
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[3]Ferrocenophanes with the bisphosphanotetryl bridge: inorganic rings on the way to tetrylenes†‡
Denis
Kargin
a,
Zsolt
Kelemen
b,
Kristijan
Krekić
a,
Martin
Maurer
a,
Clemens
Bruhn
a,
László
Nyulászi
*b and
Rudolf
Pietschnig
*a
aUniversität Kassel, Institut für Chemie und CINSaT, Heinrich-Plett-Straße 40, 34132 Kassel, Germany. E-mail: pietschnig@uni-kassel.de
bBudapest University of Technology and Economics, Department of Inorganic and Analytical Chemistry, Szent Gellért tér 4, H-1111 Budapest, Hungary
First published on 29th October 2015
Abstract
A series of [3]ferrocenophanes with functional P–E–P motifs (E = group 14 fragments) is reported. Out of these, the silicon compounds with the general formula Fe(C5H4PtBu)2SiXY (XY = Cl2, Br2, I2, H2, HCl) have been characterized by spectroscopic means and the bonding situation was analyzed using X-ray crystallography and quantum chemical calculations. Despite the two stereogenic phosphanyl centers, most of the [3]ferrocenophanes have been obtained as single isomers in the course of stereospecific reactions. The corresponding stannylene Fe(C5H4PtBu)2Sn has been obtained in the form of its dimeric adduct.
Introduction
[n]Ferrocenophanes, in which n atoms connect the two cyclopentadienyl rings within a ferrocene molecule, are attractive monomers for the ring opening polymerization (ROP) to ferrocene based polymers.1–4 The ring strain present in ferrocenophanes with short bridging units (n = 1) leads to structural peculiarities usually described as ring tilt which may involve direct interaction of the iron center with the bridging atom.5,6 For bridging units with n = 2, 3 some ring tilts may still be present but the tendency for thermal ROP is significantly reduced.7–9 Recently the unique setting in [3]ferrocenophanes has been used to prepare ferrocenylene bridged carbenes where for the corresponding cation direct interactions between iron and the cationic carbon atom have been suggested.10,11 Owing to our own interest in the low-coordinate chemistry of group 14 elements we wondered whether it might be feasible to embed heavier tetrylenes into a bridging unit of a [3]ferrocenophane with flanking phosphanyl groups.12–17 Few years ago, Power et al. have shown that a low-valent silylene center can be stabilized using two adjacent third row elements.18 Therefore, we set out to investigate the possibility to access bisphosphano substituted tetrylenes embedded into a [3]ferrocenophane unit.Results and discussion
Synthesis of P–Si–P [3]ferrocenophanes
A straightforward approach to P–Si–P bridged [3]ferrocenophanes starts from secondary 1,1′-ferrocenylene bisphosphanes in which the bridging unit is closed by attachment of the group 14 elements. We selected bisphosphane 2 as the starting point of this investigation because the t-butyl groups should provide some steric protection and furthermore may serve as useful probes in proton NMR spectroscopy. Phosphane 2 is readily prepared with lithium aluminium hydride from the corresponding chlorophosphane 1 which has been reported earlier in the literature (Scheme 1).8,19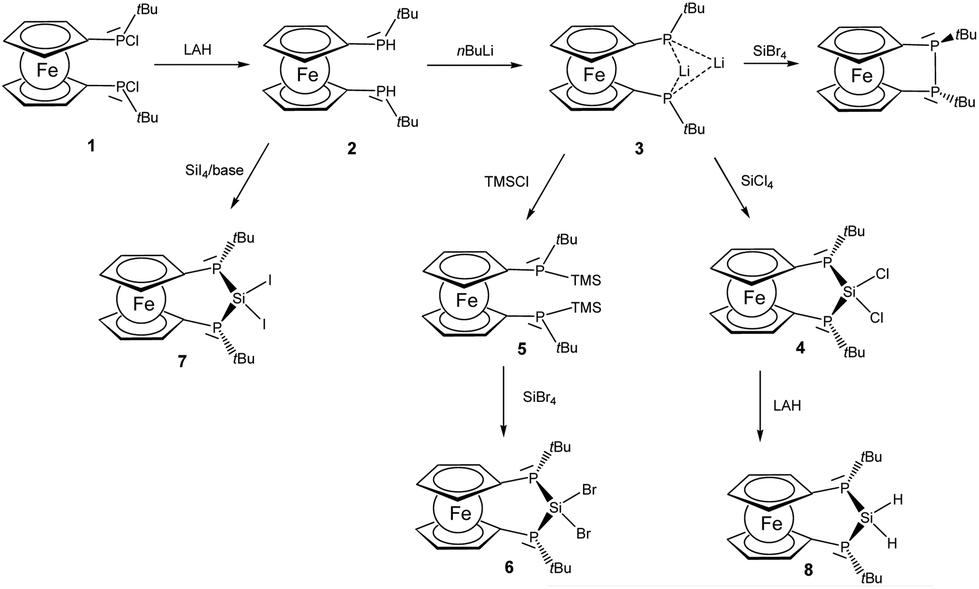 | ||
| Scheme 1 Formation of [3]ferrocenophanes 4, 6–8 (TMSCl: trimethylsilyl chloride, LAH: lithium aluminium hydride). | ||
Lithiation with n-BuLi affords dilithiophosphanide 3 which was identified by multinuclear NMR spectroscopy. The salient feature of compound 3 is the symmetric bridging of the two lithium atoms between the phosphorus atoms in solution which is evident from the 31P and 7Li NMR spectra showing identical coupling from each lithium to both phosphorus nuclei (Fig. 1) in analogy to related dimeric lithium and alkali phosphanides in the literature.20,21 Therefore, the stereochemical situation in 2 consisting of two diastereomers (rac and meso) is reset by lithiation which converts the phosphorus atoms to prochiral centers. We have also optimized the possible structures of 3 using B3LYP/6-311+G** DFT calculations, starting from asymmetric structures, with the tBu groups either at the same (“cis”) or at the opposite side (“trans”) of the phosphorus atoms. During the optimization of the “trans” structure the fully symmetrical 3 (Fig. S1 of the ESI‡) was obtained, which is in accordance with the NMR data. The “cis” structure 3′ (Fig. S1 of the ESI‡) could also be located, however, it is by 17.2 kcal mol−1 less stable.
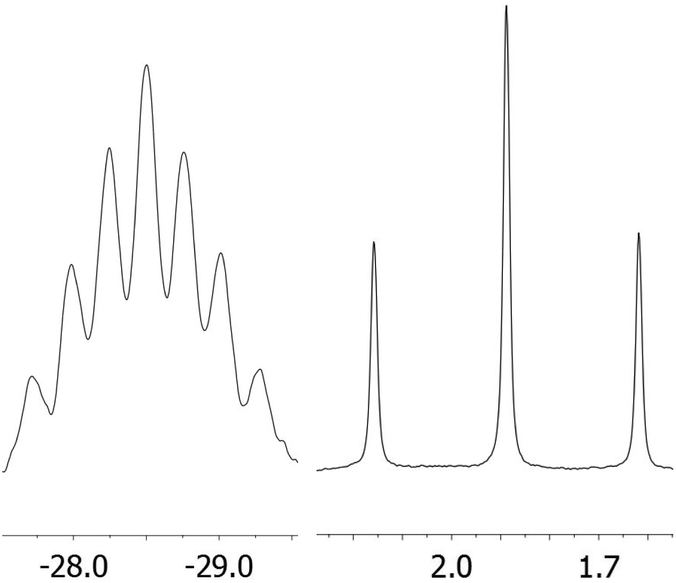 | ||
| Fig. 1 31P (left) and 7Li (right) NMR spectra of 3 in pentane solution. | ||
The reactivity of dilithiated 3 towards electrophilic silanes reveals a delicate balance between substitution and reduction where the electrophilicity of the silane is crucial. Thus the reaction of 3 with silicon tetrachloride furnishes P–Si–P bridged [3]ferrocenophane 4.
The 29Si resonance of compound 4 at 21.0 ppm indicates the presence of a dichlorosilane and the 1JSiP coupling pattern shows a triplet with a coupling constant of 88 Hz. Also the 31P NMR data indicate the presence of only one isomer in which the phosphanyl units are chemically and magnetically equivalent (δ(31P) = −38.8 ppm). By contrast the reaction of 3 with silicon tetrabromide is unselective and indicates predominant oxidation of 3 to the corresponding [2]ferrocenophane (CpPtBu)2Fe which is known from the literature.8 The connection of the two phosphanide units in 3 with a dibromosilanyl unit can, however, be accomplished by switching from a LiCl to a silyl halide elimination strategy. For this purpose phosphanide 3 is smoothly converted to the corresponding silylphosphane 5. From the multinuclear NMR data of 5 it is evident that the prochiral situation in 3 changes to a stereochemically unbiased situation in 5 where the ratio of rac and meso diastereomers occurs in a statistical mixture. For the meso form of 5 the molecular structure has been determined by X-ray diffraction and agrees well with the constitution established by spectroscopic methods (for details see ESI‡). The reaction of silylphosphane 5 with silicon tetrabromide then cleanly affords dibromosilane 6. Multinuclear NMR data of the bulk material show that during the formation of 6, just as in the case of 4, a stereospecific reaction occurs, in the course of which only a single isomer is formed. Since the reactivity of the silicon halide bond in these ferrocenophanes competes with that of the P–Si bonds we considered also the corresponding diiodosilane 7 which can be prepared by reacting 2 with SiI4 in the presence of a base. Again only a single diastereomer can be observed and the 29Si chemical shift at −63.5 ppm is the most shielded one observed for this series of bisphosphano dihalosilane bridged [3]ferrocenophanes, 4, 6 and 7. Within this series a clear trend can be derived from the heteronuclear NMR data (Table 1). With increasing electronegativity of the halide the silicon nuclei are deshielded, while in turn the corresponding phosphorus resonances appear at higher field. The value of the coupling constant 1JSiP continuously increases on moving from Cl to Br and I.
| P–E–P | δ(31P) [ppm] | δ(29Si) [ppm] | 3 J SiP [Hz] | |
|---|---|---|---|---|
| SiCl2 | 4 | −38.8 | +21.0 | 88 |
| SiBr2 | 6 | −30.8 | +1.6 | 102 |
| SiI2 | 7 | −18.4 | −63.5 | 112 |
Bonding situation in P–Si–P [3]ferrocenophanes 4, 6 and 7
For bisphosphanodihalosilyl bridged [3]ferrocenophanes 4 (Fig. 2), 6 (Fig. 3) and 7 (Fig. 4) single crystal structure determinations could be performed. All compounds are isostructural and reveal a stereochemical situation in which both t-butyl groups of the phosphanyl units point to the same side of the molecule which is in contrast to cyclic bisphosphano dichlorosilanes from the literature.22 Therefore the silyl units are shifted towards the opposite side where the lone pairs of the phosphanyl groups are located.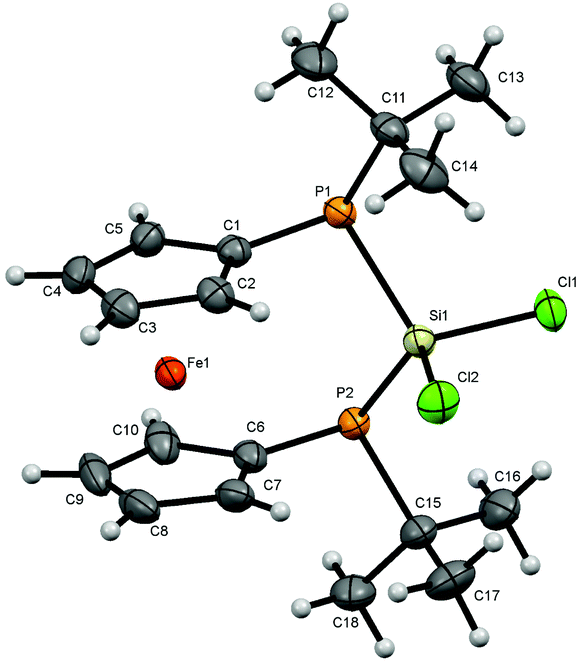 | ||
| Fig. 2 Molecular structure of 4. Ellipsoids are drawn at 30% probability level. | ||
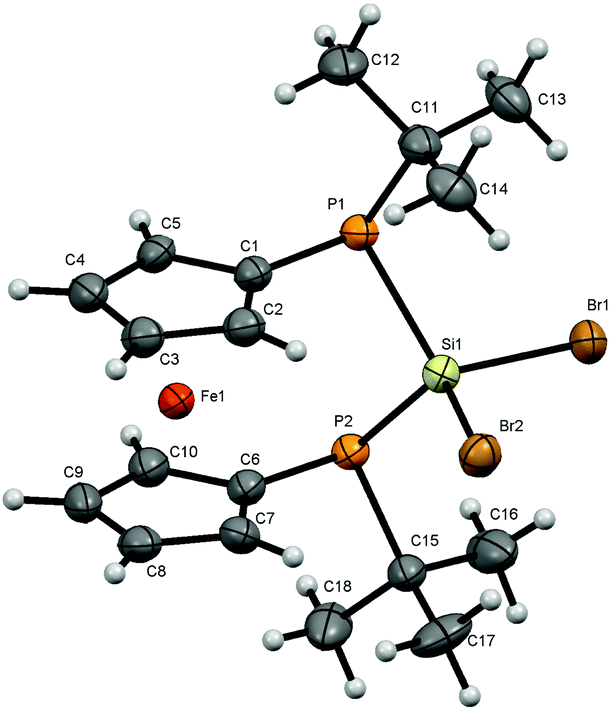 | ||
| Fig. 3 Molecular structure of 6. Ellipsoids are drawn at 30% probability level. | ||
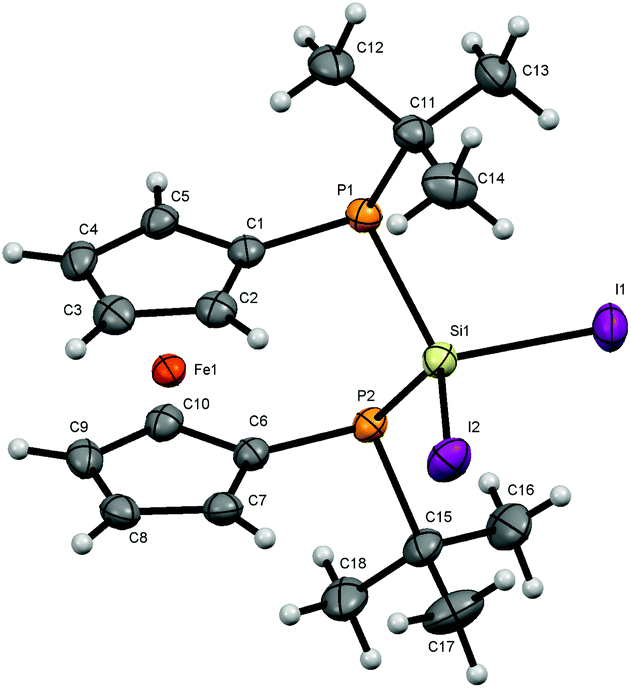 | ||
| Fig. 4 Molecular structure of 7. Ellipsoids are drawn at 30% probability level. | ||
This stereochemical arrangement of the reaction product is understandable by considering the structure of the reactant 3, for which no stable “trans”-like arrangement of the tBu groups could be located. While we did not investigate the mechanism of the reaction in detail here, it is apparent that any incoming silyl-halide reactant will interact at one side of the molecule and accordingly the tBu groups should be situated at the other side resulting in the observed stereochemical situation.
All bond lengths and angles for the halogenated compounds are without peculiarities. For each of the compounds the Cp-rings deviate slightly from coplanarity with angles between the normal to the ring plane being 3.52(6)° (4), 3.10(16)° (6) and 2.73(8)° (7) which are equal to the corresponding interplanar angles of the Cp-rings within a molecule. These [3]ferrocenophanes display almost unstrained structures in an eclipsed conformation with the ferrocene backbone opening towards the phosphorus lone pairs of the ansa bridge. The Fe⋯Si distance increases, corresponding to the size of the group 17 element (4: 3.7233(6) Å, 6: 3.7400(11) Å, 7: 3.7624(7) Å). The halogen atoms X show two positions, one with an almost linear Fe⋯Si–X arrangement (Fe⋯Si–Cl1 = 167.37(2)°, Fe⋯Si–Br1 = 167.73(4)°, Fe⋯Si–I1 = 165.09(2)°) and the other one with an almost perpendicular geometry (Fe⋯Si–Cl2 = 88.90(2)°, Fe⋯Si–Br2 = 89.83(3)°, Fe⋯Si–I2 = 90.63(2)°). In the case of substitution with chlorine and bromine, the bond length of the linearly arranged halogen (Si–Cl1 = 2.0831(5) Å, Si–Br1 = 2.2431(10) Å) is slightly longer than the perpendicular one (Si–Cl2 = 2.0755(5) Å, Si–Br2 = 2.2305(10) Å). In the case of iodine it is vice versa with the perpendicularly arranged halogen being further away (Si–I1 = 2.4736(7) Å, Si–I2 = 2.4835(6) Å). It may be anticipated that the difference in bond length could entail a gradual reactivity of the halogen atoms. The dihedral angle at the phosphorus atoms C1P1P2C6 and P1P2Si shows almost identical values for the halogenated compounds (4: 71.85(3)°, 6: 71.95(7)°, 7: 71.12(4)°) but varies in 8. Regarding the sum of the angles at the phosphorus atoms which are in the range between 314° and 317° for all compounds a pyramidal structure can be assumed (328° for tetrahedral conformation, 280° in PH3).23
The smallest angle is found in between the ipso-CPSi atoms with roughly 100°, indicating a small amount of ring-strain, and the largest angle is between the t-butyl groups and the silicon atom amounting to roughly 110°. The angle at the silicon atom between the geminal halogen atoms for the three derivatives 4, 6 and 7 shows values of Cl1SiCl2 = 103.73(2)°, Br1SiBr2 = 102.42(4)°, I1SiI2 = 104.28(2)°.
Our DFT calculations revealed that the other possible stereoisomers of 4, 6 and 7 in which the two t-butyl units at the two phosphorus atoms point to different sites of the molecule (Fig. S1 in the ESI‡) exhibit somewhat lower stability (by 3.5 kcal mol−1 for 4 and by 3.7 kcal mol−1 for 6 at the B3LYP/6-311+G** and 3.8 kcal mol−1 for 7 at the B3LYP/Def2-TZVP//B3LYP/6-311+G** levels of theory). Since the presence of the other isomer was not observed by NMR spectroscopy even after heating 6 in toluene to 100 °C for 10 hours, we calculated the barrier between the two isomers, to see whether the isomerisation is kinetically feasible. We could locate a transition state for the interconversion of the two isomers of 4 in which one of the phosphorus atoms is planarized (see Fig. S1 in the ESI‡). The energy of this transition structure is 19.2 kcal mol−1 at the B3LYP/6-311+G** and 21.6 kcal mol−1 at the M06-2X/6-311+G** levels of theory. Similarly, a 19.1 kcal mol−1 B3LYP/6-311+G** barrier was obtained for 6. This value is significantly smaller than the usual inversion barrier for phosphorus in phosphanes (for PH3 35 kcal mol−1 was reported).24,25 However, a similarly low (18 kcal mol−1 at B3LYP/6-311+G**)26 barrier was obtained for phosphole, where the reduction of the inversion barrier was attributed to the aromaticity of the planar transition structure.27 The lowering of the inversion barrier should at least partly be attributed to the neighbouring silyl group, since the calculated inversion barrier of silyl-phosphane (H3Si-PH2) is 23.8 kcal mol−1 at B3LYP/6-311+G**. Altogether this suggests that the interconversion at higher temperatures might be possible, and very probably the energy difference between the two isomers is large enough to prevent the observation of the isomer shown in Fig. S2 of the ESI.‡
To reveal any suggested direct interaction between the bridging Si atom and the iron center, the atoms in molecules (AIM) theory was applied but no bond critical point was observed between the Fe and the Si atoms in 4, 6 and 7.
Reactivity of P–Si–P [3]ferrocenophanes
Dihalosilanes 4, 6 and 7 are promising precursors for the reduction to the corresponding silylene. We tried several reagents to accomplish the dehalogenation including alkali metals, C8K, Collman's reagent, Na-naphthalenide and [(DipNacnac)Mg]2.28 While we obtained no reaction in the case of Na, K, Collman's and Jones reagents, we observed cleavage of the adjacent P–Si bond in the case of sodium-naphthalenide as indicated by the formation of P–P coupled products and further unidentified products with no 1JPSi-coupling in the 29Si NMR spectra. By contrast, the straightforward reduction of dihalosilane 4 to hydrosilane 8 is easily achieved by a reaction with lithium aluminium hydride. Again only a single diastereomer is obtained in which the two Si–H protons are chemically inequivalent resonating at 4.77 and 4.88 ppm. The 2JHH coupling of 9 Hz is augmented with 2JHP couplings which differ substantially for the two protons. Thus the hydrogen atom resonating at lower field shows a 2JHP coupling constant of 22 Hz while this coupling is <1 Hz for the Si–H resonating at higher field. Owing to the predominant s-character of the lone pair at phosphorus it seems plausible to assume that the Si–H oriented towards the phosphorus lone-pairs should show a higher JHP which corresponds to the more deshielded proton.The pronounced chemical and magnetic inequivalence of the Si–H protons indicates a markedly different local molecular environment for both hydrogen atoms as a consequence of the fixed stereo-chemical situation at the phosphorus centers. Although the latter is also the case for halogenated derivatives 4, 6 and 7 based on their crystal structures, in solution it is most easily visible for 8 using NMR spectroscopy. X-ray diffraction on single crystals of 8 allowed the determination of the molecular structure of 8 in the solid state (Fig. 5). Compound 8 crystallizes in a triclinic system and together with one molecule of solvent (benzene) (Fig. 5).
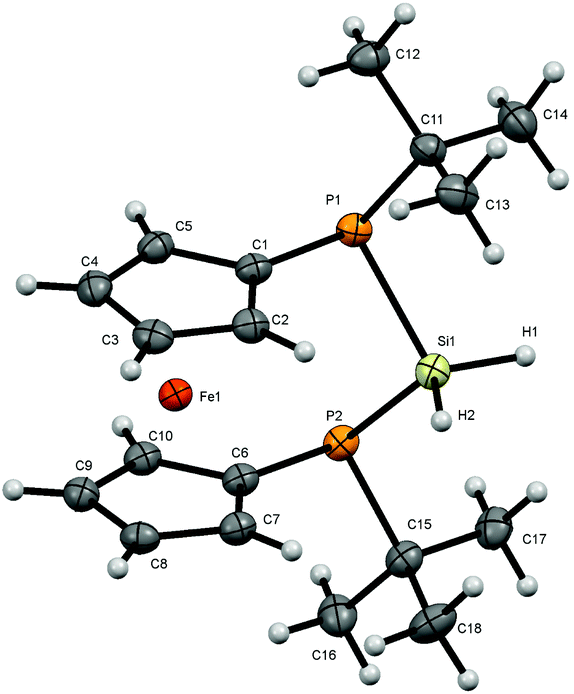 | ||
| Fig. 5 Molecular structure of 8. Ellipsoids are drawn at 30% probability level. One molecule of solvent (benzene) is omitted for clarity. | ||
The conformation of [3]ferrocenophane 8 is very similar to the ones found for halogenated derivatives 4, 6 and 7. Again the Cp-rings in 8 deviate slightly from coplanarity with an angle between the normal to the ring plane being 4.44(8)° which fits into the trend with the halogenated compounds, while the angles at the silicon center vary slightly. Both hydrogen atoms adjacent to the silicon atom have been located and refined. The deviation for an almost linear Fe⋯Si–H arrangement compared to the halogenated derivatives reaches 4° towards a higher angle (Fe⋯Si–H1 = 172.34(90)°) and for the corresponding perpendicular geometry 9° towards a smaller angle (Fe⋯Si–H2 = 80.17(91)°). The Si–H bond lengths show the same behavior as mentioned before with the linear arrangement with respect to the iron center having a slightly longer distance to the silicon atom. Regarding the sum of the angles at the phosphorus atoms, smaller angles in the range between 310° and 311°, combined with the smaller angle at the silicon atom PSiP = 98.54(3)° suggest a slightly higher ring strain within the bridge of [3]ferrocenophane 8 compared with the previously discussed compounds in which this angle increases with the electronegativity of the halide (4: P1–Si1–P2 = 102.46(2)°, 6: P1–Si1–P2 = 100.73(5)°, 7: P1–Si1–P2 = 100.21(3)°). At the same time the angle at the silicon atom in between the hydrogen atoms is 4° to 6° larger than in the halogenated structures (H1SiH2 = 108(1)°). Considering the smaller electronegativity of hydrogen compared to halogens, the increased bond angle is understandable according to the VSEPR theory.29 A pyramidal structure at the phosphorus centers can be confirmed as well.
The dihedral angle between the planes C1P1P2C6 and P1P2Si differs by about 4° to 5° towards a bigger angle (75.38(4)°) compared with the halogenated derivatives. The distance Fe–X for the almost perpendicular geometry is the shortest for the displayed [3]ferrocenophanes (Fe–H2 = 3.7305(195) Å, Fe–Cl2 = 4.2277(6), Fe–Br2 = 4.3490(7), Fe–I2 = 4.5319(7) Å). Moreover, the Fe⋯Si distance with 3.7139(7) Å is also the shortest among the compounds presented here.
Since the Si–H protons in 8 serve as spectroscopic probes for the difference in the chemical environment of the faces of the ferrocenophane ring, the spectroscopic findings suggest a potentially gradual reactivity for these two hydrogen atoms. This situation is more pronounced for different substituents at silicon for instance in hydrochlorosilane 9 which is readily available from silicochloroform and lithiophosphanide 3. Owing to the difference in facial orientation above and below the ferrocenophane ring which originates from the orientation of the phosphanyl lone pairs and substituents, two isomers are obtained in the formation of 9 (Scheme 2).
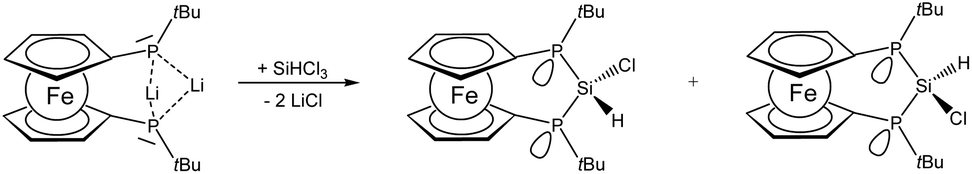 | ||
| Scheme 2 Formation of [3]ferrocenophane 9 as two diastereomers. | ||
The isomers of 9 mainly differ in the relative orientation of the substituents at silicon which has substantial impact on the NMR spectroscopic data of the nuclei involved. Again the Si–H proton can be used as a spectral probe to assign the resonances to the respective isomers based on the plausible assumption that orientation towards the phosphorus lone-pairs results in an increased JHP. Indeed one isomer of 9 shows a markedly higher value for 2JHP of 26 Hz than the other one (<1 Hz), which was also in good agreement with our M06-2X/6-311+G** 2JHP values (1.9 and 27.1 Hz for the two isomers, the latter value indeed belonging to hydrogen pointing towards the P lone pair). The larger 2JHP coupling is accompanied by a deshielded resonance of the corresponding proton at 6.54 ppm while for the isomer with hydrogen pointing away from the phosphorus lone pairs a higher field resonance at 5.91 ppm is observed. Also in the 31P and 29Si NMR spectra shift differences of roughly 20 ppm are found for these two isomers. The isomer with the deshielded Si–H proton resonance shows also more deshielded resonances in the 31P (δ = −41.7 ppm) and 29Si (δ = +5.8 ppm) NMR spectra compared with the other isomer showing higher field resonances (δ(31P) = −62.3 ppm, δ(29Si) = −13.2 ppm). For the isomer of 9 with the Si–H pointing towards the phosphorus lone-pairs in turn the lone-pairs should adopt an anti periplanar arrangement with respect to chlorine. The interaction of σ* states associated with the Si–Cl bond may become populated and the corresponding bond order reduced which would explain the general trend of deshielding, as in related cases.15 The marked difference in the chemical environment for the two exocyclic substituents at silicon in the two isomers of 9 is evident from the spectral data and potentially may be even used to differentiate between the two isomers in terms of reactivity. The ratio of the isomers of 9 slightly varies depending on the reaction conditions employed. At room temperature a ratio of 1.00![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.75 is obtained, while at lower temperatures a ratio of 1.00:0.54 was observed at −20 °C and −80 °C, respectively. The B3LYP/6-311+G** and M06-2X/6-311+G**//B3LYP/6-311+G** energy difference between the two isomers was only 1.2 and 0.6 kcal mol−1, respectively. This small computed energy difference is in agreement with the observation of both isomers. However, we have not been able to achieve the formation of only one out of the two isomers of 9 and also their separation from the mixture remained unsuccessful so far. According to our DFT calculations the interconversion barrier is similar (21.2 kcal mol−1 at the B3LYP/6-311+G** level of theory) to that of 4. Given, however, that in this case the reaction temperature was rather low, it is likely that the two isomers were already formed during the reaction of 3 with SiHCl3.
:0.75 is obtained, while at lower temperatures a ratio of 1.00:0.54 was observed at −20 °C and −80 °C, respectively. The B3LYP/6-311+G** and M06-2X/6-311+G**//B3LYP/6-311+G** energy difference between the two isomers was only 1.2 and 0.6 kcal mol−1, respectively. This small computed energy difference is in agreement with the observation of both isomers. However, we have not been able to achieve the formation of only one out of the two isomers of 9 and also their separation from the mixture remained unsuccessful so far. According to our DFT calculations the interconversion barrier is similar (21.2 kcal mol−1 at the B3LYP/6-311+G** level of theory) to that of 4. Given, however, that in this case the reaction temperature was rather low, it is likely that the two isomers were already formed during the reaction of 3 with SiHCl3.
Formal dehydrohalogenation of 9 should lead to the envisaged silylene species, for which higher reactivity compared with N-heterocyclic silylenes can be anticipated owing to the limited π-donor properties of the phosphanyl unit. However, compound 9 does not react with neutral or anionic nitrogen bases like DBU, DABCO, NEt3 and LiNTMS2 or NaNTMS2. This is in marked contrast to strained [1]ferrocenophanes with a bridging >SiHCl unit.30 Also N-heterocyclic carbenes (NHCs) induced no reaction of compound 9 and did not lead to the envisaged silylene–NHC adducts.31
Synthesis of P–E–P [3]ferrocenophanes (E = Sn, Ge)
Since the generation of a bisphosphano substituted silylene turned out to be difficult, we wondered whether the heavier congeners in group 14 would be more feasible owing to the higher stability of the divalent state for these elements.The reaction of 3 with SnCl2 furnishes a product where the heteronuclear NMR spectra reveal the formation of the P–Sn–P unit. However, the two phosphorus centers have become inequivalent showing substantially different 31P chemical shift values of −32.5 ppm and −94.1 ppm with a much larger coupling of 1JSnP 1179 Hz for the phosphorus nucleus resonating at higher field compared with 1JSnP 702 Hz for the more deshielded phosphorus nucleus (Fig. 6).
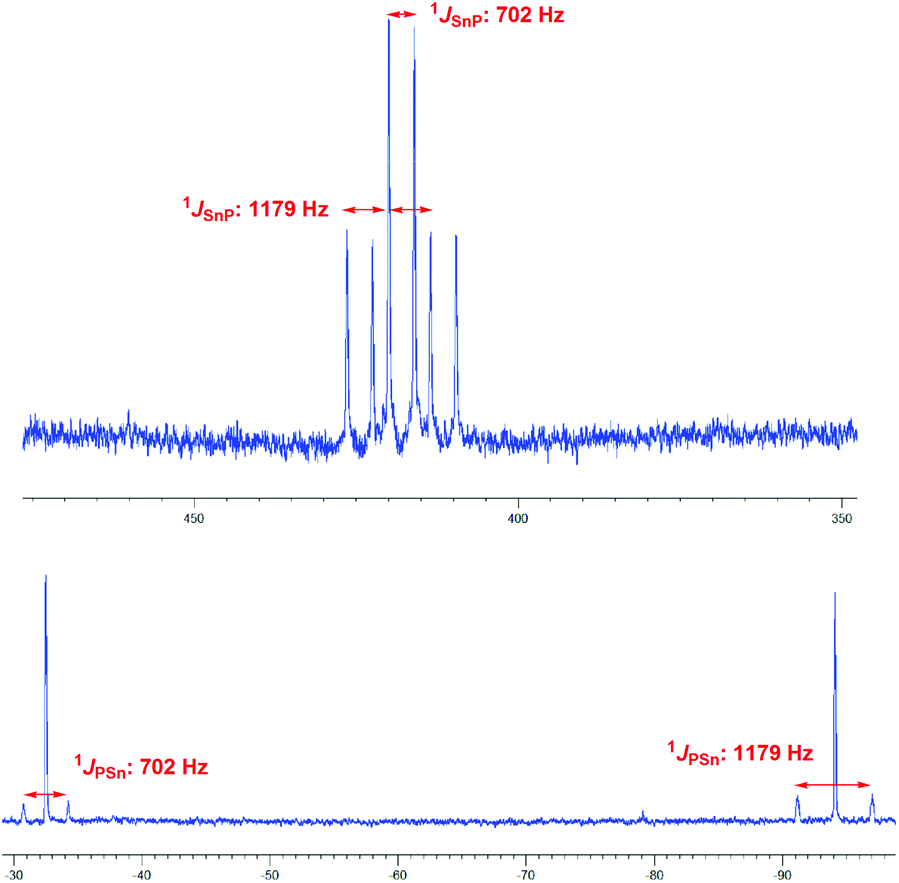 | ||
| Fig. 6 119Sn-NMR (top) and 31P-NMR (bottom) resonances of 10 in pentane/tetrahydrofuran solution. | ||
Based on these data and the splitting pattern for the 119Sn resonance at +418 ppm the formation of stannylene dimers or oligomers seems plausible in which the stannylene units mutually form donor adducts with one of the phosphane units of each other. Owing to its high symmetry the absence of other isomers and the presence of only two resonances for tert-butyl groups in the proton NMR spectra, the dimeric form 10 would be consistent with the observed data (Scheme 3). The instability of the compound precluded further characterization. The bond motif present in 10 has been first established by Du Mont et al. for acyclic bisphosphano stannylenes several years ago and the NMR data are in excellent agreement with the reported data.32 The reaction of 3 with GeCl2 furnishes a comparable product with 31P chemical shift resonances −30.8 ppm and −90.9 ppm, however, the product is prone to decomposition and could not be isolated. These findings suggest that the >P–E–P< motif (E = group 14 elements) can be achieved, however, the steric protection exerted by the t-butyl groups at phosphorus is obviously insufficient to prevent dimerization.
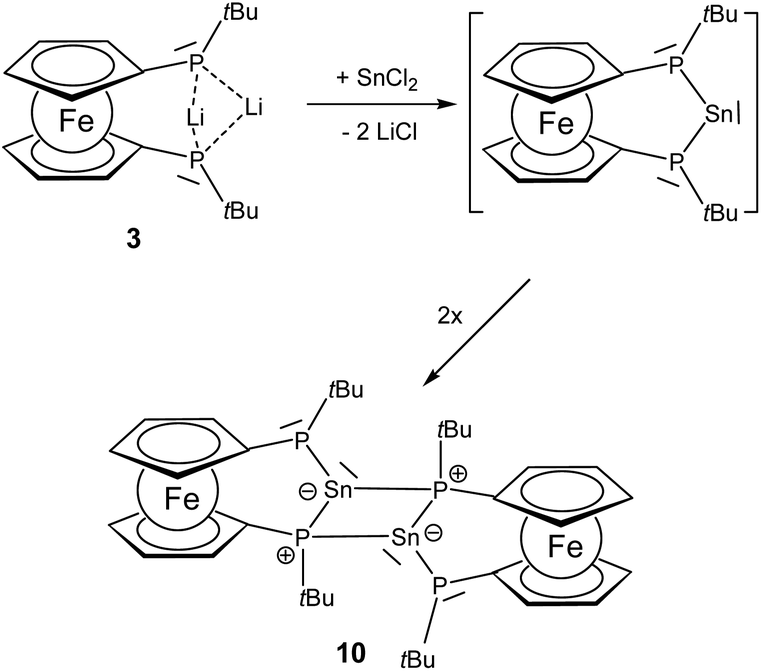 | ||
| Scheme 3 Illustration of Sn based [3]ferrocenophanes with the tentative formation of dimeric 10. | ||
Our DFT calculations showed that the dimerization energy to form the four membered ring is −29.8 kcal mol−1 for the germylene at the M06-2X/cc-pvDZ level of theory, while for the stannylene a slightly smaller value (−28.6 kcal mol−1) was obtained at the M06-2X/cc-pvDZ level of theory (further methods and computational details are given in Table S1 in the ESI‡). The isomeric dimeric structure with an E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E bond has also been optimized, exhibiting the expected trans-bent structure (Fig. S3 in the ESI‡), which has similar, but somewhat reduced stability both for E: Ge and Sn (E: Ge ΔEEE bonded–EP bonded = −2.0 kcal mol−1 and for E: Sn ΔEEE bonded–EP bonded = −3.5 kcal mol−1 at the B3LYP/cc-pvDZ level of theory). Similar relative stability was reported for aminosilylenes.33 Since the energy of the dimerization is small, the entropy contribution has a significant effect and the room temperature calculated gas phase Gibbs free energy of the dimerization process is not extremely high (ΔG = −12.3 for E: Ge and ΔG = −28.6 for E: Sn). Thus, we predict that with bigger substituents at the phosphorus atoms this dimerization process can be blocked, or alternatively, an equilibrium between the monomeric and dimeric structures might be feasible for both compounds.
E bond has also been optimized, exhibiting the expected trans-bent structure (Fig. S3 in the ESI‡), which has similar, but somewhat reduced stability both for E: Ge and Sn (E: Ge ΔEEE bonded–EP bonded = −2.0 kcal mol−1 and for E: Sn ΔEEE bonded–EP bonded = −3.5 kcal mol−1 at the B3LYP/cc-pvDZ level of theory). Similar relative stability was reported for aminosilylenes.33 Since the energy of the dimerization is small, the entropy contribution has a significant effect and the room temperature calculated gas phase Gibbs free energy of the dimerization process is not extremely high (ΔG = −12.3 for E: Ge and ΔG = −28.6 for E: Sn). Thus, we predict that with bigger substituents at the phosphorus atoms this dimerization process can be blocked, or alternatively, an equilibrium between the monomeric and dimeric structures might be feasible for both compounds.
Conclusions
In summary, we have prepared a series of [3]ferrocenophanes with functional P–E–P motifs (E = group 14 fragments). Although these compounds are promising precursors for the generation of the corresponding tetrylenes, only in the case of tin a stannylene has been obtained in the form of a dimeric mutual donor acceptor adduct. With respect to the stereogenic phosphanyl centers, compounds 4, 6–8 are formed in a surprisingly stereospecific reaction from prochiral 3 as well as from rac–meso mixed 2 and 5. The molecular structures based on X-ray diffraction indicate no relevant ring strain in these [3]ferrocenophanes which suggests a low propensity to undergo thermal ROP. Future work will focus on increasing the steric congestion at the flanking phosphanyl units to prevent dimerization and to tame the reactivity of the related tetrylenes.Experimental
All manipulations were carried out under strict exclusion of moisture and air under an inert argon atmosphere. All used solvents were dried over sodium potassium alloy or CaH2 and distilled prior to use. Starting materials (CpP(Cl)tBu)2Fe, SiBr4 and DipNHC were synthesized according to the literature procedures.8,34,351H, 13C, 31P, 29Si and 119Sn-NMR spectra were recorded on a Varian VNMRS-500 MHz or MR-400 MHz-spectrometer at room temperature using TMS as the external reference for 1H, 13C and 29Si. ESI-MS spectra have been recorded on a microTOF (Bruker Daltonics). LIFDI-MS spectra have been recorded on an AccuTOF GCv (Jeol).X-ray diffraction measurements
X-ray diffraction measurements were performed on a Stoe 4CD or a Bruker-AXS SMART APEX 2 CCD diffractometer using graphite-monochromated Mo Kα radiation or a Stoe StadiVari with a Dectris Pilatus 200 K detector using monochromated Cu Kα radiation (Table 2). The structures were solved using direct methods and refined by full-matrix least-squares techniques against F2 (SHELXL-2014/6).36 Details of the structure determinations and refinement for 4, 5, 6, 7 and 8 are summarized in Table 2. Further programs used for analysis and visualisation of structural information include WinGX, Parst and Mercury.37–39 Supplementary crystallographic data for this paper can be obtained by quoting CCDC 1417298–1417301 and 1434408.| 4 | 6 | 8 | 7 | |
|---|---|---|---|---|
| CCDC code | 1417298 | 1417299 | 1417300 | 1417301 |
| Empirical formula | C18H26Cl2FeP2Si | C18H26Br2FeP2Si | C18H28FeP2Si·C6H6 | C18H26FeI2P2Si |
| Formula weight | 459.17 | 548.07 | 468.39 | 642.07 |
| Crystal description | Yellow block | Yellow plate | Yellow plate | Yellow block |
| Crystal size [mm] | 0.49 × 0.22 × 0.20 | 0.30 × 0.27 × 0.04 | 0.380 × 0.350 × 0.120 | 0.290 × 0.183 × 0.110 |
| Crystal system, space group | Monoclinic, P21/n | Monoclinic, P21/n | Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Monoclinic, P21/n |
| Radiation and λ [Å] | Mo Kα, 0.71073 | Mo Kα, 0.71073 | Mo Kα, 0.71073 | Mo Kα, 0.71073 |
| Monochromator | Plane graphite | Plane graphite | Plane graphite | Graded multilayer mirror |
| Temperature [K] | 143(2) | 100(2) | 100(2) | 173(2) |
| Unit cell dimensions: | ||||
| a [Å] | 10.5219(10) | 6.4147(3) | 6.1180(5) | 10.7839(8) |
| b [Å] | 12.3405(6) | 17.8803(6) | 12.6634(11) | 11.3855(6) |
| c [Å] | 16.7560(14) | 18.9078(10) | 16.8515(14) | 18.8031(14) |
| α [°] | 90.00 | 90.00 | 109.022(6) | 90.00 |
| β [°] | 99.201(7) | 99.599(4) | 94.610(6) | 90.907(6) |
| γ [°] | 90.00 | 90.00 | 102.104(7) | 90.00 |
| Volume [Å3] | 2147.70(25) | 2138.30(15) | 1191.11(40) | 2308.36(3) |
| Z | 4 | 4 | 2 | 4 |
| Calculated density | 1.420 | 1.703 | 1.306 | 1.848 |
| F(000) | 952 | 1096 | 496 | 1240 |
| Linear absorption coefficient μ [mm−1] | 1.155 | 4.647 | 0.826 | 3.518 |
| Absorption correction | Integration | Integration | Integration | Integration |
| Unit cell determination | STOE X-area | STOE X-area | STOE X-area | STOE X-area |
| Diffractometer | STOE IPDS 2 | STOE IPDS 2 | STOE IPDS 2 | STOE StadiVari |
| Radiation source | Genix Mo HF | Genix Mo HF | Genix Mo HF | Mo Genix |
| Scan type | Omega scan | Omega scan | Omega scan | Omega scan |
| θ Range for data collection | 1.65–25.33 | 1.58–25.96 | 1.30–25.64 | 1.78–28.75 |
| Index ranges | −11 < h < 12 | −7 < h < 7 | −7 < h < 7 | −13 < h < 13 |
| −13 < k < 14 | −21 < k < 21 | −15 < k < 15 | −14 < k < 8 | |
| −19 < l < 19 | −18 < l < 22 | −20 < l < 20 | −23 < l < 23 | |
| Refl. collected/unique | 10922/3709 |
11865/3772 |
9043/4438 | 28242/5013 |
| Significant unique refl. | 3203 | 3283 | 4178 | 4439 |
| R(int), R(σ) | 0.0221, 0.0206 | 0.0803, 0.0512 | 0.0470, 0.0342 | 0.0493, 0.0242 |
| Completeness to θ = 26.0° | 0.984 | 1.000 | 0.985 | 0.997 |
| Refinement method | SHELXL-2014 | SHELXL-2014 | SHELXL 2014 | SHELXL-2014 |
| Data/parameters/restraints | 3709/223/0 | 3772/223/0 | 4438/265/0 | 5013/223/0 |
| Goodness-of-fit on F2 | 0.954 | 1.0340 | 1.066 | 1.0470 |
| Final R indices [I > 2σ(I)] | 0.0200 | 0.0374 | 0.0345 | 0.0227 |
| R indices (all data) | 0.0251 | 0.0439 | 0.0357 | 0.0277 |
| Largest difference peak/hole [e Å−3] | 0.2700/−0.1900 | 0.6200/−0.9000 | 0.364/−0.655 | 0.7400/−0.3200 |
Quantum chemical calculations
Quantum chemical calculations were performed using the Gaussian 09 suite of programs.40 Geometry optimizations were carried out using the M06-2X or B3LYP functional with the 6-31G*, 6-31+G*, and 6-311+G** cc-pVDZ basis sets. For the Sn and I containing systems Def2-TZVP basis was used for the Sn and I atoms. For the energy difference between the isomeric/dimeric structures, further single point energy calculations were carried out at the B3LYP/Def2-TZVP level. The nature of the stationary point obtained by geometry optimization has been verified by a subsequent analysis of the second derivatives, which have been found all positive in the case of minima, and exhibited a single negative value in the case of transition states. The structures were visualized by the MOLDEN package.41 The topological analysis of the electron density was carried out by the AIMAll package.42Synthesis of 2
To a suspension of LiAlH4 (637 mg, 16.8 mmol) in 20 mL of Et2O, a solution of 1 (7.26 g, 16.8 mmol) in 100 mL Et2O was added. The reaction mixture was stirred for 15 minutes and degassed water was added until the evolution of gas stopped. The residue was extracted with Et2O and the organic layer was washed with water and brine. The organic layer was dried with MgSO4 and the solvent was removed in vacuo. Recrystallization from pentane yielded 85% product (5.15 g, 14.2 mmol) as orange crystals.31P NMR (202 MHz, C6D6) δ: −26.9 (d, 1JPH = 206 Hz), −27.4 (d, 1JPH = 208 Hz) ppm; 1H NMR (400 MHz, C6D6) δ: 4.32–4.26 (pseudo d, J = 6.1 Hz, 2H Cp), 4.20–4.14 (pseudo d, J = 7.7 Hz, 4H Cp), 4.10–4.04 (pseudo d, J = 10.4 Hz, 2H Cp), 3.95 (d, 1JPH = 207.6 Hz, 1H PH), 3.93 (d, 1JPH = 204.3 Hz, 1H PH), 1.09–1.02 (pseudo d, 3JPH = 12.0 Hz, 18H CH3) ppm; 13C NMR (101 MHz, C6D6) δ: 78.0–77.0 (m Cp), 76.8–76.3 (m Cp), 72.7–72.4 (m Cp), 71.8–71.5 (m Cp ipso C), 30.1–29.6 (m tBu CH3), 29.0–28.8 (m tBu C q) ppm. Elemental analysis calculated: C: 59.69, H: 7.79, found: C: 59.97, H: 8.02. MS (ESI-HR) m/z 831.110591 ([2M + Ag]+ 100%) calculated: 831.108243.
Synthesis of 3
To a solution of 2 (362 mg, 1 mmol), a solution of n-BuLi in hexane (0.88 mL, 2 mmol, 2.5 M) and TMEDA (0.33 mL, 2 mmol) were added at room temperature. This mixture was stirred for 12 h forming a yellow solid within a red solution. This slurry was used in situ for further reactions.31P NMR (202 MHz, C6D6) δ: −28.5 (sept) ppm; 7Li NMR (194 MHz, C6D6) δ: 1.89 (t, 1JPLi = 52.4 Hz) ppm.
Synthesis of 4
SiCl4 (0.88 ml, 2 mmol) was added to the stirred slurry of 3 at room temperature and stirring was continued for 10 minutes. All volatile components were removed in vacuo, the residue was extracted with 30 mL of pentane and the solid was removed and discarded. The pentane extract was evaporated to dryness yielding the raw product. Recrystallization from pentane afforded 91% yield (417 mg, 0.91 mmol) as orange crystals.31P NMR (202 MHz, C6D6) δ: −38.8 (1JPsi = 88 Hz) ppm; 29Si NMR (99 MHz, C6D6) δ: 21.0 (t, 1JPsi = 88 Hz) ppm; 1H NMR (500 MHz, C6D6) δ: 4.64–4.61 (m, 2H Cp), 4.13–4.10 (m, 2H Cp), 4.07–4.04 (m, 2H Cp), 4.01–3.98 (m, 2H Cp), 1.34–1.29 (m, 18H CH3) ppm; 13C NMR (126 MHz, C6D6) δ: 79.6 (pseudo t, JPC = 22.2 Hz, Cp), 76.2 (pseudo t, JPC = 3.8 Hz, Cp), 73.2 (m Cp), 72.2 (pseudo t, JPC = 5.4 Hz, Cp), 66.0 (pseudo t, JPC = 10.0 Hz, Cp ipso C), 34.0–33.8 (m tBu C q), 31.2 (pseudo t, 6.9 Hz, tBu CH3) ppm. Elemental analysis (%) for C18H26Cl2FeP2Si·0.15C18H28FeP2 calculated: C: 48.624, H: 5.961, found: C: 48.778, H: 5.820. MS (LIFDI-HR): m/z 362.10107 ([M − SiCl2 + 2H]+ 100%), 396.06346 ([M − SiCl + H]+ 3%) calculated: 396.06259.
Synthesis of 5
TMSCl (0.25 ml, 2 mmol) was added to the stirred slurry of 3 at room temperature and stirring was continued for 10 minutes. All volatile components were removed in vacuo, the residue was extracted with 30 mL of pentane and the solid was removed and discarded. The pentane extract was evaporated to dryness yielding the raw product. Recrystallization from pentane afforded an orange-brown powder in 80% yield (407 mg, 0.8 mmol).31P NMR (202 MHz, C6D6) δ: −48.4 (1JPsi = 32 Hz), −48.6 (1JPsi = 32 Hz) ppm; 29Si NMR (99 MHz, C6D6) δ: −1.5 (t, 1JPsi = 32 Hz); −1.4 (t, 1JPsi = 32 Hz) ppm; 1H NMR (400 MHz, C6D6) δ: 4.34–4.30 (m, 2H Cp), 4.30–4.27 (m, 2H Cp), 4.27–4.22 (m, 2H Cp), 4.14–4.09 (m, 2H Cp), 1.28–1.21 (m, 18H tBu CH3), 0.35–0.26 (m, 18H TMS CH3) ppm; 13C NMR (101 MHz, C6D6) δ: 77.2–76.7 (m ipso C Cp), 74.6–74.2 (m Cp), 73.9–73.5 (m Cp), 72.6–72.3 (m Cp), 72.3–71.9 (m Cp), 32.1–31.8 (m tBu CH3), 31.5–31.3 (m tBu C q), 2.1–1.8 (m TMS CH3) ppm.
Synthesis of 6
To a solution of 5 (2.02 g, 4 mmol) in 15 mL toluene, SiBr4 (0.56 mL, 4.5 mmol) was added at room temperature and the reaction mixture was heated to 100 °C for 4 hours. All volatile components were removed in vacuo, the residue was extracted with 40 mL of pentane and the solid was removed and discarded. The pentane extract was evaporated to dryness yielding the raw product. Recrystallization from pentane afforded 6 in 63% yield (1.37 g, 2.5 mmol) as orange crystals.31P NMR (202 MHz, C6D6) δ: −30.8 (1JPSi = 102 Hz) ppm; 29Si NMR (99 MHz, C6D6) δ: 1.6 (t, 1JSiP = 102 Hz) ppm; 1H NMR (500 MHz, C6D6) δ: 4.81–4.71 (m, 2H Cp), 4.13–4.08 (m, 2H Cp), 4.08–4.03 (m, 2H Cp), 4.01–3.93 (m, 2H Cp), 1.50–1.18 (m, 18H tBu CH3) ppm; 13C NMR (101 MHz, C6D6) δ: 79.68 (pseudo t, JPC = 22.1 Hz, Cp), 75.69 (pseudo t, JPC = 3.7 Hz, Cp), 73.17 (m Cp), 72.05 (pseudo t, JPC = 5.4 Hz, Cp), 67.48 (pseudo t, JPC = 10.5 Hz, Cp ipso C), 34.63 (pseudo t, JPC = 5.7 Hz, tBu C q), 31.27 (pseudo t, JPC = 6.8 Hz, tBu CH3) ppm. Elemental analysis (%) C18H26Br2FeP2Si calculated: C: 39.446, H: 4.781, found: C: 39.211, H: 4.648. MS (LIFDI-HR): m/z 362.10292 ([M − SiBr2 + 2H]+ 100%), 519.92085 (M+ 1%) calculated: 519.92054.
Synthesis of 7
To a suspension of SiI4 (550 mg, 1.02 mmol) in 10 mL of pentane, a solution of 2 (363 mg, 1 mmol) in 10 mL of pentane was added at room temperature. To the reaction mixture NEt3 (0.29 mL, 2.1 mmol) was added and stirred for 48 hours and sonicated with ultrasound few times. All volatile components were removed in vacuo, the residue was extracted with 40 mL of pentane and the solid was removed and discarded. The pentane extract was evaporated to dryness yielding the raw product. Recrystallization from pentane afforded 7 in 45% yield (290 mg, 0.45 mmol) as yellow crystals.31P NMR (202 MHz, C6D6) δ: −18.4 (s) ppm. 29Si NMR (99 MHz, C6D6) δ: −63.5 (t, 1JSiP 113 Hz) ppm; 1H NMR (500 MHz, C6D6) δ: 4.98–4.93 (m, 2H Cp), 4.11–4.08 (m, 2H Cp), 4.08–4.05 (m, 2H Cp), 3.94–3.91 (m, 2H Cp), 1.42–1.34 (m, 18H CH3) ppm; 13C NMR (126 MHz, C6D6) δ: 79.79 (pseudo t, JPC = 21.8 Hz, Cp), 74.12 (pseudo t, JPC = 3.4 Hz, Cp), 72.96 (m Cp), 71.72 (pseudo t, JPC = 5.2 Hz, Cp), 70.56 (pseudo t, JPC = 11.6 Hz, Cp ipso C), 35.34 (pseudo t, JPC = 6.3 Hz, tBu C q), 31.47 (pseudo t, JPC = 6.7 Hz, tBu CH3) ppm. Elemental analysis (%) C18H26I2FeP2Si calculated: C: 33.671, H: 4.082, found: C: 33.852, H: 3.848. MS (LIFDI-HR): m/z 641.86511 (M+ 70%) calculated: 641.87178.
Synthesis of 8
A solution of 4 (487 mg, 1 mmol) in 20 mL pentane was added to a suspension of LiAlH4 (52 mg, 1.37 mmol) in 15 mL Et2O at 0 °C. The reaction mixture was stirred for 2 hours, and all volatile components were removed in vacuo. The residue was extracted with 30 mL of pentane and the solid was removed and discarded. The pentane extract was evaporated to dryness yielding the raw product. Recrystallization from benzene afforded the crystalline product.31P NMR (202 MHz, C6D6) δ: −62.2 ppm. 29Si NMR (99 MHz, C6D6) δ: −51.6 (ddt, 1JSiH = 196 Hz, 187 Hz, 1JSiP = 40 Hz) ppm. 1H NMR (500 MHz, C6D6) δ: 4.88 (td, 2JPH = 21.8 Hz, 2JHH = 9.2 Hz, 1H), 4.77 (d, 2JHH = 9.2 Hz), 4.38–3.92 (m, 8H Cp), 1.26–1.16 (m, 18H CH3) ppm. MS (LIFDI-HR): m/z 362.10156 ([M − Si]+ 100%), 390.07870 (M+ 20%) calculated: 390.07849.
Synthesis of 9
SiHCl3 (0.21 mL, 2.1 mmol) was added to the reaction mixture of 3 and stirred for 10 minutes. All volatile components were removed in vacuo, and the residue was extracted with 30 mL of pentane. After removal of the solid 9 was obtained as a mixture of two stereo isomers (major/minor, ratio 1.00:0.75).
9 major: 31P NMR (202 MHz, C6D6) δ: −41.7 (1JPSi = 67 Hz) ppm; 29Si NMR (99 MHz, C6D6) δ: 5.8 (dt, 1JSiH = 220 Hz, JSiP = 67 Hz) ppm; 1H NMR (500 MHz, C6D6) δ: 6.54 (t, 2JHP = 26.3 Hz, 1JHSi = 219.6 Hz, 1H SiH), 4.80–3.94 (m, 8H Cp), 1.33–1.22 (m, 18H tBu CH3) ppm; 13C NMR (126 MHz, C6D6) δ: 80.2 (pseudo t, JPC = 21.8 Hz, Cp), 77.1 (pseudo t, JPC = 3.9 Hz, Cp), 72.6 (m, Cp), 70.9 (pseudo t, JPC = 10.5 Hz, Cp ipso C), 70.8 (pseudo t, JPC = 5.4 Hz, Cp), 32.2 (pseudo t, JPC = 2.6 Hz, tBu C q), 31.3 (pseudo t, JPC = 7.0 Hz, tBu CH3) ppm.
9 minor: 31P NMR (202 MHz, C6D6) δ: −62.3 (1JPSi = 55 Hz) ppm; 29Si NMR (99 MHz, C6D6) δ: −13.2 (dt, JSiH = 223 Hz, JSiP = 55 Hz) ppm; 1H NMR (500 MHz, C6D6) δ: 5.91 (m, 1JHSi = 222.8 Hz, 1H SiH), 4.80–3.94 (m, 8H Cp), 1.33–1.22 (m, 18H tBu CH3) ppm; 13C NMR (126 MHz, C6D6) δ: 79.0 (pseudo t, JPC = 21.5 Hz, Cp), 74.9 (pseudo t, JPC = 4.1 Hz, Cp), 72.4 (m, Cp), 72.1 (pseudo t, JPC = 5.0 Hz, Cp), 66.3 (pseudo t, JPC = 12.1 Hz, Cp ipso C), 30.9 (pseudo t, JPC = 6.9 Hz, tBu CH3), 30.9–30.8 (m, tBu C q) ppm. Elemental analysis (%) C18H27ClFeP2Si·0.15C18H28FeP2 calculated: C: 52.051, H: 6.588, found: C: 52.151, H: 6.536. MS (LIFDI-HR): m/z 362.10107 ([M − SiCl + H]+ 100%), 396.06247 ([M − Si + H]+ 20%) calculated: 396.06259.
Acknowledgements
The authors thank Prof. Gudat for helpful discussions and the Deutsche Forschungsgemeinschaft (PI 353/8-1 and PI 353/9-1) and OTKA NN 113772 within the framework of the ERA-Chemistry program for financial support. We also thank the EU-COST network CM1302 “Smart Inorganic Polymers” (SIPs) for funding an STSM. Moreover, we are grateful to Dr Linne and his mass spectrometry team at the University of Marburg.Notes and references
- D. E. Herbert, U. F. J. Mayer and I. Manners, Angew. Chem., Int. Ed., 2007, 46, 5060–5081 CrossRef CAS PubMed.
- I. Manners and U. Vogel, in Modern Cyclophane Chemistry, ed. R. H. Gleiter, Henning, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2004, pp. 415–433 Search PubMed.
- I. Manners, Science, 2001, 294, 1664–1666 CrossRef CAS PubMed.
- P. Nguyen, P. Gomez-Elipe and I. Manners, Chem. Rev., 1999, 99, 1515–1548 CrossRef CAS PubMed.
- M. Herberhold, Angew. Chem., Int. Ed. Engl., 1995, 34, 1837 CrossRef CAS.
- R. Broussier, A. Da Rold, B. Gautheron, Y. Dromzee and Y. Jeannin, Inorg. Chem., 1990, 29, 1817–1822 CrossRef CAS.
- C. Moser, F. Belaj and R. Pietschnig, Chem. – Eur. J., 2009, 15, 12589–12591 CrossRef CAS PubMed.
- Y. Tanimoto, Y. Ishizu, K. Kubo, K. Miyoshi and T. Mizuta, J. Organomet. Chem., 2012, 713, 80–88 CrossRef CAS.
- C. Moser, F. Belaj and R. Pietschnig, Phosphorus Sulfur Relat. Elem., 2015, 190, 837–844 CrossRef CAS.
- D. M. Khramov, E. L. Rosen, V. M. Lynch and C. W. Bielawski, Angew. Chem., Int. Ed., 2008, 47, 2267–2270 CrossRef CAS PubMed.
- U. Siemeling, C. Färber, M. Leibold, C. Bruhn, P. Mücke, R. F. Winter, B. Sarkar, M. von Hopffgarten and G. Frenking, Eur. J. Inorg. Chem., 2009, 2009, 4607–4612 CrossRef.
- S. Spirk, F. Belaj, J. H. Albering and R. Pietschnig, Organometallics, 2010, 29, 2981–2986 CrossRef CAS.
- R. Pietschnig and A. Orthaber, Eur. J. Inorg. Chem., 2006, 22, 4570–4576 CrossRef.
- C. Moser, A. Orthaber, M. Nieger, F. Belaj and R. Pietschnig, Dalton Trans., 2006, 3879–3885 RSC.
- M. Bender, E. Niecke, M. Nieger and R. Pietschnig, Eur. J. Inorg. Chem., 2006, 380–384 CrossRef CAS.
- R. Pietschnig, J. Chem. Soc., Chem. Commun., 2004, 5, 546–547 RSC.
- D. Lange, E. Klein, H. Bender, E. Niecke, M. Nieger, R. Pietschnig, W. W. Schoeller and H. Ranaivonjatovo, Organometallics, 1998, 17, 2425–2432 CrossRef CAS.
- B. D. Rekken, T. M. Brown, J. C. Fettinger, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2012, 134, 6504–6507 CrossRef CAS PubMed.
- D. Kargin, Diploma Thesis, University of Kassel, 2013.
- H. J. Reich and R. R. Dykstra, Organometallics, 1994, 13, 4578–4585 CrossRef CAS.
- R. Wolf, A. Schisler, P. Lönnecke, C. Jones and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2004, 16, 3277–3286 CrossRef.
- B. Riegel, A. Pfitzner, G. Heckmann and H. Binder, Z. Anorg. Allg. Chem., 1995, 621, 1989–1994 CrossRef CAS.
- R. T. Boere and Y. Zhang, J. Organomet. Chem., 2005, 690, 2651–2657 CrossRef CAS.
- R. E. Weston, J. Am. Chem. Soc., 1954, 76, 2645 CrossRef CAS.
- P. Schwerdtfeger, L. Laakkonen and P. Pyykkö, J. Chem. Phys., 1992, 96, 6807 CrossRef CAS.
- L. Nyulaszi, Tetrahedron, 2000, 56, 79–84 CrossRef CAS.
- L. Nyulaszi, J. Phys. Chem., 1995, 99, 586–591 CrossRef CAS.
- M. Ma, A. Stasch and C. Jones, Chem. – Eur. J., 2012, 18, 10669–10676 CrossRef CAS PubMed.
- R. J. Gillespie and I. Hargittai, The VSEPR Model of Molecular Geometry, Allyn and Bacon, Boston, 1991 Search PubMed.
- R. A. Musgrave, A. D. Russell, G. R. Whittell, M. F. Haddow and I. Manners, Organometallics, 2015, 34, 897–907 CrossRef CAS.
- R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 121, 5793–5796 CrossRef.
- W.-W. du Mont and M. Grenz, Chem. Ber., 1985, 118, 1045–1049 CrossRef CAS.
- M. Takahashi, S. Tsutsui, K. Sakamoto, M. Kira, T. Müller and Y. Apeloig, J. Am. Chem. Soc., 2001, 123, 347–348 CrossRef CAS PubMed.
- L. Jafarpour, E. D. Stevens and S. P. Nolan, J. Organomet. Chem., 2000, 606, 49–54 CrossRef CAS.
- G. Becker, M. Gekeler, H.-M. Hartmann and O. Mundt, in Synthetic Methods of Organometallic and Inorganic Chemistry: Groups 1, 2,13, and 14, ed. N. Auner and U. Klingebiel, Thieme, Stuttgart, 1996, vol. 2, p. 156 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidkock, G. P. Shields, R. Taylor, M. Towler and J. van der Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian Inc., Wallingford, CT, 2010 Search PubMed.
- G. Schaftenaar and J. H. Noordik, J. Comput.-Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS PubMed.
- A. Todd and T. K. Keith, AIMAll 11.10.16, Gristmill Software, Overland Park, 2011, http://aim.tkgristmill.com Search PubMed.
Footnotes |
| † Dedicated to the memory of Andreas Kirchmeier. |
| ‡ Electronic supplementary information (ESI) available: Details and selected data of quantum chemical calculations. CCDC 1417298–1417301 and 1434408. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03107a |
| This journal is © The Royal Society of Chemistry 2016 |