
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced ring-opening polymerisation of L-lactide via a photocaged superbase†
P.
K. Kuroishi
 ab and
A. P.
Dove
*b
ab and
A. P.
Dove
*b
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: a.dove@bham.ac.uk
First published on 17th April 2018
Abstract
The phototriggered ring-opening polymerisation of L-lactide is demonstrated for the first time using a photocaged tetramethylguanidine. The catalytic activity of the free guanidine was also investigated, showing it to be active for the polymerisation of δ-valerolactone and ε-caprolactone in the presence of a thiourea cocatalyst.
Aliphatic polyesters have been widely investigated as biodegradable polymers in a range of industries (e.g. fibers, food packaging) as well as the biomedical field (e.g. medical implants, drug delivery etc.).1,2 Within this context, the ring-opening polymerisation (ROP) of cyclic (di)esters is the typical technique employed when polymers with controlled molecular weight and low dispersity are desirable, with organometallic catalysts being most commonly utilised for this process.3,4 However, these catalysts are generally sensitive to water and/or oxygen, are toxic and can be difficult or costly to remove from the polymeric product.5,6 Hence, the first report of an organocatalyst for the ROP of lactide by Hedrick and coworkers7 triggered great advances in the area of organocatalysis,8–11 which has been proven to be a versatile alternative to metal based catalysts, displaying low toxicity, long shelf life, and being easily removed from the resulting polymers.8,12
Beyond controlling the molecular weight and dispersity of polyesters, externally regulating the polymerisation initiation has the potential to determine the polymer structure and the properties of the resulting materials.13 This regulation can be promoted by light, which is a non-invasive stimulus able to provide spatiotemporal resolution by inducing chemical reactions through the selection of appropriate wavelengths.14 This approach has been previously implemented to prepare sophisticated materials where the precise control of the space is essential, such as microelectronic devices15 and DNA microarrays.16,17 In addition, the use of light as a stimulus has been widely investigated in the preparation of non-biodegradable polymers by controlled radical polymerisations (CRP) to provide specific surface modifications and create complex structures for various applications.18–20 Metters and coworkers, for example, reported the use of photo-CRP to graft poly(methacrylic acid) (PMAA) into silicon wafers with spatially defined gradient of density by varying the irradiation exposure time across the surface.21 The PMAA films were then functionalised with arginine–glycine–aspartic acid (RGD) peptide sequence that allowed to control the degree of specific cell adhesion by changing the RGD density. Although numerous examples of photo-CRP have been reported so far, the utilisation of photoinduced processes in the preparation of biodegradable polymers such as aliphatic polyesters and polycarbonates has been hardly explored.
In this context, a few examples of photocaged organocatalysts (i.e. passive organocatalysts able to be activated upon irradiation) were reported for the photoinduced ROP of lactones. Wang and coworkers demonstrated the use of a tetraphenylborate salt of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) which, under UV irradiation at 254 nm, generated a free base (TBD) that was able to catalyse the polymerisation of ε-caprolactone (ε-CL) in a controlled manner.22 However, this system required high-energy light source, which can be harmful to biological systems, in addition to presenting limited penetration depth.23 Following this work, our group demonstrated the use of triarylsulfonium hexafluorophosphate salts to photogenerate acid (365 nm UV light), that in turn promoted the cationic polymerisation of ε-CL, δ-valerolactone (δ-VL), and trimethylenecarbonate (TMC).24 However, the polymerisation was shown to be slow, with a loss of control being observed at monomer conversions above 50%. In addition, this catalyst was not active to produce polylactide (PLA), a bio-based polymer that is of high importance in the industrial and biomedical areas.25
Herein, we describe the application of 2-(nitrophenyl)propoxycarbonyl-1,1,3,3-tetramethylguanidine (NPPOC-TMG), a photobase generator (PBG) that releases TMG upon 320–400 nm light, in the phototriggered ROP of L-LA (Scheme 1). The photoremovable protecting group NPPOC has been efficiently and widely used in the photo-controlled release of amines and alcohols under low energy UV light,26–28 whereas the use of TMG as a ROP catalyst has been only reported for N-butyl N-carboxyanhydride using alcohol initiators.29 However, we anticipated that this commercially-available superbase would also catalyse the ROP of cyclic (di)esters in a similar fashion as other acyclic guanidines30,31 as a consequence of its high pKa (TMG pKa = 23.4).32 Therefore, we report the use of a new organocatalyst for the ROP of cyclic (di)esters that, once attached to a photo-protecting group, efficiently provide temporal control over the polymerisation initiation of L-LA, which opens space for further exploration in the preparation of complex macromolecular architecture and function for advanced applications.33
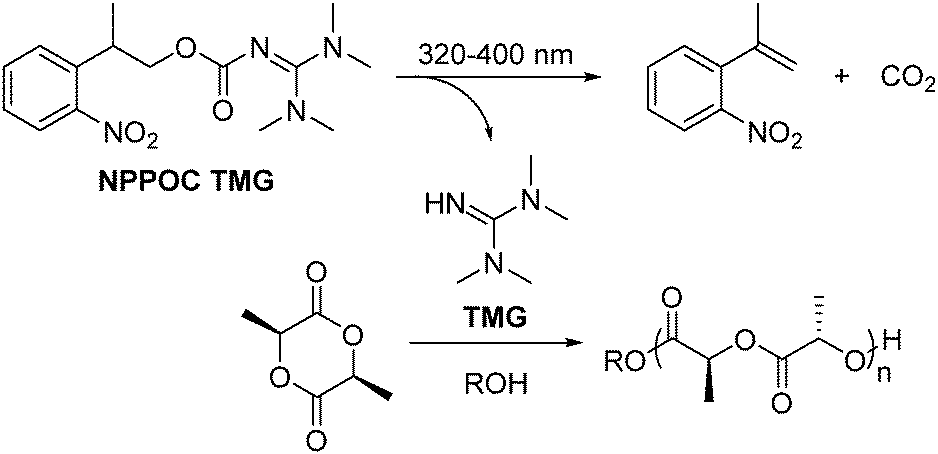 | ||
| Scheme 1 Phototriggered ROP of L-lactide using NPPOC-TMG under 320–400 nm irradiation. | ||
Prior to investigating the photocaged TMG in the polymerisation of cyclic (di)esters, we evaluated the catalytic activity of the free guanidine. For the polymerisation of L-LA, a monomer conversion of 93% was achieved in 2.5 h when 1 mol% of TMG was used ([L-LA]0 = 2 M, [L-LA]0/[BnOH]0 = 50), comparable to that mediated by amidines such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) but lower in activity compared to the most active guanidines such as TBD or phosphazene bases (Table 1).34–37 The linear increase of ln([L-LA]0/[L-LA]) with time (Fig. S1, ESI†), as well as the linear relationship between the number-average molecular weight (Mn) and the monomer conversion (Fig. 1), and the degree of polymerisation (Fig. S2, ESI†), with the dispersity (ĐM) remaining low throughout the polymerisations, demonstrated the high control over this polymerisation system. However, although the ROP of δ-VL and ε-CL catalysed by TMG was investigated in a similar way, TMG was not active enough for these polymerisations even at 5 mol% of catalyst loading. To overcome this, a thiourea (TU) cocatalyst was utilised as it has shown to activate the carbonyl group in the monomer unit by hydrogen bonding, which has proved to increase the polymerisation rate when used in combination with a tertiary amine.34,38 Similarly to the case of DBU-catalyzed ROP,34 δ-VL polymerised considerably faster when a catalyst system of TMG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TU (5/5) was utilised in comparison to TMG alone, achieving a monomer conversion of 93% in 23 h ([δ-VL]0 = 2 M, [δ-VL]0/[BnOH]0 = 30). Nevertheless, for ε-CL, this system required 15 days to promote a monomer conversion of 96% ([ε-CL]0 = 2 M, [ε-CL]0/[BnOH]0 = 25). Finally, the polymerisation of both δ-VL and ε-CL was shown to proceed in a controlled manner even at high monomer conversions, affording polymers with low dispersities and high end-group fidelity, as evidenced by 1H NMR spectroscopy and matrix assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-ToF MS) (Fig. S5–S8, ESI†). These findings indicated that the polymerisation utilising TMG involved a general-base, alcohol activation mechanism in a similar manner to that postulated for DBU.34
:TU (5/5) was utilised in comparison to TMG alone, achieving a monomer conversion of 93% in 23 h ([δ-VL]0 = 2 M, [δ-VL]0/[BnOH]0 = 30). Nevertheless, for ε-CL, this system required 15 days to promote a monomer conversion of 96% ([ε-CL]0 = 2 M, [ε-CL]0/[BnOH]0 = 25). Finally, the polymerisation of both δ-VL and ε-CL was shown to proceed in a controlled manner even at high monomer conversions, affording polymers with low dispersities and high end-group fidelity, as evidenced by 1H NMR spectroscopy and matrix assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-ToF MS) (Fig. S5–S8, ESI†). These findings indicated that the polymerisation utilising TMG involved a general-base, alcohol activation mechanism in a similar manner to that postulated for DBU.34
| Entry | Monomer | Catalysta (loading (mol%)) | [M]0/[I]0b | Time (h) | Conversionb (%) | M n (kg mol−1) | Đ M |
|---|---|---|---|---|---|---|---|
| a Loading relative to monomer. b Determined by 1H NMR spectroscopy. c Obtained from SEC analysis in CHCl3, calibrated against polystyrene standards. d After 15 minutes under 320–400 nm UV irradiation. e After 3 hours under 320–400 nm UV irradiation. | |||||||
| 1 | L-LA | TMG (1) | 50 | 2.5 | 93 | 10.6 | 1.05 |
| 2 | δ-VL | TMG/TU (5/5) | 30 | 23 | 93 | 3.7 | 1.10 |
| 3 | ε-CL | TMG (5/5) | 25 | 360 | 96 | 4.7 | 1.12 |
| 4 | L-LA | NPPOC-TMG (1)d | 50 | 3 | 90 | 9.9 | 1.05 |
| 5 | δ-VL | NPPOC-TMG/TU (5/5)e | 20 | 76 | 34 | — | — |
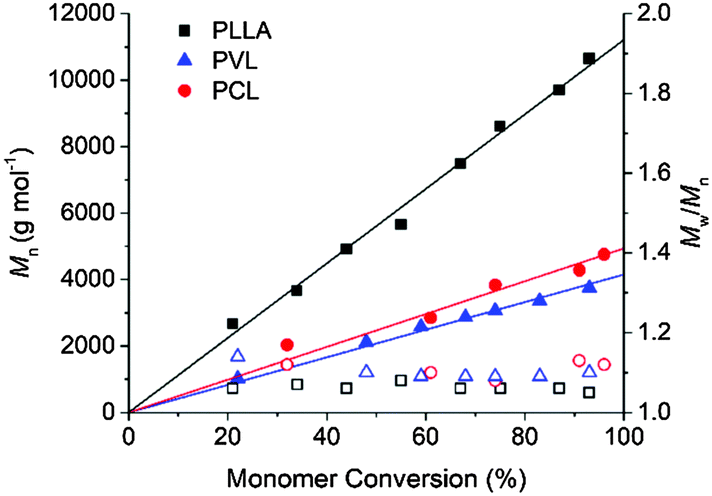 | ||
Fig. 1 Number-average molecular weight (Mn, (filled symbols)) and dispersity (ĐM = Mw/Mn; empty symbols) against monomer conversion for the ROP of L-LA (■,□; [L-LA]0/[BnOH]0/[TMG]0 = 50/1/0.5), δ-VL ( , , , ([δ-VL]0/[BnOH]0/[TMG]0/[TU]0 = 30/1/1.5/1.5)), and ε-CL ( , ([δ-VL]0/[BnOH]0/[TMG]0/[TU]0 = 30/1/1.5/1.5)), and ε-CL ( , , , [ε-CL]0/[BnOH]0/[TMG]0/[TU]0 = 25/1/1.25/1.25). , [ε-CL]0/[BnOH]0/[TMG]0/[TU]0 = 25/1/1.25/1.25). | ||
Following the demonstration that the superbase TMG provided good control over the ROP of cyclic (di)esters, we turned our attention to exploiting NPPOC as a photoprotecting group for TMG. The photocatalyst was prepared in a single step via the amidation of 2-(2-nitrophenyl)propyl chloroformate with TMG.39 The photolysis of NPPOC-TMG was then investigated under 320–400 nm UV irradiation, using concentrations similar to those applied for the polymerisation studies using free TMG. The release of TMG, which was monitored by 1H NMR spectroscopy, reached 80% in 45 min for a 20 mM solution of NPPOC-TMG in either CDCl3 or C6D6, whilst the photolysis of a 100 mM solution was considerably slower, providing the same conversion in 3 h. These results showed that the use of either CDCl3 or C6D6 had no effect on the photodegradation rate, which was very dependent on the concentration of the PBG (Fig. S9, ESI†). This can be attributed to undesirable effects, such as scattering and absorption that decrease the efficiency of the photochemical process when high concentrations of the chromophore are used.14,40
Initial investigation of the phototriggered ROP of L-LA comprised on using a polymeric solution of L-LA, benzyl alcohol and NPPOC-TMG in CDCl3 ([L-LA]0 = 2 M, [L-LA]0/[BnOH]0 = 50, NPPOC-TMG = 1 mol%) and maintaining it in the dark. Analysis by 1H NMR spectroscopy showed that no polymerisation occurred after 24 h in the absence of light, whereas, upon 15 min irradiation, the superbase was released, which triggered the ROP of L-LA that then proceeded to 90% monomer conversion after 3 h in the dark (Fig. S10, ESI†). Even though the polymerisation rate was lower compared to the one achieved using the guanidine alone, probably as a result of the incomplete release of the active catalyst into the solution, the resulting polymer showed a Mn of 9.9 kg mol−1 with a narrow dispersity (ĐM = 1.05), which was in good agreement with the polymers prepared with free TMG as catalyst. To further demonstrate that the reaction only occurred upon UV irradiation, four polymeric solutions were maintained in the dark for a determined number of days (1, 2, 5 and 9 days), after which they were individually irradiated with UV light for 15 minutes and quenched after 3 h. In all cases, the monomer conversion, and Mn and ĐM values were very similar, which confirmed that no polymerisation occurred even after 9 days in the dark (Fig. 2 and Table S1, ESI†). This finding indicated that UV light irradiation was essential to trigger the ROP of L-LA. Besides, no polymerisation took place after irradiation in the absence of NPPOC-TMG, which further demonstrated the requirement of both PBG and UV light to induce the polymerisation. The photoinduced ROP of L-LA displayed a linear increase of the ln([L-LA]0/[L-LA]) with the time, which indicated that no side reactions occurred by the presence of the nitrostyrene main byproduct formed during the photodeprotection of TMG and therefore supported that the mechanism for the polymerisation remained the same as the one using free TMG (Fig. S11, ESI†). Hence, the polymerisation was as well controlled as the ROP catalysed by free TMG, with a high end-group fidelity being evidenced by MALDI-ToF MS analysis (i.e. a single distribution attributed to sodium cationised PLLA initiated from benzyl alcohol was observed (Fig. 3)).
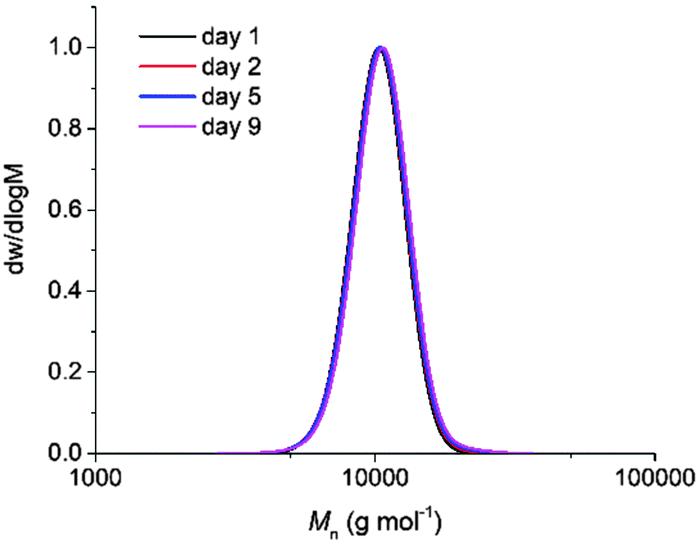 | ||
| Fig. 2 Size exclusion chromatograms of PLLA prepared by irradiating the polymeric solutions containing NPPOC-TMG that were initially kept in the dark over 1, 2, 5 and 9 days (CHCl3, RI). | ||
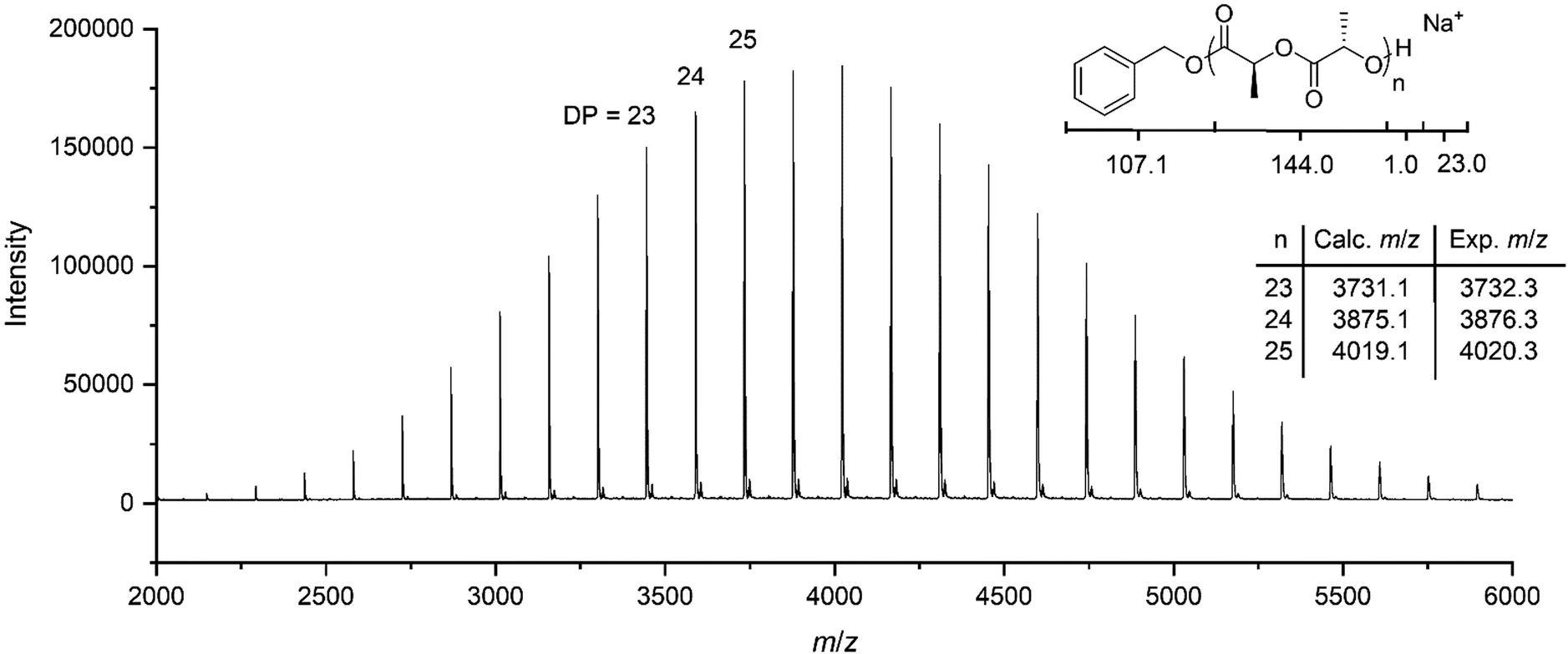 | ||
| Fig. 3 MALDI-ToF MS spectrum of PLLA. Reaction conditions: [L-LA]0/[BnOH]0/[NPPOC-TMG]0 = 25/1/0.25, 15 minutes under 320–400 nm irradiation. | ||
Finally, although the irradiation time necessary to release TMG was relatively high for a 100 mM solution of NPPOC-TMG (>3 h), the phototriggered ROP of δ-VL was also investigated in similar conditions to those described for L-LA. Accordingly, a solution of δ-VL, benzyl alcohol and NPPOC-TMG in C6D6 ([δ-VL]0 = 2 M, [δ-VL]0/[BnOH]0 = 20, NPPOC-TMG/TU = 5:5 mol%) was monitored by 1H NMR spectroscopy, which showed no polymerisation occurring after 24 h in the dark. The polymeric solution was then maintained under UV irradiation for 3 h, thus inducing the ROP of δ-VL although in a considerably lower rate than that observed with the free base (34% monomer conversion after 76 h). TU showed to be stable when exposed to UV light for 3 h, which indicated that the slower polymerisation was caused by the incomplete release of TMG.
In conclusion, we have demonstrated both the first application of the commercially-available TMG superbase for the ROP of L-LA and other cyclic ester monomers and the use of a photocaged analogue, NPPOC-TMG, to induce the ROP upon UV irradiation. The polymerisation is conducted with a high level of control over the molecular weight with narrow dispersities resulting in well defined polymers. The ability to generate polyesters by ROP on-demand after exposure to UV/visible light presents opportunities to expand the utility of spatiotemporal control by light-initiated polymerisation to this important class of renewably-source polymers for a diverse range of advanced applications.
P. K. K. acknowledges the Brazilian National Council for Scientific and Technological Development (CNPq) for the financial support (203127/2014-5). We also thank Corbion-Purac for the donation of lactide.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737 RSC.
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9 RSC.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef CAS PubMed.
- T. Fuoco and D. Pappalardo, Catalysts, 2017, 7, 64 CrossRef.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466 CrossRef CAS PubMed.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813 CrossRef CAS PubMed.
- F. Nederberg, E. F. Connor, M. Möller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712 CrossRef CAS PubMed.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409 CrossRef CAS.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687 RSC.
- S. Naumann and A. P. Dove, Polym. Chem., 2015, 6, 3185 RSC.
- S. Naumann and A. P. Dove, Polym. Int., 2016, 65, 16 CrossRef CAS.
- A. Nachtergael, O. Coulembier, P. Dubois, M. Helvenstein, P. Duez, B. Blankert and L. Mespouille, Biomacromolecules, 2015, 16, 507 CrossRef CAS PubMed.
- R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054 CrossRef CAS PubMed.
- R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982 RSC.
- D. Bratton, D. Yang, J. Dai and C. K. Ober, Polym. Adv. Technol., 2006, 17, 94 CrossRef CAS.
- D. Wöll, J. Smirnova, M. Galetskaya, T. Prykota, J. Bühler, K.-P. Stengele, W. Pfleiderer and U. E. Steiner, Chem. – Eur. J., 2008, 14, 6490 CrossRef PubMed.
- D. Wöll, S. Walbert, K.-P. Stengele, T. J. Albert, T. Richmond, J. Norton, M. Singer, R. D. Green, W. Pfleiderer and U. E. Steiner, Helv. Chim. Acta, 2004, 87, 28 CrossRef.
- S. Yamago and Y. Nakamura, Polymer, 2013, 54, 981 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167 CrossRef CAS PubMed.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73 CrossRef CAS.
- B. P. Harris, J. K. Kutty, E. W. Fritz, C. K. Webb, K. J. L. Burg and A. T. Metters, Langmuir, 2006, 22, 4467 CrossRef CAS PubMed.
- X. Sun, J. P. Gao and Z. Y. Wang, J. Am. Chem. Soc., 2008, 130, 8130 CrossRef CAS PubMed.
- K. Suyama and M. Shirai, Prog. Polym. Sci., 2009, 34, 194 CrossRef CAS.
- I. A. Barker and A. P. Dove, Chem. Commun., 2013, 49, 1205 RSC.
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367 CrossRef CAS PubMed.
- H. Yi, S. Maisonneuve and J. Xie, Org. Biomol. Chem., 2009, 7, 3847 CAS.
- F. Yang, B. Dong, K. Nie, H. Shi, Y. Wu, H. Wang and Z. Liu, ACS Comb. Sci., 2015, 17, 608 CrossRef CAS PubMed.
- N. F. König, S. Telitel, S. Poyer, L. Charles and J.-F. Lutz, Macromol. Rapid Commun., 2017, 38, 1700651 CrossRef PubMed.
- B. A. Chan, S. Xuan, M. Horton and D. Zhang, Macromolecules, 2016, 49, 2002 CrossRef CAS.
- L. Zhang, R. C. Pratt, F. Nederberg, H. W. Horn, J. E. Rice, R. M. Waymouth, C. G. Wade and J. L. Hedrick, Macromolecules, 2010, 43, 1660 CrossRef CAS.
- F. Eisenreich, P. Viehmann, F. Müller and S. Hecht, Macromolecules, 2015, 48, 8729 CrossRef CAS.
- M. Eckert-Maksić, Z. Glasovac, P. Trošelj, A. Kütt, T. Rodima, I. Koppel and I. A. Koppel, Eur. J. Org. Chem., 2008, 5176 CrossRef.
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 199 CrossRef CAS PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574 CrossRef CAS.
- L. Zhang, F. Nederberg, J. M. Messman, R. C. Pratt, J. L. Hedrick and C. G. Wade, J. Am. Chem. Soc., 2007, 129, 12610 CrossRef CAS PubMed.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Macromolecules, 2007, 40, 4154 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556 CrossRef CAS PubMed.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798 CrossRef CAS PubMed.
- W. Xi, H. Peng, A. Aguirre-Soto, C. J. Kloxin, J. W. Stansbury and C. N. Bowman, Macromolecules, 2014, 47, 6159 CrossRef CAS PubMed.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedure and additional kinetic and spectral data are included. See DOI: 10.1039/c8cc01913d |
| This journal is © The Royal Society of Chemistry 2018 |