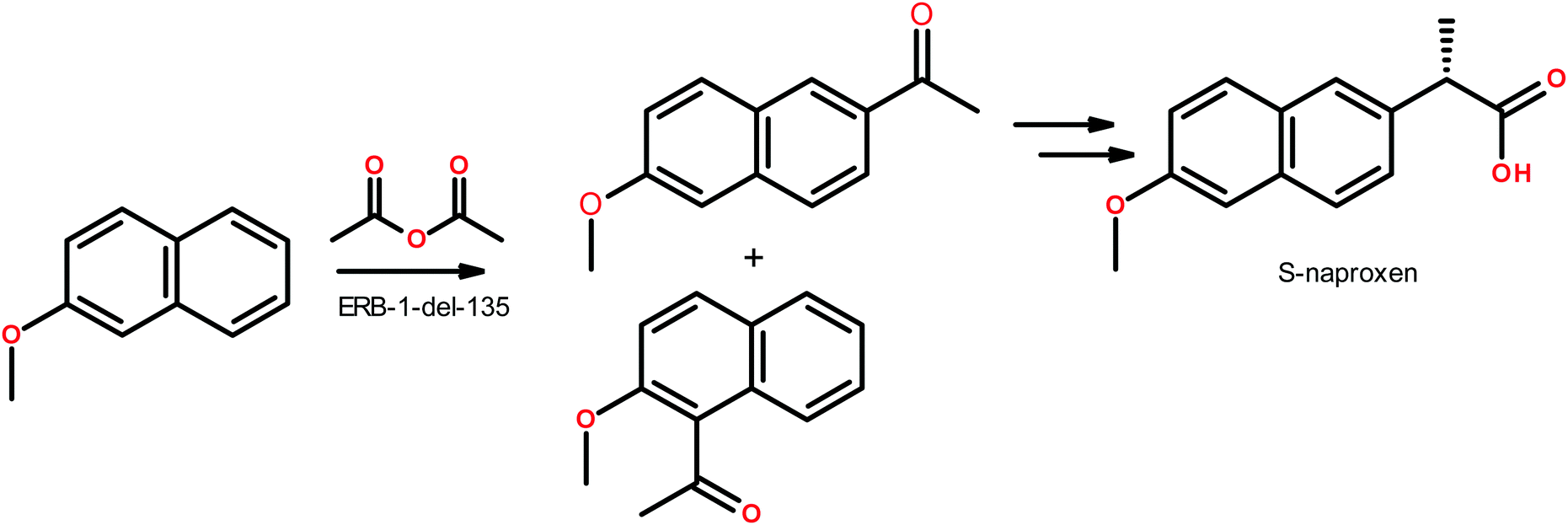 | ||
| Scheme 4 Acylation of 2-methoxynaphthalene with acetanhydride and illustration of the route to S-naproxen. | ||
 | ||
| Scheme 5 Claisen–Schmidt condensation of 2-hydroxyacetophenone with benzaldehyde. | ||
Abilities of 2D catalysts can be demonstrated by protection of aldehydes (e.g. benzaldehyde) with pentaerythritol forming bulky spiro-diacetal.203 In contrast, an opposite reaction (deprotection of carbonyl group) is used, when it is desired to perform a cascade reaction involving several types of active sites. In this way, for instance, organic pillared MFI was used for one-pot hydrolysis of benzaldehyde dimethylacetal to benzaldehyde and subsequent Knoevenagel condensation of benzaldehyde with malononitrile yielding benzylidene malononitrile (Scheme 6).247 In this case, acid sites of the zeolite were responsible for the deprotection, while basic sites located on the organic pillars catalysed the condensation.
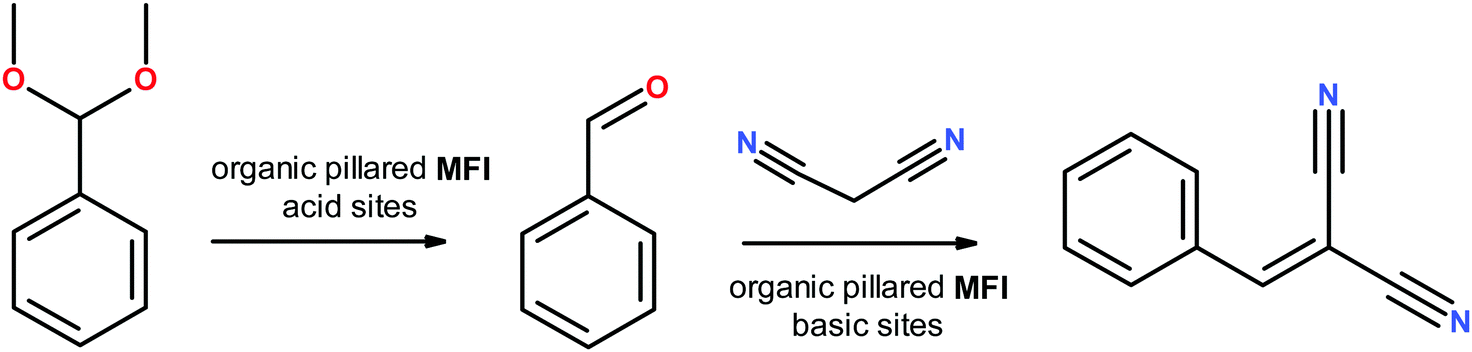 | ||
| Scheme 6 Cascade deprotection of benzaldehyde dimethylacetal and subsequent Pechmann condensation with malononitrile yielding benzilidene malononitrile. | ||
 | ||
| Scheme 7 Quaternary ammonium salt catalysed cycloaddition of CO2 on epoxides. | ||
The nanosponge MFI (and also self-pillared-pentasil catalyst) exhibited remarkable activity (in comparison with 3D zeolites MFI, *BEA and FAU) also in Pechmann condensation of pyrogallol and resorcinol with ethylacetoacetate in the liquid phase yielding corresponding dihydroxy-4-methylcoumarins.207
Taking into account the number of various transformations that can be catalysed by 2D zeolite-based catalysts, we believe that appropriately shaped 2D zeolite catalysts might become, for organic chemists, a common tool for acid catalysed reactions as, for instance, 4A molecular sieve (LTA zeolite) for solvent drying.
7. Conclusions and perspectives
This review covers the current state-of-the-art of synthesis and catalytic properties of zeolites highlighting the most important achievements in recent years and discussing the progress in the research on conventional zeolites, nanozeolites, hierarchical zeolites up to two-dimensional ones.Synthetic zeolites are typically prepared by crystallization of gels containing water, alkali cations, silica and alumina sources under hydrothermal conditions, basic pH, temperatures in the range 60–200 °C and autogenous pressure. Incorporation of organic compounds into the synthesis gel enabled discoveries of new zeolite structures, mainly with high silica compositions, since the organics act as structure-directing agents.
Seeding has been widely applied in zeolite synthesis, not just to accelerate the crystallization process, but also as a green route for the organotemplate-free synthesis of zeolites, to decrease reagent cost and environmental impact. Synthetic protocols to minimize the water content during the zeolite crystallization, such as dry-gel conversion, vapour-phase transport, steam-assisted conversion and hydrothermal treatment of wetness-impregnated xerogels have been described recently.
Fluoride-containing media have been employed for the crystallization of zeolites at neutral or even slightly acidic pH to provide large crystals with a low concentration of defects and highly hydrophobic properties. Similarly, microwave heating and ultrasound have been used in zeolite synthesis to reduce the synthesis times allowing a better control of the size and morphology of the crystals.
Although a great number of zeolitic structures have been theoretically predicted, in practice, just a relatively small number has been identified due to the limitations existing for their real synthesis. Thus, by April 2018 the International Zeolite Association recognized 235 different zeolite structures (represented by three-letter framework codes). Recently, the ADOR (assembly–disassembly–organisation–reassembly) methodology has been developed for the preparation of novel zeolite structures that could not be obtained by conventional hydrothermal synthesis. This approach started from zeolite UTL, which has been subjected to a number of post-synthesis transformations (hydrolysis, organisation and condensation) in order to generate different zeolite structures. This strategy has been very effective for the development of new zeolite structures, affording in many cases topologies, which are not feasible by direct hydrothermal synthesis.
Classical zeolites are very well known for their acidic and cation-exchange properties, which derives from the extra negative charge associated with the Al tetrahedral sites. Acidity in zeolites arises from Brønsted acid sites associated with framework Al atoms, although they may also contain significant amounts of Lewis acids sites, formed by dehydroxylation of the former or linked to extra-framework Al species. Zeolites may also present basic properties, which are generated by ion-exchange with alkali cations or by incorporation of basic metals and metal oxides.
Increasing the Si/Al ratio has been commonly employed as a method for enhancing the zeolite stability. While this can be achieved in high silica zeolites just by varying the gel composition, for low silica zeolites it is usually accomplished by post-synthesis treatments via extraction and removal of Al species. Isomorphous substitution of the Al3+ or Si species by other cations leads to different families of zeolitic materials, such as metallosilicates, aluminophosphates and silicoaluminophosphates. These modifications have a strong effect on the zeolite properties, making possible the synthesis of materials with controllable acidity in terms of both strength and type. In this way, replacement of Al by tetravalent cations has yielded materials showing very specific Lewis acidity and redox properties.
Other fundamental features of zeolites are shape selectivity and molecular sieving properties. Since zeolite micropores present diameters very close to the size of many molecules, they are able to discriminate between compounds having just a small variation in their size, so the largest ones cannot penetrate the zeolite structure or diffuse very slowly through the zeolite channels. This fact has a strong effect on the product distribution and, therefore, on the outstanding selectivity exhibited by zeolites in many reactions.
However, at the same time, the small pore size of zeolites strongly limits the application of these materials in processes involving large substrates, which is the case of many transformations within the biomass conversion and fine chemical synthesis. Accordingly, in recent years, an enormous research effort has been devoted to tailoring zeolite properties directly related to their porosity and active site accessibility. New zeolitic materials showing enhanced accessibility and external/mesopore surface area have been developed, which include nano-sized, hierarchical and two-dimensional zeolites. A sequential process can be envisaged in the evolution of conventional 3D zeolites, passing by the reduction of their crystal size below 100 nm (nanozeolites), i.e. introduction of a secondary porosity in the mesopore range (hierarchical zeolites) to finally reach lamellar particles (2D zeolites).
A number of relevant advantages have been widely reported when using zeolitic materials with enhanced accessibility as catalysts: possibility of converting bulky compounds that could not enter the micropores, enhanced transport of reactants, intermediates and products, and less pronounced deactivation effects by formation of coke deposits. The overall result coming out is that in many cases these new classes of zeolitic materials show quite better catalytic properties than the conventional zeolites. Moreover, the presence of a high proportion of external/mesopore surface is a very positive aspect when the zeolite is used as a support for other catalysis phases, as it leads to an improved dispersion of the latter with a stronger interaction with the zeolite. The resulting materials present bifunctional or even multifunctional properties, which opens the way for their use in many sectors, beyond the typical fields of zeolite applications.
One example of new catalytic applications of zeolites is the search for sustainable routes to use biomass as a raw material, according to the “biorefinery” concept. Catalytic pyrolysis, hydrotreatment, isomerizations, dehydrations, etc., to produce either biofuels or bio-based chemicals, are promoted by acid zeolite catalysts. However, biomass transformation is a complex issue as there are difficulties intrinsic to the features of the raw materials: (a) complex composition; (b) presence of heteroatoms interfering with the catalytic processes and the quality of the final product; (c) high water content and (d) high tendency to deactivate zeolites by coke deposition. Both acidity and porosity are the key features of zeolites influencing activity, selectivity and deactivation resistance by coke deposition in biomass transformation processes. Higher amounts and stronger acid sites are usually related to higher activities and selectivities to target products. This beneficial effect is counterbalanced by larger amounts of coke and, hence, faster catalyst deactivation. Regarding the porous properties, zeolites with very small micropore sizes exhibit poor activities. To reduce the coke formation and to increase both the activity and selectivity in reactions involving large molecules, different modifications of zeolites have been attempted. Acidic properties can be modulated by variation of the Si/Al ratio, cation exchange and second phase (metals, metal oxides) dispersion. The accessibility of acidic sites for bulky compounds and transport properties are enhanced by increasing the external surface/particle volume ratio or by introducing secondary mesoporosity. The first approach is mainly provided by nanozeolites and, more recently, by 2D zeolites, whereas secondary mesoporosity is typical of hierarchical zeolites. Nevertheless, to attain real beneficial catalytic effects with these porosity-tailored zeolites in reactions of biomass conversion it is necessary to reach a proper balance with their acidic properties. Thus, high density of strong or Brønsted acid sites on the accessible surface should be avoided as they are associated with coke formation or are even poorly active in some interesting reactions, like glucose dehydration to 5-HMF. In this sense, the preparation of bifunctional catalysts by dispersing active phases on zeolites with enhanced textural properties is a very rational way to overcome the aforementioned limitations. Although they have been just scarcely explored at present, 2D zeolites are especially good candidates, so more intensive research work is expected in this area in the near future.
Although zeolites have currently very strong positions in industrial catalytic applications, there are still many challenges that can be improved with regard to their synthesis and possible future applications. Some of them are mentioned here:
(a) Synthesis of important zeolites without organic structure-directing agents. A lot of successful syntheses without organics have been recently performed but some of the interesting zeolites still need organics to be prepared.
(b) Only very few zeolites can be directly synthesized in the whole range of Si/Al ratios from 1 to infinity. In most cases, we need to apply various post-synthesis methods to adjust the chemical composition.
(c) Location of active sites in zeolite frameworks, inside of the channels versus on the external surface, in different pores for zeolites having more than one type of channels, is still a matter of accidental synthesis than a scientific concept.
(d) Less than 20 zeolites have been prepared as two-dimensional analogues; however, we can believe that maybe all the zeolites will be prepared in 2D forms, which could substantially enhance the possibility for their applications.
These four points clearly indicate that despite the very strong position of zeolites in laboratories and industry, there are many possible improvements. Hopefully, this review will help in achieving them soon.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. P. and J. Č. acknowledge the Charles University Centre of Advanced Materials (CUCAM) (OP VVV Excellent Research Teams, project number CZ.02.1.01/0.0/0.0/15_003/0000417) and the Czech Science Foundation (P106/12/G015). J. Č., P. P., and D. S. gratefully acknowledge the financial support from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 604307 (CASCATBEL project). P. P. and D. S. also acknowledge the funding received from the Spanish Ministry of Economy and Competitiveness through the CATPLASBIO project (Ref. CTQ2014-602209-R).References
- S. Al-Khattaf, S. A. Ali, A. M. Aitani, N. Zilkova, D. Kubicka and J. Cejka, Catal. Rev.: Sci. Eng., 2014, 56, 333–402 CrossRef
.
- A. Burton, Catal. Rev.: Sci. Eng., 2018, 60, 132–175 CrossRef
- A. Corma, J. Catal., 2003, 216, 298–312 CrossRef
- J. L. Sun, C. Bonneau, A. Cantin, A. Corma, M. J. Diaz-Cabanas, M. Moliner, D. L. Zhang, M. R. Li and X. D. Zou, Nature, 2009, 458, U1154–U1190 CrossRef PubMed
- J. Perez-Ramirez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC
- J. Perez-Ramirez, S. Mitchell, D. Verboekend, M. Milina, N. L. Michels, F. Krumeich, N. Marti and M. Erdmann, ChemCatChem, 2011, 3, 1731–1734 CrossRef
- L. Gueudre, M. Milina, S. Mitchell and J. Perez-Ramirez, Adv. Funct. Mater., 2014, 24, 209–219 CrossRef
- W. J. Roth and J. Čejka, Catal. Sci. Technol., 2011, 1, 43–53 RSC
- W. J. Roth, P. Nachtigall, R. E. Morris and J. Čejka, Chem. Rev., 2014, 114, 4807–4837 CrossRef PubMed
- M. V. Opanasenko, W. J. Roth and J. Cejka, Catal. Sci. Technol., 2016, 6, 2467–2484 RSC
- F. S. O. Ramos, M. K. De Pietre and H. O. Pastore, RSC Adv., 2013, 3, 2084–2111 RSC
- L. Tosheva and V. P. Valtchev, Chem. Mater., 2005, 17, 2494–2513 CrossRef
- S. Maheshwari, C. Martínez, M. Teresa Portilla, F. J. Llopis, A. Corma and M. Tsapatsis, J. Catal., 2010, 272, 298–308 CrossRef
- D. P. Serrano, R. Sanz, P. Pizarro and I. Moreno, Appl. Catal., A, 2012, 435-436, 32–42 CrossRef
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–702 CrossRef PubMed
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756–768 CrossRef
-
K. G. Strohmaier, in Zeolites in Catalysis. Properties and Applications ed. J. Čejka, R. E. Morris and P. Nachtigall, 2017, pp. 73–97 Search PubMed
- D. P. Serrano and R. van Grieken, J. Mater. Chem., 2001, 11, 2391–2407 RSC
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef
- Y. Ji, Y. Wang, B. Xie and F.-S. Xiao, Comments Inorg. Chem., 2016, 36, 1–16 CrossRef
- Y. Wang, Q. Wu, X. Meng and F.-S. Xiao, Engineering, 2017, 3, 567–574 CrossRef
- X. Meng, Q. Wu, F. Chen and F.-S. Xiao, Sci. China: Chem., 2015, 58, 6–13 CrossRef
- D. P. Serrano, M. A. Uguina, G. Ovejero, R. Van Grieken and M. Camacho, Chem. Commun., 1996, 1097–1098 RSC
- D. P. Serrano, M. A. Uguina, G. Ovejero, R. Van Grieken and M. Camacho, Microporous Mater., 1996, 7, 309–321 CrossRef
- X. Meng and F.-S. Xiao, Chem. Rev., 2014, 114, 1521–1543 CrossRef PubMed
- M. A. Camblor, A. Corma and S. Valencia, J. Mater. Chem., 1998, 8, 2137–2145 RSC
- D. P. Serrano, R. Van Grieken, P. Sánchez, R. Sanz and L. Rodríguez, Microporous Mesoporous Mater., 2001, 46, 35–46 CrossRef
- S. I. Zones, R. J. Darton, R. Morris and S.-J. Hwang, J. Phys. Chem. B, 2005, 109, 652–661 CrossRef PubMed
- Y. Li and W. Yang, J. Membr. Sci., 2008, 316, 3–17 CrossRef
- S. Askari, S. Miar Alipour, R. Halladj and M. H. Davood Abadi Farahani, J. Porous Mater., 2013, 20, 285–302 CrossRef
- P. Eliášová, M. Opanasenko, P. S. Wheatley, M. Shamzhy, M. Mazur, P. Nachtigall, W. J. Roth, R. E. Morris and J. Cejka, Chem. Soc. Rev., 2015, 44, 7177–7206 RSC
- R. Pophale, P. A. Cheeseman and M. W. Deem, Phys. Chem. Chem. Phys., 2011, 13, 12407–12412 RSC
-
R. Millini and G. Bellussi, in Zeolites in Catalysis: Properties and Applications, ed. J. Čejka, R. E. Morris and P. Nachtigall, 2017, pp. 1–28 Search PubMed
-
J.-P. Gilson and M. Guisnet, Zeolites For Cleaner Technologies, Imperial College Press, 2002 Search PubMed
- E. G. Derouane, J. C. Védrine, R. R. Pinto, P. M. Borges, L. Costa, M. A. N. D. A. Lemos, F. Lemos and F. R. Ribeiro, Catal. Rev., 2013, 55, 454–515 CrossRef
- L.-E. Sandoval-Díaz, J.-A. González-Amaya and C.-A. Trujillo, Microporous Mesoporous Mater., 2015, 215, 229–243 CrossRef
- D. Barthomeuf, Catal. Rev., 1996, 38, 521–612 CrossRef
- R. J. Davis, J. Catal., 2003, 216, 396–405 CrossRef
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef
- S. M. Csicsery, Zeolites, 1984, 4, 202–213 CrossRef
- P. B. Weisz and V. J. Frilette, J. Phys. Chem., 1960, 64, 382 CrossRef
- N. Van-Den-Begin, L. V. C. Rees, J. Caro and M. Bülow, Zeolites, 1989, 9, 287–292 CrossRef
- S. L. Burkett and M. E. Davis, Chem. Mater., 1995, 7, 920–928 CrossRef
- M. A. Camblor, A. Corma and S. Valencia, Chem. Commun., 1996, 2365–2366 RSC
- G. T. Kerr, J. Phys. Chem., 1969, 73, 2780–2782 CrossRef
- A. Corma, M. Faraldos and A. Mifsud, Appl. Catal., 1989, 47, 125–133 CrossRef
- L. Brabec, M. Jeschke, R. Klik, J. Nováková, L. Kubelková, D. Freude, V. Bosáček and J. Meusinger, Appl. Catal., A, 1998, 167, 309–320 CrossRef
- T. Inui, S. Iwamoto, S. Kojo and T. Yoshida, Catal. Lett., 1992, 13, 87–93 CrossRef
- B. Kraushaar and J. H. C. Van Hooff, Catal. Lett., 1988, 1, 81–84 CrossRef
- A. Tuel, S. Moussa-Khouzami, Y. B. Taarit and C. Naccache, J. Mol. Catal., 1991, 68, 45–52 CrossRef
- G. Wu, Y. Wang, L. Wang, W. Feng, H. Shi, Y. Lin, T. Zhang, X. Jin, S. Wang, X. Wu and P. Yao, Chem. Eng. J., 2013, 215-216, 306–314 CrossRef
- H. Y. Luo, J. D. Lewis and Y. Román-Leshkov, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 663–692 CrossRef PubMed
- R. Bai, Q. Sun, N. Wang, Y. Zou, G. Guo, S. Iborra, A. Corma and J. Yu, Chem. Mater., 2016, 28, 6455–6458 CrossRef
-
W. M. H. Sachtler and Z. Zhang, in Advances in Catalysis, ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1993, vol. 39, pp. 129–220 Search PubMed
- J. Guzman and B. C. Gates, Dalton Trans., 2003, 3303–3318 RSC
- P. Mériaudeau and C. Naccache, Catal. Rev., 1997, 39, 5–48 CrossRef
- T. Barakat, J. C. Rooke, H. L. Tidahy, M. Hosseini, R. Cousin, J. F. Lamonier, J. M. Giraudon, G. De Weireld, B. L. Su and S. Siffert, ChemSusChem, 2011, 4, 1420–1430 CrossRef PubMed
- R. S. Gomez, X. Li, R. L. Yson and H. H. Patterson, Res. Chem. Intermed., 2011, 37, 729–745 CrossRef
- A. Kumbhar, Top. Curr. Chem., 2016, 375, 2 CrossRef PubMed
- E. Koohsaryan and M. Anbia, Chin. J. Catal., 2016, 37, 447–467 CrossRef
- V. Valtchev and L. Tosheva, Chem. Rev., 2013, 113, 6734–6760 CrossRef PubMed
- T. Tago, H. Konno, Y. Nakasaka and T. Masuda, Catal. Surv. Asia, 2012, 16, 148–163 CrossRef
- Y. Yan, X. Guo, Y. Zhang and Y. Tang, Catal. Sci. Technol., 2015, 5, 772–785 RSC
- S. Mintova, J. Grand and V. Valtchev, C. R. Chim, 2016, 19, 183–191 CrossRef
- N. N. Feoktistova, S. P. Zhdanov, W. Lutz and M. Büllow, Zeolites, 1989, 9, 136–139 CrossRef
- S. Mintova, V. Valtchev, E. Vultcheva and S. Veleva, Zeolites, 1992, 12, 210–215 CrossRef
- V. P. Valtchev and K. N. Bozhilov, J. Phys. Chem. B, 2004, 108, 15587–15598 CrossRef
- Y. Huang, K. Wang, D. Dong, D. Li, M. R. Hill, A. J. Hill and H. Wang, Microporous Mesoporous Mater., 2010, 127, 167–175 CrossRef
- D. Hu, Q. H. Xia, X. H. Lu, X. B. Luo and Z. M. Liu, Mater. Res. Bull., 2008, 43, 3553–3561 CrossRef
- S. N. Azizi, S. Ghasemi and S. Kavian, Biosens. Bioelectron., 2014, 62, 1–7 CrossRef PubMed
- S. Kavian, S. N. Azizi and S. Ghasemi, J. Mol. Liq., 2016, 218, 663–669 CrossRef
- E.-P. Ng, H. Awala, K.-H. Tan, F. Adam, R. Retoux and S. Mintova, Microporous Mesoporous Mater., 2015, 204, 204–209 CrossRef
- H. Zhang, H. Zhang, P. Wang, Y. Zhao, Z. Shi, Y. Zhang and Y. Tang, RSC Adv., 2016, 6, 47623–47631 RSC
- B. Xie, J. Song, L. Ren, Y. Ji, J. Li and F.-S. Xiao, Chem. Mater., 2008, 20, 4533–4535 CrossRef
- G. Reding, T. Mäurer and B. Kraushaar-Czarnetzki, Microporous Mesoporous Mater., 2003, 57, 83–92 CrossRef
- G. Majano, A. Darwiche, S. Mintova and V. Valtchev, Ind. Eng. Chem. Res., 2009, 48, 7084–7091 CrossRef
- Z. Chen, S. Li and Y. Yan, Chem. Mater., 2005, 17, 2262–2266 CrossRef
- M. Mehdipourghazi, A. Moheb and H. Kazemian, Microporous Mesoporous Mater., 2010, 136, 18–24 CrossRef
- Y. Pan, M. Ju, J. Yao, L. Zhang and N. Xu, Chem. Commun., 2009, 7233–7235 RSC
- C.-X. Zhao, L. He, S. Z. Qiao and A. P. J. Middelberg, Chem. Eng. Sci., 2011, 66, 1463–1479 CrossRef
- Y. Huang and A. J. Hill, AIChE Annual Meeting, Conference proceedings, 2008.
- X. Ke, C. Zeng, L. Zhang and N. Xu, Shiyou Huagong/Petrochemical Technology, 2005, 34, 486–490 Search PubMed
- Z. L. Sun, Y. G. Wang, J. Z. Gui, X. T. Zhang and G. Su, Shiyou Huagong Gaodeng Xuexiao Xuebao/Journal of Petrochemical Universities, 2005, 18, 15–19 Search PubMed
- A. Charkhi, H. Kazemian and M. Kazemeini, Powder Technol., 2010, 203, 389–396 CrossRef
- A. Nezamzadeh-Ejhieh and S. Khorsandi, J. Ind. Eng. Chem., 2014, 20, 937–946 CrossRef
- T. Kurniawan, O. Muraza, A. S. Hakeem and A. M. Al-Amer, Cryst. Growth Des., 2017, 17, 3313–3320 CrossRef
- H. Wang, L. Huang, Z. Wang, A. Mitra and Y. Yan, Chem. Commun., 2001, 1364–1365 RSC
- Y. Wang, Y. Tang, A. Dong, X. Wang, N. Ren and Z. Gao, J. Mater. Chem., 2002, 12, 1812–1818 RSC
- M. Hartmann, Angew. Chem., Int. Ed., 2004, 43, 5880–5882 CrossRef PubMed
- C. Schomburg, D. Wöhrle and G. Schulz-Ekloff, Zeolites, 1996, 17, 232–236 CrossRef
- C. J. H. Jacobsen, C. Madsen, J. Houzvicka, I. Schmidt and A. Carlsson, J. Am. Chem. Soc., 2000, 122, 7116–7117 CrossRef
- Y. Wang, L. Ma, N. Zhu, F. Chen and X. Zhan, Prog. Chem., 2009, 21, 1722–1733 Search PubMed
- D. P. Serrano, J. Aguado and J. M. Escola, Catalysis, 2011, 23, 253–283 Search PubMed
- D. Verboekend, S. Mitchell and J. Pérez-Ramírez, Chimia, 2013, 67, 327–332 CrossRef PubMed
- D. P. Serrano, J. M. Escola and P. Pizarro, Chem. Soc. Rev., 2013, 42, 4004–4035 RSC
- Y. Wei, T. E. Parmentier, K. P. De Jong and J. Zečević, Chem. Soc. Rev., 2015, 44, 7234–7261 RSC
- W. Schwieger, A. G. Machoke, T. Weissenberger, A. Inayat, T. Selvam, M. Klumpp and A. Inayat, Chem. Soc. Rev., 2016, 45, 3353–3376 RSC
- M. Hartmann, A. G. Machoke and W. Schwieger, Chem. Soc. Rev., 2016, 45, 3313–3330 RSC
- M. L. Occelli and P. O'Connor, Stud. Surf. Sci. Catal., 2001, 134, 1 CrossRef
- W. J. Roth, O. V. Shvets, M. Shamzhy, P. Chlubná, M. Kubů, P. Nachtigall and J. Čejka, J. Am. Chem. Soc., 2011, 133, 6130–6133 CrossRef PubMed
- W. J. Roth, P. Nachtigall, R. E. Morris, P. S. Wheatley, V. R. Seymour, S. E. Ashbrook, P. Chlubna, L. Grajciar, M. Polozij, A. Zukal, O. Shvets and J. Cejka, Nat. Chem., 2013, 5, 628–633 CrossRef PubMed
- P. Chlubná, W. J. Roth, H. F. Greer, W. Z. Zhou, O. Shvets, A. Zukal, J. Čejka and R. E. Morris, Chem. Mater., 2013, 25, 542–547 CrossRef
- M. Ogura, S. Y. Shinomiya, J. Tateno, Y. Nara, E. Kikuchi and M. Matsukata, Chem. Lett., 2000, 882–883 CrossRef
- J. Peréz-Ramírez, D. Verboekend, A. Bonilla and S. Abelló, Adv. Funct. Mater., 2009, 19, 3972–3979 CrossRef
- D. Verboekend and J. Pérez-Ramírez, Catal. Sci. Technol., 2011, 1, 879–890 RSC
- Z. Qin, L. Lakiss, J. P. Gilson, K. Thomas, J. M. Goupil, C. Fernandez and V. Valtchev, Chem. Mater., 2013, 25, 2759–2766 CrossRef
- L. Brabec and M. Kočiřík, Mater. Chem. Phys., 2007, 102, 67–74 CrossRef
- Z. Qin, G. Melinte, J. P. Gilson, M. Jaber, K. Bozhilov, P. Boullay, S. Mintova, O. Ersen and V. Valtchev, Angew. Chem., Int. Ed., 2016, 55, 15049–15052 CrossRef PubMed
- I. Schmidt, A. Boisen, E. Gustavsson, K. Ståhl, S. Pehrson, S. Dahl, A. Carlsson and C. J. H. Jacobsen, Chem. Mater., 2001, 13, 4416–4418 CrossRef
- S. S. Kim, J. Shah and T. J. Pinnavaia, Chem. Mater., 2003, 15, 1664–1668 CrossRef
- Z. Yang, Y. Xia and R. Mokaya, Adv. Mater., 2004, 16, 727–732 CrossRef
- M. Y. Kustova, P. Hasselriis and C. H. Christensen, Catal. Lett., 2004, 96, 205–211 CrossRef
- M. Y. Kustova, A. L. Kustov and C. H. Christensen, Stud. Surf. Sci. Catal., 2005, 158A, 255–262 CrossRef
- H. Chen, J. Wydra, X. Zhang, P. S. Lee, Z. Wang, W. Fan and M. Tsapatsis, J. Am. Chem. Soc., 2011, 133, 12390–12393 CrossRef PubMed
- H. S. Cho and R. Ryoo, Microporous Mesoporous Mater., 2012, 151, 107–112 CrossRef
- J. B. Koo, N. Jiang, S. Saravanamurugan, M. Bejblova, Z. Musilova, J. Čejka and S. E. Park, J. Catal., 2010, 276, 327–334 CrossRef
- J. Zhou, Z. Hua, X. Cui, Z. Ye, F. Cui and J. Shi, Chem. Commun., 2010, 46, 4994–4996 RSC
- C. Li, Y. Wang, B. Shi, J. Ren, X. Liu, Y. Wang, Y. Guo, Y. Guo and G. Lu, Microporous Mesoporous Mater., 2009, 117, 104–110 CrossRef
- M. B. Yue, L. B. Sun, T. T. Zhuang, X. Dong, Y. Chun and J. H. Zhu, J. Mater. Chem., 2008, 18, 2044–2050 RSC
- Y. Wang, Y. Tang, X. Wang, W. Shan, A. Dong, N. Ren and Z. Gao, Chem. Lett., 2002, 862–863 CrossRef
- H. Yang, Z. Liu, H. Gao and Z. Xie, Appl. Catal., A, 2010, 379, 166–171 CrossRef
- F. S. Xiao, L. Wang, C. Yin, K. Lin, Y. Di, J. Li, R. Xu, D. S. Su, R. Schlögl, T. Yokoi and T. Tatsumi, Angew. Chem., Int. Ed., 2006, 45, 3090–3093 CrossRef PubMed
- B. T. Holland, L. Abrams and A. Stein, J. Am. Chem. Soc., 1999, 121, 4308–4309 CrossRef
- F. Liu, T. Willhammar, L. Wang, L. Zhu, Q. Sun, X. Meng, W. Carrillo-Cabrera, X. Zou and F. S. Xiao, J. Am. Chem. Soc., 2012, 134, 4557–4560 CrossRef PubMed
- J. Zhao, Y. Yin, Y. Li, W. Chen and B. Liu, Chem. Eng. J., 2016, 284, 405–411 CrossRef
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodríguez and A. Peral, Chem. Mater., 2006, 18, 2462–2464 CrossRef
- H. Wang and T. J. Pinnavaia, Angew. Chem., Int. Ed., 2006, 45, 7603–7606 CrossRef PubMed
- M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D. H. Choi and R. Ryoo, Nat. Mater., 2006, 5, 718–723 CrossRef PubMed
- V. N. Shetti, J. Kim, R. Srivastava, M. Choi and R. Ryoo, J. Catal., 2008, 254, 296–303 CrossRef
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodriguez and A. Peral, J. Mater. Chem., 2008, 18, 4210–4218 RSC
- J. Aguado, D. P. Serrano and J. M. Rodríguez, Microporous Mesoporous Mater., 2008, 115, 504–513 CrossRef
- D. P. Serrano, J. Aguado, G. Morales, J. M. Rodríguez, A. Peral, M. Thommes, J. P. Epping and B. F. Chmelka, Chem. Mater., 2009, 21, 641–654 CrossRef
- D. P. Serrano, unpublished work.
- J. García-Martínez, M. Johnson, J. Valla, K. Li and J. Y. Ying, Catal. Sci. Technol., 2012, 2, 987–994 RSC
- I. I. Ivanova and E. E. Knyazeva, Chem. Soc. Rev., 2013, 42, 3671–3688 RSC
- A. Sachse and J. García-Martínez, Chem. Mater., 2017, 29, 3827–3853 CrossRef
- A. Sachse, A. Grau-Atienza, E. O. Jardim, N. Linares, M. Thommes and J. García-Martínez, Cryst. Growth Des., 2017, 17, 4289–4305 CrossRef
- N. Linares, A. Sachse, E. Serrano, A. Grau-Atienza, E. De Oliveira Jardim, J. Silvestre-Albero, M. A. L. Cordeiro, F. Fauth, G. Beobide, O. Castillo and J. García-Martínez, Chem. Mater., 2016, 28, 8971–8979 CrossRef
- D. Verboekend, R. Caicedo-Realpe, A. Bonilla, M. Santiago and J. Pérez-Ramírez, Chem. Mater., 2010, 22, 4679–4689 CrossRef
- D. Verboekend, T. C. Keller, M. Milina, R. Hauert and J. Pérez-Ramírez, Chem. Mater., 2013, 25, 1947–1959 CrossRef
- D. P. Serrano, R. Sanz, P. Pizarro, I. Moreno and S. Shami, Microporous Mesoporous Mater., 2014, 189, 71–82 CrossRef
- D. P. Serrano, J. M. Escola, R. Sanz, R. A. Garcia, A. Peral, I. Moreno and M. Linares, New J. Chem., 2016, 40, 4206–4216 RSC
- J. M. Escola, D. P. Serrano, R. Sanz, R. A. Garcia, A. Peral, I. Moreno and M. Linares, Catal. Today, 2018, 304, 89–96 CrossRef
- D. P. Serrano, T. J. Pinnavaia, J. Aguado, J. M. Escola, A. Peral and L. Villalba, Catal. Today, 2014, 227, 15–25 CrossRef
- A. Boisen, I. Schmidt, A. Carlsson, S. Dahl, M. Brorson and C. J. H. Jacobsen, Chem. Commun., 2003, 958–959 RSC
- M. H. F. Kox, E. Stavitski, J. C. Groen, J. Pérez-Ramírez, F. Kapteijn and B. M. Weckhuysen, Chem. – Eur. J., 2008, 14, 1718–1725 CrossRef PubMed
- Y. Liu, W. Zhang, Z. Liu, S. Xu, Y. Wang, Z. Xie, X. Han and X. Bao, J. Phys. Chem. C, 2008, 112, 15375–15381 CrossRef
- J. Jagiello, M. Sterling, P. Eliasova, M. Opanasenko, A. Zukal, R. E. Morris, M. Navaro, A. Mayoral, P. Crivelli, R. Warringham, S. Mitchell, J. Perez-Ramirez and J. Cejka, Phys. Chem. Chem. Phys., 2016, 18, 15269–15277 RSC
- S. Lee, C. Jo and R. Ryoo, J. Mater. Chem. A, 2017, 5, 11086–11093 RSC
- J. Aguado, D. P. Serrano, J. M. Escola and J. M. Rodríguez, Microporous Mesoporous Mater., 2004, 75, 41–49 CrossRef
-
D. P. Serrano, J. M. Escola and P. Pizarro, Mesoporous Zeolites: Preparation, Characterization and Applications, 2015, pp. 157–198 Search PubMed
- C. H. Christensen, K. Johannsen, E. Törnqvist, I. Schmidt, H. Topsøe and C. H. Christensen, Catal. Today, 2007, 128, 117–122 CrossRef
- J. C. Groen, W. Zhu, S. Brouwer, S. J. Huynink, F. Kapteijn, J. A. Moulijn and J. Pérez-Ramírez, J. Am. Chem. Soc., 2007, 129, 355–360 CrossRef PubMed
- R. Valiullin, J. Kärger, K. Cho, M. Choi and R. Ryoo, Microporous Mesoporous Mater., 2011, 142, 236–244 CrossRef
- H. Zhao, J. Ma, Q. Zhang, Z. Liu and R. Li, Ind. Eng. Chem. Res., 2014, 53, 13810–13819 CrossRef
- M. S. Holm, S. Svelle, F. Joensen, P. Beato, C. H. Christensen, S. Bordiga and M. Bjørgen, Appl. Catal., A, 2009, 356, 23–30 CrossRef
- K. Suzuki, Y. Aoyagi, N. Katada, M. Choi, R. Ryoo and M. Niwa, Catal. Today, 2008, 132, 38–45 CrossRef
- D. P. Serrano, R. A. Garcia, G. Vicente, M. Linares, D. Prochazkova and J. Cejka, J. Catal., 2011, 279, 366–380 CrossRef
- D. P. Serrano, R. A. García, M. Linares and B. Gil, Catal. Today, 2012, 179, 91–101 CrossRef
- F. Thibault-Starzyk, I. Stan, S. Abello, A. Bonilla, K. Thomas, C. Fernandez, J. P. Gilson and J. Perez-Ramirez, J. Catal., 2009, 264, 11–14 CrossRef
- K. Sadowska, K. Góra-Marek and J. Datka, J. Phys. Chem. C, 2013, 117, 9237–9244 CrossRef
- R. Srivastava, M. Choi and R. Ryoo, Chem. Commun., 2006, 4489–4491 RSC
- J. Kim, M. Choi and R. Ryoo, J. Catal., 2010, 269, 219–228 CrossRef
- L. Lakiss, F. Ngoye, C. Canaff, S. Laforge, Y. Pouilloux, Z. Qin, M. Tarighi, K. Thomas, V. Valtchev, A. Vicente, L. Pinard, J. P. Gilson and C. Fernandez, J. Catal., 2015, 328, 165–172 CrossRef
- M. Kustova, K. Egeblad, C. H. Christensen, A. L. Kustov and C. H. Christensen, Stud. Surf. Sci. Catal., 2007, 170, 267–275 CrossRef
- B. Liu, K. Xie, S. C. Oh, D. Sun, Y. Fang and H. Xi, Chem. Eng. Sci., 2016, 153, 374–381 CrossRef
- D. Verboekend, K. Thomas, M. Milina, S. Mitchell, J. Pérez-Ramírez and J. P. Gilson, Catal. Sci. Technol., 2011, 1, 1331–1335 RSC
- A. Martínez, M. A. Arribas, M. Derewinski and A. Burkat-Dulak, Appl. Catal., A, 2010, 379, 188–197 CrossRef
- J. M. Escola, J. Aguado, D. P. Serrano, L. Briones, J. L. Díaz De Tuesta, R. Calvo and E. Fernandez, Energy Fuels, 2012, 26, 3187–3195 CrossRef
- X. Peng, K. Cheng, J. Kang, B. Gu, X. Yu, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 4553–4556 CrossRef PubMed
- B. Puértolas, L. García-Andújar, T. García, M. V. Navarro, S. Mitchell and J. Pérez-Ramírez, Appl. Catal., B, 2014, 154-155, 161–170 CrossRef
- C. H. Christensen, I. Schmidt, A. Carlsson, K. Johannsen and K. Herbst, J. Am. Chem. Soc., 2005, 127, 8098–8102 CrossRef PubMed
- X. Shen, J. Kang, W. Niu, M. Wang, Q. Zhang and Y. Wang, Catal. Sci. Technol., 2017, 7, 3598–3612 RSC
- N. Jiang, H. Jin, E. Y. Jeong and S. E. Park, J. Nanosci. Nanotechnol., 2010, 10, 227–232 CrossRef PubMed
- W. Fu, L. Zhang, D. Wu, M. Xiang, Q. Zhuo, K. Huang, Z. Tao and T. Tang, J. Catal., 2015, 330, 423–433 CrossRef
- A. Berenguer, J. A. Bennett, J. Hunns, I. Moreno, J. M. Coronado, A. F. Lee, P. Pizarro, K. Wilson and D. P. Serrano, Catal. Today, 2018, 304, 72–79 CrossRef
- Y. Zhao, W. Lv, N. Lu, X. Shi, B. Fan and R. Li, Microporous Mesoporous Mater., 2018, 257, 35–41 CrossRef
- X. F. Guo and G. J. Kim, Top. Catal., 2010, 53, 510–516 CrossRef
- K. Zhu, J. Hu, X. She, J. Liu, Z. Nie, Y. Wang, C. H. F. Peden and H. K. Ja, J. Am. Chem. Soc., 2009, 131, 9715–9721 CrossRef PubMed
- S. Mitchell and J. Pérez-Ramírez, Catal. Today, 2011, 168, 28–37 CrossRef
- L. Zhou, Z. Liu, M. Shi, S. Du, Y. Su, X. Yang and J. Xu, Carbohydr. Polym., 2013, 98, 146–151 CrossRef PubMed
- C. J. Heard, J. Čejka, M. Opanasenko, P. Nachtigall, G. Centi and S. Perathoner, Adv. Mater., 2018, 1801712 CrossRef PubMed
- W. J. Roth, B. Gil, W. Makowski, B. Marszalek and P. Eliasova, Chem. Soc. Rev., 2016, 45, 3400–3438 RSC
- B. Marler, Y. Wang, J. Song and H. Gies, Dalton Trans., 2014, 43, 10396–10416 RSC
-
R. Ravishankar, T. Sen, V. Ramaswamy, H. S. Soni, S. Ganapathy and S. Sivasanker, in Studies in Surface Science and Catalysis, ed. J. Weitkamp, H. G. Karge, H. Pfeifer and W. Hölderich, Elsevier, 1994, vol. 84, pp. 331–338 Search PubMed
-
L. Puppe and J. Weisser, US Pat., 4439409, Bayer Aktiengesselschaft, 1984 Search PubMed
-
S. I. Zones, EP0231860, Chevron Research Company, 1987
-
M. K. Rubin and P. Chu, US Pat., 4954325, ExxonMobil Oil Corp., 1990 Search PubMed
- R. Millini, G. Perego, W. O. Parker Jr, G. Bellussi and L. Carluccio, Microporous Mater., 1995, 4, 221–230 CrossRef
- M. E. Leonowicz, J. A. Lawton, S. L. Lawton and M. K. Rubin, Science, 1994, 264, 1910–1913 CrossRef PubMed
- W. J. Roth, C. T. Kresge, J. C. Vartuli, M. E. Leonowicz, A. S. Fung and S. B. McCullen, Stud. Surf. Sci. Catal., 1995, 94, 301–308 CrossRef
- S. L. Lawton, A. S. Fung, G. J. Kennedy, L. B. Alemany, C. D. Chang, G. H. Hatzikos, D. N. Lissy, M. K. Rubin, H.-K. C. Timken, S. Steuernagel and D. E. Woessner, J. Phys. Chem., 1996, 100, 3788–3798 CrossRef
- W. J. Roth, Stud. Surf. Sci. Catal., 2005, 158A and B, 19–26 CrossRef
- L. Xu, X. Ji, S. Li, Z. Zhou, X. Du, J. Sun, F. Deng, S. Che and P. Wu, Chem. Mater., 2016, 28, 4512–4521 CrossRef
-
R. P. D. Graham, Proceedings and transactions of the Royal Society of Canada. Délibérations et mémoires de la Société royale du Canada, Royal Society of Canada, Ottawa, 1918, vol. 12, ser. 3, pp. 185–190 Search PubMed
- L. Schreyeck, P. Caullet, J. C. Mougenel, J. L. Guth and B. Marler, Microporous Mater., 1996, 6, 259–271 CrossRef
- T. Ikeda, Y. Akiyama, Y. Oumi, A. Kawai and F. Mizukami, Angew. Chem., Int. Ed., 2004, 43, 4892–4896 CrossRef PubMed
- D. L. Dorset and G. J. Kennedy, J. Phys. Chem. B, 2004, 108, 15216–15222 CrossRef
- Z. Zhao, W. Zhang, P. Ren, X. Han, U. Müller, B. Yilmaz, M. Feyen, H. Gies, F.-S. Xiao, D. De Vos, T. Tatsumi and X. Bao, Chem. Mater., 2013, 25, 840–847 CrossRef
- W. J. Roth, B. Gil, W. Makowski, A. Sławek, J. Grzybek, M. Kubu and J. Čejka, Chem. Mater., 2016, 28, 3616–3619 CrossRef
- U. Díaz and A. Corma, Dalton Trans., 2014, 43, 10292–10316 RSC
-
A. W. Burton and S. I. Zones, in Studies in Surface Science and Catalysis, ed. J. Čejka, H. van Bekkum, A. Corma and F. Schüth, Elsevier, 2007, vol. 168, pp. 137–179 Search PubMed
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 828 CrossRef
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef
- L. Meng, B. Mezari, M. G. Goesten and E. J. M. Hensen, Chem. Mater., 2017, 29, 4091–4096 CrossRef PubMed
- J.-C. Kim, R. Ryoo, M. V. Opanasenko, M. V. Shamzhy and J. Čejka, ACS Catal., 2015, 5, 2596–2604 CrossRef
- X. Zhang, D. Liu, D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684–1687 CrossRef PubMed
- W. Park, D. Yu, K. Na, K. E. Jelfs, B. Slater, Y. Sakamoto and R. Ryoo, Chem. Mater., 2011, 23, 5131–5137 CrossRef
- J. G. Wang, L. Xu, K. Zhang, H. G. Peng, H. H. Wu, J. G. Jiang, Y. M. Liu and P. Wu, J. Catal., 2012, 288, 16–23 CrossRef
- K. Na, M. Choi, W. Park, Y. Sakamoto, O. Terasaki and R. Ryoo, J. Am. Chem. Soc., 2010, 132, 4169–4177 CrossRef PubMed
- J. Jung, C. Jo, K. Cho and R. Ryoo, J. Mater. Chem., 2012, 22, 4637–4640 RSC
- K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, R. J. Messinger, B. F. Chmelka and R. Ryoo, Science, 2011, 333, 328–332 CrossRef PubMed
- A. G. Machoke, I. Y. Knoke, S. Lopez-Orozco, M. Schmiele, T. Selvam, V. R. R. Marthala, E. Spiecker, T. Unruh, M. Hartmann and W. Schwieger, Microporous Mesoporous Mater., 2014, 190, 324–333 CrossRef
- R. Wei, H. Yang, J. A. Scott, K.-F. Aguey-Zinsou and D. Zhang, Appl. Mater. Today, 2018, 11, 22–33 CrossRef
- W. Kim, J.-C. Kim, J. Kim, Y. Seo and R. Ryoo, ACS Catal., 2013, 3, 192–195 CrossRef
- F. Marques Mota, P. Eliasova, J. Jung and R. Ryoo, Catal. Sci. Technol., 2016, 6, 2653–2662 RSC
- C. Jo, K. Cho, J. Kim and R. Ryoo, Chem. Commun., 2014, 50, 4175–4177 RSC
- S. W. Han, J. Kim and R. Ryoo, Microporous Mesoporous Mater., 2017, 240, 123–129 CrossRef
- C. Jo, Y. Seo, K. Cho, J. Kim, H. S. Shin, M. Lee, J.-C. Kim, S. O. Kim, J. Y. Lee, H. Ihee and R. Ryoo, Angew. Chem., Int. Ed., 2014, 53, 5117–5121 Search PubMed
- H. L. Chen, S. W. Li and Y. M. Wang, J. Mater. Chem. A, 2015, 3, 5889–5900 RSC
- J. Přech, P. Eliášová, D. Aldhayan and M. Kubů, Catal. Today, 2015, 243, 134–140 CrossRef
- V. Kasneryk, M. Opanasenko, M. Shamzhy, Z. Musilova, Y. S. Avadhut, M. Hartmann and J. Cejka, J. Mater. Chem. A, 2017, 5, 22576–22587 RSC
- V. Kasneryk, M. Shamzhy, M. Opanasenko, P. S. Wheatley, S. A. Morris, S. E. Russell, A. Mayoral, M. Trachta, J. Čejka and R. E. Morris, Angew. Chem., Int. Ed., 2017, 56, 4324–4327 CrossRef PubMed
- D. S. Firth, S. A. Morris, P. S. Wheatley, S. E. Russell, A. M. Z. Slawin, D. M. Dawson, A. Mayoral, M. Opanasenko, M. Položij, J. Čejka, P. Nachtigall and R. E. Morris, Chem. Mater., 2017, 29, 5605–5611 CrossRef
- J. X. Jiang, J. H. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 3120–3145 CrossRef PubMed
- J. X. Jiang, J. L. Jorda, M. J. Diaz-Cabanas, J. H. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 4986–4988 CrossRef PubMed
- M. Mazur, P. S. Wheatley, M. Navarro, W. J. Roth, M. Položij, A. Mayoral, P. Eliášová, P. Nachtigall, J. Čejka and R. E. Morris, Nat. Chem., 2016, 8, 58–62 CrossRef PubMed
- S. A. Morris, G. P. M. Bignami, Y. Tian, M. Navarro, D. S. Firth, J. Čejka, P. S. Wheatley, D. M. Dawson, W. A. Slawinski, D. S. Wragg, R. E. Morris and S. E. Ashbrook, Nat. Chem., 2017, 9, 1012–1018 CrossRef PubMed
- R. E. Morris and J. Cejka, Nat. Chem., 2015, 7, 381–388 CrossRef PubMed
- E. Verheyen, L. Joos, K. Van Havenbergh, E. Breynaert, N. Kasian, E. Gobechiya, K. Houthoofd, C. Martineau, M. Hinterstein, F. Taulelle, V. Van Speybroeck, M. Waroquier, S. Bals, G. Van Tendeloo, C. E. A. Kirschhock and J. A. Martens, Nat. Mater., 2012, 11, 1059–1064 CrossRef PubMed
- M. Mazur, A. Arevalo-Lopez, P. S. Wheatley, G. P. M. Bignami, S. E. Ashbrook, A. Morales-Garcia, P. Nachtigall, J. P. Attfield, J. Cejka and R. E. Morris, J. Mater. Chem. A, 2018, 6, 5255–5259 RSC
- M. Opanasenko, W. O. N. Parker, M. Shamzhy, E. Montanari, M. Bellettato, M. Mazur, R. Millini and J. Čejka, J. Am. Chem. Soc., 2014, 136, 2511–2519 CrossRef PubMed
- X. Li and M. W. Deem, J. Phys. Chem. C, 2014, 118, 15835–15839 CrossRef
- M. Mazur, M. Kubu, P. S. Wheatley and P. Eliasova, Catal. Today, 2015, 243, 23–31 CrossRef
- M. V. Shamzhy, C. Ochoa-Hernandez, V. I. Kasneryk, M. V. Opanasenko and M. Mazur, Catal. Today, 2016, 277, 37–47 CrossRef
- M. V. Shamzhy, P. Eliasova, D. Vitvarova, M. V. Opanasenko, D. S. Firth and R. E. Morris, Chem. – Eur. J., 2016, 22, 17377–17386 CrossRef PubMed
- Y. Wang, Y. Liu, L. Wang, H. Wu, X. Li, M. He and P. Wu, J. Phys. Chem. C, 2009, 113, 18753–18760 CrossRef
- W. J. Roth and C. T. Kresge, Microporous Mesoporous Mater., 2011, 144, 158–161 CrossRef
- M. Opanasenko, M. Shamzhy, F. Yu, W. Zhou, R. E. Morris and J. Cejka, Chem. Sci., 2016, 7, 3589–3601 RSC
- P. Wu, J. F. Ruan, L. L. Wang, L. L. Wu, Y. Wang, Y. M. Liu, W. B. Fan, M. Y. He, O. Terasaki and T. Tatsumi, J. Am. Chem. Soc., 2008, 130, 8178–8187 CrossRef PubMed
- M. E. Landis, B. A. Aufdembrink, P. Chu, I. D. Johnson, G. W. Kirker and M. K. Rubin, J. Am. Chem. Soc., 1991, 113, 3189–3190 CrossRef
- M. Kubů, W. J. Roth, H. F. Greer, W. Zhou, R. E. Morris, J. Přech and J. Čejka, Chem. – Eur. J., 2013, 19, 13937–13945 CrossRef PubMed
- D. E. W. Vaughan, Catal. Today, 1988, 2, 187–198 CrossRef
- J. Přech and J. Čejka, Catal. Today, 2016, 277, 2–8 CrossRef
- J. Přech, M. A. Carretero and J. Čejka, ChemCatChem, 2017, 9, 3063–3072 CrossRef
- B. Liu, C. Wattanaprayoon, S. C. Oh, L. Emdadi and D. Liu, Chem. Mater., 2015, 27, 1479–1487 CrossRef
- H. Gies, M. Feyen, T. De Baerdemaeker, D. E. De Vos, B. Yilmaz, U. Müller, X. Meng, F.-S. Xiao, W. Zhang, T. Yokoi, T. Tatsumi and X. Bao, Microporous Mesoporous Mater., 2016, 222, 235–240 CrossRef
- L. Liu, U. Díaz, R. Arenal, G. Agostini, P. Concepción and A. Corma, Nat. Mater., 2016, 16, 132 CrossRef PubMed
- Z. Zhao, Y. Li, M. Feyen, R. McGuire, U. Müller and W. Zhang, ChemCatChem, 2018, 10, 2254–2259 CrossRef
- A. Corma, V. Fornes, S. B. Pergher, T. L. M. Maesen and J. G. Buglass, Nature, 1998, 396, 353–356 CrossRef
- J. Přech, unpublished work.
- A. J. Jacobson, Mater. Sci. Forum, 1994, 152-153, 1–12 Search PubMed
- R. Ma and T. Sasaki, Adv. Mater., 2010, 22, 5082–5104 CrossRef PubMed
- T. Maluangnont, Y. Yamauchi, T. Sasaki, W. J. Roth, J. Čejka and M. Kubů, Chem. Commun., 2014, 50, 7378–7381 RSC
- K. Varoon, X. Zhang, B. Elyassi, D. D. Brewer, M. Gettel, S. Kumar, J. A. Lee, S. Maheshwari, A. Mittal, C.-Y. Sung, M. Cococcioni, L. F. Francis, A. V. McCormick, K. A. Mkhoyan and M. Tsapatsis, Science, 2011, 334, 72–75 CrossRef PubMed
- K. Na, C. Jo, J. Kun, W. S. Ahn and R. Ryoo, ACS Catal., 2011, 1, 901–907 CrossRef
- J. Kim, J. Chun and R. Ryoo, Chem. Commun., 2015, 51, 13102–13105 RSC
- C. A. Fyfe, G. C. Gobbi, J. S. Hartman, J. Klinowski and J. M. Thomas, J. Phys. Chem., 1982, 86, 1247–1250 CrossRef
- G. Majano, L. Delmotte, V. Valtchev and S. Mintova, Chem. Mater., 2009, 21, 4184–4191 CrossRef
- Y. Seo, K. Cho, Y. Jung and R. Ryoo, ACS Catal., 2013, 3, 713–720 CrossRef
- A. Lacarriere, F. Luck, D. Świerczyński, F. Fajula and V. Hulea, Appl. Catal., A, 2011, 402, 208–217 CrossRef
- J.-P. Gilson, G. C. Edwards, A. W. Peters, K. Rajagopalan, R. F. Wormsbecher, T. G. Roberie and M. P. Shatlock, J. Chem. Soc., Chem. Commun., 1987, 91–92 RSC
- C. O. Arean, M. R. Delgado, P. Nachtigall, H. V. Thang, M. Rubes, R. Bulanek and P. Chlubna-Eliasova, Phys. Chem. Chem. Phys., 2014, 16, 10129–10141 RSC
- K. Góra-Marek, K. Tarach and M. Choi, J. Phys. Chem. C, 2014, 118, 12266–12274 CrossRef
- S. Laforge, P. Ayrault, D. Martin and M. Guisnet, Appl. Catal., A, 2005, 279, 79–88 CrossRef
- N. S. Nesterenko, F. Thibault-Starzyk, V. Montouilliout, V. V. Yushchenko, C. Fernandez, J.-P. Gilson, F. Fajula and I. I. Ivanova, Kinet. Catal., 2006, 47, 40–48 CrossRef
- C. Jo, R. Ryoo, N. Zilkova, D. Vitvarova and J. Cejka, Catal. Sci. Technol., 2013, 3, 2119–2129 RSC
- E. F. Rakiewicz, A. W. Peters, R. F. Wormsbecher, K. J. Sutovich and K. T. Mueller, J. Phys. Chem. B, 1998, 102, 2890–2896 CrossRef
- Q. Zhao, W.-H. Chen, S.-J. Huang, Y.-C. Wu, H.-K. Lee and S.-B. Liu, J. Phys. Chem. B, 2002, 106, 4462–4469 CrossRef
- W. E. Farneth and R. J. Gorte, Chem. Rev., 1995, 95, 615–635 CrossRef
- M. Niwa and N. Katada, Chem. Rec., 2013, 13, 432–455 CrossRef PubMed
- S. Bordiga, C. Lamberti, F. Bonino, A. Travert and F. Thibault-Starzyk, Chem. Soc. Rev., 2015, 44, 7262–7341 RSC
- G. Busca, Chem. Rev., 2007, 107, 5366–5410 CrossRef PubMed
- N. Katada, Mol. Catal., 2017 DOI:10.1016/j.mcat.2017.12.024
- M. Niwa, K. Suzuki, N. Katada, T. Kanougi and T. Atoguchi, J. Phys. Chem. B, 2005, 109, 18749–18757 CrossRef PubMed
- L. P. Hammett and A. J. Deyrup, J. Am. Chem. Soc., 1932, 54, 2721–2739 CrossRef
- L. P. Hammett and M. A. Paul, J. Am. Chem. Soc., 1934, 56, 827–829 CrossRef
- M. W. Anderson and J. Klinowski, Zeolites, 1986, 6, 150–153 CrossRef
-
M. Taramasso, G. Perego and B. Notari, US Pat., 4410501, Snam Pregetti SpA, 1983 Search PubMed
-
P. Ratnasamy, D. Srinivas and H. Knözinger, Advances in Catalysis, Academic Press, 2004, vol. 48, pp. 1–169 Search PubMed
- Q. Guo, K. Sun, Z. Feng, G. Li, M. Guo, F. Fan and C. Li, Chem. – Eur. J., 2012, 18, 13854–13860 CrossRef PubMed
- J. Přech, D. Vitvarová, L. Lupínková, M. Kubů and J. Čejka, Microporous Mesoporous Mater., 2015, 212, 28–34 CrossRef
- M. Sasaki, Y. Sato, Y. Tsuboi, S. Inagaki and Y. Kubota, ACS Catal., 2014, 4, 2653–2657 CrossRef
- G. Ricchiardi, A. Damin, S. Bordiga, C. Lamberti, G. Spanò, F. Rivetti and A. Zecchina, J. Am. Chem. Soc., 2001, 123, 11409–11419 CrossRef PubMed
- S. Bordiga, F. Bonino, A. Damin and C. Lamberti, Phys. Chem. Chem. Phys., 2007, 9, 4854–4878 RSC
- A. Corma, U. Diaz, V. Fornes, J. L. Jorda, M. Domine and F. Rey, Chem. Commun., 1999, 779–780 RSC
-
S. Valencia and A. Corma, US Pat., 09407652, Honeywell UOP LLC, 1999 Search PubMed
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423–425 CrossRef PubMed
- M. Renz, T. Blasco, A. Corma, V. Fornés, R. Jensen and L. Nemeth, Chem. – Eur. J., 2002, 8, 4708–4717 CrossRef
- A. Al-Nayili, K. Yakabi and C. Hammond, J. Mater. Chem. A, 2016, 4, 1373–1382 RSC
- R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, Y. Román-Leshkov, S.-J. Hwang, A. Palsdottir, D. Silverman, R. F. Lobo, L. A. Curtiss and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727–9732 CrossRef PubMed
- L. Ren, Q. Guo, P. Kumar, M. Orazov, D. Xu, S. M. Alhassan, K. A. Mkhoyan, M. E. Davis and M. Tsapatsis, Angew. Chem., Int. Ed., 2015, 54, 10848–10851 CrossRef PubMed
- J. W. Harris, M. J. Cordon, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro and R. Gounder, J. Catal., 2016, 335, 141–154 CrossRef
- G. Liu, J.-G. Jiang, B. Yang, X. Fang, H. Xu, H. Peng, L. Xu, Y. Liu and P. Wu, Microporous Mesoporous Mater., 2013, 165, 210–218 CrossRef
- H. Y. Luo, L. Bui, W. R. Gunther, E. Min and Y. Román-Leshkov, ACS Catal., 2012, 2, 2695–2699 CrossRef
- K. Chaudhari, T. K. Das, P. R. Rajmohanan, K. Lazar, S. Sivasanker and A. J. Chandwadkar, J. Catal., 1999, 183, 281–291 CrossRef
- N. K. Mal, V. Ramaswamy, S. Ganapathy and A. V. Ramaswamy, J. Chem. Soc., Chem. Commun., 1994, 1933–1934 RSC
- N. K. Mal and A. V. Ramaswamy, J. Mol. Catal. A: Chem., 1996, 105, 149–158 CrossRef
- M. Boronat, P. Concepción, A. Corma, M. Renz and S. Valencia, J. Catal., 2005, 234, 111–118 CrossRef
- Z. Zhu, H. Xu, J. Jiang, X. Liu, J. Ding and P. Wu, Appl. Catal., A, 2016, 519, 155–164 CrossRef
- P. R. Hari Prasad Rao, A. V. Ramaswamy and P. Ratnasamy, J. Catal., 1992, 137, 225–231 CrossRef
- A. Corma, M. a. T. Navarro and M. Renz, J. Catal., 2003, 219, 242–246 CrossRef
- R. Bermejo-Deval, M. Orazov, R. Gounder, S.-J. Hwang and M. E. Davis, ACS Catal., 2014, 4, 2288–2297 CrossRef
- C. Martínez, D. Verboekend, J. Pérez-Ramírez and A. Corma, Catal. Sci. Technol., 2013, 3, 972–981 RSC
- A. Ishihara, K. Kimura, T. Hashimoto and H. Nasu, J. Jpn. Pet. Inst., 2014, 57, 34–46 CrossRef
- A. Ishihara, D. Kawaraya, T. Sonthisawate, K. Kimura, T. Hashimoto and H. Nasu, J. Mol. Catal. A: Chem., 2015, 396, 310–318 CrossRef
- V. P. S. Caldeira, A. Peral, M. Linares, A. S. Araujo, R. A. Garcia-Muñoz and D. P. Serrano, Appl. Catal., A, 2017, 531, 187–196 CrossRef
-
The Development of Catalysis: A History of Key Processes and Personas in Catalytic Science and Technology, ed. A. Zecchina and S. Califano, John Wiley & Sons, 2017 Search PubMed
-
S. F. Abdo and S. T. Wilson, Zeolites in Catalysis: Properties and Applications, The Royal Society of Chemistry, 2017, pp. 310–350 Search PubMed
-
F. Di Renzo and F. Fajula, in Studies in Surface Science and Catalysis, ed. J. Ĉejka and H. van Bekkum, Elsevier, 2005, vol. 157, pp. 1–12 Search PubMed
- C. Perego, A. Carati, P. Ingallina, M. A. Mantegazza and G. Bellussi, Appl. Catal., A, 2001, 221, 63–72 CrossRef
- M. N. Akhtar, N. Al-Yassir, S. Al-Khattaf and J. Čejka, Catal. Today, 2012, 179, 61–72 CrossRef
- C. Fernandez, I. Stan, J. P. Gilson, K. Thomas, A. Vicente, A. Bonilla and J. Pérez-Ramírez, Chem. – Eur. J., 2010, 16, 6224–6233 CrossRef PubMed
- R. A. García-Muñoz, D. P. Serrano, G. Vicente, M. Linares, D. Vitvarova and J. Čejka, Catal. Today, 2015, 243, 141–152 CrossRef
- M. Linares, C. Vargas, A. Garcia, C. Ochoa-Hernandez, J. Cejka, R. A. Garcia-Munoz and D. P. Serrano, Catal. Sci. Technol., 2017, 7, 181–190 RSC
- J. A. Martens, D. Verboekend, K. Thomas, G. Vanbutsele, J. Pérez-Ramírez and J.-P. Gilson, Catal. Today, 2013, 218-219, 135–142 CrossRef
- T. Tang, C. Yin, L. Wang, Y. Ji and F.-S. Xiao, J. Catal., 2008, 257, 125–133 CrossRef
- A. P. Vyas, J. L. Verma and N. Subrahmanyam, Fuel, 2010, 89, 1–9 CrossRef
- A. Carrero, G. Vicente, R. Rodríguez, M. Linares and G. L. del Peso, Catal. Today, 2011, 167, 148–153 CrossRef
- Q. Zhang, W. Ming, J. Ma, J. Zhang, P. Wang and R. Li, J. Mater. Chem. A, 2014, 2, 8712–8718 RSC
- Y. Sun and R. Prins, Angew. Chem., Int. Ed., 2008, 47, 8478–8481 CrossRef PubMed
- Q. Huo, T. Dou, Z. Zhao and H. Pan, Appl. Catal., A, 2010, 381, 101–108 CrossRef
- Q. Yu, L. Zhang, R. Guo, J. Sun, W. Fu, T. Tang and T. Tang, Fuel Process. Technol., 2017, 159, 76–87 CrossRef
- M. Y. Kustova, S. B. Rasmussen, A. L. Kustov and C. H. Christensen, Appl. Catal., B, 2006, 67, 60–67 CrossRef
- A. L. Kustov, T. W. Hansen, M. Kustova and C. H. Christensen, Appl. Catal., B, 2007, 76, 311–319 CrossRef
- A. Śrebowata, K. Tarach, V. Girman and K. Góra-Marek, Appl. Catal., B, 2016, 181, 550–560 CrossRef
- K. A. Sashkina, V. S. Labko, N. A. Rudina, V. N. Parmon and E. V. Parkhomchuk, J. Catal., 2013, 299, 44–52 CrossRef
- H.-G. Chen and Y. H. P. Zhang, Renewable Sustainable Energy Rev., 2015, 47, 117–132 CrossRef
- IEA, Internation Energy Agency, 2011.
-
B. Balagurumurthy, R. Singh and T. Bhaskar, Recent Advances in Thermo-Chemical Conversion of Biomass, Elsevier, Boston, 2015, pp. 109–132 Search PubMed
-
D. P. Serrano, J. A. Melero, J. M. Coronado, P. Pizarro and G. Morales, Zeolites in Catalysis: Properties and Applications, The Royal Society of Chemistry, 2017, pp. 441–480 Search PubMed
- D. P. Serrano, J. A. Melero, G. Morales, J. Iglesias and P. Pizarro, Catal. Rev., 2018, 60, 1–70 CrossRef
- IEA, Internation Energy Agency, 2008.
- R. Rinaldi and F. Schuth, Energy Environ. Sci., 2009, 2, 610–626 RSC
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 RSC
- D. Kubička and O. Kikhtyanin, Catal. Today, 2015, 243, 10–22 CrossRef
- P. S. Rezaei, H. Shafaghat and W. M. A. W. Daud, Appl. Catal., A, 2014, 469, 490–511 CrossRef
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef
- M. Asadieraghi, W. M. Ashri Wan Daud and H. F. Abbas, RSC Adv., 2015, 5, 22234–22255 RSC
- V. Sukumar, V. Manieniyan and S. Sivaprakasam, Int. J. ChemTech Res., 2015, 8, 196–206 Search PubMed
- J. Xu, J. Jiang and J. Zhao, Renewable Sustainable Energy Rev., 2016, 58, 331–340 CrossRef
- X. Zhao, L. Wei, S. Cheng and J. Julson, Catalysts, 2017, 7, 83 CrossRef
- Y. Li, B. Li, X. Zhang, L. Chen, Q. Zhang, T. Wang and L. Ma, Energy Convers. Manage., 2016, 122, 1–9 CrossRef
- Z. Ma, E. Troussard and J. A. van Bokhoven, Appl. Catal., A, 2012, 423-424, 130–136 CrossRef
- S. Kelkar, C. M. Saffron, K. Andreassi, Z. Li, A. Murkute, D. J. Miller, T. J. Pinnavaia and R. M. Kriegel, Appl. Catal., B, 2015, 174-175, 85–95 CrossRef
- Y.-K. Park, M. L. Yoo, S. H. Jin and S. H. Park, Renewable Energy, 2015, 79, 20–27 CrossRef
- E. L. Schultz, C. A. Mullen and A. A. Boateng, Energy Technol., 2017, 5, 196–204 CrossRef
- S. Wang, Q. Cai, J. Chen, L. Zhang, X. Wang and C. Yu, Ind. Eng. Chem. Res., 2014, 53, 13935–13944 CrossRef
- H. J. Park, K.-H. Park, J.-K. Jeon, J. Kim, R. Ryoo, K.-E. Jeong, S. H. Park and Y.-K. Park, Fuel, 2012, 97, 379–384 CrossRef
- X. Ji, B. Liu, W. Ma, G. Chen, B. Yan and Z. Cheng, J. Anal. Appl. Pyrolysis, 2017, 123, 278–283 CrossRef
- H. J. Park, J. I. Dong, J.-K. Jeon, K. S. Yoo, J. S. Yim, J. M. Sohn and Y.-K. Park, J. Ind. Eng. Chem., 2007, 13, 182 Search PubMed
- M. Asadieraghi and W. M. A. Wan Daud, Energy Convers. Manage., 2015, 101, 151–163 CrossRef
- Z. Wang, R. Ma and W. Song, J. Anal. Appl. Pyrolysis, 2016, 122, 183–190 CrossRef
- K. Uemura, S. Appari, S. Kudo, J.-i. Hayashi, H. Einaga and K. Norinaga, Fuel Process. Technol., 2015, 136, 73–78 CrossRef
- Y.-M. Kim, B.-S. Kim, K.-S. Chea, T. S. Jo, S. Kim and Y.-K. Park, Appl. Chem. Eng., 2016, 27, 407–414 CrossRef
- G. T. Neumann and J. C. Hicks, ACS Catal., 2012, 2, 642–646 CrossRef
- D. P. Gamliel, H. J. Cho, W. Fan and J. A. Valla, Appl. Catal., A, 2016, 522, 109–119 CrossRef
- L. Y. Jia, M. Raad, S. Hamieh, J. Toufaily, T. Hamieh, M. M. Bettahar, G. Mauviel, M. Tarrighi, L. Pinard and A. Dufour, Green Chem., 2017, 19, 5442–5459 RSC
- Y. Wang, Y. Zu and S. Wang, Abstracts of papers of the American Chemical Society, 2014, vol. 247.
- B. Ma, X. Yi, L. Chen, A. Zheng and C. Zhao, J. Mater. Chem. A, 2016, 4, 11330–11341 RSC
- H. Hernando, J. Fermoso, C. Ochoa-Hernández, M. Opanasenko, P. Pizarro, J. M. Coronado, J. Čejka and D. P. Serrano, Catal. Today, 2018, 304, 30–38 CrossRef
- H. W. Lee, S. H. Park, J.-K. Jeon, R. Ryoo, W. Kim, D. J. Suh and Y.-K. Park, Catal. Today, 2014, 232, 119–126 CrossRef
- S. R. Naqvi, Y. Uemura, S. Yusup, Y. Sugiur, N. Nishiyama and M. Naqvi, Energy Procedia, 2015, 75, 793–800 CrossRef
- S. R. Naqvi, Y. Uemura, S. Yusup, N. Nishiyama and M. Naqvi, Energy Procedia, 2017, 105, 557–561 CrossRef
- S. R. Naqvi, Y. Uemura, S. Yusup, Y. Sugiura and N. Nishiyama, J. Anal. Appl. Pyrolysis, 2015, 114, 32–39 CrossRef
- S. R. Naqvi and M. Naqvi, Int. J. Energy Res., 2018, 42, 1352–1362 CrossRef
- J. Fermoso, H. Hernando, P. Jana, I. Moreno, J. Přech, C. Ochoa-Hernández, P. Pizarro, J. M. Coronado, J. Čejka and D. P. Serrano, Catal. Today, 2016, 277, 171–181 CrossRef
-
J. Fermoso, P. Pizarro, J. M. Coronado and D. P. Serrano, in Encyclopedia of Sustainability Science and Technology, ed. R. A. Meyers, Springer New York, New York, NY, 2017, pp. 1–33 Search PubMed
- Y. Zhu, Z. Hua, J. Zhou, L. Wang, J. Zhao, Y. Gong, W. Wu, M. Ruan and J. Shi, Chem. – Eur. J., 2011, 17, 14618–14627 CrossRef PubMed
- A. Veses, B. Puértolas, J. M. López, M. S. Callén, B. Solsona and T. García, ACS Sustainable Chem. Eng., 2016, 4, 1653–1660 CrossRef
- C. J. Barrett, J. N. Chheda, G. W. Huber and J. A. Dumesic, Appl. Catal., B, 2006, 66, 111–118 CrossRef
- R. M. West, Z. Y. Liu, M. Peter, C. A. Gärtner and J. A. Dumesic, J. Mol. Catal. A: Chem., 2008, 296, 18–27 CrossRef
- R. Xing, A. V. Subrahmanyam, H. Olcay, W. Qi, G. P. van Walsum, H. Pendse and G. W. Huber, Green Chem., 2010, 12, 1933–1946 RSC
- E. Dumitriu, V. Hulea, I. Fechete, A. Auroux, J.-F. Lacaze and C. Guimon, Microporous Mesoporous Mater., 2001, 43, 341–359 CrossRef
-
T. Komatsu, M. Mitsuhashi and T. Yashima, in Studies in Surface Science and Catalysis, ed. R. Aiello, G. Giordano and F. Testa, Elsevier, 2002, vol. 142, pp. 667–674 Search PubMed
- A. Ungureanu, S. Royer, T. V. Hoang, D. Trong On, E. Dumitriu and S. Kaliaguine, Microporous Mesoporous Mater., 2005, 84, 283–296 CrossRef
- O. Kikhtyanin, V. Kelbichová, D. Vitvarová, M. Kubů and D. Kubička, Catal. Today, 2014, 227, 154–162 CrossRef
- B. Puértolas, T. C. Keller, S. Mitchell and J. Pérez-Ramírez, Appl. Catal., B, 2016, 184, 77–86 CrossRef
- O. Kikhtyanin, P. Chlubna, T. Jindrova and D. Kubicka, Dalton Trans., 2014, 43, 10628–10641 RSC
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef PubMed
- C. Moreau, R. Durand, A. Roux and D. Tichit, Appl. Catal., A, 2000, 193, 257–264 CrossRef
- M. Moliner, Dalton Trans., 2014, 43, 4197–4208 RSC
- Y. Román-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef PubMed
- C. M. Lew, N. Rajabbeigi and M. Tsapatsis, Microporous Mesoporous Mater., 2012, 153, 55–58 CrossRef
- S. Caratzoulas, M. E. Davis, R. J. Gorte, R. Gounder, R. F. Lobo, V. Nikolakis, S. I. Sandler, M. A. Snyder, M. Tsapatsis and D. G. Vlachos, J. Phys. Chem. C, 2014, 118, 22815–22833 CrossRef
- S. Saravanamurugan, M. Paniagua, J. A. Melero and A. Riisager, J. Am. Chem. Soc., 2013, 135, 5246–5249 CrossRef PubMed
- V. Choudhary, A. B. Pinar, S. I. Sandler, D. G. Vlachos and R. F. Lobo, ACS Catal., 2011, 1, 1724–1728 CrossRef
- M. Paniagua, S. Saravanamurugan, M. Melian-Rodriguez, J. A. Melero and A. Riisager, ChemSusChem, 2015, 8, 1088–1094 CrossRef PubMed
- I. Graça, M. C. Bacariza and D. Chadwick, Microporous Mesoporous Mater., 2018, 255, 130–139 CrossRef
- G. Li, E. A. Pidko and E. J. M. Hensen, Catal. Sci. Technol., 2014, 4, 2241–2250 RSC
- P. Y. Dapsens, C. Mondelli, J. Jagielski, R. Hauert and J. Perez-Ramirez, Catal. Sci. Technol., 2014, 4, 2302–2311 RSC
- J. Zhang, L. Wang, G. Wang, F. Chen, J. Zhu, C. Wang, C. Bian, S. Pan and F.-S. Xiao, ACS Sustainable Chem. Eng., 2017, 5, 3123–3131 CrossRef
- K. Y. Nandiwale, A. M. Pande and V. V. Bokade, RSC Adv., 2015, 5, 79224–79231 RSC
- A. Feliczak-Guzik, M. Sprynskyy, I. Nowak, M. Jaroniec and B. Buszewski, J. Colloid Interface Sci., 2018, 516, 379–383 CrossRef PubMed
- L. Ren, Q. Guo, M. Orazov, D. Xu, D. Politi, P. Kumar, S. M. Alhassan, K. A. Mkhoyan, D. Sidiras, M. E. Davis and M. Tsapatsis, ChemCatChem, 2016, 8, 1274–1278 CrossRef
- X. Ma, D. Zhou, X. Chu, D. Li, J. Wang, W. Song and Q. Xia, Microporous Mesoporous Mater., 2017, 237, 180–188 CrossRef
- E. Nikolla, Y. Román-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408–410 CrossRef
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC
- S. Lima, M. Pillinger and A. A. Valente, Catal. Commun., 2008, 9, 2144–2148 CrossRef
- R. O’Neill, M. N. Ahmad, L. Vanoye and F. Aiouache, Ind. Eng. Chem. Res., 2009, 48, 4300–4306 CrossRef
- P. L. Dhepe and R. Sahu, Green Chem., 2010, 12, 2153–2156 RSC
- E. I. Gürbüz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1270–1274 CrossRef PubMed
- J. Iglesias, J. A. Melero, G. Morales, M. Paniagua and B. Hernández, ChemCatChem, 2016, 8, 2089–2099 CrossRef
- O. A. Abdelrahman, D. S. Park, K. P. Vinter, C. S. Spanjers, L. Ren, H. J. Cho, D. G. Vlachos, W. Fan, M. Tsapatsis and P. J. Dauenhauer, ACS Sustainable Chem. Eng., 2017, 5, 3732–3736 CrossRef
- M. V. Rodrigues, C. Vignatti, T. Garetto, S. H. Pulcinelli, C. V. Santilli and L. Martins, Appl. Catal., A, 2015, 495, 84–91 CrossRef
- C. S. Carriço, F. T. Cruz, M. B. dos Santos, D. S. Oliveira, H. O. Pastore, H. M. C. Andrade and A. J. S. Mascarenhas, J. Catal., 2016, 334, 34–41 CrossRef
- J. S. Yoon, T. Lee, J.-W. Choi, D. J. Suh, K. Lee, J.-M. Ha and J. Choi, Catal. Today, 2017, 293-294, 142–150 CrossRef
- W. Xu, S. J. Miller, P. K. Agrawal and C. W. Jones, Appl. Catal., A, 2013, 459, 114–120 CrossRef
- K. S. Arias, M. J. Climent, A. Corma and S. Iborra, Energy Environ. Sci., 2015, 8, 317–331 RSC
- M. Osman, S. Al-Khattaf, U. Diaz, C. Martinez and A. Corma, Catal. Sci. Technol., 2016, 6, 3166–3181 RSC
- C. Xing, W. Shen, G. Yang, R. Yang, P. Lu, J. Sun, Y. Yoneyama and N. Tsubaki, Catal. Commun., 2014, 55, 53–56 CrossRef
- C. Xing, G. Yang, M. Wu, R. Yang, L. Tan, P. Zhu, Q. Wei, J. Li, J. Mao, Y. Yoneyama and N. Tsubaki, Fuel, 2015, 148, 48–57 CrossRef
- J. Kang, X. Wang, X. Peng, Y. Yang, K. Cheng, Q. Zhang and Y. Wang, Ind. Eng. Chem. Res., 2016, 55, 13008–13019 CrossRef
- Q.-Q. Hao, C.-Y. Lei, Y.-H. Song, Z.-T. Liu and Z.-W. Liu, Catal. Today, 2016, 274, 109–115 CrossRef
- J.-C. Kim, S. Lee, K. Cho, K. Na, C. Lee and R. Ryoo, ACS Catal., 2014, 4, 3919–3927 CrossRef
- Global and China Ethylene Oxide (EO) Industry Report, 2017–2021, ReportBuyer Ltd., 2017.
- Eni, Titanium Silicalite (TS-1) zeolite based proprietary catalyst, https://www.scribd.com/document/208913719/TS1-Flyer-Lug09, accessed 13.5.2017.
- Eni, Cyclohexanone Oxime Ammoximation of Cyclohexanone with Titanium Silicalite (TS-1) proprietary catalyst, https://www.versalis.eni.com/irj/go/km/docs/versalis/Contenuti%20Versalis/EN/Documenti/La%20nostra%20offerta/Licensing/Catalizzatori/ESE_Tecniche_Cyclohexanone_180214.pdf, accessed 13.5.2017.
- F. S. Xiao, B. Xie, H. Y. Zhang, L. Wang, X. J. Meng, W. P. Zhang, X. H. Bao, B. Yilmaz, U. Muller, H. Gies, H. Imai, T. Tatsumi and D. De Vos, ChemCatChem, 2011, 3, 1442–1446 CrossRef
- Z. Deng, Y. Yang, X. Lu, J. Ding, M. He and P. Wu, Catal. Sci. Technol., 2016, 6, 2605–2615 RSC
- P. Wu, T. Tatsumi, T. Komatsu and T. Yashima, Chem. Lett., 2000, 774–775 CrossRef
- P. Wu, T. Tatsumi, T. Komatsu and T. Yashima, J. Phys. Chem. B, 2001, 105, 2897–2905 CrossRef
- J. Přech, Catal. Rev., 2018, 60, 71–131 CrossRef
- L. L. Wang, Y. Wang, Y. M. Liu, H. H. Wu, X. H. Li, M. Y. He and P. Wu, J. Mater. Chem., 2009, 19, 8594–8602 RSC
- H. Xu, L. Fu, J.-G. Jiang, M. He and P. Wu, Microporous Mesoporous Mater., 2014, 189, 41–48 CrossRef
- C.-G. Li, Y. Lu, H. Wu, P. Wu and M. He, Chem. Commun., 2015, 51, 14905–14908 RSC
- X. Fang, Q. Wang, A. Zheng, Y. Liu, Y. Wang, X. Deng, H. Wu, F. Deng, M. He and P. Wu, Catal. Sci. Technol., 2012, 2, 2433–2435 RSC
- M. B. Yue, M. N. Sun, F. Xie and D. D. Ren, Microporous Mesoporous Mater., 2014, 183, 177–184 CrossRef
- D. P. Serrano, R. Sanz, P. Pizarro and I. Moreno, Top. Catal., 2010, 53, 1319–1329 CrossRef
- M. Yoshioka, T. Yokoi and T. Tatsumi, Microporous Mesoporous Mater., 2014, 200, 11–18 CrossRef
- M. Moliner and A. Corma, Chem. Mater., 2012, 24, 4371–4375 CrossRef
- D. Serrano, R. Sanz, P. Pizarro and I. Moreno, Chem. Commun., 2009, 1407–1409 RSC
- F. Jin, C.-C. Chang, C.-W. Yang, J.-F. Lee, L.-Y. Jang and S. Cheng, J. Mater. Chem. A, 2015, 3, 8715–8724 RSC
- A. Kumar and D. Srinivas, Catal. Today, 2012, 198, 59–68 CrossRef
- N. Tsunoji, M. Opanasenko, M. Kubů, J. Čejka, H. Nishida, S. Hayakawa, Y. Ide, M. Sadakane and T. Sano, ChemCatChem, 2018, 10, 2536–2540 CrossRef
- A. Baeyer and V. Villiger, Chem. Ber., 1899, 32, 3625–3633 CrossRef
- J. Fischer and W. F. Hölderich, Appl. Catal., A, 1999, 180, 435–443 CrossRef
- R. Llamas, C. Jiménez-Sanchidrián and J. R. Ruiz, Tetrahedron, 2007, 63, 1435–1439 CrossRef
- A. Bhaumik, P. Kumar and R. Kumar, Catal. Lett., 1996, 40, 47–50 CrossRef
- H. Xu, J. Jiang, B. Yang, H. Wu and P. Wu, Catal. Commun., 2014, 55, 83–86 CrossRef
- S. Conrad, P. Wolf, P. Müller, H. Orsted and I. Hermans, ChemCatChem, 2017, 9, 175–182 CrossRef
- M. Boronat, P. Concepción, A. Corma and M. Renz, Catal. Today, 2007, 121, 39–44 CrossRef
- X. Ouyang, Y.-J. Wanglee, S.-J. Hwang, D. Xie, T. Rea, S. I. Zones and A. Katz, Dalton Trans., 2014, 43, 10417–10429 RSC
- B. Yang and Q. Zou, Chem. Lett., 2017, 46, 1781–1784 CrossRef
- X. Liu, H. Xu, L. Zhang, L. Han, J. Jiang, P. Oleynikov, L. Chen and P. Wu, ACS Catal., 2016, 6, 8420–8431 CrossRef
- W. Ponndorf, Angew. Chem., 1926, 39, 138–143 CrossRef
- H. Meerwein and R. Schmidt, Justus Liebigs Ann. Chem., 1925, 444, 221–238 CrossRef
- A. Verley, Bull. Soc. Chim. Fr., 1925, 37, 537–542 Search PubMed
- R. V. Oppenauer, Recl. Trav. Chim. Pays-Bas, 1937, 56, 137–144 CrossRef
- P. J. Kunkeler, B. J. Zuurdeeg, J. C. van der Waal, J. A. van Bokhoven, D. C. Koningsberger and H. van Bekkum, J. Catal., 1998, 180, 234–244 CrossRef
- E. J. Creyghton, S. D. Ganeshie, R. S. Downing and H. van Bekkum, J. Chem. Soc., Chem. Commun., 1995, 1859–1860 RSC
- J. C. van der Waal, K. Tan and H. van Bekkum, Catal. Lett., 1996, 41, 63–67 CrossRef
- A. Corma, M. E. Domine, L. Nemeth and S. Valencia, J. Am. Chem. Soc., 2002, 124, 3194–3195 CrossRef PubMed
- A. Corma and M. Renz, Angew. Chem., Int. Ed., 2007, 46, 298–300 CrossRef PubMed
- J. Wang, K. Okumura, S. Jaenicke and G.-K. Chuah, Appl. Catal., A, 2015, 493, 112–120 CrossRef
- A. Corma, M. E. Domine and S. Valencia, J. Catal., 2003, 215, 294–304 CrossRef
- Y. Matsunaga, H. Yamazaki, H. Imai, T. Yokoi, T. Tatsumi and J. N. Kondo, Microporous Mesoporous Mater., 2015, 206, 86–94 CrossRef
- Y. Zhu, Z. Hua, X. Zhou, Y. Song, Y. Gong, J. Zhou, J. Zhao and J. Shi, RSC Adv., 2013, 3, 4193–4198 RSC
- D. P. Serrano, R. Sanz, P. Pizarro, I. Moreno and S. Medina, Appl. Catal., B, 2014, 146, 35–42 CrossRef
- J. Přech, R. E. Morris and J. Čejka, Catal. Sci. Technol., 2016, 6, 2775–2786 RSC
-
H. van Bekkum and H. W. Kouwenhoven, Zeolite Manual for the Organic Chemist, Mijnbestseller.nl, Delft, 2015 Search PubMed
- S. V. d. Vyver and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2015, 54, 12554–12561 CrossRef PubMed
- Y. Sun and R. Prins, Appl. Catal., A, 2008, 336, 11–16 CrossRef
- K. Y. Nandiwale, P. Thakur and V. V. Bokade, Appl. Petrochem. Res., 2015, 5, 113–119 CrossRef
- D. Wang, X. Li, Z. Liu, Y. Zhang, Z. Xie and Y. Tang, J. Colloid Interface Sci., 2010, 350, 290–294 CrossRef PubMed
- J. Čejka, G. A. Kapustin and B. Wichterlová, Appl. Catal., A, 1994, 108, 187–204 CrossRef
- J. i.
![[C with combining breve]](https://www.rsc.org/images/entities/char_0043_0306.gif) ejka, A. Vondrová, B. Wichterlová, G. Vorbeck and R. Fricke, Zeolites, 1994, 14, 147–153 CrossRef
ejka, A. Vondrová, B. Wichterlová, G. Vorbeck and R. Fricke, Zeolites, 1994, 14, 147–153 CrossRef - J.-C. Kim, K. Cho and R. Ryoo, Appl. Catal., A, 2014, 470, 420–426 CrossRef
- L. Emdadi, S. C. Oh, Y. Wu, S. N. Oliaee, Y. Diao, G. Zhu and D. Liu, J. Catal., 2016, 335, 165–174 CrossRef
- M. V. Opanasenko, M. V. Shamzhy, C. Jo, R. Ryoo and J. Cejka, ChemCatChem, 2014, 6, 1919–1927 CrossRef
- B. Yang, H. Wu and P. Wu, J. Phys. Chem. C, 2014, 118, 24662–24669 CrossRef
- X. Ouyang, S.-J. Hwang, R. C. Runnebaum, D. Xie, Y.-J. Wanglee, T. Rea, S. I. Zones and A. Katz, J. Am. Chem. Soc., 2014, 136, 1449–1461 CrossRef PubMed
- L. Červený, K. Mikulcová and J. Čejka, Appl. Catal., A, 2002, 223, 65–72 CrossRef
- P. J. Harrington and E. Lodewijk, Org. Process Res. Dev., 1997, 1, 72–76 CrossRef
- C.-G. Li, L. Xu, P. Wu, H. Wu and M. He, Chem. Commun., 2014, 50, 15764–15767 RSC
| This journal is © The Royal Society of Chemistry 2018 |