
Chemistry and engineering of cyclodextrins for molecular imaging
Wing-Fu
Lai
 *ab,
Andrey L.
Rogach
c and
Wing-Tak
Wong
*b
*ab,
Andrey L.
Rogach
c and
Wing-Tak
Wong
*b
aSchool of Pharmaceutical Sciences, Health Science Centre, Shenzhen University, Shenzhen, China. E-mail: rori0610@graduate.hku.hk
bDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hong Kong. E-mail: w.t.wong@polyu.edu.hk
cDepartment of Materials Science and Engineering, and Centre for Functional Photonics (CFP), City University of Hong Kong, Hong Kong
First published on 20th September 2017
Abstract
Cyclodextrins (CDs) are naturally occurring cyclic oligosaccharides bearing a basket-shaped topology with an “inner–outer” amphiphilic character. The abundance of hydroxyl groups enables CDs to be functionalized with multiple targeting ligands and imaging elements. The imaging time, and the payload of different imaging elements, can be tuned by taking advantage of the commercial availability of CDs with different sizes of the cavity. This review aims to offer an outlook of the chemistry and engineering of CDs for the development of molecular probes. Complexation thermodynamics of CDs, and the corresponding implications for probe design, are also presented with examples demonstrating the structural and physiochemical roles played by CDs in the full ambit of molecular imaging. We hope that this review not only offers a synopsis of the current development of CD-based molecular probes, but can also facilitate translation of the incremental advancements from the laboratory to real biomedical applications by illuminating opportunities and challenges for future research.
 Wing-Fu Lai | Wing-Fu Lai received his MSc degree in Materials Engineering and Nanotechnology from City University of Hong Kong, and earned his PhD in Chemistry from the University of Hong Kong. He joined the faculty of School of Pharmaceutical Sciences at Shenzhen University in 2016, and is also holding an adjunct assistant professorship in the Department of Applied Biology and Chemical Technology at the Hong Kong Polytechnic University. His research interests cover the design of molecular probes for bioimaging, synthesis of polymeric materials for gene delivery and controlled drug release, and the development of nanoparticulate systems for theranostic applications. |
 Andrey L. Rogach | Andrey L. Rogach is a Chair Professor of Photonics Materials and the founding director of the Centre for Functional Photonics at City University of Hong Kong. He received his PhD in Physical Chemistry (1995) from the Belarusian State University in Minsk, and worked as a staff scientist at the University of Hamburg, Germany, from 1995 to 2002. During 2002–2009, he held a tenured position of lead staff scientist at the Department of Physics of the Ludwig-Maximilians-University in Munich, Germany, where he completed his habilitation in Experimental Physics. His research focuses on light-emitting materials, in particular for bioimaging application. |
 Wing-Tak Wong | Wing-Tak Wong, PhD, ScD, is a Chair Professor of Chemical Technology and Dean of the Faculty of Applied Science and Textiles at the Hong Kong Polytechnic University. His research interests include lanthanide chemistry, nanomaterials and bio-imaging. He has published more than 480 research papers and contributed two book chapters in Comprehensive Organometallic Chemistry III (Elsevier 2006), and a book chapter in Rare Earth Coordination Chemistry (Wiley 2010). He is also a co-editor of the book The Chemistry of Molecular Imaging (Wiley 2015). |
1. Introduction
Compared to conventional diagnostic imaging which is intended to visualize late consequences of disease processes, molecular imaging interrogates molecular events that mediate pathological alterations at the cellular and tissue levels.1 Clinically, it facilitates early disease detection,2 guides therapeutic decisions for personalized medicine,3 and can noninvasively monitor the pharmacokinetic and pharmacodynamic profiles of a candidate drug in a living body.4–6 To make molecular imaging executable, proper probe design is a prerequisite. On the whole, a molecular probe is comprised of two components. The first one is an imaging element. Signals emitted from this element can be magnetic, echogenic, radioactive, or luminescent in nature. The second component is a targeting moiety, which enables the probe to target molecular biomarkers in relation to disease processes, making the probe different from the conventional nonspecific one. An ideal probe for molecular imaging is the one which shows high target specificity, high in vivo stability, high selectivity for the molecular event, high biocompatibility, and low toxicity. To meet these criteria during probe development, cyclodextrins (CDs) have been playing a critical role (Fig. 1).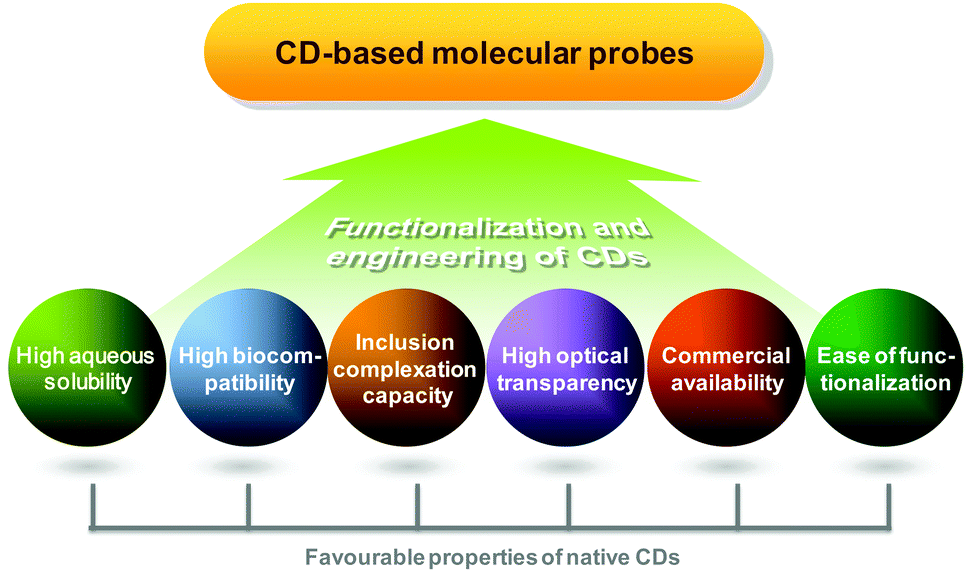 | ||
| Fig. 1 Important properties of CDs for probe design and development. | ||
CDs are naturally occurring cyclic oligosaccharides. They possess a basket-shaped topology with an “inner–outer” amphiphilic character.7 The application potential of CDs has been proven in diverse areas spanning drug delivery,8–15 nucleic acid transfer,7,16,17 chiral separation of basic drugs,18 and solubilisation of lipophilic drugs.19 Since the turn of the last century, the emerging role of CDs in probe development has begun to take centre stage in molecular imaging.20–22 In practice, CDs can be functionalized with multiple ligands to enhance the imaging specificity by enabling target-specific cellular internalization. The abundance of hydroxyl groups also allows CD molecules to be functionalized with different imaging elements for multimodal imaging. CDs with different sizes of the cavity are commercially available. This makes the tuning of the excretion behaviour and imaging time, as well as the optimization of the payload of imaging elements, possible.
Notwithstanding the application potential of CDs in molecular imaging, most of the reviews in the literature have only focused on the structural chemistry and pharmaceutical use of these oligosaccharides. Reviews on CD-based molecular imaging are lacking, leaving a strong demand for an article filling this gap. This review presents a critical view on the key chemical features of CDs for the design and development of molecular probes, and summarizes the requirements and engineering strategies adopted in this nascent field of research. The objective of this review is to position the latest advances in the field as the kernel of an ongoing collection of contributions giving structure to the chemistry and engineering of CDs for molecular imaging. It is also hoped that with the synopsis of the state of the art of CD-based molecular probes, insights into challenges and opportunities could be illuminated for translating the accumulated advancements from the laboratory to future biomedical applications.
2. Structural features of CDs for molecular imaging
CDs are cyclic (α-1,4)-linked oligosaccharides of α-D-glucopyranose.23 As early as the 1890's, Villiers isolated a crystalline substance, namely “cellulosine”, from a bacterial digest of starch. That substance, which was found to be resistant to acid hydrolysis but to have negligible reducing properties,24 was later known as “cyclodextrin”. This is regarded as the earliest reference to CDs. Somewhat later, the ring structures of CDs were resolved by Freudenberg and colleagues,25,26 with the physicochemical properties of these oligosaccharides (including solubility, cavity size, chemical structures, and reactivity) being documented by Cramer in his book “Einschlussverbindungen”.27 CDs can exist in α, β and γ forms. They are comprised of six, seven and eight α-D-glucopyranose units, respectively (Fig. 2). CDs have abundant hydroxyl groups, and are generally water-soluble. Owing to their comparatively high crystal energy,28 CD molecules in the crystal state are usually strongly bound. Compared to their linear dextrin counterparts, CDs have lower aqueous solubility. Moreover, β-CD is less capable of forming hydrogen bonds with surrounding water molecules, due to the formation of internal hydrogen bonds among secondary hydroxyl groups.28 This partly explains its lower aqueous solubility than α- and γ-CDs, whose aqueous solubility is 7- and 14-fold as much as that achieved by β-CD, respectively.29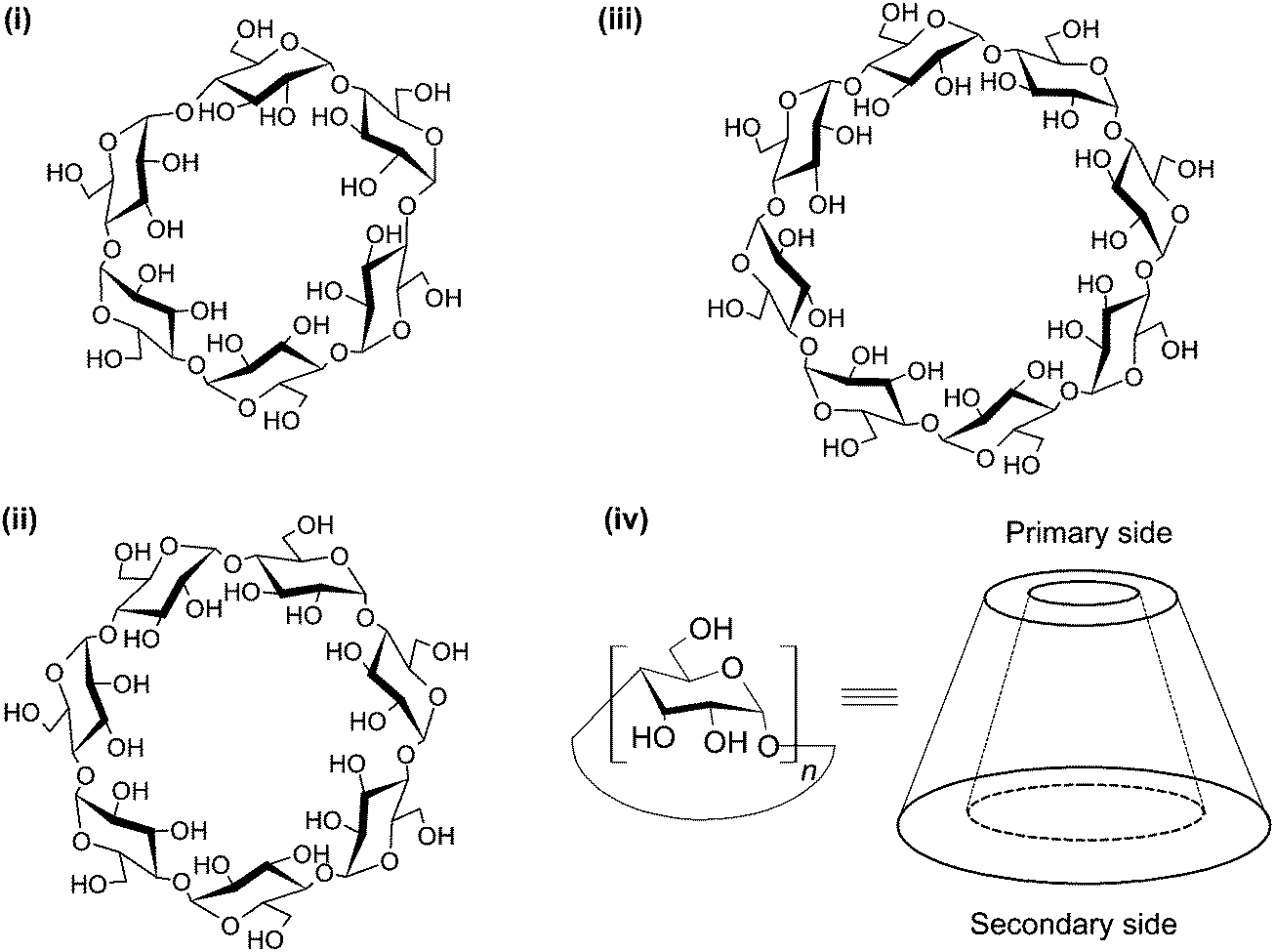 | ||
| Fig. 2 The structures of (i) α-, (ii) β- and (iii) γ-CDs, and the torus-shaped morphology of the CD molecule. The morphology of the molecule is not drawn to scale. | ||
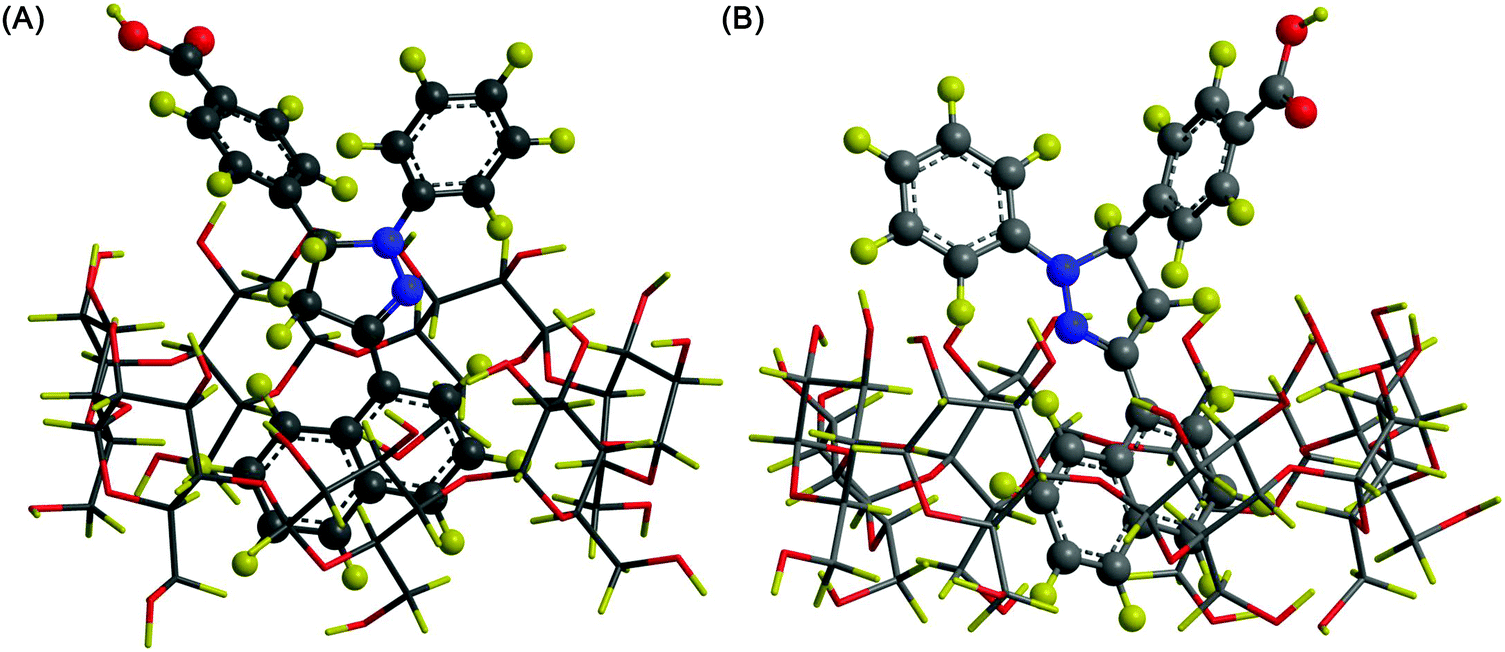
One distinctive feature of CD molecules is the torus-shaped structure, which possesses an apolar cavity interior and a hydrophilic cavity exterior.30,31 This structure creates a micro-environment for inclusion complexation with compounds such as 1,5-dihydroxyanthraquinone (1,5-DHAQ), which exists in a dianionic form (1,5-DHAQ−2H) after deprotonation of the first and second phenolic hydroxyl groups in an alkaline environment. Inclusion complexation with CDs has been shown to stabilize 1,5-DHAQ−2H for molecular imaging in response to ionic species, in particular Cu2+ and Cr2O72− ions.32 The stoichiometry ratio of the inclusion complex has been estimated, based on the Benesi–Hildebrand relation, to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.32,33 As suggested by Fourier transform infrared spectroscopy (FTIR) studies, although the three condensed benzene rings of 1,5-DHAQ−2H are, theoretically, too bulky for total penetration into the β-CD cavity, partial inclusion has occurred.32 This has been evidenced by nuclear magnetic resonance (NMR) analysis, in which an upfield shift of the chemical shift (Δδ) values of H3 and H5 protons of β-CD has been observed after the complexation process.32 The binding of ions with the 1,5-DHAQ−2H/β-CD complex has been modelled by molecular docking,32 and by geometry optimizations followed by density functional theory (DFT) calculations using the B3LYP function (Fig. 3).32 In addition to 1,5-DHAQ, there are other molecules (e.g., adamantane,34 pyrazoline derivatives,35 and 3,3′-dihydroxybenzidine36) that form inclusion complexes with CDs for molecular imaging purposes. Some of them will be discussed in detail in the subsequent sections.
:1.32,33 As suggested by Fourier transform infrared spectroscopy (FTIR) studies, although the three condensed benzene rings of 1,5-DHAQ−2H are, theoretically, too bulky for total penetration into the β-CD cavity, partial inclusion has occurred.32 This has been evidenced by nuclear magnetic resonance (NMR) analysis, in which an upfield shift of the chemical shift (Δδ) values of H3 and H5 protons of β-CD has been observed after the complexation process.32 The binding of ions with the 1,5-DHAQ−2H/β-CD complex has been modelled by molecular docking,32 and by geometry optimizations followed by density functional theory (DFT) calculations using the B3LYP function (Fig. 3).32 In addition to 1,5-DHAQ, there are other molecules (e.g., adamantane,34 pyrazoline derivatives,35 and 3,3′-dihydroxybenzidine36) that form inclusion complexes with CDs for molecular imaging purposes. Some of them will be discussed in detail in the subsequent sections.
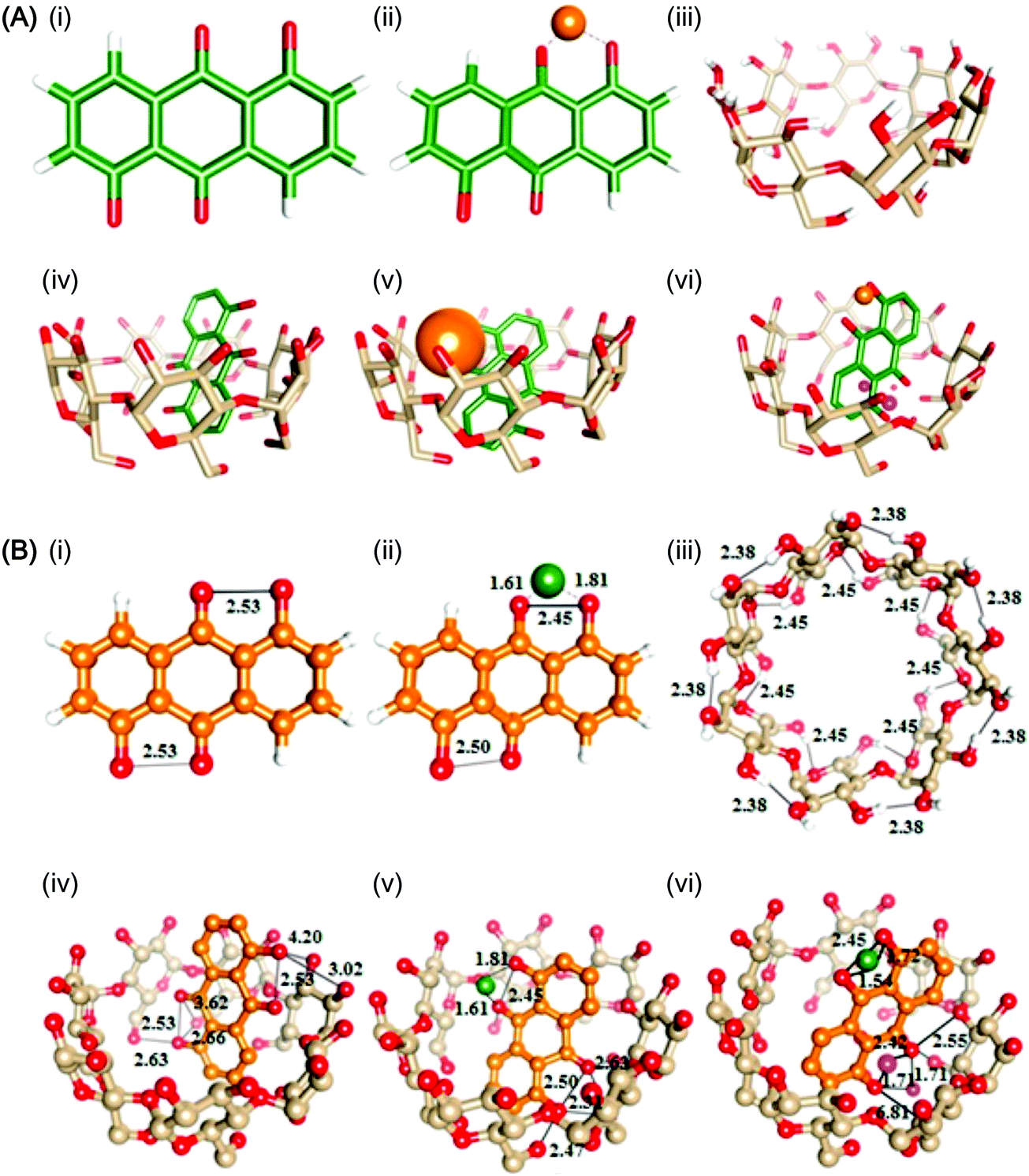 | ||
| Fig. 3 (A) Ball and stick representations of the structures of (i) 1,5-DHAQ−2H, (ii) 1,5-DHAQ−2H·Cu2+, (iii) β-CD, (iv) 1,5-DHAQ−2H/β-CD complex, (v) 1,5-DHAQ−2H/β-CD complex·Cu2+, and (vi) 1,5-DHAQ−2H/β-CD complex·Cu2+ + Cr2O72−. Cu2+ and Cr2O72− ions are represented by orange and magenta spheres, respectively. (B) The DFT optimized structures of (i) 1,5-DHAQ−2H, (ii) 1,5-DHAQ−2H·Cu2+, (iii) β-CD, (iv) 1,5-DHAQ−2H/β-CD complex, (v) 1,5-DHAQ−2H/β-CD complex·Cu2+, and (vi) 1,5-DHAQ−2H/β-CD complex·Cu2+ + Cr2O72−·Cu2+ and Cr2O72− ions are represented by green and magenta spheres, respectively. The distance is given in Å (reproduced from ref. 32 with permission from Elsevier). | ||
CDs are indeed only one of the many compounds investigated in supramolecular chemistry for host–guest interactions.37–42 Other widely studied macrocyclic compounds include cucurbiturils, pillararenes, crown-ether, and calixarenes.43–45 CDs, however, have distinctive features that make them attractive for applications in molecular imaging (Table 1).21 Firstly, while many other host molecules in their native forms display poor aqueous solubility and thus have limited potential for biomedical applications, native CDs are highly water soluble. This makes direct use of native CDs feasible. Secondly, CDs are optically transparent in the ultraviolet and visible regions of the spectrum, avoiding interference with light signals during imaging. Thirdly, in contrast to many other macrocyclic compounds whose attainment necessitates multi-step synthetic procedures, CDs are commercially available and are ideal “ready-made” molecular entities. Finally, CDs in their native forms are recognized to be safe and biocompatible.7 They have a proven record of being directly used to boost the intensity of imaging signals.32 Due to the favourable properties of CDs, CD-based “host–guest chemistry” has attracted extensive interests in imaging research from both clinicians and scientists.
| Compound | Structure | Pros | Cons |
|---|---|---|---|
| CDs | A cyclic compound consisting of glucose units |
• Highly biocompatible
• Non-toxic • Reversible in host–guest complex formation to enable flexibility in drug delivery • Highly water soluble for biomedical use • Optically transparent for imaging applications • Commercially available |
• Poor in intestinal permeation
• Possible to form crystalline precipitates in kidneys when β-CD is administered parentally in a high dose • Comparatively narrow in the choice of sizes and shapes of native CDs for probe development |
| Calixarenes | A macrocyclic oligomer made up of benzene units |
• Chemically stable
• Highly biocompatible • Bioactive due to the presence of antiviral, antibacterial, antifungal, and anticancer effects • Reversible in host–guest complex formation to enable flexibility in drug delivery |
• Poor in water solubility
• Technically demanding when molecules with odd numbers (n = 5, 7, and 9) are synthesized • Well-dispersed in some nonpolar organic solvents, but the strong competition between organic solvents and guest molecules has hampered the process of host–guest complexation |
| Cucurbiturils | A macrocyclic glycouril having two portals lined with ureido-carbonyl groups |
• Highly biocompatible
• Reversible in host–guest complex formation to enable flexibility in drug delivery |
• Poor in aqueous solubility
• Insoluble in practically all organic solvents, making chemical manipulation highly challenging |
| Pillararenes | A pillar-shaped macrocyclic compound made up of hydroquinone units linked by methylene bridges |
• Conformationally stable with a symmetrical and rigid structure
• Easy to functionalize with different substituents, as compared to macrocyclic compounds such as cucurbiturils • Soluble in many organic solvents, making functionalization technically feasible |
• Comparatively new in the literature, with the chemistry being less well-established
• Poor in aqueous solubility • Challenging and time-consuming when pillar[n]arene homologs are purified after the synthetic process |
| Crown-ether | A cyclic oligomer consisting of ethylene oxide units |
• Possessing good complex selectivity for diverse metal ions
• Stable for high temperature and radiolysis |
• Difficult to synthesize large crown ethers because of the competition of intermolecular reactions with intramolecular cyclization
• Highly toxic • Poor in adsorbing nonpolar organic molecules • Poor in aqueous solubility |
In spite of this, CDs are not without shortcomings. Partly because of their hydrophilic exterior, native CDs usually have poor intestinal permeability.19 This renders the less invasive oral route of administration inefficient. Furthermore, upon parenteral administration in a high dose, due to the comparatively low aqueous solubility of β-CD, crystalline precipitates may be formed in kidneys, resulting in nephrotoxicity.46,47 Because of these shortcomings, along with the motives to improve the flexibility of probe design, derivatizations are usually performed prior to probe fabrication. Over the years, a myriad of CD derivatives have been fabricated from chemical modification of hydroxyl groups located in the upper rim (primary side) or lower rim (secondary side), including the secondary hydroxyl groups at the 2- and 3-positions and the primary hydroxyl group at the 6-position of the glucopyranose ring.48 In addition to monofunctionalized CDs, generation of disubstituted derivatives using specific disulfonyl chlorides, whose geometry is employed to control the regiochemistry and to generate diverse isomers (Fig. 4), has been reported.48 This versatility in CD chemistry has partly been brought about by advances in high-field NMR methods, which have made detailed characterization of glucose substitution sites in CDs and their derivatives viable.49–53 Among all hydroxyl groups available in CD molecules, 3-OHs are least accessible; whereas 2-OHs are most acidic. Compared to the hydroxyl groups at these two positions, 6-OHs are most nucleophilic. Such differences in the reactivity of the hydroxyl groups at distinct positions enable functionalization of specific hydroxyl groups, facial selective functionalization (primary or secondary sides), or even functionalization of precise sets of hydroxyls (e.g., pairs, triad, and quartets).48,54 There is no doubt that one of the impetus of CD-based probe development arises from such advances in selective synthetic manipulation of CD molecules. Related chemistry has recently been reviewed elsewhere.55,56 Readers may refer to those published articles for details. The accumulated knowledge and growing sophistication of CD chemistry have laid a foundation for the molecular design and engineering, as reviewed in the subsequent sections of this article, of CD-based imaging probes.
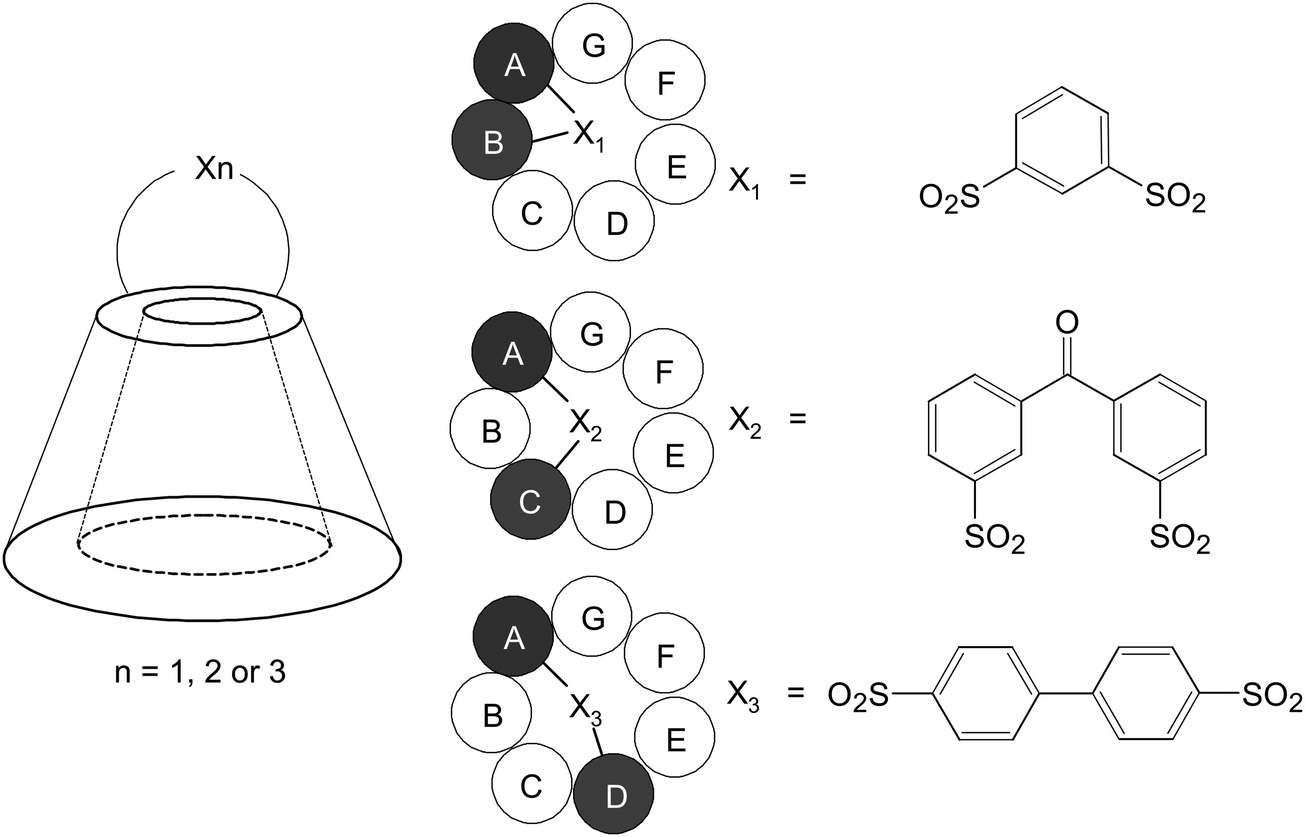 | ||
| Fig. 4 Schematic representations of the structures of the AB, AC, and AD isomers of 6-disulfonates of β-CD. | ||
3. Guest inclusion for molecular imaging applications
As far as probe design based on CDs is concerned, the truncated cone-shaped structure of the CD cavity is one of the engineerable features that have been extensively exploited. Previous studies have revealed that the reduced polarity and restricted space provided by the CD cavity can increase the blue light emission efficiency of fluorescent probes fabricated from some pyrazoline derivatives, such as 3-naphthyl-1-phenyl-5-(5-fluoro-2-nitrophenyl)-2-pyrazoline57 and 5-(1′-(4-bromophenyl)-3a′,4′,5′,6′,6a′-hexahydrocyclopentapyrazoline)-3-methyl-1-phenyl-1H-pyrazole-4-carbonitrile.58 Inclusion of meso-tetraphenylporphyrin into the cavity of β-CD has also been reported to boost fluorescence emission of meso-tetraphenylporphyrin and to increase the porphyrin metalation rate.59 The latter has enabled the macromolecular construct to display fluorescence changes upon interactions with zinc ions,59 rendering the construct promising to be developed as a selective molecular probe to monitor zinc ion concentrations at the cellular and tissue levels.Apart from the aforementioned, the process of host–guest binding is able to improve the target specificity of a probe,60 or to mediate probe self-assembly.61 When a theranostic probe is designed, inclusion complexation may enable the drug loading process as well.62 All these have pointed to the importance of a proper understanding of the complexation thermodynamics of CDs when CD-based molecular probes are constructed. Over the years, efforts in the literature have been devoted to elucidating the electrostatic potential inside the CD cavity63 and to deciphering the molecular interactions in inclusion complexes.64 It has been postulated that the complexation thermodynamics is contributed primarily by three mechanisms: (i) penetration of the hydrophobic moiety of an organic guest molecule into the CD cavity,65 (ii) degradation of the guest molecule,65–67 and (iii) conformational changes or strain release experienced by the CD molecule upon complexation.68,69 Apart from these, the solvation of the chemical species, the use of different buffers, and the release of water molecules from the cavity to bulk water may play ancillary roles in determining the thermodynamic quantities of CD-based inclusion complexation.66,70 To engineer the complexation process, one of the viable routes is to modify the structure and properties of the guest molecule, owing to the availability of sophisticated techniques in materials manipulation.71,72 Discussions in this section will focus primarily on the effects of the structural and electronic features (including molecular flexibility, chemical structures, and structural chirality) of organic guest molecules to the thermodynamics of host–guest binding for probe design and fabrication.73
3.1 Principles of complexation thermodynamics
Though different forms of CDs have similar torus heights,29 the cavity volumes of α-, β- and γ-CD are very different. The cavity volume increases with the number of α-D-glucopyranose units comprising the CD molecule, from 174 Å3 for α-CD, 262 Å3 for β-CD to 427 Å3 for γ-CD.29 Other structural dimensions of α-, β-, and γ-CD molecules are provided in Table 2.7,74,75 Taking the dimensional variations of different forms of CDs into account, the size-fit concept is the simplest way to understand the general trend in the complexation thermodynamics of these oligosaccharides. This concept has been attested by Varghese et al.,35 who have investigated how the photophysical characteristics of the pyrazoline dye, namely 3-naphthyl-1-phenyl-5-(4-carboxyphenyl)-2-pyrazoline (NPCP), have been modulated when the dye has drifted from bulk water into the CD cavity. The intensity of fluorescence emission from NPCP has been found to be increased, along with a shift to the lower wavelength side, when the concentrations of both β-CD and γ-CD in aqueous solutions increase.35 This has been proposed to be partly caused by the deep inclusion of the pyrazoline fragment into the CD cavity (Fig. 5), thereby protecting the dye from influences of hydrogen bonding from bulk water. Contributions have also been made by the restricted rotational and vibrational motions of the dye upon inclusion complexation. Such restrictions reduce the occurrence of non-radiative decay processes.76 Under the stoichiometry of 1:1, the association constants K for complexes with β-CD and γ-CD have been estimated to be 5.8 × 103 mM−1 and 1.3 × 104 mM−1, respectively. This suggests that compared to that of β-CD, the cavity of γ-CD binds to NPCP more strongly due to size fitting between the cavity and the guest molecule.35 This hypothesis has been substantiated by semi-empirical quantum mechanics calculations, which have indicated that the binding energy of the inclusion process for γ-CD is much more negative than that for β-CD, though the process for both types of CDs is exothermic in nature.35
| Molecular weight (Da) | Height of torus (Å) | Cavity volume (Å3) | Upper rim | Lower rim | |||
|---|---|---|---|---|---|---|---|
| Outer diameter (Å) | Cavity diameter (Å) | Outer diameter (Å) | Cavity diameter (Å) | ||||
| α-CD | 972 | 7.9 | 174 | 13.2 | 4.5 | 13.7 | 5.7 |
| β-CD | 1135 | 7.9 | 262 | 14.9 | 6.1 | 15.3 | 7.8 |
| γ-CD | 1297 | 7.9 | 427 | 16.1 | 7.7 | 16.9 | 9.5 |
 | ||
| Fig. 5 The most probable structure of the NPCP complex formed with (A) β-CD and (B) γ-CD. The carbon, nitrogen, hydrogen, and oxygen atoms are coloured in grey, blue, yellow, and red, respectively (reproduced from ref. 35 with permission from Elsevier). | ||
As a matter of fact, the complexation thermodynamics of CDs has provided a foundation to guide the process of probe design. For instance, α-CD has a cavity with a diameter much smaller than that of β-CD (4.7–5.3 Å for α-CD vs. 6.0–6.5 Å for β-CD),29 together with the fact that induction of van der Waals forces is largely affected by the distance of separation,73 it is anticipated that forces induced by complexation of a straight-chain guest molecule with α-CD will be larger (more negative ΔH°) than that with β-CD; whereas the situation will be reversed when the straight-chain guest is replaced by an adamantyl guest. However, it is worth noting that the van der Waals interactions may be altered by steric hindrance if the guest molecule fails to be fully accommodated by the CD cavity. Such a steric effect has been shown by ortho derivatization of aromatic compounds during complexation with β-CD.73 Based on this fact, if the guest molecule is a cyclic aliphatic compound, β-CD will be a better host; whereas if an acyclic guest is involved, α-CD will be a more preferable choice. Nonetheless, there are always exceptions. One exception to this is imidazole, which possesses a five-membered ring and can fit better into the α-CD cavity than into the β-CD cavity.73 There is also a global trend that molecules containing a phenyl moiety have a stronger affinity to β-CD than to α-CD, although the affinity of the guest molecule to CDs may vary if the phenyl moiety has undergone substitution.73
Besides van der Waals forces, hydrogen bonding may influence the stability of an inclusion complex. The involvement of hydrogel bonding in host–guest binding, however, depends largely on the type of functional groups present in the guest molecule. This has been shown in an earlier study by using a series of structurally related aromatic compounds, either with or without a phenolic hydroxyl group, as guest molecules.65 Except phenolic hydroxyl groups, any charged and hydrophilic groups present in the guest molecule tend to stay in the bulk solution even after inclusion complexation. This is suggested by the phenomenon that although 1-hexanol and 1-O-hexyl-β-D-glucopyranoside possess different types of hydrophilic groups, the equilibrium constants and standard molar enthalpies for complexation with α-CD are almost the same.77 This phenomenon points to the possibility that only the hexyl group of these guest molecules can successfully penetrate into the CD cavity, from which both the hydroxyl group of 1-hexanol as well as the glucopyranose moiety of hexyl glucopyranoside are excluded due to their hydrophilic nature.
Last but not least, the flexibility of a guest molecule may affect the thermodynamics of host–guest chemistry. This is particularly true for aliphatic chain residues, whose conformational degrees of freedom are lower than those of their saturated counterparts when a double bond is introduced, thereby making the complexation process less favourable in the entropic term. Such a phenomenon has been illustrated by using hexanoate and heptanoate, whose equilibrium constants for complexation with α-CD have been estimated to be remarkably higher than those achieved by trans-3-hexenoate and 6-heptenoate.67 Apart from structural flexibility, the stereoisomerism of a guest molecule may influence the process of inclusion complexation, whose enantioselectivity is thought to be governed by conformational freedom and desolvation.73 All these indicate that when a CD-based molecular probe is designed, the structures of other components (e.g., imaging elements and ligands) that form an integral part of the probe shall be taken into consideration, especially when host–guest binding plays a predominate role in probe fabrication and application.
3.2 Inclusion complexation in molecular imaging
One notable role played by CD-based inclusion complexation in molecular imaging is to suppress quenching of chromophores. This has been demonstrated in an earlier study, in which calcein-based supramolecular fluorescent nanoparticles have been fabricated using the “bricks and mortar” strategy.78 The nanoparticles consist of four components: (1) adamantane-functionalized calcein, (2) β-CD-grafted branched poly(ethylenimine) (PEI), (3) adamantane-functionalized poly(ethylene glycol) (PEG) derivative, and (4) adamantane-functionalized folate. The size of the nanoparticles can be tuned by changing the molar ratio of adamantane-functionalized calcein and β-CD-grafted branched PEI. Due to the host–guest interaction between β-CD and adamantane (β-CD/adamantane = 1/1), the π–π stacking and fluorescence self-quenching of calcein chromophores in water are suppressed,78 leading to an increase in fluorescence emission. Moreover, the nanoparticles exhibit a targeting capacity in vitro due to the presence of folate,78 whose receptor is frequently over-expressed in malignancies but not in normal tissues.79–82The capacity of inclusion complexation to control optical emission has also been observed in a CD-based probe synthesized by complexation between β-CD and 1,5-DHAQ−2H.32 The probe exhibits a selective colorimetric change towards Cu2+, and such a colour change is resulted from ion interactions with the phenolic oxygen of the anthraquinone derivative.32 During the process, the acceptor strength of the quinone moiety is increased by withdrawing the electron density to the cations. This facilitates intramolecular charge transfer in the probe, leading to a colour change due to a red shift of the wavelength absorption maximum.83 Compared to 1,5-DHAQ−2H, the sensitivity of β-CD:1,5-DHAQ−2H towards Cu2+ is higher. One possible mechanism is that β-CD increases the aqueous solubility of 1,5-DHAQ−2H, thereby facilitating the binding of the derivative to Cu2+. Moreover, the probe experiences photoinduced electron transfer quenching of fluorescence in the presence of Cu2+, even at an ion concentration of 10 nM.32 Fluorescence emission from the quenched probe is recovered only when Cr2O72− is added.32 This fluorescence recovery effect towards Cr2O72− has been shown to be highly selective. Similar effects have not been observed when other anions (e.g., AcO−, Br−, Cl−, CO32−, CN−, I−, MnO4−, NO3−, OH−, SCN−, and SO42−) are applied.32 These selective responses towards specific ions not only enable the probe to display XNOR (exclusive NOR) logic gate behaviour, but also allow the probe to give selective optical signals based on molecular recognition of ionic species in human alveolar basal epithelial cells.32 This example is a tribute to the value of CD-based inclusion complexation in molecular probe development.
More recently, similar concepts of monitoring ionic species of interest have been applied to the development of a selective fluorescent probe, based on an 3,3′-dihydroxybenzidine (DHB):α-CD solid inclusion complex, for molecular recognition of Hg2+ and Fe3+ ions.36 The complex can be prepared using either the co-precipitation method or the kneading method. FT-IR studies have revealed that during host–guest binding, new hydrogen bonds are formed between DHB and α-CD, while some existing hydrogen bonds between the hydroxyl groups of α-CD are broken. After the complexation process, DHB has been suggested by powder X-ray diffraction (XRD) analysis to adopt a new solid phase with a loss of the degree of crystallinity. Molecular docking has shown that the amine group (NH2) of DHB tends to interact with O7 and O121 atoms of α-CD with a distance of 2.13 Å and 2.01 Å, respectively. In addition, the hydroxyl group of DHB interacts with O28 atom of α-CD with a distance of 2.19 Å. In an acetonitrile/water system, the complex produces characteristic fluorescence emission spectra in the presence of Hg2+ and Fe3+ ions.36 Interference with such emission has not been observed upon addition of other metal ions (viz., K+, Zn2+, Mg2+, Cu2+, Cd2+, Mn2+, Cr3+, Ga3+, Al3+, and Sn4+).36In vitro studies have demonstrated that the probe emits green fluorescence only in HepG2 cells pre-treated with Hg2+ and Fe3+ ions.36 This, together with the modest cytotoxicity of the probe,36 has supported the applicability of the probe in molecular imaging to monitor the presence of selected ions in biological systems.
Another role played by inclusion complexation in molecular imaging is to mediate the self-assembly of a molecular probe. This has been illustrated in a previous report, in which a supramolecular polymer has been constructed through complexation of bridged bis(permethyl-β-CD) with Mn(III)-porphyrin bearing PEG side chains.61 As each molecule of bridged bis-CDs possesses two cavities, it can function as monomeric units of the polymer.84 Ultraviolet-visible (UV/Vis) spectroscopic analysis has shown that native Mn(III)-5,10,15,20-tetrakis(phenyl)-porphyrin (TPP) and Mn(II)-TPP give a signal at 466 nm and 434 nm, respectively.85,86 After addition of sodium ascorbate to Mn(III)-TPP, no change in the absorption spectrum has been observed.61 This suggests that sodium ascorbate fails to reduce Mn(III) to Mn(II) in TPP. However, the supramolecular polymer displays a new signal at 434 nm upon addition of sodium ascorbate,61 while the intensity of the absorbance at 466 nm is reduced.61 This substantiates the capacity of CDs to stabilize the low valent Mn(II)-TPP.
Finally, CDs can stabilize the active component of other probes via their inclusion complexation capacity. This is exemplified by the case of a single photon emission computed tomography (SPECT) radiotracer, namely [99mTcN(mpo)(PNP5)]+, which is a cationic 99mTc-nitrido complex in which PNP5 and mpo represent N-ethoxyethyl-N,N-bis[2-(bis(3-methoxypropyl)phosphino)ethyl]amine and 2-mercaptopyridine oxide, respectively.87 Here CDs have been utilized as a stabilizer for PNP5. Results have shown that for the single-vial formulation, in which all reaction components are lyophilized in a single vial, β-CD functions as a PNP5 stabilizer more effectively than γ-CD,87 with the radiochemical purity of the complex being 95–98% for the case of β-CD and only 90–93% for the case of γ-CD.87 This study has confirmed the significance of size fitting between the cavity and the guest molecule in determining the working efficiency of CDs.
4. CDs as modulators of probe properties
To mediate molecular imaging, CDs can be used directly as a carbon source for the production of imaging probes such as multicolour carbon nanoparticles.88 These nanoparticles can be obtained simply by alkali-assisted hydrothermal oxidation of CDs.88 They show excitation-dependent emission behaviour and bathochromic emission properties under UV excitation,88 and have been reported as a fluorescent probe for imaging of mouse melanoma cells.88 Lately, a diethylenetriamino-styryl-bridged bis(β-CD) dimer has been generated for imaging purposes.89 The synthetic route entails the use of 6-O-monotosyl-β-CD as a starting material, whose β-CD moiety is first functionalized with diethylenetriamine using KI and N-methyl-2-pyrrolidone (NMP). In the presence of N,N′-dicyclohexylcarbodiimide (DCC) and N-hydroxysuccinimide (HOSu), the amine group of the product undergoes a condensation reaction with the carboxyl group of 2-vinylterephthalic acid (VPA) in N,N′-dimethyl formamide (DMF) (Fig. 6). The dimer produced emits strong blue photoluminescence upon excitation with UV.89 The intensity of emission is boosted upon dimer polymerization.89 The strong emission is hypothesized to be induced by the intermolecular hydrogen bonding of amide and hydroxyl groups in the aggregation state of the dimer.89 All these examples have demonstrated the feasibility of using CDs and their derivatives as imaging probes without incorporating additional chromophores.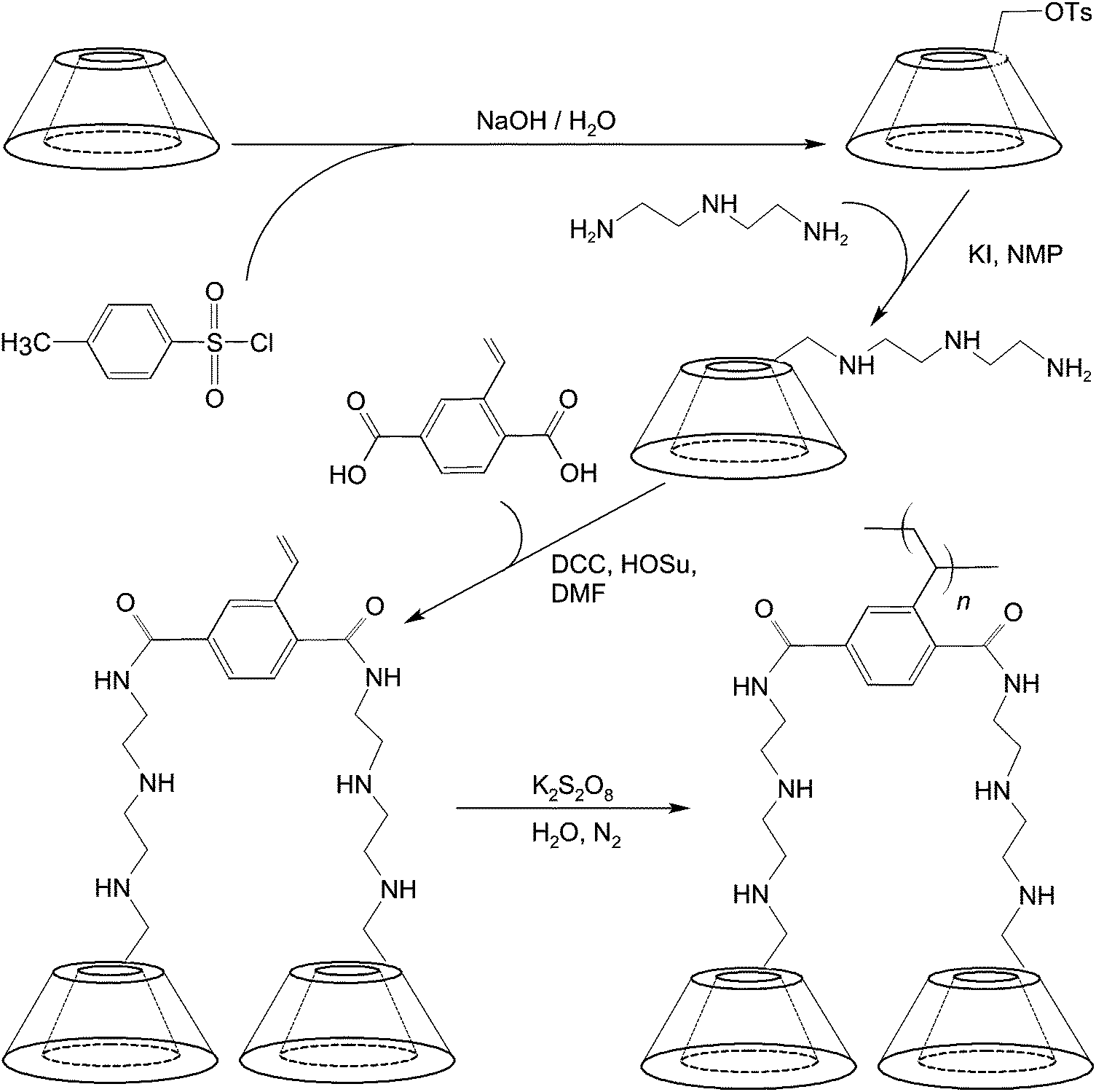 | ||
| Fig. 6 The chemical routes for the synthesis and polymerization of a β-CD dimer. | ||
In spite of this, CDs are not inherently chromophoric. Conversion of these oligosaccharides from the non-chromophoric state to the chromophoric one involves careful planning of the chemistry and engineering of the CD structure. In comparison with the direct use of CDs and their derivatives without prior incorporation of chromophores, more efforts in the literature have been devoted to using CDs to engineer other structures that contain chromophoric units, or to form macromolecular constructs with various functional units for different molecular imaging modalities (Fig. 7). Parts of this have been illustrated in the preceding section, in which the roles played by CD-based inclusion complexation in providing sites for probe self-assembly or modulating optical properties of guest species have been discussed. In this section, we will explore how CDs can modify the probe structure by functioning as structural and physiochemical modulators.
 | ||
| Fig. 7 Manipulation of CDs and their derivatives for molecular imaging. The images obtained from different imaging modalities are reproduced from ref. 193 and 232 with permission from Elsevier; from ref. 61 with permission from American Chemical Society; and from ref. 246 with permission from Wolters Kluwer Health, Inc. | ||
4.1 CDs as structural modulators
CDs can modulate the structural and functional properties of an imaging probe in two ways. The first way is to function as a direct structural modifier. This can be epitomized by the case of coumarin-3-carboxylic acid (CCA). It has been modified with CDs to form a fluorescent hepta(2I–VII,3I–VII,6I–VII-tri-O-methyl)-β-CD (MCD) derivative, namely coumarin-methyl-β-CD (CCA-MCD).90 A parallel example is cyanine dyes, which contain two aromatic or heterocyclic rings linked via a polymethine chain with conjugated carbon–carbon double bonds.91 These compounds are endowed with both colorimetric and fluorescent properties. Not only can they cover all wavelengths in the visible spectrum,92 but they also have high molar absorptivity and narrow absorption bands.92 They have emerged as promising probes for optical imaging.93,94 As early as the 1990's, synthesis of cyanine-β-CD derivatives has already been reported in an attempt to develop a fluorescent labelling reagent with enhanced photostability.95 These compounds have later been used as spectroscopic probes enabling molecular recognition of colourless guest molecules (e.g., 1-adamantanol, and vitamin B6).96 More recently, Carmona et al. have modified cyanine with CDs to generate derivatives that can carry doxorubicin.97 Molecular mechanics and molecular dynamics simulations have revealed that, during the drug loading process, enhanced system stabilization is resulted from both electrostatic and van der Waals binding energy contributions.97 Compared to the complex formed between native β-CD and doxorubicin, the stability constants for those formed with the β-CD derivatives are 4 orders of magnitude larger.97The second way for CDs to modulate probe structures is to function as a threading device in a polyrotaxane. Since the emergence of the first CD[2]rotaxane in the 1980's,98 CDs have been widely incorporated into the structural design of rotaxanes and polyrotaxanes for imaging applications.99,100 A good example is the polyrotaxane reported by Fredy and co-workers.101 It is generated by incubating α-CD molecules, pre-functionalized with bodipy fluorescent tags or Gd(III) complexes, with poly((N,N-dimethylammonio)undecamethylene chloride) in water at 80 °C for 72 hours (Fig. 8). During the threading process, each bulky dimethylammonium unit can function as a pseudo stopper.102 Such incorporation of CDs into the modular probe design offers several advantages in the practical sense. Firstly, it makes the ratio of different imaging elements present in a probe tuneable.101 Secondly, when the fluorescence properties of a probe have to be optimized, it can be done individually and separately on each CD moiety, with the size of the polyrotaxane being maintained.101 Finally, by simple manipulation of individual CD units, probes with multiple functionalities can be fabricated.101 All these can max the structural flexibility of a probe out during the design and application processes. Nonetheless, typical chemical routes for synthesis of polyrotaxanes often generate a statistical mixture of orientational mechanoisomers. This makes the probe structure less controllable. A solution to this problem has been illuminated by a current study, which has produced functionalized CD[3]rotaxanes as single isomers using a mechanostereoselective synthetic method.21 The head-to-head orientational mechanoisomery of the CD[3]rotaxanes has been attested using the transverse rotating-frame overhauser effect spectroscopy (T-ROESY).21 With the continuous advances in materials chemistry and engineering,103–112 the future development of molecular probes is anticipated to be facilitated by more precise structural and process control of CD-based polyrotaxanes.
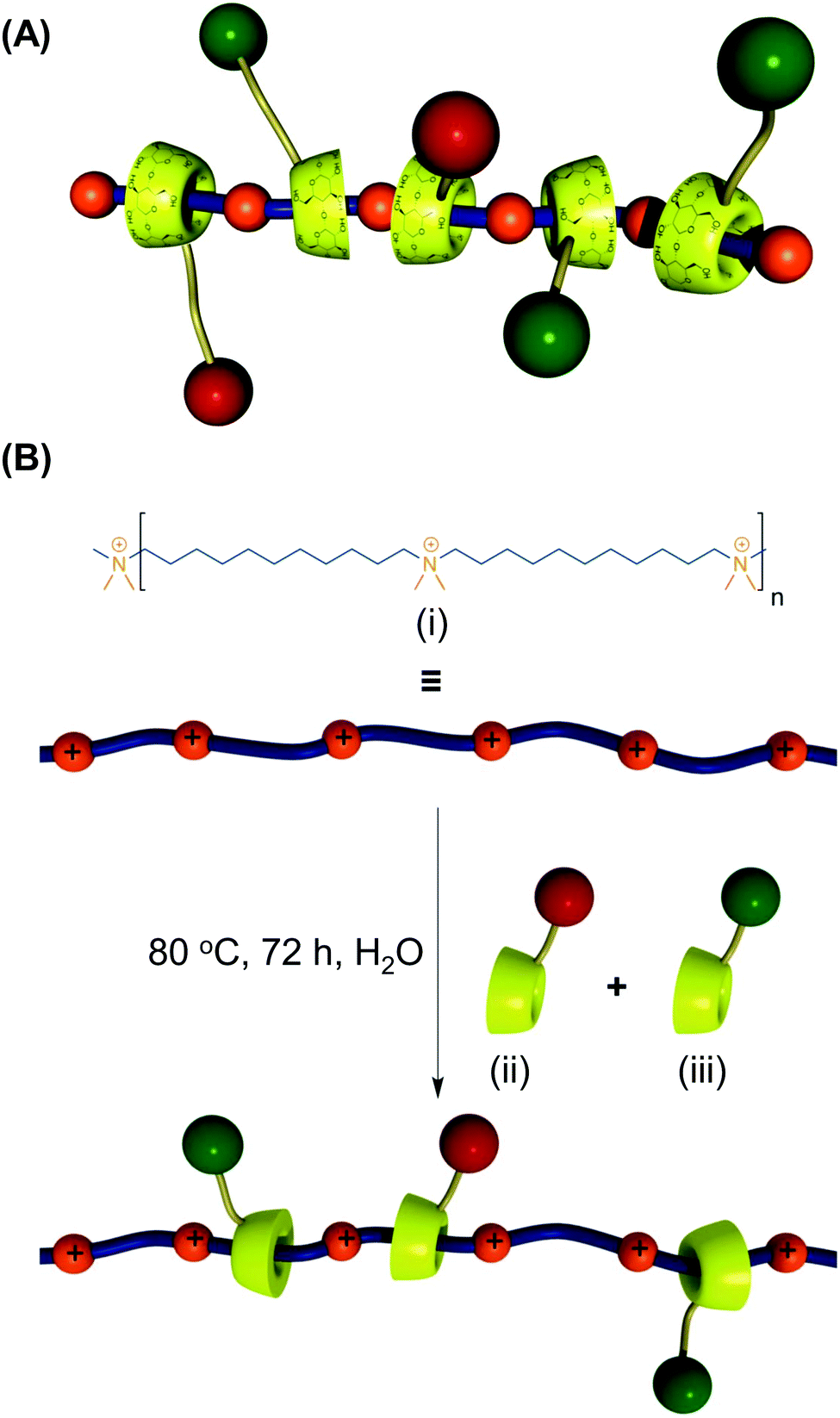 | ||
| Fig. 8 (A) A schematic representation of the polyrotaxane functionalized with fluorescent tags (red) and Gd(III) complexes (green). (B) A schematic representation of the synthesis of the bimodal polyrotaxane. In the figure, (i) represents poly((N,N-dimethylammonio)undecamethylene chloride). (ii) and (iii) are α-CD molecules functionalized with the bodipy fluorescent tag and those functionalized with the Gd(III) complex, respectively (reproduced from ref. 101 with permission from John Wiley and Sons, Inc.). | ||
4.2 CDs as physiochemical modulators
CDs can modulate not only the structural but also the physiochemical properties of a molecular probe. This has been demonstrated in an earlier study,113 in which a series of linear cationic β-CD-based polymers have been synthesized via condensation of diamino-CD monomers with diimidate comonomers. In BHK-21 cells, the IC50 values of the polymers have been shown to be much higher than those of the CD-lacking polyamidines.113 Similar toxicity attenuating effects of CDs have been applied to other contrast agents such as Mn(III)-TPP, whose anti-proliferative activity on mouse embryonic fibroblasts has been reduced upon intermolecular inclusion complexation with bridged bis(permethyl-β-CD).61 Another example is the trans-4-[p-(N,N-diethylamino)styryl]-N-methylpyridinium iodide (DEASPI)/β-CDP nanomicelles. Results have shown that incubation of the HeLa cells and MCF-7 cells with DEASPI alone (∼50 μM) causes a drop of cell viability to 68% and 78%, respectively. However, when the DEASPI molecules form a nanomicelle with β-CDP, the cytotoxicity becomes negligible, even at a similar overall concentration of DEASPI.114CDs are able to increase the internalization of a molecular probe at the cellular and tissue levels, too. This is confirmed by the case of CCA-MCD, in which the MCD moiety has been shown to improve the cell permeability of CCA for intracellular monitoring of the hydroxyl radical level.90 At the tissue level, the uptake-enhancing capacity of CDs has been documented by Schipper et al.115 They have successfully elevated the permeability of the nasal mucosa to an intranasally administered neurotrophic peptide, Org2766, by using dimentyl-β-CD, which leads to a 1–2 fold increase in absorption in rabbits and a 5-fold increase in absorption in rats.115 As a matter of fact, CDs possess multiple hydrogen donors and acceptors in their structures. Together with their modest octanol/water partition coefficients and large molecular weights (>972 Da), these oligosaccharides are unlikely to be permeable to lipophilic biological membranes such as skin and gastrointestinal mucosa.116–119 The permeation-enhancing ability of CDs is, therefore, thought to be mediated by the capacity of CDs to complex with membrane components (including phospholipids and cholesterol) to increase the membrane permeability.120 This is supported by an earlier study, which has incubated erythrocytes from different animal species in suspensions of lecithin liposomes to deplete membrane cholesterol.121 When the level of cholesterol removal has gone beyond 30%, the transfer rates of nonelectrolytes and organic acids penetrating the membrane have been found to increase significantly.121 This evidences that complexation with cholesterol is one of the possible mechanisms to enhance the membrane permeability. Despite this, the biphasic response of cholesterol depletion on the permeability of cell membranes seems not to be reproducible in artificial lipid membranes.121 Further investigations are required to fully unravel the mechanism of CD-mediated permeability enhancement.
Besides the toxicity and internalization efficiency, surface properties of a probe play a profound role in determining the efficiency of CD-based molecular imaging. This is because the first point of contact between a living body and a probe is the probe surface. The importance of manipulating properties of the probe surface has been revealed by Jain and colleagues,122 who have noted that compared to the uncoated counterparts, chitosan-based nanoparticles coated with alginate can be absorbed by the intestine more effectively, resulting in more profound anticoagulant activity upon oral administration to a venous thrombosis rat model. This reveals the relevance of surface properties to biological performance. In probe design, CD moieties can function as sites for incorporation of targeting ligands60 or other functional moieties (such as peptides123 and chemical drugs62). This approach has previously been applied to incorporate adamantane-modified GRRRDEVDK-BHQ2 (black hole quencher 2) peptide (denoted as Ad-DEVD-BHQ2) onto the surface of DEASPI/poly β-CD (β-CDP) nanomicelles.123 Attributed to the overlap between the fluorescence emission spectrum of DEASPI/β-CDP and the absorption band of BHQ2, the one-photon excitation (OPE) and two-photon excitation (TPE) fluorescence emission signals from the nanomicelles have been substantially quenched upon incorporation of Ad-DEVD-BHQ2,123 due to the energy transfer from fluorescence emission of the nanomicelles to BHQ2. This example evidences that the energy donor and acceptor have been brought into close proximity by host–guest binding, and confirms the success of surface modification of the nanomicelles.
A similar method of surface functionalization has been exploited in the design of the CD-incorporated graphene nanosheet.34 The presence of CD moieties enables incorporation with diverse functionalities (including amantadine, 5(6)-carboxyfluorescein, and adamantane-conjugated RGD peptide) via inclusion complexation. Results have indicated that the functionalized nanohybrid is capable of targeting HeLa cells upon surface incorporation of the RGD peptide.34 Along with its imaging potential after incorporation of fluorescent dyes,34 the nanohybrid warrants exploitation as a probe for future molecular imaging. More recently, the concept of using CDs as incorporation sites has also been applied to the design of a fluorescent molecular probe, which is constructed of water-dispersible CD-based conjugated polymer nanoparticles (CPNs).124 To fabricate the probe, β-CD-terminated polyfluorene (CD-PF-CD) is synthesized by functionalizing the hydroxyl groups of polyfluorene (PF-OH) using ethylenediamine-modified CD (EDA-β-CD) in the presence of 1,6-diisocyanatohexane (HDI) (Fig. 9).124 After that, CD-PF-CD in pyridine is injected into water to induce the self-assembly of CPNs.124 Those nanoparticles can subsequently be surface-functionalized via inclusion complexation between β-CD and adamantine with a binding constant of around 1 × 104 M−1.125 By incorporating adamantine-modified glycopolymers into the nanoparticle surface, the probe has been reported to show specific cell binding behaviour for targeted imaging.124
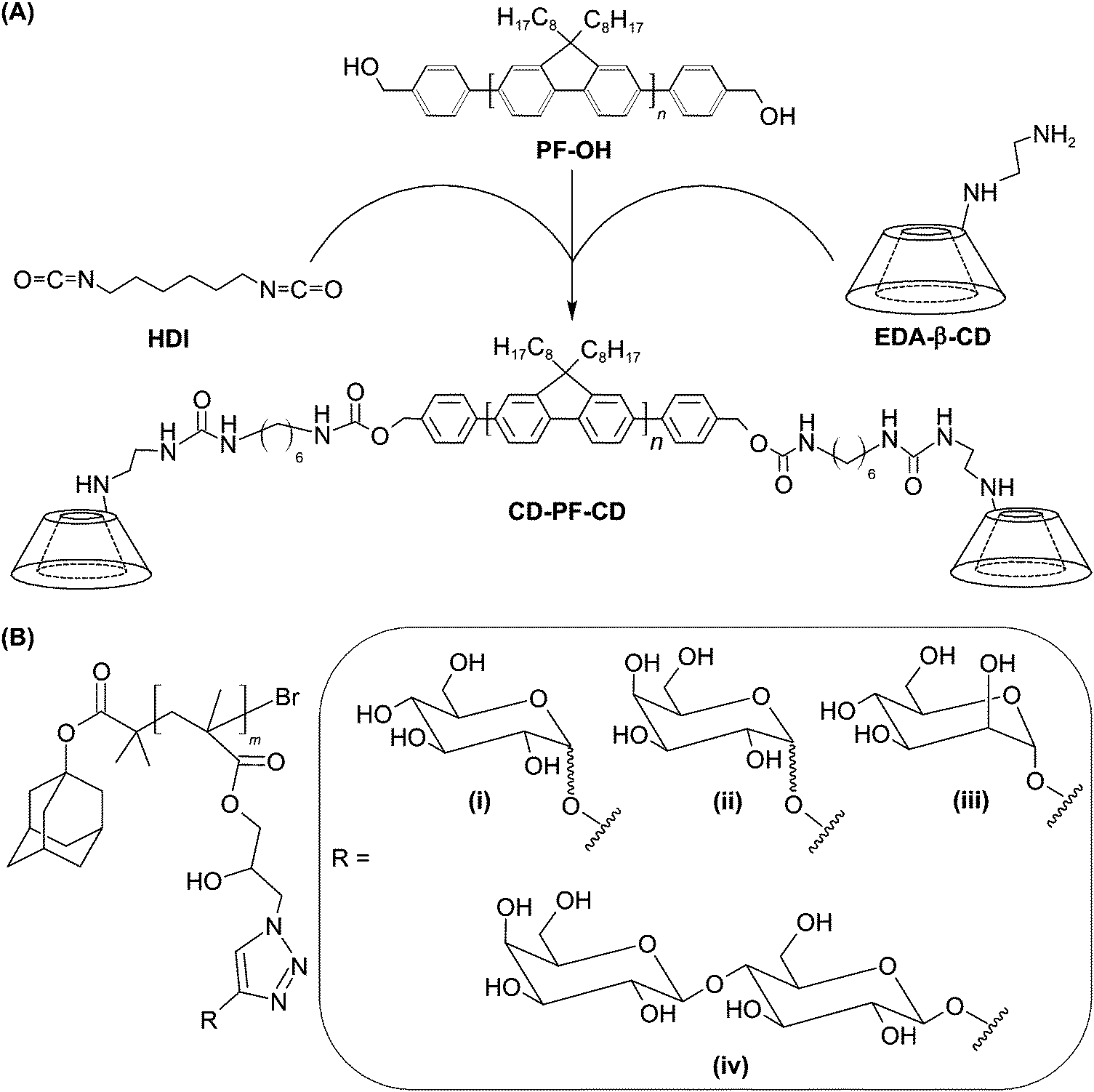 | ||
| Fig. 9 (A) The chemical route for the synthesis of CD-PF-CD. (B) The chemical structure of the adamantine-modified glycopolymer. In the figure, (i), (ii), (iii) and (iv) designate the R groups for the adamantine-modified glycopolymers formed with glucose, galactose, mannose, and lactose, respectively. | ||
5. Advances in the development of CD-based molecular probes
Having high target specificity is an important criterion for an ideal CD-based probe, which is expected to give measurable differences between normal and pathological tissues with high reproducibility. The target specificity of CD-based probes can be attained by direct or indirect methods.126 Direct targeting is achieved by designing a probe de novo, with the probe possessing a targeting element that can interact with a specific molecular event or marker directly; whereas in the indirect approach, a pre-targeting molecule, which is usually a protein or a gene, is used. That molecule is activated to give an imaging signal when a specific molecular event occurs, with the signal being captured during the imaging process.127Direct targeting is the most widely used strategy in CD-based molecular imaging. This approach benefits from the availability of sophisticated techniques for ligand incorporation and from the large variety of targeting ligands discovered by the accumulated efforts in the literature. This approach, however, is time-consuming because each probe that is designed de novo has to be validated individually before practical use.126 Proper selection of a molecular marker is also required, or high background noises result. The imaging efficiency mediated by the indirect targeting method, on the other hand, is largely dependent on how well the pre-targeting molecule can be delivered.126 In addition, the pre-targeting molecule may perturb the underlying biological process being examined, and hence requires extensive prior validation.126 The indirect method, however, enables easy production of probes, by simply altering the pre-targeting moiety, to interrogate different molecular events.126 Such a diversity of targeting methodologies enables molecular probes to be tuned to meet different biological and imaging needs. This fuels the rapid development of CD-based molecular probes, and facilitates related research to progress to preclinical trials (Fig. 10). Presented in the remaining parts of this section are more detailed descriptions of the state of the art of molecular probes based on CDs for different imaging modalities.
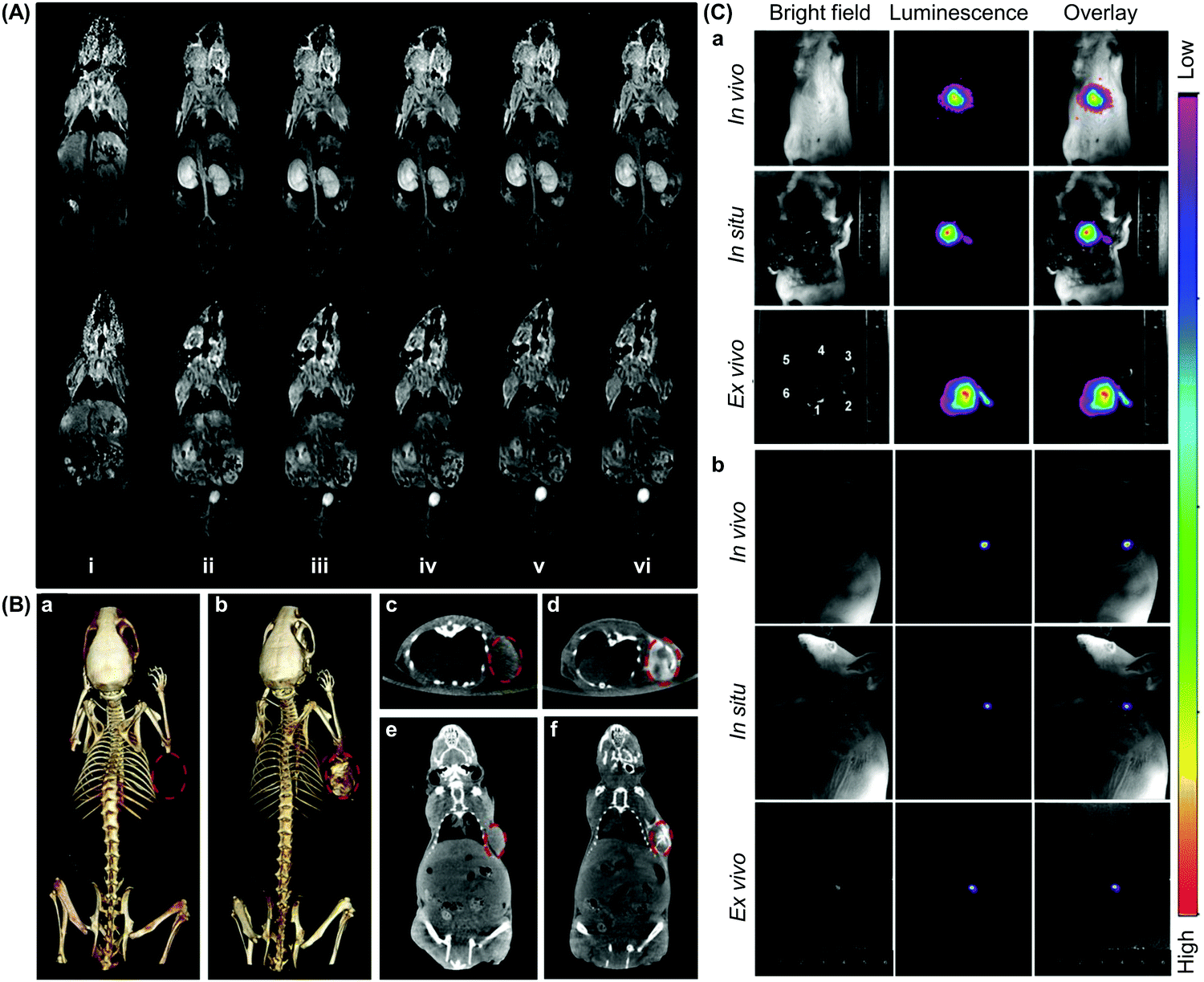 | ||
| Fig. 10 Representative in vivo images obtained from molecular imaging mediated by CD-based probes. (A) 2D coronal T1-weighted magnetic resonance images of the mice at different time points after intravenous administration of a CD-based contrast agent at a dose of 0.03 mmol Mn per kg: (i) pre-injection, (ii) 2 min, (iii) 5 min, (iv) 10 min, (v) 20 min, and (iv) 25 min. The contrast agent is generated from bridged bis-CDs, whose cavities can stabilize the bivalent state of Mn in porphyrins (reproduced from ref. 61 with permission from American Chemical Society). (B) (a and b) 3D computed tomography (CT) volume-rendered images of the tumour-bearing mice, and (c–f) the maximum intensity projections of (c and d) coronal and (e and f) transversal images of the tumour. The images are obtained (a, c and e) before and (b, d and f) after intratumoural administration of a CD-based probe (100 μL, 3 mg mL−1), which is fabricated from oleic acid (OA)-coated NaDyF4 nanoparticles. The nanoparticles self-assemble with α-CD for hydrophilic conversion before further modification with other functionalities. The position of the tumour is indicated using red circles (reproduced from ref. 232 with permission from Elsevier). (C) (a) In vivo, in situ, and ex vivo upconversion luminescence images of the mice acquired at 5 min after intravenous injection of 200 μL of CD-modified UCNPs, namely UCNP(Tm)-OA-CDT (0.5 mg mL−1). In the figure, 1, 2, 3, 4, 5, and 6 denote the liver, spleen, stomach, kidney, intestines, and heart, respectively. (b) In vivo, in situ, and ex vivo luminescence lymphatic imaging at 30 min post-injection of 20 μL of UCNP(Tm)-OA-CDT (200 μg mL−1) (reproduced from ref. 157 with permission from Elsevier). | ||
5.1 Probes for luminescence-based molecular imaging
Luminescence-based imaging is one of the major imaging modalities for non-invasive tissue visualization. Its development has profoundly facilitated the progress in biomedical research,128 enabling attainment of increasing understanding of the pathogenesis and progression of diseases such as choroidal melanomas, whose microcirculation has been successfully imaged in vivo upon administration of an indocyanine green (ICG) fluorescent probe.129 In addition to target specificity, the molecular recognition ability is a desirable add-on as it may extend the efficiency of luminescence-based molecular imaging when interrogating molecular events that link to pathological alterations. To date, sundry mechanisms of molecular recognition have emerged for probe design (Table 3).114,130–136 Many of them show potential to be applied to the construction of CD-based optical probes for tracking molecular events such as reactive oxygen species (ROS) generation.137,138 In an earlier study, CCA-MCD has been utilized as a cell-permeable probe for molecular recognition of hydroxyl radicals.90 The probe is generated by first incubating a mixture of coumarin-3-carboxylic acid and hydroxybenzotriazole in DMF at 0 °C, followed by addition of DCC and 6I-amino-6I-deoxy-2I,3I-di-O-methyl-hexakis(2II–VII,3II–VII,6II–VII-tri-O-methyl)-β-CD (6-amino-PMCD).90 The fluorescence intensity of CCA-MCD has been found to exhibit a positive relationship with the concentration of Cu2+, which leads to changes in the concentration of hydroxyl radicals generated by the CuSO4/H2O2/ascorbic acid system in an aqueous solution (Fig. 11).90 No remarkable change in the fluorescence intensity has been detected upon addition of ROS (including H2O2, superoxide ion, nitric oxide, alkylperoxyl radical, and hypochlorite) other than hydroxyl radicals.90 The probe has emerged as a selective probe for molecular imaging in pathological situations (such as oxidative damage and DNA fragmentation) where hydroxyl radicals are one of the key players.| Strategy | Underlying principle | Practical example | |||
|---|---|---|---|---|---|
| Form of CDs | Working principle | Molecular target | Ref. | ||
| Host–guest binding | Inclusion complexation with a guest molecule can modulate the intensity of light emission | β-CD-CuInS2 quantum dots (QDs) | Inclusion complexation occurs between the adenosine-5′-triphosphate (ATP)-binding aptamer and β-CD-CuInS2 quantum dots to generate a near-infrared (NIR) fluorescent probe. In the presence of ATP, G-quadruplexes are formed between ATP and the aptamer. The complex enters into the hydrophobic cavity of β-CD, leading to enhancement of the fluorescence intensity. | ATP | 130 |
| Per-O-methylated β-CD | A fluorescent probe is constructed of three moieties: a DNA strand for dimerization, pyrene for optical signal emission, and per-O-methylated β-CD for molecular recognition. In the presence of hydrophobic guest molecules such as porphyrin derivatives, inclusion complexation with the CD moiety occurs. This promotes DNA hybridization, subjecting the probe to emission switching. | Porphyrin derivatives | 131 | ||
| Probe aggregation | Aggregation of the probe in response to the presence of a molecular biomarker causes changes in the optical properties of the probe | β-CD-grafted citrate | Silver nanoparticles are prepared using β-CD-grafted citrate, which functions as both a stabilizer and reducer. The presence of riboflavin causes severe aggregation of the nanoparticles, thereby inducing a colour change from yellow to red. | Riboflavin | 132 |
| Surface sensing | Cellular uptake of the probe is driven by receptor-mediated endocytosis | β-CD polymer | A fluorescent probe generated from the β-CD polymer is anchored with adamantane-modified cyclic RGD peptide via inclusion complexation with the β-CD moiety. As the peptide targets αvβ3 and αvβ5 integrin receptors on the cell surface, fluorescence signals are observable only in integrin-positive but not integrin-negative cells. | Integrin receptor | 114 |
| β-CD | Nanoparticles containing ZnSe/ZnS QDs are fabricated using β-CD/suberoylanilide hydroxamic acid complexes and folate-conjugated chitosan. The nanoparticles display long-term optical properties, and can preferentially accumulate in tumours in which folic acid receptors are overexpressed. | Folic acid receptor | 133 | ||
| Förster resonance energy transfer (FRET) | The energy transfer between a donor chromophore to an acceptor can modulate emission of the probe | Dansyl-linked β-CD | A supramolecular β-CD/dye complex, consisting of dansyl-linked β-CD and spirolactam rhodamine-linked adamantane, is fabricated as a FRET probe. The dansyl moiety serves as the donor. In the presence of ferric ions, a ring-opening reaction occurs in the spirolactam-rhodamine B derivative, generating a long-wavelength rhodamine B fluorophore which functions as an energy acceptor. | Ferric ions | 134 |
| β-CD modified with Cy5 (or QSY9) | β-CD modified with Cy5 (or QSY9) binds to the dye-conjugated E. coli maltose binding protein (MBP) to form a FRET complex. In the presence of maltose, the complex is disrupted, resulting in changes in fluorescence emission. | Maltose | 135 | ||
| β-CD modified with both the probe dye and the donor dye | The thiocarbamido-containing probe dye is covalently linked onto the hydrophilic β-CD rim, while the donor dye is anchored inside the β-CD cavity via the adamantly moiety. The probe gives intracellular green fluorescence in the absence of mercury ions, and gives red fluorescence in cells pretreated with mercury ions. | Mercury ions | 136 | ||
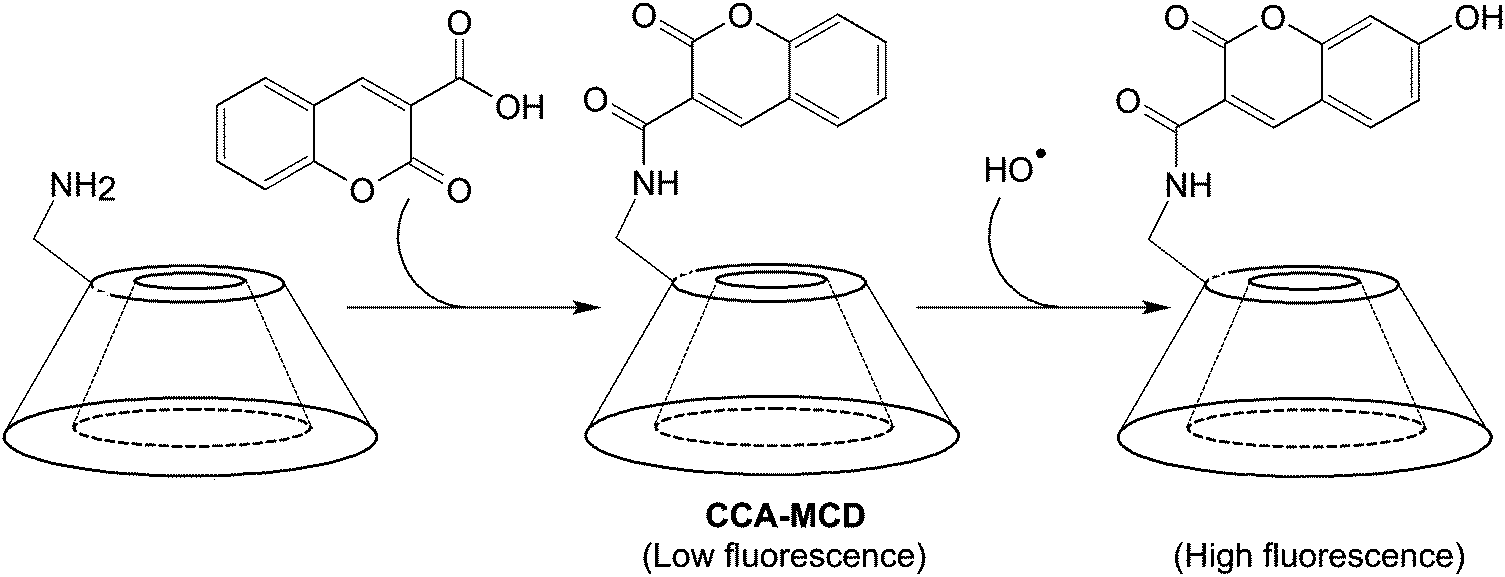 | ||
| Fig. 11 The chemical route for the synthesis of CCA-MCD and the subsequent reaction with a hydroxyl radical. | ||
Lately, another probe has been developed based on Förster resonance energy transfer (FRET) for molecular recognition of trypsin in serum.139 To generate the probe, p-tosyl chloride is first reacted with β-CD in dry pyridine to attain mono-(6-p-toluenesulfonyl-6-deoxy)-β-CD.139 After that, an aqueous solution of lysine containing K2CO3 is added to mono-(6-p-toluenesulfonyl-6-deoxy)-β-CD in DMF to form lysine-bridged-bis(β-CD) (Fig. 12). Under 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC)/HOSu conditions, the bridged bis(β-CD) is linked to mercaptoethylamine-modified gold nanoparticles (MGNPs), which can be obtained by stirring mercaptoethylamine and citrate-stabilized gold nanoparticles in an aqueous environment, to generate a bridged β-CD dimer–dye complex as a molecular imaging probe.139 In the probe, mercaptoethylamine-modified-gold nanoparticles function as an energy acceptor; whereas the lysine-bridged-bis(β-CDs)-fluorescein complex functions as an energy donor. If trypsin is present in a biological environment, it cleaves the probe and recovers quenched fluorescence.139 The molecular recognition capacity of the probe has been shown to be free from the interference of diverse cations (e.g., K+, Na+, Ca2+, Mg2+, and NH4+), sugars (e.g., glucose, and sucrose), and proteins (e.g., lipase, α-amylase, bovine serum albumin, glutathione reductase, thioredoxin, and α-chymotrypsin) commonly found in serum.139 As trypsin plays an important role in regulating pancreatic exocrine functions,140 along with the clinical association of an elevated trypsin level with pathological changes in the pancreas,141,142 the probe may be useful in molecular imaging for diagnosis based on the serum trypsin level in the future.
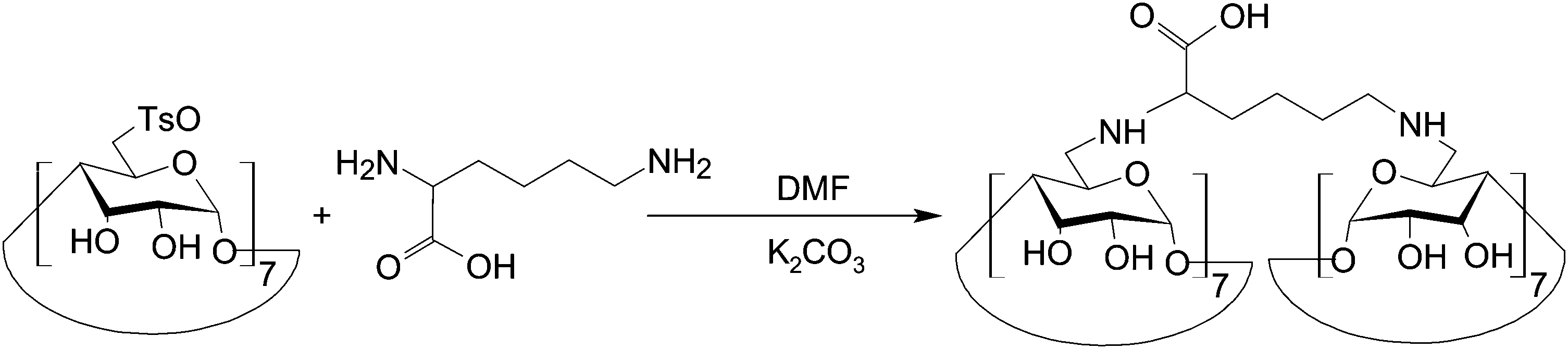 | ||
| Fig. 12 The chemical route for the synthesis of lysine-bridged-bis(β-CD). | ||
During the fabrication of the probe mentioned above, Tang's group has applied bridged bis-CDs rather than native CDs in their probe design.139 Compared to native CDs (and those mono- or di-functionalized CD derivatives), bridged bis-CDs are more favourable candidates for probe development because they exhibit a higher binding capacity and molecular selectivity due to cooperative binding of two adjacent CD units.143 These properties are what an ideal molecular probe is expected to possess. Cooperative binding in bis-CDs can be at the intramolecular or intermolecular levels.84 Intramolecular cooperative binding allows the molecules to form stable inclusion complexes with guest molecules; whereas intermolecular cooperative binding enables the formation of molecular assemblies. The latter has been adopted in an earlier study, in which bridged bis(β-CD)s have been used as monomeric units, with which a supramolecular polymer has been constructed through intermolecular inclusion complexation with Mn(III)-porphyrin bearing PEG side chains.61In vitro and in vivo studies have revealed that the supramolecular polymer enables enhancement of imaging signals while showing little cytotoxicity.61 As a matter of fact, choosing a suitable bridge chain is critical to the functioning of bis-CDs because the separation and orientation of the two CDs units can be modulated by the use of different chains. Regarding the structural flexibility of bis-CDs, apart from modulating the spatial size effect that governs host–guest interactions, using bis-CDs with appropriate bridging chains can be a future direction to fine-tune the donor/acceptor assembly process during probe development, thereby extending the tunability of the properties of CD-based FRET probes.
In addition to FRET, another mechanism widely adopted to mediate CD-based luminescence-based molecular imaging is inclusion complexation. By and large, fluorescence emission from an organic fluorophore can be modulated upon inclusion into a CD cavity, partly due to changes in the rotational freedom of the fluorophore. One class of CDs potentially applicable to probe development based on this process is metallocyclodextrins, which are obtained by the coordination of a metal ion either to the hydroxyl groups of native CDs or to other metal-binding moieties present in a CD derivative.48,144 When metallocyclodextrins are used in probe design, careful selection of a metal ion or complex as the centre is required. The excited state of the centre is expected to be sufficiently long-lived either to function as a donor or acceptor in energy or electron transfer processes, or to release its energy as luminescence. In addition to metallocyclodextrins, a multitude of CD-based imaging probes for molecular recognition of transition metals,145 amino acids145 and anionic guests146 have been reported. One example is 6-deoxy-6-N-(N′′-dansyldiethylenetriamino)-β-CD, which possesses dansyl fluorescent groups that are linked to the primary side of CDs via an ethylene diamine linker.145 Upon addition of Cu2+, the fluorescence emission can be quenched, but this can be reversed in the presence of copper-binding amino acids or their derivatives (such as alanine, tryptophan and thyroxine).145 To confirm possible translation of the probe into biomedical use, it would be instructive to further evaluate the physiochemical performance. Yet, the plausibility of attaining selective fluorescence emission through the mechanism of inclusion complexation has already been evidenced. This has laid a foundation from which further development of molecular probes, on the principle of CD-based inclusion complexation, may be launched.
Till now all aforementioned examples of CD-based probes are designed mainly for molecular imaging based on fluorescence emission. This type of imaging allows for multiplex detection, and has practical advantages in terms of high cost-effectiveness and high sensitivity.147 However, due to the short excitation wavelengths and the possible induction of photodamage, along with the high tissue autofluorescence and the photon loss caused by self-absorption and scattering, applications of imaging based on fluorescence are often restricted to visualization of superficial lesions with a known origin. To image deep-lying lesions or lesions with an undefined location, more established technologies for deep-tissue molecular imaging are required. One technology for this is near-infrared (NIR) imaging. Using NIR photons as an excitation source for TPE offers several advantages over conventional OPE, including lower tissue autofluorescence, higher spatial resolution, lower self-absorption, reduced photodamage, reduced photobleaching, and deeper photon penetration.148
To mediate TPE in molecular imaging, the use of a two-photon probe is needed. The technical plausibility of constructing such a probe has been illustrated by Yan et al.,114 who have generated two-photon absorption (TPA) fluorescent nanomicelles that show high cell-permeability, high stability, and good biocompatibility. The nanomicelles are constructed based on the host–guest interactions between polymerized CDs and DEASPI. Polymerized CDs can be synthesized by reacting CD molecules with epichlorohydrin in an aqueous solution of NaOH.114 The synthetic pathway involves two steps. In the first step, the CD molecules are stirred in an alkaline solution to form alcoholate sites. Epichlorohydrin here functions as a bifunctional agent, and reacts with the hydroxyl groups of CD molecules. In the second step, the epoxy ring in the side chain can either be hydrolyzed, or react with the hydroxyl group of another CD molecule to form a glyceryl bridge connecting the two molecules together (Fig. 13A). In the former, the hydrolyzed epoxy ring can be re-activated by reacting with epichlorohydrin. The re-activated epoxy group can then undergo reactions with another CD molecule to generating a glyceryl bridge (Fig. 13B). By fine-tuning the initial molar ratio of epichlorohydrin and β-CD, as well as the reaction time and the NaOH concentration, the molecular weight of the resulting polymerized CDs can be modulated,149 enabling the stability, biocompatibility, and cell permeability of the TPA fluorescence nanomicelles to be fine-tuned. Furthermore, the cavities of polymerized CDs can provide the nanomicelles with a protective microenvironment to decrease the effect of the twisted intra-molecular charge transfer (TICT) process on the embedded DEASPI.114 Compared to those given by free DEASPI molecules, TPE fluorescence emission and one photon-induced emission given by the nanomicelles are more intense.114 By using rhodamine 6G as a reference, the maximal TPA action cross-section (δΦ) of the embedded DEASPI molecule (44.5 GM) has been found to be substantially higher than that of the free DEASPI molecule (1.0 GM).114 Upon anchoring adamantine-modified cyclic RGD onto the nanomicelle surface, intense fluorescence signals from the nanomicelles are observed only in integrin-positive HeLa cells but not in integrin-negative MCF-7 cells. Similar targeting effects have been noted at the tissue level, in which fluorescence emission is detected in cervical tumour tissues (which are characterized by over-expression of αvβ3/αvβ5 integrins) but not in normal cervical tissues.114 Intriguingly, bright TPE fluorescence signals from the nanomicelles in a 2.0 mm-thick cervical tumour tissue slice can be detected until a penetration depth of 300 μm.114 This is much more powerful than conventional OPE, which can hardly be detected when the penetration depth is over 100 μm.114 This study vividly demonstrates the possibility of increasing the diagnostic power of luminescence-based imaging by replacing OPE with TPE, in which NIR photons are used as an excitation source.
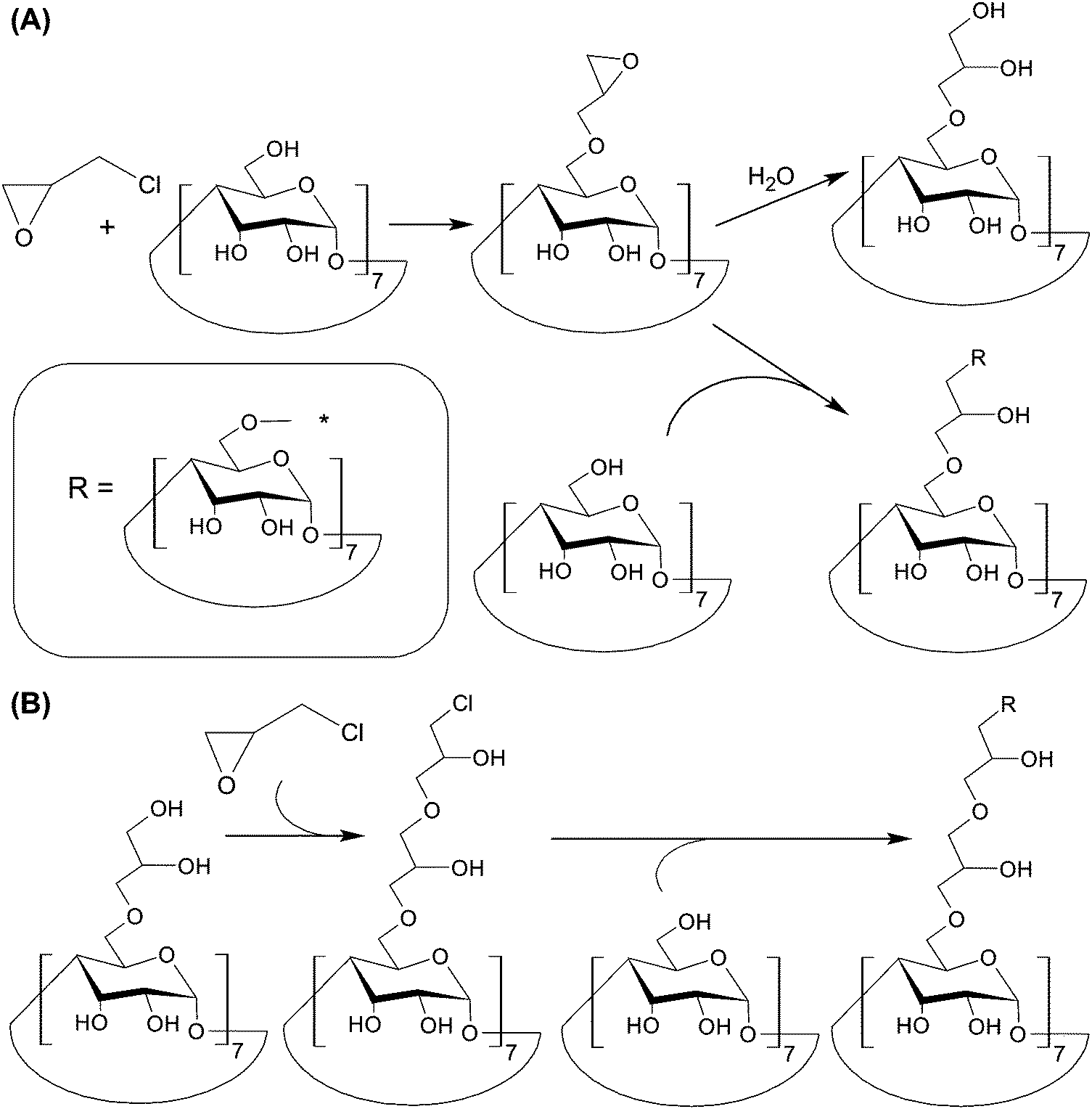 | ||
| Fig. 13 (A) Reactions between β-CD and epichlorohydrin in a sodium hydroxide solution. (B) The chemical routes for the reactivation of the hydrolyzed epoxy ring. | ||
More recently, the aforementioned nanomicelles have been anchored with the Ad-DEVD-BHQ2 peptide for caspase-3 activation imaging.123 By examining the action of different endocytosis inhibiting agents (viz., chlorpromazine, sucrose, filipin, and cytochalasin D) on cellular entry of the nanomicelles, clathrin-mediated endocytosis has been found to be the major route of cellular uptake.123 BHQ2 (with maximum absorption at 550 nm) in the nanomicelles can function as a FRET quencher of DEASPI; whereas GRRRDEVDK is a caspase-3 cleavable peptide linker possessing a cleavage site between D and K. The working principle of the nanomicelles is based on the fact that when cells undergo apoptosis, caspase-3 is activated.150 The activated caspase-3 cleaves the peptide linker to remove the quencher, triggering fluorescence emission from the nanomicelles. In vitro studies have shown that fluorescence emission from the nanomicelles is observed in HeLa cells only after the cells are treated with staurosporine (STS) (Fig. 14).123 In a doxorubicin-treated cervical tumour tissue slice, the nanomicelles give intense TPE signals with a penetration depth of around 360 μm. This penetration depth is much larger than that reachable by conventional single-photon imaging.123 Despite such encouraging results, the success of deep tissue imaging using the nanomicelles has been tested only in the ex vivo context. The efficiency of the nanomicelles in molecular imaging in a living body cannot be conclusively confirmed at this stage. Yet, the nanomicelles have already been demonstrated to enable high-contrast imaging based on capase-3 activities and to allow for quantitative analysis of caspase-3 activation.123 If more stringent tests could be performed to evaluate their biological performance upon in vivo administration, optimization and potential applications of the nanomicelles for monitoring the progress of apoptosis-associated diseases are expected to be greatly facilitated.
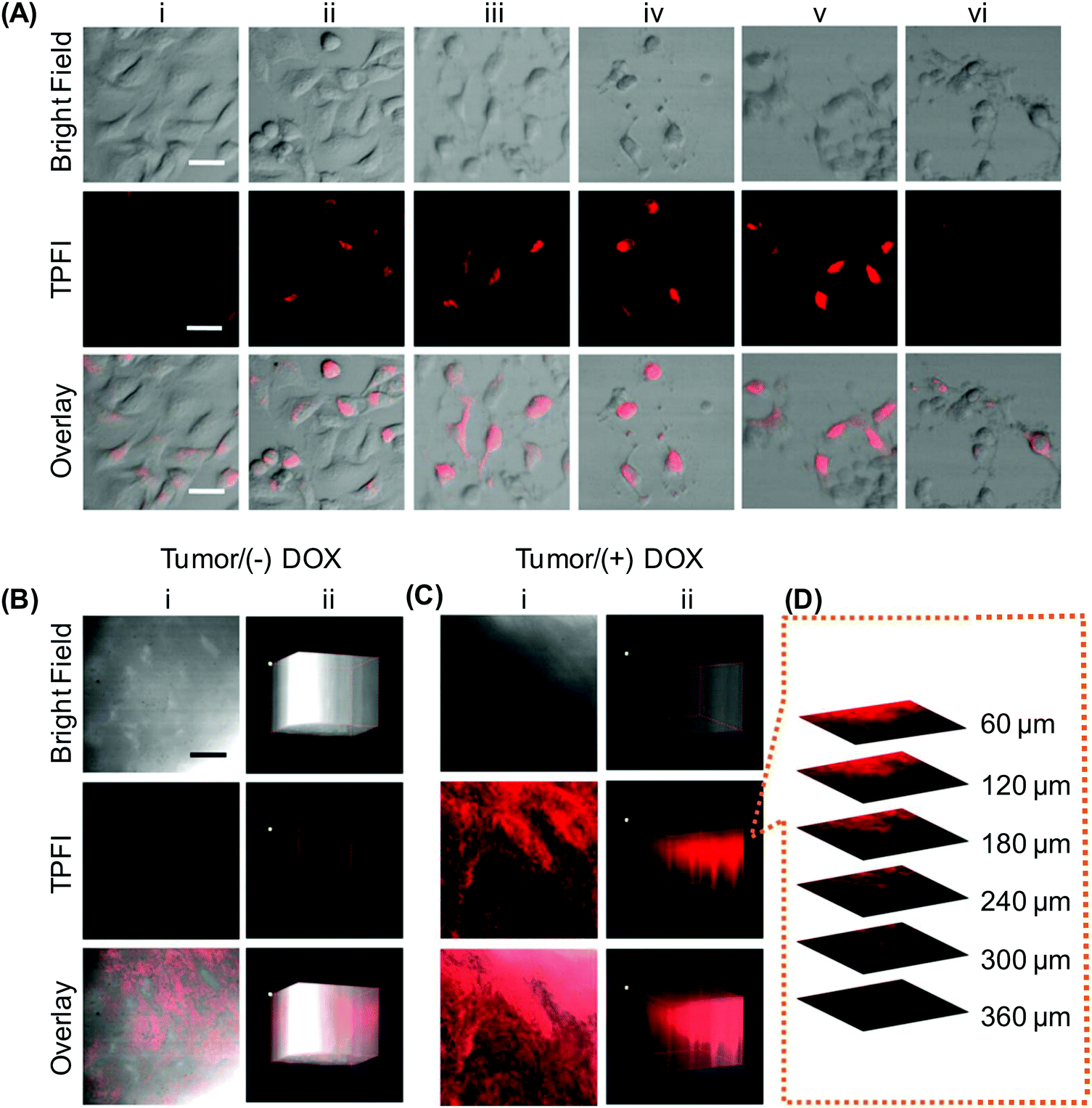 | ||
| Fig. 14 (A) The tip position modulation (TPM) images of HeLa cells treated with STS for different time periods after probe incubation: (i) 0 h, (ii) 0.5 h, (iii) 1 h, (iv) 2 h, and (v) 3 h. The TPM images of HeLa cells treated with STS and a caspase-3 inhibitor, namely Z-DEVD-FMK, for 3 h before incubation with the probe for 1 h at 37 °C were shown in (vi). (B and C) (i) The TPM images of a cervical tumour tissue slice at a depth of 120 μm, and (ii) the corresponding 3D images accumulated along the Z-direction at a depth of 60–360 μm. The slice in (C) was treated with doxorubicin (DOX) prior to probe incubation; whereas that in (B) was incubated with the probe directly without any pre-treatment. (D) Confocal Z-scan two-photon excited fluorescence imaging (TPFI) sections of a probe-incubated apoptotic tumour tissue slice at various penetration depths. The scale bar in (A) represents 30 μm; whereas those in (B) and (C) represent 200 μm (reproduced from ref. 123 with permission from American Chemical Society). | ||
Besides using epichlorohydrin as a bifunctional linker as what the design of the aforementioned nanomicelles does, a variety of other methods have been reported for preparation of polymerized CDs. Examples include radical polymerization of acryloyl CD monomers (which may also copolymerize with other monomers such as acrylic acid, N-vinylpyrrolidone, and acrylamide),151,152 and covalent linkage of mono-6-(p-tolylsulfonyl)-β-CD or CD derivatives, by nucleophilic substitution, to existing polymers such as PEI153 and poly(allylamine).154 A more detailed comparison of different categories of polymerized CDs in molecular imaging is provided in Table 4. In regard of the pros and cons of each category, a combination of both covalent and non-covalent interactions in one single system may be adopted. For instance, after the synthesis of a CD-based polymer via epichlorohydrin-mediated covalent cross-linking,114 the polymer can be functionalized with an appropriate adamantane–peptide conjugate using the self-assembly approach for the fabrication of a two-photon fluorescent probe for molecular imaging.114 Such a combination of fabrication strategies has substantially extended the versatility and flexibility in probe development, and is expected to continue to be the trend in the future.
| Category | Subclass | Chemical route | Strengths | Drawbacks |
|---|---|---|---|---|
| Covalently cross-linked polymer networks | Cross-linked CDs (or CD derivatives) | Crosslinking in alkaline media using epichlorohydrin as a bifunctional cross-linker |
• The synthetic procedure is relatively simple and straightforward
• Multiple structural components can be incorporated into the cross-linked system during the cross-linking process. This may extend the tunability of the probe properties |
• Chemical crosslinkers are involved. They may interact with the imaging element and reduce the intensity of the imaging signal.
• The rotational freedom, as well as the accessibility towards guest molecules, shown by the CD molecules may be reduced if the degree of cross-linking is too high |
| Cross-linked systems combining CDs with other polymers | Epichlorohydrin-, diepoxide-, or diisocyanate-mediated cross-linking of CDs in the presence of other water-soluble polymers, such as poly(vinyl alcohol) (PVA) or hydroxypropyl methylcellulose (HPMC). | |||
| Copolymerization of vinyl- or (meth)acryloyl-modified CD monomers with other monomers, such as N-isopropylacrylamide (NIPAAm), 2-hydroxyethyl methacrylate (HEMA), and acrylic acid. | ||||
| Reactions of CDs with the reactive end groups of the end-modified polymers | ||||
| Self-assembled polymer systems | Self-assembled particles | Self-assembly is driven by host–guest interactions between the guest molecules and CDs (or their derivatives) |
• The avoidance of using chemical cross-linkers can prevent side reactions being imposed on the imaging element present in the CD-based probe.
• The reversibility of the assembly process enables dislodging of the sensing component in a molecular probe, enabling the use of one single probe to monitor multiple molecular events |
• The physical stability of the system may be easily affected by host–guest interactions, complexation thermodynamics, or threading methods
• A proper understanding of the chemical and physical fundamentals of the fabrication process is required |
| Poly(pseudo)rotaxanes | CDs are threaded onto a polymer chain. The process is driven by van der Waals forces, hydrophobic interactions, and hydrogen bonds. |
• The approach enables fast and effective fabrication of complex molecular objects.
• By modulating the types of rings threaded on a common axle, the system can show high modularity and flexibility in composition and structure. |
||
Since the turn of the last century, advances in NIR imaging have been facilitated by the emergence of upconversion nanoparticles (UCNPs). Upconversion is a non-linear anti-Stokes process that can be achieved by excited-state absorption, energy transfer upconversion, cooperative sensitization upconversion, cross relaxation, or photon avalanche. With the use of ladder-like long-lived energy levels of lanthanide ions embedded in an inorganic matrix host, the energy of the outcome photons can be higher than that of the incident ones. Compared to the conventional fluorophores, UCNPs have several merits including deeper tissue penetration, less photobleaching and photoblinking, higher spatial resolution, lower auto-fluorescence from surrounding tissues, and less photodamage caused by the excitation light to fragile biomolecules (e.g., nucleic acids and proteins). Despite this, clinical translation of UCNP-based imaging modalities has been partly limited by a lack of simple methods to obtain uniform, biocompatible, and hydrophilic UCNPs.60 This problem has recently been tackled in multiple studies using CDs.155–157 One study has complexed β-CD with adamantaneacetic acid-capped UCNPs to enhance the hydrophilicity of the nanoparticles.156 The validity of this approach is supported by the observation that in a chloroform/water biphasic system, the adamantaneacetic acid-capped UCNPs in the organic phase show three upconversion luminescence signals under continuous-wave (CW) excitation at 980 nm.156 Two of them locate at 521 and 540 nm, and are attributed to the 4H11/2–4I15/2 and 4S3/2–4I15/2 transitions, respectively.156 One signal is detected at 654 nm, and is assigned to the 4F9/2–4I15/2 transition.156 Upon addition of β-CD, the UCNPs undergo phase transfer, rendering the upconversion luminescence signals detectable in the aqueous layer. As revealed by transmission electron microscopy (TEM), the nanoparticles retain their spherical morphology before and after host–guest binding with β-CD. No significant aggregation or change in the crystalline lattice is observed.156 In the in vitro context, cells internalizing the UCNPs show intense upconversion luminescence emission. With further incorporation of the molecular recognition capacity and target specificity, the nanoparticles may have potential to be developed as a deep-tissue imaging probe to interrogate disease-linked molecular and biochemical events.
Lately, β-CD derivatives have also been used to modify the surface of oleic acid (OA)-capped NaYF4:Yb,Er UCNPs to produce water-dispensable nanoparticles.60 During the process, OA-capped UCNPs first undergo a ligand exchange process,60 in which 2-azidoethylphosphonic acid ligands are adopted to replace the original OA ligands. This process is thought to be driven by the higher affinity of the phosphate of the azidoethylphosphonate ligand to the lanthanide ions on the nanoparticle surface.158,159 The azide of the azidoethylphosphonate ligand provides a site for subsequent conjugation of 6-propargylamino-6-deoxy-β-CD to UCNPs via the cycloaddition “click” reaction.60 From the chemical point of view, incorporation of CD moieties into the nanoparticles on one hand enhances the stability, hydrophilicity, and biocompatibility of the UCNPs,60 and on the other hand provides a convenient way to incorporate other functional elements.60 Emission from the nanoparticles is predominately green in colour under CW excitation at 980 nm, due to green light emission from the doped Er3+ ions. Upon incorporation of cyclic RGD-conjugated adamantane (Ad-RGD) onto the UCNP surface via CD-based inclusion complexation, the nanoparticles successfully mediate targeted cellular imaging.60 At the tissue level, luminescence signals are observed only in cervical tumour tissue slices but not in normal cervical tissues.60 This points to the excellent capability of the UCNPs for both targeted live cell imaging and deep-tissue imaging. Despite the prospect of mediating molecular imaging by UCNPs, 980 nm laser irradiation has been used as the major source of excitation to date because this wavelength matches with the absorption of the sensitizer (Yb3+). Light at this wavelength, however, can be absorbed by water, producing heat that can cause damage to biological tissues. Over the years, numerous advances have been underway to modulate the excitation wavelength for UCNPs,160,161 with the excitation wavelength being successfully blue-shifted by sensitizing Yb3+ using Nd3+ as the second sensitizer. If the excitation wavelength can be tuned to the one that is transparent to tissues, clinical translation of CD-based molecular imaging mediated by UCNPs is anticipated to be substantially facilitated.
5.2 Probes for magnetic resonance molecular imaging
An alternative to NIR molecular imaging for deep-tissue visualization is magnetic resonance imaging (MRI), which enables rapid and precise diagnostic imaging of soft tissues. The main challenges to this type of imaging arise from the low magnetic signal intensity and low sensitivity. These challenges, however, seem hard to sustain because of the possibility of using contrast agents to enhance signal differences. This has been further contributed by recent advances in CD chemistry, which enables CDs to be applied as polyfunctionalizable scaffolds to bear diethylenetriaminepentaacetic acid (DTPA) or 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) units,162–165 forming paramagnetic complexes to shorten the relaxation time of the surrounding water protons and to boost the image contrast.166,167 Over the years, diverse CD-based MRI contrast agents have been reported.168–173 For instance, CD-based complexes possessing three to seven Gd(III) chelates have been generated by click reactions.174 Due to the formation of a triazole ring linkage during the robust reaction between heptakis-6-azido-6-deoxy-β-CD and the DOTA-Gd-alkyne derivative, the local rotation of the Gd(III) complexes is hindered by the rigid nature of the triazole linker, resulting in high molecular relaxivities.174 The use of multifunctionalized CDs in MRI has also been reported by Idriss et al.,175 who have generated a contrast agent based on β-CD that bears seven carboxylate functions on the primary side. The agent has been shown to have a positive effect on the MRI signal due to the effect of the secondary hydration sphere.175More recently, head-to-head [3]rotaxanes of α-CD selectively functionalized with gadolinium 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid monoamide (DO3A-MA) complexes have been evaluated for MRI enhancement.21 Generation of these rotaxanes adopts copper-catalyzed azide–alkyne cycloaddition as a stoppering reaction (Fig. 15). Before synthesis, α-CD molecules are first mono- or di-functionalized with the gadolinium DO3A-MA complex. The functionalized molecules are then mixed with the diazidododecane axle (designated as 3) in water to generate a pseudorotaxane via hydrophobic interactions.21 Finally, the end-capping reaction is performed using the cycloaddition reaction.21 Owing to its large size, dicarboxylic acid (designated as 5) is used to prevent the dethreading of α-CD molecules.176 The availability of carboxylate groups can also elevate the aqueous solubility of the rotaxane at physiological pH for in vivo applications.21 The reaction entails regioselective deprotection of perbenzylated CDs,177 and click reactions between propargyl-functionalized DO3A-MA and azido CDs.178–182In vitro and in vivo studies have corroborated that the [3]rotaxanes lead to higher contrast and retention in the kidney than the gadolinium-DOTA (Gd-DOTA) complex.21 This suggests that CDs enhance the relaxation properties and biological properties of the gadolinium complex for MRI. Apart from this, CDs can modulate the relaxivity value via host–guest coupling. This has been reported by Cravotto and colleagues,183 who have confirmed the correlation between relaxivity and host–guest interactions by studying the complexation capacity of bridged CD dimers and trimers in the presence of DTPA functionalized with cyclohexyl groups.183 This correlation has later been observed in three CD derivatives that have been designated as 1(Gd), 2(Gd) and 3(Gd) (Fig. 16).184 Contrary to 1(Gd), the secondary sides of 2(Gd) and 3(Gd) are permethylated. 3(Gd) also possesses a flexible pendant introduced into the secondary side via an ether function at the C-2 position. As revealed by the differences in the inclusion complexation capacity of the derivatives, relaxivity can be enhanced by self-inclusion complex formation.184
 | ||
| Fig. 15 The chemical routes for the synthesis of head-to-head CD [3]rotaxanes functionalized with (A) one or (B) two Gd-DO3A-MA complexes (abbreviations: rt, room temperature; PMDETA, N,N,N′,N′,N′′-pentamethyldiethylenetriamine) (reproduced from ref. 21 with permission from American Chemical Society). | ||
 | ||
| Fig. 16 Structures of the CD derivatives: (A) 1(Gd), (B) 2(Gd) and (C) 3(Gd). | ||
Although many of the reported gadolinium complexes can result in MRI enhancement, most of them are not specific to biomarkers. They are by no means as robust as are generally believed when they are applied to high-resolution molecular imaging with MRI. To surmount this obstacle, one possible solution is to use carriers to deliver a larger amount of Gd(III) chelates to the target site. Examples of carriers developed in the literature include polymeric micelles,185 mesoporous silica nanoparticles,186 dendrimers,187 and gold nanoparticles.188 Using this approach may elevate the relaxivity and signal intensity;187,189,190 however, the low excretion rate of many of these carriers raises safety concerns.191 A proposal with widespread support to address these concerns is to carry the chelates using CDs or their derivatives, which on one hand have good biocompatibility and on the other hand enable reversible host–guest interactions to facilitate the excretion of the contrast agent.7,192 This concept has been brought forward by Lu and colleagues, who have fabricated a CD-based carrier by first conjugating β-CD molecules to polyhedral silsesquioxane (POSS).193 Inclusion complexation of β-CD with adamantane-modified cyclic RGDfK peptide is then carried out to target the carrier to αvβ3 intergrins,193 which are often overexpressed in tumour vascular endothelial cells and in cells of cancers such as mammary carcinoma.194–197 After that, a macrocyclic Gd(III) chelate and cyanine 5 (Cy5) are incorporated into the carrier via host–guest chemistry to generate a contrast agent (Fig. 17A).193 Upon inclusion of the Gd(III) chelate into the CD cavity, the rotational motion of the chelate can be slowed down. This increases the relaxivity value.193 In mice bearing 4T1-GFP-Luc2 flank tumours, the contrast agent leads to strong and prolonged contrast enhancement in tumour tissues, as compared to the non-targeting counterpart and ProHance®.193 The targeting capacity of the agent has been substantiated by high-resolution fluorescence imaging, with which intense Cy5 fluorescence has been detected in tumour tissues from mice injected with the contrast agent.193 On the contrary, no significant fluorescence emission has been detected in tissues obtained from those injected with the non-targeting control (Fig. 17B).193 The excellent performance depicted above illustrates the power of harnessing both CD chemistry and ligand conjugation chemistry in the development of contrast agents for high-resolution magnetic resonance molecular imaging.
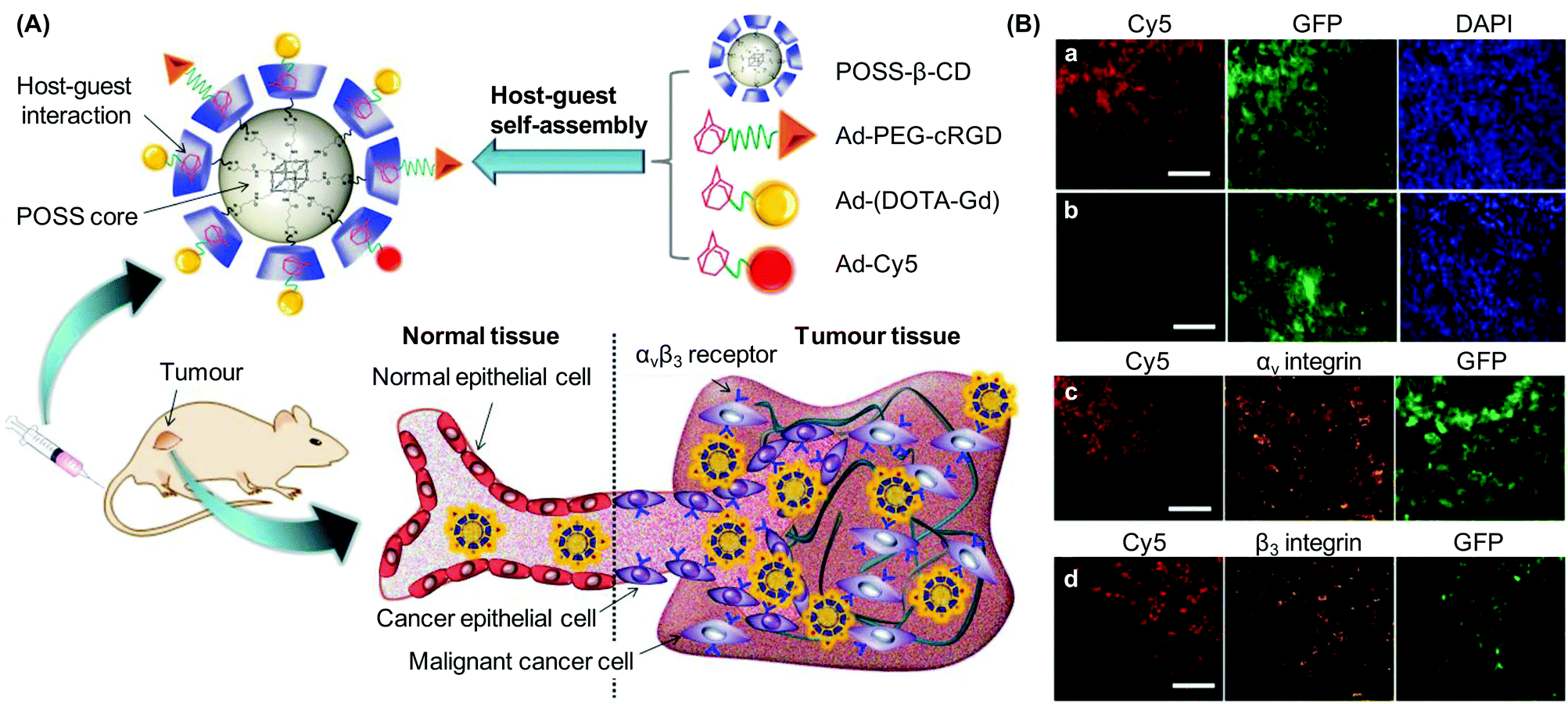 | ||
| Fig. 17 (A) A contrast agent self-assembled from multiple components via CD-based inclusion complexation: POSS-β-CD, adamantane-modified cyclic RGDfK peptide (Ad-PEG-cRGD), 2,2′,2′′-(10-(2-(1-adamantylamino)-2-oxoethyl)-1,4,7,10-tetraazacyclo-dodecane-1,4,7-triyl)triacetate gadolinium(III) (Ad-(DOTA-Gd)), and adamantyl sulfo-cyanine 5 (Ad-Cy5). This agent can preferentially bind to αvβ3 receptors on tumour vascular endothelial cells and malignant breast cancer cells, enhancing MRI signals in tumour tissues. (B) Fluorescence images of 4T1-luc2-GFP tumour histological sections obtained from mice injected with the (a) molecularly targeted contrast agent and the (b) non-targeting control agent. Sections obtained from mice injected with the molecularly targeted agent have been further stained for (c) αv and (d) β3 integrins. The tumour slices have been imaged using confocal microscopy. The scale bar represents 100 μm (reproduced from ref. 193 with permission from Elsevier). | ||
Although CD-based carriers can enhance the target specificity of gadolinium complexes, the risk of nephrogenic systemic fibrosis caused by the complexes still cannot be eliminated.198 This propels a search of alternative approaches for T1-weighted imaging. One of these approaches is to employ non-lanthanide metals, in particular manganese, to replace gadolinium. Due to its high electronic spins (5/2) and long electronic relaxation time, along with its role in cells as an essential metal element and an enzyme cofactor, Mn(II) has great potential to be developed as an emerging contrast agent.199,200 The physicochemical properties of the Mn-based contrast agent can be further improved upon chelation.198 Compared to Mn(II) dipyridoxal diphosphate, which is a clinically approved Mn-based contrast agent for liver imaging, Mn porphyrins have higher transmembrane permeability.201–203 Upon chelation to porphyrins, the bivalent state of manganese, however, rapidly undergoes oxidation to Mn(III). The electronic spin also decreases from 5/2 to 4/2, reducing the MRI efficiency of the agent.204 This problem can be circumvented via inclusion complexation with bridged bis-CDs, whose cavity can stabilize the bivalent state of Mn in porphyrins, to form a supramolecular construct.61 Results have shown that the relaxivity value r1 of the construct is approximately 4 times higher than that of the commercially available Gd-based contrast agent.61 Upon injection of the construct into mice, strong contrast enhancement in blood, kidney, and urinary bladder has been observed in the time-dependent 2D coronal images (Fig. 10A).61 This reveals that the construct undergoes rapid circulation in the bloodstream, and can be excreted via renal infiltration. Meanwhile, very low signal enhancement has been detected in the liver, implying that liver accumulation and hence hepatotoxicity of the construct are minimal.61 This study has opened new possibilities for CD-based Mn(II) probes to map physiopathological changes at the organ level and to become promising alternatives to the Gd(III) counterparts in future MRI.
MRI in molecular imaging cannot only image target tissues, but can also map hemodynamic parameters over a large volume of interest. One of the important hemodynamic parameters is the blood volume fraction (BVf).205 Conventionally, measurement of cerebral BVf using MRI necessitates intravenous injection of a blood-pool contrast agent, which has to be confined to the intravascular space in order to get accurate measurements. Such confinement is not achievable in high-grade glioma tissues, whose leaky vasculature causes systematic measurement errors.206 A means of addressing this problem has lately been proposed by Lahrech and co-workers, who have generated an inclusion complex of Gd(III) ions with hexakis (2-O-carboxymethyl-3,6-anhydro)-α-CD.207 The complex has the shape of a flat disc with a hydrophilic cavity for binding several kinds of metal cations.208 It has been used to quantify, by using the rapid steady-state T1 method, the cerebral BVf in the presence of blood–brain barrier lesions in a C6 glioma model.207 Analysis of the T1-weighted images has indicated that extravasation of the complex into the tumour tissue is not significaant.207 Based on the results of the BVf measurements, while both the complex and the contrast agent Gd-DOTA can be used to quantify cerebral BVf in healthy brain tissues and in the contralateral hemisphere of the tumour-bearing rat; only the complex can be applicable to cerebral BVf quantification in the tumour region.207 This work has signified the versatility of CDs and their derivatives in extending the applicability of magnetic resonance molecular imaging.
5.3 Probes for ionizing radiation-based molecular imaging
The third most commonly used modality for molecular imaging is radionuclide molecular imaging, which is a form of diagnostic imaging based on ionizing radiation. The signalling component in radionuclide molecular imaging can be introduced into a probe by small isotopic substitution (e.g., substituting 12C with 11C), nonisotopic substitution (e.g., substituting 1H with 18F), or incorporation with larger isotopes (e.g., 99mTc and 64Cu).209–211 Compared to luminescence-based imaging and MRI in which the probe is usually administered in a large quantity (e.g., in a range from μm to mg), a low dose is sufficient for most of the radiolabelled probes to function.127 Probe radiolabelling is often performed using γ-emitting isotopes (e.g., 131I, 111In, and 99mTc), whose signals can be detected by SPECT to produce tomographic images.210,211 Probe development for SPECT can be facilitated by CD chemistry. This has been documented in an earlier report, in which CDs are incorporated into poly(anhydride) nanoparticles that are subsequently radiolabelled with 99mTc.212 Compared to the native poly(anhydride) nanoparticles, those incorporated with CDs interact more effectively with the gastrointestinal mucosa.212 This is attributed to the higher content of hydroxyl groups upon CD incorporation, which enhances the formation of hydrogen bonds between the nanoparticles and the mucosa components to increase the efficiency of bioadhesion.212 To facilitate further development of CD-based SPECT, emergence of more sophisticated methods to manipulate the biological half-life of CD-based probes is required. If a probe can be designed in a way that it can be eliminated in a timeframe shorter than the half-life of the radionuclide adopted, this can ensure more complete removal of non-specific signals within the imaging timeframe and can strengthen the diagnostic power of CD-based SPECT.An alternative to γ-emitting isotopes in radionuclide molecular imaging is positron-emitting isotopes, which produce positrons that can combine with nearby electrons to produce photons of γ-ray radiation.213,214 Commonly used isotopes include 11C, 15O, 13N, and 18F, although positron emitters such as 14O, 64Cu, 62Cu, 68Ga, 76Br, 82Rb, and 124I have also been used. Detection of signals from positron-emitting isotopes is often achieved by positron emission tomography (PET). Compared to other modalities such as γ scintigraphy, PET has the merits of deeper tissue penetration and superior spatial resolution.2 Due to its higher detection efficiency and sensitivity, PET is also recognized as a more robust imaging tool than SPECT for molecular imaging.210,211,213 Over the years, different PET tracers have been reported as clinical research tools and diagnostic agents. For instance, 2-[18F]fluoro-2-deoxyglucose (FDG), synthesized via either electrophilic or nucleophilic radiofluorination,215,216 is a tracer for imaging malignancies.217 [18F]-1-Deoxy-1-fluoro-scyllo-inositol is also a PET imaging agent for examining breast cancer and Alzheimer's disease.218 Other examples include [18F]fluorothymidine (FLT) for examining cell proliferation,217 and [11C]raclopride for examining Huntington's disease, Parkinson's disease, and schizophrenia.219 More recently, the efficiency of PET has been boosted by the integration of CD chemistry into tracer development. This is exemplified by the work of Davis’ team,220 which has conjugated DOTA to the 5′ end of an siRNA molecule, followed by 64Cu labelling. The labelled siRNA molecule can undergo electrostatic interactions with CD-containing polycations for formation of nanoparticles,221 whose surface can be functionalized with various adamantane-containing molecules (such as adamantane-modified PEG for steric stabilization, and adamantane-modified PEG-linked transferrin for cell-specific targeting).221 After intravenous administration of the nanoparticles to mice, the tumour localization of 64Cu-labeled molecules can be determined using micro-PET. Results have shown that targeting with transferrin successfully reduces nonspecific interactions,220 allowing more 64Cu-labelled molecules to be internalized by tumour cells.220 Taken together, the integration of CD chemistry into the design of PET tracers has provided a route for easy functionalization of the tracer, and has enhanced the imaging efficiency and quality.
Here it is worth noting that although CD-based PET tracers can help to interrogate the biodistribution kinetics and molecular events,222,223 more advances in radiochemistry are required before the flexibility in the design and construction of those tracers can be extended. At this moment, applications of CD-based PET are largely limited by the comparatively short half-lives of some commonly used PET isotopes such as 18F and 11C. As synthesis and purification of isotopically labelled CD-based molecular tracers are expected to be in a timeframe that is no more than two to three times the half-life of the radionuclide adopted,224 this makes the radiolabelling protocols technically challenging, not to mention the need of an on-site cyclotron. Overcoming these challenges is underway, with diverse strategies (such as microwave heating225 and microfluidic technologies226,227) proposed to escalate the efficiency of the tracer generation process. Unfortunately, all these achievements still fail to completely eradicate the above-mentioned limitations of radiochemistry. In addition, careful planning of the imaging procedure is required when multiple molecular events are to be examined using PET. This is because the γ-ray photons produced from different positron-emitting isotopes are of the same energy. Even if two CD-based tracers, each with a separate isotope for a separate molecular event, are administered, signals from the two tracers will not be distinguishable during PET measurements. To circumvent this problem, the two tracers have to be administered at different time points. Alternatively, SPECT rather than PET has to be used, because emission of photons with distinct energies is possible when using γ-emitting isotopes.
Apart from γ-ray radiation and positron emission, X-rays can mediate the execution of ionizing radiation-based molecular imaging. An example of this is computed tomography (CT), whose working principle is based on the detection of differential attenuation of X-rays in different component tissues. To elevate the soft tissue contrast in CT, administration of contrast agents containing elements with high atomic numbers (e.g., I, Au, Bi, Dy, or Ta) is needed.228,229 In an earlier study, polymer–lipid nanoparticles incorporated with hydroxypropyl-β-CD (HP-β-CD) have been assembled using microfluidic chips,230 and have enabled CT to locate the tumour site.230 More recently, by taking advantage of Dy which has a high X-ray mass attenuation coefficient (3.36 cm2 g−1 at 100 keV) and a comparatively large K-edge energy value (53.8 keV),231 a CT probe has been generated from OA-coated NaDyF4 nanoparticles.232 The nanoparticles self-assemble with α-CD via inclusion complexation between α-CD and OA for hydrophilic conversion, before further modification with other functionalities. At a mass concentration of 10 mg mL−1, the Hounsfield unit (HU) offered by the CD-based probe has been found to be much higher than that achieved by NaGdF4 nanoparticles.232,233 After intratumoral administration of the probe, significant tumour signals can be observed in the 3D CT volume-rendered images (Fig. 10B),232 with the HU increasing from 109 for the control (to which no contrast agent has been administered) to 212 after contrast enhancement.232 In spite of this promising performance, so far the tumour specificity of most of the CD-based CT probes is achieved either by intratumoral injection or by passive targeting via the enhanced permeation and retention (EPR) effect. Incorporation of more active targeting components may maximize the transferability of these probes to interrogate molecular events linked to cancers.
5.4 Probes for ultrasound-based molecular imaging
Other than the modalities based on luminescence, magnetic resonance and ionizing radiation, molecular imaging can be performed based on the evaluation of reflected echoes.234 Compared to PET or SPECT, ultrasound-based molecular imaging is a cost-effective, non-invasive and reliable quantitative technique that has the merits of higher sensitivity and better spatiotemporal resolution. During the process of image collection, a transducer is first placed against the skin to emit high-frequency (>20 kHz) sound waves, which are reflected back from the internal organs under examination.235 Variations in imaging algorithms, sound attenuation effects, backscattering coefficients, and sound speeds can affect the contrast in the image collected.236,237 One of the most widely used contrast agents is acoustically active microbubbles, whose development has rapidly advanced since the first report of enhanced ultrasound reflections in the aortic root after injection of air bubbles in 1969.238 These microbubbles consist of a gas core stabilized by a shell designed to reduce gas dissolution in blood.236 The flexibility in their structural design can be remarkably increased by techniques of CD chemistry. This is illustrated by the development of CD-coated poly(vinyl alcohol) (PVA)-based microbubbles, whose surface functionalization is achieved by the reaction of acetalization between the carbonyl ends of the telechelic PVA chains projecting from the microbubble surface and the hydroxyl groups of β-CD.239 With the inclusion complexation capacity of CDs, these microbubbles can be incorporated with targeting ligands either during or after microbubble synthesis, and be developed into molecularly targeted microbubbles for recognition of specific cell-surface receptors. Antibodies have been one of the extensively used ligands for this purpose. The current trend, however, is emphasizing on the use of small-molecule compounds as ligands, which can reduce the immunogenicity and display better binding kinetics in vessels with high shear forces.For future research, optimization of microbubble adhesion to target sites is highly desirable as it can increase the signal-to-noise ratio of imaging mediated by CD-based contrast agents. Development of new detection algorithms, which can better model changes in acoustic properties of microbubbles during target binding, can also help to improve the imaging quality.240,241 Microbubbles used as contrast agents are typically in the size range of 1–5 μm.236 They can be retained effectively within the vascular compartment upon administration to living bodies, thereby minimizing background signals by preventing unspecific accumulation of the microbubbles in the interstitial space. This renders CD-based ultrasound-based imaging inherently advantageous when pathological processes characterized by a differential expression of molecules in the vasculature are monitored.236 However, also because of the lack of extravasation from the vasculature, applications of molecularly targeted microbubbles are largely limited to cases where intravascular targets are involved. Although more understanding of the molecular pathophysiology of different diseases may enable identification of more disease-specific targets and may widen the clinical spectrum of applications of CD-based ultrasound molecular imaging, the physical confinement of the microbubbles within the vasculature is one of the challenges to be tackled when the diagnostic option of this imaging modality is to be extended.
Finally, until now works on ultrasound-based molecular imaging have been conducted predominately in 2D, with only a small anatomical region being sampled in most of the time.242 2D imaging sometimes, however, may not be sufficient when certain pathological processes are monitored. One example lies in the case of tumour growth monitoring, which may have errors when 2D imaging is implemented due to the heterogeneity of tumours with regard to vascularity, neoangiogenesis, as well as hemorrhagic and necrotic manifestations. This problem has been documented by Wang et al., who have discerned that the anti-angiogenic treatment effects in human colon cancer xenograft models are overestimated when 2D imaging is adopted.243 This demonstrates the practical need of achieving ultrasound-based molecular imaging in 3D. This need can potentially be fulfilled by the recent emergence of a ultrasound-based modality, namely photoacoustic imaging. During the imaging process, the photoacoustic tracer in the target tissue is first excited by laser pulses at a specific wavelength. The excited tracer then undergoes thermo-elastic expansion to release ultrasonic pressure waves which can be detected using a photoacoustic receiver.244 This modality provides 2D information, which can be employed to construct 3D representations of the tissue being examined.245 Various CD-incorporated probes have been reported for photoacoustic imaging to date. One example is CD-conjugated ICG. The use of the CD is to enhance the uptake of the probe by bacteria so as to image the site of Staphylococcus aureus infection in a mouse model of prosthetic joint infection.246 Compared to using ICG alone, the one incorporated with β-CD has led to a higher signal percentage in the region of interest.246 A parallel example is the OA-stabilized NaYF4:Yb3+,Er3+ UCNPs surface-modified with α-CD.247 Under 980 nm laser excitation, the presence of α-CD molecules in the modified nanoparticles leads to luminescence quenching, partly due to the solvent-induced non-radiative relaxation, which results in thermal expansion and hence enhancement of photoacoustic signals.247 The CD moieties can also provide a site for easy functionalization of the nanoparticles to target specific biomarkers.247 This, along with the sharp emission bandwidth and high photostability,247 has made the CD-modified UCNPs a promising candidate for use in photoacoustic molecular imaging.
5.5 Probes for multimodal molecular imaging and theranostics
The combination of multiple modalities of molecular imaging can have synergistic advantages over a single modality by offering complementary information. Advances in CD chemistry have enabled multifunctionalization, making the integration of multiple functional moieties and even therapeutic components into one entity feasible. This has been demonstrated by Fredy et al., who have synthesized a polyrotaxane having poly((N,N-dimethylammonio)undecamethylene chloride) as the polymeric thread and CD molecules as threading devices.101 Those CD molecules have been pre-functionalized with bodipy fluorescent tags or Gd(III) complexes before the threading process.101 UV/Vis and fluorescence spectroscopic measurements have confirmed that threading the CD molecules onto the polymer chain does not affect the fluorescence properties of the bodipy tag in luminescence-based imaging.101 Compared to Gd-DOTA, the relaxivity of the Gd-bearing polyrotaxane is approximately five-fold higher.101 Upon incorporation of targeting ligands, this polyrotaxane has potential to be applied to bimodal molecular imaging.Another example of CD-based multimodal probes is the G5F2C contrast agent.248 It is generated by first incubating fluorescein isothiocyanate (FITC) in DMSO with per-6-amino-β-CD to generate a fluorescein-labeled CD core, which forms a conjugate upon incubation with 1,4,7,10-tetraazacyclododecane-4,7,10-triacetice-1-{methyl[(4-isothiocyanatophenyl)methyl]phosphinic acid} (DO3APNCS). The conjugate subsequently complexes with Gd(III) ions to obtain a bimodal fluorescence/MRI contrast agent (Fig. 18).248 Due to fast water exchange mediated by the chosen macrocyclic ligand, and the slow rotational dynamics led by the rigid CD scaffold, the probe displays high relaxivity. At 20 MHz and 25 °C, the relaxivity of the probe is as high as 150 s−1 mM−1 per molecule.248 At a high imaging field (200 MHz), the relaxivity of the probe reaches 60–80 s−1 mM−1 per molecule, and this is much higher than that of Gd-DOTA (3.6 s−1 mM−1) under the same conditions.248 During imaging of pancreatic islets, the probe has a negligible influence on the insulin producing capacity of the islets.248 After probe administration, individual islets can be visualized clearly using MRI.248 The islets have also been examined by fluorescence imaging, which has revealed that the probe tends to be taken up by macrophages, α-cells, and β-cells.248 In addition to imaging pancreatic islets, the probe enables imaging of mesenchymal stem cells (MSCs). Fluorescence in the cells can be detected even 24 hours after probe administration.248 This, along with the low toxicity of the probe,248 renders the probe applicable to multimodal molecular imaging in the biological context and to stem cell tracking, which is a cornerstone of the future development of stem cell therapy.249,250
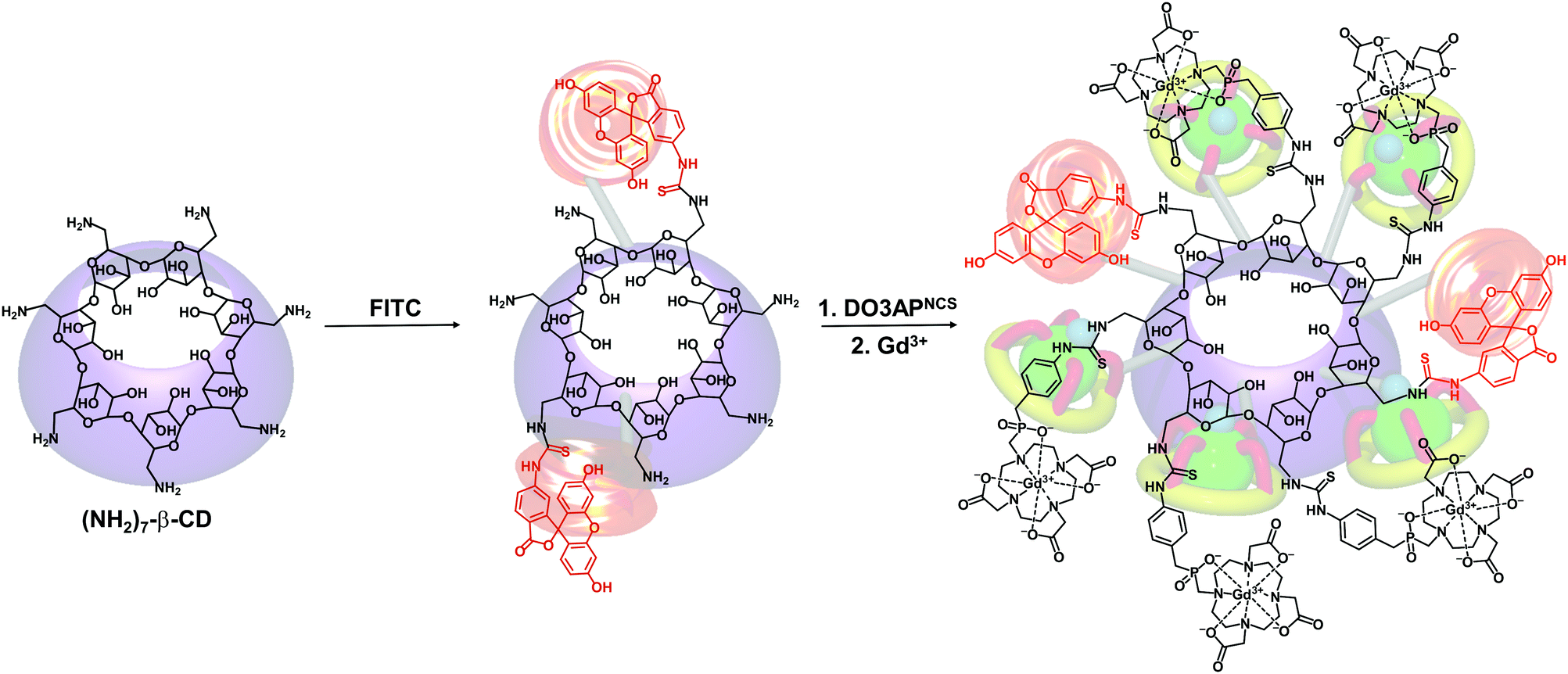 | ||
| Fig. 18 Generation of the G5F2C bimodal contrast agent. Water molecules, that are coordinated to Gd(III) ions, are omitted in this figure for the sake of clarity (reproduced from ref. 248 with permission from John Wiley & Sons, Inc.). | ||
In recent years, the versatility in probe development has been increased by the combination of CD chemistry and UCNP technologies. This has been demonstrated in a previous study, in which a multimodal probe has been developed based on CD-modified OA-capped UCNPs (Fig. 19).157 With laser scanning upconversion luminescence microscopy, the nanoparticles have been shown to be internalized by both cancer cells and normal cells. Based on the overlays of confocal luminescence and bright field images, the upconversion luminescence has been confirmed to be emitted from the intracellular region.157 After loading the osmium(II) complex into the nanoparticles, the complex enters the cells successfully, giving fluorescence signals for dual labelling of living cells.157 Tail vein injection of the nanoparticles into mice leads to nanoparticle accumulation in the spleen and liver; whereas intradermal administration of the nanoparticles to the claw results in intense upconversion luminescence emission in the lymphatic drainage basins of the oxter (Fig. 10C). Apart from enabling luminescence-based live cell imaging and lymph monitoring,157 the nanoparticles can function as a PET probe after 18F incorporation.157 Upon intravenous administration of the 18F-incorporated nanoparticles, intense radioactive signals have been detected in the liver and spleen.157 Although the target specificity of the nanoparticles relies principally on passive targeting to the spleen and liver, by taking advantage of the inclusion complexation capacity of CDs, the targeting mode can be easily converted from passive to active by incorporation of ligands, antibodies, or other functionalities that can interact with specific cell-surface receptors.251
 | ||
| Fig. 19 TEM images of (i) UCNP-OAT, (ii) UCNP-OA-CDT, (iv) UCNP-OAS, (v) UCNP-OA-CDS, (vii) UCNP-OAH, and (viii) UCNP-OA-CDH; high-resolution transmission electron microscopy (HRTEM) images of (iii) UCNP-OAT, (vi) UCNP-OAS, and (ix) UCNP-OAH. The methods of UCNP generation are denoted by superscripts, in which T, S, and H represent thermal decomposition, solvothermal synthesis, and hydrothermal synthesis, respectively (reproduced from ref. 157 with permission from Elsevier). | ||
Till now our discussions on CD-based probes have been confined to the context of imaging alone, but a vista of new opportunities may be opened up in theranostics if the therapeutics delivery capacity of CDs is integrated into probe design. Good examples of CD-based theranostic probes are the two derivatives formed by conjugating cyanine dyes with CDs (Fig. 20).97 Apart from their use in luminescence-based imaging, they can function as drug carriers due to the capacity of the CD moiety to form inclusion complexes with drugs. Another example is the one reported by Zhang and colleagues, who have first loaded resveratrol into acetylated β-CD nanoparticles, followed by encapsulation of the nanoparticles into microbubbles for both drug delivery and ultrasound imaging purposes.252 Lately, nanocomposites generated from functionalized CDs have also been reported for theranostic use.62 During the synthetic process, amino-modified magnetic Fe3O4 nanoparticles (MNPs) and carboxymethyl-β-CD are first prepared.62 Afterwards, carboxymethyl-β-CD is coupled with the 3-aminopropyltriethoxysilane (APTES) moiety of the nanoparticle through the amide bond under EDC/HOSu conditions (Fig. 21). A similar mechanism is adopted to functionalize MNPs with HP-β-CD and sulfobutyl ether-β-CD (SBE-β-CD).62 Results have shown that the drug loading efficiency of these nanocomposites is around 30–40%. After cellular internalization of the nanocomposites, fluorescence emission from the cells can be observed.62 Together with their magnetic properties and drug release sustainability, the nanocomposites are worth optimization and development for pharmaceutical intervention and live cell imaging.62
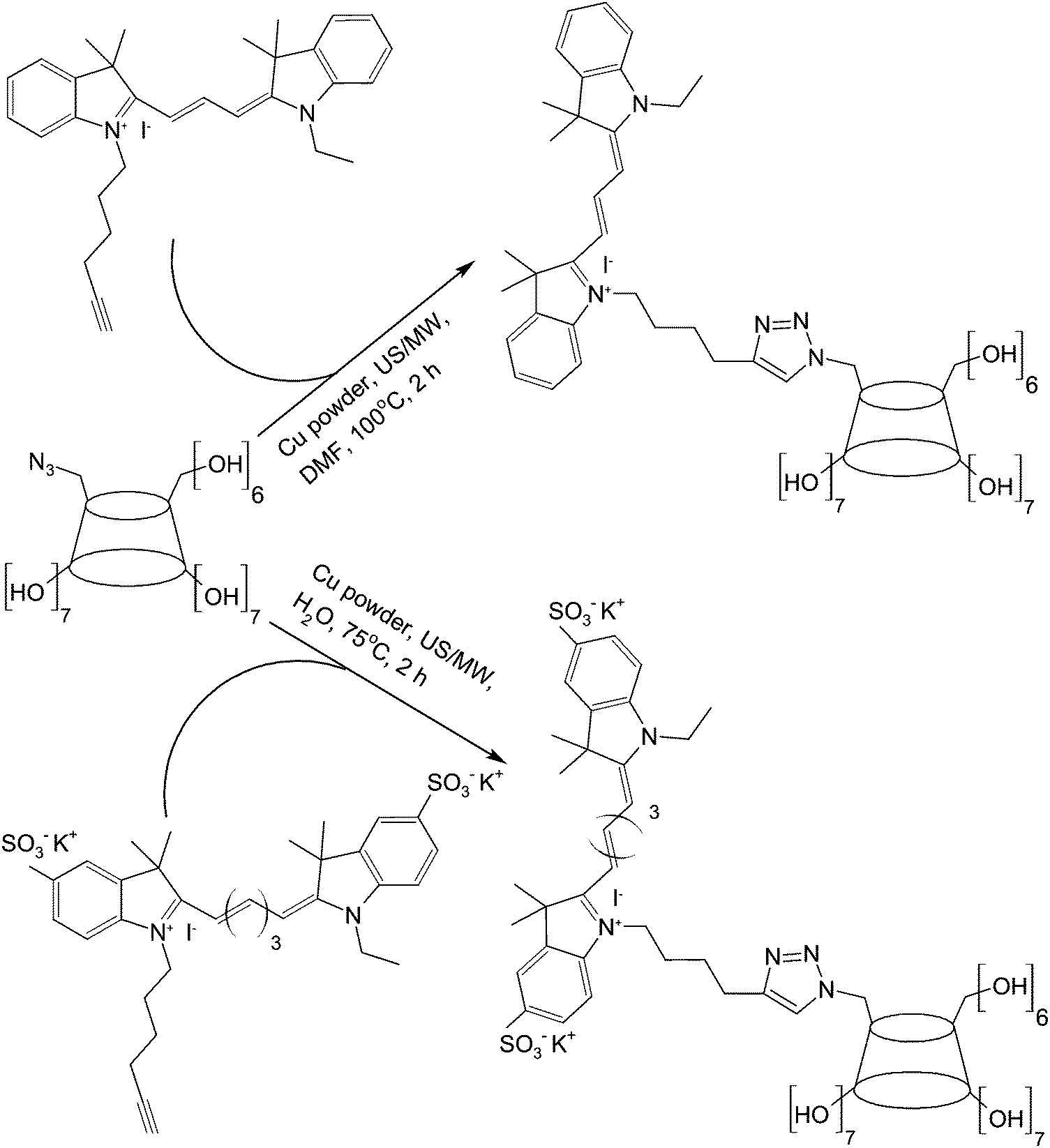 | ||
| Fig. 20 The chemical routes for the synthesis of the two cyanine dye/β-CD derivatives under simultaneous ultrasound/microwave irradiation (US/MW). | ||
 | ||
| Fig. 21 The chemical routes for the functionalization of MNPs with carboxymethyl-β-CD, HP-β-CD, and SBE-β-CD. | ||
Apart from the aforementioned examples, recently starlike polysaccharides with dextran arms have been reported as a dual-functional probe for MRI and drug delivery.253 The probe is synthesized from heptakis-6-azido-6-deoxy-β-CD, which possesses seven azide groups on the hydrophobic side of the β-CD cylinder. Dextran is attached to the CD core through click chemistry, followed by modification of dextran with stearic acid to obtain an amphiphilic polymer that can self-assemble into micelles.253 The micelles can be loaded with anticancer drugs for chemotherapy or with superparamagnetic iron oxide nanoparticles to give high T2 relaxivity.253 Although the target specificity of the micelles is limited at the moment, this problem can be easily solved by surface-functionalization. Once the target specificity is enhanced, these micelles will have potential to enable quantitative therapy under visualization of drug distribution in target regions. In addition to small-molecule drugs, nucleic acids can be delivered using CD-based probes. This has been demonstrated using a β-CD-based star copolymer,254 which consists of an asymmetrically functionalized β-CD core, poly(N,N-dimethylaminoethyl methacrylate) (PDMA) arms, and covalently conjugated Gd-DOTA complexes. The copolymer is synthesized by azide–alkyne Huisgen cycloaddition, in which atom transfer radical polymerization (ATRP) of N,N-dimethylaminoethyl methacrylate (DMA) on a β-CD derivative occurs (Fig. 22). That derivative contains 7 azide moieties at the upper rim of the rigid toroidal β-CD core for click reactions and 14 α-bromopropionate functionalities at the lower rim for ATRP initiation.254 The availability of the polycationic arms in the resulting star copolymer enables electrostatic interactions between the copolymer and plasmids. Meanwhile, the presence of Gd-DOTA complexes renders the copolymer capable of T1-weighted MRI contrast enhancement. In the same Gd(III) ion concentration range, the star copolymer is more effective in contrast enhancement than the alkynyl-DOTA-Gd complex,254 with the value of r1 achieved by the former being around 3.5 times as much as that achieved by the latter. Such enhanced T1 relaxivity is attributed to the restricted mobility of the Gd-DOTA complexes after incorporation into the star copolymer.254 Other contributing factors include the lengthening of the rotational correlation time for each Gd(III) ion, and the increase in the local concentration of the Gd-DOTA complexes.
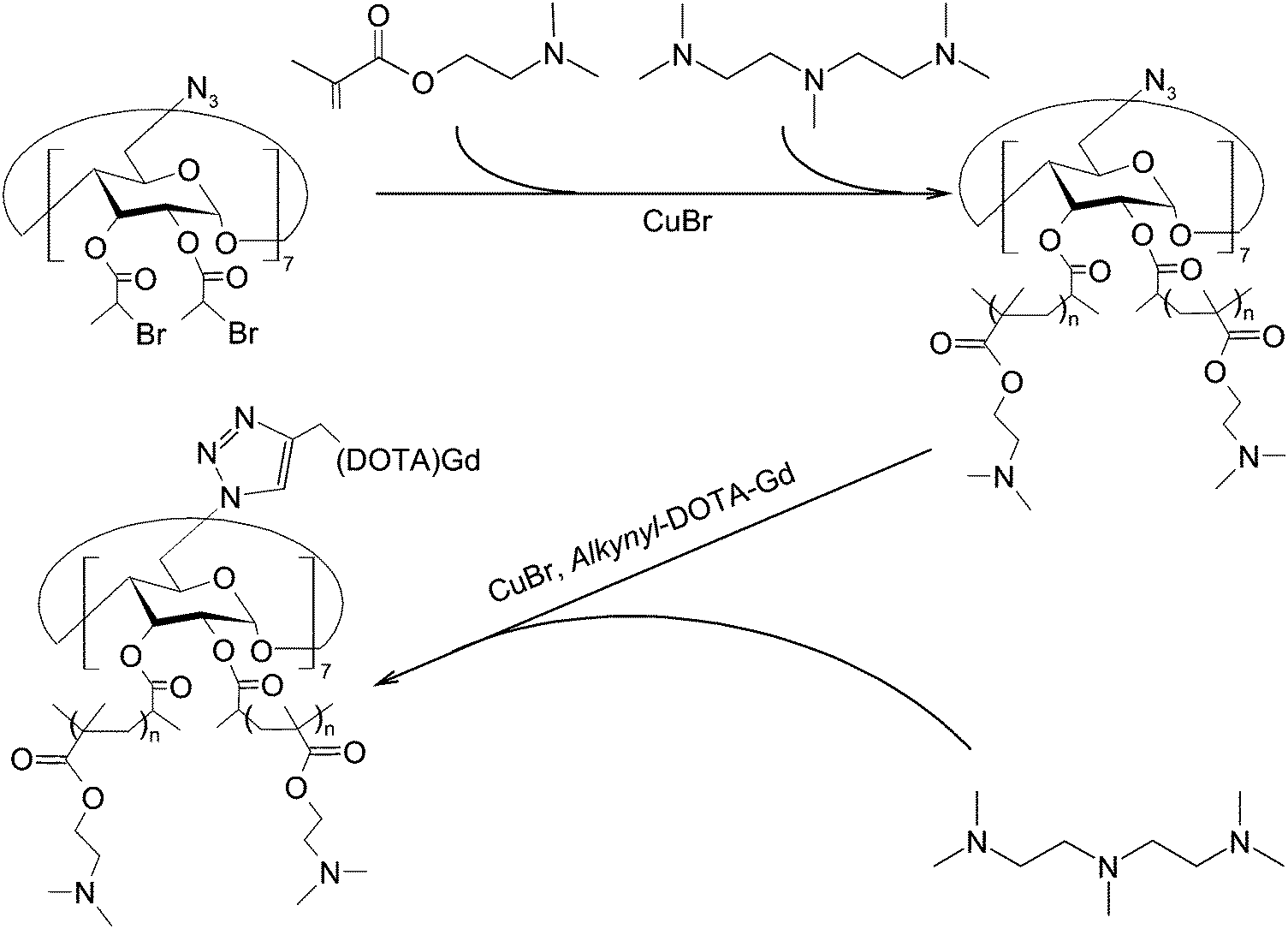 | ||
| Fig. 22 The chemical routes for the synthesis of the CD-based 14-arm star copolymer via ATRP and click reactions. | ||
Notwithstanding the promising potential as presented above, it is worth noting that compatibility problems may arise when multiple agents are co-delivered. This has previously been supported by the observation that the expression of a transgene can be severely suppressed when the transgene is co-delivered with certain chemotherapeutic drugs (e.g., cisplatin and teniposide).255 A recent study has also indicated that upon co-delivery with PEI, the photoluminescence of QDs can be quenched totally.256 To circumvent this obstacle, development of strategies to separate the co-delivered agents into different partitions is desired. One tactic, at least possible in the theoretical sense, is to incorporate CD moieties with different chemical properties into one single construct. The working principle is that the affinity of guest molecules toward CDs is largely determined by the conformity between the guest molecule and the CD cavity.73 By modulating the guest affinity of the CD moieties in different parts of a probe, the co-delivered agents can be partitioned into separate compartments. The feasibility of this approach has been hinted at by complexation thermodynamic studies with CDs. Under the 1:1 stoichiometric condition and by using 2-naphthalenesulfonate as a model guest compound, the enthalpic and entropic contributions to complex stability vary profoundly when different CD molecules are used, with the free energy (−ΔG) of the complex ranging from 3.49 kcal mol−1 for α-CD, 7.33 kcal mol−1 for β-CD to 1.58 kcal mol−1 for γ-CD.257 Such differences in the stability of complexes formed with different CD molecules have also been observed when other model guest compounds (e.g., 2-7-naphthalenedisulfonate,257 adamantane carboxylate,258 and adamantane acetate258) are adopted. To take advantage of this structure-specific guest affinity of CDs for partitioning the co-delivered agents, proper selection of CDs is necessary. Evaluation of the affinity of different CDs towards guest compounds has recently been facilitated by the use of liquid crystals, which are anisotropic fluids bearing a long-range orientational order. The method is based on the phenomenon that host–guest inclusion complexation can subject the liquid crystals to an optical transition from the dark to the bright state.259 Such a transition is ascribed to the disruption of hydrophobic interactions between the guest molecule and CDs, leading to a transition of the liquid crystals from the homeotropic orientation to the planar alignment.259 This optical approach has been successfully used to identify the higher affinity of α-CD than β-CD towards cetyltrimethyl ammonium bromide (CTAB). It shall be applicable to the selection of CD moieties with the best-tuned guest affinity for partitioning co-delivered functionalities into separate compartments.
6. Optimization and engineering of CD-based probes for molecular imaging
In the preceding section, the latest advances in chemistry underpinning the development of CD-based probes for different modalities have been reviewed. The strengths and limitations, as well as the trends for future research, for diverse molecular probes are summarized in Table 5. The promising progress in CD-based molecular imaging, in fact, has not only been revealed by the exponentially growing number of CD-based probes developed in the literature, but has also been manifested by the upsurge of granted patents on the use of CDs in imaging applications (Table 6).260–270 While chemical modification will continue to be a key player in probe optimization, the pace of probe development is anticipated to be synergistically accelerated in the forthcoming decades by advances in materials engineering, processing and fabrication.107,271–276 For instance, to interrogate a molecular event at the cellular and tissue levels, the prevalent approach is to tailor a specific molecular probe to meet the needs of the application. This may limit the pace of probe development. This obstacle may be circumvented if one probe can be used in different situations, with only the sensing component of the probe being required to be changed to recognize specific molecular species. The feasibility of this has been supported by the case of inclusion complexes (NS1, NS2, and NS3) formed between α-CD-functionalized magnetic nanoparticles (CDNPs) and rhodamine-based azobenzene derivatives (A1, A2, and A3).277 The azobenzene part can undergo photoisomerization. It can assemble into and disassemble out of the CD cavity based on actual needs (Fig. 23).277 This concept renders one single probe applicable to the interrogation of the levels of multiple metal ions,277 and is a note-worthy avenue for streamlining the development of CD-based molecular probes in the future.| Imaging modality | Current development | Trends for future research | ||
|---|---|---|---|---|
| Strengths | Limitations | Present state | ||
| Luminescence-based molecular imaging |
• High cost-effectiveness
• Possible to have multiplex detection • High sensitivity • Flexible probe design led by advances in CD chemistry • No limit to the number of repeated imaging as no ionising radiation is involved |
• High tissue autofluorescence
• Photodamage led by the short excitation wavelength • Photon loss caused by self-absorption and scattering |
• CDs are used as sites for anchoring targeting moieties
• Manipulation of signal emission (via mechanisms such as FRET and inclusion complexation) has been achieved using CDs and their derivatives • Optimization of the CD-based probe is achieved mainly by chemically modifying the probe composition and structure |
• NIR photons can be used as a promising excitation source to enable deep tissue imaging
• UCNPs will continue to extend the versatility and flexibility in the design of CD-based probes for luminescence-based molecular imaging • More stringent in vivo tests will be needed for future research • CD-based probes can be optimized not only in their chemical composition and structure, but also in their size and geometry • Advances in luminescence-based molecular imaging will be facilitated by the emergence of carriers that release signalling elements only in target regions |
| Magnetic resonance molecular imaging |
• Rapid and precise in imaging soft tissues
• Excellent anatomic resolution • No limit to the number of repeated imaging as no ionising radiation is involved |
• Long scan and post-processing time
• Involvement of expensive equipment |
• CD molecules are widely used as scaffolds for the formation of paramagnetic complexes for MRI enhancement
• Modification of the probe composition and structure is currently the prevailing method to optimize CD-based probes • CD-based carriers have been used to enhance probe specificity and to stabilize the bivalent state of Mn in porphyrins |
• CD-based Mn(II) agents will continue to be established as potential alternatives to the Gd(III) counterparts
• The toxicity potentially imposed by Mn(II)-based agents can reduced by using CD-based carriers. The carriers encapsulate Mn(II) ions, and enable ion release only when the carriers can reach the biological target • Optimization of the CD-based probe will benefit from advances in materials manipulation, which enables manipulation of probe properties via engineering methods • Development of carriers which enable targeted release of contrast agents will enhance the specificity of magnetic resonance molecular imaging |
| Ionizing radiation-based molecular imaging |
• High sensitivity
• Possible to image multiple probes simultaneously |
• Comparatively low spatial resolution (in the range of 15 mm)
• Technically demanding when CD-based probes are incorporated with short-lived isotopes • Involvement of expensive equipment • Subjects’ exposure to radiation |
• Many tracers for PET and SPECT have been developed and tested as clinical research tools
• Incorporation of CDs (and their derivatives) into tracer design has enabled modulation of the physiological properties (e.g., bioadhesive ability) of the tracer • Functional groups present in CDs have allowed for further ligand conjugation and chemical modification |
• Development of more rapid and efficient radiolabelling reactions will be needed to accommodate the short-lives of the PET and SPECT isotopes
• The precursor toxicity can be reduced by using more mature precursor design and by improving the efficiency of purifying the final product from precursors. • The biological half-life of CD-based tracers will be controlled more effectively |
| Ultrasound-based molecular imaging |
• Non-invasiveness
• Possible to have real-time imaging • High cost-effectiveness • High portability |
• Limited spatial resolution
• Low signal-to-noise ratios • Limited diagnostic options due to the lack of extravasation of the microbubbles from the vasculature |
• CDs have been used to provide sites for further functionalization to convert non-targeting microbubbles into molecularly targeted ones
• The signal-to-noise ratio has been improved by enhancing the targeting capacity of the microbubbles |
• Optimization of microbubble adhesion to target sites will be needed to improve the signal-to-noise ratio
• Identification of more disease-specific targets is desirable for future design of CD-based probes • Development of detection algorithms to better model any changes in acoustic properties of microbubbles upon target binding will be needed to improve the imaging quality • Development of carriers which enable targeted release of contrast agents will enhance the specificity of ultrasound-based molecular imaging |
| Type of imaging | Patent number | Year | Patent title | Relevance to molecular imaging | Ref. |
|---|---|---|---|---|---|
| Luminescence-based imaging | US8343463B2 | 2013 | Optical imaging agent | This patent discloses the fabrication and use of a fluorescent CD-based blood pool contrast agent. | 260 |
| EP2194068B1 | 2013 | Cyclodextrin compound modified with folic acid, process for production thereof, drug delivery agent for targeting drug delivery system, pharmaceutical composition, and imaging agent | This patent discloses the fabrication and use of folate-modified CDs as molecularly targeted imaging agents. | 261 | |
| US5661040A | 1994 | Fluorescent polymer labeled conjugates and intermediates | This patent describes a method to generate a fluorescent conjugate, which consists of a specific binding member bound to a fluorescent polymer. The polymer contains multiple signal generating groups and CD moieties. | 262 | |
| MRI | CN103301483B | 2014 | Preparation of supramolecular dendritic nano-aggregate for magnetic resonance imaging contrast | This patent discloses a method to generate supramolecular dendrimer nano-aggregates from CDs for MRI contrast enhancement. | 263 |
| US8192721 B2 | 2012 | Compositions useful for reducing toxicity associated with gadolinium-based contrast agents | The patent relates to the fabrication of a composition which consists of a gadolinium-based contrast agent and a CD derivative. | 264 | |
| US6068831A | 2000 | Pseudopolyrotaxanes | This patent relates to the development of CD-based pseudopolyrotaxanes for imaging applications. | 265 | |
| Ionizing radiation-based imaging | US6858723B1 | 2005 | Amphiphile cyclodextrins, preparation and use thereof for solubilizing organized systems and incorporating hydrophobic molecules | This patent discloses the fabrication and use of CD derivatives for delivery of hydrophobic compounds, including [123I]-16-iodo-3-methylhexadecanoic acid. | 266 |
| Ultrasound-based imaging | US6177062B1 | 2001 | Agents and methods for enhancing contrast in ultrasound imaging | This patent partly relates to the development of CD-based contrast agents for ultrasound-based imaging. Examples include β-CD-isopentane-inclusion compounds, β-CD-2-methyl-2-butene-inclusion compounds, β-CD-2-methyl-1-butene-inclusion compounds, β-CD-isoprene-inclusion compounds, β-CD-isopentane-inclusion compounds, xenon/α-CD-inclusion compounds, carbon dioxide/α-CD-inclusion compounds, and furan/hydroxypropyl-β-CD-inclusion compounds. | 267 |
| DE3803971C1 | 1989 | Ultrasound contrast medium | This patent relates to a method to generate an ultrasound contrast agent. The agent consists of gas-retaining and/or gas-releasing microparticles, which are generated from CDs or CD derivatives. | 268 | |
| Multimodal imaging | US6113880A | 2000 | Polyrotaxane derivatives for X-ray and nuclear magnetic resonance imaging | The patent discloses the fabrication and use of CD-based polyrotaxanes. The polyrotaxanes contain metal complexes, or iodine-containing benzene derivatives, as imaging components for multimodal diagnosis. | 269 |
| US5928626A | 1999 | Contrast agents, consisting of carbohydrate particles | This patent partly discloses the development of a contrast agent consisting of water-soluble microbubble-generating carbohydrate microparticles, in which the carbohydrate can be CDs. The agent is proposed to be used to enhance diagnostic ultrasonic images and magnetic resonance images of the gastrointestinal system. | 270 | |
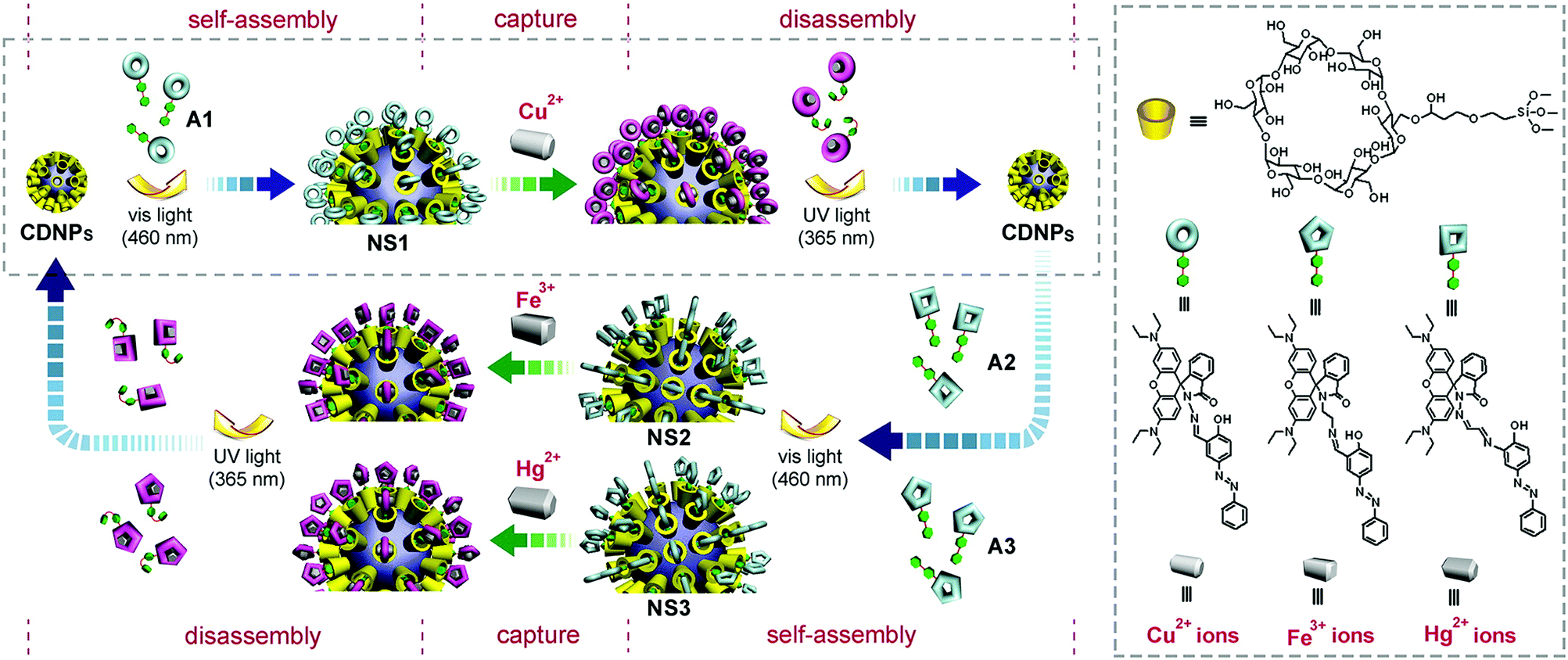 | ||
| Fig. 23 Chemical structures of different fluorescent probes, and the synthetic routes thereof, for molecular recognition of metal ions (reproduced from ref. 277 with permission from American Chemical Society). | ||
As far as probe optimization is concerned, one option is to chemically modify the probe structure and composition. This is especially common for the case of luminescence-based molecular imaging and magnetic resonance molecular imaging (Table 5). Possibilities to improve the probe performance, however, have been extended recently to post-synthetic manipulation of physical parameters using engineering methods (Fig. 24). One engineerable parameter is the zeta potential of a probe. Positive surface charge, by and large, favours cellular uptake due to the negativity of the plasma membrane.278–280 Nonetheless, CDs are electrostatically neutral. Incorporation of CDs into an already-charged probe may result in a reduction in the overall charge density,7 but reports on reversing the sign of the surface charge simply by CD incorporation is much less frequent if any. Another engineerable parameter is probe geometry. The relevance of probe geometry to probe performance has been hinted at by the observation that nanoparticles with optimal shapes display improved cellular uptake properties and a longer circulation half-life.281–284 Micelles with flexible rodlike morphology have also been reported to exhibit longer blood circulation time than spherical ones, partly due to the higher efficiency of the rodlike micelles to evade macrophage uptake.285 All these have evidenced that optimization of probe geometry is a promising non-chemical route for enhancement of probe performance. This has been substantiated by Zhou et al.,286 who have designed a multivalent Gd(III) MRI contrast agent based on a flexible rodlike polyrotaxane scaffold. During the synthetic process, HP-β-CD and Pluronic F127 are adopted to generate a polyrotaxane, with a poly(propylene oxide) block coverage of approximately 46% (Fig. 25). That polyrotaxane is further activated by 1,1′-carbonyldiimidazole (CDI), followed by modification with 1,8-diamino-3,6-dioxooctane to increase its aqueous solubility. The product is coupled with S-2-(4-isothiocyanatobenzyl)-1,4,7,10-tetraazacyclododecane tetraacetic acid (DO3A-Bn-SCN). The contrast agent, namely Gd3+-DO3A-HP-β-CD/Pluronic PR, is obtained after treatment of the product with GdCl3. As revealed by atomic force microscopy (AFM), Gd3+-DO3A-HP-β-CD/Pluronic PR has rodlike morphology with a length in the range of 30–40 nm and a diameter of around 5 nm.286 Contrary to the monomeric Gd3+-DO3A-HP-β-CD derivative which is cleared from blood shortly after its injection into Balb/c mice,286 Gd3+-DO3A-HP-β-CD/Pluronic PR significantly elevates the intensity of the signal in the blood of the heart, and also facilitates visualization of the blood vessel organization.286 This evidence supports the feasibility of modulating probe performance by varying probe geometry.
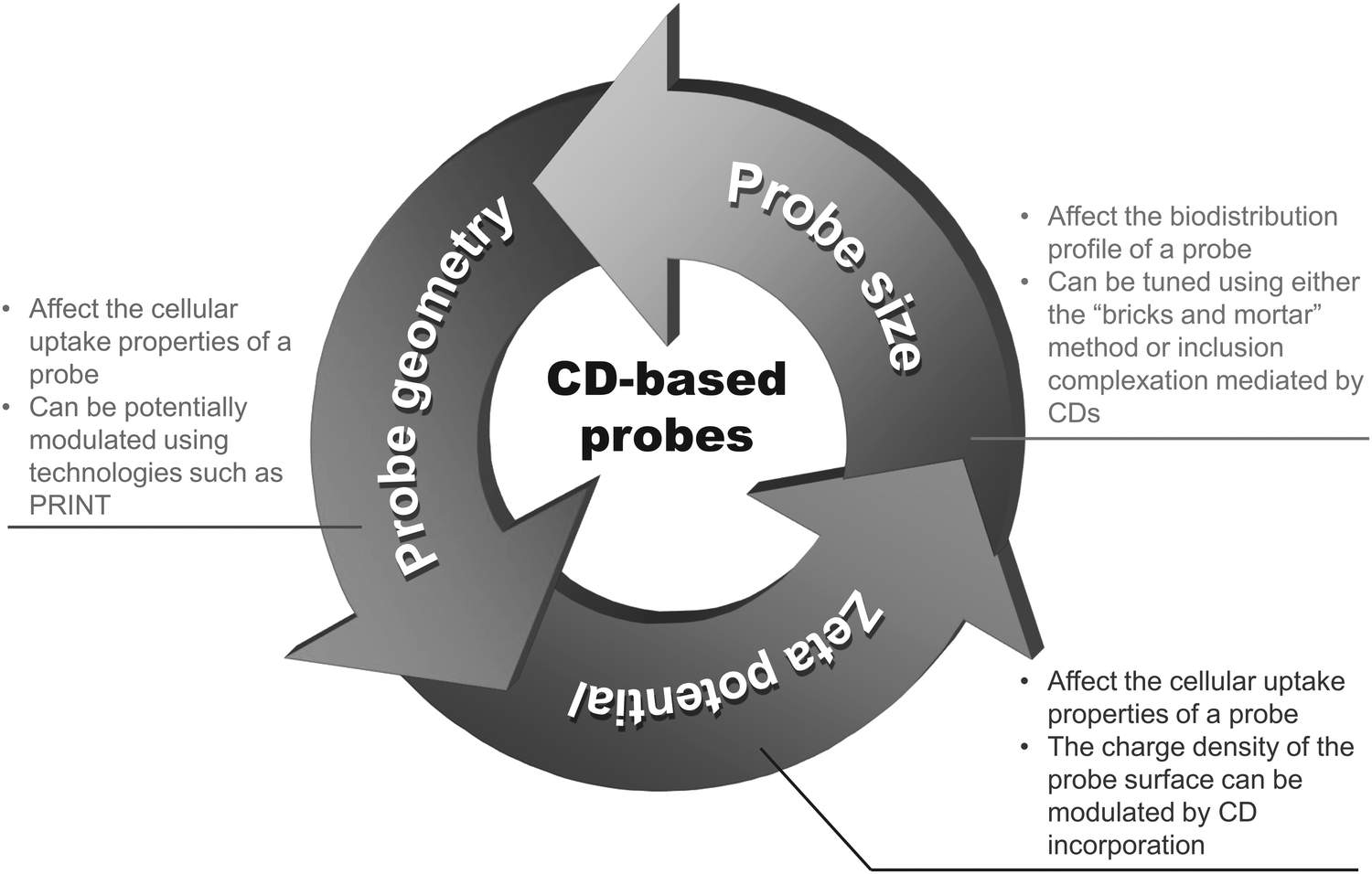 | ||
| Fig. 24 Three major engineerable parameters for optimization of CD-based molecular probes. | ||
 | ||
| Fig. 25 The chemical routes for the fabrication of Gd3+-DO3A-HP-β-CD/Pluronic PR (reproduced from ref. 286 with permission from American Chemical Society). | ||
Engineering of probe geometry, indeed, is especially critical if the probe is to be applied to theranostic applications, which entail release of drugs mediated by diffusional mass transport.287–289 In those cases, changing probe geometry may change the kinetics of drug diffusion and finally the drug release sustainability. Although efforts devoted to controlling the geometry of CD-based molecular probes are limited in the literature, related technologies have emerged continuously since the turn of the last century, with a few of them standing out as particularly prominent for manipulating the geometry of polymeric particles. One representative technology is the “particle replication in nonwetting templates” (PRINT) technology, whose development has benefited from advances in lithographic techniques and roll-to-roll processes. This technology is applicable not only to the generation of nonspherical magneto-polymer composite particles (e.g., nanorice, nanoworm, micron-sized block, and micron-sized boomerang) in which alignment of the embedded Fe3O4 nanoparticles can be precisely manipulated,290 but also to the synthesis of anisotropic rods from organic materials with tuneable multiphases.291 If being translated into the fabrication and engineering of CD-based molecular probes, it can extend the flexibility in probe optimization. Apart from the PRINT technology, other strategies (such as film stretching and template-assisted assembly) have been deployed to modulate the geometry of polymer-based particulate systems (Table 7).282,292–299 As CD-based molecular probes exist principally in the form of polymeric particles, we envision that the transferability of these approaches to probe engineering may accelerate the development of the field in the upcoming decades.
| Category | Basic principle | Strategy | Working principle | Use | Ref. |
|---|---|---|---|---|---|
| Top-down | Molecules are self-assembled into larger molecular constructs via intermolecular and noncovalent interactions | Film stretching | The spherical particles, which have been embedded into a film beforehand, either are liquefied at high temperature or are plasticized by using a plasticizer. After that, the particles are subjected to stretching in one or two dimensions so as to generate non-spherical nanoparticles. | This strategy has been used to fabricate nonspherical polystyrene and poly(lactic-co-glycolic acid) (PLGA) nanoparticles with different shapes (including wormlike particles, oblate or prolate ellipsoids, and elliptical discs) | 292 and 293 |
| Patterned wafers made of silicon are produced by photolithography. They are subsequently used as master templates for the production of nonspherical particles. | This approach has been employed to generate non-spherical nanoparticles from insulin and albumin. | 294 and 295 | |||
| Step and flash imprint lithography | By using a quartz template with relief images, photopolymerizable macromers are moulded into patterns on a substrate. The nanoparticles are revealed upon the removal of the template, and are harvested by subjecting the thin residual layer to oxygen plasma etching. | This strategy has been employed to generate square, triangular, and pentagonal hydrogel nanoparticles from PEG diacrylate | 296 | ||
| Bottom-up | Particles are fabricated by processing macroscopic materials into constructs in the micron- or nanoscale | Template-assisted assembly | Heat or chemical crosslinking is adopted to join together spherical particles, which have been put into a template beforehand | This strategy has been reported for the production of fibre-like structures from polyelectrolyte-encapsulated nanoparticles | 297 |
| Mini-emulsion technique | By using a mini-emulsion as a template and by evaporating the polymer-containing dispersed phase, polymeric nanoparticles are generated | This approach has been employed to fabricate ellipsoidal nanoparticles from main-chain liquid crystalline polymers. The ellipsoidal shape is resulted from the quasi-equilibrium shape memory of the entangled polymers. | 298 | ||
| Layer-by-layer assembly | Polymers are deposited sequentially onto a sacrificial substrate, followed by substrate dissolution | This strategy has been used to fabricate discoidal hydrogel capsules consisting of poly(N-vinyl-2-pyrrolidone) (PVP) and poly(methacrylic acid) (PMAA). The same method has been adopted to produce rigid hydrogen-bonded tannic acid/PVP hemispherical hydrogel capsules. | 282 and 299 | ||
The biological performance of a CD-based probe can be modulated by manipulating not only the zeta potential and probe geometry but also the particle size of the probe. The probe size can be tuned using either the “bricks and mortar” method300 or inclusion complexation mediated by CDs. The latter has been demonstrated by the observation that, upon addition of CDs into an aqueous solution of poly(acrylic acid) (PAA) and PEG, host–guest binding mediated by CDs complete with hydrogen bonding interactions in the system,301 changing the size of the interpolymer complexes formed. Optimization of the probe size is indeed pivotal to effective molecular imaging because it largely determines the biodistribution profile of a probe. It has been reported that particles with a size of less than 20–30 nm are, on the whole, susceptible to renal excretion.302,303 Those with a size of 30–150 nm may accumulate in the kidney,302,303 heart,304 stomach,305 and bone marrow.306 Those with a size of 150–300 nm often deposit in the spleen307 and liver.308 This suggests that based on the molecular event to be monitored, special care has to be taken when the size of a molecular probe is engineered.
Finally, as depicted in Table 5, probe specificity has been achieved in the literature predominately by exploiting the intrinsic inclusion capability of CDs to incorporate a probe with targeting ligands or related functionalities. The efficiency of molecular imaging using this ligand incorporation approach, however, may be impaired by the low target-to-background ratio, which can be caused by the low receptor density and availability, rapid efflux of the probe from cells, limited clearance kinetics from the interstitial space, and nonspecific cellular probe uptake.126,127 This problem may not be significant in the in vitro context, in which unbound molecular probes can be easily removed by changing the cell culture medium.126 Yet, in living bodies, elimination of the unbound probes relies largely on physiological mechanisms which sometimes may not be efficient, thereby resulting in high background noises.126 In the future, this problem may possibly be solved by the development of stimuli-responsive carriers (Table 5), which release the signalling elements only in a target region in which the molecular event to be interrogated occurs. The viability of this has been illustrated by the case of enzyme-responsive porous silica nanoparticles.309 The nanoparticles are comprised of surface CD gatekeepers, functional stalks, and fluorescent probes located within the porous channels. Among them, the CD gatekeepers, as well as the ester linkage in the stalk, can be hydrolyzed to release guest molecules from the channels.309 Although the work has only been exploited for controlled drug delivery, if systems like this can be engineered to be responsive to molecular events and to carry imaging elements such as Gd(III) chelates, a vista of new impetus to future advances may be opened up in CD-based molecular imaging.
7. Conclusions and perspectives
Molecular imaging can impact on disease diagnosis, monitoring, and imaging-guided biopsy. Based on the research reports covered by this review, it is not difficult to see that significant progress has already been made in synthesizing and manipulating the structures and properties of CDs and their derivatives for molecular imaging. This promising progress has been contributed, on one hand, by increasing understanding of inclusion chemistry and on the other hand, by advances in materials engineering and imaging instrumentation, which provides a big fillip to the development and optimization of CD-based molecular probes. Notwithstanding the promising progress, it is worth noting that while a plethora of CD-based molecular probes have been reported over the last several decades, the number of probes that have reached clinical trials is highly limited. This is partly resulted from the fact that a disproportionate quantum of resources has been directed towards characterization of the physical properties of a probe as an emissive material. Evaluation of the biological performance of the CD-based probe in most cases has been confined solely to cytotoxicity assays and live cell imaging studies. To extend the use of CD-based probes from the laboratory context to clinical settings, additional efforts on evaluating the pharmacokinetics and long-term safety of the probes will be the next stage to pursue. This is particularly true because the wide variations in probe attributes (e.g., size and zeta potential) may lead to highly variable absorption, distribution, metabolism, and excretion characteristics, thereby yielding unexpected outcomes upon administration to a living body.310–312 This problem may partly be solved by surface engineering of the probe for reducing nonspecific binding and for increasing the accumulation of the probe in target tissues. The use of Food and Drug Administration (FDA)-approved materials as the building blocks for probe fabrication may also be helpful to mitigate concerns on toxicity. Finally, effective cellular uptake of the probe in target regions is a precondition for proper execution of molecular imaging. The process of cellular internalization of the CD-based probes should, therefore, be investigated in parallel with the imaging potential. Future works should focus more on further deciphering the mechanism adopted by CDs and their derivatives to permeabilize biological membranes and to modulate cholesterol distribution in living cells.120Apart from probe optimization and validation as mentioned above, molecular probes are often handled as ordinary drugs by many regulatory agencies prior to the approval of clinical use. High developmental costs have, therefore, been imposed to clinical translation of CD-based molecular probes. This may be lesser a problem for CD-based agents that can be administered in “trace amounts”. A good example is those utilized in ionizing radiation-based imaging. Requirements on preclinical testing of such agents prior to clinical trials can be less demanding if the exploratory investigative new drug application (eIND) pathway in the USA can be followed.313,314 However, such a pathway is not available for most of the other types of molecular probes, rendering the process of clinical translation of CD-based molecular imaging technologies lengthy and tedious. The situation is worsened by the fact that a fair comparison of the performance of CD-based probes reported in the literature is a holy grail for experimentalists at the moment because of the incomparability of studies. This can be somewhat manifested by the example of cell imaging experiments, in which the cell confluence and incubation time vary greatly among different studies, from 70% cell confluence with a 12-hour incubation315 to 90% cell confluence with a 1-hour incubation.60 This limits the comparability of experimental results, and imposes difficulties of identifying promising probes for further pursuits in clinical trials. To rectify this situation, adoption of more standardized testing methods may be helpful; however, there is a long way to go before widespread appreciation of the legitimacy of such thinking, in relation to both the desirability and feasibility of such a practice, can be possibly attained. In this path forward, easy recognition of a probe as a general favourite among others may not necessarily be plausible, yet we envision the continuous emergence of new probes for monitoring molecular events in disease prevention, detection, diagnosis, and treatment during the process. In all, the unique attributes of CDs may enable a transformation in the ways molecular imaging is advanced and administered.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Wai-Sum Lo, Weijie Hu, Cheng-Shen Hu, Yau-Foon Tsui, Guoan Wang, Guoxing Deng, Fanyue Meng, Jinzheng Chen, and Minjian Huang for helpful comments and suggestions during the writing of this manuscript. This work is supported by the HK Polytechnic University Area of Excellent Grants (1-ZVGG), Natural Science Foundation of Shenzhen University (2017092), and the grant from the Shenzhen Science and Technology Innovation Committee (JCYJ20170302144812937).References
- H. R. Herschman, Science, 2003, 302, 605–608 CrossRef CAS PubMed.
- K. H. Jung and K. H. Lee, J. Pathol. Transl. Med., 2015, 49, 5–12 CrossRef PubMed.
- M. F. Kircher, H. Hricak and S. M. Larson, Mol. Oncol., 2012, 6, 182–195 CrossRef CAS PubMed.
- S. R. Mudd, R. A. Comley, M. Bergstrom, K. D. Holen, Y. Luo, S. Carme, G. B. Fox, L. Martarello and J. D. Beaver, Drug Discovery Today, 2017, 22, 140–147 CrossRef CAS PubMed.
- E. G. de Vries, S. de Jong and J. A. Gietema, J. Clin. Oncol., 2015, 33, 2585–2587 CrossRef CAS PubMed.
- L. Cunha, K. Szigeti, D. Mathe and L. F. Metello, Drug Discovery Today, 2014, 19, 936–948 CrossRef CAS PubMed.
- W. F. Lai, Biomaterials, 2014, 35, 401–411 CrossRef CAS PubMed.
- H. He, S. Chen, J. Zhou, Y. Dou, L. Song, L. Che, X. Zhou, X. Chen, Y. Jia, J. Zhang, S. Li and X. Li, Biomaterials, 2013, 34, 5344–5358 CrossRef CAS PubMed.
- S. Lepretre, F. Chai, J. C. Hornez, G. Vermet, C. Neut, M. Descamps, H. F. Hildebrand and B. Martel, Biomaterials, 2009, 30, 6086–6093 CrossRef CAS PubMed.
- G. Fundueanu, M. Constantin, A. Dalpiaz, F. Bortolotti, R. Cortesi, P. Ascenzi and E. Menegatti, Biomaterials, 2004, 25, 159–170 CrossRef CAS PubMed.
- F. Quaglia, L. Ostacolo, A. Mazzaglia, V. Villari, D. Zaccaria and M. T. Sciortino, Biomaterials, 2009, 30, 374–382 CrossRef CAS PubMed.
- T. R. Thatiparti, A. J. Shoffstall and H. A. von Recum, Biomaterials, 2010, 31, 2335–2347 CrossRef CAS PubMed.
- Y. Y. Liu and X. D. Fan, Biomaterials, 2005, 26, 6367–6374 CrossRef CAS PubMed.
- I. Antoniuk and C. Amiel, J. Pharm. Sci., 2016, 105, 2570–2588 CrossRef CAS PubMed.
- C. Ortiz Mellet, J. M. García Fernández and J. M. Benito, in Supramolecular Systems in Biomedical Fields, ed. H. J. Schneider, Royal Society of Chemistry, UK, 2013, pp. 94–139 Search PubMed.
- C. Ortiz Mellet, J. M. Garcia Fernandez and J. M. Benito, Chem. Soc. Rev., 2011, 40, 1586–1608 RSC.
- M. E. Davis, Mol. Pharmaceutics, 2009, 6, 659–668 CrossRef CAS PubMed.
- I. Bechet, P. Paques, M. Fillet, P. Hubert and J. Crommen, Electrophoresis, 1994, 15, 818–823 CrossRef CAS PubMed.
- A. Beig, R. Agbaria and A. Dahan, PLoS One, 2013, 8, e68237 CAS.
- D. W. Oliver, I. C. Dormehl, W. Louw, E. Kilian, V. de Beco and J. L. Morretti, Arzneim. Forsch., 2000, 50, 758–764 CAS.
- J. W. Fredy, J. Scelle, G. Ramniceanu, B. T. Doan, C. S. Bonnet, E. Toth, M. Menand, M. Sollogoub, G. Vives and B. Hasenknopf, Org. Lett., 2017, 19, 1136–1139 CrossRef CAS PubMed.
- S. Aime, I. M. Botta, F. Fedeli, E. Gianolio, E. Terreno and P. Anelli, Chemistry, 2001, 7, 5261–5269 CrossRef CAS PubMed.
- E. M. Martin Del Valle, Process Biochem., 2004, 39, 1033–1046 CrossRef.
- A. Villiers, C. R. Acad. Sci., 1891, 112, 536–538 Search PubMed.
- K. Freudenberg and M. Meyer-Delius, Ber. Dtsch. Chem. Ges. A, 1938, 71, 1596–1600 CrossRef.
- K. Freudenberg, E. Schaaf, G. Dumpert and T. Ploetz, Naturwissenschaften, 1939, 51, 850–853 CrossRef.
- F. Cramer, Einschlussverbindungen, Springer-Verlag, Berlin, 1954 Search PubMed.
- T. Loftsson and D. Duchene, Int. J. Pharm., 2007, 329, 1–11 CrossRef CAS PubMed.
- W. J. Shieh and A. R. Hedges, J. Macromol. Sci., Part A: Pure Appl. Chem., 1996, 33, 673–683 CrossRef.
- T. Loftsson, M. E. Brewster and M. Másson, Am. J. Drug Delivery, 2004, 2, 261–275 CrossRef CAS.
- J. Szejtli, Pure Appl. Chem., 2004, 76, 1825–1845 CrossRef CAS.
- S. Mohandoss, J. Sivakamavalli, B. Vaseeharan and T. Stalin, Sens. Actuators, B, 2016, 234, 300–315 CrossRef CAS.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- H. Dong, Y. Li, J. Yu, Y. Song, X. Cai, J. Liu, J. Zhang, R. C. Ewing and D. Shi, Small, 2013, 9, 446–456 CrossRef CAS PubMed.
- B. Varghese, S. N. Al-Busafi, F. O. Suliman and S. M. Z. Al-Kindy, Spectrochim. Acta, Part A, 2017, 173, 383–389 CrossRef CAS PubMed.
- M. Maniyazagan, C. Rameshwaran, R. Mariadasse, J. Jeyakanthan, K. Premkumar and T. Stalin, Sens. Actuators, B, 2017, 242, 1227–1238 CrossRef CAS.
- K. Yang, Y. X. Pei, J. Wen and Z. C. Pei, Chem. Commun., 2016, 52, 9316–9326 RSC.
- C. Sathiyajith, R. R. Shaikh, Q. Han, Y. Zhang, K. Meguellati and Y. W. Yang, Chem. Commun., 2017, 53, 677–696 RSC.
- M. Xue, Y. Yang, X. D. Chi, Z. B. Zhang and F. H. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed.
- X. Ma and Y. L. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- F. A. De, S. A. Fernandes and A. A. Sabino, Curr. Drug Discovery Technol., 2009, 6, 151–170 CrossRef.
- D. Shohat and E. Grushka, Anal. Chem., 1994, 66, 747–750 CrossRef CAS.
- K. Jie, Y. Zhou, Y. Yao and F. Huang, Chem. Soc. Rev., 2015, 44, 3568–3587 RSC.
- S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- H. W. Frijlink, A. C. Eissens, N. R. Hefting, K. Poelstra, C. F. Lerk and D. K. F. Meijer, Pharm. Res., 1991, 8, 9–16 CrossRef CAS.
- D. W. Frank, J. E. Gray and R. N. Weaver, Am. J. Pathol., 1976, 83, 367–382 CAS.
- F. Bellia, D. La Mendola, C. Pedone, E. Rizzarelli, M. Saviano and G. Vecchio, Chem. Soc. Rev., 2009, 38, 2756–2781 RSC.
- G. Impellizzeri, G. Pappalardo, E. Rizzarelli and C. Tringali, J. Chem. Soc., Perkin Trans. 2, 1996, 1435–1440 RSC.
- R. Deschenaux, M. M. Harding and T. Ruch, J. Chem. Soc., Perkin Trans. 2, 1993, 1251–1258 RSC.
- M. E. Amato, G. C. Pappalardo and B. Perly, Magn. Reson. Chem., 1993, 31, 455–460 CrossRef CAS.
- M. E. Amato, K. B. Lipkowitz, G. M. Lombardo and G. C. Pappalardo, Magn. Reson. Chem., 1998, 36, 693–705 CrossRef CAS.
- H. Cousin, P. Cardinael, H. Oulyadi, X. Pannecoucke and J. C. Combret, Tetrahedron: Asymmetry, 2001, 12, 81–88 CrossRef CAS.
- A. R. Khan, P. Forgo, K. J. Stine and V. T. D'Souza, Chem. Rev., 1998, 98, 1977–1996 CrossRef CAS PubMed.
- S. Guieu and M. Sollogoub, in Modern Synthetic Methods in Carbohydrate Chemistry: From Monosaccharides to Complex Glycoconjugates, ed. D. B. Werz and S. Vidal, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013, pp. 241–283 Search PubMed.
- M. Sollogoub, Synlett, 2013, 2629–2640 CrossRef CAS.
- B. Varghese, S. N. Al-Busafi, F. O. Suliman and S. M. Z. Al-Kindy, Spectrochim. Acta, Part A, 2015, 136, 661–671 CrossRef CAS PubMed.
- S. S. Mati, S. Sarkar, S. Rakshit, A. Sarkar and S. C. Bhattacharya, RSC Adv., 2013, 3, 8071–8082 RSC.
- R. H. Yang, K. A. Li, K. M. Wang, F. L. Zhao, N. Li and F. Liu, Anal. Chem., 2003, 75, 612–621 CrossRef CAS PubMed.
- C. Ma, T. Bian, S. Yang, C. Liu, T. Zhang, J. Yang, Y. Li, J. Li, R. Yang and W. Tan, Anal. Chem., 2014, 86, 6508–6515 CrossRef CAS PubMed.
- M. Sun, H. Y. Zhang, B. W. Liu and Y. Liu, Macromolecules, 2013, 46, 4268–4275 CrossRef CAS.
- Y. H. Zhou, C. L. Wang, F. Wang, C. Z. Li, C. Dong and S. M. Shuang, Chin. J. Chem., 2016, 34, 599–608 CrossRef CAS.
- M. Sakurai, M. Kitagawa, H. Hoshi, Y. Inoue and R. Chujo, Chem. Lett., 1988, 895–898 CrossRef CAS.
- W. Linert, L. F. Han and I. Lukovits, Chem. Phys., 1989, 139, 441–455 CrossRef CAS.
- P. D. Ross and M. V. Rekharsky, Biophys. J., 1996, 71, 2144–2154 CrossRef CAS PubMed.
- D. Hallen, A. Schon, I. Shehatta and I. Wadso, J. Chem. Soc., Faraday Trans., 1992, 88, 2859–2863 RSC.
- M. V. Rekharsky, M. P. Mayhew, R. N. Goldberg, P. D. Ross, Y. Yamashoji and Y. Inoue, J. Phys. Chem. B, 1997, 101, 87–100 CrossRef CAS.
- Y. Matsui and K. Mochida, Bull. Chem. Soc. Jpn., 1979, 52, 2808–2814 CrossRef CAS.
- W. Saenger, Angew. Chem., Int. Ed. Engl., 1980, 19, 344–362 CrossRef.
- G. Barone, G. Castronuovo, P. Delvecchio, V. Elia and M. Muscetta, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 2089–2101 RSC.
- J. Zhang, L. Zhang, W. Wang, L. H. Han, J. C. Jia, Z. W. Tian, Z. Q. Tian and D. P. Zhan, Chem. Sci., 2017, 8, 2407–2412 RSC.
- D. P. Zhan, L. H. Han, J. Zhang, Q. F. He, Z. W. Tian and Z. Q. Tian, Chem. Soc. Rev., 2017, 46, 1526–1544 RSC.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1918 CrossRef CAS PubMed.
- M. E. Brewster and T. Loftsson, Adv. Drug Delivery Rev., 2007, 59, 645–666 CrossRef CAS PubMed.
- S. Loethen, J. M. Kim and D. H. Thompson, Polym. Rev., 2007, 47, 383–418 CrossRef CAS.
- A. Samanta, N. Guchhait and S. C. Bhattacharya, J. Phys. Chem. B, 2014, 118, 13279–13289 CrossRef CAS PubMed.
- M. V. Rekharsky, R. N. Goldberg, F. P. Schwarz, Y. B. Tewari, P. D. Ross, Y. Yamashoji and Y. Inoue, J. Am. Chem. Soc., 1995, 117, 8830–8840 CrossRef CAS.
- R. J. Dong, H. Y. Chen, D. L. Wang, Y. Y. Zhuang, L. J. Zhu, Y. Su, D. Y. Yan and X. Y. Zhu, ACS Macro Lett., 2012, 1, 1208–1211 CrossRef CAS.
- H. Elnakat and M. Ratnam, Adv. Drug Delivery Rev., 2004, 56, 1067–1084 CrossRef CAS PubMed.
- P. C. Elwood, J. Biol. Chem., 1989, 264, 14893–14901 CAS.
- D. S. Theti and A. L. Jackman, Clin. Cancer Res., 2004, 10, 1080–1089 CrossRef CAS PubMed.
- G. Toffoli, C. Bevilacqua, A. Franceschin and M. Boiocchi, Int. J. Hyperthermia, 1989, 5, 163–172 CrossRef CAS PubMed.
- T. Q. Duong and J. S. Kim, Chem. Rev., 2010, 110, 6280–6301 CrossRef PubMed.
- Y. Liu and Y. Chen, Acc. Chem. Res., 2006, 39, 681–691 CrossRef CAS PubMed.
- D. P. Goldberg, A. G. Montalban, A. J. P. White, D. J. Williams, A. G. M. Barrett and B. M. Hoffman, Inorg. Chem., 1998, 37, 2873–2879 CrossRef CAS.
- K. Kano, H. Kitagishi, Y. Sone, N. Nakazawa and M. Kodera, Eur. J. Inorg. Chem., 2006, 4043–4053 CrossRef CAS.
- Y. Zheng, S. Ji, E. Tomaselli and S. Liu, J. Labelled Compd. Radiopharm., 2014, 57, 584–592 CrossRef CAS PubMed.
- H. P. Yan, M. Q. Tan, D. M. Zhang, F. S. Cheng, H. Wu, M. K. Fan, X. J. Ma and J. H. Wang, Talanta, 2013, 108, 59–65 CrossRef CAS PubMed.
- X. Guan, D. Zhang, T. Jia, Y. Zhang, L. Meng, Q. Jin, H. Ma, D. Lu, S. Lai and Z. Lei, Ind. Eng. Chem. Res., 2017, 56, 3913–3919 CrossRef CAS.
- M. Yu, G. Zhang, W. H. Wang, J. B. Niu and N. Zhang, Supramol. Chem., 2012, 24, 799–802 CrossRef CAS.
- M. Panigrahi, S. Dash, S. Patel and B. K. Mishra, Tetrahedron, 2012, 68, 781–805 CrossRef CAS.
- M. Henary and A. Levitz, Dyes Pigm., 2013, 99, 1107–1116 CrossRef CAS.
- T. Behnke, J. E. Mathejczyk, R. Brehm, C. Wurth, F. R. Gomes, C. Dullin, J. Napp, F. Alves and U. Resch-Genger, Biomaterials, 2013, 34, 160–170 CrossRef CAS PubMed.
- C. S. Huang, S. George, M. Lu, V. Chaudhery, R. M. Tan, R. C. Zangar and B. T. Cunningham, Anal. Chem., 2011, 83, 1425–1430 CrossRef CAS PubMed.
- R. Guether and M. V. Reddington, Tetrahedron Lett., 1997, 38, 6167–6170 CrossRef CAS.
- J. L. Zhao, Y. Lv, H. J. Ren, W. Sun, Q. Liu, Y. L. Fu and L. Y. Wang, Dyes Pigm., 2013, 96, 180–188 CrossRef CAS.
- T. Carmona, G. Marcelo, L. Rinaldi, K. Martina, G. Cravotto and F. Mendicuti, Dyes Pigm., 2015, 114, 204–214 CrossRef CAS.
- H. Ogino, J. Am. Chem. Soc., 1981, 103, 1303–1304 CrossRef CAS.
- A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS PubMed.
- G. Wenz, B. H. Han and A. Muller, Chem. Rev., 2006, 106, 782–817 CrossRef CAS PubMed.
- J. W. Fredy, J. Scelle, A. Guenet, E. Morel, S. A. de Beaumais, M. Menand, V. Marvaud, C. S. Bonnet, E. Toth, M. Sollogoub, G. Vives and B. Hasenknopf, Chem. – Eur. J., 2014, 20, 10915–10920 CrossRef CAS PubMed.
- G. Wenz, C. Gruber, B. Keller, C. Schilli, T. Albuzat and A. Muller, Macromolecules, 2006, 39, 8021–8026 CrossRef CAS.
- R. W. Kubiak, J. D. Mighion, S. M. Wilkerson-Hill, J. S. Alford, T. Yoshidomi and H. M. L. Davies, Org. Lett., 2016, 18, 3118–3121 CrossRef CAS PubMed.
- L. Zerkoune, A. Angelova and S. Lesieur, Nanomaterials, 2014, 4, 741–765 CrossRef CAS PubMed.
- A. Angelova, C. Ringard-Lefebvre and A. Baszkin, J. Colloid Interface Sci., 1999, 212, 275–279 CrossRef CAS PubMed.
- C. Oliveras-Gonzalez, F. Di Meo, A. Gonzalez-Campo, D. Beljonne, P. Norman, M. Simon-Sorbed, M. Linares and D. B. Amabilino, J. Am. Chem. Soc., 2015, 137, 15795–15808 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- A. Angelova, C. Fajolles, C. Hocquelet, F. Djedaini-Pilard, S. Lesieur, V. Bonnet, B. Perly, G. Lebas and L. Mauclaire, J. Colloid Interface Sci., 2008, 322, 304–314 CrossRef CAS PubMed.
- L. Zerkoune, S. Lesieur, J. L. Putaux, L. Choisnard, A. Geze, D. Wouessidjewe, B. Angelov, C. Vebert-Nardin, J. Doutch and A. Angelova, Soft Matter, 2016, 12, 7539–7550 RSC.
- A. Angelova, B. Angelov, R. Mutafchieva and S. Lesieur, J. Inorg. Organomet. Polym., 2015, 25, 214–232 CrossRef CAS.
- A. Angelova, B. Angelov, M. Drechsler and S. Lesieur, Drug Discovery Today, 2013, 18, 1263–1271 CrossRef CAS PubMed.
- S. J. Hwang, N. C. Bellocq and M. E. Davis, Bioconjugate Chem., 2001, 12, 280–290 CrossRef CAS PubMed.
- H. J. Yan, L. L. He, C. Ma, J. S. Li, J. F. Yang, R. H. Yang and W. H. Tan, Chem. Commun., 2014, 50, 8398–8401 RSC.
- N. G. Schipper, J. C. Verhoef, L. M. De Lannoy, S. G. Romeijn, J. H. Brakkee, V. M. Wiegant, W. H. Gispen and F. W. Merkus, Br. J. Pharmacol., 1993, 110, 1335–1340 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- T. Loftsson, P. Jarho, M. Masson and T. Jarvinen, Expert Opin. Drug Delivery, 2005, 2, 335–351 CrossRef CAS PubMed.
- T. Irie and K. Uekama, J. Pharm. Sci., 1997, 86, 147–162 CrossRef CAS PubMed.
- H. Matsuda and H. Arima, Adv. Drug Delivery Rev., 1999, 36, 81–99 CrossRef CAS PubMed.
- R. Zidovetzki and I. Levitan, Biochim. Biophys. Acta, 2007, 1768, 1311–1324 CrossRef CAS PubMed.
- M. Grunze and B. Deuticke, Biochim. Biophys. Acta, 1974, 356, 125–130 CrossRef CAS.
- A. P. Bagre, K. Jain and N. K. Jain, Int. J. Pharm., 2013, 456, 31–40 CrossRef CAS PubMed.
- H. Yan, L. He, W. Zhao, J. Li, Y. Xiao, R. Yang and W. Tan, Anal. Chem., 2014, 86, 11440–11450 CrossRef CAS PubMed.
- P. F. Sun, M. C. Lin, G. S. Chen and M. Jiang, Sci. China: Chem., 2016, 59, 1616–1620 CrossRef CAS.
- G. Chen and M. Jiang, Chem. Soc. Rev., 2011, 40, 2254–2266 RSC.
- T. F. Massoud and S. S. Gambhir, Genes Dev., 2003, 17, 545–580 CrossRef CAS PubMed.
- R. N. Day and F. Schaufele, Mol. Endocrinol., 2005, 19, 1675–1686 CrossRef CAS PubMed.
- P. Sharma, S. Brown, G. Walter, S. Santra and B. Moudgil, Adv. Colloid Interface Sci., 2006, 123, 471–485 CrossRef PubMed.
- A. J. Mueller, D. U. Bartsch, U. Schaller, W. R. Freeman and A. Kampik, Int. Ophthalmol., 2001, 23, 385–393 CrossRef CAS PubMed.
- T. Y. Hu, W. D. Na, X. Yan and X. G. Su, Talanta, 2017, 165, 194–200 CrossRef CAS PubMed.
- K. Fujimoto, Y. Muto and M. Inouye, Bioconjugate Chem., 2008, 19, 1132–1134 CrossRef CAS PubMed.
- Q. Ma, J. P. Song, S. F. Zhang, M. F. Wang, Y. Guo and C. Dong, Colloids Surf., B, 2016, 148, 66–72 CrossRef CAS PubMed.
- C. Shu, R. X. Li, J. Guo, L. Ding and W. Y. Zhong, J. Nanopart. Res., 2013, 15, 1927 CrossRef.
- M. Y. Xu, S. Z. Wu, F. Zeng and C. M. Yu, Langmuir, 2010, 26, 4529–4534 CrossRef CAS PubMed.
- I. L. Medintz, E. R. Goldman, M. E. Lassman and J. M. Mauro, Bioconjugate Chem., 2003, 14, 909–918 CrossRef CAS PubMed.
- G. Fang, M. Y. Xu, F. Zeng and S. Z. Wu, Langmuir, 2010, 26, 17764–17771 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- S. K. Ko, X. Chen, J. Yoon and I. Shin, Chem. Soc. Rev., 2011, 40, 2120–2130 RSC.
- P. Li, Y. Liu, X. Wang and B. Tang, Analyst, 2011, 136, 4520–4525 RSC.
- K. Ochiai, K. Kaneko, M. Kitagawa, H. Ando and T. Hayakawa, Dig. Dis. Sci., 2004, 49, 1953–1956 CrossRef PubMed.
- M. F. Byrne, R. M. Mitchell, H. Stiffler, P. S. Jowell, M. S. Branch, T. N. Pappas, D. Tyler and J. Baillie, Can. J. Gastroenterol., 2002, 16, 849–854 CrossRef PubMed.
- P. G. Lankisch, S. Burchard-Reckert and D. Lehnick, Gut, 1999, 44, 542–544 CrossRef CAS PubMed.
- R. Breslow, S. Halfon and B. L. Zhang, Tetrahedron, 1995, 51, 377–388 CrossRef CAS.
- K. Mochida and Y. Matsui, Chem. Lett., 1976, 963–966 CrossRef CAS.
- R. Corradini, A. Dossena, G. Galaverna, R. Marchelli, A. Panagia and G. Sartor, J. Org. Chem., 1997, 62, 6283–6289 CrossRef CAS.
- S. Furukawa, H. Mihara and A. Ueno, Macromol. Rapid Commun., 2003, 24, 202–206 CrossRef CAS.
- M. Longmire, P. L. Choyke and H. Kobayashi, Curr. Top. Med. Chem., 2008, 8, 1180–1186 CrossRef CAS PubMed.
- A. L. Rogach and M. Ogris, Curr. Opin. Mol. Ther., 2010, 12, 331–339 CAS.
- E. Renard, A. Deratani, G. Volet and B. Sebille, Eur. Polym. J., 1997, 33, 49–57 CrossRef CAS.
- C. Pop, Y. R. Chen, B. Smith, K. Bose, B. Bobay, A. Tripathy, S. Franzen and A. C. Clark, Biochemistry, 2001, 40, 14224–14235 CrossRef CAS PubMed.
- M. Furue, A. Harada and S. I. Nozakura, J. Polym. Sci., Part C: Polym. Lett., 1975, 13, 357–360 CrossRef CAS.
- A. Harada, M. Furue and S. Nozakura, Macromolecules, 1976, 9, 701–704 CrossRef CAS.
- J. H. Suh, S. H. Lee and K. D. Zoh, J. Am. Chem. Soc., 1992, 114, 7916–7917 CrossRef CAS.
- T. Seo, T. Kajihara and T. Iijima, Makromol. Chem., 1987, 188, 2071–2082 CrossRef CAS.
- G. Tian, W. L. Ren, L. Yan, S. Jian, Z. J. Gu, L. J. Zhou, S. Jin, W. Y. Yin, S. J. Li and Y. L. Zhao, Small, 2013, 9, 1929–1938 CrossRef CAS PubMed.
- Q. A. Liu, C. Y. Li, T. S. Yang, T. Yi and F. Y. Li, Chem. Commun., 2010, 46, 5551–5553 RSC.
- Q. Liu, M. Chen, Y. Sun, G. Y. Chen, T. S. Yang, Y. Gao, X. Z. Zhang and F. Y. Li, Biomaterials, 2011, 32, 8243–8253 CrossRef CAS PubMed.
- D. Costa, H. D. Burrows and M. da Graca Miguel, Langmuir, 2005, 21, 10492–10496 CrossRef CAS PubMed.
- L. L. Li, P. Wu, K. Hwang and Y. Lu, J. Am. Chem. Soc., 2013, 135, 2411–2414 CrossRef CAS PubMed.
- J. Shen, G. Chen, A. M. Vu, W. Fan, O. S. Bilsel, C. C. Chang and G. Han, Adv. Opt. Mater., 2013, 1, 644–650 CrossRef.
- X. Xie, N. Gao, R. Deng, Q. Sun, Q. H. Xu and X. Liu, J. Am. Chem. Soc., 2013, 135, 12608–12611 CrossRef CAS PubMed.
- Z. Kotkova, L. Helm, J. Kotek, P. Hermann and I. Lukes, Dalton Trans., 2012, 41, 13509–13519 RSC.
- D. Maffeo, M. Lampropoulou, M. Fardis, Y. G. Lazarou, I. M. Mavridis, D. A. I. Mavridou, E. Urso, H. Pratsinis, D. Kletsas and K. Yannakopoulou, Org. Biomol. Chem., 2010, 8, 1910–1921 CAS.
- J. M. Bryson, W. J. Chu, J. H. Lee and T. M. Reineke, Bioconjugate Chem., 2008, 19, 1505–1509 CrossRef CAS PubMed.
- A. Barge, G. Cravotto, B. Robaldo, E. Gianolio and S. Aime, J. Inclusion Phenom. Macrocyclic Chem., 2007, 57, 489–495 CrossRef CAS.
- Z. Zhou and Z. R. Lu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 1–18 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- F. X. Wan, T. K. Zhang, C. C. Li and L. Jiang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2017, 47, 288–293 CrossRef CAS.
- N. Rammohan, R. J. Holbrook, M. W. Rotz, K. W. MacRenaris, A. T. Preslar, C. E. Carney, V. Reichova and T. J. Meade, Bioconjugate Chem., 2017, 28, 153–160 CrossRef CAS PubMed.
- D. D. Jiang, X. P. Zhang, D. X. Yu, Y. N. Xiao, T. Q. Wang, Z. H. Su, Y. J. Liu and N. Zhang, J. Biomed. Nanotechnol., 2017, 13, 243–254 CrossRef CAS.
- F. Ye, J. Liu and H. Ouyang, Medicine, 2015, 94, e1157 CrossRef CAS PubMed.
- T. Jahanbin, H. Sauriat-Dorizon, P. Spearman, S. Benderbous and H. Korri-Youssoufi, Mater. Sci. Eng., C, 2015, 52, 325–332 CrossRef CAS PubMed.
- H. D. Cai, X. An, S. H. Wen, J. C. Li, G. X. Zhang, X. Y. Shi and M. W. Shen, Part. Part. Syst. Charact., 2015, 32, 934–943 CrossRef CAS.
- Y. Song, E. K. Kohlmeir and T. J. Meade, J. Am. Chem. Soc., 2008, 130, 6662–6663 CrossRef CAS PubMed.
- H. Idriss, F. Estour, I. Zgani, C. Barbot, A. Biscotti, S. Petit, C. Galaup, M. Hubert-Roux, L. Nicol, P. Mulder and G. Gouhier, RSC Adv., 2013, 3, 4531–4534 RSC.
- Y. L. Zhao, W. R. Dichtel, A. Trabolsi, S. Saha, I. Aprahamian and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 11294–11296 CrossRef CAS PubMed.
- T. Lecourt, A. Herault, A. J. Pearce, M. Sollogoub and P. Sinay, Chem. – Eur. J., 2004, 10, 2960–2971 CrossRef CAS PubMed.
- M. Menand, S. Adam de Beaumais, L. M. Chamoreau, E. Derat, S. Blanchard, Y. Zhang, L. Bouteiller and M. Sollogoub, Angew. Chem., Int. Ed., 2014, 53, 7238–7242 CrossRef CAS PubMed.
- E. Deunf, E. Zaborova, S. Guieu, Y. Bleriot, J. N. Verpeaux, O. Buriez, M. Sollogoub and C. Amatore, Eur. J. Inorg. Chem., 2010, 4720–4727 CrossRef CAS.
- S. Guieu and M. Sollogoub, Angew. Chem., Int. Ed., 2008, 47, 7060–7063 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- S. Aime, E. Gianolio, F. Arena, A. Barge, K. Martina, G. Heropoulos and G. Cravotto, Org. Biomol. Chem., 2009, 7, 370–379 CAS.
- I. Zgani, H. Idriss, C. Barbot, F. Djedaini-Pilard, S. Petit, M. Hubert-Roux, F. Estour and G. Gouhier, Org. Biomol. Chem., 2017, 15, 564–569 CAS.
- K. S. Kim, W. Park, J. Hu, Y. H. Bae and K. Na, Biomaterials, 2014, 35, 337–343 CrossRef CAS PubMed.
- M. H. Chan and H. M. Lin, Biomaterials, 2015, 46, 149–158 CrossRef CAS PubMed.
- M. Q. Tan, X. M. Wu, E. K. Jeong, Q. J. Chen and Z. R. Lu, Biomacromolecules, 2010, 11, 754–761 CrossRef CAS PubMed.
- F. J. Nicholls, M. W. Rotz, H. Ghuman, K. W. MacRenaris, T. J. Meade and M. Modo, Biomaterials, 2016, 77, 291–306 CrossRef CAS PubMed.
- M. A. Bruckman, S. Hern, K. Jiang, C. A. Flask, X. Yu and N. F. Steinmetz, J. Mater. Chem. B, 2013, 1, 1482–1490 RSC.
- E. Battistini, E. Gianolio, R. Gref, P. Couvreur, S. Fuzerova, M. Othman, S. Aime, B. Badet and P. Durand, Chem. – Eur. J., 2008, 14, 4551–4561 CrossRef CAS PubMed.
- Y. J. Liu and N. Zhang, Biomaterials, 2012, 33, 5363–5375 CrossRef CAS PubMed.
- J. M. Hu and S. Y. Liu, Acc. Chem. Res., 2014, 47, 2084–2095 CrossRef CAS PubMed.
- Z. Zhou, Z. Han and Z. R. Lu, Biomaterials, 2016, 85, 168–179 CrossRef CAS PubMed.
- D. A. Sipkins, D. A. Cheresh, M. R. Kazemi, L. M. Nevin, M. D. Bednarski and K. C. Li, Nat. Med., 1998, 4, 623–626 CrossRef CAS PubMed.
- T. Ke, E. K. Jeong, X. Wang, Y. Feng, D. L. Parker and Z. R. Lu, Int. J. Nanomed., 2007, 2, 191–199 CAS.
- X. L. Hu, X. G. Guan, J. Li, Q. Pei, M. Liu, Z. G. Xie and X. B. Jing, Chem. Commun., 2014, 50, 9188–9191 RSC.
- T. Meyer, J. F. Marshall and I. R. Hart, Br. J. Cancer, 1998, 77, 530–536 CrossRef CAS PubMed.
- D. P. J. Pan, A. H. Schmieder, S. A. Wickline and G. M. Lanza, Tetrahedron, 2011, 67, 8431–8444 CrossRef CAS PubMed.
- L. Tei, G. Gugliotta, M. Fekete, F. K. Kalman and M. Botta, Dalton Trans., 2011, 40, 2025–2032 RSC.
- M. Kueny-Stotz, A. Garofalo and D. Felder-Flesch, Eur. J. Inorg. Chem., 2012, 1987–2005 CrossRef CAS.
- S. Aime, M. Botta, E. Gianolio and E. Terreno, Angew. Chem., Int. Ed., 2000, 39, 747–750 CrossRef CAS PubMed.
- T. Lee, X. A. Zhang, S. Dhar, H. Faas, S. J. Lippard and A. Jasanoff, Chem. Biol., 2010, 17, 665–673 CrossRef CAS PubMed.
- A. Skjold, B. H. Amundsen, R. Wiseth, A. Stoylen, O. Haraldseth, H. B. Larsson and P. Jynge, J. Magn. Reson. Imaging, 2007, 26, 720–727 CrossRef PubMed.
- B. Drahos, I. Lukes and E. Toth, Eur. J. Inorg. Chem., 2012, 1975–1986 CrossRef CAS.
- H. J. Aronen, I. E. Gazit, D. N. Louis, B. R. Buchbinder, F. S. Pardo, R. M. Weisskoff, G. R. Harsh, G. R. Cosgrove, E. F. Halpern, F. H. Hochberg and B. R. Rosen, Radiology, 1994, 191, 41–51 CrossRef CAS PubMed.
- F. Kiessling, B. Morgenstern and C. Zhang, Curr. Med. Chem., 2007, 14, 77–91 CrossRef CAS PubMed.
- H. Lahrech, A. T. Perles-Barbacaru, S. Aous, J. F. Le Bas, J. C. Debouzy, A. Gadelle and P. H. Fries, J. Cereb. Blood Flow Metab., 2008, 28, 1017–1029 CrossRef CAS PubMed.
- F. Fauvelle, A. Gadelle, Y. Pailler, S. Aous and J. C. Debouzy, J. Inclusion Phenom. Macrocyclic Chem., 2002, 42, 203–207 CrossRef CAS.
- S. K. Imam, Cancer Biother. Radiopharm., 2005, 20, 163–172 CrossRef CAS PubMed.
- G. D. Luker and D. Piwnica-Worms, Acad. Radiol., 2001, 8, 4–14 CrossRef CAS PubMed.
- A. Kjaer, Adv. Exp. Med. Biol., 2006, 587, 277–284 CrossRef PubMed.
- P. Areses, M. T. Agueros, G. Quincoces, M. Collantes, J. A. Richter, L. M. Lopez-Sanchez, M. Sanchez-Martinez, J. M. Irache and I. Penuelas, Mol. Imaging Biol., 2011, 13, 1215–1223 CrossRef PubMed.
- V. Sharma, G. D. Luker and D. Piwnica-Worms, J. Magn. Reson. Imaging, 2002, 16, 336–351 CrossRef PubMed.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- T. Ido, C. N. Wan, V. Casella, J. S. Fowler, A. P. Wolf, M. Reivich and D. E. Kuhl, J. Labelled Compd. Radiopharm., 1978, 14, 175–183 CrossRef CAS.
- K. Hamacher, H. H. Coenen and G. Stocklin, J. Nucl. Med., 1986, 27, 235–238 CAS.
- O. Couturier, A. Luxen, J. F. Chatal, J. P. Vuillez, P. Rigo and R. Hustinx, Eur. J. Nucl. Med. Mol. Imaging, 2004, 31, 1182–1206 CrossRef CAS PubMed.
- N. Vasdev, J. Chio, E. M. van Oosten, M. Nitz, J. McLaurin, D. C. Vines, S. Houle, R. M. Reilly and A. A. Wilson, Chem. Commun., 2009, 5527–5529 RSC.
- H. Fonge, M. de Saint Hubert, K. Vunckx, D. Rattat, J. Nuyts, G. Bormans, Y. Ni, C. Reutelingsperger and A. Verbruggen, Bioorg. Med. Chem. Lett., 2008, 18, 3794–3798 CrossRef CAS PubMed.
- D. W. Bartlett, H. Su, I. J. Hildebrandt, W. A. Weber and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15549–15554 CrossRef CAS PubMed.
- D. W. Bartlett and M. E. Davis, Bioconjugate Chem., 2007, 18, 456–468 CrossRef CAS PubMed.
- J. Gallo, I. S. Alam, J. Jin, Y. J. Gu, E. O. Aboagye, W. T. Wong and N. J. Long, Dalton Trans., 2014, 43, 5535–5545 RSC.
- A. Haslop, L. Wells, A. Gee, C. Plisson and N. Long, Mol. Pharmaceutics, 2014, 11, 3818–3822 CrossRef CAS PubMed.
- S. L. Pimlott and A. Sutherland, Chem. Soc. Rev., 2011, 40, 149–162 RSC.
- M. C. Lasne, C. Perrio, J. Rouden, L. Barre, D. Roeda, F. Dolle and C. Crouzel, in Contrast Agents II. Topics in Current Chemistry, ed. W. Krause, Springer, Berlin, 2002, pp. 201–258 Search PubMed.
- C. C. Lee, G. Sui, A. Elizarov, C. J. Shu, Y. S. Shin, A. N. Dooley, J. Huang, A. Daridon, P. Wyatt, D. Stout, H. C. Kolb, O. N. Witte, N. Satyamurthy, J. R. Heath, M. E. Phelps, S. R. Quake and H. R. Tseng, Science, 2005, 310, 1793–1796 CrossRef CAS PubMed.
- J. M. Gillies, C. Prenant, G. N. Chimon, G. J. Smethurst, W. Perrie, I. Hamblett, B. Dekker and J. Zweit, Appl. Radiat. Isot., 2006, 64, 325–332 CrossRef CAS PubMed.
- H. Lusic and M. W. Grinstaff, Chem. Rev., 2013, 113, 1641–1666 CrossRef CAS PubMed.
- Y. Liu, K. Ai and L. Lu, Acc. Chem. Res., 2012, 45, 1817–1827 CrossRef CAS PubMed.
- A. J. Mieszawska, Y. Kim, A. Gianella, I. van Rooy, B. Priem, M. P. Labarre, C. Ozcan, D. P. Cormode, A. Petrov, R. Langer, O. C. Farokhzad, Z. A. Fayad and W. J. M. Mulder, Bioconjugate Chem., 2013, 24, 1429–1434 CrossRef CAS PubMed.
- N. Lee, S. H. Choi and T. Hyeon, Adv. Mater., 2013, 25, 2641–2660 CrossRef CAS PubMed.
- J. Zhou, Z. Lu, G. Shan, S. Wang and Y. Liao, Biomaterials, 2014, 35, 368–377 CrossRef CAS PubMed.
- M. He, P. Huang, C. L. Zhang, H. Y. Hu, C. C. Bao, G. Gao, R. He and D. X. Cui, Adv. Funct. Mater., 2011, 21, 4470–4477 CrossRef CAS.
- J. U. Voigt, Methods, 2009, 48, 92–97 CrossRef CAS PubMed.
- R. Gessner and P. A. Dayton, Mol. Imaging, 2010, 9, 117–127 Search PubMed.
- N. Deshpande, A. Needles and J. K. Willmann, Clin. Radiol., 2010, 65, 567–581 CrossRef CAS PubMed.
- L. Abou-Elkacem, S. V. Bachawal and J. K. Willmann, Eur. J. Radiol., 2015, 84, 1685–1693 CrossRef PubMed.
- R. Gramiak, P. M. Shah and D. H. Kramer, Radiology, 1969, 92, 939–948 CrossRef CAS PubMed.
- F. Cavalieri, A. El Hamassi, E. Chiessi, G. Paradossi, R. Villa and N. Zaffaroni, Biomacromolecules, 2006, 7, 604–611 CrossRef CAS PubMed.
- J. J. Rychak, J. R. Lindner, K. Ley and A. L. Klibanov, J. Controlled Release, 2006, 114, 288–299 CrossRef CAS PubMed.
- M. Lankford, C. Z. Behm, J. Yeh, A. L. Klibanov, P. Robinson and J. R. Lindner, Invest. Radiol., 2006, 41, 721–728 CrossRef PubMed.
- J. E. Streeter, R. C. Gessner, J. Tsuruta, S. Feingold and P. A. Dayton, Mol. Imaging, 2011, 10, 460–468 CAS.
- H. Wang, O. F. Kaneko, L. Tian, D. Hristov and J. K. Willmann, Invest. Radiol., 2015, 50, 322–329 CrossRef CAS PubMed.
- V. Ntziachristos and D. Razansky, Chem. Rev., 2010, 110, 2783–2794 CrossRef CAS PubMed.
- S. E. Bohndiek, S. Bodapati, D. Van De Sompel, S. R. Kothapalli and S. S. Gambhir, PLoS One, 2013, 8, e75533 CAS.
- Y. Wang, J. M. Thompson, A. G. Ashbaugh, P. Khodakivskyi, G. Budin, R. Sinisi, A. Heinmiller, M. van Oosten, J. M. van Dijl, G. M. van Dam, K. P. Francis, N. M. Bernthal, E. A. Dubikovskaya and L. S. Miller, J. Am. Acad. Orthop. Surg., 2017, 25, S7–S12 CrossRef PubMed.
- S. K. Maji, S. Sreejith, J. Joseph, M. J. Lin, T. C. He, Y. Tong, H. D. Sun, S. W. K. Yu and Y. L. Zhao, Adv. Mater., 2014, 26, 5633–5638 CrossRef CAS PubMed.
- Z. Kotkova, J. Kotek, D. Jirak, P. Jendelova, V. Herynek, Z. Berkova, P. Hermann and I. Lukes, Chem. – Eur. J., 2010, 16, 10094–10102 CrossRef CAS PubMed.
- A. S. Arbab and J. A. Frank, Regener. Med., 2008, 3, 199–215 CrossRef CAS PubMed.
- D. L. Kraitchman, W. D. Gilson and C. H. Lorenz, J. Magn. Reson. Imaging, 2008, 27, 299–310 CrossRef PubMed.
- W. A. Weber, A. L. Grosu and J. Czernin, Nat. Clin. Pract. Oncol., 2008, 5, 160–170 CrossRef CAS PubMed.
- Y. J. Lv, L. Hao, W. J. Hu, Y. Ran, Y. Bai and L. K. Zhang, Sci. Rep., 2016, 6, 29321 CrossRef CAS PubMed.
- H. Y. Su, Y. H. Liu, D. Wang, C. Q. Wu, C. C. Xia, Q. Y. Gong, B. Song and H. Ai, Biomaterials, 2013, 34, 1193–1203 CrossRef CAS PubMed.
- Y. Li, Y. F. Qian, T. Liu, G. Y. Zhang, J. M. Hu and S. Y. Liu, Polym. Chem., 2014, 5, 1743–1750 RSC.
- W. F. Lai and M. C. Lin, PLoS One, 2015, 10, e0126367 Search PubMed.
- W. F. Lai, A. S. Susha and A. L. Rogach, ACS Appl. Mater. Interfaces, 2016, 8, 871–880 CAS.
- Y. Inoue, T. Hakushi, Y. Liu, L. H. Tong, B. J. Shen and D. S. Jin, J. Am. Chem. Soc., 1993, 115, 475–481 CrossRef CAS.
- R. I. Gelb, S. Raso and J. S. Alper, Supramol. Chem., 1995, 4, 279–285 CrossRef CAS.
- Z. J. Liao, S. N. Du, Z. L. Qin, J. Y. Wang, F. Zuo and J. B. Luo, Chem. Phys. Lett., 2015, 637, 189–194 CrossRef CAS.
- W. Dow, US Pat., 8343463B2, 2013 Search PubMed.
- K. Hattori, Eur. Pat., 2194068B1, 2013 Search PubMed.
- J. B. Huff, C. Bieniarz and W. J. Horng, US Pat., 5661040A, 1994 Search PubMed.
- Y. Liu, M. Sun, H. Y. Zhang, X. Y. Hu and B. W. Liu, China Pat., 103301483B, 2014 Search PubMed.
- V. D. Rowe, US Pat., 8192721B2, 2012 Search PubMed.
- J. Platzek and H. Schmitt-Willich, US Pat., 6068831A, 2000 Search PubMed.
- R. Auzely-Velty, B. Perly and F. Djedaini-Pilard, US Pat., 6858723B1, 2005 Search PubMed.
- M. Stein, D. Heldmann, T. Fritzsch, J. Siegert and G. Roessling, US Pat., 6177062B1, 2001 Search PubMed.
- D. Heldmann, M. Stein, T. Fritzsch, J. Siegert, U. Speck and G. Roesling, Ger. Pat., 3803971C1, 1989 Search PubMed.
- J. Platzek and H. Schmitt-Willich, US Pat., 6113880A, 2000 Search PubMed.
- J. Klaveness, P. Rongved and L. Stubberud, US Pat., 5928626A, 1999 Search PubMed.
- A. Floris, S. Haq, M. I. Veld, D. B. Amabilino, R. Raval and L. Kantorovich, J. Am. Chem. Soc., 2016, 138, 5837–5847 CrossRef CAS PubMed.
- A. Nonat, C. F. Chan, T. Liu, C. Platas-Iglesias, Z. Y. Liu, W. T. Wong, W. K. Wong, K. L. Wong and L. J. Charbonniere, Nat. Commun., 2016, 7, 11978 CrossRef CAS PubMed.
- S. Chen, L. G. Xiao, X. J. Zhu, X. B. Peng, W. K. Wong and W. Y. Wong, Chem. Commun., 2015, 51, 14439–14442 RSC.
- C. Bakewell, G. Fateh-Iravani, D. W. Beh, D. Myers, S. Tabthong, P. Hormnirun, A. J. P. White, N. Long and C. K. Williams, Dalton Trans., 2015, 44, 12326–12337 RSC.
- A. Angelova, C. Ringard-Lefebvre and A. Baszkin, J. Colloid Interface Sci., 1999, 212, 280–285 CrossRef CAS PubMed.
- O. Penon, M. J. Marin, D. B. Amabilino, D. A. Russell and L. Perez-Garcia, J. Colloid Interface Sci., 2016, 462, 154–165 CrossRef CAS PubMed.
- W. Wang, N. K. Wong, M. D. Sun, C. Q. Yan, S. Y. Ma, Q. B. Yang and Y. X. Li, ACS Appl. Mater. Interfaces, 2015, 7, 8868–8875 CAS.
- W. F. Lai and H. C. Shum, Nanoscale, 2016, 8, 517–528 RSC.
- W. F. Lai and M. C. Lin, Curr. Gene Ther., 2015, 15, 472–480 CrossRef CAS PubMed.
- W. F. Lai and H. S. Jung, PLoS One, 2014, 9, e100258 Search PubMed.
- S. Shah, Y. Liu, W. Hu and J. Gao, J. Nanosci. Nanotechnol., 2011, 11, 919–928 CrossRef CAS PubMed.
- J. Chen, V. Kozlovskaya, A. Goins, J. Campos-Gomez, M. Saeed and E. Kharlampieva, Biomacromolecules, 2013, 14, 3830–3841 CrossRef CAS PubMed.
- H. Meng, S. Yang, Z. Li, T. Xia, J. Chen, Z. Ji, H. Zhang, X. Wang, S. Lin, C. Huang, Z. H. Zhou, J. I. Zink and A. E. Nel, ACS Nano, 2011, 5, 4434–4447 CrossRef CAS PubMed.
- O. Shimoni, A. Postma, Y. Yan, A. M. Scott, J. K. Heath, E. C. Nice, A. N. Zelikin and F. Caruso, ACS Nano, 2012, 6, 1463–1472 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- Z. X. Zhou, Y. Mondjinou, S. H. Hyun, A. Kulkarni, Z. R. Lu and D. H. Thompson, ACS Appl. Mater. Interfaces, 2015, 7, 22272–22276 CAS.
- S. Herrmann, G. Winter, S. Mohl, F. Siepmann and J. Siepmann, J. Controlled Release, 2007, 118, 161–168 CrossRef CAS PubMed.
- A. Streubel, J. Siepmann, N. A. Peppas and R. Bodmeier, J. Controlled Release, 2000, 69, 455–468 CrossRef CAS PubMed.
- Y. Zhou and X. Y. Wu, J. Controlled Release, 2003, 90, 23–36 CrossRef CAS PubMed.
- J. Nunes, K. P. Herlihy, L. Mair, R. Superfine and J. M. DeSimone, Nano Lett., 2010, 10, 1113–1119 CrossRef CAS PubMed.
- J. Y. Wang, Y. P. Wang, S. S. Sheiko, D. E. Betts and J. M. DeSimone, J. Am. Chem. Soc., 2012, 134, 5801–5806 CrossRef CAS PubMed.
- J. A. Champion, Y. K. Katare and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11901–11904 CrossRef CAS PubMed.
- C. C. Ho, A. Keller, J. A. Odell and R. H. Ottewill, Colloid Polym. Sci., 1993, 271, 469–479 CAS.
- J. Xu, D. H. C. Wong, J. D. Byrne, K. Chen, C. Bowerman and J. M. DeSimone, Angew. Chem., Int. Ed., 2013, 52, 6580–6589 CrossRef CAS PubMed.
- J. Y. Kelly and J. M. DeSimone, J. Am. Chem. Soc., 2008, 130, 5438–5439 CrossRef CAS PubMed.
- L. C. Glangchai, M. Caldorera-Moore, L. Shi and K. Roy, J. Controlled Release, 2008, 125, 263–272 CrossRef CAS PubMed.
- D. Kohler, M. Schneider, M. Kruger, C. M. Lehr, H. Mohwald and D. Y. Wang, Adv. Mater., 2011, 23, 1376–1379 CrossRef CAS PubMed.
- Z. Q. Yang, W. T. S. Huck, S. M. Clarke, A. R. Tajbakhsh and E. M. Terentjev, Nat. Mater., 2005, 4, 486–490 CrossRef CAS PubMed.
- V. Kozlovskaya, J. F. Alexander, Y. Wang, T. Kuncewicz, X. W. Liu, B. Godin and E. Khariampieva, ACS Nano, 2014, 8, 5725–5737 CrossRef CAS PubMed.
- H. Wang, S. T. Wang, H. Su, K. J. Chen, A. L. Armijo, W. Y. Lin, Y. J. Wang, J. Sun, K. Kamei, J. Czernin, C. G. Radu and H. R. Tseng, Angew. Chem., Int. Ed., 2009, 48, 4344–4348 CrossRef CAS PubMed.
- Y. Chen, Y. Pang, J. L. Wu, Y. Su, J. Y. Liu, R. B. Wang, B. S. Zhu, Y. F. Yao, D. Y. Yan, X. Y. Zhu and Q. Chen, Langmuir, 2010, 26, 9011–9016 CrossRef CAS PubMed.
- R. Nakaoka, Y. Tabata, T. Yamaoka and Y. Ikada, J. Controlled Release, 1997, 46, 253–261 CrossRef CAS.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 CAS.
- M. Gaumet, A. Vargas, R. Gurny and F. Delie, Eur. J. Pharm. Biopharm., 2008, 69, 1–9 CrossRef CAS PubMed.
- T. Banerjee, S. Mitra, A. Kumar Singh, R. Kumar Sharma and A. Maitra, Int. J. Pharm., 2002, 243, 93–105 CrossRef CAS PubMed.
- S. M. Moghimi, Adv. Drug Delivery Rev., 1995, 17, 61–73 CrossRef CAS.
- S. M. Moghimi, Adv. Drug Delivery Rev., 1995, 17, 103–115 CrossRef CAS.
- Y. Takakura, R. I. Mahato and M. Hashida, Adv. Drug Delivery Rev., 1998, 34, 93–108 CrossRef CAS PubMed.
- C. Park, H. Kim, S. Kim and C. Kim, J. Am. Chem. Soc., 2009, 131, 16614–16615 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Madler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- R. Hardman, Environ. Health Perspect., 2006, 114, 165–172 CrossRef PubMed.
- K. L. Aillon, Y. Xie, N. El-Gendy, C. J. Berkland and M. L. Forrest, Adv. Drug Delivery Rev., 2009, 61, 457–466 CrossRef CAS PubMed.
- S. Mosessian, S. M. Duarte-Vogel, D. B. Stout, K. P. Roos, G. W. Lawson, M. C. Jordan, A. Ogden, C. Matter, S. Sadeghi, G. Q. Mills, H. R. Schelbert, C. G. Radu, J. Czernin, M. Couto and M. E. Phelps, Mol. Imaging Biol., 2014, 16, 441–448 CrossRef PubMed.
- M. F. Kircher and J. K. Willmann, Radiology, 2012, 264, 349–368 CrossRef PubMed.
- X. Ai, L. Niu, Y. Li, F. Yang and X. Su, Talanta, 2012, 99, 409–414 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |