
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–organic frameworks (MOFs) bring new life to hydrogen-bonding organocatalysts in confined spaces
Juan V.
Alegre-Requena
ab,
Eugenia
Marqués-López
b,
Raquel P.
Herrera
*b and
David Díaz
Díaz
*ac
aInstitut für Organische Chemie, Universität Regensburg, Universitätsstr. 31, 93040 Regensburg, Germany. E-mail: David.Diaz@chemie.uni-regensburg.de; Tel: + 49 941 9434373
bLaboratorio de Organocatálisis Asimétrica, Departamento de Química Orgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: raquelph@unizar.es; Tel: +34 976761190
cIQAC-CSIC, Jordi Girona 18-26, 08034 Barcelona, Spain
First published on 18th January 2016
Abstract
Hydrogen-bonding organocatalysis has emerged as a promising biomimetic alternative to Lewis acid catalysis. Urea, thiourea and squaramide moieties represent the most common hydrogen-bond donors used for the preparation of these catalysts. However, their significant tendency to undergo self-quenching (self-aggregation) often decreases their solubility and reactivity. Recently, scientists have found a promising way around this problem by immobilizing the hydrogen-bonding organocatalysts on metal–organic frameworks (MOFs). Along with advantageous modular synthesis and recycling properties, the tunable porosity and topology of MOFs also allows fast mass transport and/or interactions with substrates. Herein, we highlight the existing examples dealing with the fabrication and testing of hydrogen-bonding organocatalyst-containing MOFs, providing also our vision for further advances in this area. The results derived from these studies will likely serve as inspiration for the future development of superior hydrogen-bonding organocatalysts to accomplish in confined spaces chemical transformations that are either slow or unaffordable under standard homogeneous conditions.
Introduction
Most of the chemical reactions that occur in living systems are high-energy reactions that would be unaffordable or would occur very slowly without the catalysis provided by enzymes. This has inspired scientists during many decades to develop artificial catalysts1,2 within a general quest for economic profit. As a matter of fact, catalysts are nowadays the workhorses of the chemical transformations in industry (ca. 85–90% of the products of the chemical industry are fabricated through catalytic processes).3 However, the increasing on-demand production of bulk chemicals, fine chemicals and fuels is also threatening the stability of the ecosystems in our planet due to environmentally harmful wastes. Thus, stricter environmental regulations by governments have motivated the replacement of numerous homogeneous reactions by greener and economical heterogeneous catalytic processes.4Within this context, supramolecular coordination chemistry offers a promising biomimetic platform with well-defined functional groups suitable for catalysis.5–9 Features such as numerous metal geometries, tunable bond directionality and self-correcting kinetic reversibility have allowed the development of a variety of metal–organic materials (MOMs) with major relevance in the field of catalysis.10,11 Among the vast field of MOMs,12 and supported by the gradual maturation of crystal engineering,13 nanoscale metal–organic frameworks (MOFs)14,15 have taken a pivotal role in the rational design of eco-friendly heterogeneous catalysts16 beyond their more traditional applications in gas storage and gas separation,17–22 sensing,23,24 ion exchange,25 drug delivery26,27 and light harvesting.28 MOFs are composed of inorganic nodes (metal ions or clusters) judiciously linked by polytopic functional organic struts to form uniform three-dimensional (3D) arrays of high surface area (up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 m2 g−1),29 large pores (98 Å)30 and low density (0.13 g cm−3).31 In general, the active sites of MOF-based catalysts can be either metal centers with unsaturated coordination environments, catalysts encapsulated in the pores or the sites inherent in the organic framework.32 Along with the advantages of their modular synthesis33,34 and recycling properties, the tunable porosity and topology of MOFs also allow fast mass transport and/or interactions with substrates. These features, often challenging in homogeneous states, differentiate MOFs from other nanoporous materials such as zeolites and activated carbons. Overall, MOFs constitute ideal candidates for heterogeneous catalysis if the framework crystallinity and regularity are retained under the reaction conditions or during post-synthetic modification strategies that can be used to introduce functional groups on MOFs in order to extend their catalytic activity.35,36
400 m2 g−1),29 large pores (98 Å)30 and low density (0.13 g cm−3).31 In general, the active sites of MOF-based catalysts can be either metal centers with unsaturated coordination environments, catalysts encapsulated in the pores or the sites inherent in the organic framework.32 Along with the advantages of their modular synthesis33,34 and recycling properties, the tunable porosity and topology of MOFs also allow fast mass transport and/or interactions with substrates. These features, often challenging in homogeneous states, differentiate MOFs from other nanoporous materials such as zeolites and activated carbons. Overall, MOFs constitute ideal candidates for heterogeneous catalysis if the framework crystallinity and regularity are retained under the reaction conditions or during post-synthetic modification strategies that can be used to introduce functional groups on MOFs in order to extend their catalytic activity.35,36
Despite the extensive use of MOFs in heterogeneous catalysis during the last decade,32,37 some drawbacks are usually associated to unsatisfactory yields, poor selectivities or limited substrate scope.37 Moreover, among the plethora of MOF-based catalysts, hydrogen-bonding organocatalysts38 have met MOFs only recently. Hydrogen-bonding organocatalysts have emerged as a biomimetic alternative to Lewis acid activation38–40 by means of mainly urea, thiourea or squaramide moieties. This is relevant because although a significant number of bond-forming reactions can be catalysed by homogeneous hydrogen-bonding organocatalysts,41 their tendency to undergo self-quenching (self-aggregation) often decrease their solubility and reactivity.
Herein, we highlight the recent contributions involving the fabrication and use of hydrogen-bonding organocatalysts immobilized on MOFs. We believe that these unique examples are paving the way for the development of superior hydrogen-bonding organocatalysts for complicated chemical transformations.
Urea-containing MOF catalysts
Farha, Hupp, Scheidt and co-workers reported the first urea-containing metal–organic framework (MOF) with the idea of preventing the self-association that homogeneous catalysts, such as ureas, undergo in their reactions. This self-quenching is responsible for the consequent decrease in the solubility and reactivity of the catalysts (Fig. 1, top).42,43 For this purpose, the authors envisioned that this self-quenching may be avoided if the urea catalyst is incorporated into coordination polymers using an appropriate metal ion, which possess defined reaction environments and high porosity (Fig. 1, bottom).43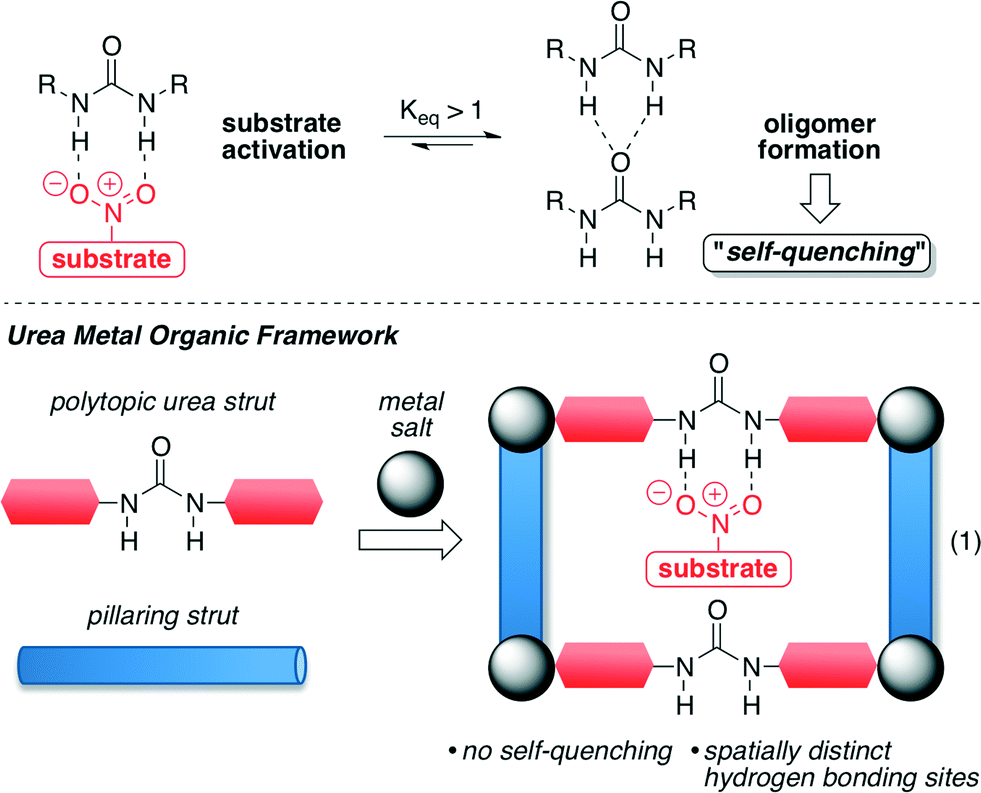 | ||
| Fig. 1 Synthetic strategy of urea MOF derivatives. Adapted with permission from ref. 43. Copyright ® American Chemical Society. | ||
Based on this pioneering idea, NU-601 MOF was synthesised under solvothermal conditions starting from urea 1 and 4,4′-bipyridine (bipy, 2) as a pillaring strut (Scheme 1).43,44
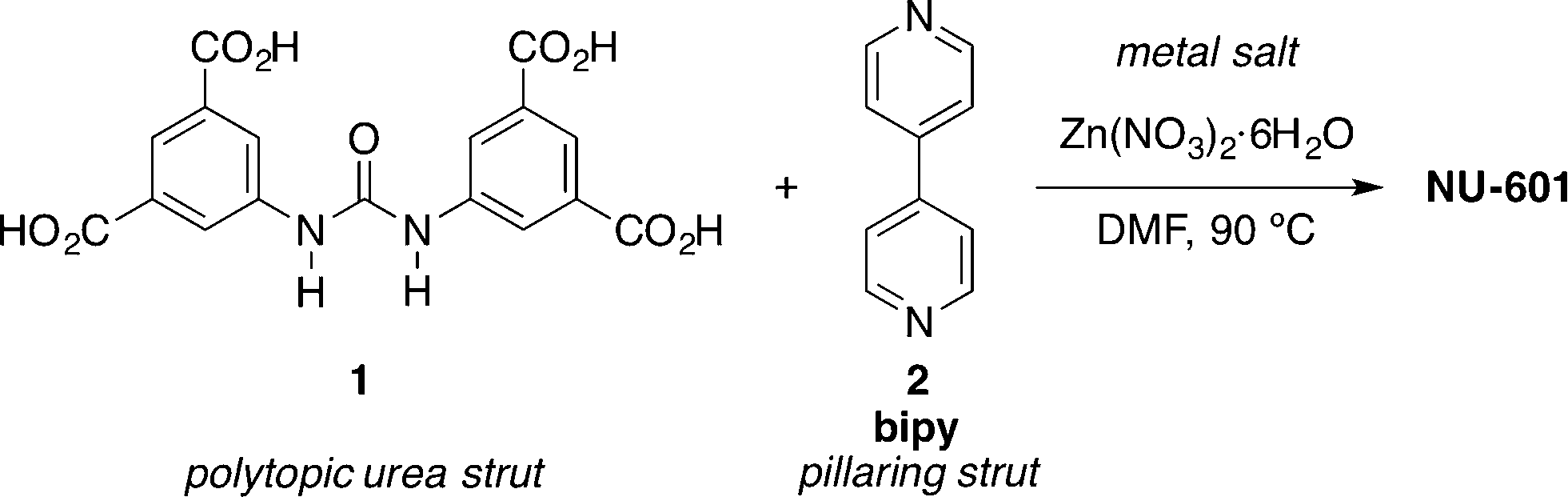 | ||
| Scheme 1 Synthesis of MOF NU-601.43 | ||
After 2 days of heating, light-yellow needle-shaped crystals of NU-601 MOF were obtained. The structure of Zn2(bipy)2(1-4H) contains 2D sheets of strut 1 pillared with 2 and Zn2 nodes, featuring large channels in all directions as disclosed in Fig. 2.
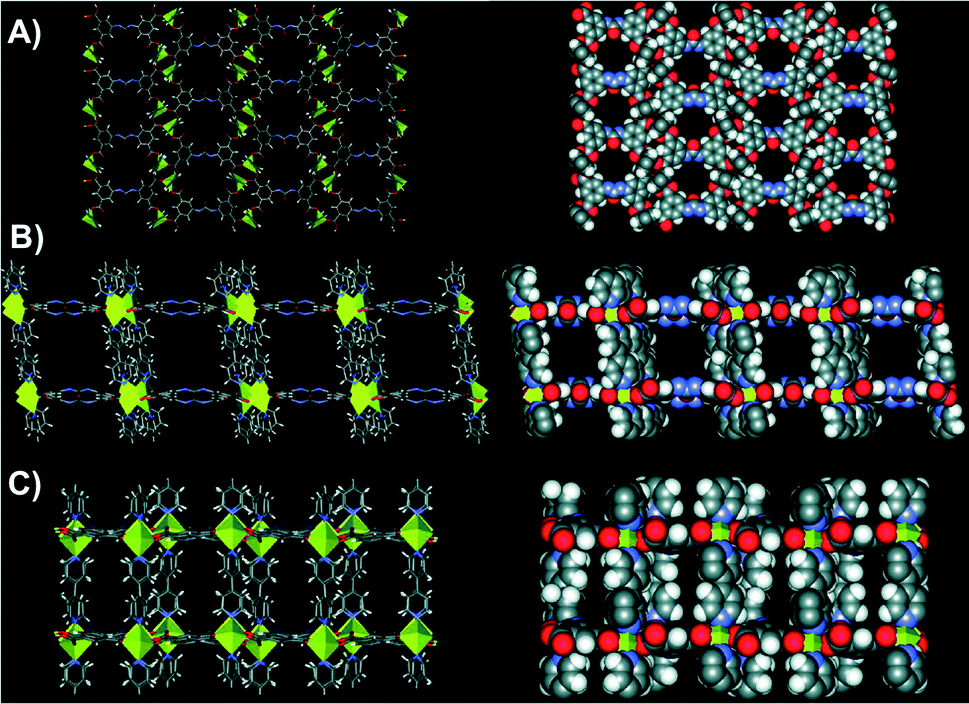 | ||
| Fig. 2 Stick and space-filling representations of NU-601 (P21/c space group). A) View down the a axis (pore size 12.05 Å × 13.95 Å). B) View down the b axis (pore size 11.38 Å × 13.69 Å). C) View down the c axis (pore size 11.38 Å × 4.87 Å). Adapted with permission from ref. 43. Copyright ® American Chemical Society. | ||
Since DMF molecules remained tightly hydrogen-bonded to the N–H groups of urea in the solid, in order to activate the structure, the exchange of the DMF with MeNO2 was carried out over 4 days. After this exchange/activation process, the effectiveness of NU-601 as a hydrogen-bond donor catalyst was explored in the Friedel–Crafts alkylation between nitroalkenes and pyrroles.45
In the screening of the reaction, a higher reactivity was observed when using NU-601 as the catalyst compared with the reactivity of diphenylurea (90% conv. at 18 h vs. 65% conv.) and the control reaction (21% conv. at 48 h). Then, in order to demonstrate if the catalysis took place on the surface of the MOF or within the pores, the authors employed larger substrates as steric probes (Scheme 2).
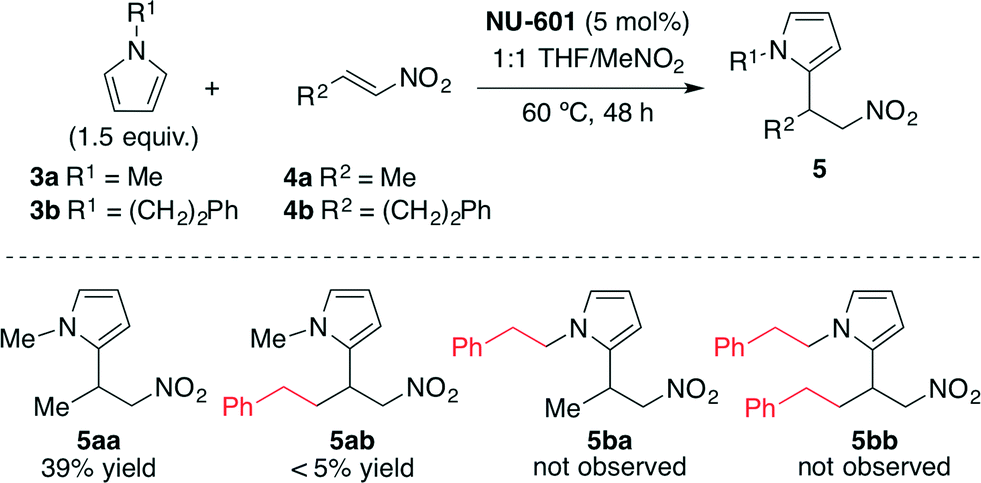 | ||
| Scheme 2 Size-exclusion catalysis experiments.43 | ||
For this aim, pyrrole 3b and nitroalkene 4b were also employed, and a rapid decrease in the reaction rate was detected. These results provided strong evidence that NU-601 exhibited reagent size selectivity since the large substrates show significantly diminished yields because they cannot get into the porous network. NU-601 also proved to be reusable, maintaining its reactivity after one cycle of use (33% vs. 39% yield after 48 h of reaction) and suffering only a slight degradation of reactivity after five cycles (29% vs. 39% yield after 48 h of reaction).
The same research group reported in 2013, as an extension of their research work,46 a new class of urea-based UiO-67 derivatives as plausible catalysts.47UiO-67 is formed by a cubic metal–organic framework of cationic Zr6O4(OH)4 nodes and biphenyl-4,4′-dicarboxylate (bpdc) 7 linkers, providing two pore sizes with diameters of 23 and 12 Å (Fig. 3).48
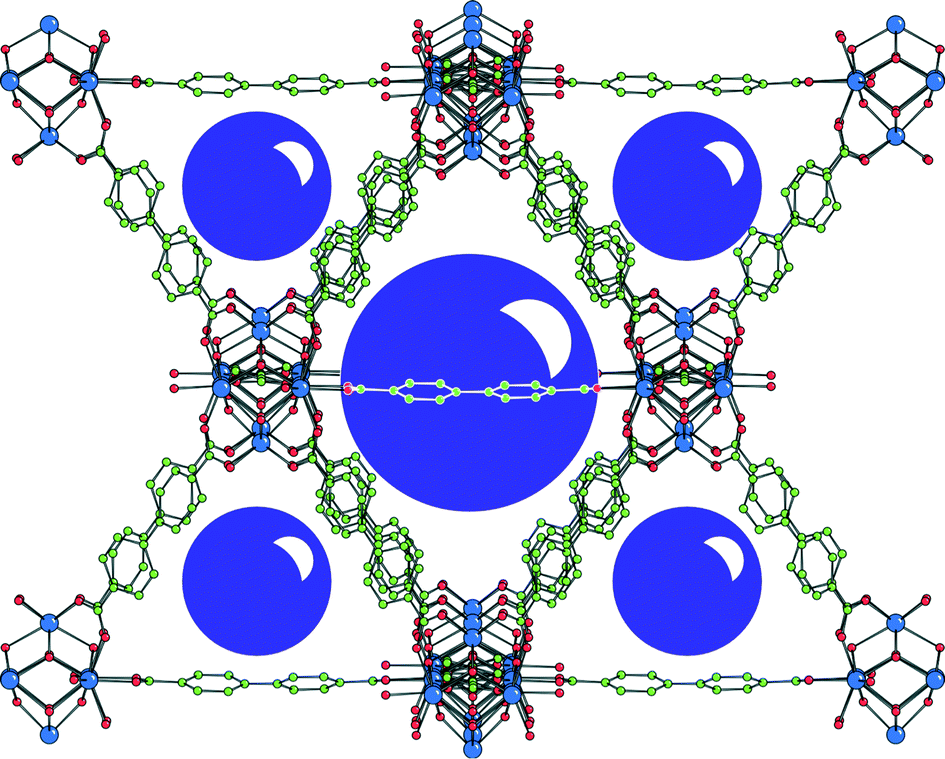 | ||
| Fig. 3 UiO-67 structure represented by a single octahedral cage (large sphere). The face of each octahedral cage is shared with eight smaller tetrahedral cages (small spheres). Reprinted with permission from ref. 46. Copyright ® Royal Society of Chemistry. | ||
In this study, the authors incorporated for the first time the urea scaffold into the UiO-67 MOF with the aim of designing plausible hydrogen-bond donating catalysts. In order to evaluate the effect of the porosity on the catalytic activity of the materials, different species were prepared by means of various H2-urea/H2-bpdc ratios during the solvothermal synthesis (Scheme 3).
 | ||
| Scheme 3 Synthesis of derivatives of MOF UiO-67.47 | ||
All prepared species were explored in the nitroaldol (Henry)49 reaction between nitromethane (9) and benzaldehyde (8a) as the model reaction. The results are disclosed in Scheme 4.
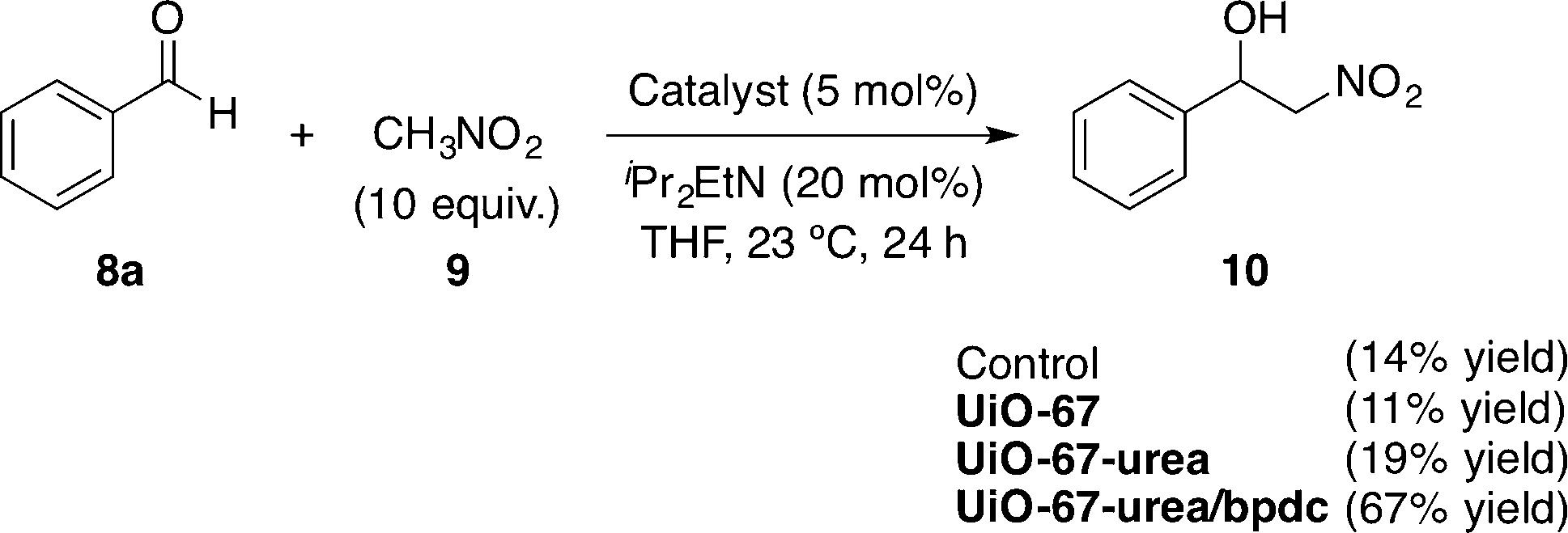 | ||
| Scheme 4 Screening of the nitroaldol (Henry) reaction.47 | ||
The mixed strut UiO-67-urea/bpdc framework was found to be the most active catalyst (67% yield after 24 h). The results were very similar in the control experiment and when using Zr6O4(OH)4 by itself (UiO-67) (14% and 11%, respectively). Interestingly, the use of UiO-67-urea afforded the same poor results (19% yield). The best catalytic activity showed by UiO-67-urea/bpdc was attributed to its high Brunauer–Emmett–Teller (BET) surface area (1550 m2 g−1) compared with that of UiO-67-urea (390 m2 g−1) as measured by the authors. It is remarkable that the pore size distribution on UiO-67-urea/bpdc did not suffer a significant decrease in the pore diameters (21.5 and 12 Å) in comparison to the precursor UiO-67 (23 and 11.5 Å). In contrast, both structures are significantly larger than that of the pure strut UiO-67-urea (12 and 9 Å).
More recently, Hu, Liu and co-workers have developed novel functionally diverse urea-derived MOF hydrogen-bond-donating heterogeneous catalysts for their application in Friedel–Crafts alkylation reactions.50 The heterogeneous catalysts were synthesised following a post-synthetic modification (PSM) approach, which has been proven to be an effective tool to introduce functional groups in MOFs.36,51,52 Thus, three different urea-containing Cr-MIL-101 (chromium(III) terephthalate MOF) heterogeneous catalysts were prepared. Cr-MIL-101-NH2 was first prepared starting from Cr-MIL-101, and then the former was submitted to react with the appropriate isocyanate in CH3CN at 120 °C in a sealed tube for 12 h. It is noteworthy that both aliphatic and aromatic isocyanates were successfully anchored onto the MIL-101 walls via covalent incorporation, leading to Cr-MIL-101-UR1–3 in nearly quantitative conversion and catalyst loading of 2.61, 2.48 and 1.85 mmol g−1, respectively (Fig. 4).50 The gas sorption behavior of these MOFs demonstrated that they were still highly porous after post-synthetic modification (i.e., Brunauer–Emmett–Teller (BET) surface areas were 1409.9 m2 g−1, 1178.6 m2 g−1 and 829.3 m2 g−1, respectively).
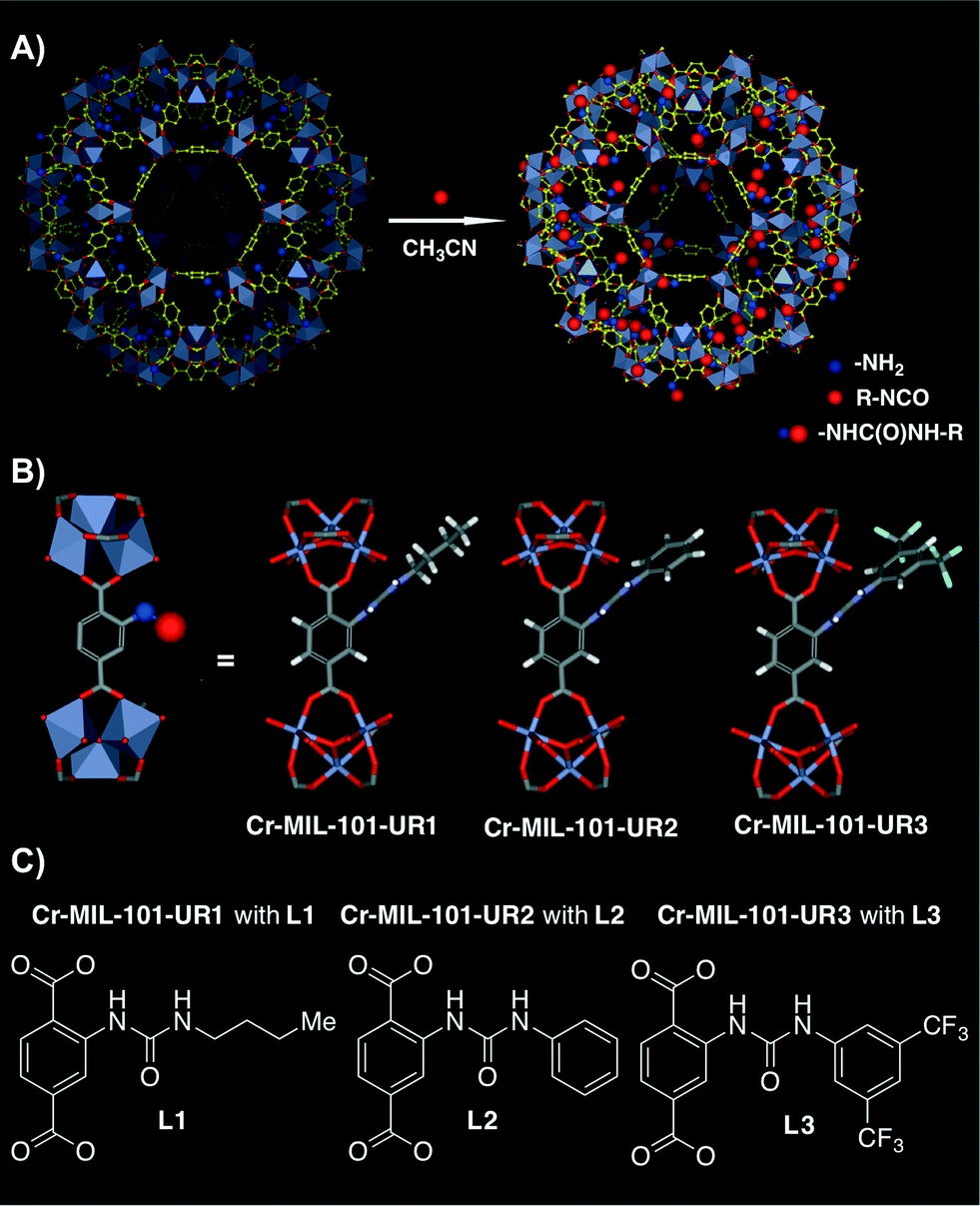 | ||
| Fig. 4 A) PSM approach of Cr-MIL-101-NH2 with isocyanates to generate Cr-MIL-101-UR1–3. B) Schematic representation of the ligand structure and [Cr3O(CO2)6] cluster nodes. C) Differently substituted ureas present in the final synthesised MOFs. Adapted with permission from ref. 50. Copyright ® Royal Society of Chemistry. | ||
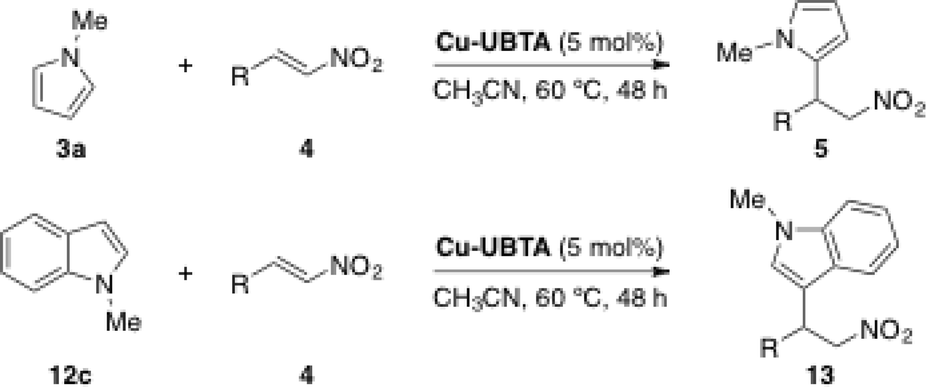
The catalytic activity of Cr-MIL-101-UR1–3 in the Friedel–Crafts alkylation reaction was tested in the model reaction between N-methylpyrrole (3a) and trans-β-nitrostyrene (4c) (Table 1).45,50
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Yieldb (%) |
|
a Reactions performed with 3a (0.2 mmol), 4c (0.1 mmol), and catalyst (15 mol%) in the solvent (0.15 mL).
b Determined using 1H NMR.
c Ratio 5:1 for 3a:4c used.
d 10 mol% catalyst loading.
e 5 mol% catalyst loading.
|
||||
| 1 | Cr-MIL-101-UR3 | DMF | 36 | Trace |
| 2 | Cr-MIL-101-UR3 | THF | 36 | 26 |
| 3 | Cr-MIL-101-UR3 | Acetone | 36 | 27 |
| 4 | Cr-MIL-101-UR3 | CH3CN | 60 | 96 |
| 5c | Cr-MIL-101-UR3 | CH3CN | 24 | 95 |
| 6d | Cr-MIL-101-UR3 | CH3CN | 24 | 90 |
| 7e | Cr-MIL-101-UR3 | CH3CN | 24 | 73 |
| 8c | Cr-MIL-101-UR1 | CH3CN | 24 | 72 |
| 9c | Cr-MIL-101-UR2 | CH3CN | 24 | 87 |
| 10 | — | CH3CN | 24 | Trace |
| 11 | Cr-MIL-101 | CH3CN | 48 | 22 |
| 12c | 11 | CH3CN | 24 | 79 |
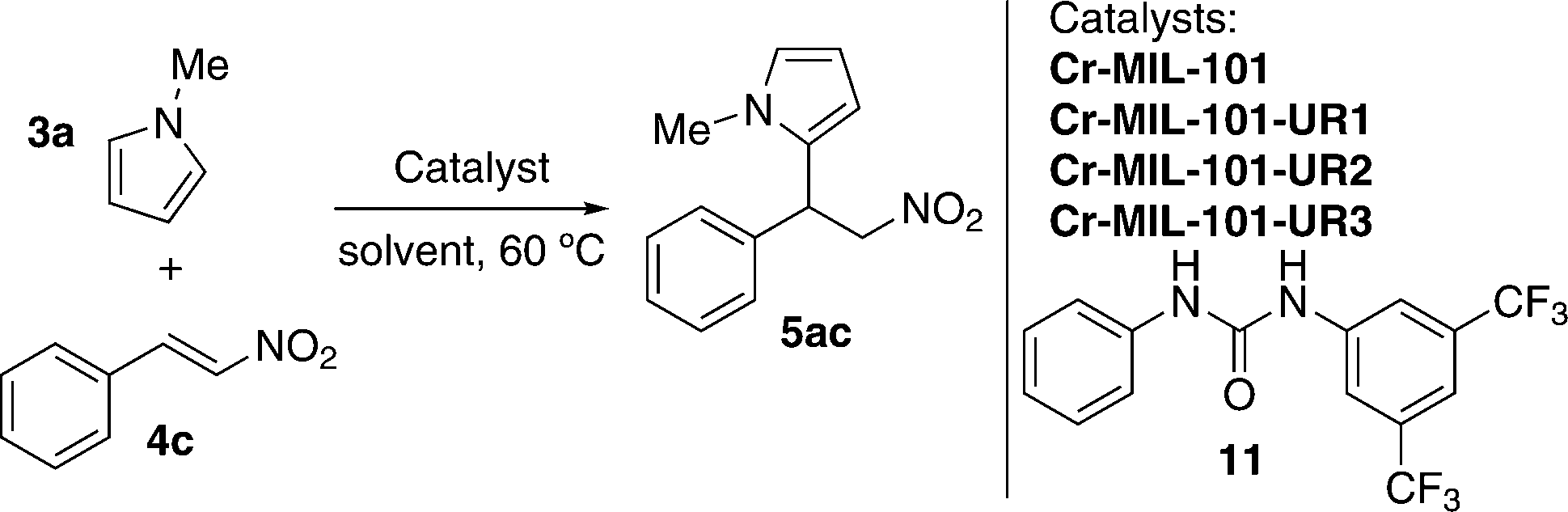
After exploring different reaction conditions and catalysts, the best results were achieved with Cr-MIL-101-UR3 (entries 4 and 5), which bears appropriate steric hindered and electron-withdrawing substituents. The catalyst loading could be reduced to 10 mol%, without a strong influence on the yield (entry 6) and better reactivity was found in comparison with urea 11, a homogeneous catalyst (95% vs. 79% yield, entries 5 and 12, respectively). The other two catalysts (Cr-MIL-101-UR1 and Cr-MIL-101-UR2) gave poorer results than Cr-MIL-101-UR3 (entries 8 and 9 compared with entry 5). The better reactivity found in the case of the latter is attributable to the spatial isolation achievable in the porous MOF environment. With the optimized conditions in hand, other electron-rich nucleophiles (12a–c) and a broad scope of different nitroalkenes (4c–m) were explored, achieving the same good reactivity (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | 4 (R) | Nu (12) | 13 | Yieldb (%) |
| a Reactions performed with 4 (0.1 mmol), 12 (0.12 mmol), and catalyst (15 mol%) in CH3CN (0.15 mL). b Isolated yield. c Isolated yield using catalyst 11. | ||||
| 1 | 4c (Ph) |
 12a
12a
|
 13ca
13ca
|
95 |
| 2 | 4c (Ph) |
 12b
12b
|
 13cb
13cb
|
93 |
| 3 | 4c (Ph) |
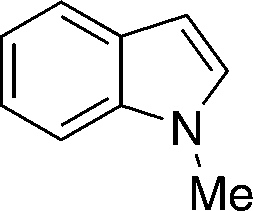 12c
12c
|
 13cc
13cc
|
94 |
| 4 | 4d (4-MeO-Ph) | 12c | 13dc | 90 |
| 5 | 4e (2-MeO-Ph) | 12c | 13ec | 81 |
| 6 | 4f (4-CF3-Ph) | 12c | 13fc | 90 |
| 7 | 4g (4-Cl-Ph) | 12c | 13gc | 88 |
| 8 | 4h (2-naphthyl) | 12c | 13hc | 91 |
| 9 | 4i (2-furyl) | 12c | 13ic | 92 |
| 10 | 4j (2-thiophenyl) | 12c | 13jc | 93 |
| 11 | 4k (Pr) | 12c | 13kc | 65 |
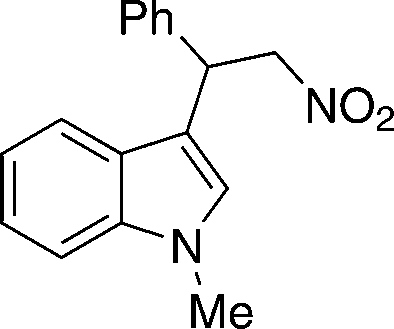
| 12 |
 4l
4l
|
12c | 13lc | 40 (80)c |
| 13 |
 4m
4m
|
12c | 13mc | 17 (76)c |
Interestingly, with long-chain or large nitroalkenes as substrates (4l–m), better results were obtained using urea 11 (entries 12 and 13). This fact supports that the heterogeneous catalysis mainly occurred within the pores of the MOF, thus exhibiting reagent size selectivity. Moreover, the study of the recyclability of Cr-MIL-101-UR3 provided that the catalyst was easily isolated from the reaction suspension by centrifugation and reused at least four times with little loss of reactivity while retaining its crystallinity as confirmed by PXRD analysis.
Recently, Morsali and co-workers have developed another example of MOFs containing urea units inside the struts of the MOF.53 Two different MOFs (TMU-18 and TMU-19) were synthesised by reacting Zn(NO3)2·6H2O, the urea-based ligand (H2-urea) 14 and either bipy (2) or 1,2-bis(4-pyridyl)ethane (bpe, 15) as pillaring struts under solvothermal conditions (Scheme 5). These MOFs are modified versions of the urea-containing MOF synthesised by Farha, Hupp, Scheidt and co-workers in 2012 (ref. 43) and exhibited different characteristics. Both MOFs showed large channels along the bc-plane with an aperture size of 13.5 × 9.9 Å for TMU-18, and along the b-axis with an aperture size of 12.1 × 10.6 Å for TMU-19.
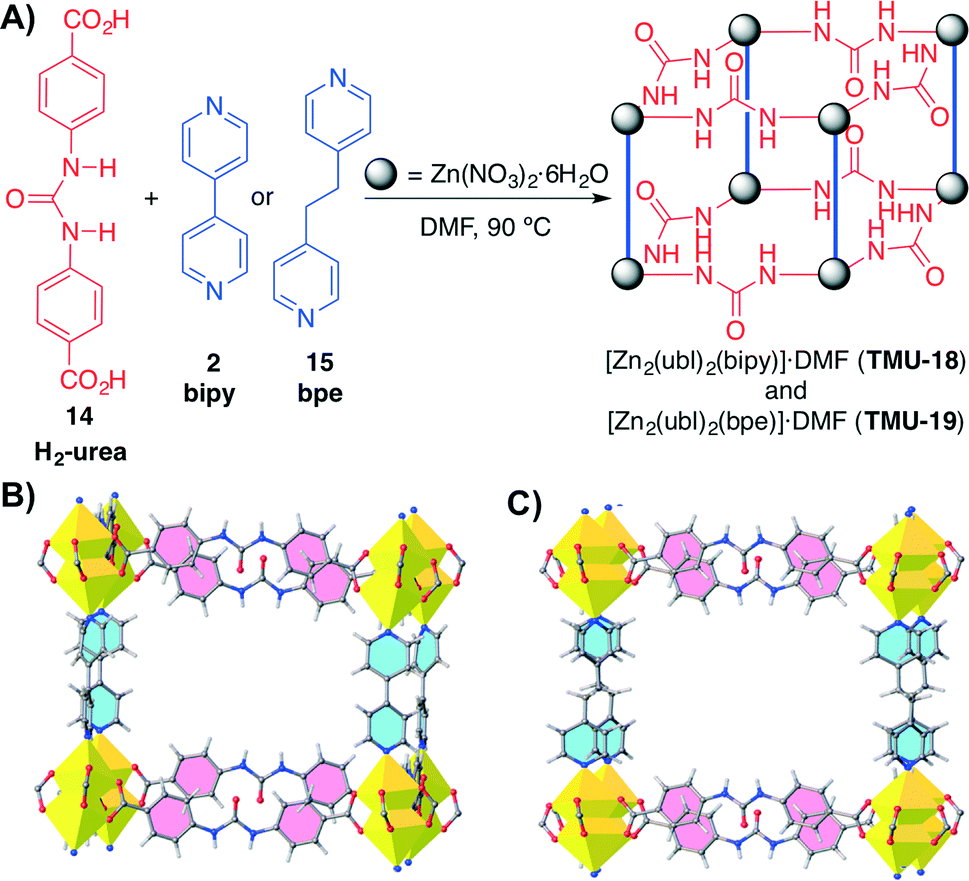 | ||
Scheme 5 A) Synthesis of catalytic urea-containing MOFs TMU-18 (triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and TMU-19 (orthorhombic Pnna). B) and C) View along the pore direction of TMU-18 and TMU-19, respectively. In contrast to TMU-18, TGA analysis indicated a lower thermal stability for TMU-19. Adapted with permission from ref. 53. Copyright ® Royal Society of Chemistry. ) and TMU-19 (orthorhombic Pnna). B) and C) View along the pore direction of TMU-18 and TMU-19, respectively. In contrast to TMU-18, TGA analysis indicated a lower thermal stability for TMU-19. Adapted with permission from ref. 53. Copyright ® Royal Society of Chemistry. | ||
These urea-based MOFs, especially TMU-18, showed good catalytic activity in the regioselective methanolysis of diverse epoxides (Fig. 5 and Table 3). In the case of styrene oxide (16a) both catalysts provided very high conversions after 140 h (100% and 95%, entry 3). However, for the other three less reactive epoxides (16b–d) poorer conversions were obtained in general, even in the presence of double the amount of catalyst (50 mol% vs. 25 mol%). In all cases, very low conversions were obtained after the same reaction time when no catalyst was used (entries 1, 5, 7 and 9 vs. 2, 6, 8 and 10, respectively). A fifth bulky epoxide (16e) was tested in order to support the happening of the reaction within the pores of the frameworks, and the low conversion reached after 140 h indicates it so (entry 11).
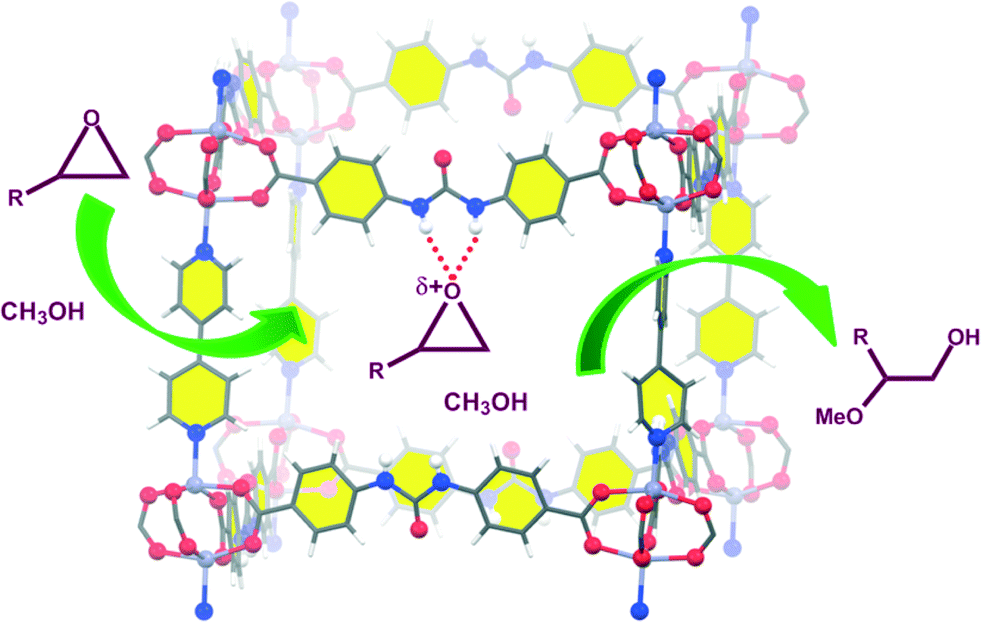 | ||
| Fig. 5 Activation of epoxides by TMU-18 for their regioselective methanolysis. Reprinted with permission from ref. 53. Copyright ® Royal Society of Chemistry. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Major product | Catalyst loading (mol%) | Conv.b,c (%) TMU-18 | Conv.b,c (%) TMU-19 |
| a Reactions performed with 16a–e (0.2 mmol) and the appropriate amount of urea catalyst in methanol (3 mL) at 60 °C. b GC yield using the internal-standard method. c The data in parentheses correspond to the selectivity for the major product. d 140 h reaction time. | |||||
| 1 |
 16a
16a
|
 17a
17a
|
— | 19 | |
| 2 | 16a | 17a | 25 | 78 (96) | 72 (97) |
| 3d | 16a | 17a | 25 | 100 (98) | 95 (98) |
| 4 | 16a | 17a | 50 | 100 (98) | 95 (98) |
| 5 |
 16b
16b
|
 17b
17b
|
— | 22 | |
| 6 | 16b | 17b | 50 | 78 (96) | 67 (91) |
| 7 | 16c | 17c | — | 26 | |
| 8 |
 16c
16c
|
 17c
17c
|
50 | 64 (81) | 53 (77) |
| 9 |
 16d
16d
|
 17d
17d
|
— | 16 | |
| 10 | 16d | 17d | 50 | 51 (78) | 48 (72) |
| 11d |
 16e
16e
|
 17e
17e
|
25 | 9 | 6 |
It is noteworthy to mention that the reaction conversion was lower when 40 mol% of urea powder was used as catalyst (15% vs. 35% and 31%, with TMU-18 and TMU-19, respectively, after 24 h), which proves the importance of including the urea moieties within the MOF structures. In addition, both catalysts were recovered, washed and used one more time, but they showed a decrease in their catalytic activity (39% vs. 45% and 29% vs. 41% for TMU-18 and TMU-19, respectively, after 40 h), in agreement with a gradual loss of crystallinity through the cycles as verified by PXRD measurements. However, these results open new possibilities for designing reusable urea-containing MOF catalysts for reactions carried out in aggressive media for the metal centers of the MOFs such as MeOH.
Contemporaneously, Wang, Zhao and co-workers, along with their research line,54 designed a novel (3,24)-connected rht-type metal organic framework (MOF Cu-UBTA). It was prepared by a solvothermal reaction between an unsymmetrical hexacarboxylate ligand H6-L (18) containing a urea group and Cu(NO3)2 in DMF at 75 °C for 3 days (Fig. 6).55
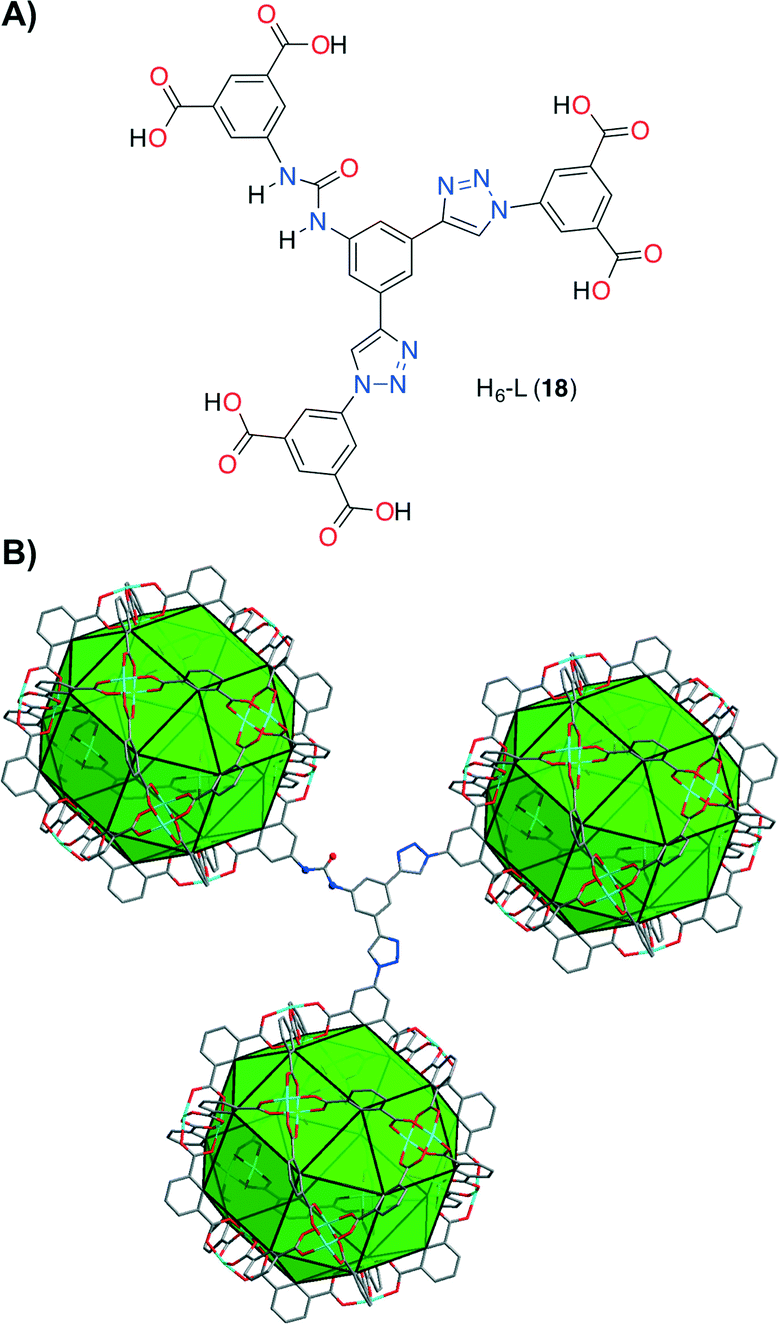 | ||
| Fig. 6 A) Structure of the hexacarboxylate acid H6-L (18). B) Single crystal structure of the (3,24)-connected rht-topological framework in Cu-UBTA. Tetragonal space group I4/m, unit cell dimensions a = b = 31.4206 Å, c = 44.8239 Å. BET surface area = 3134 m2 g−1. Adapted with permission from ref. 55. Copyright ® Royal Society of Chemistry. | ||
As a proof of its high porosity, the application of Cu-UBTA in gas storage and separation was further evaluated towards CO2, H2 and N2. The results disclosed in Fig. 7 suggested a high selectivity with CO2 and H2 against N2.
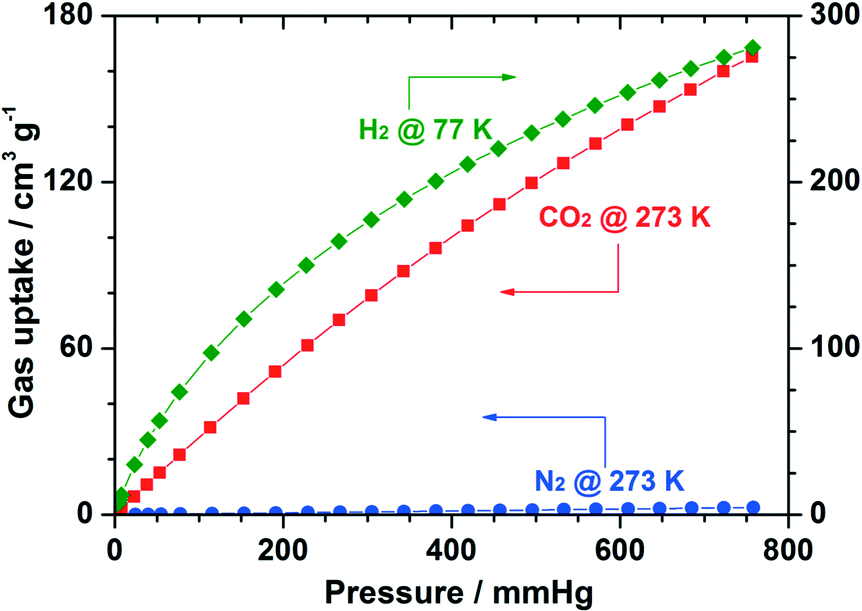 | ||
| Fig. 7 Gas sorption isotherms of Cu-UBTA for CO2 and N2 (at 273 K), and H2 (at 77 K). Reprinted with permission from ref. 55. Copyright ® Royal Society of Chemistry. | ||
The catalytic activity of Cu-UBTA was also evaluated using the Friedel–Crafts alkylation reaction between N-methylpyrrole (3a) and trans-β-nitrostyrene (4c) as a model reaction (Scheme 6).
 | ||
| Scheme 6 Model Friedel–Crafts alkylation reaction catalysed by Cu-UBTA.55 | ||
With Cu-UBTA the desired product was obtained in 85% yield after 48 h, whereas only traces of the product were observed in the control experiment without catalyst. In order to confirm the utility of the urea moiety in the catalytic system, the authors used their previous MOF NTU-105 (ref. 54) without urea as the catalyst under the same reaction conditions. Interestingly, NTU-105 exhibited a lower catalytic activity in relation to Cu-UBTA. This fact confirmed that the urea group in rht-MOF Cu-UBTA could also function as a hydrogen-bond-donating site to catalyse the Friedel–Crafts reaction in a synergic effect with the open metal sites, since the moderate catalytic activity shown by NTU-105 was attributed to the Lewis acidic open copper sites in the structure. The scope of the application of this catalyst was extended to other nucleophiles (12c) and nitroalkenes 4, leading to the desired products with good to excellent yields (Table 4). The recyclability of the MOF was also explored and the catalyst was reused after a simple centrifugation, showing similar catalytic activity in successive reaction cycles (81% for the 2nd run, 75% for the 3rd run). Moreover, PXRD measurements of the recycled catalyst revealed that the MOF retained its crystallinity after the catalytic cycles.
|
|
|||
|---|---|---|---|
| Entry | 4 (R) | Yieldb (%) 3a + 4 | Yieldb (%) 12c + 4 |
| a Reactions were carried out with 3a or 12c (0.2 mmol), 4 (0.1 mmol) and catalyst Cu-UBTA (5 mol%) in CH3CN. b Determined by 1H NMR using the internal-standard method. | |||
| 1 | 4d (4-MeO-Ph) | 92 | 76 |
| 2 | 4f (4-CF3-Ph) | 92 | 91 |
| 3 | 4g (4-Cl-Ph) | 91 | 79 |
| 4 | 4i (2-furyl) | 86 | 47 |
| 5 | 4j (2-thiophenyl) | 87 | 66 |
| 6 | 4n (4-Me-Ph) | 88 | 76 |
| 7 | 4o (4-tBu-Ph) | 93 | 63 |
| 8 | 4p (4-F-Ph) | 91 | 83 |
Thiourea-containing MOF catalysts
With the idea in mind of preventing also the self-quenching and to make possible the recycling of thiourea catalysts, Wang and co-workers have recently immobilised thiourea functionalities into IRMOF-3 following a post-synthetic modification strategy (PSM).56 The synthesis was addressed with IRMOF-3, a porous MOF with primary amine groups in its structure, which was further modified by adding an isothiocyanate under basic conditions (Scheme 7).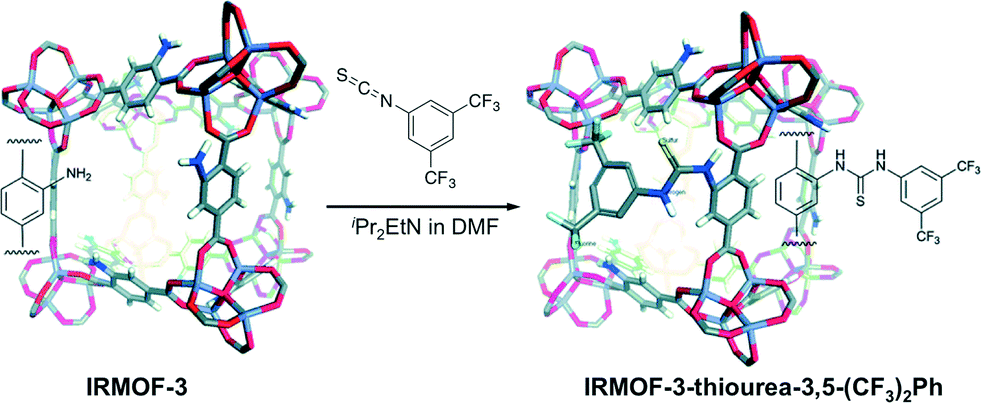 | ||
| Scheme 7 PSM approach between IRMOF-3 with 3,5-bis(trifluoromethyl)phenyl isothiocyanate. Adapted with permission from ref. 56. Copyright ® Royal Society of Chemistry. | ||
The introduction of the thiourea functionalities did not affect the nanomorphology of IRMOF-3 as shown in Fig. 8, since similar cubic crystals were produced in both cases. This indicates the high similarity of both crystalline structures. In addition, the obtained nano MOF catalyst showed good thermal stability (up to ∼344 °C) similar to IRMOF-3.
 | ||
| Fig. 8 SEM images of nanoscale A) IRMOF-3 and B) IRMOF-3-thiourea-3,5-(CF3)2Ph derivative. Reprinted with permission from ref. 56. Copyright ® Royal Society of Chemistry. | ||
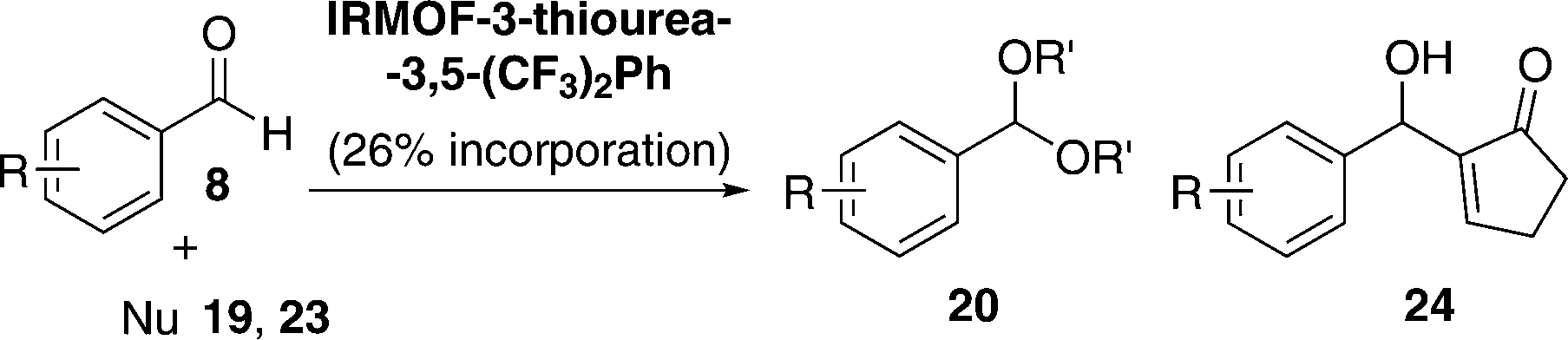
An initial screening of the acetalisation reaction of benzaldehyde (8a) at room temperature was performed testing all the catalysts synthesised (Table 5).
|
|
||||
|---|---|---|---|---|
| Entry | Catalystb | Loading (mol%) | Yieldc (%) | TON |
| a The reaction was performed using benzaldehyde (8a) (1.0 mmol), dry ethanol (19a) (4.0 mmol) and the appropriate amount of the organocatalyst at room temperature for 12 h. b MOF catalysts are used in nanoscale unless otherwise noted. c Determined by GC-MS. d 17% thiourea incorporation. e 26% thiourea incorporation. | ||||
| 1 | — | — | 0 | 0 |
| 2 | p-TsOH | 10 | 99 | 10 |
| 3 | Thiourea 21 | 1 | 81 | 81 |
| 4 | Thiourea 22 | 1 | 89 | 89 |
| 5 | IRMOF-3 | 10 | 6 | 0.6 |
| 6 | IRMOF-3-thiourea-Ph | 0.2 | 15 | 75 |
| 7 | IRMOF-3-urea-3,5-(CF 3 ) 2 Ph | 0.2 | 64 | 320 |
| 8 | IRMOF-3-thiourea-3,5-(CF 3 ) 2 Ph | 0.2 | 96 | 480 |
| 9 | Bulk (IRMOF-3-thiourea-3,5-(CF3)2Phd) | 0.2 | 82 | 410 |
| 10 | IRMOF-3-thiourea-3,5-(CF 3 ) 2 Ph | 0.2 | 98 | 490 |

As shown in Table 5, IRMOF-3 did not work after 12 h (entry 5) and this is in agreement with the fact that the thiourea moiety seems to be responsible for the acetalisation reactivity.57 The phenyl-substituted IRMOF-3-thiourea-Ph catalyst exhibited low reactivity (entry 6) in comparison to the 3,5-bis(trifluoromethyl)-phenylthiourea derivative IRMOF-3-thiourea-3,5-(CF3)2Ph (entry 8). The latter afforded higher reactivity than its urea counterpart (entry 7) and also showed better activity than its bulk partner (regular MOF instead of nano MOF) (entry 9), which suggested a higher utilization rate of the MOF surface and pore. A higher thiourea loading (26% vs. 17% thiourea incorporation) on the catalyst led to a comparable yield (98% vs. 96%, respectively), with turnover numbers (TON) of up to 490 (vs. 480) (entry 10). With the best reaction conditions in hand, IRMOF-3-thiourea-3,5-(CF3)2Ph (with 26% thiourea incorporation) was applied successfully to a range of substrates for acetalisation and Morita–Baylis–Hillman reactions (Table 6).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Nu | Product | Yield (%) |
| a Reaction conditions: for acetalisation (entries 1–4): aldehyde (8a–d) (1 mmol), dry alcohol (19a, b) (4 mmol) and catalyst (0.2 mol%) at room temperature for 12 h. Yields were determined by GC–MS. b For the Morita–Baylis–Hillman reaction (entries 5 and 6): aldehyde (8a, e) (1 mmol), 2-cyclopenten-1-one (23) (2 mmol), 1,4-diazabicyclo[2.2.2] octane (0.5 mmol) and catalyst (2 mol%) were stirred at 4 °C for 24 h. The product was purified by column chromatography on silica gel and the yield was based on the isolated product. | ||||
| 1a |
 8b
8b
|
EtOH 19a |
 20b
20b
|
92 |
| 2a |
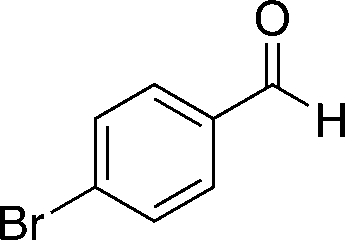 8c
8c
|
EtOH 19a |
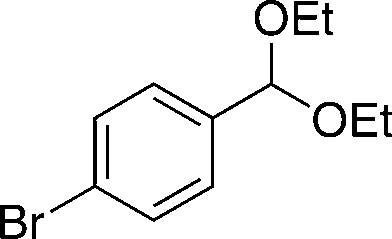 20c
20c
|
91 |
| 3a |
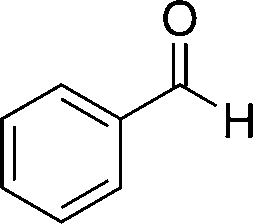 8a
8a
|
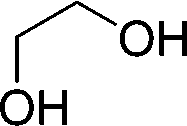 19b
19b
|
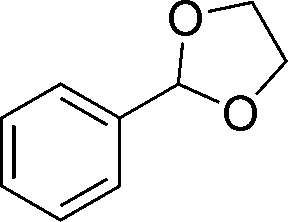 20d
20d
|
96 |
| 4a |
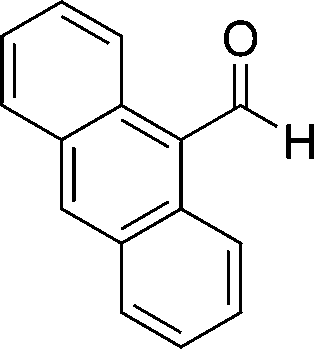 8d
8d
|
EtOH 19a |
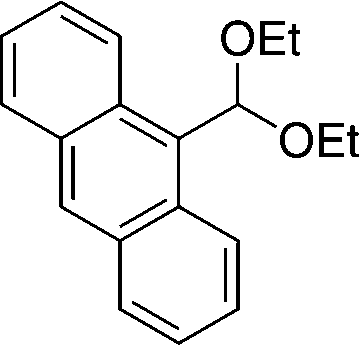 20e
20e
|
<5 |
| 5b |
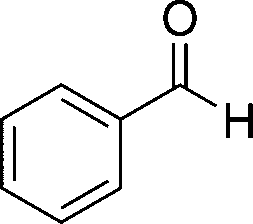 8a
8a
|
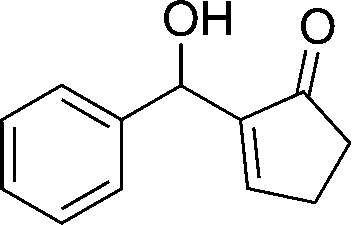
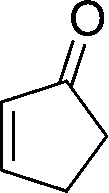 23
23
|
 24a
24a
|
73 |
| 6b |
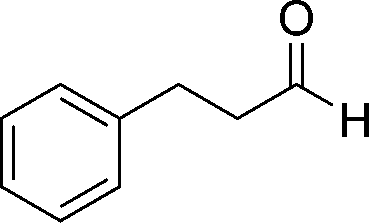 8e
8e
|
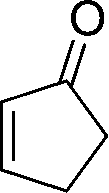 23
23
|
 24b
24b
|
81 |
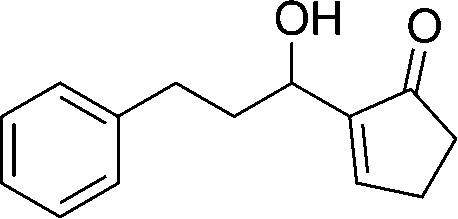
The strong covalent bond between the thiourea moiety and the amino group on IRMOF-3 ensures the stability of the catalytic functional groups, which maintains over 95% yield after 5 cycles. PXRD and FTIR analyses showed no differences before and after the recycling experiments. The mechanism depicted in Scheme 8 was proposed for the catalytic acetalisation of aldehydes. It starts with the coordination of the catalyst with the carbonyl group. Then, the nucleophilic attack of two alcohols through an ion-pair intermediate, which leads to benzaldehyde acetal and H2O binding, takes place. And finally, the dehydration of H2O through ligand exchange regenerates the catalyst. In the case of the M–B–H reaction the nucleophile is compound 23 in the presence of a base.
 | ||
| Scheme 8 Mechanistic proposal for the acetalisation reaction catalysed by IRMOF-3-thiourea-3,5-(CF3)2Ph.56 | ||
Squaramide-containing MOF catalysts
Hupp, Farha, Mirkin and co-workers have recently developed the first example of a MOF bearing acidic squaramide moieties: UiO-67-Squar/bpdc.58 In the synthesis of this material, preparation of the squaramide-functionalised strut (H2-Squar, 27) was first achieved from dimethyl 2-aminobiphenyl-4,4′-dicarboxylate (25) and 3-((3,5-bis-(trifluoromethyl)phenyl) amino)-4-methoxycyclobut-3-ene-1,2-dione (26) (Scheme 9). Then, it was incorporated into a zirconium-cluster-based UiO-67 MOF by reaction with the biphenyl chain (H2-bpdc, 7) in the presence of ZrCl4 as the source of metal. This 1:1 molar ratio mixed-strut approach was used to accommodate the relatively sterically demanding squaramide motif and to maintain open pores.
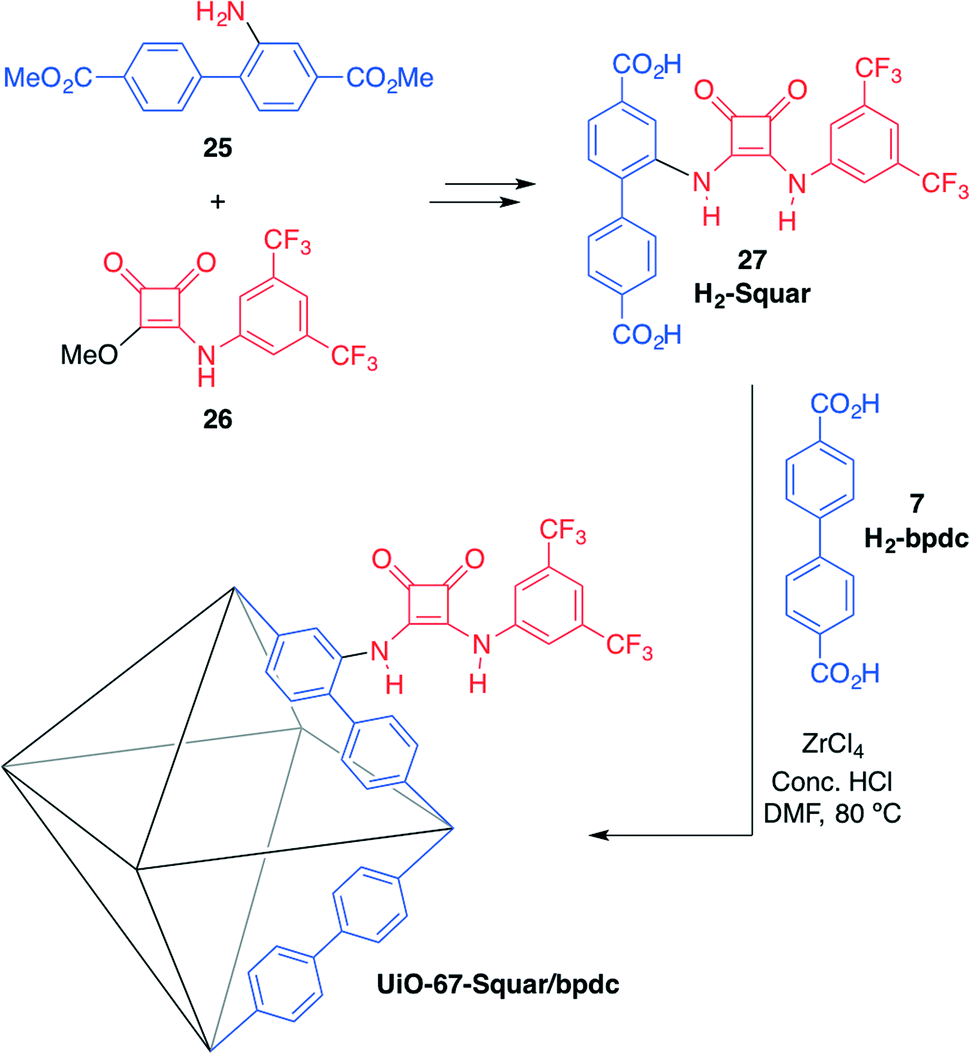 | ||
| Scheme 9 Synthesis of squaramide-based MOF UiO-67-Squar/bpdc and its structure. BET surface areas: 2500 m2 g−1 (UiO-67), 1700 m2 g−1 (UiO-67-Squar/bpdc).58 | ||
MOF UiO-67-Squar/bpdc was employed successfully as the catalyst for the Friedel–Crafts reaction between indole (12b) and nitrostyrene (4c) (Table 7, entry 6). The introduction of different amounts of squaramide groups into this MOF affected to a great extent the reaction rate. When all the biphenyl groups were substituted with squaramide units (UiO-67-Squar), the reactions showed similar outcomes compared to those obtained with the control experiment using only the non-substituted MOF (UiO-67) (entries 2 and 5). This lack of catalytic activity may be due to the formation of intermolecular bonds that block reactive squaramide centers and/or the shrinking of the pores of the MOF. However, when they employed UiO-67-Squar/bpdc with only 50% of the biphenyl groups linked to squaramide moieties, the results observed were much higher than those of the control experiment (Table 7, entry 6). The authors attributed this improvement in the reactivity to a decrease in the self-aggregation of squaramides, since any cooperative effect was observed between UiO-67 and the free squaramide (Me2-Squar (27′), the methyl ester analogues of squaramide 27) (Table 7, entry 4). Interestingly, when using low-polarity solvents at a higher temperature (50 °C), the corresponding product 5cc was obtained in nearly complete conversion (Table 7, entries 7 and 8).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Catalyst | Yieldb (%) |
| a Reactions performed with 3c or 12b (0.03 M), 4c (0.02 M) and the catalyst (10 mol%) in CD2Cl2 at r.t. b Determined by 1H NMR spectroscopy. c 10 mol% MOF and 10 mol% methyl ester analogous of squaramide 27 (Me2-Squar, 27′). d 1,2-Dichlorobenzene-d4 was used as solvent, at 50 °C. e Toluene-d8 was used as solvent, at 50 °C. | ||||
| 1 | 12b | 13b | — | 0 |
| 2 | 12b | 13b | UiO-67 | 22 |
| 3 | 12b | 13b | Me 2 -Squar (27′) | 0 |
| 4c | 12b | 13b | UiO-67 + Me 2 -Squar (27′) | 21 |
| 5 | 12b | 13b | UiO-67-Squar | 22 |
| 6 | 12b | 13b | UiO-67-Squar/bpdc | 78 |
| 7d | 12b | 13b | UiO-67-Squar/bpdc | 93 |
| 8e | 12b | 13b | UiO-67-Squar/bpdc | 95 |
| 9 | 3c | 5cc | — | 0 |
| 10 | 3c | 5cc | UiO-67 | 8 |
| 11 | 3c | 5cc | UiO-67-Squar/bpdc | 51 |
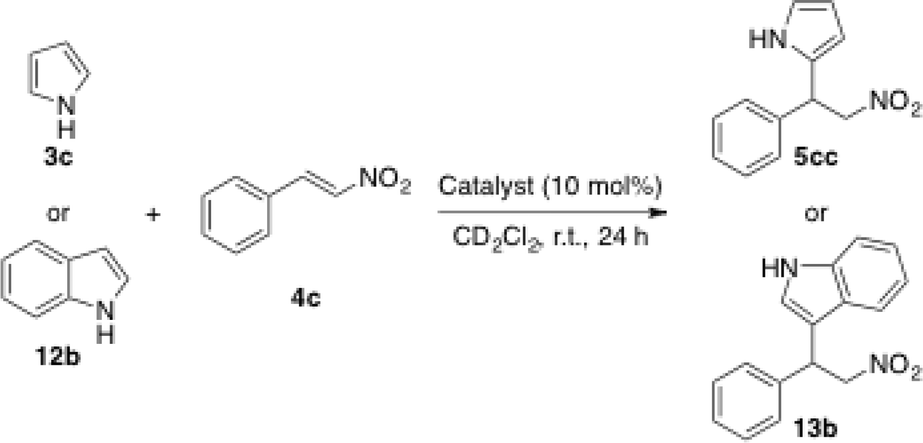
In the case of using pyrrole (3c) as the nucleophile, a lower conversion was achieved under the same conditions described for indole (compare entries 2 and 6 with 10 and 11, respectively). Remarkably, this catalyst could be reused through multiples cycles, yet a little decrease in the yield is observed over successive catalytic cycles.
Summary and outlook
In conclusion, the modular synthesis, tunable topology, large surface area and intrinsic porosity of MOFs make them very promising materials for heterogeneous catalysis. The well-defined confined space imposed by MOFs provides a unique functional microenvironment with catalytic sites uniformly arranged along the 3D supramolecular structure. The versatile field of MOF-based heterogeneous catalysis is now offering a new world of possibilities for hydrogen-bonding organocatalysts by overcoming self-aggregation and solvation issues that are usually found in homogeneous conditions. The first examples of urea-, thiourea- and squaramide-containing MOFs have demonstrated their effectiveness in relevant chemical transformations such as Friedel−Crafts reactions, nitroaldol (Henry) reactions, acetalisation, alcoholysis of epoxides and Morita–Baylis–Hillman reactions. In general, the results derived from these studies support the conclusion that catalysis occurs mainly within the pores of the functionalised MOFs, showing improved catalyst stability and substrate size selectivity in comparison to the analogous homogeneous catalysts. Moreover, the organocatalyst-containing MOFs could be easily separated and recycled several times without loss of activity and/or major leaching problems.These materials can be synthesised using either organocatalyst-containing organic struts or by post-synthetic modification of suitable functionalised MOFs. Obviously, the enhancement of the thermochemical stability of the hybrids as well as the achievement of larger size cavities constitute major aspects to consider when engineering new MOF-based organocatalytic systems that are expected to compete with other porous materials such as zeolites and mesoporous silica. However, the development of hydrogen-bonding organocatalyst-containing MOFs is still in its infancy with only the few examples summarized in this contribution. Many other examples are still necessary to demonstrate the reliability and broad use of these materials. In this sense, we anticipate intense research activities focused on asymmetric transformations via the introduction of chiral hydrogen-bonding organocatalysts,59,60 cascade or tandem reactions through the engineering of multiple catalytic sites and the activation of new reactive pathways that are otherwise unfeasible. The development of a new generation of MOF materials with the ability to respond to external stimuli that are common during a given reaction would facilitate the control of the catalytic function as an autonomous on-off unit. Moreover, appropriate comparative studies in terms of overall costs, reaction times, catalyst loading and TON/TOF numbers with the current homogeneous catalysts used in industry will be necessary to establish the real impact of these new heterogeneous organocatalysts. The report of isolated yields in future works could also provide additional credibility to the potential advantages of these heterogeneous catalysts.
We are convinced that the examples and results highlighted in this contribution will serve as an inspiration for the development of superior hydrogen-bonding organocatalysts to accomplish, in confined spaces, chemical transformations that are either slow or unaffordable under homogeneous conditions.
Acknowledgements
Universität Regensburg, Deutsche Forschungsgemeinschaft (DFG, 9209720), Universidad de Zaragoza (JIUZ-2014-CIE-07), Consejo Superior de Investigaciones Científicas (CSIC, PIE-201580I010) and Diputación General de Aragón (DGA) (Research Group E-104) are acknowledged for financial support. J. V. A.-R. thanks DGA for a predoctoral fellowship. D. D. D. thanks DFG for the Heisenberg Professorship Award.Notes and references
- J. Elemans, J. Cornelissen, M. Feiters, A. Rowman and R. Nolte, in Supramolecular Catalysis, ed. P. W. N. M. van Leeuwen, Wiley, 2008, ch. 6 Search PubMed.
- Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100 CrossRef CAS PubMed.
- J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615 CrossRef CAS PubMed.
- P. Tundo, P. Anastas, D. S. Black, J. Breen, T. Collins, S. Memoli, J. Miyamoto, M. Polyakoff and W. Tumas, Pure Appl. Chem., 2000, 72, 1207 CAS.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972 CrossRef CAS PubMed.
- D. Fiedler, D. Leung, R. Bergman and K. Raymond, Acc. Chem. Res., 2005, 38, 351 CrossRef PubMed.
- R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8794 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114 CrossRef CAS PubMed.
- J. J. Perry IV, J. A. Perman and M. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400 RSC.
- Z. Wang, G. Chen and K. Ding, Chem. Rev., 2009, 109, 322 CrossRef CAS PubMed.
- N. Ahmad, A. H. Chughtai, H. A. Younus and F. Verpoort, Coord. Chem. Rev., 2014, 280, 1 CrossRef CAS.
- M. O'Keeffe, Chem. Soc. Rev., 2009, 38, 1215 RSC.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782 CrossRef PubMed.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- E. N. Jacobsen and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20618 CrossRef CAS PubMed.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2011, 112, 782 CrossRef PubMed.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498 RSC.
- A. Carne, C. Carbonell, I. Imaz and D. Maspoch, Chem. Soc. Rev., 2011, 40, 291 RSC.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed.
- J. An and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 5578 CrossRef CAS PubMed.
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260 CrossRef CAS PubMed.
- W. Morris, W. E. Briley, E. Auyeung, M. D. Cabezas and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 7261 CrossRef CAS PubMed.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982 RSC.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. O. Z. R. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016 CrossRef CAS PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu and S. Asahina, Science, 2012, 336, 1018 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 123044 CrossRef PubMed.
- A. H. Chughtai, N. Admad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804 RSC.
- C. Volkringer and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 49, 4644 CrossRef CAS PubMed.
- T. Lescouet, J. G. Vitillo, S. Bordiga, J. Canivet and D. Farrusseng, Dalton Trans., 2013, 42, 8249 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2012, 48, 11275 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970 CrossRef CAS PubMed.
- For recent excellent reviews on MOF heterogeneous catalysts, see ref. 16 and L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC; M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed; J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC.
- P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289 RSC.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed.
- Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187 RSC.
- S. Lancianesi, A. Palmieri and M. Petrini, Chem. Rev., 2014, 114, 7108 CrossRef CAS PubMed.
- S. V. Deshapande, C. C. Meredith and R. A. Pasternak, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 1396 CrossRef CAS For X-ray studies of self-association of urea structures, see: S. Stanković and G. D. Andreetti, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3787 CrossRef; S. George, A. Nangia and V. M. Lynch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 777 Search PubMed; S. George and A. Nangia, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, 901 Search PubMed; A. M. Z. Slawin, J. Lawson, J. M. D. Storey and W. T. A. Harrison, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, 2925 Search PubMed.
- J. M. Roberts, B. M. Fini, A. A. Sarjeant, O. K. Farha, J. T. Hupp and K. A. Scheidt, J. Am. Chem. Soc., 2012, 134, 3334 CrossRef CAS PubMed.
- Different MOFs containing hydrogen-bond donors (primarily ureas) were synthesised before ref. 43; however, none of them were previously used as HBD catalysts.
- For the pioneering work using urea, see: G. Dessole, R. P. Herrera and A. Ricci, Synlett, 2004, 2374 CAS.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449 RSC.
- P. W. Siu, Z. J. Brown, O. K. Farha, J. T. Hupp and K. A. Scheidt, Chem. Commun., 2013, 49, 10920 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed.
- C. Palomo, M. Oiarbide and A. Mielgo, Angew. Chem., Int. Ed., 2004, 43, 5442 CrossRef CAS PubMed; C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2561 CrossRef; Y. Alvarez-Casao, E. Marqués-López and R. P. Herrera, Symmetry, 2011, 3, 220 CrossRef.
- X.-W. Dong, T. Liu, Y.-Z. Hu, X.-Y. Liu and C.-M. Che, Chem. Commun., 2013, 49, 7681 RSC.
- E. Dugan, Z. Wang, M. Okamura, A. Medina and S. M. Cohen, Chem. Commun., 2008, 3366 RSC.
- S. Bernt, V. Guillerm, C. Serre and N. Stock, Chem. Commun., 2011, 47, 2838 RSC.
- A. A. Tehrani, S. Abedi, A. Morsali, J. Wang and P. C. Junk, J. Mater. Chem. A, 2015, 3, 20408 CAS.
- J. Li, P.-Z. Li, Q.-Y. Li, Y. Cao, H. Lu, H. Wu, F. Li, Y. Shi, X.-J. Wang and Y. Zhao, RSC Adv., 2014, 4, 53975 RSC.
- X.-J. Wang, J. Li, Q.-Y. Li, P.-Z. Li, H. Lu, Q. Lao, R. Ni, Y. Shi and Y. Zhao, CrystEngComm, 2015, 17, 4632 RSC.
- Y. Luan, N. Zheng, Y. Qi, J. Tang and G. Wang, Catal. Sci. Technol., 2014, 4, 925 CAS.
- M. Kotke and P. R. Schreiner, Tetrahedron, 2006, 62, 434 CrossRef CAS.
- C. M. McGuirk, M. J. Katz, C. L. Stern, A. A. Sarjeant, J. T. Hupp, O. K. Farha and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 919 CrossRef CAS PubMed.
- For the use of primary and secondary amines coordinated to the MOFs and acting through covalent interactions, see: M. Banerjee, S. Das, M. Yoon, H. J. Choi, M. H. Hyun, S. M. Park, G. Seo and K. Kim, J. Am. Chem. Soc., 2009, 131, 7524 CrossRef CAS PubMed; D. J. Lun, G. I. N. Waterhouse and S. G. Telfer, J. Am. Chem. Soc., 2011, 133, 5806 CrossRef PubMed; W. Zhu, C. He, P. Wu, X. Wu and C. Duan, Dalton Trans., 2012, 41, 3072 RSC; P. Wu, C. He, J. Wang, X. Peng, X. Li, Y. An and C. Duan, J. Am. Chem. Soc., 2012, 134, 14991 CrossRef PubMed; T. Shi, Z. Guo, H. Yu, J. Xie, Y. Zhong and W. Zhua, Adv. Synth. Catal., 2013, 355, 2538 CrossRef; A. L. W. Demuynck, M. G. Goesten, E. V. Ramos-Fernandez, M. Dusselier, J. Vanderleyden, F. Kapteijn, J. Gascon and B. F. Sels, ChemCatChem, 2014, 6, 2211 CrossRef.
- M. J. Ingleson, J. P. Barrio, J. Bacsa, C. Dickinson, H. Park and M. J. Rosseinsky, Chem. Commun., 2008, 1287 RSC For examples of chiral Brønsted acid organocatalysts incorporated into the MOFs, see: M. Zheng, Y. Liu, C. Wang, S. Liu and W. Lin, Chem. Sci., 2012, 3, 2623 RSC; Z. Zhang, Y. R. Ji, L. Wojtas, W.-Y. Gao, S. Ma, M. J. Zaworotko and J. C. Antilla, Chem. Commun., 2013, 49, 7693 RSC; M. Lee, S. Min, S. N. Jeong and P. K. Thallapally, Dalton Trans., 2015, 44, 9349 RSC.
| This journal is © The Royal Society of Chemistry 2016 |