
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porous dendritic copper: an electrocatalyst for highly selective CO2 reduction to formate in water/ionic liquid electrolyte†
Tran Ngoc
Huan
a,
Philippe
Simon
a,
Gwenaëlle
Rousse
e,
Isabelle
Génois
f,
Vincent
Artero
*bcd and
Marc
Fontecave
*abcd
aLaboratoire de Chimie des Processus Biologiques, CNRS UMR 8229, Collège de France, Université Pierre et Marie Curie, 11 Place Marcelin Berthelot, 75231 Paris Cedex 05, France. E-mail: mfontecave@cea.fr; Tel: +33 0144271372
bUniversité Grenoble Alpes, 38000 Grenoble, France. E-mail: vincent.artero@cea.fr
cLaboratory of Chemistry and Biology of Metals, CNRS UMR 5249, 17 rue des Martyrs, 38054 Grenoble, France
dCommissariat à l'énergie atomique et aux énergies alternatives (CEA), Fundamental Research Division, 38000 Grenoble, France
eLaboratoire Chimie du Solide et Energie, CNRS FRE 3677, Collège de France, Université Pierre et Marie Curie, 11 Place Marcelin Berthelot, 75231 Paris Cedex 05, France
fLaboratoire de Chimie de la Matière Condensée de Paris, Collège de France, 11 place Marcelin Berthelot, 75005 Paris, France
First published on 20th September 2016
Abstract
Copper is currently extensively studied because it provides promising electrodes for carbon dioxide electroreduction. The original combination, reported here, of a nanostructured porous dendritic Cu-based material, characterized by electron microcopy (SEM, TEM) and X-ray diffraction methods, and a water/ionic liquid mixture as the solvent, contributing to CO2 solubilization and activation, results in a remarkably efficient (large current densities at low overpotentials), stable and selective (large faradic yields) electrocatalytic system for the conversion of CO2 into formic acid, a product with a variety of uses. These results provide new directions for the further improvement of Cu electrodes.
Introduction
Although reduction of CO2 into energy-dense liquids or gaseous fuels is a fascinating strategy in the context of global warming and substitution of renewable energies for fossil fuels, its practical implementation is highly challenging due to the stability of the CO2 molecule. Furthermore, CO2 transformation can generate a great variety of compounds but these reactions also involve multiple electrons and protons. Another recurring and crucial issue is competition with proton reduction into dihydrogen. Thus the development of selective and efficient catalysts and the appropriate tuning of reaction conditions (temperature, pressure, solvent, etc.) are essential.1Regarding catalysts for CO2 reduction, current research mainly focuses on coordination complexes under homogeneous conditions or on solid-state, mostly metallic, electrodes.2–4 Following pioneering reports from Hori,5,6 various metals with different structures (nanoparticles, nanorods, dendrites, etc.) have been carefully revisited as electrocatalysts for CO2 conversion with special attention paid to electrodes based on abundant and cheap non-noble metals.7–10 Among them, Cu is of specific interest as it is low cost and has a high catalytic potential for transforming CO2 into various products including methane and hydrocarbons.9,11–21 However, these systems still have a major drawback of being relatively unselective, generating a complex mixture of products often including a large proportion of hydrogen.13 Selectivity is a key issue for technological applications as it facilitates product separation. There is therefore a need to develop more selective Cu-based catalysts. For example, while Cu catalyzes the electroreduction of CO2 to formate, faradic yields never exceed 40%.9,19,22
In the future conversion of CO2 to formic acid may indeed have a number of useful industrial applications.23,24 Traditional uses for formic acid have been in the leather tanning and animal feed markets. It can also be used to replace mineral acids. Furthermore it is a suitable H2 storage material as it can be easily and selectively decomposed into H2 and CO2 in the presence of a catalyst.24 At ambient temperature and pressure formic acid stores 580 times more H2 than the same volume of gas.23 Formic acid can also be converted into syngas.23 Finally formate salts are used as effective, non-corrosive and environmentally friendly anti-icing agents. Development of new and sustainable technologies that would decrease the cost of formic acid production might lead to an increased demand which is currently still low (500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 t yr−1).23
000 t yr−1).23
Recently, CO2 electroreduction in ionic liquids (ILs) has been a matter of interest.25–29 In 2011 Rosen et al. first demonstrated stable production of CO from CO2-saturated H2O/IL solvent with high faradic yield and a very low overpotential using a silver cathode.30 Following this observation several metal-based electrodes known to catalyze CO2 electroreduction have been reevaluated under comparable conditions, with the notable exceptions of gold and copper.31–33 ILs are redox-robust, generally possess a wide electrochemical window and are better than water and organic solvents for solubilizing CO2.34 Interestingly, the imidazolium cations of ILs can act as co-catalysts during CO2 reduction by stabilizing catalytic intermediates.35,36 This interaction lowers the activation barrier, therefore significantly reducing the overpotential requirement for the reduction of CO2.
Herein, we report that a porous and dendritic Cu-based material,37,38 with high specific surface area, displays high selectivity for formic acid production when assessed in an ionic liquid electrolyte, 1-ethyl-3-methylimidazolium tetrafluoroborate, [EMIM](BF4), in the presence of water. Such an activity is sustained for hours at high current densities at a moderate operational potential without deactivation of the electrode. A comparable material has been investigated for electroreduction of CO2 in aqueous conditions but gave low faradic yields (<40%) of CO2 reduction products, with H2 being the major product.11,19 Given the general versatility of copper electrodes for producing a variety of CO2 reduction products, our study highlights the importance of the media to orient the reaction. Importantly ionic liquids specifically favor CO2 reduction to formate over H2 evolution at copper electrodes, while more complex mixtures are obtained when using aqueous electrolytes.
Results and discussion
Electrodeposition of Cu from an acidic CuSO4 solution was carried out on a Cu plate electrode. Application of a large current (0.5 A cm−2) during a short period of time (20–120 s) resulted in the intense formation of hydrogen bubbles at the Cu plate electrode which contributed to Cu deposition in the form of a nanostructured foam, consisting of three-dimensional Cu porous dendrites, as shown by electron microscopy (see below).Electrochemical characterization of the electrode obtained after 80 s electrodeposition was carried out by cyclic voltammetry (CV) and controlled potential electrolysis (CPE), using a [EMIM](BF4)–water mixture (92/8 v/v) as the electrolyte. Fig. 1A compares the CVs obtained with a standard Cu plate and the modified electrode, either under N2 or at CO2 saturation. A catalytic wave was observed in both cases upon addition of CO2. However the modified electrode gave much larger current densities (values are normalized with respect to the geometry surface area, 1 cm2) at any potential. The catalytic wave, with an onset potential of −1.2 V vs. Fc+/Fc, displayed a peak at −1.5 V vs. Fc+/Fc followed by a second peak at −1.8 V vs. Fc+/Fc, likely reflecting two different CO2 reduction mechanisms whose identification requires further investigation. A comparable behavior has previously been reported for an indium disc electrode.33Fig. 1B shows the effect of the applied potential on the current density during CPE using the same modified electrode. Increased current densities were observed as a more negative potential was applied. Product analysis showed formation of formate, CO and H2 with excellent overall faradic yields (85–95%) (Fig. 2). The best selectivity for the formation of CO2 reduction products was achieved with an applied potential of −1.55 V vs. Fc+/Fc with a total faradic yield of almost 90% (formate: 83%; CO: 6%). It was confirmed that this formate and CO originated from CO2 by 13C-NMR and mass spectrometry analysis of the CPE experiment reaction mixture using 13CO2 as the substrate (Fig. S1†). The same experiment has been carried out using a Cu plate electrode. In this case, we could not achieve more than 45% formate (3% CO) formation, however this experiment was conducted at a much more negative potential (−1.8 V vs. Fc+/Fc) and with a much lower current density (1.8 mA cm−2). The major product was H2.
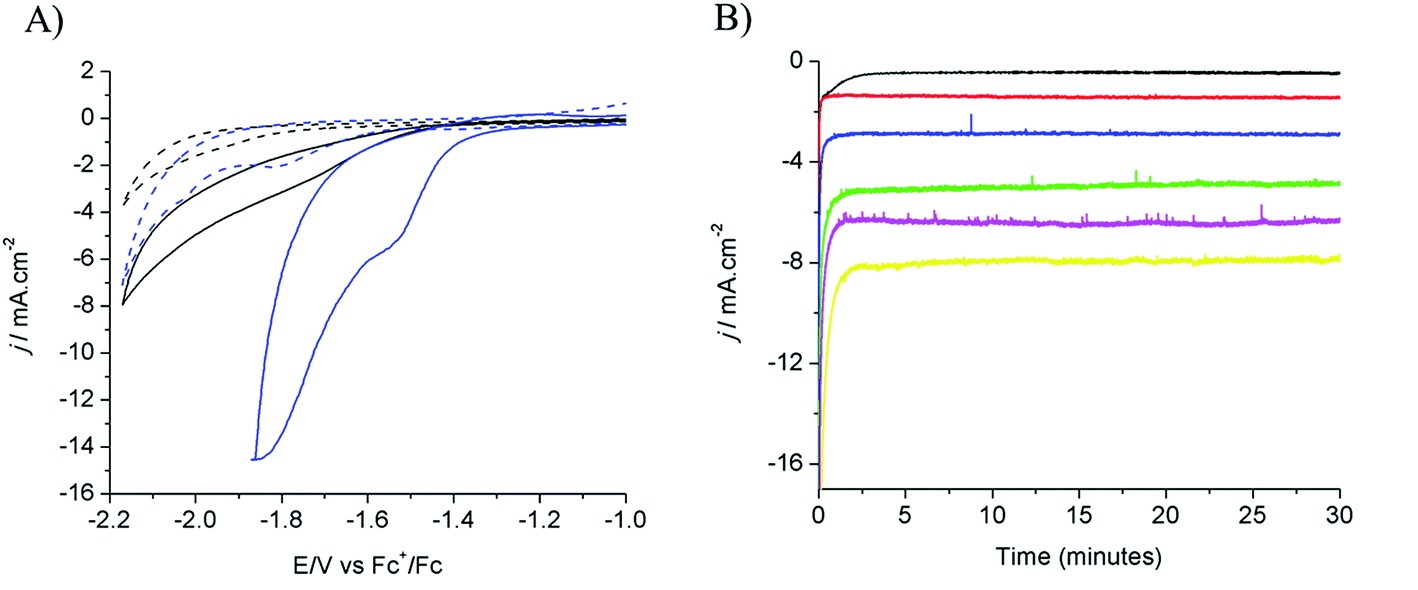 | ||
| Fig. 1 (A) Cyclic voltammograms recorded on a modified Cu electrode (80 s electrodeposition) (blue) and Cu plate electrode (black) in [EMIM](BF4)/H2O (92/8 v/v) after N2 purging (dashed lines) and CO2 purging (solid lines). (B) Current densities during electrolysis in CO2-saturated [EMIM](BF4)/H2O (92/8 v/v) at different potentials: −1.25 V (black), −1.35 V (red), −1.45 V (blue), −1.55 V (green), −1.65 V (magenta), and −1.75 V (yellow) vs. Fc+/Fc. | ||
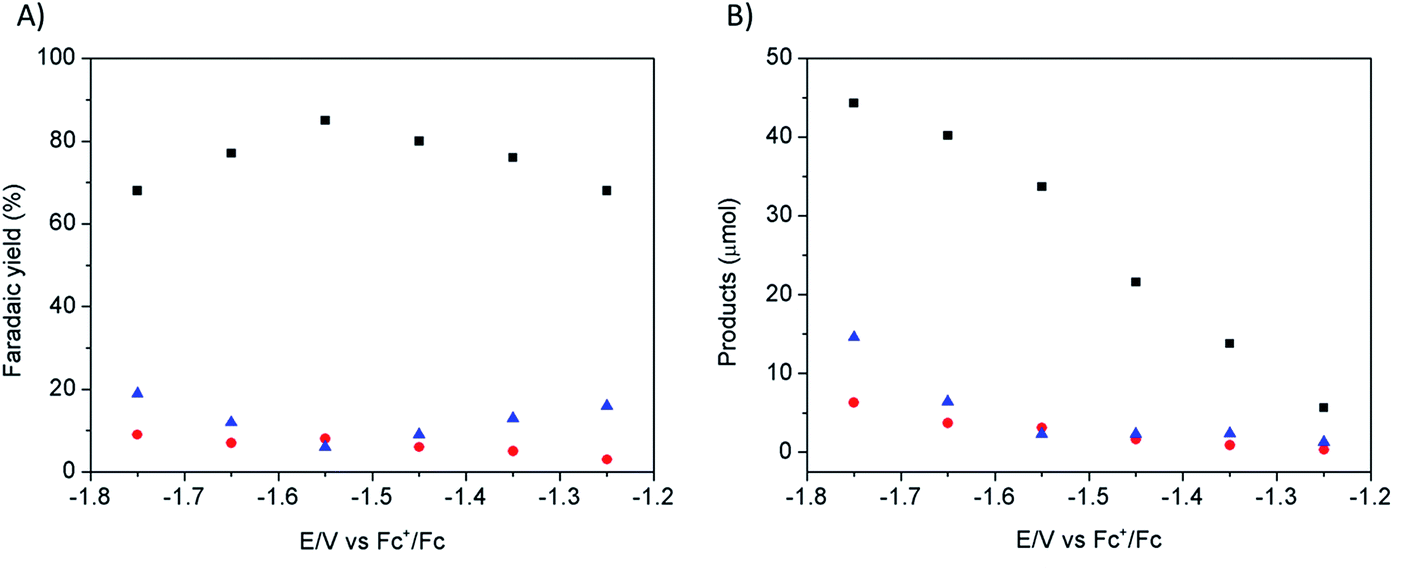 | ||
| Fig. 2 CO2 reduction products recorded on a modified Cu electrode (80 s electrodeposition) as a function of applied potential: faradic yields (A) and absolute amounts (B) of formate (black squares), CO (red circles) and H2 (blue triangles) during 30 minutes electrolysis in CO2-saturated [EMIM](BF4)/H2O (92/8 v/v). | ||
The effect of the electrodeposition time on the activity of the modified electrode was studied by CPE at −1.55 V vs. Fc+/Fc at CO2 saturation in [EMIM](BF4)/H2O (92/8 v/v). As shown in Fig. S2,† increased activity was obtained upon increasing the electrodeposition time (current intensity values of 6.5 mA cm−2 for 80 s electrodeposition as compared to 4 mA cm−2 for 40 s electrodeposition). For both 40 s and 80 s electrodeposited samples, faradic yields were above 90% with formic acid (83%) as the major product and only small amounts of CO and H2 (see Table S1†). A longer deposition time (120 s) had only a minor effect on the current density but resulted in a slightly larger production of H2 (Table S1†). As a control experiment, electrolysis under N2 saturation gave a current density of only 0.5 mA cm−2 (Fig. S2†).
The effect of water concentration was studied by CV and CPE in [EMIM](BF4)/H2O (85/15 v/v) (Fig. S3†). Under these conditions, much higher current densities correlated with a larger production of H2, with formate accounting for only 50% of the products (Table S2†).
Finally the modified electrode proved to be stable during long-term electrolysis as shown from an 8 hour experiment (current density: 6.5 mA cm−2. Faradic yields for formate: 87%; CO: 5%; H2: 7%) (Fig. S4†).
Further evidence for the 80 s electrodeposition providing an optimized electrode is presented in Fig. S5 and S6.† This evidence shows that the Cu reduction peak observed at −0.05 V vs. Ag/AgCl increases as a function of the electrodeposition time but levels off almost after 80 s (Fig. S5A†). The amount of surface-active copper, calculated from Fig. S5A,† is at least 7 times larger than that of a standard Cu plate electrode (Fig. S5B†). In addition the Randles–Sevcik equation (ESI†), when applied to the reduction of 5 mM K3[Fe(CN)]6 (Fig. S6†), was used to estimate the electrochemical diffusion surface area of both the Cu plate and the modified Cu electrode (80 s electrodeposition), giving Adiff = 2.04 cm2 and 19.8 cm2, respectively.
Fig. 3 shows the linear sweep voltammograms (LSVs) of the modified Cu electrode (80 s electrodeposition) at a scan rate of 1.0 mV s−1 in CO2- and N2-saturated [EMIM](BF4)/H2O (92/8 v/v) solutions. As shown above, the current measured under these conditions mostly corresponds to the reduction of CO2 into formic acid. The corresponding Tafel plot is shown in the inset of Fig. 3. The onset potential for this reaction is −1.2 V vs. Fc+/Fc, in good agreement with the CV measurements shown in Fig. 1A. The Tafel data are linear in the range of −1.2/−1.5 V vs. Fc+/Fc, with a slope of 130 mV per decade, a value consistent with a mechanism in which the first electron transfer to adsorbed CO2 is the rate determining step.12 At a more negative potential, the slope significantly increases to 300–400 mV per decade. By analogy with oxygen reduction reaction studies at porous electrode materials, this is indicative of current limitation by mass transport phenomena including both CO2 diffusion and proton migration within the porous material dipped in the electrolyte.39 Under such conditions, current densities already exceed 10 mA cm−2.
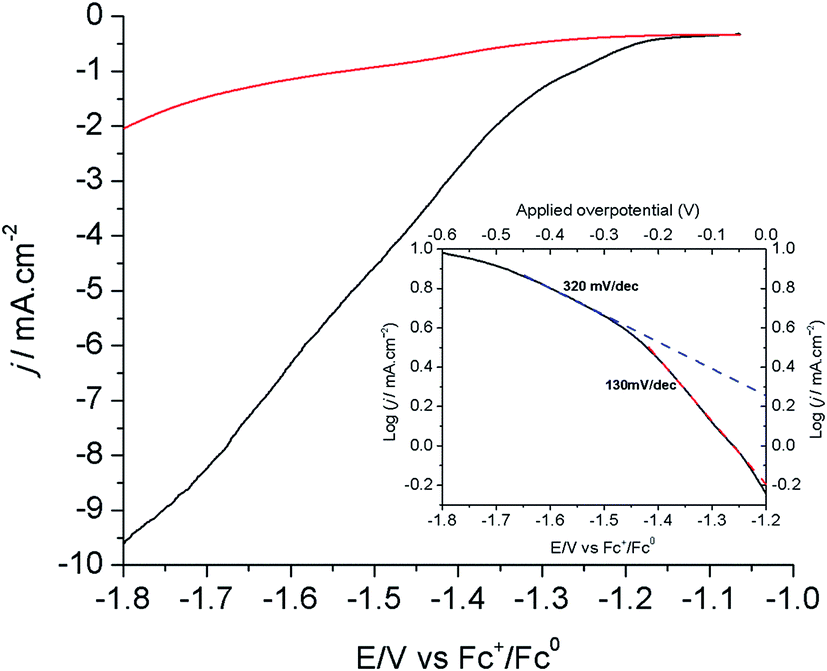 | ||
| Fig. 3 LSVs of the modified Cu electrode (80 s electrodeposition) in N2- (red) and CO2-saturated (black) [EMIM](BF4)/H2O (92/8 v/v). Inset: Tafel plot for the electrode in CO2-saturated [EMIM](BF4)/H2O (92/8 v/v). | ||
The evaluation of overpotential values is not trivial here as the equilibrium potential of the CO2/HCOOH couple in [EMIM](BF4)/H2O is unknown. However, we determined by LSV the onset potential for electroreduction of CO2 in CH3CN/H2O (92/8 v/v) + 0.1 M n-Bu4BF4 at −1.4 V vs. Fc+/Fc (Fig. S7†). The equilibrium potential of the CO2/HCOOH couple in CH3CN is −1.32 V vs. Fc+/Fc when considering hydrated CO2 as the strongest acid in solution (ESI†). Under these conditions, the modified Cu electrode shows a remarkably low overpotential requirement of 80 mV. In addition, shifting from the organic solvent to the IL/water solvent resulted in an ∼200 mV decreased onset potential. Similarly, IL-promoted decrease of the onset potentials has been observed at Ag-based electrodes and have been related to the interaction of the IL with CO2 and/or with the surface during the catalytic transformation resulting in decreased activation barriers.26,30,40 As the onset for catalysis is observed at −1.2 V vs. Fc+/Fc in [EMIM](BF4)/H2O (92/8 v/v), we conclude that the equilibrium potential in this solvent is more positive than in CH3CN. This may result from various factors including a stronger solvation energy of formic acid and a lowered pKa value of hydrated CO2 acting as the proton source.
In order to get some insights into the structure of the modified active electrodes, electron microscopy and X-ray diffraction (XRD) analysis were carried out. Scanning electron microscopy (SEM) images of the modified electrodes were obtained after different deposition times (20 s, 40 s, 60 s, 80 s, 120 s, and 160 s) at a constant applied current of 0.5 A cm−2 (Fig. 4A–C and S8†). They revealed a porous structure with pores having a diameter of 30–40 µm and the presence of a network of dendrites forming the walls within the Cu foam. Fig. S8† shows that the porosity and the nanostructure of the Cu electrodes are dependent on the deposition time, with an optimized porosity achieved after an 80 s or 120 s electrodeposition time, in agreement with the observed effect of deposition time on catalytic activity.
 | ||
| Fig. 4 (A–C) SEM images of the modified Cu electrode (80 s electrodeposition) at different magnifications; (D) TEM image of a Cu dendrite; (E) atomic resolution image taken from the area indicated (red circle) in (D); selected area electron diffraction (SAED) pattern of a Cu nanodendrite (F). | ||
A transmission electron microscopic (TEM) image of the Cu dendrites is shown in Fig. 4D, and a high resolution image allows clear observation of the Cu atomic structure (Fig. 4E). A selected area electron diffraction (SAED) image of these dendrites is shown in Fig. 4F, in which a circular diffraction pattern indicated a highly crystalline structure.
Finally, Fig. 5 shows the powder X-ray diffraction patterns obtained for the Cu porous material which indicated a major contribution from Cu0. The spectrum also displays a small peak corresponding to Cu2O and accounting for around 10 (±2)% of the mass. After 1 h CO2 electroreduction, XRD analysis confirmed that the electrode material has kept its crystalline state, while the intensities of the peaks corresponding to Cu2O significantly decreased (5 ± 2%) in agreement with reduction of CuI to Cu0 during the reaction.9 As shown from the SEM images taken after long-term CPE, the Cu material still displayed a porous and dendritic structure (Fig. S9†). Furthermore the amount of surface active copper after catalysis (0.74 µmol cm−2) is similar to that of the same electrode before catalysis.
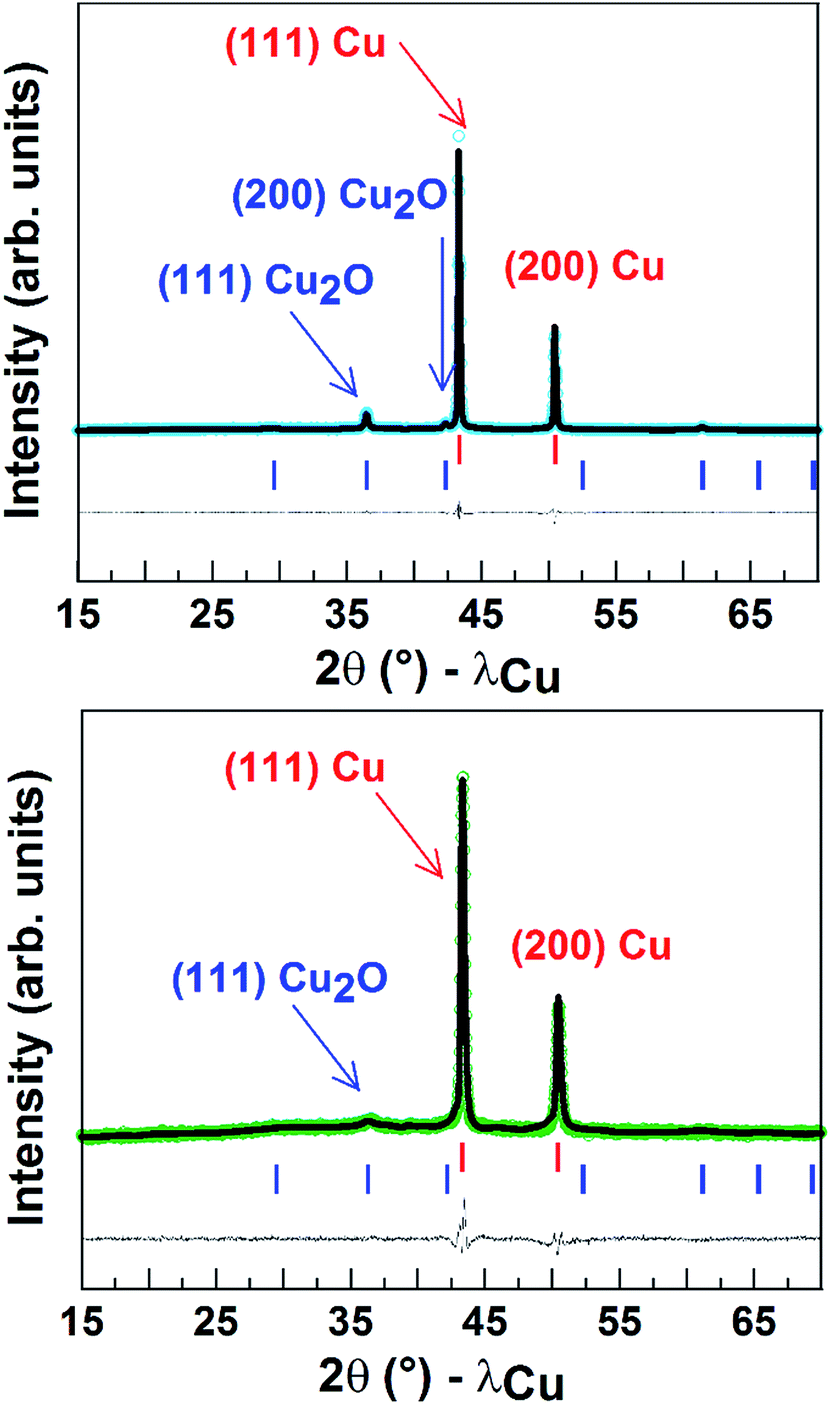 | ||
| Fig. 5 Rietveld refinement of X-ray patterns of a powder of the Cu porous material (80 s electrodeposition) before (top) and after (below) CO2 electroreduction. The observed (open circles), calculated (black line) and difference (blue line on top and green line on bottom) patterns are shown. The red and blue vertical tick bars represent the Bragg positions for Cu and Cu2O, respectively. | ||
Conclusion
We here show that a low cost Cu porous dendritic material, as shown by SEM, TEM and XRD, displays remarkable electrocatalytic activity for the reduction of CO2. It is a unique electrocatalyst as it is the first copper-based material for the catalytic conversion of CO2 into formic acid in a H2O/IL solvent with such a high faradic yield. The selectivity for formate production is the highest reported so far at low overpotentials for a Cu-based electrode. As a representative example, Cu nanoparticles, developed by Kanan et al., afforded only 33% formic acid under electrolysis at an overpotential of 250 mV with a current density of 5 mA cm−2 in aqueous KHCO3 solution. Formic acid can be obtained with a few other metallic electrodes in IL/water media. For example In and Sn electrodes are classically known as selective catalysts for formate production in aqueous media and retain their high selectivity (90%) in ionic liquid/water electrolytes, but with low current densities (<2 mA cm−2) and only at very negative potentials (−1.95 V vs. Ag/AgCl, 3 M NaCl).33 Furthermore at lower potentials the faradic yield drastically decreased as a result of H2 formation becoming predominant. During the course of the revision of this article a selective reduction of CO2 to formate (faradic yield: 90%) using Pb and Sn electrodes was reported.41A very high current density is reported, but only at extremely negative potentials, at least 300 mV more negative than those operating in the system we report here, and only using very complex solvent mixtures including ILs, water and an organic solvent. Recently a high faradic yield for formic acid, at a low overpotential was obtained using a superbase ionic liquid, however with lower current densities and using an electrode based on silver.42The increased specific active surface of our Cu electrode provided by the dendritic porous network combined with high CO2 solubility in ILs likely explains the high current densities (>10 mA cm−2) observed during electrolysis. The large selectivity for formic acid is more intriguing. Indeed, Cu electrodes are well known to produce hydrocarbons but at very negative potentials and to produce hydrogen at more positive potentials in aqueous solutions.5,6 Indeed, the ability of Cu to reduce protons has recently been supported by theoretical and experimental studies.43,44 It is tempting to suggest that the IL specifically contributes to activate CO2 enough so as to reorient the reactivity of the activated H atoms towards CO2 to produce formate. The low production of H2 and CO at low overpotentials make the system reported here unique and these characteristics are likely to be explained by the combination of the nanodendritic porous structure of the catalytic material and the use of the ionic liquid/water mixed electrolyte, illustrating the importance of the reaction medium. As a matter of fact, during the preparation of this article, a similar porous nanodendritic Cu material was reported and investigated under different solvent and electrolytic conditions. It was shown to produce a more complex mixture of products, with much larger amounts of H2 and CO and less formate, but, more interestingly, with significant amounts of ethane and ethylene (no methane) with efficiency for C2 products reaching 55%.11 This porous dendritic Cu material thus seems to show great potential as a platform for the development of even more efficient and selective Cu-based systems, as selectivity is a major issue with respect to technological applications.
Acknowledgements
We acknowledge support from the French National Research Agency (ANR, Carbiored ANR-12-BS07-0024-03; Labex program ARCANE, ANR-11-LABX-0003-01 and DYNAMO, ANR-11-LABX-0011) and from Fondation de l'Orangerie for individual Philanthropy and its donors.References
- J. L. Qiao, Y. Y. Liu, F. Hong and J. J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- C. Costentin, M. Robert and J. M. Saveant, Acc. Chem. Res., 2015, 48, 2996–3006 CrossRef CAS PubMed.
- H. R. Jhong, S. C. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- C. D. Windle and E. Reisner, Chimia, 2015, 69, 435–441 CrossRef CAS PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- L. Chen, S. X. Guo, F. Li, C. Bentley, M. Horne, A. M. Bond and J. Zhang, ChemSusChem, 2016, 9, 1271–1278 CrossRef CAS PubMed.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and F. Jiao, ACS Catal., 2015, 5, 4586–4591 CrossRef CAS.
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 CAS.
- S. Lee, D. Kim and J. Lee, Angew. Chem., Int. Ed., 2015, 54, 14701–14705 CrossRef CAS PubMed.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. R. Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS PubMed.
- S. Sen, D. Liu and G. T. R. Palmore, ACS Catal., 2014, 4, 3091–3095 CrossRef CAS.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Norskov and I. Chorkendorff, Phys. Chem. Chem. Phys., 2012, 14, 76–81 RSC.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- A. S. Agarwal, Y. M. Zhai, D. Hill and N. Sridhar, ChemSusChem, 2011, 4, 1301–1310 CrossRef CAS PubMed.
- P. Sponholz, D. Mellmann, H. Junge and M. Beller, ChemSusChem, 2013, 6, 1172–1176 CrossRef CAS PubMed.
- Y. Matsubara, D. C. Grills and Y. Kuwahara, ACS Catal., 2015, 5, 6440–6452 CrossRef CAS.
- J. Medina-Ramos, R. C. Pupillo, T. P. Keane, J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2015, 137, 5021–5027 CrossRef CAS PubMed.
- Y. Oh and X. L. Hu, Chem. Commun., 2015, 51, 13698–13701 RSC.
- L. Y. Sun, G. K. Ramesha, P. V. Kamat and J. F. Brennecke, Langmuir, 2014, 30, 6302–6308 CrossRef CAS PubMed.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55, 6771–6775 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Kral, J. Abiade and A. Salehi-Khojin, Nat. Commun., 2014, 5, 4470 CAS.
- B. C. M. Martindale and R. G. Compton, Chem. Commun., 2012, 48, 6487–6489 RSC.
- J. D. Watkins and A. B. Bocarsly, ChemSusChem, 2014, 7, 284–290 CrossRef CAS PubMed.
- C. Cadena, J. L. Anthony, J. K. Shah, T. I. Morrow, J. F. Brennecke and E. J. Maginn, J. Am. Chem. Soc., 2004, 126, 5300–5308 CrossRef CAS PubMed.
- D. C. Grills, Y. Matsubara, Y. Kuwahara, S. R. Golisz, D. A. Kurtz and B. A. Mello, J. Phys. Chem. Lett., 2014, 5, 2033–2038 CrossRef CAS PubMed.
- Y. Oh and X. L. Hu, Chem. Soc. Rev., 2013, 42, 2253–2261 RSC.
- T. N. Huan, T. Ganesh, K. S. Kim, S. Kim, S. H. Han and H. Chung, Biosens. Bioelectron., 2011, 27, 183–186 CrossRef CAS PubMed.
- H. C. Shin and M. L. Liu, Chem. Mater., 2004, 16, 5460–5464 CrossRef CAS.
- F. Jaouen, G. Lindbergh and G. Sundholm, J. Electrochem. Soc., 2002, 149, A437–A447 CrossRef CAS.
- J. Medina-Ramos, J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2014, 136, 8361–8367 CrossRef CAS PubMed.
- Q. Zhu, J. Ma, X. Kang, X. Sun, H. Liu, J. Hu, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2016, 55, 9012–9016 CrossRef CAS PubMed.
- N. Hollingsworth, S. F. R. Taylor, M. T. Galante, J. Jacquemin, C. Longo, K. B. Holt, N. H. de Leeuw and C. Hardacre, Angew. Chem., Int. Ed., 2015, 54, 14164–14168 CrossRef CAS PubMed.
- X. Liu, S. S. Cui, Z. J. Sun and P. W. Du, Chem. Commun., 2015, 51, 12954–12957 RSC.
- J. S. Yoo, R. Christensen, T. Vegge, J. K. Norskov and F. Studt, ChemSusChem, 2016, 9, 358–363 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc03194c |
| This journal is © The Royal Society of Chemistry 2017 |