
Membrane gas separation technologies for biogas upgrading
Xiao Yuan Chen
ab,
Hoang Vinh-Thang
b,
Antonio Avalos Ramirez
a,
Denis Rodrigue
*b and
Serge Kaliaguine
b
aCentre National en Électrochimie et en Technologies Environnementales, Collège Shawinigan, Shawinigan, QC G9N 6V8, Canada
bDepartment of Chemical Engineering, Université Laval, Quebec City, QC G1V 0A6, Canada. E-mail: Denis.Rodrigue@gch.ulaval.ca; Tel: +1-418-6562903
First published on 17th February 2015
Abstract
Biogas is a renewable energy source like solar and wind energies and mostly produced from anaerobic digestion (AD). The production of biogas is a well-established technology, but its commercial utilization is limited because on-site purification is needed before its transport or use. Biogas composition varies with the biomass digested and contains mainly methane (CH4) and carbon dioxide (CO2), as well as traces of hydrogen sulfide (H2S), ammonia (NH3), hydrogen (H2), nitrogen (N2), carbon monoxide (CO), oxygen (O2). In some cases dust particles and siloxanes are present. Several purification processes including pressurized water scrubbing, amine swing absorption, pressure swing adsorption, temperature swing adsorption, cryogenic separation and membrane technologies have been developed. Nevertheless, membrane technology is a relatively recent but very promising technology. Also, hybrid processes where membranes are combined with other processes are believed to have lower investment and operation costs compared with other processes. In this report, a discussion on the different materials used to produce membranes for gas separation is given including inorganic, organic and mixed matrix membranes, as well as polymer of intrinsic microporosity (PIM). Advantages and limitations for each type are discussed and comparisons are made in terms of permeability and diffusivity for a range of operating conditions.
Xiao Yuan Chen obtained her Ph.D. in 2012 on the synthesis and characterization of new polyimide membranes for gas separation from the department of chemical engineering at Université Laval, Canada. She also obtained her MSc from the same department on the production and characterization of cellular polymers. She is presently a research assistant at Centre National en Électrochimie et en Technologies Environnementales, Collège Shawinigan, Shawinigan, Canada, as well as a postdoctoral fellow (part time) at Université Laval. Her work involves the preparation of polymer hollow fibre membranes and mixed matrix membranes suitable for biogas separations. |
Vinh-Thang Hoang received his B.Sc. degree in Organic and Petrochemical Technology from Hanoi University of Science and Technology in 1993, M.Sc. degree in Physico- and Theoretical Chemistry from the Institute of Chemistry, Vietnam Academy of Sciences and Technology in 2001, and a Ph.D. degree in Chemical Engineering from Laval University in 2005. After some years of postdoctoral research in the laboratory of Prof. Serge Kaliaguine, he is currently a Research Staff in the Chemical Engineering Department, Laval University. His research interests are focused on the experimental and modeling investigations of zeolites, mesoporous materials, and metal–organic frameworks in catalysis, adsorption–diffusion, membrane separation, and electrochemistry. |
Avalos Ramirez, Chemical engineer from Universidad Nacional Autonoma de Mexico and Ph.D. from Universite de Shrbrooke. Dr Avalos Ramirez is researcher in agrienvironmental and bioprocesses engineering in the “Centre national en electrochimie et en technologies environnementales (CNETE)”. Specialist with 10 year experience in treatment of air polluted with volatile organic compounds and greenhouse gases using biological processes, such as biofilter and biotrickling filter. Other fields of expertise are: anaerobic digestion, wastewater treatment, integration and simulation of chemical and biological processes, and valorisation of biomasses in biofuels and bioproducts. |
Denis Rodrigue is a professor in the department of chemical engineering at Université Laval, Canada. He obtained a Ph.D. in chemical engineering from Université de Sherbrooke in 1996 with a specialization in rheology and non-Newtonian fluid mechanics. His main research activities involves polymer (thermoplastics and elastomers) processing, characterization and modelling. |
Professor Serge Kaliaguine received his Ph.D. from the University of Toulouse in 1967. Since then he has been teaching at Université Laval. His research interests include the development of industrial catalysts, the applications of surface science and studies of mixed matrix membranes for gas separation and PEM fuel cells. |
1. Introduction
Biogas is a renewable energy source like solar and wind energies. It is also a carbon neutral fuel produced from anaerobic digestion (AD) which is one of the most efficient ways to store energy. Solid and liquid digestates of AD are rich in nutrients for plants and soil microflora, such as nitrogen and phosphorus. Additionally, pathogens and parasites are inactivated during AD. Most of the time, the digestates simply need a stabilization post-treatment and their characteristics allow them to be used for soil amendment without sanitation risks, such as water borne diseases.1The substrates to produce biogas by means of AD are residual organic materials (ROM) issued from municipal, industrial, institutional and agricultural sectors. AD can take place in liquid or solid phase, but the most common digester operation is in liquid phase. The inlet solid concentration in the digester is usually in the 2–10% range.2 AD technology is also cheaper and simpler than others to produce bio-fuels. It can also be found in a wide range of sizes. For example, small scale application is a common way to transform house wastes into biogas for heating and cooking in several countries. The production of biogas as a fuel does not contribute to the accumulation of greenhouse gases (GHG) in the earth's atmosphere because the carbon dioxide (CO2) produced during combustion was previously captured by plants. The production of biogas from ROM represents a controlled capture of methane (CH4) produced during AD, thus avoiding the emissions of this GHG to the atmosphere like in the case of landfilled ROM.3
Biogas has a high calorific value (35–44 kJ g−1) which is similar to diesel, kerosene and LPG. It is also higher than other energy sources like coal and wood.4 Typically, biogas contains 55–60% CH4 and 38–40% CO2. It can also contain small amounts of incondensable gases like nitrogen (N2), oxygen (O2) and hydrogen (H2), as well as traces of hydrogen sulfide (H2S) and volatile organic compounds (VOC). The acid compounds in the gas and the impurities are corrosive or have the potential to produce corrosive compounds during biogas combustion. These compounds will affect the metal parts of internal combustion engines and tubing.5 Therefore, biogas purification is mandatory before corrosive compounds enter the natural gas grid or combustion engines. The purification costs can sometimes be so important that the production of upgraded biogas is economically less attractive than other biofuels. Nowadays, technological processes to clean-up biogas, as well as their optimisation, are attractive to decrease biogas upgrading costs. Examples of these technologies are absorption, high pressure scrubbing, high pressure adsorption, as well as cryogenic separation and membrane separation. Among these technologies, the latter is potentially advantageous for biogas purification, as discussed in the present document.6
2. Biogas
Biogas is produced by the biological transformation under anaerobic conditions of organic matter present in wastes like manures, sewage, sludge, municipal wastes, green wastes and plant residues.7 Most of the substrates used to produce biogas are solid wastes or wastewater issued from agri-food industry, beverage industry, alcohol distilleries, pulp and paper industry, and other miscellaneous sectors.8 The sources of biogas production are from landfill gas (LFG) and agricultural production, as well as different organic streams from municipal, residential and commercial sources. Another important source is wastewater treatment plant residuals. Moreover, biogas is commonly produced using regionally available wastes and its use decreases the consumption of fossil fuels.9 This gives biogas production and combustion its “environmentally friendly” label, and led many governments to promote its production by means of renewable energy subsidies.10 Overall, biogas is an excellent energy source for a huge applications, which can be grouped in three categories: heat and steam, generation/cogeneration of electricity, and vehicle fuel.8,11–13 Fig. 1 shows the biogas life pathway, from biological sources up to final uses.14 | ||
| Fig. 1 Pathway from biogas source, ad reaction, and clean up to distribution for utilization. | ||
2.1 History of biogas production and technology
Initially, biogas has been used cooking in Assyria as early as the 10th century B.C. AD was also applied in ancient China using solid wastes.15 Marco Polo mentions the use of covered sewage tanks going back 2000–3000 years in ancient Chinese literature. There are documents recording the use of AD by humans in the mid-nineteenth century, for example the construction of digesters in New Zealand and India, as well as the capture of biogas from a sewage sludge digester in Exeter (UK) to fuel streetlights in the 1890.16 In the Guangdong province of China, an 8 m3 hydraulic biogas tank fed with garbage was constructed as early as 1921 to commercially produce biogas for cooking and lighting.15 At the same time, the first plant of sewage treatment product biogas into the public gas supply started in Germany and the first large scale agricultural biogas plant began operation around 1950 in Germany. In the 1970, high oil prices led to the development and research of alternative energy sources, thus contributing to increased interest in biogas technology. This also resulted in many countries of Asia, Latin America and Africa to experience a rapid growth of biogas digesters construction during the 1970 and the first half of the 1980.17 The domestic use of AD digesters continued to grow in Asia. For example, in China at the end of 1988, 4.7 million biogas digesters for household were recorded, while the number grew up to 26.5 million digesters in 2007.18 But the majority of those digesters had volumes between 6 and 10 m3. Another example is India where more than three million family-sized digesters were reported in 1999. From this date to 2007, the Indian government promoted the construction of nearly four million of these family-sized digesters.19According to an Energy Barometer on Biogas published in Europe, the production of renewable fuel has high potential and is growing rapidly due to increased concerns about oil and gas prices, as well as climate changes. For example, biogas production in the EU was 5.35 million tons of oil-equivalents (mtoe) in 2006, which was 13.6% higher than the biogas production during 2005. One of the main final uses of biogas is the production of electricity, which grew up by almost 29% over the same period. Germany was ranked first in Europe for the generation of electricity from renewable gas with a 55.9% growth in 2006.20 Fig. 2 shows the trend of biogas development in Germany where the number of digesters increased from 139 to 3711 between 1992 and 2007.21
 | ||
| Fig. 2 Development of biogas plants in Germany between 1992 and 2007. | ||
The first anaerobic digester supplied exclusively with commercial food wastes in North America (BioCycle London, ON) started in 2013.22 The AD plant, designated as “Energy Garden”, has the capacity to treat about 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per year. The treated wastes are mostly food wastes (fats, used fryer oils, grease and other wastes from restaurants, grocery stores, and food processing). The CH4 produced is sent to a 6 MW generator producing electricity sold under the Ontario government's Renewable Energy Standard.
000 tons per year. The treated wastes are mostly food wastes (fats, used fryer oils, grease and other wastes from restaurants, grocery stores, and food processing). The CH4 produced is sent to a 6 MW generator producing electricity sold under the Ontario government's Renewable Energy Standard.
2.2 Anaerobic digestion (AD)
Anaerobic digestion is widely used to produce biogas as a renewable biofuel. Recently, AD attracted the attention of several countries, especially the United Kingdom, Germany and Denmark.23 This bioprocess can play an important role to solve environmental problems such as the management of residual organic wastes and increasing GHG concentration in the atmosphere. Furthermore, the liquid fraction of the digestate can be used as a fertilizer, while the solid fraction can be used for other value-added products such as ligninolytic enzymes or fuels like lignocellulosic ethanol and syngas.24Nevertheless, AD process is a complex and the biotransformation of into CH4 is performed by chemoheterotrophic/methanogenic microorganisms. This conversion follows four steps: (1) hydrolysis, (2) acidogenesis, (3) acetogenesis, and (4) methanogenesis (see Fig. 3).25 During hydrolysis, polymeric and complex compounds of organic matter are hydrolyzed to free sugars, alcohols and other simple compounds. In acidogenesis and acetogenesis, these simple compounds are transformed into volatile fatty acids (VFA), acetic acid, CO2, and hydrogen (H2). Finally, during methanogenesis, acetic acid, CO2 and H2 are converted to CH4.11 Typical parameters used to follow AD performance are VFA, alkalinity, VFA/alkalinity ratio, biogas production rate, biogas concentration of CH4 and CO2, pH, COD (chemical oxygen demand). Generally, these parameters are monitored to get complementary information.26,27
 | ||
| Fig. 3 Metabolic route for the conversion of organic matter to the methanogenic substrates (acetate, CO2 and H2) and finally to CH4 and CO2. | ||
2.3 Biogas composition and utilization requirements
As mentioned above, biogas contains mainly two molecules: CH4 and CO2. Nevertheless, traces of different common gases (H2S, NH3, H2, N2, O2, CO) and saturated or halogenated carbohydrates can be present. Also, the gas mixture is saturated with water with possible presence of solid particles and siloxanes. Biogas composition varies with the biomass digested and Table 1 presents typical compositions for three different biomasses.28 The calorific power of biogas is proportional to its CH4 content. For internal combustion engines, a CH4 concentration higher than 90% is recommended.29 CO2 concentration in biogas is however up to 50% leading to reduced engine power output for electrically driven power plants by internal combustion engines. Water causes corrosion in the distribution pipeline and the presence of H2S or CO2 may corrode metallic surfaces such as valves, gears and exhaust systems. Sulfur stress cracking (SSC) is the main corrosion mechanism when a metallic part is in contact with H2S. Gosh reported that this mechanism starts when the concentration of H2S is above 50 ppm.30 This problem increases the engine maintenance costs.| Component | Agricultural waste | Landfills | Industrial waste |
|---|---|---|---|
| Methane CH4 | 50–80 | 50–80 | 50–70 |
| Carbon dioxide CO2 | 30–50 | 20–50 | 30–50 |
| Hydrogen sulphide H2S | 0.70 | 0.10 | 0.80 |
| Hydrogen H2 | 0–2 | 0–5 | 0–2 |
| Nitrogen N2 | 0–1 | 0–3 | 0–1 |
| Oxygen O2 | 0–1 | 0–1 | 0–1 |
| Carbon monoxide CO | 0–1 | 0–1 | 0–1 |
| Ammonia NH3 | Traces | Traces | Traces |
| Siloxanes | Traces | Traces | Traces |
| Water H2O | Saturation | Saturation | Saturation |
Biogas can be upgraded to natural gas for the same applications and Table 2 shows the variation of biogas specifications and the different requirements according to final use.31 For example, H2S concentrations below 1000 ppm are required for heating boilers, while for gas engines (CHP) the H2S content should be lower to increase engine operation time with water partial pressure low enough to avoid condensation. The presence of organic silicon compounds such as siloxanes in biogas can lead to abrasion problems due to deposition of silica on metallic surfaces.
| Application | H2S | CO2 | H2O |
|---|---|---|---|
| a Yes: removal required. No: removal not required. | |||
| Gas heating (boiler) | <1000 ppm | No | No |
| Kitchen stove | Yes | No | No |
| Stationary engine (CHP) | <1000 ppm | No | No condensation |
| Vehicle fuel | Yes | Recommended | Yes |
| Natural gas grid | Yes | Yes | Yes |
When biogas is used as fuel for transport vehicles, it is injected in the same engines configured for natural gas. This means that CO2, H2S, NH3, particles, water, and other trace components must be removed to obtain a fuel with a CH4 content of 95% (v/v) for high calorific value and engine safety. The specifications related to biogas quality for use as vehicle fuel and for introduction into the natural gas grid vary in each country. Table 3 reports the specifications for upgraded biogas to be distributed in the natural gas grid in France, Germany, Austria, and USA.32,33
| Compound | Unit | France | Germany | Austria | USA |
|---|---|---|---|---|---|
| Wobbe index | kW h m−3 | 13-15.7 | 12.8–15.7 | 13.3–15.7 | — |
| Heating value | kW h m−3 | — | 8.4–13.1 | 10.7–12.8 | 9.8–11.4 |
| CO2 | mol% | <2 | <6 | <2 | <2 |
| H2O | <Dewpoint | <Dewpoint | <Dewpoint | <120 ppm | |
| H2S | mol% | <0.00052 | <0.0003 | <0.0004 | <0.00037 |
| H2 | mol% | <6 | <5 | <4 | — |
| O2 dehydrated gas networks | mol% | — | <3 | <4 | <0.2–1 |
| O2 not dehydrated gas networks | mol% | — | <0.5 | <0.5 | <0.2–1 |
3. Comparison and evaluation of upgrading technologies for biogas purification
As mentioned above, biogas is a mixture of several compounds containing CH4 (giving its calorific value) and non-combustible carbon dioxide (CO2). In order to increase its calorific value, improve biogas combustion and decrease corrosion problem, CH4 concentration must be increased and impurities must be removed. It is know that CH4 is a clean fuel, the combustion being without any soot particles or other pollutants. Apart from CO2, biogas also contains small quantity of hydrogen sulphide (H2S). When water is present, H2S is dissolved and the aqueous solution is highly corrosive, making the biogas unusable. When the biogas is burned, H2S is oxidized to sulfur oxides which react with water and form acid (H2SO3). This acid is also corrosive and attacks the metallic surfaces of gas pipeline. The nonflammable CO2 in biogas not only reduces its calorific value, but also corrodes pipelines when water is present. On average, the biogas calorific value is 21.5 MJ m−3, while that of natural gas is 35.8 MJ m−3.7The biogas calorific value is upgraded, when CO2 is removed from the biogas. Others impurities like N2, O2, ammonia, siloxanes and particles are function of the source type and environment. Siloxanes can also damage heat exchangers and pumping equipment because they react to form silicon oxides during combustion producing solid deposits eroding mobile surfaces and reducing heat transfer.
Removal of CO2, H2S and impurities from biogas is commonly named upgrading. Biogas upgrading improves gas quality, which must be composed of more than 88% CH4 to become acceptable for more advanced utilisation, especially heat efficiency. It is then suitable for use in gas grids and vehicles.28
Today, several technologies are available on the market for biogas upgrading. The main unit operations are absorption, adsorption, cryogenic separation, and membrane separation (Fig. 4) as described next.
 | ||
| Fig. 4 Current technologies for biogas upgrading. | ||
3.1 Absorption
Absorption can be a physical or chemical phenomenon. High pressure water scrubbing (HPWS)34 and organic physical scrubbing (OPS) are physical absorption. Amine scrubbing (AS) and inorganic solvents scrubbing (ISS) are chemical absorption. HPWS is a technique based on physical absorption by dissolving gases in a liquid (water). In the case of biogas upgrading, the solubility of CO2 and H2S is much higher than that of CH4. Pressure affects the solubility of all compounds. First, the biogas enters a separator at a pressure of 2 bar where water and compounds heavier than CH4 and CO2 condense. Then, the gas is compressed to 10 bar and injected into the bottom of a scrubber where water is sprayed to absorb CO2. The gas leaving the scrubber is sent to dry and CH4 concentration can reach 98%. Water is sent to a unit of desorption where the pressure decreases to 1 bar allowing water regeneration. The main advantage of HPWS is its simplicity and high efficiency of methane recovery (>97% CH4). This technique requires water and an absorption column. The main disadvantages are high investment costs, high operating costs, possible clogging because of bacterial growth, foaming, low flexibility toward variation of gas input, as well as important consumption of water and energy.29Organic physical absorption uses organic solvent solution (polyethylene glycol) in place of water. Carbon dioxide has higher solubility in these organic solvents, resulting in lower scrubbing liquid circulation and less equipment for the same raw biogas capacity. Examples of organic physical scrubbing are commercially available for biogas upgrading technologies: Genosorb, Selexol, Sepasolv, Rektisol, and Puriso. The advantages are the same as HPWS including high recovery efficiency (>97% CH4), at the same time elimination of organic components as well as H2S, NH3, HCN, and H2O. The disadvantages are high investment and operation costs, complex operation, unfinished regeneration when stripping and vacuum are used (boiling required). There is also limited performance if glycol dilution (water) is implemented.28
Amine scrubbing (AS) is a chemical scrubbing process using aqueous solutions of different alkylamines to remove H2S and CO2. The most common alkanolamines used in industry are diethanolamine (DEA), monoethanolamine (MEA), and methyldiethanolamine (MDEA). A typical amine gas treatment includes an absorption column and an amine regeneration unit. In absorption columns, amine solution absorbs the gases to be removed (H2S and CO2) producing a sweetened gas. Then, the amine solution including acid gases is sent to the regeneration unit (stripper and re-boiler) to regenerate or “lean” amine which is returned to the absorption column. The stripped overhead contains a highly concentrated H2S and CO2 stream.
The main advantages are high upgrading efficiency with CH4 concentration >99% and low operation costs. The disadvantages are high investment costs, heat is necessary for regeneration, corrosion, and decomposition and poisoning of the amines by O2 or other compounds, precipitation of salts, and foaming.28
Inorganic solvent scrubbing (ISS) can be performed using potassium, sodium carbonate, and aqueous ammonia solutions.35 Generally, these processes are done with slight solvent changes and catalytic additions.
3.2 Adsorption
Adsorption processes can be categorized as pressure swing adsorption (PSA), vacuum swing adsorption (VSA), temperature swing adsorption (TSA), and electrical swing (ESA). PSA is a process separating molecules in a gas mixture at elevated pressure. The adsorbing materials generally used are different types of activated carbon, molecular sieves or zeolites, depending on the gas molecular characteristics and affinity of the adsorbing material. These adsorbing materials can preferably adsorb CO2 and H2S from the biogas, thus methane concentration increases in the gas. The higher the pressure, the more gas is adsorbed. When the pressure is reduced, the gas is freed or desorbed. This process produces a separation since different molecules in a gas mixture tend to be more or less strongly attracted by different solid surfaces. When the adsorbed bed is close to saturation, the regeneration reaction takes place by reducing pressure, thereby freeing the adsorbed gases. It is then ready to cycle again. The advantages of PSA are: high CH4 concentration (95–99%) and the gas can be directly delivered at high pressure (no need of compression). PSA disadvantages are high investment costs, high operation costs and extensive process control needed.36The process of VSA is a special case of PSA where the pressure is reduced to near-vacuum condition.37
In the case of TSA, adsorbent regeneration is achieved by an increase in temperature as increasing temperature at constant partial pressure decreases the amount adsorbed in the gas phase (or concentration in the liquid phase).38 A very important characteristic of TSA is that it is used exclusively for treating low adsorbate concentration feeds. Temperature increase alone is not used in commercial processes, but passage of a hot purge gas or steam through the bed to release the desorbed components is almost always used in conjunction with increasing temperature. TSA disadvantages are low energy efficiency and thermal aging of the adsorbent.
In ESA, a voltage is applied to heat the adsorbent and release the adsorbed gas. This technique is not very common in industrial practice.39
3.3 Cryogenic separation
Cryogenic process is based on the principle that different gases liquefy under different temperature-pressure conditions. It is a distillation process operated under very low temperatures (close to −170 °C) and high pressure (around 80 bar). Therefore, the production of very pure CH4 can use this technology. The process consists of cooling and compressing the raw biogas in order to liquefy CO2, which is then easily separated from the biogas. This process allows treating high flow rates of raw biogas reaching CH4 concentration in the range from 90% to 99%. Cryogenic processes require the use of a large amount of equipment and instruments such as compressors, turbines, heat exchangers, and distillation columns. It also requires high capital and operating costs.313.4 Membrane separation
Membrane separations are particularly appealing for biogas upgrading due to their lower energy consumption, good selectivity, easily engineered modules, and therefore lower costs. High CH4 recovery efficiency can be reached (>96%), while pure CO2 can be obtained. The main disadvantage of membrane separation is that multiple steps are required to reach high purity.28 This technology for biogas upgrading is based on gas dissolution and diffusion into polymer materials (membranes). When a differential pressure is applied on opposing sides of a polymer film, gas transport across the film (permeation) occurs. The gas rate of permeation is controlled by the solubility coefficient and diffusion coefficient of the gas-membrane system. Polysulfone, polyimide or polydimethylsiloxane are the common membrane materials for biogas upgrading. In the mid-1980, Cynara (Natco), Separex (UOP), and Grace Membrane Systems were already selling membranes made from cellulose acetate to remove CO2 from CH4 in natural gas. Thereafter, the largest membrane plant for natural gas processing (CO2/CH4 separation) was installed in Pakistan in 1995 with spiral wound modules.40 Medal (Air Liquide) polyimide hollow fiber membrane modules were available almost at the same time, in 1994.333.5 Comparison and evaluation of biogas upgrading technologies
A comparison between different biogas upgrading technologies can be difficult because several important parameters are strongly dependent on local conditions and requirements. The development of most biogas upgrading technologies is presently enough to satisfy any potential plant operation. For a rapid assessment of the main parameters and costs of the different biogas upgrading systems, a “Biomethane Calculator” was developed.36 Table 4 presents these parameters and the most important biogas upgrading technologies used for typical raw biogas composition and small plant capacity (below 1000 m3 h−1). The values for the different parameters represent means of upgrading plants taken from the literature data. The cost basis used is March 2012.| Parameter | Water scrubbing | Organic physical scrubbing | Amine scrubbing | PSA | Membrane technology |
|---|---|---|---|---|---|
| Typical methane content in biomethane [vol%] | 95.0–99.0 | 95.0–99.0 | >99.0 | 95.0–99.0 | 95.0–99.0 |
| Methane recovery [%] | 98.0 | 96.0 | 99.96 | 98.0 | 80–99.5 |
| Typical delivery pressure [bar(g)] | 4–8 | 4–8 | 0 | 4–7 | 4–7 |
| Electric energy demand [kWhel m−3 biomethane] | 0.46 | 0.49–0.67 | 0.27 | 0.46 | 0.25–0.43 |
| Heating demand and temperature level | — | Medium 70–80 °C | High 120–160 °C | — | — |
| Desulphurization requirements | Process dependent | Yes | Yes | Yes | Yes |
| Consumables demand | Antifouling agent, drying agent | Organic solvent (non-hazardous) | Amine solution (hazardous, corrosive) | Activated carbon (non-hazardous) | |
| Partial load range [%] | 50–100 | 50–100 | 50–100 | 85–115 | 50–105 |
| Number of reference plants | High | Low | Medium | High | Low |
|
|||||
| Typical investment costs [€/(m3 h−1) biomethane] | |||||
| For 100 m3 h−1 biomethane | 10100 |
9500 | 9500 | 10400 |
7300–7600 |
| For 250 m3 h−1 biomethane | 5500 | 5000 | 5000 | 5400 | 4700–4900 |
| For 500 m3 h−1 biomethane | 3500 | 3500 | 3500 | 3700 | 3500–3700 |
|
|||||
| Typical operational costs [ct m3 h−1 biomethane] | |||||
| For 100 m3 h−1 biomethane | 14.0 | 13.8 | 14.4 | 12.8 | 10.8–15.8 |
| For 250 m3 h−1 biomethane | 10.3 | 10.2 | 12.0 | 10.1 | 7.7–11.6 |
| For 500 m3 h−1 biomethane | 9.1 | 9.0 | 11.2 | 9.2 | 6.5–10.1 |
Table 4 shows that membrane technology presents several advantages. For example, it has the possibility to adjust the plant layout to local particularities like low demand of electric energy, low investment and operating costs. The lower methane recovery (80%) could be improved to 99.5% using multiple membrane steps and multiple compressors or efficient membrane configurations. It is also clear that both investment and operational costs are lower for membrane separation processes. This comparison is however only true for low capacity equipment (below 1000 m3 h−1).
4. Commercial polymer membranes for clean biogas
Although membrane technologies were shown to have remarkable performance for biogas upgrading, the number of commercially operated plants is limited. In general, biogas upgrading plants are classified into commercial plants (high capacity) and research installations (low capacity). Table 5 presents a list of biogas upgrading installations using membrane separation.| Country | Location | Operating since | Product stream (m3 h−1) |
|---|---|---|---|
| a Raw gas flow rate. | |||
| The Netherlands | Collendoorn | 1990 | 25 (Today 375) |
| USA | Los Angeles (CA) | 1993 | 2600a |
| Canada | Berthierville, (QC) | 2003 | NA |
| USA | Pittsburgh – Monroeville (PA) | 2004 | 5600a |
| The Netherlands | Beverwijk | 2006 | 80 |
| USA | Raeger (PA) | 2006 | 4721a |
| USA | Johnson city (TN) | 2006 | 2361a |
| Austria | Bruck an der Leitha | 2007 | 100 |
| Austria | Margarethen am Moos | 2007 | 70 |
| USA | Kersey (PA) | 2007 | 14164a |
| USA | Imperial (PA) | 2007 | 7082a |
| USA | Cairnbrook (PA) | 2007 | 4721a |
| USA | Davidsville (PA) | 2007 | 2361a |
| USA | Oklahoma city (OK) | 2008 | 2361a |
| US | Church hill (TN) | 2008 | 2361a |
| USA | Winder (GA) | 2008 | 7082a |
| USA | Atlanta (GA) | 2009 | 8263a |
| USA | Seattle (WA) | 2009 | 18886a |
| Germany | Kisslegg–Rahmhaus | 2010 | 300 |
| The Netherlands | Witteveen | 2010 | 200 |
| USA | Pittsburgh (PA) | 2010 | 4721a |
| USA | New Orleans (LA) | 2010 | 10623a |
| Austria | Wiener Neustadt | 2011 | 120 |
| Austria | Neunkirchen | 2011 | 10 |
| USA | Athens (TN) | 2011 | 3541a |
| USA | San Diego (CA) | 2011 | 2361a |
| USA | Fresno (CA) | 2011 | 2361a |
| Norway | Lillehammer | 2012 | 30 |
The first biogas upgrading plant was built in 1990 in Collendoorn (Netherlands) for commercial use. CH4 concentration can reach 88% with a rate of 25 m3 (STP)/h from landfill and uses hollow fiber membranes (Cirmac). The raw gas flow rate can reach 375 m3 (STP) per h today.
The first plant in USA to upgrade biogas using membrane separation (UOP, SeparexTM) was situated in 1993 in Los Angeles County. The biogas was produced by landfill and the plant had a capacity of 2600 m3 of raw gas per h and a gas containing 97.5% of CH4 was obtained.
Bebra Biogas established an upgrading setup in Kisslegg-Rahmhaus (Germany) to treat 300 m3 h−1 and produce a gas with a CH4 concentration of 98.7%. The feed pressure was 5–7 bar. Previous treatment was necessary in this case, such as dehydratation by condensation and H2S removal via activated carbon. If not, the gas permeation step to remove CO2 from the remaining mixture is affected. In recent years, upgrading biogas plants based on membranes increased substantially in USA and Europe since biogas is now believed to be a competitive renewable energy indicating great potential in the world energy market.
5. Types of materials and mechanisms of polymeric membrane for gas separation
5.1 Membrane materials
Table 6 presents the most important materials used for gas separation.42 Inorganic membranes are based on different materials like metal (alumina, cobalt, copper, iron, nickel, niobium, palladium, platinum, tantalum and vanadium), zeolites, carbon, and ceramic, etc. Generally, these membranes show higher gas separation performances combined with substantial chemical and thermal stability. Nevertheless, these materials have poor mechanical properties and are difficult to process. This is why their fabrication is expensive.43 Furthermore, they are easily cracked (fragile), therefore conversion into high surface area modules is very difficult.43 Porous or dense ceramic membranes can resist high temperatures due to their chemical stability.42 They can also offer good selectivity and high permeability. At commercial scale, only palladium alloys used for ultra-pure hydrogen generation are still used. From the last decade, some inorganic membranes have been exploited with excellent selectivity for specific gas separation and were described in the scientific literature, with some applications close to commercialization. Table 6 presents the main materials for membrane gas separation.| Organic polymers | Inorganic materials |
|---|---|
| Polysulfone, polyethersulfone | Carbon molecular sieves |
| Cellulose acetate | Nanoporous carbon |
| Polyimide, polyetherimide | Zeolites |
| Polycarbonate (brominated) | Ultramicroporous amorphous silica |
| Polyphenylene oxide | Palladium alloys |
| Polymethylpentene | Mixed conducting perovskites |
| Polydimethylsiloxane | Metal organic frameworks (MOFs) |
| Polyvinyltrimethylsilane |
MOF (metal organic frameworks) are a newer class of crystalline and porous materials and are now used to overcome the limitations of inorganic membranes. Today, several investigations of mixed matrix membranes (MMM) technology combining the advantages of inorganic fillers with the acceptable mechanical properties and cost-effective processability of polymers were performed.44–46 Therefore, most of the membrane materials used today for gas separation are organic polymers. Many polymers can be easily processed into high surface area modules membranes giving reasonable separation property. The main polymers used are polycarbonate (PC), cellulose acetate (CA), polyesters (PE), polysulfone (PSf), polyimide (PI), polyetherimide (PEI) and polypyrrolones. Cellulose acetate, polysulfone and polyimide are used for industrial level utilization. At the present time, companies producing these membranes for industrial uses are: Air Products, Air Liquide, Cynara, GKSS Licensees, MTR, PermSelect, Praxair, UBE and UOP.47,48 Commercial membrane suppliers for CO2 removal are reported in Table 7.
| Company | Principal natural gas separation | Membrane module type | Membrane material |
|---|---|---|---|
| Medal (Air Liquide) | CO2 | Hollow fiber | Polyimide |
| W.R. Grace | CO2 | Spiral-wound | Cellulose acetate |
| Separex (UOP) | CO2 | Spiral-wound | Cellulose acetate |
| Cynara (Natco) | CO2 | Hollow fiber | Cellulose acetate |
| ABB/MTR | CO2, N2, C3+ hydrocarbons | Spiral-wound | Perfluoro polymers, silicon rubbers |
| Permea (Air Products) | Water | Hollow fiber | Polysulfone |
Cellulose acetate (CA) is the first commercial membrane material used to remove CO2 and H2S.49 These were then mainly used for desalination with high surface area asymmetric structure.50 CA membranes (spiral wound modular configuration) displayed much lower selectivity for gas mixture than ideal gas selectivity calculated for neat gas because of CO2 or heavier hydrocarbons plasticization.51,52 Until 1983, polydimethylsiloxane (PDMS), a silicone rubber, was regarded as an excellent candidate because of higher gas permeability compared to other synthetic polymers.53 Table 8 shows that the permeability of CO2 and CH4 in PDMS is higher than others due to the presence of several configuration and composition of the side chain and backbone. Obviously, disadvantages of this type of materials are poor mechanical properties and lower separation factor.54
Later, scientist interests shifted from rubbery to glassy polymers to improve permeation. For example, polysulfones (PSF) have lower selectivity leading to polyethersulfones (PES) investigations. PES selectivity (PCO2/PCH4) was slightly higher (28) than PSF (26).55 On the other hand, polycarbonates (PC) which are another type of polyesters with reasonably low CO2 permeability, was combined with flexible soft polymers like silicone rubber, to reach CO2 permeability of 970 Barrer. Also, poly(vinyl alcohol) (PVA) can be modified with methyl vinyl sulfone (MVSF) or t-butyl vinyl sulfone (BVSF) to remove SO2 from air.56 Commercial modules of PPO produced by Parker Filtration and Separation B.V. (The Netherlands) have been used for CO2/CH4 separation.57 Table 9 shows the gas separation properties of polymer membranes. PEI (polyetherimide, Ultem®1000) displays low CO2 permeability (1.4 Barrer) and moderate selectivity for CO2/CH4 (40).58 Among these polymers, polyimides are believed to be excellent membrane materials. Polyimides (PI) are particularly suited for the separation of CO2 from CH4. First, polyimides have excellent thermal, chemical, and mechanical properties. They are also easily produced into films. Second, polyimides exhibit better gas separation performances than most commonly used glassy polymers like PSF and PC. Third, they are relatively easy to prepare into a series of different chemical structures, because a wide range of acid dianhydrides and diamines is possible.
| Polymer | Permeability at 30 °C (Barrer) | Selectivity (—) | |||
|---|---|---|---|---|---|
| H2 | CH4 | CO2 | H2/CO2 | CO2/CH4 | |
| a 1 Barrer = 10−10 [cm3(STP) cm cm−2 s−1 cmHg−1]. | |||||
| CA | 2.63 | 0.21 | 6.3 | 0.41 | 30.0 |
| EC | 87 | 19 | 26.5 | 3.33 | 1.39 |
| PC | — | 0.13 | 4.23 | — | 32.5 |
| PDMS | 550 | 800 | 2700 | 0.20 | 3.38 |
| PI | 28.1 | 0.25 | 10.7 | 2.63 | 42.8 |
| PMP | 125 | 14.9 | 84.6 | 1.49 | 5.75 |
| PPO | 113 | 11 | 75.8 | 1.49 | 6.89 |
| PSf | 14 | 0.25 | 5.6 | 2.5 | 22.4 |
Polyimide membranes were applied in various gas separations and the first application was for separating helium in 1962 by DuPont laboratories. In 1987, the first commercial application of industrial polyimide (PI) membranes to purify hydrogen (hollow fibers membranes) was performed in USA. PI hollow fiber membranes were used to separate CO2 from CH4 and have been installed in 1994. Polyimide membranes can be grouped in two categories: 6FDA-based and non 6FDA-base polyimides. The first series are composed of 6FDA–DAT, 6FDA–ODA, 6FDA–BAPAF, 6FDA–DAP, 6FDA–DABA, 6FDA–TrMPD, 6FDA–DAM, 6FDA–mPD, 6FDA–4mPD, and their co-polyimide. The second series are composed of Matrimid® 5218 (BTDA–DAPI), Kapton® (PMDA–ODA) and P84 (BTDA–TDI/MDI) which are three common commercial polyimides with their respective chemical structures listed in Fig. 5.
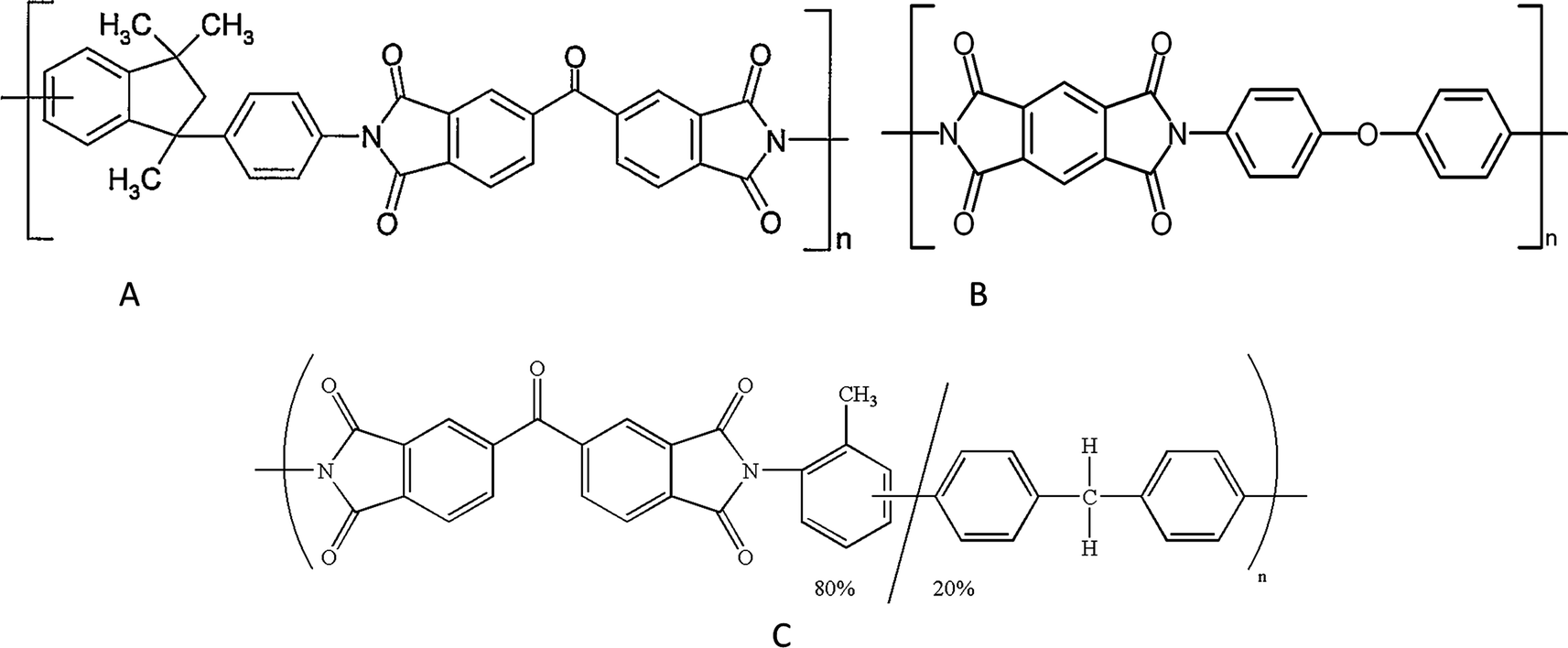 | ||
| Fig. 5 Chemical structure of commercial polyimides: (a) Matrimid® 5218, (b) Kapton®, and (c) p84. Chemical structure of commercial polyimides: (a) Matrimid® 5218, (b) Kapton®, and (c) p84. | ||
From Table 10, all three commercial polyimide membranes have smaller permeation and moderate selectivity for CO2/CH4 separation than 6FDA-based membranes (see Table 11 and 12). Among these three materials, Matrimid has the largest permeability but lowest selectivity, whereas Kapton and P84 have very low permeabilities. Therefore, these three materials do not have sufficient performances for gas separation. It is thus necessary to develop other polyimides with better properties.
| Polymer material | Configuration | CO2 permeability/permeance | CH4 permeability/permeance | Selectivity | Operation conditions | Ref. |
|---|---|---|---|---|---|---|
| a 1 GPU = 10−6 cm3(STP) cm−2 s−1 cmHg−1. | ||||||
| Matrimid® | Flat | 6.5 Barrer | 0.19 Barrer | 34 | Pure gas, 35 °C, 10 atm | 59 |
| Matrimid® | Hollow fibre | 14.7 GPU | 0.24 GPU | 59.6 | Pure gas, 20 bar, 30 °C | 60 |
| Matrimid® | Hollow fiber | 11.2 GPU | 0.26 GPU | 47 | 15 bar, 20 °C, 20/80 CO2/CH4 | 61 |
| Matrimid®/PES | Dual layer hollow fiber | 9.5 GPU | 0.24 GPU | 40 | 10 bar, 22 °C, 40/60 CO2/CH4 | 62 |
| Matrimid®/p84 | Hollow fiber | 11 GPU | 0.26 GPU | 42 | 10 bar, 35 °C, 20/80 CO2/CH4 | 63 |
| Kapton | Flat | 1.5 Barrer | 0.03 Barrer | 50.8 | 30 °C, 40 bar, 2–5/98–95 CO2/CH4 | 60 |
| Kapton | Flat | 0.866 Barrer | 0.026 Barrer | 33.3 | Pure gas, 35 °C, 10 bar | 64 |
| P84 | Flat | 1.2 Barrer | 0.02 Barrer | 50 | Pure gas, 35 °C, 10 atm | 59 |
| Sl. no. | Membrane | Operating conditions | Permeability (Barrer) | Selectivity α(CO2/CH4) | ||
|---|---|---|---|---|---|---|
| Temp. (°C) | Pressure (bar) | CO2 | CH4 | |||
| a 1 Barrer = 10−10 cm3(STP) cm cm−2 s−1 cmHg−1. | ||||||
| 1 | 6FDA–TAD PO | 30 | 1 | 27.4 | 0.52 | 52.2 |
| 6FDA–TABP | 63.6 | 1.37 | 46.2 | |||
| 2 | 6FDA-DAM DABA (2:1 membrane) |
35 | 20 | 121 | 4.48 | 27 |
| CHDM cross-linked | 22 | 0.73 | 30 | |||
| BG cross-linked 6FDA–DAM DABA (2:1 membrane) |
46 | 1.35 | 34 | |||
| 3 | 6FDA-mPD | 35 | 3.7 | 11.03 | 0.19 | 58 |
| 6FDA–mPDBA (9:1) |
6.53 | 0.10 | 65.3 | |||
| X-6FDA–mPD/DABA (9:1) |
9.50 | 0.15 | 63.3 | |||
| X-6FDA–DABA | 10.40 | 0.12 | 87.0 | |||
| 4 | 6FDA–durene | 35 | 10 | 455.8 | 28.4 | 16.05 |
| 5 | 6FDA–TAPA (amine terminated) | 35 | 1 | 65 | 1.59 | 41 |
| 6FDA–TAPA (anhydride terminated) | 6.7 | 0.11 | 61 | |||
| DSDA–TAPA (amine terminated) | 4.0 | — | — | |||
| DSDA–TAPA (anhydride terminated) | 1.0 | — | — | |||
| 6 | 6FDA–DATPA | 30 | 10 | 23 | 0.68 | 34 |
| 7 | 6FDA–PFDAB | 25 | 5 | 17.77 | 0.44 | 40.4 |
| 6FDA–m-PDA | 9.73 | 0.21 | 46.3 | |||
| ODPA–PFDAB | 11.03 | 0.36 | 30.6 | |||
| ODPA–m-PDA | 0.301 | 0.0064 | 47 | |||
| BTDA–PFDAB | 10.10 | 0.29 | 34.8 | |||
| BTDA–m-PDA | 0.428 | 0.0086 | 49.8 | |||
| 8 | 6FDA–6PDA-ceramic composite before irradiation | 35 | 3.5 | 47.27 | 3.65 | 12.94 |
| 6FDA–6FPDA-ceramic composite after irradiation | 71.52 | 1.75 | 40.9 | |||
| 9 | 6FDA–1,5-NDA | 35 | 10 | 22.6 | 0.46 | 49 |
| 10 | 6FDA–HAB | RT | 55 | 6 × 102 | 10 | 60 |
| 11 | 6FDA–durene/mPDA cross-linked with DMEA (6 h) | RT | 1 | 49.1 | 1.63 | 30.1 |
| 12 | 6FDA–NDA | 2 | 7 | 22.6 | 0.47 | 48.1 |
| 6FDA–NDA/durene (75:25) |
70.0 | 1.65 | 42.4 | |||
| 6FDA–NDA/durene (50:50) |
96.4 | 3.93 | 24.5 | |||
| 6FDA–NDA/durene (25:75) |
274 | 12.9 | 21.2 | |||
| 6FDA–NDA/durene | 423 | 28 | 15.1 | |||
| 13 | 6FDA–DDS | 30 | 5 | 35 | 0.37 | 94.6 |
| 14 | 6FDA–TAB | 30 | 10 | 54 | 0.9 | 60 |
| 6FDA–TAB/DAM(75:25) |
73.7 | 1.67 | 44 | |||
| 6FDA–TAB/DAM(50:50) |
155 | 7.38 | 21 | |||
| 6FDA–DAM | 370 | 17.6 | 21 | |||
| 15 | 6FDA–terphenyl | 31 | 2 | 21.48 | 0.747 | 28.76 |
| 6FDA–biphenyl | 12.97 | 0.358 | 36.23 | |||
| 6FDA–phenyl | 11.89 | 0.353 | 33.68 | |||
| 16 | 6FDA-zero generation amino terminated PAMAM dendrimer (100 mm) | 30 | 10 | 0.5 | 0.4 | 25 |
| 6FDA-first generation amino terminated PAMAM dendrimer (100 mm) | 0.7 | 0.6 | 20 | |||
| 6FDA-second generation amino terminated PAMAM dendrimer (100 mm) | 0.9 | 0.8 | 18 | |||
| Sl. no. | Membrane | Operating conditions | Permeance (GPU) | Selectivity α(CO2/CH4) | ||
|---|---|---|---|---|---|---|
| Temp. (°C) | Pressure (bar) | CO2 | CH4 | |||
| a 1 GPU = 10−6 cm3(STP)/cm2 sec cmHg. | ||||||
| 17 | 6FDA–BAPAF | 30 | 21 | 24.6 | 1.10 | 22.78 |
| 6FDA–DAP | 38.57 | 0.49 | 78.82 | |||
| 6FDA–DABA | 26.30 | 0.56 | 46.96 | |||
| 18 | 6FDA–3BDAF | 25 | 2 | 42.45 | 0.88 | 48 |
| 19 | 6FDA–IPDA | 31 | 0.6 | 14.8 | 0.38 | 43 |
| 20 | 6FDA–DAFO | 30 | 3.5 | 26.5 | 0.44 | 60 |
| 21 | 6FDA–APPS | 35 | 2 | 36.7 | 0.94 | 39 |
| 22 | 6FDA–durene/1,3-phenylene diamine (mPDA) copolyimide dense film coated with silicone rubber | 19.5 | 14 | 53.3 | 1.24 | 42.9 |
| 23 | 6FDA–DAT (3900 Å) | 20 | 7 | 300 | 4.60 | 65 |
6FDA-based polyimides have better gas separation performance than others with higher permeabilities.65–67 It was shown that –C(CF3)2– in dianhydride and diamine moieties can induce high selectivity and permeability. Tables 11 and 12 show the performance of 6FDA-based polyimide flat (Table 11) and hollow fiber (Table 12) membranes.
From Tables 11 and 12, flat membranes of 6FDA–HAB thermally treated at temperatures up to 450 °C show the best results in terms of CO2 permeability (600 Barrer) and CO2/CH4 selectivity (60). However, this thermal treatment was never used with hollow fibres because high treatment temperature (450 °C) makes the membrane very fragile. Hollow fibres of 6FDA–3BDAF (PCO2/αCO2/CH4 42.5/48), 6FDA–DAP (38.57/78.82) and 6FDA–DABA (26.3/46.96) have the best gas separation performance for CO2/CH4.
The selection of polymer materials to make membrane for gas separation applications depends on the polymer's chemical resistance, as well as sorption capacity and mechanical resistance. Other important requirements are: (a) intrinsic polymer permselectivity, (b) swelling resistance to membrane plasticization, and (c) film processability into asymmetric morphology.53
As reported in Table 13, Baker and Lokhandwala presented typical membrane materials that can be used for impurities separation from natural gas.48 The selectivities reported seem lower than the values in the literature because the latters are usually presented as the ratio of pure gas permeabilities, which is ideal selectivity. Here, the reported selectivities are the separation factors measured at high pressure, especially natural gas containing plasticizing molecules like CO2, water, BTEX aromatics (benzene, toluene, ethylbenzene, and xylene), and other heavy hydrocarbons. So separation factors are usually significantly lower than ideal selectivities. The selectivities reported in Table 13 can be reasonable for commercial/industrial membranes under “real” operations because they were determined under real gas mixture conditions. These typical membrane materials can also be used as good quality membrane for biogas purification.
| Component tobepermeated |
Categoryof preferred polymer material |
Typical polymerused |
Typical selectivities over methanea(%) |
|---|---|---|---|
| a Selectivities are typical of those measured with high-pressure natural gas. | |||
| CO2 | Glassy | Celluloseacetate, polyimide, perfluoropolymer |
10–20 |
| H2S | Rubbery | Ether-amideblock co-polymer |
20–30 |
| N2 | Glassy | Perfluoropolymer | 2–3 |
| Rubbery | Siliconerubber |
0.3 | |
| Water | Rubberyor glassy |
Several | >200 |
| C3+hydrocarbons |
Rubbery | Siliconerubber |
5–20 |
5.2 Mechanisms of polymer membrane gas separations
Studies on the limitations of inorganic materials for polymeric membranes used for biogas upgrading are based on the ‘solution-diffusion’ theory.68,69 Graham in 1866 (ref. 70) indicated that gases were able to permeate through non-porous rubber films because they can dissolve and diffuse in rubber films. He concluded that microscopic interconnecting pores or capillaries (open porosity) were not necessary for mass transfer to occur in polymers, but permeation consists of two steps: sorption and diffusion. Gas molecules are absorbed by the rubber depending on some ‘chemical affinity’. These sorbed gas molecules then can then diffuse.70 Gas sorption in polymers can be thermodynamically classified in two stages which are gas condensation and mixing with the polymer. This indicates that the solubility coefficient depends on gas condensability and interactions between gas molecules and polymers. Generally, diffusion coefficients in a polymer depend on the gas molecular sizes.71 Kinetic diameter (dk) is widely used as the penetrant size for gas diffusion. For CH4, dk is 0.38 nm, while the kinetic diameter of CO2 is 0.33 nm, which are very close to each other.72
 | (1) |
 | ||
| Fig. 6 Representation of gas or vapor transport through a non-porous polymeric membrane. | ||
 | ||
| Fig. 7 Literature data for CO2/CH4 ideal selectivity versus CO2 permeability (1991). | ||
 | ||
| Fig. 8 Robeson upper bound correlation for CO2/CH4 separation 2008 (alpha CO2/CH4 is selectivity of CO2/CH4; tr, thermally rearranged data reference). | ||
At steady state, the gas flux is calculated by Fick's first law:74
 | (2) |
 | (3) |
The gas equilibrium solubility coefficient is the ratio between gas concentration (gas molecules dissolved in the polymer at equilibrium) and the partial pressure of individual gas in the gas phase:74
| S = C/P | (4) |
When eqn (4) is introduced into eqn (3) and (5) simplifies to:
| P = DS | (5) |
It is clear the permeability coefficient (P) is determined by two elements: (1) a thermodynamic part which is the solubility coefficient (S) and determined by the number of gas molecules absorbed into and onto the polymer, and (2) a kinetic or mobility part which is the diffusion coefficient (D) determined by the mobility of gas molecules as they diffuse through the polymer. This means that permeability represents a pressure and thickness normalized gas flux (eqn (1)). It also determines the number of gas molecules dissolved and their flux through the polymer.
The values of the parameters D, P, and S can be determined by several method, which have been thoroughly reviewed.75,76 A more accurate procedure relies on independent measurements of P (steady state permeation) and S (equilibrium sorption).77–79 In this case, D is calculated via the solution-diffusion model of eqn (5) as the ratio between P and S. Another method to determine D is the “time-lag” method and solubility S can also be obtained from eqn (5). A widely used and accepted unit for P is:
| 1 Barrer = 10−10 cm3(STP) cm cm−2 s−1 cmHg−1. |
Permeance is generally used to characterize asymmetric or composite membranes, while permeability is typically used for dense film. For industrial applications, a focus on permeance or flux instead of permeability should be made since one could make a very dense film and have high permeability, however permeance could be very low. Permeance is defined through the steady state permeation flux via the pressure difference across the membrane as:
 | (6) |
| 1 GPU = 1 Barrer/1 micron = 10−6 cm3(STP) cm−2 s−1 cmHg−1 |
| αAB = PA/PB | (7) |
When gas mixtures permeate across a membrane, the separation factor (α*AB), which represents the ability of a membrane to separate a binary gas mixture, is defined as:74
| α*AB = (yA/yB)(xA/xB) | (8) |
 | (9) |
Thus, the separation factor not only depends on the gas-polymer membrane system, but also on a driving force which is the pressure difference (p2 − p1) between upstream and downstream, as well as feed composition (xA, xB) and permeate gas (yA, yB).80 When p2 is much higher than p1, eqn (9) simplifies to:
| α*AB = αAB | (10) |
6. Polymeric membranes
As mention above, polymers are dominating materials because they are more easily processed into hollow fibers for commercial gas separation (biogas upgrading). Nevertheless, polymer membranes have two major problems: the permeability/selectivity trade-off (Robeson plots), and the effect of plasticization at high pressure or long time period (because of CO2 or C3+ heavy hydrocarbons in biogas).6.1 Robeson's upper bound
The first Robeson curve in 1991 was proposed for the CO2/CH4 separation factor (ideal selectivity of pure gases) versus CO2 permeability in glassy and rubber polymers at 10 atm.81 Generally, glassy polymers have higher selectivity and lower permeability compared to rubbery polymers.Then, Robeson established another CO2/CH4 upper bound relationship in 2008.82 The new data also included thermally rearranged (TR) polymers that were synthesized from 6FDA and bisAPAF via thermal imidization up to 300 °C and thermally rearranged at 350, 400 and 450 °C.83 The latter comprises benzoxazole-phenylene or benzothiazole-phenylene groups on the backbone and were found to show high CO2/CH4 separation abilities. These polymers are unique because they have free-volume elements such as pores and channels influencing molecular sieving as produced via thermal reactions leading to insoluble and infusible polymers. Therefore, 6FDA-based PI-membranes with hexafluoro substituted carbon –C(CF3)2 in their backbone could improve performances and are widely used for CO2/CH4 separation.
From 1991 to 2008, it was clear that improvements in CO2/CH4 selective membranes occurred compared to the previous few decades because these new modified membranes surpassed the 1991 upper bound. Carbon molecular sieve (CMS) membranes formed by the pyrolysis of polyimide precursors can also perform beyond the 2008 limit.
6.2 Plasticization
Glassy polymer membranes have higher permselectivity, higher chemical resistance, as well as good thermal stability and mechanical strength compared to rubbery polymers, giving them an edge over other polymers.84 On the other hand, glassy polymers suffer from plasticization effects at high pressure or long period of biogas upgrading. Plasticization is defined as the increase of polymer chains motion due to the presence of one or several molecules (CO2 or C3+ heavy hydrocarbons). As a result, the permeability of each component increases while selectivity decreases.85 In CO2/CH4 membrane separation, CO2 sorption in glassy polymers can improve local molecular organization leading to decreased permselectivity. Plasticization of PI membranes may have three negative effects on gas separation. First, as observed in previous studies,86–88 most of the glassy polymer membranes display a decreased permeability with increasing pressure. Permeability increases rapidly if the pressure is higher than the plasticization pressure. Second, the separation factor decreases sharply with increasing feed pressure in CO2/CH4 separation.89 The highly sorbed molecules (carbon dioxide or heavy hydrocarbons) because free volume increase and methane can start permeate. Hence, the polymer chains are “solved” by penetrant molecules leading to matrix swelling, as well as increasing free volume and segmental mobility of the polymer matrix. Third, the permeability of a gas pair is not constant for a plasticized glassy polymer; i.e. it increases slowly and continuously with time above the plasticization pressure.In the work of Donohue et al.,90 the ideal selectivity of CO2 over CH4 was around 3–5 times the separation factor of the mixed gases for cellulose acetate membranes at CO2 concentrations higher than 50% in feed gas and feed pressure up to 54 bar. This causes swelling and plasticization since CO2 is more soluble in CA than CH4. However, Schell et al.91 used CA membranes to remove both CO2 and H2S and were able to attain the US pipeline specifications in terms of sour gas concentrations. In another example, Sridhar et al.60 used different PI membranes (Matrimid, P84 and Kapton) to separate CO2/CH4 and varied the CO2 feed content between 0 and 20 mol%. The results showed different CO2 and CH4 permeabilities and selectivities for pure gas and mix gases. Matrimid membranes gas separation factor was 76% lower than their ideal selectivity, as well as 40% lower than the other two PI membranes. This was caused by coupling effects between CO2 and CH4 and plasticization effect at higher CO2 concentration in the feed, both leading to selectivity loss.
6.3 Co-polyimide
Co-polyimides used as membrane materials, are expected to go beyond the Robson upper bound curve and were studied in recent years. 6FDA-based polyimides are the main polymers with examples such as 6FDA–TMPDA/DAT92 6FDA–TMPDA/MOCA93 and 6FDA–TeMPD/ODA.67 These copolyimides were synthesized systematically with different diamine ratio (1/0, 0.75/0.25, 0.5/0.5, 0.25/0.75, and 0/1). The results showed that these 6FDA-based co-polyimide have lower pure gas permeability for CO2 and CH4, but ideal selectivity increased with DAT, MOCA or ODA content. Furthermore, 6FDA–ODA with nine different diamines: DBSA, DABA, DAPy, DANT, DDS, MDA, BADS, BABP and DABN copolymers were produced by one-step polymerization. Diamine monomers, having different reactivities with respect to polycondensation, will produce a wide range of molecular weight and CO2 permeability varied with diamine content.94,95 6FDA–DDS/6FAP copolymers were tested with different diamine ratios (1/0, 0.75/0.25, 0.67/0.33, 0.5/0.5, 0.33/0.67, 0.25/0.75, and 0/1). Their pure gas CO2 permeability increased and the CO2/CH4 ideal selectivity decreased with increasing 6FAP content.96 Pebax® is a commercial polyether–polyamide copolymer and Lillepärg97 used Pebax® MH1657 blended with poly(ethylene glycol) (PEG) of low molecular weight for gas separation. The best results for CO2 permeability was from 79 to 378 Barrer, and ideal selectivity from 16.8 to 14.3. Until now, no significant improvements in gas separation properties have been shown for co-polymerization modified membranes.6.4 Suppression of plasticization for polymeric membranes
Numerous researchers investigated ways to suppress plasticization and develop different polymer membrane gas transport properties, including novel polymer synthesis, blending polymers, thermal treatment,98,102 UV cross-linking, and chemical cross-linking.99–101 These methods are summarized in Table 14 and presented below.| Reference | Material | Structures | Methods of suppression | Years |
|---|---|---|---|---|
| Bos et al.102 | Matrimid 5218 | Dense flat sheet | Thermal treatment | 1998 |
| Bos et al.103 | Matrimid 5218 | Dense, flat sheet | Semi-interpenetrating | 1999 |
| Staudt-Bickel and Koros109 | 6FDA-based polyimides | Dense, flat sheet | Chemical crosslinking | 2001 |
| Bos et al.104 | Matrimid 5218 | Dense, flat sheet | Matrimid blend with polysulfone and copolyimide P84 | 2001 |
| Wind110,111 | 6FDA-based polyimides | Dense, flat sheet | Covalent and ionic crosslinking | 2002 |
| Chen et al.101 | 6FDA-based polyimides | Dense, flat sheet | Chemical crosslinking | 2012 |
Commercial polyimide Matrimid® 5218 membranes were thermally annealed at 350 °C for 15–30 min and shown a great decrease of CO2 plasticization for pure gas and mixed gas permeation tests.102 Suppression of CO2 plasticization by the formation of a semi-interpenetrating polymer network (s-ipn) was investigated by the same authors.103 The mixture of Matrimid® 218 and Thermid FA-700 (oligomer) was made at three different ratios (70/30, 80/20, 90/10) with thermal treatment and at different curing times (15, 30, 60, 120 min). The results show that suppression of CO2-induced plasticization can be up to 40 atm. Blending polymers such as Matrimid® 5218 with polysulfone (PSf) (50:50 w/w), and Matrimid with P84 (60:40) showed that resistance to plasticization was improved from 18 atm for neat Matrimid up to 25 atm for PI blends at 55/45 mol% mixture of CO2/CH4 and 35 °C.104
It is difficult to use thermal treatments for commercial membrane manufacturing processes because they need more energy to produce. Currently, chemical cross-linking to suppress plasticization is a simple method without heating and is believed to be more efficient.101,105–112 6FDA–durene polyimides (PI) and Matrimid were selected to study the effect of chemical cross-linking in solution.91 EDA, PDA, BuDA and CHBA were chosen as chemical cross-linking agents. Other cross-linking agents are p-xylenediamine,106,107 diol reagents (ethylene glycol),108 1,4-butylene glycol,109 1,4-cyclohexanedimethanol,110 1,4-benzenedimethanol,111 and 1,3-propanediol.112 APTMDS, a diamino organosilicone, as well as a chemical cross-linking agent was used to prepare membranes for CO2/CH4 separation. The results showed that plasticization can almost be totally eliminated by immersion in aqueous methanol or via methanol addition during synthesis.101 Also, the modified membranes were shown to sustain pressures as high as 40 atm. Among all the methods available to suppress plasticization, chemical cross-linking is easier, more efficient, and also more fitted for industrial application.
7. Mixed matrix membranes
7.1 Inorganic membranes
Inorganic membranes are generally made using metals, ceramics, zeolites or carbon molecular sieves (CMS).113 These membrane have excellent thermal and chemical stabilities. Some of these inorganic membranes show much higher gas fluxes and selectivity compared with polymer membranes. For example, zeolites and carbon molecular sieve membranes have much higher diffusivity and selectivity than polymer membranes. Precise size and shape discrimination led to the narrow pore size distribution leading to excellent selectivity.114 For example, zeolite T (ERI-OFF) (0.41 nm pore size) which have small-pore, as well as zeolite of DDR (0.36 nm × 0.44 nm), and SAPO-34 (0.38 nm) which have small-pore very similar in size to CH4 (0.38 nm), but larger than CO2 (0.33 nm). Those membranes displayed high CO2/CH4 selectivity due to a molecular sieve effect. In the case of T-type membranes (ERI-OFF), Cui et al.115 reported a separation factor α = 400 with a CO2 permeance of P = 4.6 × 10−8 mol m−2 s−1 Pa−1 (138 GPU) at 35 °C. Tomita et al.116 reported a CO2/CH4 separation factor of α = 220 with a CO2 permeance of P = 7 × 10−8 mol m−2 s−1 Pa−1 (210 GPU) at 28 °C on a DDR membrane.Saracco and Speccia summarized the advantages and disadvantages of inorganic membranes.117 As shown in Table 15, inorganic membranes have some advantages such as long-term high temperatures stability and solvents resistance at high pressure. But they also have some disadvantages such as extremely high cost, brittleness, as well as lack of continuous fabrication technology to produce defect-free membranes. Other disadvantages are low surface area per unit volume and difficulty to be transformed into module with large surface area for industrial application.
| Advantages of inorganic membranes | Disadvantages of inorganic membranes |
|---|---|
| Long-term stability at high temperatures | High capital costs |
| Resistance to harsh environments | Brittleness |
| Resistance to high pressure drops | Low membrane surface per module volume |
| Easy cleanability after fouling | Difficulty in achieving high selectivities in large scale microporous membranes |
| Easy catalytic activation | Generally low permeability of the highly selective (dense) membranes at medium temperatures |
| Difficult membrane-to-module sealing at high temperatures | |
| Low membrane surface per module volume |
7.2 Mixed matrix membranes
Mixed matrix membranes (MMM) consist of an organic polymer combined with an inorganic (or sometimes organic) particles. The dispersed phase may be zeolites, carbon molecular sieves (CMS), carbon nanotubes (CNT) or other nano-size particles.118 Recently, MMM were prepared using metal–organic frameworks (MOF) with polymers matrices for CO2/CH4 gas separation.119 Therefore, MMM are desirable and present potential for high selectivity, high permeability or both, compared to actual polymer and inorganic membranes. Performances of MMM are however not the sum of the intrinsic properties of each individual component. Complex interactions between all the parameters can seriously affect the performance of MMM. Transport properties of MMM are highly function of membrane morphology at the nano-scale, which is critical for the overall membrane properties.Fig. 9 displays the different nano-scale structures of the interface between the polymer and the particles.120 Case I is an ideal morphology and difficult to get with perfect adhesion at the filler–polymer interface. Case II is a situation where the pores have been partially blocked at the surface of the particles by polymer chains. Case III shows that polymer molecules in direct contact with the zeolite surface are rigidified (limited mobility) compared to the bulk polymer. Case IV represents the detachment of polymer molecules from the particles surface producing voids at the interface.
 | ||
| Fig. 9 Schematic of different morphologies at the nano-scale in mmms. | ||
These four cases were reported in the literature. For example, Duval observed voids (case IV) at the interface between the polymer (polydimethylsiloxane, PDMS) and zeolites (silicalite-1, 13X and K–Y).121 He proposed that important stresses are produced on the external surface of the adsorbent during solvent evaporation leading to polymer dewetting. Other possible reasons for voids formation include repulsive forces at the polymer/filler interfaces, as well as different coefficients of thermal expansion.122
Polymer chains mobility in the contact region with the particles can be inhibited, an effect called rigidification (case III). In addition, increasing permeation activation energy can also reveal lower chain mobility (rigidification). As a result, selectivity increase leads to permeability decreasing quickly in MMM. The glass transition temperature (Tg) can be used to determine whether rigidification in the MMM occurs or not. It is well-known that Tg can qualitatively be used to estimate polymer chains flexibility. Therefore, MMM with rigidified polymer chains, have higher Tg compared to the base polymer.123
Reduction of surface area of porous fillers can be associated to pore obstruction by polymer molecules (case II) in MMM.122,124–126 Depending on pore size, polymer chains can enter the pores at different levels or even make complete blockage. Pore blockage always causes a decrease in gas permeability, the selectivity relying on the type of particles used. Smaller particles give more interfacial area between the polymer and particles, potentially making better MMM. Moreover, thinner MMM can be made by using smaller particles.127
Particle agglomeration due to sedimentation and migration to the surface is an important problem for the manufacture of MMM. Differences in density and other physical properties between the zeolite and the polymer can lead to spatial distribution problems. Zeolite precipitation may even occur. Agglomeration of zeolites may also cause pitting and forming non-selective defects in MMM.128 To solve this problem, increased solution viscosity, use of ultra-thin crystallites, and control of drying conditions are applied during membrane manufacture.
It is necessary to choose both materials for the same gas pairs. Inorganic particles usually have high selectivity compared to neat polymers. The Maxwell model states that inorganic fillers at low volume fraction in a polymer phase leads to important increases of the overall separation efficiency.129 The Maxwell model for MMM composed of a dilute suspension of spherical particles is given by:
 | (11) |
In order to have ideal fillers and polymer for MMM, two cases must be considered. One is to allow the smaller molecular gas (CO2) to go through. Therefore, MMM should combine inorganic fillers with molecular sieving properties and economical processability of polymers. For more condensable molecules, solubility selectivity is dominant for gas transport. Therefore, MMM must be produced to get selective adsorption and/or surface diffusion for the most condensable molecule, while limiting the less condensable one.
The general procedure to make flat MMM is as follow: (1) making an homogeneous polymer/solvent mixture, (2) preparation of a slurry of inorganic fillers/solvent mixture by sonication, (3) evaporation of the solvent mixture, (4) casting the solution, (5) membrane annealing at a specific temperature for residual solvent removal. This procedure is very similar to make dense polymer membranes. This is another advantage of MMM over more complex approaches for inorganic membrane production.45,46,118–120 It is however function of the polymer, solvents and fillers characteristic used.
7.3 MMM composed of polymers and zeolites
A zeolite is a crystalline microporous aluminosilicate having large cations and water molecules with high freedom of motion. This can allow good ion-exchange and reversible dehydration properties.130 Over 150 different zeolite crystal structures are known today. Most of them are synthetic materials, but some of these structures also occur as natural geological materials. Many type and families of zeolites have been made and used for gas separation.131 Structure type, structural dimension and pore size of some commonly used zeolites are summarized in Table 16.| Zeolite | Structural type | Structural dimension | Pore size (A°) |
|---|---|---|---|
| 3A | LTA | 3D | 3.0 |
| 4A | LTA | 3D | 3.8 |
| 5A | LTA | 3D | 4.3 |
| ITQ-29 | LTA | 3D | 4.0 |
| 13X | Faujasite | 3D | 7.4 |
| NaY | Faujasite | 3D | 7.4 |
| ZSM-2 | Faujasite | 3D | 7.4 |
| L | LTL | 2D | 7.1 |
| Beta | BEA | 3D | (5.5–5.5) and (6.4–7.6) |
| Silicalite-1 | MFI | 2D | (5.1–5.5) and (5.3–5.6) |
| ZSM-5 | MFI | 2D | (5.1–5.5) and (5.3–5.6) |
| SSZ-13 | CHA | 3D | 3.8 |
| SAPO-34 | CHA | 3D | 3.8 |
For adsorption, interaction with highly polar surface within the pores is the main driving force in zeolites. CO2 adsorbs more strongly than H2, CH4, and N2 on zeolites because of electrostatic quadrupole moment and molecular weight of CO2 are higher than others light gases. LTA, FAU, CHA, and MOR zeolites have high CO2 heat of adsorption.133,134 This unique property results in high CO2 adsorption capacity even at low concentration. Adsorption on zeolites is dependent on the following physical molecular properties:
Size and shape: most zeolites can be divided into three categories.135 First, small pore size (0.30–0.45 nm). These zeolites have 8 membered-rings pore apertures with free diameters like zeolite NaA. Second, medium pore size (0.45–0.60 nm). These zeolites have 10 membered-ring apertures, within free diameter like zeolite ZSM-5. Third, large pore size (0.6–0.8 nm). These zeolites have 12 membered-ring apertures or more within free diameter like zeolite faujasite (X, Y). Gas molecules smaller than the pore size can adsorb on zeolites, whereas larger gas molecules cannot.
Molecular polarity: gas molecules with higher polarity can be better adsorbed than non-polar gas for the majority of zeolites under identical conditions.
Counter-ion: the type of cation controls the electric field inside the pores, basicity, and the available pore volume, which offers a convenient means for tuning adsorption properties.135
Earlier research on MMM focused on zeolites dispersed in rubbery polymers for gas separation. As reported by Paul and Kemp in 1973, the diffusion time lag was increased quickly for CO2/CH4, but only small effects on steady-state permeation were observed, especially for polydimethylsiloxane (PDMS) with 5A zeolite.136
Then in 1991, the permeation properties of MMM from PDMS with silicalite-1 fillers was investigated for various gases (He, H2, O2, CO2, N2 and CH4) by Jia et al.137 Silicalite was considered as a molecular sieve: shape-selective effect for equilibrium adsorption of gas molecules and the kinetics of adsorption and diffusion into zeolites. However, the kinetic diameters of all gases was smaller than the zeolite pore size under study.
In 1993, Duval et al.138 investigated zeolites (silicalite-1, zeolites 13X, 3A, 4A, 5A and KY) addition to rubber polymer (PDMS, EPDM, PCP and NBR). The results indicated that zeolites 3A, 4A and 5A could not however improve the rubbery polymers permeation properties. Others zeolites improved the gas separation properties due to both CO2 sorption increase as well as the molecular sieving effect. Unfortunately, rubbery polymers have low mechanical strength, but good inherent transport properties compared to more rigid (glassy) polymers. These results were actually not very attractive and researchers started to study zeolite-filled glassy polymer membranes. But it was found difficult to improve the gas separation performance due to void formation at the filler–polymer interface or particle agglomeration. The main reason is poor polymer–zeolite compatibility. Therefore, Duval et al.139 used different silane coupling agents to modify the zeolite surface and improve adhesion. SEM micrographs of the membranes showed good adhesion between the silane and zeolite. Unfortunately, they did not obtained good permselectivity improvement.
Yong et al.140 used 2,4,6-triaminopyrimidine (TAP) as a compatibilizer to get rid of interfacial voids between polyimide (Matrimid) and filler (zeolite L) in MMM. They concluded that forming hydrogen bonding between zeolite particles and polyimide chains enhanced their contact. They also compared permeability and selectivity of composite membranes of PI, PI/TAP, PI/4A (pore size 0.38 nm), PI/13X (0.74 nm), PI/4A/TAP, and PI/13X/TAP for CO2/CH4 separation. PI/4A/TAP membrane had CO2/CH4 selectivity of α = 617 and CO2 permeability of P = 0.185 Barrer. But these type of zeolite-filled with polyimide mix matrix membranes did not improve CO2/CH4 separation.
Pechar et al. combined MMM using zeolite L with pore opening size of 0.71 nm as inorganic fillers and co-polyimide of 6FDA–6FpDA–DABA as the polymer phase.141 Aminopropyl-triethoxysilane (APTES) was used as a coupling agent to modify the zeolite surface, resulting in carboxylic acid groups on the polyimide backbone which were covalently linked with these groups. CH4 and CO2 permeability decreased with feed pressure both in zeolite-PI membrane and pure co-polyimide membrane. They concluded that both gases could not enter the zeolite pores, however the pore size was larger than both gas molecules because the effect of partial blockage was formed by the APTES surfactant. As expect, the MMM ideal selectivity increased from 39.2 to 61 with increasing feed pressure.141 This effect might also be associated to the lower sorption capacity of zeolite L for CH4 than for CO2 with increasing pressure.
Pechar et al.142 also fabricated MMM from 6FDA–6FpDA–DABA, a similar co-polyimide and zeolite ZSM-2. They found that for a zeolite content of 20 wt%, the ideal selectivity for CO2/CH4 mixture increased from 30.2 for the neat polymer membrane to 24.2. Also, at the same zeolite concentration, the CO2 diffusion coefficient was reduced by 38%, but the solubility coefficient was increased by 17%. Hence, the authors concluded that the CO2/CH4 ideal selectivity was decreased because ZSM-2 did not separate the molecules by size exclusion (pore size = 0.79 nm), but by preferential adsorption of CO2 on the cation sites. This is why CO2 showed a larger increase in solubility.
Hillock et al.143 prepared MMM from SSZ-13 and a crosslinkable polymer, 6FDA–DAM–DABA (1:0.6:0.4), for CO2/CH4 separation. SSZ-13 is a specialty alumino-silicate chabazite zeolite having a pore size of 0.38 nm. 6FDA–DAM–DABA were chemically crosslinked using 1,3-propanediol (PDMC polymer). The authors fabricated three kinds of MMM with different filler surface modifications, namely SSZ-13 primed, SSZ-13 grafted with APDMES with PDMC crosslinked polymer, and SSZ-13 primed with unmodified PDMC polymer (6FDA–DAM–DABA). They concluded that crosslinked MMM using PDMC and SSZ-13 grafted with APDMES as fillers had excellent CO2/CH4 selectivities up to 47 (mixed gas) and CO2 permeabilities of up to 89 Barrer. At the same time, this type of MMM can resist CO2 plasticization up to 450 psia.
Chen et al.45,46 used intergrowth zeolites (FAU/EMT) grafted and 6FDA–ODA polyimide cross-linked to make MMM. Zeolite was grafted using APTES, APMDES, and APDMES in different polarity solvents: isopropanol, isopropanol/water mixture (95/5 v/v), and toluene. APTMDS was selected as crosslinking agent for polyimide modification. MMM gas properties were studied for pure gas and CO2/CH4 mixtures at 35 °C and 10 atm of feed pressure. The results of Fig. 10 show that the performance of MMM, which were prepared from 6FDA–ODA and 25 wt% zeolite in isopropanol, were best for CO2/CH4 separation. Fig. 11 shows SEM images (cryogenically fractured MMM), which are Matrimid combined with non-grafted and grafted FAU/EMT zeolites. Poor distribution are observed in Fig. 11A, C, and E which are non-grafted zeolite MMM. Sedimentation surely occurred since zeolite concentration gradually increased from top to bottom. Also, zeolite agglomeration at the bottom of the membrane is obvious and very small voids between zeolites particles can be seen. The authors concluded that zeolite sedimentation occurred due to the difference of density between the solid particles (density close to 1.91 g cm−3) and the PI solution (PI and chloroform density around 1.48 g cm−3). Particle agglomeration led to whole formation and non-selective defects.45 On the contrary, Fig. 11B, D, and F are better cases (case I) of MMM, named M-IPA-3ET-2% (Matrimid mixed with amine-grafted zeolite in isopropanol solvent) since zeolite particles dispersion is more homogeneous. It is seen that each particle is located in the center of a polymer alveolus, and the interface between both phases is good.
 | ||
| Fig. 10 Performance of neat polyimide (6FDA-ODA and Matrimid 5218), cross-linked modified pi and their mixed matrix membranes with amine-grafted FAU/EMT zeolite. | ||
 | ||
| Fig. 11 SEM micrographs of m-zo Matrimid/non-amine-grafted zeolite (a, c and e) and m-IPA-3ET Matrimid/amine-grafted zeolite (b, d and f). | ||
In zeolite filled mixed matrix membranes, the zeolites exhibit higher penetrant sorption capacities and improved penetrant size-based selectivities for gas molecules than polymers. This is due to the large micropore volume and to the molecular sieving effect of the pore windows. For different types of zeolite-polymer MMM, separation objective and operation conditions are presented in Table 17 where the majority are flat dense MMM.144 In order to enlarge the application range of MMM, a more efficient membrane geometry was developed which is the asymmetric hollow fiber membranes. Miller et al.,145 Ekiner and Kulkarni,146 and Koros et al.147 first presented works on MMM hollow fibers with zeolite for hydrocarbon separations. The hollow fiber structure is preferred due to: (1) large membrane surface area per unit volume, (2) good flexibility, (3) easy handling, and (4) easier module fabrication.148 Several factors influence hollow fiber membranes properties. The main limitation in MMM hollow fibers fabrication are: (1) making thin selective MMM layer since zeolite particle size is not small being usually in micron size, (2) reducing defects in the fiber selective skin, and (3) to take advantage of the highly selective molecular sieving effects of zeolites.
| Polymer | Zeolite | Zeolite particle size | Zeolite loading | Solvent | Polymer concentration | Additives | Type of effect of additive |
|---|---|---|---|---|---|---|---|
| PC | 4A | Average size: 3 μm | Z/P b: 5–30% (w/w) | DCM | P/S: 12% (w/v) | pNA | LMWA |
| PDMS | Silicalite-1, NaX, NaA, graphite | 1.7, 2.3, 4, 1.5 μm | Z/Z + P: 15, 30, 50 wt% | DCM | P/S: 1:10 wt% |
— | — |
| PDMS | Silicalite | 0.1, 0.4, 0.7, 0.8, 1.5, 8 μm | 20 and 40 wt% | Iso-octane, chloroform | — | — | — |
| Crosslinked PDMS | SSZ-13 | — | 15 wt% | — | — | APDMES | Silanation |
| PEBA | ZSM-5 | 1–5 μm | Z/P: 10, 30 wt% | n-Butanol and n-propanol (4:1 wt%) |
P/S: 15 wt% | — | — |
| PEEK-WC | 4A | 3 μm | Z/Z + P: 30 wt% | DCM, DMA | 15 wt% | APDEMS, DEA | Silanation |
| PES | 3A, 4A, 5A | 1–2 μm | 10, 20, 30, 40, 50 wt% | NMP | 30 wt% | APDEMS | Silanation |
| PES | 4A | Nano size 4A: 50–140 nm, commercial 4A: 1–5 μm | Z/P: 20 wt% | NMP | — | — | — |
| PES | NaA, AgA | 1–2 μm | 20, 30, 40, 50 wt% | NMP | 30 wt% | — | — |
| PES | 4A | Average size: 100 nm | 20 wt% | NMP | 30 wt% | — | — |
| PES | 3A, 4A, 5A | 1–5 μm | 20 wt% | NMP | 30 wt% | — | — |
| PES | SAPO-34 | 0.5–1 μm | 20% (w/w) | Dimethyl sulfoxide | 20% (w/v) | HMA | LMWM |
| PES/PI (20/80 wt%) | 4A | Less than 2 μm | Z/(Z + P): 20 wt% | NMP | P/S: 25/75 wt% | — | — |
| PES/PI | BEA | 0.1–0.3 μm | 10, 20, 30 wt% | NMP | — | — | — |
| PES/PI (50/50 wt%) | 4A | — | 10–50 wt% | NMP | 20 wt% | — | — |
| PI | Nu-6(2) | 60 nm × 1000 nm × 1000 nm | 4, 8, 15 wt% | DCM | P/S: 88 wt% | — | — |
| PI | 4A | — | Z/total materials: 15% vol% | — | — | 1. RDP fyroflex; 2. Di-butyl phthalate; 3. 4-Hydroxy benzophenone | Plasticizer |
| PI | 4A, 5A, 13X, NaY | 0.64–4.33 μm | 43 wt% | DMSO | P/S: 1/(5.71) wt% | TAP | LMWM |
| PI | L | 250–300 nm | Z/P: 20/80 wt% | THF | P/S: 24 g l−1 | APTES | Silanation |
| PI | Sodalite | Average size: 105 nm | Z/P: 15, 25, 35 wt% | DMF | — | ADMS | Silanation |
| PI | ZSM-5 | 200 nm | Z/P + S: 10, 20, 30% (w/w) | TCE | 10% (w/w) solution | — | — |
| PI | FAU/EMT | 500–800 nm | 25 wt% | NMP | P/S: 10 wt% | APDEMS | Silanation |
| Fluorinated PI | ZSM-2 | — | 20 wt% | THF | — | APTES | Silanation |
| Poly(imide siloxane) | L | — | 0–20 wt% | THF | — | — | — |
| PI, PSF | Silicalite-1 | Hollow zeolite sphere: 4 μm; crystals: 0.3 μm × 1 μm × 2.0 μm | 4, 8, 16 wt% | Chloroform | 90 wt% of solvent,10 wt% polymer + zeolite | — | — |
| PI/PSF (0/100, 30/70, 50/50, 70/30, 100/0 wt%) | ZSM-5 | — | 0, 10, 20 wt% of total | DCM | 15 wt% polymer blend, 85 wt% DCM | — | — |
| Crosslinkable PI-PDMC | (SM) SSZ-13 | — | 25 wt% | — | — | — | — |
| Crosslinkable PI | FAU/EMT | 700–900 nm | 25 wt% | Chloroform | P/S: 10–12 wt% | APTES, APMDES, APDMES | Silanation |
| Crosslinkable PI | SSZ-13 | 500 nm | 25 wt% | THF | — | — | — |
| PMMA | 4A | — | 20, 33 wt% | THF | 15 wt% | TMOPMA | Silanation |
| PPO | SBA-15 | 2–3 μm | 0, 1, 5, 10, 15 wt% | Chloroform | P/S: 10 wt% | — | — |
| PPZ | SAPO-34, AlPO | AlPO: 10–20 nm | Z/P: 25%, 22% wt% | THF | — | — | — |
| PSF | 4A | Average size: 100 nm | Z/P: 0, 15, 25, 30, 35 wt% | NMP | P/S: 8 wt% | — | — |
| PSF | ITQ-29 | 2.5 μm | Z/P: 4, 8, 12 wt% | DCM, THF | — | — | — |
| PSF | Nu-6(2) | 5–10 μm | Z/P + S + Z: 0.47, 0.87, 1.92 wt% | DCM | — | — | — |
| PSF-Ac | 3A, 5A | — | 40 wt% | TCM | Additive/P: 25 wt%, P/S: 6.5 wt% | APTMS | Silanation |
| PVA | 4A | 2–2.5 μm | Z/P: 15, 25, 40 vol% | Toluene, DCM | P + Z: 20–25 wt% | — | — |
| PVA | 4A, SSZ-13 | — | Z/P: 15 vol% | 4A:DCM SSZ-13:isopropanol |
(Z + P)/solvent: 1:4 wt% |
— | — |
| PVA | 4A | 0.5–1.5 μm | 50 vol% | Toluene | 25 wt% | — | — |
| PVA, Ultem | 4A | 5 μm | 15, 30, 40 wt% | DCM, toluene | P/S: 20 wt% | — | — |
| PVA/Ultem® 1000 polyetherimide | 4A | 5 μm | 15, 30, 40 wt% sieve loading film | DCM or toluene | P/S: 20 wt% | — | — |
| Crosslinked PVA/PEG: 36/64 wt% | 5A | 3–5 μm | Z/P: 18.7, 33.2, 58 wt% | Deionized (DI) water | — | — | — |
| Glycerinedimethacrylaturethanetriethoxysilane (GUS)-based ORMOCER | BEA | 200 nm to 1 μm | 20–40 wt% | Ethyl acetate | 2 g of ORMOCER resin was added into the solvent (0.5 g of ethyl acetate) | Irgacure-184 (1-hydroxy-cyclohexyl-phenyl-ketone) | — |
| SEBS-29S | BEA | Average size: 0.62 μm | 10 wt% of the dry polymer | Toluene | 20 wt% in toluene | Various organosilane | Silanation |
| Teflon AF 1600 | Silicalite-1 | 0.35, 0.080 μm | 350 nm: 29 wt%, 80 nm: 30.0, 40.2 wt% | Galden HT 110 | — | — | — |
| PES | 4A | 2 μm | 20 wt% of zeolite in total solid | NMP | P/S: 25/75 wt% | Dynasylan Ameo (DA) | Silanation |
| PES | Outer layer: BEA inner layer: Al2O3 | BEA: 300 nm; Al2O3: 0.2 μm | Outer layer: 20 wt% of solution; inner layer: 0–60 wt% | NMP | Outer layer: PES/NMP/EtOH: 35/50/1 wt%; inner layer: PES/NMP/EtOH: 25/61/14 wt% | — | — |
| Outer layer: PES; inner layer: PI | Outer layer: BEA; inner layer: — | 0.3 μm | Z/(Z + P): 20 wt% | NMP | Outer layer: PES/NMP/EtOH: 35/50/15 wt%; inner layer: PI/NMP/EtOH: 20/67/13 wt% | — | — |
| Outer layer: PSF; inner layer: PI | Outer layer: BEA | BEA: 0.4 μm | Z/(Z + P): 10, 20, 30 wt% | NMP, EtOH as a solvent and nonsolvent | Outer layer: 30 wt%; inner layer: 23 wt% | p-Xylenediamine/methanol: 2.5/100 (w/v) | Hydrogen bonding between Z&P |
| PI | BEA | 0.3–0.5 μm | Z/P: 20% wt% | NMP | 20 wt% | — | — |
| Outer layer: PSF; inner layer: PI | Outer layer: BEA; inner layer: — | Average size: 0.4 μm | Z/P: 20 wt% | NMP and EtOH as the solvent and nonsolvent | Outer layer: P/S:30 wt%; inner layer: P/S: 23 wt% | — | — |
| Ultem® 1000 polyetherimide | Hssz-13 | — | Z/S: 10 wt% | NMP THF lithium nitrate | 26–30 wt% | APDMES | Silanation |
| Ultem, Matrimid | MFI | 2, 5 μm; 100, 300 nm | 20, 30, 35 wt% | — | — | — | — |
These MMM have the potential to supply high selective molecular sieves of zeolites and good mechanical/economical properties of polymers. But generally, MMM have three main problems. The first one is poor compatibility between the zeolites and polymer phase, leading to voids or other interfacial defects between both phases. Second, large particles (micron range) have lower specific surface areas. In general, smaller particles (nano-size) can provide higher polymer/filler interfacial area and improve polymer-filler interfacial contact. Finally, the fabrication of hollow fiber MMM is difficult at large industrial scales (large gas separation systems).
Zeolite-based mixed-matrix membrane performances for CO2/CH4 separation are summarized in Table 18. Ideal selectivities higher than 50 are reported for MMM from different ref. 45, 46, 142, 150, 152 and 160 using ZIF 8, ZSM-5, Zeolite L and FAU/EMI with Matrimid and others polyimides. Among these, MMM made from ZIF-8 with Matrimid without modification show excellent results (50 and 60 wt% PCO2 = 5 or 8 Barrer, ideal selectivity of 125 or 81).150 ZIF are a sub-group of MOF having a wide range of pore sizes and chemical functionalities. ZIF-8 is available commercially and has exceptional chemical stability with a wide structural diversity compared to zeolites.
| MMMs | Membrane type | Major application | Operating condition | Example performance | Ref. | ||
|---|---|---|---|---|---|---|---|
| Zeolite (loading, wt%) | Polymer | Permeability (Barrer) | Selectivity | ||||
| Zeolite 4A 44 | PES | Hollow fiber | CO2/CH4 | 25 °C 10 atm | PCO2 = 1.6–6.7 | αCO2/CH4 = 46.3–28.7 | 149 |
| ZIF-8 (up to 60) | Matrimid | Plane | CO2/CH4 | 35 °C, 2.6 atm | PCO2 = 2.6–25 | αCO2/CH4 = 42–125 | 150 |
| NaA AgA (0–50) | PES | Plane | CO2/CH4 | 35 °C, 10 atm | PCO2 = 1.2–1.8 | αCO2/CH4 = 31.4–59.6 | 151 |
| ZSM-5 (up to 30) | Matrimid | Plane | CO2/CH4 | 35 °C 2 atm | PCO2 = 7.3–15 | αCO2/CH4 = 35–66 | 152 |
| MCM-41 (up to 30) | Matrimid | Plane | CO2/CH4 | 25 °C 10 atm | PCO2 = 7.5–10 | αCO2/CH4 = 35–38 | 153 |
| zeolite L (up to 20) | 6FDA–6FpDA–DABA | Plane | CO2/CH4 | 35 °C 4–12 atm | PCO2 = 21–18.3 | αCO2/CH4 = 40–60 | 141 |
| Zeolite 3A 4A 5A (up to 50) | PES | Plane | 35 °C 10 atm | PCO2 = 2.6–1.8 | αCO2/CH4 = 32–44 | 154 | |
| SAPO-34 (up to 20) | PES | Plane | CO2/CH4 | 35 °C 3 atm | PCO2 = 0.8–5.5 | αCO2/CH4 = 44–38 | 155 |
| 4A (50 vol%) | PVAc | Plane | CO2/CH4 | 35 °C, 3 atm, 30 atm | PCO2 = 4.33 PCO2 = 11.5 | αCO2/CH4 = 49.4 αCO2/CH4 = 40.6 | 156 |
| HSSZ-13 (13.3 vol) | Ultem® 1000 | Hollow fiber | CO2/CH4 | 35 °C 7.8 atm | PCO2 = 6.8 | αCO2/CH4 = 46.9 | 148 |
| Zeolite 3A 4A 5A (to 30) | PI and PEI | Plane | CO2/CH4 | 25 °C 8 atm | PCO2 = 5.31–7.93 PCO2 = 20.3–36.3 | αCO2/CH4 = 15.2–27.3 αCO2/CH4 = 6.50–11.2 | 157 |
| Zeolite up to 30 | PES Matrimid | Dual-layer hollow fiber | CO2/CH4 | 35 °C 5 atm | PCO2 = 2–2.5GPU | αCO2/CH4 = 25–40 | 158 |
| ZSM-5 (up to 30) | Matrimid | Plane | CO2/CH4 | 1.4 atm | PCO2 = 8–15 | αCO2/CH4 = 35–66 | 159 |
| zeolite 5 (up to 58) | PVA/PEG blend | Plane | CO2/CH4 | 30 °C 10 atm | PCO2 = 80.2 | αCO2/CH4 = 33 | 160 |
| FAU/EMI (up to 25) | 6FDA–ODA | Plane | CO2/CH4 | 35 °C 10 atm | PCO2 = 15–40 | αCO2/CH4 = 20–60 | 45 and 46 |
7.4 MMMs composed of polyimides, carbon molecular sieves and carbon nanotubes
The most common polymers used as membrane precursors of CMS are polyimides (PI), polyacrylonitrile (PAN), phenolic resins (PR) and poly(furfuryl alcohol) (PFA). Owing to the good mechanical and permeation properties of the resulting CMS membranes, polyimides are considered the best matrix precursor. PFA and PR are cheaper than PI, but these polymers need to be coated on supports before pyrolyzing since they are liquids.161,163,165 CMS membranes are usually used in four different geometries including flat sheet, supported on a tube, capillary, and hollow fiber membranes. The first CMS hollow fibers were produced from pyrolyzed cellulose, which is particularly convenient because of their lower cost compared to PI.166 Jones and Koros produced CMS membranes by pyrolyzing asymmetric hollow fiber PI precursors at different temperatures.167 In their study, polyimides were derived from 2,4,6-trimethyl-1,3-phenylenediamine (TrMPD), 5,5-[2,2,2-trifluoro-1-(trifluoromethyl)ethylidene-1,3-isobenzofurandione (BPDA), and 3,3′,4,4′-biphenyl tetracarboxylic acid dianhydride (6FDA). The resulting CMS membranes demonstrated exceptional gas separation performance for CO2/CH4 separation. The CO2 and CH4 permeabilities were 53 GPU and 0.38 GPU, respectively, while the selectivity for mixed gas (50/50) was 140 at room temperature and 150 psi of feed pressure. Compared to the conventional polymer membranes with typical CO2/CH4 selectivities of 15 to 40, a large increase in CO2/CH4 selectivity was achieved.
Vu and Koros used the same method to fabricate CMS hollow fibers by pyrolysis under vacuum of two integrally skinned asymmetric polyimide fibers such as 6FDA/BPDA-DAM and 6FDA/Matrimid.168 The resulting CMS hollow fibers showed excellent permeation properties for CO2/CH4 separation at high-pressure (up to 1000 psi), giving them more attention in many industrial applications (CO2 removal from natural gas) (Table 19). Moreover, their mechanical and selective stabilities at high pressures, which can be modified upon optimized pyrolysis procedure, are especially desirable.
Addition of CMS particles into a polymer matrix has been suggested as an alternative method to produce MMMs, owing to their permeation performances exceeding the Robeson limit trade-off bound. Low cost CMS particles are expected to have better affinity with glassy polymers, and improving interfacial adhesion without introducing processing problems.
Vu et al.43,168–170 focused on CMS membranes with different polyimides to form MMMs for CO2/CH4 separation. In these studies, CMS materials were formed by pyrolyzing Matrimid® and Ultem® polyimides at controlled temperatures up to 800 °C under vacuum. The pyrolyzed CMS solids were then ball-milled into particle sizes ranging from submicron to 2 μm before undergoing a membrane casting protocol. As a result, the CMS films could attain a relatively high CO2/CH4 selectivity of 200 at a CO2 permeability of 44 Barrer. For Ultem®/CMS MMMs, pure gas permeation results showed a 40% increase in CO2/CH4 selectivity compared to neat Ultem®. Similarly for Matrimid®/CMS MMMs, a slight increase in CO2/CH4 selectivity (45%) was observed (Table 20). Based on their permeation results, the authors concluded that fine CMS particles showed an effective affinity with glassy polymers, yielding good polymer–CMS adhesion upon minimal modifications in MMM preparation protocol. Similar enhancement trend in CO2/CH4 separation performance was also reported for hybrid MMM materials made from 19 wt% CMS particles and glassy Matrimid® 5218 polyimide with a low concentration of toluene impurity in the gas feed.170 In this case, toluene as a larger-sized impurity may block/occupy the large non-selective pores of CMS particles.
| Type of membrane | PCO2 (Barrer) | PCH4 (Barrer) | PCO2/PCH4 |
|---|---|---|---|
| CMS 800-2 membrane | 44 | 0.22 | 200 |
| Matrimid® 5218 membrane | 10 | 0.28 | 35.3 |
| 17 vol% CMS with Matrimid® | 10.3 | 0.23 | 44.4 |
| 19 vol% CMS with Matrimid® | 10.6 | 0.23 | 46.7 |
| 33 vol% CMS with Matrimid® | 11.5 | 0.24 | 47.5 |
| 36 vol% CMS with Matrimid® | 12.6 | 0.24 | 51.7 |
| Ultem® 1000 membrane | 1.45 | 0.037 | 38.8 |
| 16 vol% CMS with Ultem® | 2.51 | 0.058 | 43 |
| 20 vol% CMS with Ultem® | 2.9 | 0.060 | 48.1 |
| 35 vol% CMS with Ultem® | 4.48 | 0.083 | 53.7 |
As the common limitations mentioned for zeolite-based MMMs, the rigid nature of CMS materials could cause some restrictions to form continuous and defect-free membranes. To improve matrix-CMS interfacial adhesion, several approaches have been proposed. For example, dispersing or priming CMS particles in a polymer solution with high viscosity could avoid the sieve-in-cage or void membrane defect.43 Rafizah and Ismail modified CMS particles in polyvinylpyrrolidone Kollidone-15 (PVP K-15) as coupling agent was used before embedding in polysulfone (PSF) Udel® P-1700. As a result, CMS/PSF-PVP MMM exhibited O2/N2 selectivity 1.7 times higher than unmodified MMMs, owing to a significant improvement in CMS-PSF adhesion.171 Das et al.172 evaluated the effects of casting method and annealing temperature on the permeance properties of the hybrid MMMs made from CMS particles and 6FDA–6FpDA polymer (6FDA = 4,4′-(hexafluoroisopropylidene)diphthalic anhydride; 6FpDA = 4,4′-(hexafluoroisopropylidene)-dianiline). By modifying the solvent-evaporation process with a continuous sonication technique, the formation of agglomerates can be suppressed until the polymer viscosity is high enough to sufficiently limit particle mobility and prevent agglomeration. Moreover, an improvement in CMS-polymer adhesion was observed for CMS-polymer MMMs at different annealing temperatures, resulting in higher CO2/CH4 and O2/N2 selectivities compared to those of the neat polymer membrane. The authors also verified their experimental results with the Maxwell model prediction.
 | ||
| Fig. 12 (a) Flat sheet of graphite, (b) partially rolled sheet of graphite, (c) SWCNTs and (d) MWCNTs. | ||
The extraordinary inherent smoothness of their potential energy surfaces offer CNT good mechanical, electrical, thermal, and mass transport properties. They can be incorporated as fillers in MMM domains. Since the last decade, a number of studies, as summarized in Table 21, focused on the use of CNT as inorganic phase dispersed in polymer matrices to prepare MMM. The pioneering report on the incorporation of aligned MWCNT into polystyrene (PS) for ionic Ru(NH3)63+ transport process showed the ability of small gas molecular transport through the nanotube inner core and their potential applications in chemical separation and sensing.177
| MMMs | Functionalization/modification | Major application | Type of membrane | Example performance | Ref. | ||
|---|---|---|---|---|---|---|---|
| CNTs (loading wt%) | Polymer | Permeability (Barrer) | Selectivity (−) | ||||
| a AFS = Asymmetric flat sheet, APTES = 3-aminopropyltriethoxylsilane; BPPOdp = brominated poly(2,6-diphenyl-1,4-phenylene oxide); CA = cellulose acetate; CTAB = cetyl trimethyl ammonium bromide; ODA = octadecylammonium; PBNPI = poly(bisphenol A-co-4-nitrophthalic anhydride-co-1,3-phenylene diamine); PC = polycarbonate; PDMS = polydimethylsiloxane; PEG = poly ethylene glycol; PEI = polyetherimide; PES = polyethersulfone; PI = polyimide; PIS = poly(imide siloxane); PS = polystyrene; PSF = polysulfone; PVC = polyvinylchloride; SBR = styrene butadiene rubber; SDF = symmetric dense film; SDS = sodium dodecyl sulfate. | |||||||
| SWCNTs (2 and 10) | PIS | H2SO4:HNO3 |
CO2, O2, N2, CH4 | SDF | PCO2 = 36.6–39.8 PN2 = 14.4–17.8 PCO2 = 190.7–191.3 PCH4 = 34.2–36.7 | — | 179 |
| MWCNTs (–) | PS | — | H2/CO2 | SDF | PCO2 = 47 ± 5 PH2 = 290 ± 5 | αH2/CO2 = 6.17 | 180 |
| MWCNTs (1 to 10) | PDMS | — | H2/CH4 | SDF | — | — | 181 |
| MWCNTs (up to 5) SWCNTs (up to 5) | BPPOdp | HNO3 | CO2/N2 | SDF SDF | PCO2 = 134–153 PN2 = 4.0–5.4 PCO2 = 123 PN2 = 4.3 | αCO2/N2 = 28 ÷ 34 αCO2/N2 = 29 | 182 |
| MWCNTs (up to 15) | PBNPI | H2SO4:HNO3 |
H2/CH4 CO2/CH4 | SDF | PCO2 = 2.18–6.00 PCH4 = 0.74–1.78 PH2 = 4.95–14.31 | αCO2/CH2 = 3.7 αH2/CH4 = 8.0 | 183 |
| MWCNTs (1 to 10) | PC/PEG | H2SO4/HNO3 | CO2/CH4 | AFS | PCO2 = 8.35–20.3 PCH4 = 0.28–0.57 | αCO2/CH4 = 29.8–35.6 | 184 |
| MWCNTs (0.5 to 5) | PVC/SBR | H2SO4:HNO3 |
CO2/CH4 CO2/N2 | AFS | PCO2 = 2.3–16.3 PCH4 = 0.04–0.26 PN2 = 0.09–0.47 | αCO2/CH2 = 56.5–63.5 αCO2/N2 = 25.1–34.7 | 185 |
| MWCNTs (up to 10) | PES | H2SO4:HNO3 |
CO2/N2 | SDF | PCO2 = 2.7–4.5 PN2 = 0.12–0.20 | αCO2/N2 = 19–22.5 | 186 |
| MWCNTs (up to 10) | PES | H2SO4:HNO3/Ru metal H2SO4:HNO3/Fe metal |
CO2/N2 | SDF | PCO2 = 3.56 PCO2 = 4.23 | αCO2/N2 = 26.5 αCO2/N2 = 12.9 | 187 |
| SWCNTs (up to 15) | PSF/ODA | H2SO4:HNO3/ODA |
O2/N2 CH4/N2 CO2/CH4 | SDF | PO2 = 5.45–6.48 PN2 = 1.70–1.94 PCO2 = 1.97–2.10 PCH4 = 0.54–0.55 | αCO2/N2 = 5.04–5.35 αCH4/N2 = 1.17–1.27 αCO2/CH4 = 16.1–18.8 | 188 |
| SWCNTs (1) | PES | H2SO4/Dynasylan ameo | CO2/CH4 | AFS | PCO2 = 22.18 PCH4 = 0.18 | αCO2/CH2 = 30.42 | 189 |
| MWCNTs (0.5 to 3) | PES | H2SO4:HNO3/APTES |
CO2/CH4 O2/N2 | SDF | PCO2 = 2.8–13.6 (GPU) PCO2 = 3.06–4.36 (GPU) | αCO2/CH2 = 5–250 αO2/N2 = 3.7–10.7 | 190 |
| MWCNTs (0.5 to 3) | PIM-1 | HNO3/SOCl2/PEG | CO2/N2 CO2/CH4 O2/N2 | SDF | PCO2 = 12274 PCH4 = 1483 PO2 = 1680 | αCO2/CH4 = 8.3–16.3 αCO2/N2 = 17.2–23.9 αO2/N2 = 2.4–3.7 | 191 |
| MWCNTs (1) | PEI | SDS CTAB Triton-X100 | O2/N2 | AFS | PO2 = 3.84 (GPU) PO2 = 7.36 (GPU) PO2 = 2.29 (GPU) | αO2/N2 = 1.10 αO2/N2 = 1.58 αO2/N2 = 3.75 | 192 |
| MWCNTs (1) | PI | Chitosan | CO2/CH4 | AFS | PCO2 = 14.27 PCH4 = 1.43 | αCO2/CH4 = 10 | 193 |
| MWCNTs (0.5 to 1) | PI | Beta-cyclodextrin | CO2/CH4 | SDF | PCO2 = 10.5 (GPU) | αCO2/CH4 = 8.5 | 194 |
| MWCNTs (up to 6) | PI | Beta-cyclodextrin | CO2/CH4 | AFS | PCO2 = 2.2–3.1 (GPU) | αCO2/CH4 = 38.9–62.9 | 195 |
| MWCNTs (up to 0.2) | CA | Beta-cyclodextrin | CO2/N2 | SDF | PCO2 = 138 ÷ 741 (GPU) PN2 = 16 ÷ 18.5 | αCO2/CH4 = 8.4 ÷ 40.1 | 196 |
Because of their relatively smooth surfaces, weak interfacial bonds between CNT and polymers occurs. Hydrophobicity of both the nanotube graphitic walls and core entrances need to be modified by chemical treatment. The most common chemical method used is surface modification with strong inorganic acids containing hydroxyl or carboxyl functional groups to improve polymer–CNT compatibility, as well as to modify the CNT hydrophobic nature.178
Kim et al.179 reported the transport of CO2, O2, N2, and CH4 inside nano-composites consisting of 2 and 10 wt% SWCNT dispersed in poly(imide siloxane) (PIS) copolymer matrix. At low CNT loading (2 wt%), an improvement in dispersion of the acid-modified CNT particles within the copolymer matrix was observed, because their siloxane segment enhanced the interfacial contact between both phases. For high CNT loading (10 wt%), O2, N2, and CH4 permeabilities increased, while CO2 permeability was almost constant. However, an increase of CO2 permeability by 15% compared to the neat polymer membrane was reported for SWCNT/PIS MMMs with 2 and 10 wt% CNT.
Kumar et al.180 dispersed MWCNT into polystyrene (PS) to prepare CNT/PS MMMs by solution casting for hydrogen gas permeation application. Using a similar technique, Nour et al.181 synthesized polydimethylsiloxane composites with different amounts of MWCNT for the separation of H2 from CH4. MMMs with low CNT loading (1 wt%) showed an increase of CH4/H2 selectivity by 94.8% compared to neat PS membranes. Unfortunately, a reduction in separation performance due to the presence of interfacial voids, which became more important at higher MWCNT loading, was reported.
Cong et al.182 reported the formation of CNT/BPPOdp MMMs from pristine SWCNT or MWCNT dispersed in brominated poly(2,6-diphenyl-1,4-phenylene oxide) (BPPOdp) polymer matrix. The results showed increasing CO2 permeability and similar CO2/N2 selectivity in comparison with neat BPPOdp membranes. A significant increase of CO2 permeability with increasing CNT loading was reported with a maximum of 155 Barrer for CNT/BPPOdp membrane filled with 9 wt% SWCNT, and a maximum of 148 Barrer for CNT/BPPOdp composite containing 5 wt% MWCNT. The authors pointed out that the incorporation of CNTs into a polymer matrix could enhance membrane mechanics without decreasing MMM gas separation performance.
Weng et al.183 prepared a series of CNT/PBNBI MMMs with high MWCNT loading (up to 15 wt%) embedded in poly(bisphenol A-co-4-nitrophthalic anhydride-co-1,3-phenylenediamine) (PBNBI) for H2/CH4 separation. After a H2SO4:HNO3 mixed acid treatment, the obtained CNT/PBNBI MMMs were quite homogeneous, even at high loading of 15 wt%, resulting in high dispersion of small CNT fragments in the PBNPI matrix. Although at low MWCNT loading (1–5 wt%), the H2, CO2 and CH4 permeabilities were almost unchanged, at higher MWCNT content (15 wt%), maximum CO2 and CH4 diffusivities were reached, due to the high diffusivity of the CNT tunnels as well as the CNT–PBNBI interfaces.
To improve both the separation performance and mechanical properties of CNT/polycarbonate (PC) MMMs, Moghadassi et al.184 used polyethylene glycol (PEG) as second rubbery polymer to prepare MWCNT-PC MMMs for CO2/N2 and CO2/CH4 separations. Their results showed that the use of carboxyl-functionalized MWCNT instead of raw MWCNT (at 5 wt% CNT loading) in MMM made from PC provides better CO2/N2 and CO2/CH4 separation performances as compared to the neat polymer membrane. In carboxyl-functionalized MWCNT-PC/PEG blend MMM, an improvement of CO2/CH4 selectivity at 2 bar of pressure to 35.6 compared to a value of 27.2 for the nano-composite membranes using PC alone as the polymer matrix was reported. Using a similar approach, Rajabi et al.185 modified MWCNT-PVC MMM with styrene butadiene rubber (SBR) for CO2/CH4 separation. As expected, the MMM showed a significant improvement of tensile modulus (from 4.65 to 4.90 MPa) with blending.
Ge et al.186 reported an improvement in CO2/N2 gas selectivity over a series of nano-composite membranes consisting of 1–10 wt% carboxyl-functionalized MWCNT embedded in polyethersulfone (PES), because of better affinity between carboxyl functional groups with polar CO2, while hindering the nonpolar N2 solubility. Similar observations have been found for CNT/PES MMMs containing carboxyl-functionalized MWCNT further modified with Ru or Fe.187 Metal-modified sites on the external CNT wall strongly adsorb non-polar N2 molecules, hence increased diffusion resistance of N2, and consequently enhanced CO2/N2 selectivity were obtained.
Kim et al.188 treated SWCNT with a concentrated H2SO4:HNO3 solution, followed by functionalizing with a long-chain alkyl octadecylammonium (ODA) amine to enhance dispersion in polysulfone. H2, O2, CH4, and CO2 permeabilities and diffusivities of these CNT/PSF membranes increased with CNT content. However, SWCNTs were well dispersed in the PSF phase only at 5 wt%, while the CNT formed two different domains at high CNT content (15 wt%): homogeneous and dense regions. This was attributed to the presence of interfacial voids between SWCNT and PSF at high CNT loading.
Before introducing MWCNT into polyethersulfone (PES) for biogas purification, Mustafa et al.189 functionalized their surface by chemical modification using Dynasylan Ameo (DA) silane agent. As expected, the modified MWCNT-PES MMM showed improvements of the mechanical properties, productivity and biogas purity. With 1 wt% CNT, the maximum selectivity achieved for CO2/CH4 was 36.8.
After purification with acid mixtures (H2SO4:HNO3) to remove carbonaceous impurities, Ismail et al.190 used 3-aminopropyltriethoxylsilane (APTES) as another silane agent to enhance MWCNT dispersion in N-methylpyrrolidone (NMP) during the preparation of MWCNT-PES suspension. Khan et al.191 first oxidized MWCNT in HNO3, then chlorinated them in presence of SOCl2 and finally esterified with poly(ethylene glycol) (PEG). For both cases, the mechanical properties and gas performance of modified CNT-based MMM were enhanced.
It is clear that chemical modification via acid treatment may damage the CNT structures and eventually limit their intrinsic separation properties. To overcome these drawbacks, many efforts focused on physical modifications through non-covalent functionalization such as surfactant dispersion and polymer wrapping. Using surfactants with different charges may improve physical surfactant molecules adsorption on CNT, thus lowering surface tension and limiting CNT aggregation. For example, Goh et al.192 used SDS, CTAB, and Triton X100 as surfactants to disperse MWCNT in polyetherimide (PEI). The resulting surfactant-dispersed MMMs exhibited better thermal stability and mechanical strength compared to neat PEI membranes. Among these surfactants, Triton X100 showed the highest improvement of O2 and N2 permeabilities.
On the other hand, the polymer wrapping technique is based on van der Waals interactions and π–π bonding between CNTs and polymer molecules containing aromatic rings to create supermolecular complexes of CNTs, hence also limiting CNT agglomeration. For example, to improve the interfacial contact between CNT and polyimide in the preparation of MWCNT/polyimide flat sheet MMMs, Aroon et al.193 used chitosan as a functional agent to wrap around the MWCNT, preventing void formation. CO2/CH4 separation results revealed that 1 wt% MWCNT into the polymer phase was enough to increase both CO2 and CH4 permeabilities. The CO2/CH4 selectivity increased by 51% (from 10.9 to 16.5) over chitosan-functionalized MWCNT/polyimide MMMs compared to the neat PI membrane.
Another non-covalent polymer wrapping agent is beta-cyclodextrin (beta-CD).194–196 After grinding of beta-CD with CNTs, the graphite walls of CNTs can be functionalized with hydroxyl group, which allow improving the solubility of CO2 molecules resulting in increased CO2 permeability. For example, beta-CD treatment resulted in a homogeneous dispersion of MWCNTs in cellulose acetate (CA).196 With 0.1 wt% beta-CD functionalized MWCNT, the composite membranes exhibited excellent CO2/N2 selectivity of 40.
From the above listed reports, CNT have a great potential as fillers in MMM. The main interest of CNT/polymer MMMs is related to the smoothness of the interior channels which allows the rapid transport of gases through MMMs. Up to now, the use of CNT as filler is, however, still limited due to dispersion difficulties in a polymer matrix during MMM preparation, poor CNT-polymer interfacial interaction, and high production cost.
7.5 MMM composed of polyimide and MOF
During the last decade, metal–organic hybrids have emerged as a new class of porous crystalline materials from the self-assembly of complex subunits containing transition metals connected by multifunctional organic ligands to create 1D, 2D and 3D structures. These hybrids are usually labeled as metal–organic frameworks (MOF). These materials have interesting properties such as structure regularity, high surface area, high porosity, low density, and a wide range of pore size, shape and geometry. Compared to other porous materials, MOF accept almost all tetravalent cations except metals. MOF also have disadvantages because they are generally only stable up to 200 °C, which does not allow high temperature applications. In addition, some MOF are weakly stable in an aqueous environment, causing some limitations to their use in membrane synthesis.197 All these MOF properties make them promising candidates for the production of mixed matrix membranes. The interface morphology between MOF and polymer matrices is easily controlled because of their organic linkers having better affinity with polymer chains. Their surface properties can also be modified by functionalization and particles with small sizes may be used.Up to now, many type of MOF have been used for the preparation of MMM, including divalent (Zn2+, Cu2+) or trivalent (Al3+, Cr3+) metal cations interconnected with several organic linkers such as Cu-BPDC-TED, {[Cu2(PF6)(NO3)-(4,4′-bpy)4]·2PF6·2H2O}n, Mn(HCOO)2, MOF-5 or IRMOF-1, HKUST-1 or Cu3(BTC)2, Fe-BTC, Zn(BDC)(TED)0.5, Cu(hfipbb)(H2hfipbb)2, Cu–BPY–HFS, Cu-TPA, MOP-18, MIL-47, MIL-53, MIL-101, ZIF-7, ZIF-8, ZIF-90, UiO-66. Generally, MOF-based MMM combine the high sorption properties of MOF with good permeability and mechanical properties of polymers. MMM based on MOF have strong MOF-polymer interaction, but their gas separation performance has great potential to be improved.198 These new materials have been invented by Yaghi's research team.199
MOF are structures with specific cavity sizes with high CO2 storage capacity by adsorption. For example, surface area of zeolite Y is around 904 m2 g, but some MOFs have values over 3000 m2 g−1. For example, MOF-177 has an estimated surface area of 4500 m2 g−1 which is the highest surface area reported until now.200
Yehia et al.201 first explored the incorporation of MOF into a polymer matrix to make MMM for gas separations in 2004. MMM produced from copper(II) biphenyl dicarboxylate-triethylenediamine and poly(3-acetoxyethylthiophene) were shown to have better CH4 selectivity. Then, Car et al.202 fabricated two MMMs from Cu3(BTC)2 and Mn(HCOO)2 with poly-dimethylsiloxane (PDMS) and polysulfone (PSf). They synthesized MOF under hydrothermal conditions in such a way to obtain porous materials with high sorption properties. They found that CO2 and CH4 permeabilities for PDMS/Cu3(BTC)2 membranes increased and the selectivity remained unchanged. For membranes of PSF/Cu3(BTC)2 and PSF/Mn(HCOO)2, increased CO2 and CH4 permeability was reported, while decreased CO2/CH4 selectivity was found comparable to neat polymer membranes.
Zhang et al.203 used Matrimid® and Cu–BPY–HFS to form freestanding films with microporous metal–organic framework having surface area of 2000 m2 g−1 and pore diameter of 0.8 nm. The structure of Cu–BPY–HFS is built with 2D copper bipyridine complexes pillared with SiF62− ions. The gas transport properties of these membranes were not significantly improved for pure gases (CH4 and CO2) and their mixtures.
Perez et al.119 used MOF-5 as filler for the fabrication of MMM. They synthesized MOF-5 nano-crystals with 100 nm particle size, high surface area (3000 m2 g−1) and good thermal stability (up to 400 °C), then introduced them into Matrimid (PI) as the continuous polymer phase to form MMM for gas separation.
Up to now, several MOF were used to prepare MMM.202–221 Even if these MOF-based MMM were shown to have good MOF-polymer interactions, gas separation properties are still below desirable values. Details on the synthesis and performances of MOF-based MMM are given in Table 22.
| MMMs | Synthesis procedure (solvent) | Major application | Operating condition | Measurement method | Example performance | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| MOF (loading, wt%) | Polymer | Permeability (Barrer) | Selectivity | |||||
| Zn-IRMOF-1 (up to 20) | Matrimid 5218 | Solvent evaporation (dioxolane, NMP) | CO2/CH4 | 50 °C, 100 psig | Constant-volume | PCO2 = 38.8 | αCO2/CH4 = 29.2 | 205 and 206 |
| PCH4 = 1.33 | αH2/CH4 = 86.4 | |||||||
| H2/CH4 | PH2 = 114.9 | αH2/CH4 = 86.4 | ||||||
| Zn-IRMOF-1 (10 and 20) | Ultem 1000 | Solvent evaporation | CO2/CH4 | 50 °C, 100 psig | Constant-volume | PCO2 = 2.97 | αCO2/CH4 = 26.3 | 205 and 206 |
| H2/CH4 | PCH4 = 0.11 | αH2/CH4 = 149.3 | ||||||
| PH2 = 16.9 | ||||||||
| Zn-MOF-5 (10 ÷ 30) | Matrimid 5218 | Solvent evaporation (chloroform) | CO2/CH4 | 35 °C, 2 atm | αCO2/CH4 = 44.7 | 119 | ||
| CH4/N2 | ||||||||
| CuTPA (15) | Poly(vinyl acetate) | Solvent evaporation, (toluene) | CO2/CH4 | 35 °C, 1.35 psig | Constant-volume | PCO2 = 3.26 | αCO2/CH4 = 40.4 | 207 |
| PCH4 = 0.08 | ||||||||
| Cu(hfipbb)–(H2hfipbb)2 (up to 30) | Matrimid 5218 | — | CO2/CH4 | 35 °C, 2 atm | Maxwell and Bruggeman models | PCO2 = 9–20 | αCO2/CH4 = 95 | 208 |
| Zn(bdc)(ted)0.5 (up to 30) | Polytrimethylsilypropyne, Hyflon AD60X, teflon AF-2400, Sulfonated polyimide, 6FDA-mMPD, 6FDA-DDBT | — | CH4/H2 | 35 °C, 2 atm | Maxwell model | PH2 = 187–426.8 | — | 209 |
| PH2 = 52–118.8 | ||||||||
| Cu3(BTC)2 (up to 40) | Polydimethylsiloxane polysulfone | — | CO2/CH4 | — | — | PCO2 = 2000–3000 | αCO2/CH4 = 3–3.6 | 202 |
| PCO2 = 6–8 | αCO2/CO4 = 7–21 | |||||||
| Cu3(BTC)2 (up to 30) | Matrimid 5218 | Solvent evaporation, (dioxolane, NMP) | CO2/CH4 | 50 °C, 100 psig | Constant-volume | PCO2 = 22.1 | αCO2/CO4 = 29.8 | 206 |
| H2/CH4 | PCH4 = 0.74 | αH2/CH4 = 90.3 | ||||||
| PH2 = 66.9 | ||||||||
| Cu3(BTC)2 (up to 30) | Matrimid/Polysulfone | Phase inversion, (NMP, dioxolane) | CO2/CH4 | 35 °C, 10 bar | Constant-volume | PCO2 = 10–18 | αCO2/CO4 = 19.5–28 | 210 |
| CO2/N2 | PCH4 = 7–19 | αCO2/N2 = 13–27 | ||||||
| Cu3(BTC)2 (up to 20) | Poly(amic acid) | Solvent evaporation (PMDA, DMAc and ODA) dry/wet spinning | H2/CH4 | 0.5–5 atm | Constant-volume | PH2 = 1266 | αH2/CH4 = 240 | 211 |
| H2/N2 | αH2/N2 = 163 | |||||||
| H2/CO2 | αH2/CO2 = 28 | |||||||
| H2/O2 | αO2/H2 = 42 | |||||||
| Cu3(BTC)2 (25) | 6FDA-ODA | Solvent evaporation | CO2/CH4 | 35 °C, 10 atm | Constant-volume | PCO2 = 22–27 | αCO2/CO4 = 51–60 | 212 |
| PCH4 = 0.43–0.45 | ||||||||
| Mn(HCOO)2 (up to 40) | Polysulfone | — | H2/CH4 | — | — | PCO2 = 10–10.5 | αH2/CH4 = 14–26 | 213 |
| Cu-BPY-HFS (up to 30) | Matrimid 5218 | Solvent evaporation (chloroform) | CO2/CH4 | 35 °C, 2 bar | Constant-volume | PCO2 = 7.81–15.06 | αCO2/CO4 = 25.55–31.93 | 203 |
| H2/CH4 | PCH4 = 0.24–0.59 | αH2/CH4 = 45.38–69.15 | ||||||
| CH4/N2 | PH2 = 16.75–26.74 | |||||||
| MOP-18 (up to 60) | Matrimid 5218 | Solvent evaporation (chloroform) | CO2/CH4 | 35 °C, 1000 torr | Constant-volume | PCO2 = 9.4–15.6 | αCO2/CO4 = 16.47–23.19 | 213 and 214 |
| H2/CH4 | PCH4 = 0.41–0.95 | αH2/CH4 = 23.52–44.55 | ||||||
| PH2 = 17.8–22.3 | ||||||||
| ZIF-8 (up to 80) | Matrimid 5218 | Solvent evaporation (chloroform) | CO2/CH4 | 35 °C, 2000 torr | Constant-volume time-lag | PCO2 up to 24.55 | αCO2/CO4 up to 124 | 150 and 215 |
| H2/CH4 | PCH4 up to 0.89 | αH2/CH4 up to 427 | ||||||
| CO2/C3H8 | PH2 up to 71.22 | αCO2/C3H8 up to 50 | ||||||
| H2/C3H8 | PC3H8 up to 0.52 | |||||||
| ZIF-8 (up to 30) | Poly(1,4-phenylene ether-ether-sulfone) | Solvent evaporation (chloroform) | CO2 | 5 °C 0–10 bar | Constant-volume time-lag PFG NMR | PCO2 = 6–26 | — | 216 |
| ZIF-90 (15) | Ultem 1000, Matrimid 5218, 6FDA-DAM polyimide | Solvent evaporation (dichloromethane) | CO2/CH4 | 25 °C, 2 atm | Constant-volume | PCO2 = 590–720 | αCO2/CO4 = 34–37 | 217 |
| Zr-BDC (25) | 6FDA-ODA | Solvent evaporation | CO2/CH4 | 35 °C, 10 atm | Constant-volume | PCO2 = 50.4 | αCO2/CO4 = 46 | 212 |
| PCH4 = 1.1 | ||||||||
| Zr-ABDC (25) | 6FDA-ODA | Solvent evaporation | CO2/CH4 | 35 °C, 10 atm | Constant-volume | PCO2 = 13.7 | αCO2/CO4 = 51.6 | 212 |
| PCH4 = 0.27 | ||||||||
| Zr-BPDC (25) | 6FDA-ODA | Solvent evaporation | CO2/CH4 | 35 °C, 10 atm | Constant-volume | PCO2 = 20.8 | αCO2/CO4 = 15 | 212 |
| PCH4 = 1.4 | ||||||||
| Al-MIL-53 (up to 32) | 6FDA-ODA | Solvent evaporation | CO2/CH4 | 35 °C, 10 atm | Constant-volume | PCO2 = 13–15 | αCO2/CO4 = 45–78 | 218 |
| PCH4 = 0.24 | ||||||||
| Al-MIL-53 (up to 35) | 6FDA-ODA-DAM copolymer, Matrimid, Ultem | Solvent evaporation | CO2/CH4 | 35 °C, 10 atm | Constant-volume | PCO2 = 50–66 | αCO2/CO4 = 20–37 | 58 |
| Al-MIL-53 (up to 25) | Polysulfone | Solvent evaporation | CO2/CH4 | 35 °C, 4 bar | PCO2 = 4.8–5 | αCO2/CO4 = 25–45 | 219 | |
| ZIF-8 (up to 30) | Matrimid | Solvent evaporation | CO2/CH4 | 22 °C, 4 bar | Constant-volume | PCO2 = 8.8–29 | αCO2/CO4 = 25–39 | 220 |
| H2/CH4 | PH2 = 33–112 | αH2/CH4 = 97–148 | ||||||
| CO2/N2 | PN2 = 0.4–1.7 | αCO2/N2 = 17–22 | ||||||
Detailed CO2/CH4 gas separation properties are reported in Table 22. The CO2/CH4 selectivity of MMM from ZIF-8/Matrimid® with contents up to 80 wt% by Balkus's group150,215 is up to 124. ZIF-8 is the most studied ZIF compounds because of its large pore (11.6 Å) connected via small openings (3.4 Å), high surface areas (1300–1600 m2 g−1), and good thermal stability (up to 400 °C). The authors suggested that ZIF-8 pore opening (3.4 Å) allows to directly adsorb small molecules like H2 and CO2. Therefore, the sieving effect of ZIF-8 improved CO2/CH4 permselectivity (Table 23).
| ZIF-8 loading (w/w) | PCO2 (Barrer) | Ideal CO2/CH4 | 10:90 mol% CO2/CH4 |
|---|---|---|---|
| Matrimid® (0%) | 9.5 | 43 | 42 |
| 20 | 9.0 | 50 | — |
| 30 | 15.5 | 40 | — |
| 40 | 24.5 | 33 | — |
| 50 | 4.7 | 126 | 89 |
| 60 | 7.8 | 81 | 80 |
Another interesting MOF is amine-functionalized nano-size (100–150 nm) Al-MIL-53 (NH2–Al-MIL-53) which can be combined with 6FDA–ODA polyimide to make MMM without addition of any compatibilizing agent.218 These MMM display excellent CO2/CH4 gas separation capability, with high ideal selectivity (up to 77) and good separation factor (up to 53). Moreover, NH2–Al-MIL is a “breathing” material leading to enhanced CO2/CH4 separation factor with increasing feed pressure. The “breathing” effect of NH2–Al-MIL-53/6FDA-ODA MMM compared to neat 6FDA-ODA membrane is shown in Fig. 13. CO2 permeability of neat polymer and MOF-PI membranes displays similar trends; i.e. both values decrease with increasing feed pressure (Fig. 13A). Generally, the separation factor decreases with increasing feed pressure. However, the separation factor of 6FDA–ODA–MIL–NH2–25% increases with feed pressure from 150 to 300 psi (Fig. 13B). At the same time, CO2 adsorption capacity improved rapidly for CO2 pressure between 150 and 400 psi (Fig. 13C).219 This behavior was associated to MOF breathing leading to important increase in CO2 adsorption content at equilibrium. This is a great advantage for high-pressure gas separation applications, such as natural gas and biogas upgrading.
 | ||
| Fig. 13 CO2 permeability (a) and separation factor (α*) (b) for gas mixture (CO2:CH4 = 50:50) as a function of feed pressure for 6FO (6FDA-ODA) and 6FO-mil-NH2-25% membrane at 308 K. CO2 adsorption isotherm of pure Al-mil-53-NH2 MOF (c). | ||
Mixed matrix membranes with MOF-based particles embedded in a continuous polymer matrix have enormous potential in biogas separation applications. A great number of MOF are known, but only a little more than 10 have been used to make MMM for biogas separation (see Table 21). Then, more research and development are still required to explore MOF-based MMM for gas separation. Vinh-Thang and Kaliaguine198 proposed the following topics for future research on MOF-based MMM:
(i) Understanding the basic interactions between the polymers and MOF particles,
(ii) Synthesizing nano-sized MOF particles without agglomeration,
(iii) Understanding the intrinsic separation performances of MOF,
(iv) Synthesizing new MOF with excellent separation and storage properties,
(v) Functionalizing MOF with halogeno or amino groups to improve the adhesion and compatibility between the surface of MOF particles and the polymers,
(vi) Developing novel approaches to uniformly and easily disperse MOF particles in continuous polymer matrices,
(vii) Developing new prediction models to guide the selection of both MOF and polymers with good MMM separation performance,
(viii) Developing new applications of MMM not only for gas separations, but also for other industrial processes like dry bio-ethanol production for bio-fuels, etc.
8. Polymers of intrinsic microporosity (PIM)
PIM received a great deal of attention for gas separation since McKeown and Budd introduced these new polymers designated as “polymers of intrinsic microporosity” in 2004.221,222 PIM have potential for gas separation, heterogeneous catalysis and hydrogen storage.223–226 PIM are not only used as the polymer phase mixed with inorganic fillers such as zeolitic imidazolate framework ZIF-8,227 silica nanoparticles,228 and multi-walled carbon nanotubes (MWCNT)229 in mixed matrix membranes, but also as an organic filler blend with Matrimid,230,231 Ultem polyetherimide232,233 and PI (6FDA–m-PDA233 to enhance gas separation performances. This is the reason why PIM are included in this review.Intrinsic microporosity is defined as “a successive network of interconnected intermolecular voids, which results from the form and rigidity of the element macromolecules”.226 Conventional polymers have enough conformational flexibility, which permits to organize their conformation to maximize intermolecular cohesive interactions and fill the space. Nevertheless, PIM are highly rigid and contorted molecular structures. In particular, due to their fused ring structures, PIM do not have rotational freedom in their backbone so that macromolecules cannot restructure their conformation leading to a rigid structure fixed by their synthesis.234
The history of PIM was developed from McKeown's work on phthalocyanine materials during the 1990. The concept behind their design is simple: by preventing efficient packing of polymer chains and restricting rotation around the backbone, microporosity can be built into ladder polymers.234 Up to 2008, ten structures have been reported (PIM-1 to PIM-10)225 which can be seen in Table 24 and Fig. 14. The most relevant monomers are 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethyl-1,1′-spirobisindane (TTSBI is low cost commercial monomer A1 in Fig. 14) and 2,3,5,6-tetrafluoroterephthalonitrile (TFTPN, monomer B1 in Fig. 14). Each monomer must have a functionality of at least 2 for the reaction to proceed successfully. First, PIM-1 was synthesized from A1 and B1 leading to some initial interest due to its distinct green fluorescence and was sent for testing to Covion, a company specializing in organic light emitting diodes (OLED). But, the results were disappointing and the polymer was tested for nitrogen adsorption (77 K) giving a very respectable apparent BET surface area of around 800 m2 g−1. Then, at the University of Manchester, Peter Budd saw the potential of what they then termed PIM-1 for making membranes and performed some initial testing for phenol extraction from water via pervaporation.222
| Monomers | Solubility | Name | Surface area (BET; m2 g−1) |
|---|---|---|---|
| A1 + B1 | THF, CHCl3 | PIM-1 | 760–850 |
| A1 + B2 | THF | PIM-2 | 600 |
| A1 + B3 | THF | PIM-3 | 560 |
| A2 + B1 | THF | PIM-4 | 440 |
| A2 + B2 | THF | PIM-5 | 540 |
| A3 + B2 | THF | PIM-6 | 430 |
| A1 + B4 | CHCl3 | PIM-7 | 680 |
| A4 + B4 | CHCl3 | PIM-8 | 677 |
| A1 + B5 | CHCl3 | PIM-9 | 661 |
| A4 + B5 | m-Cresol | PIM-10 | 680 |
 | ||
| Fig. 14 Structure of monomers for the synthesis of pims 1–10. | ||
In 2005, Budd et al.235 choose PIM-1 and PIM-7 to make membranes for gas separation. The membranes properties were shown to go beyond Robeson's 1991 upper bound for O2/N2 and CO2/CH4 gas pairs. The authors concluded that the excellent properties of PIM are associated to their rigid but contorted macromolecular structures, which limits packing and establish free volume, combined with chemical functionality allowing strong intermolecular interactions. PIM-1 and PIM-7 structures with their detailed synthesis route are presented in Fig. 15. Gas permeation data specific for CO2 and CH4 are reported in Table 25. PIM-1 has higher CO2 permeability and selectivity for CO2/CH4 pairs. Note that the values are very close to the 2008 Robeson upper bound.
 | ||
| Fig. 15 Preparation and structures of polymers pim-1 and pim-7. reagents and conditions: (i) K2CO3, dmf, 65 °C; (ii) conc. HNO3, hoac; (iii) hoac, reflux; (iv) 18-crown-6, K2CO3, dmf, 150 °C. | ||
| PIMs | Gas | P (Barrer) | D (10−8 cm2 s−1) | S (10−3 cm3 cm−3 cmHg−1) | αCO2/CH4 |
|---|---|---|---|---|---|
| PIM-1 | CO2 | 2300 | 26 | 880 | 18.4 |
| PIM-1 | CH4 | 125 | 6.8 | 180 | — |
| PIM-7 | CO2 | 1100 | 21 | 520 | 17.7 |
| PIM-7 | CH4 | 62 | 5.1 | 125 | — |
PIM-1 has received the most attention, as it contains the contorted spirobisindane unit which led to easily make it (synthesis) and obtain high molecular weights. PIM-1 combines exceptional permeability with moderate selectivity for CO2/CH4 separations. In 2008, Budd et al.236 continued to study PIM-1 membrane gas permeation and performed measurements of thermodynamic properties and free volume, using gas chromatographic and barometric methods. PIM-1 gas permeability and free volume was very strongly sensitive to post-treatment by methanol immersion. From Table 26, the membranes of state 1 have relatively low gas permeability for O2, N2, CO2 and CH4, while that of state 3 have a great increase in gas permeability and improved CO2/CH4 ideal selectivity. The free volume sizes varied and led to gas permeability changes as determined by positron annihilation lifetime spectroscopy (PALS).
| State | P (O2) | P (N2) | P (CO2) | P (CH4) | O2/N2 | CO2/N2 | CO2/CH4 |
|---|---|---|---|---|---|---|---|
| 1 (Wet) | 150 | 45 | 1550 | 114 | 3.3 | 34.4 | 13.6 |
| 2 (Reprecipitated) | 584 | 180 | 4390 | 310 | 3.2 | 24.2 | 14.2 |
| 3 (MeOH treated) | 1610 | 500 | 12 600 | 740 | 3.3 | 25.5 | 17.0 |
Khan et al.237 used PIM-1 thermally crosslinked (250 °C at 1 °C min−1 and kept for a period of 1 h) for CO2/N2, CO2/CH4, and propylene/propane (C3H6/C3H8) gas separation. Polyethylene glycol biazide (PEG-biazide) was selected as a crosslinking agent in nitrene reaction. PIM-1 and crosslinked PIM-1/biazide (not methanol treated) membranes with different crosslinker ratio for pure gas permeation were tested 30 °C and 1 bar feed pressure (Fig. 16). N2 permeability decreased from 548 to 14 Barrer, CO2 permeability decreased from 10667 to 433 Barrer as biazide content increased from 0 to 20 wt%, while the FFV decreased from 0.208 to 0.153. CO2/N2 selectivities increased from 21 to 31, that of CO2/CH4 from 11 to 19.5 as the crosslinker content increased from 0 to 20 wt%. The feed pressure of CO2/CH4 and CO2/N2 gas pair was up to 30 atm for additional experience. The results showed that crosslinked PIM-1 could suppress penetrant-induced plasticization for condensable gases.
 | ||
| Fig. 16 Permeability of N2, CH4, O2, He, H2, Co2 and gas pair selectivity of CO2/N2, CO2/CH4 and O2/N2 in pim-1 and crosslinked pim-1 peg-biazide membranes as 1/FFV and ratio of crosslinker. | ||
 | ||
| Fig. 17 Correlation of permeability and permselectivity for CO2/CH4 gas pair. The solid line is the Robeson upper bound of 2008. triangles indicate pim-1/zif-8 films after ethanol treatment, squares pim-1/zif-8 films as cast. | ||
Thomas et al.238 synthesized PIM-1 and compared with PTMSP (poly(1-trimethylsilyl-1-propyne). PTMSP is a linear chain microporous glassy acetylene-based polymer with very high pure-gas permeability, combined with very low ideal selectivities (for example PO2 = 7500 Barrer, αO2/N2 = 1.2). In this regard, polymers with average PLAS chain spacing of 7–20 Å are defined as microporous materials by some authors. In this case, polycarbonates and polysulfones are not microporous because their average PLAS chain spacing is less than 7 Å based on wide-angle X-ray diffraction (WAXD) data.239,240 On the contrary, PIM-1 and PTMSP with values of 5.8–10.6 Å and 5.1–12.4 Å respectively, are and their volumetric physical properties are reported in Table 27.
| Polymer | PIM-1 | PTMSP |
|---|---|---|
| BET surface area (m2 g−1) | 760, 830 | 780 |
| PALS chain spacing (Å) | 5.8–10.6 | 5.1–12.4 |
| Fractional free volume (%) | 24–26 | 29 |
The results of Table 28 indicate that PIM-1 has excellent performance for n-butane/methane separation with a selectivity of 24 and n-butane permeability of 4200 Barrer. PIM-1 selectivity is similar to microporous PTMSP, but around 2.5 times higher than PDMS. Permeability is nevertheless lower for both. Presently, PTMSP is the only commercial rubbery membrane material for n-butane/methane separation. PIM-1 however has higher hydrocarbons chemical resistance compared to PTMSP, so that this novel microporous polymeric membrane has great potential for hydrocarbon/methane separation like biogas and natural gas upgrading.
| Material | Permeability coefficient × 1010 (cm3(STP) cm cm−2 s−1 cmHg) | Selectivity n-C4H10/CH4 | |
|---|---|---|---|
| n-C4H10 | CH4 | ||
| PIM-1 | 4200 | 175 | 24 |
| PTMSP | 53500 |
1800 | 30 |
| PDMS | 12900 |
1250 | 10 |
MMM based on PIM-1 and zeolitic imidazolate framework ZIF-8 were prepared by Bushell et al.227 ZIF-8 up to 43 vol% was used and excellent results are shown in Fig. 16 for CO2 and CH4. With increasing ZIF-8 content, permeability, diffusion coefficients and separation factors increased. CO2 permeability increases from 4390 to 6300 Barrer and CO2/CH4 selectivity slightly increased from 14.2 to 14.7.
Ahn et al.228 reported the gas transport properties of PIM-1/silica nano-composite membranes for O2/N2. In Fig. 18 open stars are results for nano-composites with different fumed silica contents (0, 6.7, 13.0, 19.1, 23.5 vol%). O2 permeability increased from 1340 to 3730 Barrer, and O2/N2 selectivity decreased from 3.3 to 2.1, while CO2 permeability increased from 6000 to 13400 Barrer. Unfortunately, there are no CH4 permeation data available.
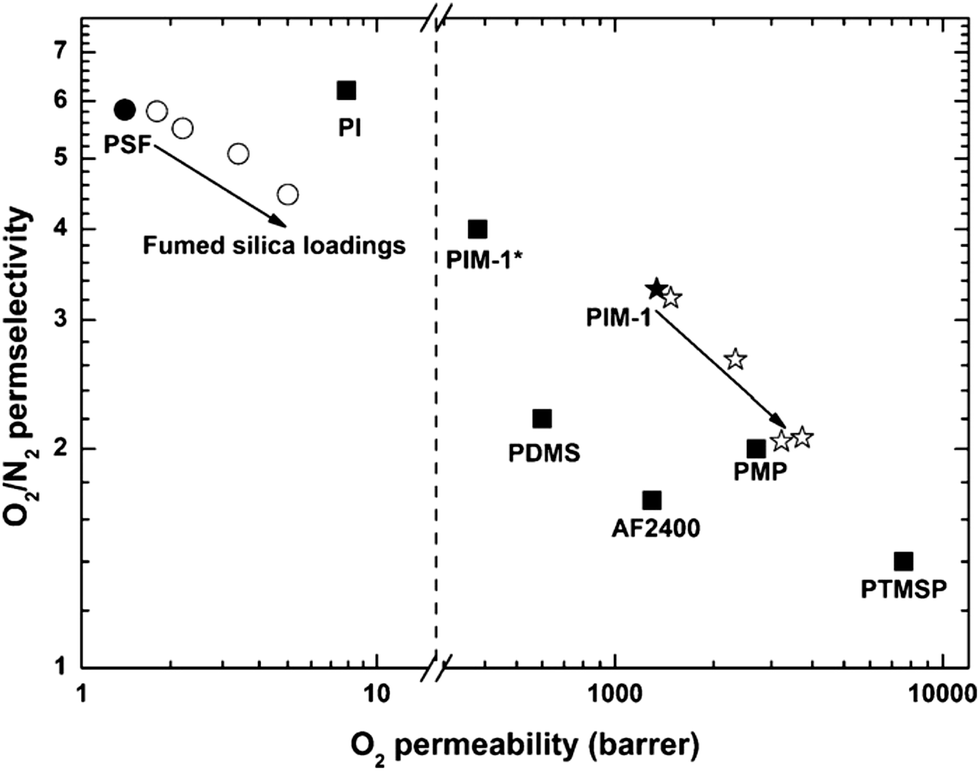 | ||
| Fig. 18 The trade-off performance for gas permeability and selectivity through conventional, low-free-volume glassy polysulfone (psf), polyimide (pi), high free-volume glassy poly(4-methyl-2-pentyne) (pmp), poly(1-trimethylsilyl-1-propyne) (ptmsp), poly(2,2-bis(trifluoromethyl)-4,5-difluoro-1,3-dioxole-cotetrafluoroethylene) (teflon af2400), rubbery polydimethylsiloxane (pdms), and pim-1 value. Open circles and stars show the results of nanocomposites with fumed silica at various loadings. | ||
MMM were fabricated using functionalized multi-walled carbon nanotubes (f-MWCNT) as inorganic particles and PIM-1 for the polymer matrix. Some homogeneity improvement for MWCNT in MMM was obtained by covalent functionalization of MWCNT with poly(ethylene glycol).229 Due to good interfacial adhesion and the absence of voids between f-MWCNT and polymer matrix, the MMM had higher permeabilities, as well as improved CO2/N2 and O2/N2 selectivities. MMM with 0.5 to 3 wt% f-MWCNT had CO2 permeabilities increase, but ideal selectivities decrease. Table 29 and 30 show gas permeation results versus f-MWCNT loading.
| Membrane | Permeance (N m3 m−2 h−1 bar−1) | Permeability (Barrer) | ||||||
|---|---|---|---|---|---|---|---|---|
| O2 | N2 | CO2 | CH4 | O2 | N2 | CO2 | CH4 | |
| PIM-1 | 1.93 | 0.69 | 19.4 | 1.26 | 533 | 190 | 5360 | 348 |
| 0.5 wt% f-MWCNTs/PIM-1 | 2.07 | — | — | — | 571 | 204 | 6830 | 604 |
| 1 wt% f-MWCNTs/PIM-1 | 2.19 | 0.78 | 25.7 | 2.47 | 605 | 215 | 7090 | 682 |
| 2 wt% f-MWCNTs/PIM-1 | 3.49 | 0.89 | 29.8 | 3.69 | 964 | 245 | 8230 | 1020 |
| 3 wt% f-MWCNTs/PIM-1 | 2.66 | 0.79 | 29.9 | 3.20 | 734 | 218 | 8250 | 883 |
| Membrane | Selectivity | ||
|---|---|---|---|
| O2/N2 | CO2/N2 | CO2/CH4 | |
| PIM-1 | 2.80 | 28.2 | 15.4 |
| 0.5 wt% f-MWCNTs/PIM-1 | 2.79 | 33.5 | 11.3 |
| 1 wt% f-MWCNTs/PIM-1 | 2.81 | 32.9 | 10.4 |
| 2 wt% f-MWCNTs/PIM-1 | 3.93 | 33.5 | 8.08 |
| 3 wt% f-MWCNTs/PIM-1 | 3.37 | 37.8 | 9.32 |
Chung's group230–232 made polymer blend membranes, PIM-1 being used as an organic filler to enhance the gas separation performance with two commercial polymers: Matrimid® 5218 and Ultem® 1010 polyetherimide. Matrimid was chosen as the polymer phase because of its good thermal stability and processability. On the other hand, Matrimid has relatively low CO2 permeability (6.5) and moderate gas-pair selectivity of around 35 for CO2/CH4. PIM-1 has superior CO2 permeability of 4030 Barrer and CO2/CH4 selectivity of 11.5. The PIM-1 ratio in the blend was varied over a wide range (5, 10, 30, 50, 70, 90 to 95 wt%) in Matrimid to make flat membranes and the CO2/CH4 separation performance (35 °C and 3.5 atm) was compared to Robeson's 2008 upper bound in Fig. 19. It is clear that all the blends have separation performance below the upper bound. Then, hollow fiber membranes from PIM-1/Matrimid were used for CO2/CH4, O2/N2 and CO2/N2 separation.231 From Table 31, the newly developed fibers have exceptional gas separation performances surpassing other polyimide blend membranes for these separations. These new materials have potential for industrial use of hollow fiber membranes.
 | ||
| Fig. 19 Comparison with Robeson upper bound of pim-1/Matrimid polymer blends of CO2/CH4. | ||
| Hollow fibers ID | Permeance (P/L) (GPUa) | Ideal selectivity (—) | |||||
|---|---|---|---|---|---|---|---|
| O2 | N2 | CH4 | CO2 | O2/N2 | CO2/CH4 | CO2/N2 | |
| a 1 GPU = 1 × 10−6 cm3 (STP)/cm2 s cmHg = 7.5005 × 10−12 m s−1 Pa−1. | |||||||
| After silicon rubber coating for 3 min | |||||||
| PIM-1/Matrimid (15/85)-A | 59.9 | 9.9 | 7.1 | 243.2 | 6.1 | 34.3 | 24.6 |
| PIM-1/Matrimid (15/85)-B | 57.1 | 9.2 | 7.3 | 234.6 | 6.2 | 32.1 | 25.5 |
| PIM-1/Matrimid (15/85)-C | 50.1 | 8.0 | 6.8 | 217.1 | 6.2 | 32.0 | 27.1 |
PIM-1/Matrimid hollow fiber membranes by solution spinning were studied and conditions were: polymer composition with PIM-1 of 15 wt% and Matrimid of 85 wt%; dope composition with polymer of 11 wt%, NMP solvent of 44.5 wt% and THF solvent of 44.5 wt%; dope flow rate of 2.8 ml min−1; bore fluid flow rate of 1 ml min−1; the distance of air gap of 2.5 cm; take-up speed of 6.3 m min−1; composition of bore fluid of 95% water with 5% NMP (A), 80% NMP with 20% water (B) and 50% NMP with 50% water (C). Because Matrimid is an expensive polyimide material and has a low plasticization pressure against CO2,232 as well as relatively high gas permeability, polyetherimide (PEI) Ultem®, 1010 which is very cheap and has better chemical resistance to common solvents, is suitable for applications under harsh environments. The pure gas permeability of Ultem to CO2 and O2 are 1.33 and 0.41 Barrer respectively, while the ideal selectivity of CO2/CH4, CO2/N2 and O2/N2 gas pairs are 37, 25 and 7.5, respectively. Hao et al.232 summarized the results (see Table 32) of gas separation performance of different polymers with PIM-1 blend systems.
| MMMS | Permeability (Barrer) | Ideal selectivity (—) | |||
|---|---|---|---|---|---|
| CO2 | O2 | O2/N2 | CO2/N2 | CO2/CH4 | |
| a Ref. 234 test was conducted at 35 °C and 3.5 bar.b Ref 235 test was conducted at 50 °C and 100 psig.c Ref 231 test was conducted at 35 °C and 3.5 bar. | |||||
| Ultem/PIM-1a | |||||
| Pristine Ultem | 1.48 | 0.38 | 7.1 | 27.4 | 37.0 |
| 90:10 |
3.95 | 1.1 | 6.8 | 25.2 | 33.8 |
| 80:20 |
6.58 | 1.6 | 6.1 | 25.7 | 34.6 |
| 70:30 |
9.27 | 2.2 | 5.8 | 24.8 | 34.7 |
| Ultem/PIM-1b | |||||
| 90:10 |
2.89 | — | — | — | 31.6 |
| 80:20 |
5.69 | — | — | — | 31.2 |
| 70:30 |
5.77 | — | — | — | 30.2 |
| Matrimid/PIM-1c(b) | |||||
| Pristine Matrimid | 9.6 (10.0) | 2.1 | 6.4 | 30 | 36 (28.2) |
| 95:5 |
12 (-) | 2.6 | 6.6 | 29 | 35 (-) |
| 90:10 |
17 (20.3) | 3.4 | 6.1 | 30 | 34 (27.1) |
| 70:30 |
56 (35.9) | 11 | 5.8 | 28 | 31 (24.8) |
| 6FDA-m-PDA/PIM-1b | |||||
| Pristine 6FDA-m-PDA | 14.8 | — | — | — | 48.4 |
| 92.5:7.5 |
22.3 | — | — | — | 48.7 |
Bezzu et al.241 described the synthesis of PIM-SBF using 1,1′-spirobisindane instead of the more rigid 9,9′-spirobifluorene (SBF) unit. Table 33 shows PIM-SBF membranes gas permeability data compared to PIM-1. This work gives a direct demonstration that gas permeation may be enhanced by increasing polymer chain rigidity.
| Membrane | PCO2 (Barrer) | PCH4 (Barrer) | αCO2/CH4 (−) | DCO2 (10−12 m2 s−1) | DCH4 (10−12 m2 s−1) | DCO2/DCH4 (−) | SCO2 (cm3 (STP)/cm3 bar) | SCH4 [cm3 (STP)/cm3 bar] | SCO2/SCH4 (−) |
|---|---|---|---|---|---|---|---|---|---|
| PIM-SBF (180 μm) | 13900 |
1100 | 12.6 | 181 | 42 | 4.3 | 53.2 | 19.6 | 2.7 |
| PIM-SBF (81 μm) | 10400 |
754 | 13.8 | 147 | 33 | 4.5 | 53.0 | 17.0 | 3.1 |
| PIM-1 (128 μm) | 13600 |
1360 | 10.0 | 226 | 79 | 2.9 | 45.2 | 12.9 | 3.5 |
8.1 Conclusion
1. PIM-1 as polymer phase to make MMM, fumed silica, functionalized multi-walled carbon nanotubes (f-MWCNT) and ZIF-8 as organic fillers with various levels were studied for gas transport performances. For PIM-1/ZIF-8 MMM, it was shown that both permeabilities and separation factors: α(H2/N2), α(H2/CH4), α(O2/N2) were improved with ZIF-8 loading in MMM. The gas separation performances were above the 2008 Robeson upper bound. Silica nanoparticles combined with PIM-1 improved the overall gas permeability of O2 and decreased O2/N2 selectivity. MMM based on f-MWCNT/PIM-8 have increased CO2 permeabilities, but CO2/CH4 selectivities decreased with increasing loading.2. PIM-1 as an organic filler blend with PI and PEI enhanced the permeability of low permeability materials for industrial gas separation applications. The combination of PIM-1 and Matrimid have a higher gas permeability and a minimum in gas-pair selectivity compared to Matrimid membranes. For CO2/CH4 separation, the gas separation performance varies like the 2008 Robeson upper bound as PIM-1 content increases, but stay below this upper bound. It is expected that adding 5-10 wt% PIM-1 in Matrimid could make the resultant blends more suitable for CO2/CH4 separation without compromise in CO2/CH4 selectivity. PIM-1 is easily dispersed in the polymer without any other agent addition. For PIM-1 in Ultem 1010, the permeability of CO2 increased by 47% and 167% when PIM-1 loadings were 5 and 10 wt%, respectively. For 6FDA-m-PDA with PIM-1, CO2 permeability was changed from 14.8 to 22.3 Barrer (0–7.5% PIM-1) and ideal selectivity was almost constant (from 48.4 to 48.7). Compared to pristine Matrimid hollow fiber, the CO2 permeance of the spun blend fibers with 5 and 10 wt% PIM-1 increased by 78% and 146% (from 86.3 GPU to 153.4 GPU and 212.4 GPU) respectively, without showing much loss in CO2/CH4 selectivity.
3. Future developments for PIM will be to modify or create new types of PIM, blend them with other polymers and use them with organic fillers.
9. Hollow fibers
When membrane gas separation is applied, large surface area is highly needed for high process capacity. In the present, hollow fiber, spiral wound, and envelope type modules are three common types of configuration used for industrial applications.242 The properties of these three types of gas permeation modules are presented in Table 34. Since the surface area of the envelope has lower packing density, lower surface per module and higher cost for module requirement, commonly used membranes are hollow fiber and spiral wound modules. Because hollow fibers produces higher effective surface area per unit volume of membrane module compared with the others, this configuration has attracted greater interest. Additionally, hollow fibers provide mechanical support (module) and are easier to handle in fabrication, as well as gas separation process operation. Table 35 lists the commercial suppliers involved in gas separation, the majority using hollow fiber membranes.| Property | Unit | Hollow fiber | Spiral wound | Envelope |
|---|---|---|---|---|
| Packing density | m2 m−3 | <10000 |
200–1000 | 30–500 |
| Approximate area per module | m2 | 300–600 | 20–40 | 5–20 |
| Supplier | Module type | Polymer |
|---|---|---|
| Air Liquide medal | Hollow fiber | Polyimide, polyaramide |
| Air products | Hollow fiber | Polysulfone |
| GMT membrantecnik | Envelope | Poly(ethylene oxide)poly(butylene terephthalate) |
| Evonik | Hollow fiber | Polyimide |
| IGS generon membrane technology | Hollow fiber | Tetrabrome polycarbonate |
| Kvaerner membrane systems (no longer active) | Spiral wound | Cellulose acetate |
| MTR Inc. | Spiral wound | Perfluoro polymer, silicon rubber |
| Parker | Hollow fiber | Polyphenylene oxide |
| Praxair (no longer active) | Hollow fiber | Polyimide |
| UBE membranes | Hollow fiber | Polyimide |
| UOP former grace | Spiral wound | Cellulose acetate |
The (outside) diameter of hollow fibers can change depending on application, and varies from 50 to 3000 μm. Fibers can be made from one or two materials. Two layers are used for the outside or inside surface. The selective layer can be integrated with the fiber or a separate one (coating) put on a porous support (fiber). Outside diameter of 50 to 200 μm is normally named fine hollow fibers. High-pressure gas separations usually need these fine fibers because they can resist very high outside hydrostatic pressures up to 1000 psig. For low-pressure gas separations, a fluid is generally put inside the fiber, while the permeate is placed in the outer shell. The fiber diameter is usually greater than 200–500 μm. The fibers are called capillary fibers if the diameter is above 500 μm.
Hollow fibers preparation can be done via melt spinning (free-solvent spinning) and solution spinning (dry-wet spinning).244 Melt spinning equipment consists of an extruder, spinneret, water cooling tank and take-up unit. A hot polymer melt is extruded from the spinneret and the fiber is cooled and solidified when immersed in a cooling tank. Fibers by this melt-spun process can reach high take-up speeds and be very fine depending on take-up speed (force). These fibers are normal denser since they do not have a porous surface giving lower gas fluxes in separation applications compared to asymmetric hollow fibers from solution-spun fibers. Due to the fact that the materials used in CO2/CH4 separation, such as polyimides, polysulfones and polyetherimides, often have high glass transition temperatures, it is difficult to use melt-spinning technology for hollow fiber membranes. So solution-spinning is the most common process for membrane fabrication used in biogas separation. Generally, 20–30 wt% polymer solutions are used leading to high viscosity and flow rate is controlled by a gear pump. The polymer is precipitated into a non-solvent (generally water) leading to an anisotropic structure.
9.1 Hollow fiber solution spinning
Loeb and Sourirajan245 were the first to produce asymmetric cellulose acetate for reverse osmosis (RO) via phase inversion. Since then, many papers focused on hollow fiber membranes production and applications.246–259 The formation mechanism based on solution-spinning to produce hollow fiber via phase inversion is however very complex.Fig. 20 illustrates a simple production line of solution-spinning via phase inversion. Evidently, the spinneret plays an important role. Two metering pumps transfer precise quantities of the polymer solution dope and the bore fluid to the spinneret. Then, the polymer solution and non-solvent fluid go from the spinneret and solvent evaporation in the air-gap region. Finally, the take-up unit collects the fibers from the coagulation bath. A magnification of the zone near the spinneret is presented in Fig. 21.254 When the dope polymer solution is degassed and prepared, the process includes: (1) feeding at constant flow rate (may be carried out by pressurized nitrogen) the spinning polymer solution dope and bore fluid simultaneously, (2) introducing the spinning solution through a spinneret, (3) internal coagulation between the bore fluid and the polymer solution dope, (4) solvent evaporation on the outer surface of fiber in the gap region, (5) extension by gravity or elongation by the take-up unit, and (6) if necessary, residual solvents are evaporated by post-treatments which can partially control pore sizes.
 | ||
| Fig. 20 Schematic diagram of the spinning apparatus for hollow fiber membrane. | ||
 | ||
| Fig. 21 Schematic diagram of area nearby the spinneret and the formation of nascent hollow fiber during phase inversion. | ||
The important factors for hollow fiber spinning are illustrated in Fig. 20 and 21. First, the polymer concentration is a very important parameter playing a key role on the overall hollow fiber process. Generally, the spinning dope is very viscous due to relatively high polymer concentrations (20–30%), higher than that of casting solutions (5–10%) which are used to make flat-sheet membranes. This is because hollow fiber membranes should have the capacity to separate gases and withstand high pressure without collapsing. Second, the solvent molecules size controls the precipitation path and fiber morphology when nascent hollow fibers precipitated in coagulating process. Generally, solvent molecules are smaller and faster diffusing, solvent exchange is also faster, or vice versa. Dimethyl formamide, N-methyl pyrrolidone and dimethyl acetamide are the most common casting (aprotic) solvents. Third, the non-solvent choice is also important. Water is the best precipitation phase, but methanol or isopropanol, which are organic-based solvent can also be used. Nevertheless, the latter precipitate slowly the casting polymer solution, so these membranes are often denser and less anisotropic leading to lower mass flux.
Solution spinning is complex and asymmetric hollow fibers performance depends on polymer solution and bore fluid compositions, dope and bore flow rates, spinneret design, air-gap length, and take-up speed. Parameters such as spinneret and coagulation bath temperature also have a great effect on asymmetric fiber morphology. Peng et al. used Torlon® polyamideimide to make hollow fibers.255 Fig. 22a presents different fiber morphology depending on spinneret temperatures. An increase of spinneret temperature or a decrease of polymer solution viscosity, causes more macrovoids in the fiber morphology. Another example shown in Fig. 22b is the effect of the coagulation bath temperature.256 A more porous structure is formed when the external coagulant temperature increases because of delayed demixing for 6FDA/6FDAM polyimides fibers.
 | ||
| Fig. 22 The effect of spinning temperature on fiber morphology. (a) SEM cross-section images of Torlon® polyamideimide membranes spun at different spinneret temperatures (copyright 1997 John Wiley & Sons, Inc.). (b) SEM external surface images of 6FDA/6FDAM polyimide membranes spun at different coagulation bath temperatures (copyright 2008, Elsevier B.V.). | ||
The drawing force is also a key element in the process, which can be of two type. One is from gravity due to the fiber weight, while another force is from the take-up unit. If air-gaps is long enough and high take-up speeds are used, the draw force can affect fiber surface roughness and cross-section morphology. For example, in 6FDA/6FDAM fibers, a larger air-gap led to longer coagulation time, and therefore more macrovoids (Fig. 23a).257 Fig. 23b shows that the presence of macrovoids decreased with increasing take-up speed.258 Higher elongation forces applied by the take-up device produced smaller fiber diameters. If the air-gap distance was too long, macrovoids can be created on the fiber surface. This is similar when too high elongation is applied leading to tearing the chains apart due to excessive elongational stresses. Therefore, the air-gap length, take-up speed, temperature of the spinneret and coagulation bath, composition of bore fluid, dope and bore flow rates all depend on the polymer/solvent selection. Obviously, the spinneret design is also very important to control molecular orientation, polymer morphology and fiber dimensions.
 | ||
| Fig. 23 The effect of air-gap distance and take-up speed on hollow fiber morphology. (a) SEM external surface images of 6FDA/6FDAM polyimide membranes with various air-gap lengths, (b) SEM cross-section images of p84 polyimide membranes with various take-up speeds. | ||
9.2 Membrane modules
Industrial or commercial application needs hundreds to thousands of square meters to carry out at a useful scale a gas separation process. Therefore, membrane separation process must be economical and efficient in volume optimization (high areas) to be used industrially. The configuration of choice is membrane module where several formats are available: plate-and-frame, tubular, spiral-wound, and hollow fiber membrane modules.259For hollow fiber modules, certain quantities of fibers of a certain length are assembled into bundles to make these modules. The bundle of several hollow fiber is placed inside a stainless steel tube and secured by gluing the ends of the fibers into the tube ends. Epoxy resins, polyurethanes, or silicone resins are used to close the ends. Generally, the gas flow direction are shell-side and bore-side feed into modules (Fig. 24259). The first type (Fig. 24a) is shell-side feed: the gas mixture supports the outside of the fiber bundle, and the permeated gas is received from the hollow fibers. The second type is bore-side feed (Fig. 24b): the gas mixture supports the hollow fibers, and the permeated gas is received from the fiber bundle. A suitable module type (bore-side or shell-side feed) choice is determined by the gas pressure, pressure drop, composition, and permeance in the membrane. Shell-side feed modules are used for high-pressure applications due to fiber wall can support high pressure up to 1000 psig. The fibers used in this configuration have small diameters and thick walls; i.e. 50 μm inside diameter and 100–200 μm outside diameter. With this type of feed, the feed stream however should be free of particles before entering the module since fouling may be a problem. Bore-side feed modules can reduce fouling and concentration polarization on the outside the fiber, but they can be used for feed pressure up to 150 psig.
 | ||
| Fig. 24 Two types of hollow-fibre modules used for gas separation applications. | ||
Hollow fiber membrane sheet are fixed in a module so that the gas concentration gradient in the gas flow direction is almost constant and to limit short-cuts. The gas is generally compressed to a certain pressure before entering the module. The feed gas must be free of particles and liquids because these undesired components must not enter compression devices. Hence, gas mixture separation process by hollow fiber modules does not required further pretreatment.
10. Designs of membrane systems for biogas upgrading
Generally, in the process of upgrading biogas, raw biogas from the bioreactor contains CH4 and CO2, as well as H2S, H2O, O2, N2, H2, ammonia, siloxanes, and particles. First, the raw biogas is passed through mechanical filters to remove solid particles. Oxygen is normally completely consumed by the reaction of aerobic microorganisms in the digester. Hydrogen has no restriction or requirement to apply for grid injection or for use as vehicle fuel. H2 separation is therefore not necessary. Nitrogen in the biogas can be eliminated via membranes or low temperature (PSA), but this is costly. N2 presence in the biogas means that air was sucked in. Therefore air should not be allowed inside to limit nitrogen in the biogas.28For H2O, water will condense when pressure is increased or temperature decreases; it can thereby be separated from biogas. Cooling can be simply realized by burying the gas line equipped with a condensate trap underground. Water is also easily eliminated from biogas by both rubbery and glassy polymer membranes gas permeation (see Table 13).18
Siloxanes used in products such as deodorants and shampoos, contain a silicon–oxygen bond. They can be found in biogas from sewage sludge treatment plants and in landfill gas. Glassy microcrystalline silica is a white powder produced by siloxanes when burned. This white powder can create a problem in gas engines.18 They are considered to be the most important contaminant. Therefore, it is necessary to remove siloxane to increase processing equipments lifespan.260 Polydimethylsiloxane (PDMS) has been proposed as a potential membrane material to eliminate siloxanes and other trace of volatile compounds.261
Ammonia (NH3) is formed during the degradation of proteins. Ammonia is formed by the anaerobic digestion of some molecules. It is highly corrosive and a health risk from common contaminants. It is not considered as harmful as H2S because its combustion only slightly increases nitrogen oxides (NOx) emissions. A separate cleaning step for ammonia is not recommended due to elimination during biogas drying or upgrading.28.
The H2S content can vary with the organic being composted, but typical values of 10–10000 ppmv (0.0001–1 vol%) are reported. H2S has a very bad smell and can produce highly corrosive, unhealthy and environmentally hazardous sulfur dioxide (SO2) and sulfuric acid H2SO4. It is necessary to remove H2S for any eventual biogas use.262 Polyimide membranes were used for biogas purification and enrichment by Harasimowicz et al.29 It was possible to achieve CH4 enrichment from 55–85% up to 91–94%. At the same time, H2S concentration was reduced from 2 mol% to 0.95 mol%.
Halogenated hydrocarbons are mainly found in landfill gas and lead to the corrosion of CHP engines. They can be eliminated via pressurized tube exchangers filled with specific activated carbon. Finally, CO2 is sometimes considered to be a nuisance because of large quantities (10–65%), and is inert in combustion, thus decreasing the biogas calorific value. Therefore, removal of CO2 is very important for biogas upgrading. The separation of CO2/CH4 by membrane is based on CO2 and CH4 having different solubility and diffusivity in the membrane and various types of membranes were discussed in Section 7.
Generally, biogas upgrading consists of two steps. Removal of carbon dioxide to increase the gas calorific value, and elimination of undesired molecules such as water, hydrogen sulfide, ammonia, as well as potential TOC (trace organic components) to improve biogas performance.
Fig. 2541 gives an example of a biogas upgrading generating system on a farm with an integrated membrane separation unit with a typical raw gas flow rates of less than 4000 m3 h−1.263 The pressure of the raw gas is about 1.1 bar when it leaves the fermenter at 30 °C. Generally, the raw gas containing CH4, CO2 and others impurities is first compressed to 20 bar, and then filtered at ambient temperature to remove any liquids. It is then fed into the membrane separation unit. Afterwards, the temperature is controlled by a heat exchanger to keep the process under high enough temperatures. The retentate is mostly CH4, which is compressed to 40 bar to be delivered to a natural gas grid. The permeate stream (CO2, H2O, H2S) is sent to a gas treatment unit to eliminate these contaminants not to be released into the environment. The CO2 enriched gas (higher than 80% content) is then compressed to 10 bar to send to a CH4 recovery unit.
 | ||
| Fig. 25 The process equipment for a membrane-based upgrading process. | ||
10.1 Single step gas permeation processes
Scholz and Wessling described two single step membrane permeation processes for biogas upgrading (Fig. 26).41 On the left, the main part is the process to remove CO2 from CH4. However, CH4 loss in this gas permeation process is high and limited by membrane selectivity. In these plants, methane loss can reach 10–15% on the permeate side. If the application is not fuel use, it must be flared leading to revenue loss. Nonetheless, this one-step membrane unit, due to low capital and operating costs, can be used for gas wells producing 1–2 MMscfd.48 The right side process is a single membrane module process coupled with a partial permeate stream recycling and CH4 recovery substantially increases using this process. Nevertheless, CH4 recovery cannot reach more than 95% while a CH4 purity of 96% is necessary for grid injection. Furthermore, the flow rate passing through the compressor increases because of partial recycling and therefore the energy for driving the separation process increases.41 | ||
| Fig. 26 Single stage membrane-based biogas upgrading process using feed compression. Process (a) the permeate flows to the ambient. Process (b) the permeate is partially recycled to enhance the CH4 recovery. | ||
10.2 Two-step gas permeation processes
In order to improve CH4 recovery and simultaneously get CH4 purity, scientists applied various membrane modules in the upgrading system. Four different two-stage upgrading processes are depicted in Fig. 27. | ||
| Fig. 27 Two stage processes for biogas upgrading. | ||
Process (a) only needs one compressor and recycles the permeate of the second step. Process (b), which was investigated by Deng and Hägg,263 needs two compressors and recycles the retentate of the second step. In process (c), which is related to process (b), two compressors are needed and the retentate of the second step is mixed with the one coming from the first step. In process (d) the feed gas is used as a sweep gas on the permeate side of the second module. Process (d) configuration is similar to process (a). When the CO2 mole fraction in the second module permeate is higher than that of the feed stream, the sweep stream is applied as to decrease the CO2 mole fraction on the permeate side of the second module. Here, only one compressor is required.
Deng and Hagg evaluated CH4 recovery, specific energy, specific membrane area, specific upgrading costs of single stage and three two-stage processes. The results are shown in Table 36. It is seen that process (b) has the lowest upgrading costs, as well as the highest CH4 recovery and module specific membrane area and specific energy with 66.67 GPU of CO2 permeability and 2.08 GPU of CH4 permeability.
| Process | CH4 recovery | Specific energy (kW h m−3) | Specific area (m2 h m−3) | Upgrading costs (Euro ct h m−3) | Supply pressure (bar) |
|---|---|---|---|---|---|
| Single stage | 0.855 | 0.277 | 1.70 | 0.228 | 20 |
| Two stage (process (a)) | 0.957 | 0.318 | 1.92 | 0.220 | 20 |
| Two stage (process (b)) | 0.997 | 0.286 | 1.69 | 0.201 | 20 |
| Two stage (process (c)) | 0.973 | 0.295 | 1.57 | 0.206 | 20 |
10.3 Three-step gas permeation processes
Makaruk and Harasek proposed a three-step biogas upgrading process (Fig. 28) which is similar to process (d) in Fig. 27.264 It is important to note that the unpressurized raw gas is mixed with the permeate of module 3 and sent to module 2 on the permeate side as to dilute the CO2 concentration on the permeate stream of module 2. Hence, lower membrane area is needed without increasing the recycle stream. | ||
| Fig. 28 Three stage gas permeation process for biogas upgrading. | ||
 | ||
| Fig. 29 Flow chart diagram of the pilot plant. | ||
10.4 Hybrid gas permeation processes
Rautenbach and Welsh265 described a pilot plant for the treatment of 200 m3(STP) h−1 landfill feed gas which was operated on a landfill dumpsite in Germany (Fig. 28). The composition of landfill gas is 54% of CH4, 40% of CO2, 4% of N2, 1% of O2 1% of water vapor, and 100 mg m−2 of H2S, 200 mg m−2 of C2–Cn, 100 mg m−2 of CFC. The process was composed of two steps: an adsorption step for removal of the toxic trace components (CFC) and another step which is the membrane unit for CO2 removal. Almost pure methane was produced with only traces of contaminants. It was fed into the local naturel gas network or stored at high pressure as an engine fuel in distribution points. The residual concentrations of trace components such as H2S and CFC are both less than 2 mg m−3(STP), activated carbon was chosen to remove these components. Polyamide membranes from UBE-Industries were installed in 5 hollow fiber modules giving a total membrane surface area of 700 m2. Unfortunately, the membrane properties could not be found in the text. According to the author's calculations, gas permeation was favorable, especially for small sites producing less than 1000 m3 (STP) h−1.Another hybrid process combining membrane separation and diethanolamine (30% aqueous DEA) absorption was used for raw natural gas having up to 40 mol% CO2 and up to 1 mol% H2S.266 In this case, “asymmetric” cellulose acetate (CA) membrane modules were used for acid gas removal, in particular for removal CO2. The product could reach the final requirement of US pipeline specifications (<2 mol% CO2 and <4 ppm H2S). In this case, CA membrane with a CO2/CH4 selectivity of 21 and H2S/CH4 selectivity of 19, a feed pressure of 800 psia, a permeate pressure of 20 psia, and effective membrane thickness of 0.394 mm and a membrane life of 3 years, was used to remove high concentration of acid gases, and the rest of acid gases was removed by aqueous DEA. The authors estimated the total costs of independent membrane separation and gas absorption processes and hybrid process for the following conditions: feed flow of 35 MMSCFD [991100 m3(STP) per day] and two feed stream compositions: A (not containing H2S) 73 mol% CH4, 25 mol% CO2, 1 mol% N2, 1 mol% C2H6, and B (containing H2S) 73 mol% CH4, 24.5 mol% CO2, 0.5 mol% (5000 ppm) H2S, 1 mol% N2, 1 mol% C2H6.
It was found (Table 37) that the total cost depends on feed stream H2S content. So the membrane process not having H2S (stream A) was more efficient compared with other processes. On the other hand, the membrane process with H2S (stream B) was more costly than the hybrid process and membrane process not having H2S. This is due to increased membrane area and operating costs to remove H2S. Total capital investment of the hybrid process was less than gas absorption because 78% of CO2 was removed by the membrane and lower DEA solvent circulation rate.
| Membrane area, methane losses and the cost items | Membrane process (stream A) | Membrane process (stream B) | Gas absorption process (stream A and B) | Hybrid process |
|---|---|---|---|---|
| Membrane area (102 m2) | 104.1 | 150.7 | — | 43.3 |
| Methane losses (MM% per year) | 1.137 | 1.431 | 0.144 | 0.983 |
| Total capital investment (MM%) | 2.836 | 3.688 | 6.226 | 4.196 |
| Operating expenses (MM% per year) | 1.033 | 1.318 | 2.853 | 1.516 |
| Total separation cost (% per MSCF of feed) | 0.244 | 0.311 | 0.373 | 0.296 |
Recently, a hybrid of temperature-swing-adsorption (TSA) and membrane process was investigated for biogas upgrading.267 The feed flow rate was 200 N m3 h−1, with a composition of 40% CO2 and 60% CH4. The H2S, H2O, VOCs, and siloxanes were removed by the first step, (top part in Fig. 30). The gas blower, with a pressure of 1.4 bar (absolute pressure), was used to overcome the total losses associated to the adsorption beds in series. A high-efficiency iron-oxide adsorbent in the temperature-swing-adsorption (TSA) tower, then removed water vapor, H2S, VOC and siloxanes via packed adsorbents (activated alumina and activated carbon).267 The second step consists of three membranes (A1-A3). The A1 membrane generated a CO2-enriched permeate, further enriched to 99 vol.% by the A3 membrane. The gas permeate leaving A1 is not compressed before entering the membrane in A3. The retentate of A1 membrane goes to the A2 membrane to enrich CH4, further upgraded to 97 vol.% as pipeline methane in A2. The A1-A3 membranes were made from blends of polyetherimide-biomaleimide (PEI-BMI) with CO2 permeability of 25 GPU and CO2/CH4 selectivity of 55.
 | ||
| Fig. 30 Flow-chart of a membrane process designed for upgrading biogas from wastewater plant digesters, with the adsorption and temperature-swing-adsorption as the pre-treatment. | ||
Membrane processes can also be combined with heat and power engines (CHP). 268 CH4 drives the combined heat and power engine, which is from the permeate of the membrane stage. A description of this investigation was recently reported by Makaruk et al.268
Makaruk et al.269 evaluated hybrid membrane system composed of two steps of membrane separation for biogas desulfurization and upgrading: a rubbery membrane for separate H2S/CH4, and another is a glassy membrane for selective CO2/CH4. The raw biogas are composed of 60% (v/v) methane, and 2500 ppmv or 4% (v/v), two hydrogen sulfide concentrations in the feed gas were chosen for this work, the rest is carbon dioxide (39% or 36% v/v). The rubbery polymer PDMS [poly(dimethyl siloxane)], and Pebax® [poly(amide-6-b-ethylene oxide)] were used as the rubbery polymer, which exhibit higher H2S/CH4 selectivities (Pebax® of 54) than polyimide (12) because the polymer contains blocks with polar groups. In the second stage, polyimide glassy membrane is responsible for the removal of bulk carbon dioxide (see Table 38). Fig. 31 presents a simplified biogas upgrading process, which integrate rubbery and glassy membranes. The raw biogas was sent to rubbery membranes after compression. The retentate of the first step goes to the second stage without hydrogen sulfide and steam. Glassy membranes equipped in second step, separates CO2 and provides high-quality methane.
| Polymer | Selectivity (—) | |
|---|---|---|
| H2S/CH4 | CO2/CH4 | |
| Poly(dimethyl siloxane) | 10.5 | 3.4 |
| Poly(amide-6-b-ethylene oxide) | 54 | 12 |
| Poly(ether urethane) | 21 | 7.0 |
| Poly(ether urethane urea) | 74 | 17 |
| Cellulose acetate | 19 | 22 |
| Generic polyimide | 12 | 37 |
 | ||
| Fig. 31 A simplified process scheme including process integration of the hybrid membrane system for the desulfurization and upgrading of biogas. | ||
The authors conclude that this system can be effective for biogas upgrading. If relatively high H2S/CH4 selectivities of rubbery membranes are applied in the system, relatively low energy consumption and acceptable methane recovery will be reached and the final product will be able to satisfy natural gas grid standards (CH4 > 97% (v/v), CO2 < 2%, and H2S < 3.3 ppmv).
Scholz et al.270 used commercial polyimide membranes (Evonik Industries) which have CH4 permeance of 1 GPU, CO2 of 60 GPU, H2O of 300 GPU and H2S of 100 GPU combined with pressurized water scrubbing (in Fig. 32A and B), amine absorption (Fig. 32C and D), cryogenic separation (in Fig. 32E), and a combined heat and power engine (Fig. 32 CHP–F) at a pressure of 1 bar and a temperature of 20 °C. The raw gases were CH4 mole fraction of 60%, CO2 of 36.7%, H2O of 3% and H2S of 0.2%. The raw gas flow rates were 150–2000 m3 (STP) h−1. The product gas mole fractions were CH4 of 96%, H2O of 0.8% and H2S of 3 ppm at a pressure of 16 bar. Moreover, an individual three-step gas permeation process was installed (Fig. 32G). The specific upgrading costs were compared to conventional separation processes (Fig. 33).
 | ||
| Fig. 32 Membrane hybrid processes for which gas permeation technology is combined with pressurized water scrubbing (PWS1-2: a and b), amine scrubbing (amine 1-2: c and d) equipment, a cryogenic separation (cryogen: e), and a combined heat and power engine (CHP: f), three stage gas permeation process (g). | ||
 | ||
| Fig. 33 Annual operation costs splitted in annual operation costs and annual costs for investment (a) and CH4 recovery for the different hybrid processes at a feed flow rate of 1000 m3(STP) h−1 (b). The CH4 recovery of the hybrid combined heat and power process (CHP) is significantly lower compared to the other process configuration. | ||
From Fig. 33, it is clear that only three processes (PWS1:A, PWS2:B and CHP:F) have lower upgrading costs (total of operation and investment) than the three-stage membrane process. However, high CH4 losses was observed in the operating PWS 1. In general, PWS hybrid processes have higher investment costs than the three-stage membrane system because they include several different equipments. For heat and power process (CHP) hybrid processes, the CH4 recovery was very low. Therefore building such plant was not proposed. The non-hybrid three-stage gas permeation process (G) has low upgrading costs (investment and operation) and high CH4 recovery. Because of its simple design (a single compressor is needed), this configuration is highly attractive.
11. Conclusion
Biogas production is an effective and developed technology. However, its commercial use is limited because the biogas needs to be upgraded on-site before transportation or combustion. Membrane technology is a technology competing with other biogas purification processes. But, membrane separation may be combined with pressurized water scrubbing (PWS), amine swing absorption (AS), pressure swing adsorption (PSA), temperature swing adsorption (TSA), and cryogenic separation to clean up the biogas. These hybrid processes have lower investment and operation costs compared with single-step processes. Another interesting option is multi-membrane stage process which shows low investment and operation costs with high CH4 recovery.Membrane-based technology will likely be largely and frequently used in the future for biogas upgrading. It is therefore significant that scientists continue working on membrane development to obtain higher performance membranes. There are several avenues for this search of new membrane materials including neat polymers, neat inorganics and MMM. The following aspects should be more investigated.
First, a larger membrane material choice in needed. Only 8 or 9 polymers were discussed in the literature for 90% of the total gas separations. Further search towards new materials must include improvement of membrane materials with separation factor higher than 60 and adequate permeance, suppression of plasticization at high CO2 partial pressures, and enhanced long term stability of gas permeation systems. Current MMM consist of an organic polymer with inorganic (or organic) dispersed particles which may be a zeolite, carbon molecular sieve (CMS), carbon nanotubes (CNT), nano-size inorganic particles or metal–organic frameworks (MOF). MMM are interesting as they present higher selectivity, higher permeability or both, compared to existing polymer membranes. MMM also have increased mechanical properties and resistance to plasticization compared to neat polymer membranes because of the dispersed particles. MMM are thus believed to be a new type of membrane suitable for biogas upgrading.
Second, biogas may contain H2S, siloxanes or other volatile organics after filtration and condensation. This is why membrane-based biogas upgrading systems should separate simultaneously CO2/CH4 and H2S/CH4 using membranes based on different types of materials. Siloxanes may be removed by polydimethylsiloxane (PDMS), while CO2 can be removed by polyimides (PI).
Finally, membrane-based biogas upgrading systems must be further explored to provide easy operation and increased energy efficiency, using for example multi-stage membrane or hybrid processes which are more efficient and less costly for biogas upgrading.
References
- J. A. Alburquerque, C. de la Fuente, A. Ferrer-Costa, L. Carrasco, J. Cegarra, M. Abad and M. P. Bernal, Biomass Bioenergy, 2012, 40, 181 CrossRef CAS PubMed.
- http://www.ecovillage.org.in/, accessed on August, 2013.
- V. Uusitalo, J. Havukainen, K. Manninen, J. Höhn, E. Lehtonen, S. Rasi, R. Soukka and M. Horttanainen, Renewable Energy, 2014, 66, 90 CrossRef CAS PubMed.
- S. Rasi, A. Veijanen and J. Rintala, Energy, 2007, 32, 1375 CrossRef CAS PubMed.
- http://home.att.net/Bcat6a/fuels-VII.htm, accessed on August, 2013.
- S. Basu, A. L. Khan, A. Cano-Odena, C. Liu and I. F. J. Vankelecom, Chem. Soc. Rev., 2010, 39, 750 RSC.
- National Non-Food Crops Centre, NNFCC Renewable Fuels and Energy Factsheet: Anaerobic Digestion, Retrieved on 16 February 2011 Search PubMed.
- L. H. Pol, J. van Lier and G. Zeeman, in Proceedings de la Energía Sostenible en Andalucía: Nuevas Fuentes Sostenibles, Biogas, Seville, Spain, 2008 Search PubMed.
- D. Andrews, Biomethane fueled vehicles the carbon neutral option, Claverton Energy Conference, Bath, UK, 24 October 2009 Search PubMed.
- B. Bluemling, A. P. J. Mol and Q. Tu, Energy Policy, 2013, 63, 10–17 CrossRef PubMed.
- J. Coombs, in Critical Reports on Applied Chemistry, Anaerobic Digestion: A Waste Treatment Technology Applied Science, ed. A. Wheatley, Elsevier, London, 1990, Vol. 31, pp. 1–42 Search PubMed.
- L. Appels, J. Baeyens, J. Degrève and R. Dewil, Prog. Energy Combust. Sci., 2008, 34, 755 CrossRef CAS PubMed.
- Biogas & Engines, www.clarke-energy.com, (Accessed on November, 2013).
- A. Jalalzadeh-Azar, G. Saur and A. Lopez, Biogas Resources Characterization, FY 2010 Annual Progress Report, pp. 1191–1195 Search PubMed.
- P. J. He, Waste Manage., 2010, 30, 549 CrossRef CAS PubMed.
- University of Adelaide, http://www.adelaide.edu.au/biogas/history/, (Accessed on November, 2013).
- J. Q. Ni and E. J. Nyns, Energy Convers. Manage., 1996, 37, 1525 CrossRef.
- Y. Chen, G. Yang, S. Sweeney and Y. Feng, Energy Rev., 2010, 14, 545 Search PubMed.
- T. Bond and M. R. Templeton, Energy Sustainable Dev., 2011, 15, 347 CrossRef PubMed.
- http://news.mongabay.com/bioenergy/2007/07/study-eu-biogas-production-grew-136-in.html, (accessed on September 2013).
- K. Kayser and K. Backes, BioCycle, 2009, 50, 40 Search PubMed.
- Anaerobic Digest, BioCycle, 2013, 54, 18 Search PubMed.
- Anaerobic Digestion Strategy and Action Plan, http://defra.gov.uk. (accessed on September 2013).
- Z. Yue, C. Teater, Y. Liu, J. MacLellan and W. Liao, Biotechnol. Bioeng., 2010, 105, 10319 Search PubMed.
- D. R. Kashyap, K. S. Dadhich and S. K. Sharma, Bioresour. Technol., 2003, 87, 147 CrossRef CAS.
- P. Fongsatitkul, Ph.D. Thesis, The University of British Columbia, 1992.
- M. S. Switzenbaum, E. Giraldo-Gomez and R. F. Hickey, Enzyme Microb. Technol., 1990, 12, 722 CrossRef CAS.
- J. I. Huertas, N. Giraldo and S. Izquierdo, in Mass transfer in chemical engineering processes, ed. J. Markoš, Intech, 2011, pp. 133–135 Search PubMed.
- M. Harasimowicz, P. Orluk, G. Zakrzewska-Trznadel and A. G. Chmielewski, J. Hazard. Mater., 2007, 144, 698 CrossRef CAS PubMed.
- D. P. Gosh, Wet H2S cracking problem in oil refinery processes – Material selection and operation control issues, Tri-Service Corrosion Conference, Denver, Colorado, USA, 2007 Search PubMed.
- A. Wellinger and A. Lindberg, Biogas upgrading and utilization, In: IEA Bioenergy, Task 24: Energy from biological conversion of organic waste, IEA Bioenergy, 2000 Search PubMed.
- R. W. Baker, Membrane technology and applications, John Wiley & Sons Ltd., 2nd edn, 2004, pp. 302–304 Search PubMed.
- A. Petersson and A. Wellinger, Biogas upgrading technology-developments and innovations. In: IEA Bioenergy, Task 37: Energy from biogas and landfill gas, IEA Bioenergy, 2009 Search PubMed.
- P. Cozm, C. Ghinea, I. Mamaliga, W. Wukovits, A. Friedl and M. Gavrilescu, Clean: Soil, Air, Water, 2013, 41(9), 917 CrossRef.
- H. P. Huang, S. G. Chang and T. Dorchak, Energy Fuels, 2002, 16, 904 CrossRef CAS.
- Biogas to biomethane technology review, Task 3.1.1. Institute of chemical engineering research, Divison thermal process engineering and simulation, Vienna University of Technology. Intelligent Energy Europe: 2012.
- J. G. Jee, J. H. Jung, J. W. Lee, S. H. Suh and C. H. Lee, Rev. Roum. Chim., 2006, 51, 1095 CAS.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030 CAS.
- H. Emamipour, Z. Hashisho, D. Cevallos, M. J. Rood, D. L. Thurston and K. J. Hay, et al., Environ. Sci. Technol., 2007, 41, 5063 CrossRef CAS.
- P. Bernardo and G. Clarizia, Chem. Eng. Trans., 2013, 32, 1999 Search PubMed.
- M. Scholz, T. Melin and M. Wessling, Renewable Sustainable Energy Rev., 2013, 17, 199 CrossRef CAS PubMed.
- S. P. Nunes and K. V. Peinemann, in Membrane technology in the chemical industry, Wiley-VCH Verlag GmbH & Co. KGaA, 2nd edn, 2001, pp. 39–67 Search PubMed.
- D. Q. Vu, W. J. Koros and S. J. Miller, J. Membr. Sci., 2003, 211, 311 CrossRef CAS.
- T. S. Chung, L. Y. Jiang, Y. Li and S. Kulprathipanja, Prog. Polym. Sci., 2007, 32, 483 CrossRef CAS PubMed.
- X. Y. Chen, O. G. Nik, D. Rodrigue and S. Kaliaguine, Polymer, 2012, 53, 3269 CrossRef CAS PubMed.
- O. G. Nik, X. Y. Chen and S. Kaliaguine, J. Membr. Sci., 2011, 379, 468 CrossRef CAS PubMed.
- R. W. Baker, Ind. Eng. Chem. Res., 2002, 41, 1393 CrossRef CAS.
- R. W. Baker and K. Lokhandwala, Ind. Eng. Chem. Res., 2008, 47, 2109 CrossRef CAS.
- I. Pinnau and B. D. Freeman, in Membrane formation and modification, ACS Symposium Series, ed. I. Pinnau and B. D. Freeman, American Chemical Society, 1999, vol. 744, pp. 1–22 Search PubMed.
- J. P. Agrawal and S. Sourirajan, J. Appl. Polym. Sci., 1970, 14, 1303 CrossRef CAS.
- W. J. Koros and I. Pinnau, in Polymeric gas separation membranes, ed. D. R. Paul and Y. P. Yampolskii, CRC Press, Florida, 1994 Search PubMed.
- O. M. Ekiner, R. A. Hayes and P. Manos, US Pat., 5 085 676, 1992.
- C. E. Rogers, in Polymer permeability, ed. J. Comyn, Elsevier, London, 1985, ch. 2, pp. 11–73 Search PubMed.
- S. Sridhar, B. Smitha and T. M. Aminabhavi, Sep. Purif. Rev., 2007, 36, 113 CrossRef CAS.
- J. S. Chiou, Y. Maeda and D. R. Paul, J. Appl. Polym. Sci., 1987, 33, 1823 CrossRef CAS.
- K. Imai, T. Shiomi, Y. Tezuka and M. Takada, J. Appl. Polym. Sci., 1988, 35, 1817 CrossRef CAS.
- M. P. Chenar, M. Soltanieh, T. Matsuura, A. Tabe-Mohammadi and C. Feng, Sep. Purif. Technol., 2006, 51, 359 CrossRef CAS PubMed.
- X. Y. Chen, H. Vinh-Thang, D. Rodrigue and S. Kaliaguine, RSC Adv., 2013, 3, 24266 RSC.
- P. S. Tin, T. S. Chung, Y. Liu and R. Wang, Carbon, 2004, 42, 3123 CrossRef CAS PubMed.
- S. Sridhar, R. S. Veerapur, M. B. Patil, K. B. Gudasi and T. M. Aminabhavi, J. Appl. Polym. Sci., 2007, 106, 1585 CrossRef CAS.
- G. Dong, H. Li and V. Chen, J. Membr. Sci., 2010, 353, 17 CrossRef CAS PubMed.
- L. Jiang, T.-S. Chung, D. F. Li, C. Cao and S. Kulprathipanja, J. Membr. Sci., 2004, 240, 91 CrossRef CAS PubMed.
- T. Visser, N. Masetto and M. Wessling, J. Membr. Sci., 2007, 306, 16 CrossRef CAS PubMed.
- M. Khoshkam, M. Sadeghi, M. P. Chenar and M. J. NamaziFard, in Proceedings of the Polymer Processing Society Asia/Australia Regional Meeting, PPS-2011, Kish Island, Iran, 2011 Search PubMed.
- K. J. Kim, S. H. Park, W. W. So, D. J. Ahn and S. J. Moon, J. Membr. Sci., 2003, 211, 41 CrossRef CAS.
- C. Cao, R. Wang, T. S. Chung and Y. Liu, J. Membr. Sci., 2002, 209, 309 CrossRef CAS.
- X. Y. Chen and S. Kaliaguine, J. Appl. Polym. Sci., 2013, 128, 380 CrossRef CAS.
- D. R. Paul, Sep. Purif. Rev., 1976, 5, 33 CrossRef CAS.
- J. G. Wijmans and R. W. Baker, J. Membr. Sci., 1995, 107, 1 CrossRef CAS.
- T. Graham, Philos. Mag., 1866, 32, 401 Search PubMed.
- A. R. Berens and H. B. Hopfenberg, J. Membr. Sci., 1982, 10, 283 CrossRef CAS.
- S. Matteucci, Y. Yampolskii, B. D. Freeman and I. Pinnau, in Materials science of membranes for gas and vapor separation, ed. Y. Yampolskii, B. D. Freeman and I. Pinnau, John Wiley & Sons, Ltd, 2006, pp. 1–47 Search PubMed.
- S. von Wroblewski, J. Phys. Theor. Appl., 1879, 8, 418 Search PubMed.
- K. Ghosal and B. D. Freeman, Polym. Adv. Technol., 1994, 5, 673 CrossRef CAS.
- R. M. Felder and G. S. Huvard, in Methods of experimental physics, ed. R. A. Fava, Academic Press, New York, NY, USA, 1980, vol. 16C, pp. 315–377 Search PubMed.
- H. Lin and B. D. Freeman, in Springer-Handbook of Materials Measurement Methods, ed. H. Czichos, T. Saito and L. E. Smith, Springer-Verlag, Berlin, 2006, vol. 16C, ch. 7, pp. 371–387 Search PubMed.
- V. I. Bondar, B. D. Freeman, Y. P. Yam and H. polskii, Macromolecules, 1999, 32, 6163 CrossRef CAS.
- V. I. Bondar, B. D. Freeman and I. Pinnau, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 2463 CrossRef CAS.
- S. N. Dhoot, B. D. Freeman and M. E. Stewart, Ind. Eng. Chem. Res., 2004, 43, 2966 CrossRef CAS.
- D. T. Coker, B. D. Freeman and G. K. Fleming, AIChE J., 1998, 44, 1289 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390 CrossRef CAS PubMed.
- H. B. Park, C. H. Jung, Y. M. Lee, A. J. Hill, S. J. Pas, S. T. Mudie, E. van Wagner, B. D. Freeman and D. J. Cookson, Science, 2007, 318, 254 CrossRef CAS PubMed.
- W. J. Koros and M. W. Hellums, in Encyclopedia of polymer science and engineering, John Wiley & Sons, Inc., 1990, pp. 724–802 Search PubMed.
- A. Bos, I. G. M. Pünt, M. Wessling and H. Strathmann, J. Membr. Sci., 1999, 155, 67 CrossRef CAS.
- E. S. Sanders, J. Membr. Sci., 1988, 37, 63 CrossRef CAS.
- A. J. Erb and D. R. Paul, J. Membr. Sci., 1981, 8, 11 CrossRef CAS.
- G. S. Huvard, V. T. Stannett, W. J. Koros and H. B. Hopfenberg, J. Membr. Sci., 1980, 6, 185 CrossRef CAS.
- J. D. Wind, C. Staudt-Bickel, D. R. Paul and W. J. Koros, Ind. Eng. Chem. Res., 2002, 41, 6139 CrossRef CAS.
- M. D. Donohue, B. S. Minhas and S. Y. Lee, J. Membr. Sci., 1989, 42, 197 CrossRef CAS.
- W. J. Schell, C. D. Houston and W. L. Hopper, Separation of CO2 from mixtures by membrane permeation, 61st Gas Conditioning Conference, University of Oklahoma, Norman, 1983 Search PubMed.
- L. Wang, Y. Cao, M. Zhou, Q. Liu, X. Ding and Q. Yuan, Eur. Polym. J., 2008, 44, 225 CrossRef CAS PubMed.
- L. Wang, Y. Cao, M. Zhou, S. J. Zhou and Q. Yuan, J. Membr. Sci., 2007, 305, 338 CrossRef CAS PubMed.
- S. Xiao, X. Feng and R. Y. M. Huang, Polym. Eng. Sci., 2008, 48, 795 CAS.
- S. Xiao, R. Y. M. Huang and X. Feng, Polymer, 2007, 48, 5355 CrossRef CAS PubMed.
- M. Mikawa, S. Nagaoka and H. Kawakami, J. Membr. Sci., 1999, 163, 167 CrossRef CAS.
- J. Lillepärg, P. Georgopanos and S. Shishatskiy, J. Membr. Sci., 2014, 467, 269 CrossRef PubMed.
- K. Vanherck, I. Vankelecom and T. Verbiest, J. Membr. Sci., 2011, 373, 5 CrossRef CAS PubMed.
- L. Shao, L. Liu, S. X. Cheng, Y. D. Huang and J. Ma, J. Membr. Sci., 2008, 312, 174 CrossRef CAS PubMed.
- K. Vanherck, I. Vankelecom, A. Cano-Odena, G. Koeckelberghs and T. Dedroog, J. Membr. Sci., 2010, 353, 135 CrossRef CAS PubMed.
- X. Y. Chen, D. Rodrigue and S. Kaliaguine, Sep. Purif. Technol., 2012, 86, 221 CrossRef CAS PubMed.
- A. Bos, I. G. M. Punt, M. Wessling and H. Strathmann, Sep. Purif. Technol., 1998, 14, 27 CrossRef CAS.
- A. Bos, I. G. M. Punt, M. Wessling and H. Strathmann, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 1547 CrossRef CAS.
- A. Bos, I. G. M. Punt, H. Strathmann and M. Wessling, AIChE J., 2001, 47, 1088 CrossRef CAS.
- C. Zhou, T. S. Chung, R. Wang, Y. Liu and S. H. Goh, J. Membr. Sci., 2003, 225, 125 CrossRef CAS PubMed.
- H. Kita, T. Inada, K. Tanaka and K. Okamoto, J. Membr. Sci., 1994, 87, 139 CrossRef CAS.
- Y. Liu, R. Wang and T. S. Chung, J. Membr. Sci., 2001, 189, 231 CrossRef CAS.
- P. S. Tin, T. S. Chung, Y. Liu, R. Wang, S. L. Liu and K. P. Pramoda, J. Membr. Sci., 2003, 225, 77 CrossRef CAS PubMed.
- C. Staudt-Bickel and W. J. Koros, J. Membr. Sci., 1999, 155, 145 CrossRef CAS.
- J. D. Wind, C. Staudt-Bickel, D. R. Paul and W. J. Koros, Macromolecules, 2003, 36, 1882 CrossRef CAS.
- J. D. Wind, S. M. Sirard, D. R. Paul, P. F. Green, K. P. Johnston and W. J. Koros, Macromolecules, 2003, 36, 6433 CrossRef CAS.
- A. M. W. Hillock and W. J. Koros, Macromolecules, 2007, 40, 583 CrossRef CAS.
- S. Morooka and K. Kusakabe, Mater. Res. Soc. Bull., 1999, 24(3), 25 CAS.
- J. E. Koresh and A. Soffer, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 2472 RSC.
- Y. Cui, H. Kita and K. I. Okamoto, J. Mater. Chem., 2004, 14, 924 RSC.
- T. Tomita, K. Nakayama and H. Sakai, Microporous Mesoporous Mater., 2004, 68, 71 CrossRef CAS PubMed.
- G. Saracco and V. Speccia, Chem. Ind., 1998, 71, 463 CAS.
- Y. Dai, J. R. Johnson, O. Karvan, D. S. Sholl and W. J. Koros, J. Membr. Sci., 2012, 401–402, 76 CrossRef CAS PubMed.
- E. V. Perez, J. K. Balkus, J. P. Ferraris and I. H. Musselman, J. Membr. Sci., 2009, 328, 165 CrossRef CAS PubMed.
- H. Vinh-Thang and S. Kaliaguine, J. Membr. Sci., 2014, 452, 271 CrossRef CAS PubMed.
- J. M. Duval, Ph.D Thesis, University of Twente, 1995.
- Y. Li, T. S. Chung, C. Cao and S. Kulprathipanja, J. Membr. Sci., 2005, 260, 45 CrossRef CAS PubMed.
- M. Moaddeb and W. J. Koros, J. Membr. Sci., 1997, 125, 143 CrossRef CAS.
- R. Mahajan and W. J. Koros, Polym. Eng. Sci., 2002, 42, 1420 CAS.
- R. Mahajan and W. J. Koros, Polym. Eng. Sci., 2002, 42, 1432 CAS.
- G. Clarizia, C. Algieri and E. Drioli, Polymer, 2004, 45, 5671 CrossRef CAS PubMed.
- S. B. Tantekin-Ersolmaz, C. Atalay-Orala, M. Tather, A. Erdem-Senatalar, B. Schoeman and J. Sterte, J. Membr. Sci., 2000, 175, 285 CrossRef CAS.
- C. I. Chaidou, G. Pantoleontos, D. E. Koutsonikolas, S. P. Kaldis and G. P. Sakellaropoulos, Sep. Sci. Technol., 2012, 47, 950 CrossRef CAS.
- R. H. B. Bouma, A. Checchetti, G. Chidichimo and E. Drioli, J. Membr. Sci., 1997, 128, 141 CrossRef CAS.
- J. Caro and M. Noack, Microporous Mesoporous Mater., 2008, 115, 215 CrossRef CAS PubMed.
- A. Corma, Adv. Mater., 1995, 7, 137 CrossRef CAS.
- C. Baerlocher, L. B. McCusker and D. H. Olson, Atlas of zeolite framework types, 6th edn, Elsevier, Amsterdam, 2007 Search PubMed.
- M. Hong, Ph.D. Thesis, University of Colorado, 2007.
- D. W. Breck, Zeolite molecular sieves, structure, chemistry and use, John Wiley & Sons Inc., New York, 1974, pp. 593–724 Search PubMed.
- O. Talu, S. Y. Zhang and D. T. Hayhurst, J. Phys. Chem., 1993, 97, 12894 CrossRef CAS.
- D. R. Paul and D. R. Kemp, J. Polym. Sci., Polym. Symp., 1973, 41, 79 CrossRef.
- M. Jia, K. V. Peinemann and R.-D. Behling, J. Membr. Sci., 1991, 57, 289 CrossRef CAS.
- J. M. Duval, B. Folkers, M. H. V. Mulder, G. Desgrandchamps and C. A. Smolders, J. Membr. Sci., 1993, 80, 189 CrossRef CAS.
- J. M. Duval, A. J. B. Kemperman, B. Folkers, M. H. V. Mulder, G. Desgrandchamps and C. A. Smolder, J. Membr. Sci., 1994, 54, 409 CAS.
- H. H. Yong, H. C. Park, Y. S. Kang, J. Won and W. N. Kim, J. Membr. Sci., 2001, 188, 151 CrossRef CAS.
- T. W. Pechar, S. Kim and B. Vaughan, J. Membr. Sci., 2006, 277, 195 CrossRef CAS PubMed.
- T. W. Pechar, M. Taspatsis, E. Marand and R. Davis, Desalination, 2002, 146, 3 CrossRef CAS.
- A. M. W. Hillock, S. J. Miller and W. J. Koros, J. Membr. Sci., 2008, 314, 193 CrossRef CAS PubMed.
- D. Bastani, N. Esmaeili and M. Asadollahi, J. Ind. Eng. Chem., 2013, 19, 375 CrossRef CAS PubMed.
- S. J. Miller, C. L. Munson, S. S. Kulkarni, J. Hasse and J. David, US Pat., 6 500 233, 2002.
- O. M. Ekiner and S. S. Kulkarni, US Pat., 6 663 805, 2003.
- W. J. Koros, D. Wallace, J. D. Wind, S. J. Miller, C. S. Bickel and D. Q. Vu, US Pat., 20 030 140 789, 2003.
- S. Husain and W. J. Koros, J. Membr. Sci., 2007, 288, 195 CrossRef CAS PubMed.
- A. F. Ismail, T. D. Kusworo and A. Mustafa, J. Membr. Sci., 2008, 319, 306 CrossRef CAS PubMed.
- J. M. C. Ordonnez Jr, K. J. Balkus, J. P. Ferraris and I. H. Musselman, J. Membr. Sci., 2010, 361, 28 CrossRef PubMed.
- Y. Li, T. S. Chung and S. Kulprathipanja, AIChE J., 2007, 53, 610 CrossRef CAS.
- Y. Zhang Jr, K. J. Balkus, I. H. Musselman and J. P. Ferraris, J. Membr. Sci., 2008, 325, 28 CrossRef PubMed.
- A. LaeeqKhan, C. Klaysom, A. Gahlaut, A. UllahKhan and I. J. Vankelecom, J. Membr. Sci., 2013, 447, 73–79 CrossRef PubMed.
- Y. Li, T. S. Chung, Z. Huang and S. Kulprathipanja, J. Membr. Sci., 2006, 277, 28 CrossRef CAS PubMed.
- U. Cakal, L. Yilmaz and H. Kalipcilar, J. Membr. Sci., 2012, 417–418, 45 CrossRef CAS PubMed.
- R. T. Adams, J. S. Lee, T. H. Bae, J. K. Ward, J. R. Johnson, C. W. Jones, S. Nair and W. J. Koros, J. Membr. Sci., 2011, 367, 197 CrossRef CAS PubMed.
- B. Ozturk and F. Demirciyeva, Chem. Eng. J., 2013, 222, 209 CrossRef CAS PubMed.
- L. Y. Jiang, T. S. Chung and S. Kulprathipanja, AIChE J., 2006, 52, 2898 CrossRef CAS.
- N. Widjojo, T.-S. Chung and S. Kulprathipanja, J. Membr. Sci., 2008, 325, 326 CrossRef CAS PubMed.
- R. Xing and W. S. W. Ho, J. Taiwan Inst. Chem. Eng., 2009, 40, 654 CrossRef CAS PubMed.
- A. F. Ismail and L. I. B. David, J. Membr. Sci., 2001, 193, 1 CrossRef CAS.
- M. B. Hägg, J. A. Lie and A. Lindbråthen, Ann. N. Y. Acad. Sci., 2003, 984, 329 CrossRef PubMed.
- P. J. Williams and W. J. Koros, in Advanced membrane technology and applications, ed. N. N. Li, A. G. Fane, W. S. WinstonHo and T. Matsuura, John Wiley & Sons, Inc., New Jersey, 2008, pp. 599–631 Search PubMed.
- M. G. Buonomenna, W. Yave and G. Golemme, RSC Adv., 2012, 2, 10745 RSC.
- S. M. Saufi and A. F. Ismail, Carbon, 2004, 42, 241 CrossRef CAS PubMed.
- J. E. Koresh and A. Sofer, Sep. Sci. Technol., 1983, 18, 723 CrossRef CAS.
- C. W. Jones and W. J. Koros, Carbon, 1994, 32, 1419 CrossRef CAS.
- D. Q. Vu, W. J. Koros and S. J. Miller, Ind. Eng. Chem. Res., 2001, 41, 367 CrossRef.
- D. Q. Vu, W. J. Koros and S. J. Miller, J. Membr. Sci., 2003, 211, 335 CrossRef CAS.
- D. Q. Vu, W. J. Koros and S. J. Miller, J. Membr. Sci., 2003, 221, 233 CrossRef CAS.
- W. A. W. Rafizah and A. F. Ismail, J. Membr. Sci., 2008, 307, 53 CrossRef CAS PubMed.
- M. Das, J. D. Perry and W. J. Koros, Ind. Eng. Chem. Res., 2010, 49, 9310 CrossRef CAS.
- A. I. Skoulidas, D. M. Ackerman, J. K. Johnson and D. S. Sholl, Phys. Rev. Lett., 2002, 89, 185901 CrossRef.
- P. M. Ajayan and O. Z. Zhou, in Carbon nanotubes, ed. M. Dresselhaus, G. Dresselhaus and P. Avouris, Springer, Berlin Heidelberg, 2001, pp. 391–425 Search PubMed.
- A. Aqel, K. M. M. A. El-Nour, R. A. A. Ammar and A. Al-Warthan, Arabian J. Chem., 2012, 5, 1 CrossRef CAS PubMed.
- A. F. Ismail, P. S. Goh, S. M. Sanip and M. Aziz, Sep. Purif. Technol., 2009, 70, 12 CrossRef CAS PubMed.
- B. J. Hinds, N. Chopra, T. Rantell, R. Andrews, V. Gavalas and L. G. Bachas, Science, 2004, 303, 62 CrossRef CAS PubMed.
- K. MacKenzie, O. Dunens and A. T. Harris, Sep. Purif. Technol., 2009, 66, 209 CrossRef CAS PubMed.
- S. Kim, T. W. Pechar and E. Marand, Desalination, 2006, 192, 330 CrossRef CAS PubMed.
- S. Kumar, S. Srivastava and Y. K. Vijay, Int. J. Hydrogen Energy, 2012, 37, 3914 CrossRef CAS PubMed.
- M. Nour, K. Berean, S. Balendhran, J. Z. Ou, J. Du Plessis, C. McSweeney, M. Bhaskaran, S. Sriram and K. Kalantar-zadeh, Int. J. Hydrogen Energy, 2013, 38, 10494 CrossRef CAS PubMed.
- H. Cong, J. Zhang, M. Radosz and Y. Shen, J. Membr. Sci., 2007, 294, 178 CrossRef CAS PubMed.
- T. H. Weng, H. H. Tseng and M. Y. Wey, Int. J. Hydrogen Energy, 2009, 34, 8707 CrossRef CAS PubMed.
- A. R. Moghadassi, Z. Rajabi, S. M. Hosseini and M. Mohammadi, Sep. Sci. Technol., 2013, 48, 1261 CrossRef CAS.
- Z. Rajabi, A. R. Moghadassi, S. M. Hosseini and M. Mohammadi, J. Ind. Eng. Chem., 2013, 19, 347 CrossRef CAS PubMed.
- L. Ge, Z. Zhu and V. Rudolph, Sep. Sci. Technol., 2011, 78, 76 CAS.
- L. Ge, Z. Zhu, F. Li, S. Liu, L. Wang, X. Tang and V. Rudolph, J. Phys. Chem. C, 2011, 115, 6661 CAS.
- S. Kim, L. Chen, J. K. Johnson and E. Marand, J. Membr. Sci., 2007, 294, 147 CrossRef CAS PubMed.
- A. Mustafa, T. D. Kusworo, A. Busairi, A. F. Ismail and P. Budiyono, Int. J. Sci. Eng., 2010, 1, 15 Search PubMed.
- A. F. Ismail, N. H. Rahim, A. Mustafa, T. Matsuura, B. C. Ng, S. Abdullah and S. A. Hashemifard, Sep. Purif. Technol., 2011, 80, 20 CrossRef CAS PubMed.
- M. M. Khan, V. Filiz, G. Bengtson, S. Shishatskiy, M. M. Rahman, J. Lillepaerg and V. Abetz, J. Membr. Sci., 2013, 436, 109 CrossRef CAS PubMed.
- P. S. Goh, B. C. Ng, A. F. Ismail, S. M. Sanip, M. Aziz and M. A. Kassim, Sep. Sci. Technol., 2011, 46, 1250 CrossRef CAS.
- M. A. Aroon, A. F. Ismail, M. M. Montazer-Rahmati and T. Matsuura, J. Membr. Sci., 2010, 364, 309 CrossRef CAS PubMed.
- S. M. Sanip, A. F. Ismail, P. S. Goh, T. Soga, M. Tanemura and H. Yasuhiko, Sep. Purif. Technol., 2011, 78, 208 CrossRef CAS PubMed.
- M. A. Aroon, A. F. Ismail and T. Matsuura, Sep. Purif. Technol., 2013, 115, 39 CrossRef CAS PubMed.
- A. L. Ahmad, Z. A. Jawad, S. C. Low and S. H. S. Zein, J. Membr. Sci., 2014, 451, 55 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed.
- H. Vinh-Thang and S. Kaliaguine, in Coordination Polymers and Metal Organic Frameworks: Properties, Types and Applications, Chemical Engineering Methods and Technology Series, ed. O. L. Ortiz and L. D. Ramírez, Nova Science Publishers, Hauppauge, New York, 2011, Ch. 4 Search PubMed.
- O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703 CrossRef CAS.
- H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523 CrossRef CAS PubMed.
- H. Yehia, T. J. Pisklak, J. P. Ferraris, K. J. Balkus and I. H. Musselman, Abstr. Pap., Jt. Conf. - Chem. Inst. Can. Am. Chem. Soc., 2004, 227, U351 Search PubMed.
- A. Car, C. Stropnik and K.-V. Peinemann, Desalination, 2006, 200, 424 CrossRef CAS PubMed.
- Y. F. Zhang, I. H. Musselman, J. P. Ferraris and K. J. Balkus, J. Membr. Sci., 2008, 313, 170 CrossRef CAS PubMed.
- S. Keskin and D. S. Sholl, Langmuir, 2009, 25, 11786 CrossRef CAS PubMed.
- C. Liu, S. Kulprathipanja and S. T. Wilson, US pat., 020 950 5A1, 2007.
- C. Liu, B. McCulloch, S. T. Wilson, A. I. Benin and M. E. Schott, US pat., 763 798 3B1, 2009.
- R. Adams, C. Carson, J. Ward, R. Tannenbaum and W. Koros, Microporous Mesoporous Mater., 2010, 131, 13 CrossRef CAS PubMed.
- S. Keskin and D. S. Sholl, Energy Environ. Sci., 2010, 3, 343 CAS.
- S. Keskin, J. Phys. Chem. C, 2010, 114, 13047 CAS.
- S. Basu, A. Cano-Odena and I. F. J. Vankelecom, J. Membr. Sci., 2010, 362, 478 CrossRef CAS PubMed.
- J. Hu, H. Cai, H. Ren, Y. Wei, Z. Xu, H. Liu and Y. Hu, Ind. Eng. Chem. Res., 2010, 49, 12605 CrossRef CAS.
- O. G. Nik, X. Y. Chen and S. Kaliaguine, J. Membr. Sci., 2012, 412–413, 48 CrossRef PubMed.
- I. H. Musselman Jr, K. J. Balkus and J. P. Ferraris, Mixed-matrix membranes for CO2 and H2 gas separations using metal–organic frameworks and mesoporous hybrid silica, Report DE-FG26-04NT42173, The University of Texas at Dallas, 2008 Search PubMed.
- E. V. Perez, Ph.D Thesis, University of Texas at Dallas, 2009.
- M. J. C. Ordoñez, M.S Thesis, University of Texas at Dallas, 2009.
- K. Díaz, L. Garrido, M. López-González, L. F. del Castillo and E. Riande, Macromolecules, 2009, 43, 316 CrossRef.
- T. H. Bae, J. S. Lee, W. Qiu, W. J. Koros, C. W. Jones and S. Nair, Angew. Chem. Int. Ed., 2010, 49, 9863 (Angew. Chem., 2010, 122, 10059) CrossRef CAS PubMed.
- X. Y. Chen, H. Vinh-Thang, D. Rodrigue and S. Kaliaguine, Ind. Eng. Chem. Res., 2012, 51, 6895 CrossRef CAS.
- B. Zornoza, A. Martinez-Joaristi, P. Serra-Crespo, C. Tellez, J. Coronas, J. Gascon and F. Kapteijn, Chem. Commun., 2011, 47, 9522 RSC.
- Q. Song, S. K. Nataraj, M. V. Roussenova, J. C. Tan, D. J. Hughes, W. Li, P. Bourgoin, M. A. Alam, A. K. Cheetham, S. A. Al-Muhtaseb and E. Sivaniah, Energy Environ. Sci., 2012, 5, 8359 CAS.
- P. M. Budd, E. S. Elabas, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib, C. E. Tattershall and D. Wang, Adv. Mater., 2004, 16, 456 CrossRef CAS.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 2, 230 RSC.
- P. M. Budd, N. B. McKeown and D. Fritsch, Macromol. Symp., 2006, 245, 403 CrossRef.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675 RSC.
- B. S. Ghanem, N. B. McKeown, P. M. Budd and D. Fritsch, Macromolecules, 2008, 41, 1640 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163 CrossRef CAS.
- A. F. Bushell, M. P. Attfield, C. R. Mason, P. M. Budd, Y. Yampolskii, L. Starannikova, A. Rebrov, F. Bazzarelli, P. Bernardo, J. C. Jansen, M. Lanc, K. Friess, V. Shantarovich, V. Gustov and V. Isaeva, J. Membr. Sci., 2013, 427, 48 CrossRef CAS PubMed.
- J. Ahn, W. J. Chunga, I. Pinnau, J. Song, N. Du, G. P. Robertson and M. D. Guiver, J. Membr. Sci., 2010, 346, 280 CrossRef CAS PubMed.
- M. M. Khan, V. Filiz, G. Bengtson, S. Shishatskiy, M. Rahman and V. Abetz, Nanoscale Res. Lett., 2012, 7, 504 CrossRef PubMed.
- W. F. Yong, F. Y. Li, Y. C. Xiao, P. Li, K. P. Pramoda, Y. W. Tong and T. S. Chung, J. Membr. Sci., 2012, 407–408, 47 CrossRef CAS PubMed.
- W. F. Yong, F. Y. Li, Y. C. Xiao, T. S. Chung and Y. W. Tong, J. Membr. Sci., 2013, 443, 156 CrossRef CAS PubMed.
- L. Hao, P. Li and T. S. Chung, J. Membr. Sci., 2014, 453, 614 CrossRef CAS PubMed.
- C. Liu and S. T. Wilson, US Pat., 7 410 525, 2008.
- N. B. McKeown, ISRN Mater. Sci., 2012, 513986 Search PubMed.
- P. M. Budd, K. J. Msayib, C. E. Tattershall, B. S. Ghanem, K. J. Reynolds, N. B. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263 CrossRef CAS PubMed.
- P. M. Budd, N. B. McKeown, B. S. Ghanem, K. J. Msayib, D. Fritsch, L. Starannikova, N. Belov, O. Sanfirova, Y. Yampolskii and V. Shantarovich, J. Membr. Sci., 2008, 325, 851 CrossRef CAS PubMed.
- M. M. Khan, G. Bengtson, S. Shishatskiy, B. N. Gacal, M. M. Rahman, S. Neumann, V. Filiz and V. Abetz, Eur. Polym. J., 2013, 49, 4157 CrossRef CAS PubMed.
- S. Thomas, I. Pinnau, N. Du and M. D. Guiver, J. Membr. Sci., 2009, 333, 125 CrossRef CAS PubMed.
- L. A. Pessan, W. J. Koros, J. C. Schmidhauser and W. D. Richards, J. Polym. Sci., Polym. Phys. Ed., 1995, 33, 487 CrossRef CAS.
- J. S. McHattie, W. J. Koros and D. R. Paul, Polymer, 1991, 32, 840 CrossRef CAS.
- C. G. Bezzu, M. Carta, A. Tonkins, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Adv. Mater., 2012, 24, 5930 CrossRef CAS PubMed.
- M. Scholz, M. Wessling and J. Balster, in Membrane engineering for the treatment of gases, ed. E. Drioli and G. Barbieri, The Royal Society of Chemistry, 2011, vol. 1, ch. 5, pp. 125–149 Search PubMed.
- E. Drioli and L. Giorno, in Comprehensive membrane science and engineering, ITM-CNR, University of Calabria, Rende (CS), Italy: Elsevier Science, 2010 Search PubMed.
- I. Moch Jr, in Kirk-Othmer Encyclopedia of Chemical Technology, 4th edn, John Wiley-InterScience Publishing, New York, 1995, vol. 13, p. 312 Search PubMed.
- S. Loeb and S. Sourirajan, US Pat., 3 133 132, 1964.
- I. Mahon, US Pat., 3 228 876, 1966.
- D. R. Lloyd, K. E. Kinzer and H. S. Tseng, J. Membr. Sci., 1990, 52, 239 CrossRef CAS.
- W. S. W. Ho and K. K. Sirkar, Membrane Handbook, Van Nostrand Reinhold, New York, 1992 Search PubMed.
- J. Koros and I. Pinnau, in Polymeric gas separation membranes, ed. D. R. Paul and Y. P. Yampol’skii, CRC Press, Boca Raton, FL, USA, 1994, ch. 5, pp. 209–272 Search PubMed.
- G. G. Lipscomb, Polym. Adv. Technol., 1994, 5, 745 CrossRef CAS.
- I. M. Wienk, R. M. Boom, M. A. M. Beerlage, A. M. W. Bulte, C. A. Smolders and H. Strathmann, J. Membr. Sci., 1996, 113, 361 CrossRef CAS.
- T. S. Chung, Polym. Polym. Compos., 1996, 4, 269 CAS.
- D. J. Stookey, in Membrane technology in the chemical industry, ed. S. P. Nunes and K.V. Peinemann, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006, ch. 2, pp. 119–147 Search PubMed.
- N. Peng, N. Widjojo, P. Sukitpaneenit, M. M. Teoh, G. G. Lipscomb, T.-S. Chung and J.-Y. Lai, Prog. Polym. Sci., 2012, 37, 1401 CrossRef CAS PubMed.
- N. Peng, T.-S. Chung and J.-Y. Lai, J. Membr. Sci., 2009, 326, 608 CrossRef CAS PubMed.
- T. S. Chung and E. R. Kafchinski, J. Appl. Polym. Sci., 1997, 65, 1555 CrossRef CAS.
- T. S. Chung, J. Membr. Sci., 1997, 126, 19–34 CrossRef CAS.
- N. Peng, T. S. Chung and K. Y. Wang, J. Membr. Sci., 2008, 318, 363 CrossRef CAS PubMed.
- R. W. Baker, in Advanced membrane technology and applications, ed. N. N. Li, A. G. Fane, W. S. Winston Ho and T. Matsuura, John Wiley & Sons, Inc., New Jersey, 2008, pp. 557–580 Search PubMed.
- R. Dewil, L. Appels and J. Baeyens, Energy Convers. Manage., 2006, 47, 1711 CrossRef CAS PubMed.
- M. Ajhar and T. Melin, Desalination, 2006, 200, 234 CrossRef CAS PubMed.
- N. Abatzoglou and S. Boivin, Biofuels, Bioprod. Biorefin., 2009, 3, 42 CrossRef CAS.
- L. Deng and M.-B. Hägg, Int. J. Greenhouse Gas Control, 2010, 4, 638 CrossRef CAS PubMed.
- A. Makaruk and M. Harasek, J. Membr. Sci., 2009, 344, 258 CrossRef CAS PubMed.
- R. Rautenbach and K. Welsch, J. Membr. Sci., 1994, 87, 107 CrossRef CAS.
- B. D. Bhide, A. Voskericyan and S. A. Stern, J. Membr. Sci., 1998, 140, 27 CrossRef CAS.
- P. Shao, M. Dal-Cin, A. Kumar, H. Li and D. P. Singh, J. Membr. Sci., 2012, 413–414, 17 CrossRef CAS PubMed.
- A. Makaruk, M. Miltner and M. Harasek, Sep. Purif. Technol., 2010, 74, 83 CrossRef CAS PubMed.
- A. Makaruk, M. Miltner and M. Harasek, Water Sci. Technol., 2013, 67, 326 CrossRef CAS PubMed.
- M. Scholz, B. Frank, F. Stockmeier, S. Falß and M. Wessling, Techno-economic analysis of hybrid processes for biogas upgrading, Ind. Eng. Chem. Res., 2013, 52, 16929 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |