
Sorption of radionuclides from aqueous systems onto graphene oxide-based materials: a review
Shujun
Yu
ab,
Xiangxue
Wang
ab,
Xiaoli
Tan
*acd and
Xiangke
Wang
*cdef
aInstitute of Plasma Physics, Chinese Academy of Sciences, P.O. Box 1126, Hefei, 230031, P.R. China. E-mail: tanxl@ipp.ac.cn
bUniversity of Science and Technology of China, Hefei, 230026, P.R. China
cSchool for Radiological and Interdisciplinary Sciences (RAD-X), Soochow University, Suzhou, 215123, P.R. China
dCollaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, P.R. China
eFaculty of Engineering, King Abdulaziz University, Jeddah 21589, Saudi Arabia
fSchool of Environment and Chemical Engineering, North China Electric Power University, Beijing, 102206, P.R. China. E-mail: xkwang@ipp.ac.cn; xkwang@suda.edu.cn
First published on 30th March 2015
Abstract
Graphene oxide (GO), one of the most important graphene derivatives, has many oxygen-containing functional groups on its basal plane and on the edges in the form of epoxy, hydroxyl and carboxyl groups. It has attracted increasing interest in multidisciplinary research because of its unique structure and exceptional physicochemical properties. In particular, GO-based materials have great potential in environmental remediation and energy applications. Herein, we review the recent advances in GO-based materials for the sorption of radionuclides, mainly from the last decade. This review summarizes the preparation of GO-based materials and their application in the sorption of radionuclides (such as U(VI), Eu(III), Sr(II), etc.) from aqueous systems. The main sorption mechanisms are investigated using kinetic analysis, thermodynamic analysis, surface complexation models, spectroscopic techniques and theoretical calculations. It is evident that GO-based materials have good potential for the removal of radionuclides from aqueous systems. However, it is necessary to carry out more research focusing on the development of lower cost, higher efficiency and more environmentally friendly GO-based materials, either for scientific interest or practical applications.
 Shujun Yu | Shujun Yu received her BE in 2012 and ME degree in 2014 from the University of Jinan, P. R. China. She is currently a PhD candidate at the Institute of Plasma Physics, Chinese Academy of Sciences. Her interests include the synthesis of graphene-based materials, and their application in radioactive contamination treatment. |
 Xiangxue Wang | Xiangxue Wang received his ME degree in 2010 from Xi'an University of Architecture and Technology, P. R. China. He is currently a PhD candidate at the Institute of Plasma Physics, Chinese Academy of Sciences. His interests include the synthesis of graphene-based nanomaterials, and their application in pollution management. |
 Xiaoli Tan | Xiaoli Tan is an associate professor of the Institute of Plasma Physics, Chinese Academy of Sciences. She received her BS from Lanzhou University in 2001 and PhD from the institute in 2009. She focuses on the application of nanomaterials in the field of nuclear waste management, and also radionuclide physicochemical behavior in the environment. She has published over 30 papers in peer-review journals. |
 Xiangke Wang | Xiangke Wang is a professor of the School of Environment and Chemical Engineering, North China Electric Power University. He received his BS in 1995 and his PhD in 2000 from Lanzhou University. Then he joined the SUBATECH Laboratory (France) as a research fellow and the Karlsruhe Research Center (Germany) as an Alexander von Humboldt research fellow. He focuses on the synthesis of nanomaterials and their applications in energy and environmental pollution management, and also radionuclide physicochemical behavior in the environment. He has published over 180 papers in peer-review journals, and his H-index is now 61. |
1. Introduction
Nuclear energy is currently undergoing rapid development and attracting broad research effort with the increase in energy demand. It has also been regarded as a reasonable solution for global climate change and the shortage of fossil fuels. In the meanwhile, large amounts of radioactive waste are worryingly released into the environment due to the nuclear fuel cycle and mining operations, which cause a long-term threat to the surface and subsurface environments.1 As a result of the earthquake and tsunami on the east coast of Japan on 11 March 2011, water as high as 15 m inundated the Fukushima Daiichi Nuclear Power Plant, resulting in one of the most important releases of artificial radionuclides into the environment.2 Leakage of radionuclides, such as 90Sr, 137Cs, 235U, and 129I, has aroused serious public concern because the fission products can enter the food chain from fresh water systems. Such radionuclides in contaminated water with high mobility would move into soils and enter plants, ultimately becoming a part of animals and human beings.3 For work safety in the nuclear industry and for human health, investigations into fast and efficient removal of long-lived radionuclides from nuclear wastewater have great scientific and practical value.4A variety of technologies have been investigated as potential means of attenuating radionuclide concentration, such as precipitation, membrane filtration, sorption, ion exchange, etc.5–9 Among these methods, the sorption technique has been widely used because it is easily carried out, economical and can be applied on a large scale for practical applications. Some adsorbents, such as clay minerals10–15 and oxides16–20 have been studied intensively for the removal radionuclides from aqueous systems. However, the low sorption capacities or efficiencies of these materials have obviously restricted their applications.
Carbon materials, with their high specific surface areas, have been one of the most effective materials for environmental treatment, and have attracted great attention recently. Mauter and Elimelech reviewed the properties and possible environmental applications of carbon nanomaterials, including carbon nanotubes, nanodiamonds, fullerenes, graphene and other carbon materials.21 Among them, graphene was well-studied and proved to be non-toxic and biodegradable.22–26 The remarkable properties of graphene reported so far include high values of its Young's modulus (∼1100 GPa),27 fracture strength (125 GPa),27 thermal conductivity (∼5000 W (m K)−1),28 mobility of charge carriers (200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 (V s)−1),29 specific surface area (theoretical calculated value of 2630 m2 g−1)30 and fascinating transport phenomena such as the quantum hall effect.31 Graphene oxide (GO), one of the most important graphene derivatives, has many oxygen-containing functional groups on its basal plane and on the edges in the form of epoxy, hydroxyl, and carboxyl groups.32 These oxygen-containing groups can bind metal ions and organic pollutants through coordination, electrostatic interaction, hydrogen bonding, etc., which ensures its potential application in environmental pollution control and remediation.33–37
000 cm2 (V s)−1),29 specific surface area (theoretical calculated value of 2630 m2 g−1)30 and fascinating transport phenomena such as the quantum hall effect.31 Graphene oxide (GO), one of the most important graphene derivatives, has many oxygen-containing functional groups on its basal plane and on the edges in the form of epoxy, hydroxyl, and carboxyl groups.32 These oxygen-containing groups can bind metal ions and organic pollutants through coordination, electrostatic interaction, hydrogen bonding, etc., which ensures its potential application in environmental pollution control and remediation.33–37
GO-based materials provide a high surface area, abundance of surface functional groups (such as epoxides, carboxyls and hydroxyls) and a low specific mass, especially when compared to low cost materials such as clay minerals and oxides.38 This in turn provides GO-based materials with an optimal sorption ability for pollutants. The special functional groups can provide particular sorption sites for selective removal of radionuclides. Compared with biomaterials, viz. algae, fungi, bacteria, plant biomass, etc., the GO-based materials are easier to process and easily functionalized. In addition, GO tends to be more hydrophilic than graphene due to GO having oxygen-containing functional groups that may form stable complexes with many contaminants. A large number of research articles have reported the sorption of small molecules,39–41 heavy metal ions,42–44 radionuclides45–47 and organic chemicals48–50 on GO-based materials. Wang's group has done lots of useful work to develop GO-based materials. They successfully synthesized many kinds of GO-based materials (functionalized GO51–55 and GO-based composites56–58) and used them for environmental remediation. Chai's group studied the interaction mechanisms of radionuclides and GO-based materials using spectroscopy technology combined with theoretical calculations.59–61 There are also some patents on the application of GO-based materials in environmental remediation.62,63 Tour et al. investigated the kinetics, pH sorption edges and isotherms of sorption of radionuclides (such as U(VI), Am(III), Sr(II) and Pu(IV)) onto GO and applied for a patent for the study.62 The CO2 sorption capacity of layered double hydroxide (LDH) was increased by supporting LDH with graphene or GO aerogels and xerogels. Gallastegui et al. claimed a patent on the preparation of graphene or graphene oxide aerogels and xerogels containing LDH.63
Since the first review of graphene in 2005, there have been more than 400 reviews published in this field, revealing the growing enthusiasm for this important and significant material.64 For example, Wang et al. reviewed predominantly the recent developments of graphene-based materials and demonstrated their enhanced performance in the sorption of organic compounds, metal ions, and solid phase extraction as well as in separation science since 2012.65 Kemp et al. reviewed how graphene and its derivatives were applied in pollution management with an emphasis on gas sorption and water remediation.66 Wang et al. discussed the research into graphene-based materials as adsorbents for the removal of various types of contaminants in air and water systems.67 Wang et al. reviewed the recent development of plasma synthesis of graphene-based materials and their electrochemical application in fuel cells.68
Pollution caused by radionuclides is a serious problem throughout the world. To solve this problem, many researchers have studied the sorption of radionuclides.69–74 Kepák reviewed the sorption and isotopic exchange of I2 and CH3I on activated charcoal, inorganic adsorbents, amorphous silicic acid and aluminium oxide.69 Macášek reported the composites of natural microporous materials (bentonite clays) and their application in the removal of radionuclides.70 Li et al. studied the sorption coefficients and molecular mechanisms of Pu, U, Np, Am and Tc of sorption by Fe (hydr)oxides (hematite, magnetite, goethite and ferrihydrite).71 Tan et al. evaluated the speciation of lanthanides, mainly Eu(III), on natural or synthetic mineral surfaces, and the available analytical techniques.74
Although there are many reviews on graphene and the removal of radionuclides respectively,64–77 a review of GO-based materials as adsorbents for the removal of radionuclides is still lacking. This review introduces the commonly used GO-based materials, followed by placing the emphasis on recent advancements in GO-based materials as adsorbents in the decontamination of radionuclides, mainly from the last 10 years. Meanwhile, the sorption mechanisms for the removal of radionuclides have been highlighted. Finally, challenges and outlooks are offered to inspire more exciting developments in this promising field.
2. Preparation of GO-based materials
The discovery of graphene by Andre Geim and co-workers at the University of Manchester by using the deceptively simple scotch tape method has caused a revolution in the scientific community.78,79 Graphene has attracted great attention due to its intrinsic 2 dimensional structure, outstanding physical and chemical properties, and its promising potential applications in the areas of materials science, electronic science, biomedicine, catalyst carriers, etc.80–83 However, the practical applications of graphene were greatly hampered by its preparation and dispersity in solvents. Many strategies, including mechanical exfoliation,78 liquid-phase exfoliation,84 chemical vapor deposition,85 and reduction of GO,86 have been developed for the production of graphene. The first three methods can produce graphene with a relatively perfect structure and excellent properties. In comparison, GO has two important characteristics: (I) it can be produced by cost-effective chemical methods, and (II) it is highly hydrophilic and can form stable aqueous colloids to facilitate the assembly of macroscopic structures by simple and cheap solution processes, both of which are important in the large-scale usage of graphene.87 Furthermore, GO has abundant oxygen-containing functional groups, such as hydroxyl, epoxy, carbonyl and carboxyl groups (Fig. 1), which make it a good candidate for usage in polymer composites, energy-related materials, sensors and “paper-like” materials (free-standing carbon-based membrane materials). The synthesis and surface functional groups of different GO-based materials are listed in Table 1.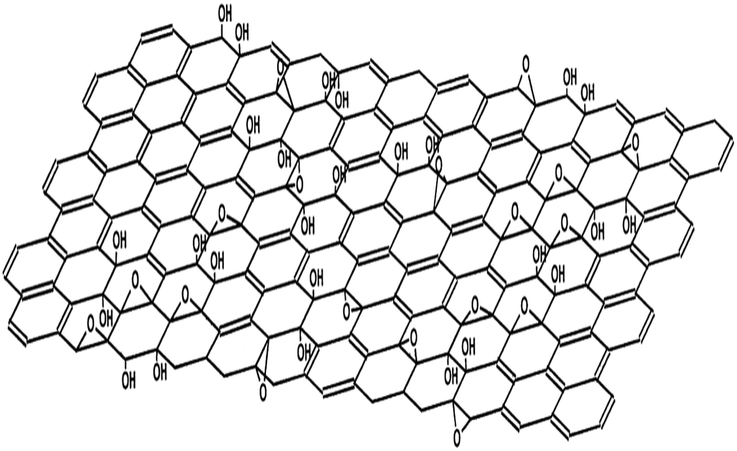 | ||
| Fig. 1 Structural model of graphene oxide.81 | ||
| GO-based materials | Preparation | Surface functional group | Ref. |
|---|---|---|---|
| GO | Hummers method | Epoxy, hydroxyl, carboxyl and carbon sp2 | 90 |
| GO | Improved Hummers method | Epoxy, hydroxyl, carbonyl, carboxyl and carbon sp2 | 26 |
| BEGO | Exfoliation of GO was in DMF, then BE was added into as-prepared GO/DMF, followed by the esterification reaction. | Epoxy, hydroxyl, carbonyl | 91 |
| Oligothiophene functionalized graphene | Mixing the GO, amine functionalized oligothiophene, 1,3-diisopropylcarbodiimide and o-dichlorobenzene for 48 h under Ar at 80 °C. | Amido group, hydroxyl, carbonyl | 93 |
| Amine-terminated ionic liquid functionalized graphene | Mixing GO and 1-(3-aminopropyl)-3-methylimidazolium bromide in KOH solution. After ultrasonication, the homogeneous solution was vigorously stirred at 80 °C for 24 h. | Epoxy, ether group, carbonyl, C bound to nitrogen | 94 |
| Amines and amino acids modified GO | Mixing GO, CnH2n+1NH2 (n = 2, 4, 8, 12) and N-(β-aminoethyl)-γ-aminopropyltrimethoxyl silane, washing with 1:1 H2O–ethanol mixture and acetone. |
Amido group, hydroxyl, carbonyl | 95 |
| Sodium azide modified GO | Mixing GO and NaN3 in water–AcCN (1:1) solution for one week under nitrogen. |
Amido group, hydroxyl | 96 |
| Isocyanate-treated GO | Mixing GO and anhydrous DMF under nitrogen. Adding organic isocyanate and stirring under nitrogen. | Amides, carbamate ester | 97 |
| GO-TDI-AO | Mixing GO-TDI and amphiphilic oligoester in anhydrous DMF at 80 °C under nitrogen. | Amide, carbamate ester, isocyanate group, epoxy | 98 |
| GO–Ag nanocomposites | In situ reduction method using GO sheets as substrates and low-temperature plasma as reductant. | Ag, epoxy, hydroxyl, carboxyl | 121 |
| RGO/Ag/CeO2 | Mixing poly(vinylpyrrolidone), GO and CeO2 in water with ultrasonication. Adding AgNO3 and NaOH successively, and stirring for 1 h at 95 °C. | Ag, CeO2, hydroxyl, carboxyl | 104 |
| Pt/GH and Pt/GHA | Mixing H2 or NH3 plasma treated GO with H2PtC6 solution. Treating the mixture with hydrogen plasma. | Pt, hydroxyl, carboxyl | 125 |
| GO/TiO2 | Mixing GO and sodium dodecylsulfate in water. Adding TiCl3, Na2SO4 and H2O2 successively, and stirring 16 h at 90 °C. | TiO2, hydroxyl, carboxyl | 129 |
| GO–iron oxides | Coprecipitation method by mixing GO, FeCl3·6H2O and FeSO4·7H2O in water under N2. | Fe3O4, hydroxyl, carboxyl | 141 |
| GO–CdS | Mixing GO and CdS in toluene solution and stirring for 24 h. | CdS, hydroxyl, carboxyl | 138 |
| PANI@GO | PANI@GO composites were synthesized by the polymerization of aniline monomer on the amine-terminated GO surface in the presence of (NH4)2S2O8. | Quinonoid group, amine group, hydroxyl, carboxyl | 142 |
| GO/PPy | Dielectric barrier discharge plasma treatment of GO and pyrrole monomer. | Hydroxyl, carboxyl | 144 |
| MOF-5–GO | Dispersing GO powder in a zinc nitrate–1,4-benzenedicarboxylate mixture, the mixture was heated at 115–120 °C for 24 h. | ZnO4, hydroxyl, carboxyl | 146 |
| LaMOF–GO | Mixing GO and La(NO3)3. Afterward, adding 1,3,5-benzenetricarboxylic acid and N,N-dimethyl formamide solution and stirring the mixture. | Carbonyl, hydroxyl, carboxyl | 114 |
2.1 Pristine GO
Brodie first demonstrated the synthesis of GO in 1859 by adding a portion of potassium chlorate to a slurry of graphite in fuming nitric acid.88 In 1898, Staudenmaier improved this protocol by using concentrated sulfuric acid as well as fuming nitric acid and adding the chlorate in multiple aliquots over the course of the reaction. This small change in the procedure made producing the highly oxidized GO in a single reaction vessel more practical.89 In 1958, Hummers introduced the now often-used method, with KMnO4 and NaNO3 in concentrated H2SO4.90 Recently, Marcano et al.26 reported an improved method for the preparation of GO. The improved GO (IGO) synthesis was evaluated in comparison to Hummers’ method or Hummers’ method with additional KMnO4 (Fig. 2). For clarity, they named the GO produced using these methods as IGO, HGO, and HGO+, respectively. This improved method provided a greater amount of GO compared to Hummers’ method or Hummers’ method with additional KMnO4. Moreover, even though the GO produced using this method contained many more oxygen-containing functional groups than that produced using Hummers’ method, when reduced in the same chamber with hydrazine, they both formed chemically converted graphene (CCG) with equivalent electrical conductivity. In contrast to Hummers’ method, the new method did not generate toxic gas and the temperature was easier to control. The improved synthesis method for GO may be important for large-scale production of GO as well as the construction of devices composed of the subsequent CCG.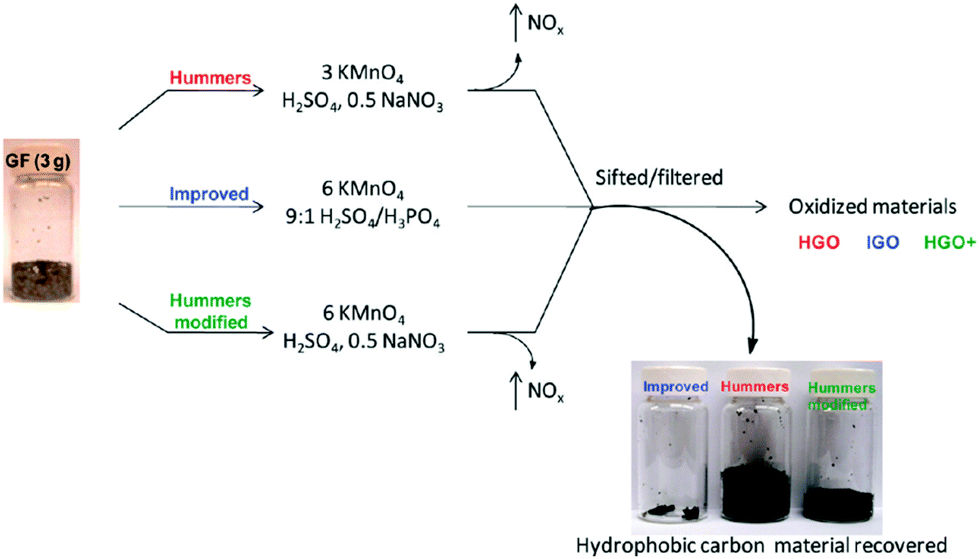 | ||
| Fig. 2 Representation of the procedures followed, starting with graphite flakes (GF). Under-oxidized hydrophobic carbon material is recovered during the purification of IGO, HGO, and HGO+. The increased efficiency of the IGO method is indicated by the very small amount of under-oxidized material produced.26 | ||
2.2 Functionalization of GO
The functionalization of GO using various chemical reactions allows for either covalent or non-covalent attachment to the resulting chemically modified GO. Such approaches, which add functionality to groups already present on the GO surfaces, make GO a more versatile precursor for a wide range of applications.87GO has various oxygen-containing functional groups, mainly epoxides and hydroxyls on their basal planes and carboxyls on the edges, which can facilitate the dispersion of GO in water. Through the noncovalent strategy, chemical reduction of GO can still be stably suspended in water in the presence of dispersants or stabilizers. A kind of bio-based polyester (BE) was synthesized by Tang et al., and the BE was grafted onto GO via an esterification between the hydroxyls of BE and carboxyls of GO. Subsequently, the grafted GO was reduced to graphene using vitamin C.91 The final BE/graphene can be dispersed in chloroform, acetone, or tetrahydrofuran. In a similar way, GO was functionalized with poly(vinyl alcohol) (PVA) polymer through a carbodiimide-activated esterification reaction between the carboxylic acid moieties on the GO and hydroxyl groups on PVA.92 Liu et al. reported an organic solution-processable functionalized graphene hybrid material with oligothiophene (6THIOP-NH-SPFGraphene) (Fig. 3).93 The covalent functionalization of GO with oligothiophenes changes GO from hydrophilic to hydrophobic and the hybrid can be dissolved in organic solvents such as o-dichlorobenzene (ODCB). This makes the graphene hybrid material homogeneously dispersed (together with other organic materials) in organic solvents, meeting the needs of various organic electronic applications.
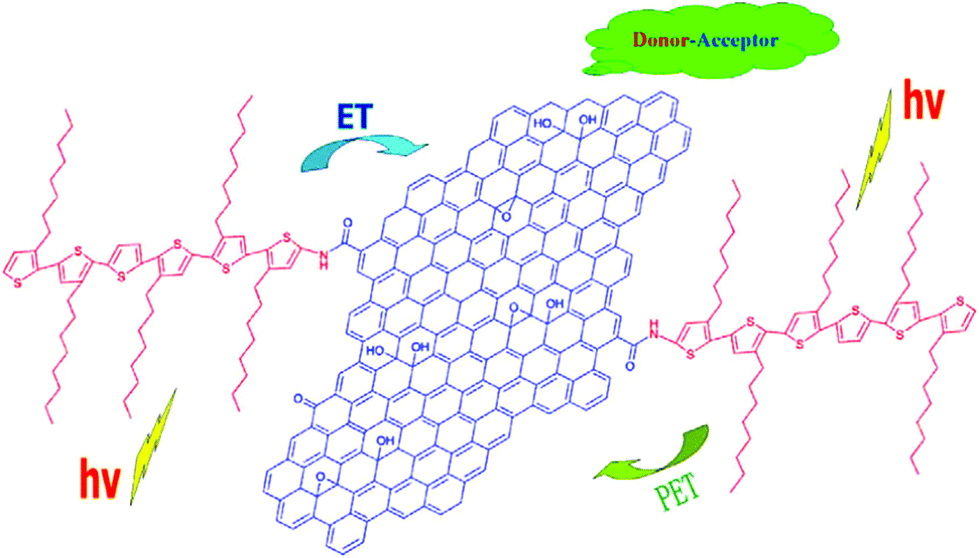 | ||
| Fig. 3 Structure of 6THIOP-NH-SPFGraphene.93 | ||
GO contains plentiful and reactive epoxy groups, and therefore, the nucleophilic ring-opening reaction between epoxy groups and amine groups may be easy. Chemically-converted graphene functionalized by amine-terminated ionic liquid (IL-NH2) has been reported by Yang et al.94 The cations of the IL-NH2 were introduced into the graphene, contributing to a stabilization of graphene dispersions via electrostatic repulsion. Moreover, the polydispersity in water and several organic solvents makes graphene an ideal candidate for various applications. Bourlinos et al. further confirmed that, on account of the surface-exposed epoxy groups present in the GO solid, the modification of GO with neutral molecules, primary aliphatic amines or amine-containing molecules (amino acids and amino siloxanes) could take place easily through the corresponding nucleophilic substitution reactions.95 GO functionalized with sodium azide was reported by Salvio and co-workers, which was then reduced to a new graphene material with amino groups (GO-NH2) that can be used for chemical reactions (Fig. 4).96 The azide derivative of graphene can also be functionalized with long alkylic chains through click chemistry, which allow for further exfoliation down to a single sheet.
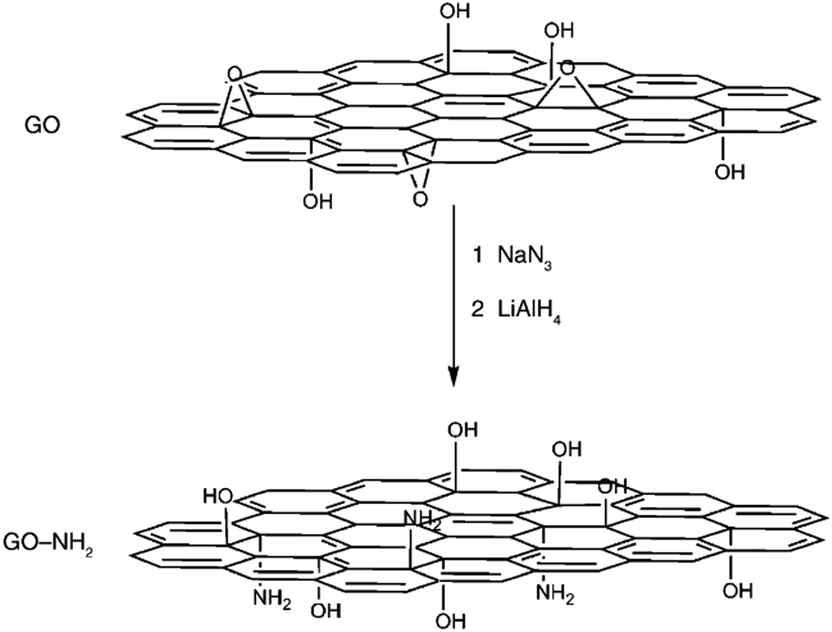 | ||
| Fig. 4 Proposed reactions during the treatment of GO with NaN3 and then with LiAlH4, resulting in the amino-functionalized material (GO-NH2).96 | ||
The treatment of GO with organic isocyanates can lead to the derivatization of the edge carboxyl and surface hydroxyl functional groups via the formation of amides or carbamate esters, respectively (Fig. 5).97 This work was an early demonstrated example of complete exfoliation of chemically derivatized GO in organic solvents. In addition, the use of isocyanates containing orthogonal functionalities, such as cyano, keto, and azidosulfonyl, allows for further modification of the surface properties and chemistry of GO.97 In a similar way, Xu et al. applied toluene-2,4-diisocyanate (TDI) to create anchor sites on GO and coupled them with an amphiphilic oligoester through a ‘grafting to’ approach.98 Surfactant-wrapped chemically converted graphene sheets obtained from reduction of GO with hydrazine were functionalized by treatment with aryl diazonium salts.99 The graphene nanosheets can be dispersed readily in polar aprotic solvents, allowing alternative avenues for simple incorporation into different polymer matrices. Highly dispersed sulfonated graphene sheets were synthesized, and the water-soluble graphene existed in the form of a single carbon sheet with an electrical conductivity comparable with graphite.100,101
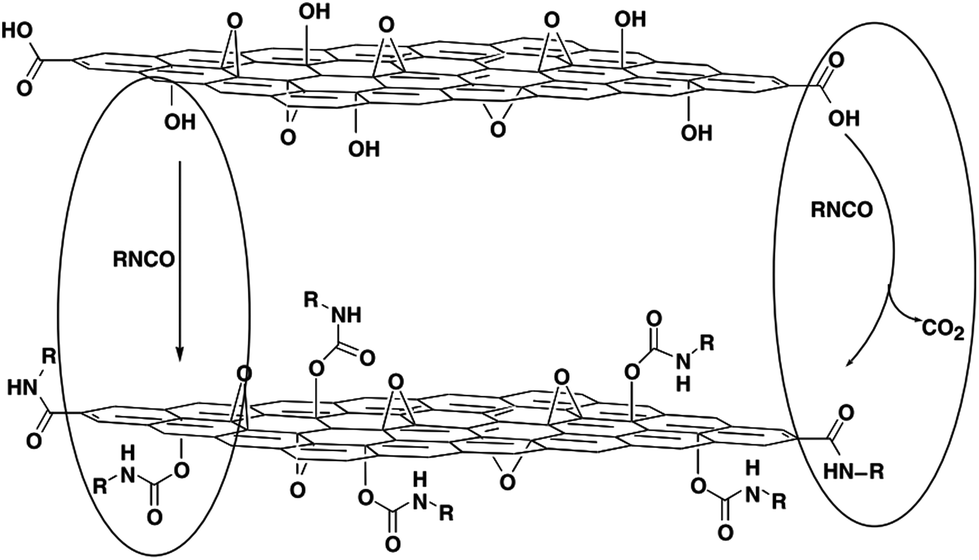 | ||
| Fig. 5 Proposed reactions during the isocyanate treatment of GO, where organic isocyanates react with the hydroxyl (left oval) and carboxyl groups (right oval) of graphene oxide sheets to form carbamate and amide functionalities, respectively.97 | ||
The low-temperature plasma technique has been widely used for many applications and has become one of the most popular treatments for nanomaterials, such as nanomaterial synthesis, structuring and processing, in the last two decades.68 Wang et al. developed a simple, low cost, and green approach to prepare reduced graphene using H2 plasma.102 Zhao et al. synthesized few-layered graphene by etching the graphite using the H2O2 plasma technique.51 Dato et al. synthesized graphene nanosheets using a substrate-free, atmospheric-pressure microwave plasma method.103 They produced graphene directly in the gas phase, and the synthesis took place within a second, in an atmospheric-pressure environment.
2.3 GO-based composites
To date, GO-based composites have been successfully synthesized with inorganic nanostructures,104–106 organic crystals,107,108 polymers,109–111 metal–organic frameworks (MOFs),112–114 and biomaterials.115–117 They were intensively explored in applications such as batteries, fuel cells, photovoltaic devices, photocatalysis, sensing platforms, adsorbents, and so on.In the last few decades, huge efforts have been made to synthesize inorganic nanostructures with controlled shape, size, crystallinity and functionality. A great number of inorganic nanostructures have been hybridized with GO and its derivatives, which include metals like Au,118–120 Ag121–123 and Pt;124–126 oxides like TiO2,127–129 MnO2130–132 and Fe3O4;133–136 and chalcogenides like CdS137–139 and CdSe.140 Wei et al. presented a room temperature dry reduction approach, i.e., a low pressure dielectric barrier discharge plasma jet, to synthesize highly dispersed GO–Ag nanocomposites.121 Furthermore, a three-component composite composed of reduced graphene oxide (RGO), CeO2 and Ag nanoparticles was designed and synthesized by Ji et al.104 Wang et al. reported a one-step in situ plasma approach to synthesize highly dispersed Pt nanoparticles on graphene under mild conditions.124 The Ar plasma was highly effective in activating both the Pt ions and GO. Furthermore, they presented a novel and facile mild plasma approach to synthesize Pt nanoparticles on H-doped graphene (GH) and N-doped graphene (GHA) in the gas phase (Fig. 6).125 Zhao et al. investigated the efficacy of graphene@TiO2, which could reduce charge recombination and enhance reactivity.128 The graphene@TiO2 composites can absorb significantly more light in the region of 420–800 nm compared with pure TiO2 and graphene. This different behavior defines the graphene@TiO2 as a “dyade” structure, where the combination of TiO2 and graphene gives rise to synergistic properties arising from the beneficial interaction between the two.128 GO/TiO2 composites were prepared using TiCl3 and GO as reactants.129 The GO–iron oxide and RGO–iron oxide hybrid materials were synthesized by Yang and co-workers, which can be easily separated by magnetic separation.141 In a similar way, a composite of porous Fe3O4 hollow microspheres/GO was fabricated through a facile self-assembly approach.135 GO–CdS composites were synthesized successfully via a novel two-phase mixing method by Gao et al.138
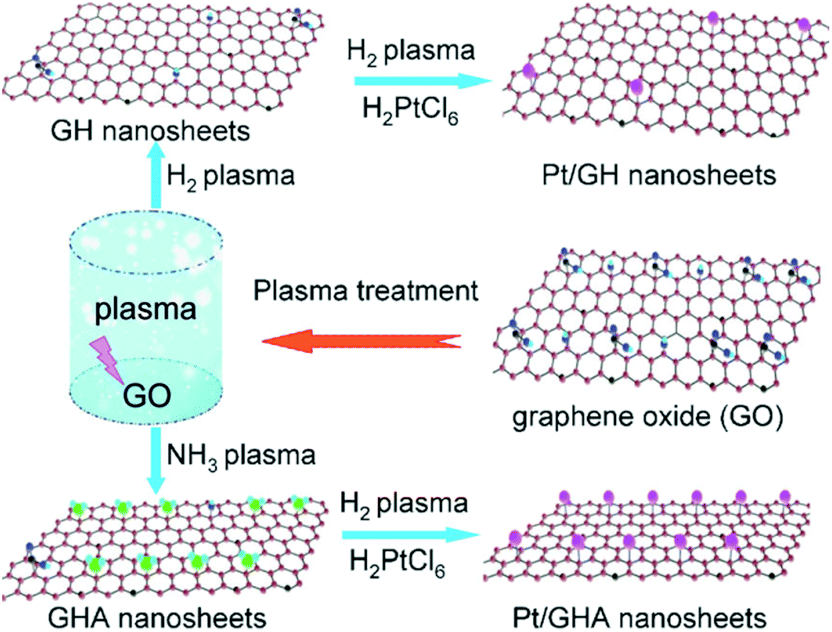 | ||
| Fig. 6 Synthesis of Pt/GH and Pt/GHA using the plasma approach.125 | ||
The conducting polymers have good prospects in sorption applications due to the positively charged nitrogen atoms in the polymer chains. Therefore, conducting polymers and their combination with GO have shown an enhanced ability to remove pollutants from water.142,143 GO-supported polyaniline (PANI@GO) composites were synthesized by chemical oxidation.142 Zhang et al. developed a dilute polymerization method for the synthesis of hierarchical PANI/GO nanocomposites using 1-dimensional PANI and 2-dimensional GO nanosheets (Fig. 7).143 Hu et al. prepared GO/polypyrrole (GO/PPy) composites using a dielectric barrier discharge plasma technique under nitrogen.144 The presence of graphene in polypyrrole enhanced the charge density of the pyrrolic nitrogen, thus increasing the electrostatic interaction between the adsorbate and adsorbent. Chen et al. used an in situ polymerization reaction to fabricate poly (amidoxime) grafted graphene (PAO-g-RGO) composites.145
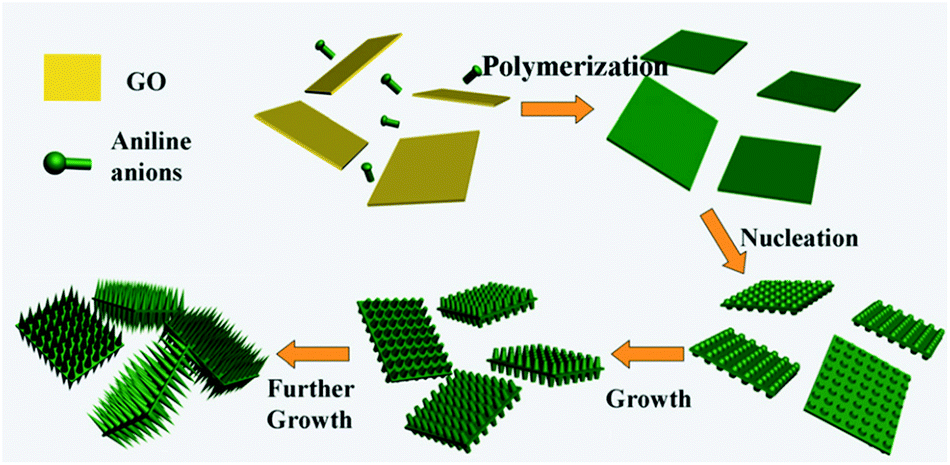 | ||
| Fig. 7 Schematic illustration of the nucleation and growth mechanism of aligned PANI nanorods on GO nanosheets.143 | ||
MOFs are novel inorganic–organic hybrid materials that have aroused great interest in recent years. The unique properties of MOFs include permanent nanoscale porosity, high surface area, uniform but tunable pore size, in-pore functionality, and out-surface modification.119 A lot of effort has been directed towards the preparation of GO–MOF composites by combining the unique properties of both GO layers and MOFs for various applications. Zn4O(H-1,4-benzenedicarboxylate)3 (MOF-5)–GO composites prepared through a solvothermal process illustrated that MOF-5 blocks were attached to GO through the replacement of oxygen atoms in the ZnO4 tetrahedra by epoxy groups on the surface of GO.146 Liu et al. synthesized a novel GO–La(BTC)(H2O)6 (H3BTC = 1,3,5-benzenetricarboxylic acid) metal–organic framework (LaMOF–GO) composite through a simple and large-scale method at room temperature.114 Bao et al. successfully prepared mesoporous MOF reduced graphene oxide (MIL-101(Cr)@RGO) materials using in situ chemical oxidation polymerization. Subsequently, sulfur (S) was loaded using a two-step liquid method to fabricate MOF@RGO/S multi-composites (Fig. 8).147 Well-intergrown nanocrystals of zeolitic imidazolate frameworks (ZIF-8) supported by 3-dimensional graphene were prepared using a counter diffusion technique.112
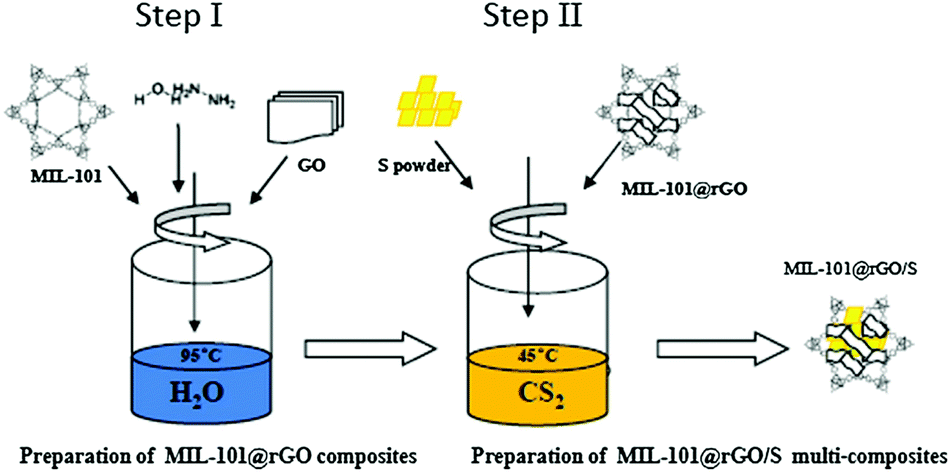 | ||
| Fig. 8 Synthesis procedure of the MIL-101(Cr)@RGO/S multi-composites.147 | ||
Other than inorganic nanostructures, polymers and MOFs, materials such as organic crystals107,108 and biomaterials,115–117 have also been hybridized with graphene derivatives for various applications. For example, N,N′-dioctyl-3,4,9,10-perylenedicarboximide (PDI)–graphene core/shell nanowires were prepared through π–π interactions.107 In addition, peptide–graphene core/shell nanowires were prepared by mixing an aqueous system of graphene and an organic peptide solution (diphenylalanine in 1,1,1,3,3,3-hexafluoro-2-propanol).108 Moreover, biomaterials like deoxyribonucleic acid hybridized with GO or RGO were used in fluorescent sensing platforms based on fluorescence resonance energy transfer (FRET),115,116 and biocompatible aptamer–GO composites can be applied in probing living cells.117
3. Radionuclide sorption
The nuclear industry generates large amounts of radioactive wastewater that must be effectively treated before it is discharged into the environment.72 More radioactive elements, viz. thorium (Th), uranium (U), neptunium (Np), europium (Eu), strontium (Sr), etc., are highly toxic and hazardous to the human body.142 U, Eu and Sr are deeply discussed in this review as they are the representative elements of radionuclides. Uranium, a representative actinide element, is of fundamental importance in the nuclear fuel cycle and is the main component of nuclear waste.144 Strontium, as one of the most abundant radionuclides in nuclear fission products, poses a hazardous threat to human safety due to its long half life (T1/2 = 28.8 years).148 Europium is a trivalent lanthanide, which exhibits similar sorption properties to both trivalent lanthanides and actinides.4In recent years, the removal of radionuclides from aqueous systems has been extensively studied using types of adsorbents such as clay minerals,148,149 metal oxides,150,151 and biomaterials.152,153 However, these materials have some disadvantages, such as poor sorption capacities and low efficiencies. On the other hand, GO-based materials are potentially excellent candidates for the enrichment of radionuclides from radioactive wastewater because of their unique properties, such as chemical stability, hydrophilicity and large surface area, which make them able to form strong chemical bonds with radionuclides. The sorption properties of several crucial radionuclides on GO-based materials are listed in Table 2.
| GO-based materials | Adsorbates | Adsorbent dosage (g L−1) | Initial concentration of radionuclides (mg L−1) | pH | Maximum sorption capacity (mg g−1) | Type of Sorption | Ref. |
|---|---|---|---|---|---|---|---|
| GO | U(VI) | 0.06 | 98.056 | 5.0 | 97.5 | Inner-sphere surface complexation | 154 |
| U(VI) | 0.4 | 119 | 4.0 | 299 | Inner-sphere surface complexation | 59 | |
| U(VI) | 0.2 | 10 | 4.0 | 208.33 | Inner-sphere surface complexation | 45 | |
| Eu(III) | 28.7 | ||||||
| Th(IV) | 0.36 | 120 | 2.6 | 214.6 | Inner-sphere surface complexation | 60 | |
| Sr(II) | 0.038 | 1.09 × 10−2 | 6.5 | 23.83 | Coagulation | 155 | |
| Am(III) | 9.58 × 10−5 | 3.4 | 8.51 | ||||
| Eu(III) | 5.21 × 10−7 | 5.0 | 115.49 | ||||
| U(VI) | 0.038 | 5.12 × 10−2 | 5.0 | 27.608 | — | 62 | |
| Sr(II) | 1.09 × 10−2 | 6.5 | 23.833 | ||||
| Am(III) | 9.58 × 10−5 | 3.5 | 0.486 | ||||
| RGO | Th(IV) | 0.5 | 10 | 3.0 | 48.72 | — | 156 |
| GO–ACF | U(VI) | 0.2 | 50 | 5.5 | 298 | — | 157 |
| AOMGO | U(VI) | 0.2 | 47.6 | 5.0 | 284.92 | Inner-sphere surface complexation | 158 |
| GO@sepiolite composites | U(VI) | — | — | 5.0 | 161.29 | At low pH: ion exchange; at high pH: surface complexation | 159 |
| MGO | Eu(III) | — | 8.0 | — | — | Inner-sphere surface complexation | 160 |
| M/GO | Sr(II) | 0.4 | 6.0 | 8.5 | 9.81 | Surface complexation | 161 |
| Fe3O4/GO | U(VI) | 0.3 | 26.66 | 5.5 | 69.49 | At low pH: inner-sphere surface complexation; at high pH: precipitation and inner-sphere surface complexation | 162 |
| MnO2–Fe3O4–RGO | U(VI) | 1.0 | 120 | 6.0 | 108.7 | Surface complexation, cation exchange and electrostatic attraction | 163 |
| PB/Fe3O4/GO | Cs(I) | 1.7 | 10 | 7.0 | 55.56 | H+-exchange and/or ion trapping | 164 |
| Al2O3/EG | Eu(III) | 1.2 | 10 | 6.0 | 5.14 | At low pH: outer-sphere surface complexation; at high pH: inner-sphere surface complexation | 165 |
| GO–HAp | Sr(II) | 0.5 | 20 | 7.0 | 702.18 | Ion-exchange and surface complexation | 3 |
| GO/PPy | U(VI) | 0.3 | 27 | 5.0 | 147.06 | Ion exchange and outer-sphere surface complexation | 144 |
| PANI/GO | U(VI) | 0.05 | 50 | 5.0 | 1960 | Electrostatic interactions and hydrogen bonds | 166 |
| PANI@GO | U(VI) | 0.25 | 15 | 3.0 | 245.17 | Complexation with nitrogen- and oxygen-containing functional groups | 142 |
| Eu(III) | 250.74 | ||||||
| Sr(II) | 147.20 | ||||||
| Cs(I) | 184.74 | ||||||
| PAO/RGO | U(VI) | 0.05 | 50 | 4.0 | 872 | — | 167 |
| PAO-g-RGO | Eu(III) | 0.2 | 60 | 5.5 | 296.4 | Surface complexation and cationic exchange | 145 |
| Sr(II) | 99.4 | ||||||
| CD/GO | U(VI) | 0.1 | 24 | 5.0 | 97.33 | Electrostatic interaction and surface complexation | 168 |
| G/AgCS | U(VI) | 0.5 | — | — | 13.31 | — | 169 |
| nZVI/RGO | U(VI) | 2.5 | 119 | 5.0 | — | Multimeric surface complexation | 46 |
| RGO/NiAl-LDH | U(VI) | 0.5 | 130 | 4.0 | 277.8 | Surface complexation | 170 |
| GONS | Eu(III) | 0.2 | 50.91 | 6.0 | 175.44 | At low pH: mononuclear monodentate complexation; at high pH: binuclear bidentate complexation | 4 |
| GO-Ch | U(VI) | 0.75 | 40.46 | 4.0 | 225.78 | Inner-sphere surface complexation and surface coprecipitation | 153 |
| GTiP | Eu(III) | 1 | 100 | 5.5 | 64.33 | — | 171 |
| RAFT-IIP | Sr(II) | 0.4 | 5 | 6.0 | 145.77 | — | 172 |
| PFGM | Cs(I) | — | 10 | 7.0 | 43.52 | K+/H+-exchange and ion trapping | 173 |
| PAM/GO | U(VI) | 0.2 | 47.6 | 5.0 | 166.12 | Surface complexation | 174 |
| Eu(III) | 30.4 | 189.20 |
3.1 U(VI) sorption
Uranium (U), a representative actinide element of radionuclides with a number of relatively radioactive products, has attracted great concern due to its extensive distribution, long half-life and carcinogenicity.155 U can be reused after enrichment because of the high U content in nuclear waste management.144 Excessive amounts of U have entered the environment through the activities of the nuclear industry.175 It usually exists in the hexavalent form (U(VI)) in the environment. U(VI) disposed into the environment can eventually reach the top of the food chain and be ingested by humans, causing severe kidney or liver damage and even death.176 The U.S. environmental protection agency (EPA) has recommended a drinking water standard of 20 μg L−1 for 238U.177 Therefore, it is necessary to treat wastewater containing U(VI) in order to prevent radioactive contamination of the environment.Zhao et al. used few-layered GO nanosheets for the removal of U(VI) ions from an aqueous system.154 The maximum sorption capacity of U(VI) was 97.5 mg g−1. They reported that GO had good potential for the disposal of U(VI) from wastewater. However, the aggregation and accumulation of GO nanosheets in the aqueous systems significantly decreased their sorption performance. Functionalization of GO nanosheets was helpful in overcoming this limitation. Cheng et al. synthesized GO-supported sepiolite (GO@sepiolite) composites and applied them for the removal of U(VI).159 The maximum sorption capacity of the GO@sepiolite composites was 161.29 mg g−1. Chen and co-workers prepared GO-activated carbon felt (GO–ACF) composites by an electrophoretic deposition and subsequent thermal annealing.157 The maximum sorption capacity of GO–ACF for U(VI) was 298 mg g−1, much higher than that of ACF (173 mg g−1). They demonstrated that the carboxyl functional groups of GO–ACF played an important role in the sorption. Tan et al. presented a facile route for the fabrication of hierarchical 3-dimensional composites (layered double hydroxide/graphene).170 These composites were obtained via in situ growth of layered double hydroxide (LDH) nanosheet arrays onto graphene sheets. The maximum sorption capacity of U(VI) on the LDH/graphene was 277.8 mg g−1. Cyclodextrin-modified GO nanosheets (CD/GO) were synthesized using an in situ polymerization method.168 The mutual effects on the simultaneous removal of U(VI) and humic acid from an aqueous system by CD/GO were investigated by Song et al.168 Their results indicated that U(VI) and humic acid (HA) sorption on CD/GO were greatly affected by the pH and ionic strength. The presence of HA enhanced U(VI) sorption at low pH and reduced U(VI) sorption at high pH, while the presence of U(VI) enhanced HA sorption. Sun et al. found that the removal of U(VI) on nanoscale zero-valent iron (nZVI) and reduced graphene oxide-supported nanoscale zero-valent iron (nZVI/RGO) was influenced by the distribution of U(VI) species (Fig. 9).46 The enhanced removal of U(VI) was attributed to the positively charged U(VI) species (i.e., UO22+, UO2OH+ and (UO2)3(OH)5+) in acidic solution. The decreased sorption of U(VI) in alkaline solution was due to the negatively charged U(VI) species (such as UO2(OH)3−, (UO2)3(OH)7− and UO2(OH)42−).
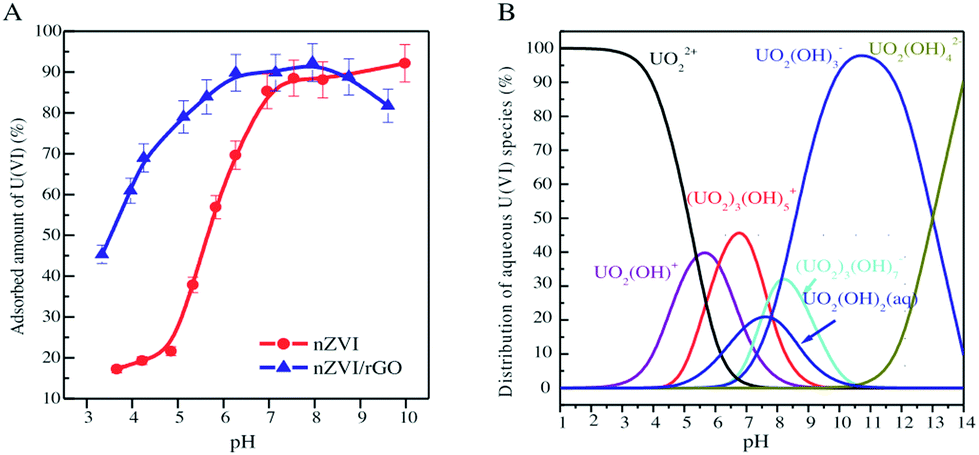 | ||
| Fig. 9 A: The effect of pH on U(VI) removal by nZVI and nZVI/rGO composites; initial concentration C0 = 0.5 mmol L−1, solid to liquid ratio (m/V) = 2.5 g L−1, ionic strength (I) = 0.01 mol L−1 NaClO4, temperature (T) = 25 ± 1 °C. B: The distribution of U(VI) species in an aqueous system.46 | ||
Due to their thermal stability, facile synthesis, non-toxicity, relatively low cost, and unique electronic and redox properties, polymers have received increasing attention.178 Hu et al. investigated the sorption of U(VI) ions on GO/PPy composites in an aqueous system.144 The sorption capacity of U(VI) on GO/PPy composites was much higher than that of U(VI) on either GO or PPy (Fig. 10). The highly selective sorption capacity toward U(VI) was attributed to the strong coordination between the U(VI) ions and nitrogen donor atoms on the GO/PPy composites in solution. In addition, the regeneration of the adsorbent is likely to be a key factor for improving wastewater process economics.172 GO/PPy composites could be regenerated for reuse using 1.0 mol L−1 HCl solution. Poly(amidoxime)–reduced GO (PAO/RGO) composites with excellent sorption capability for U(VI) were synthesized by in situ polymerization of acrylonitrile monomers on GO surfaces, followed by an amidoximation treatment with hydroxylamine.167 The sorption capacity of PAO/RGO composites for U(VI) reached as high as 872 mg g−1.
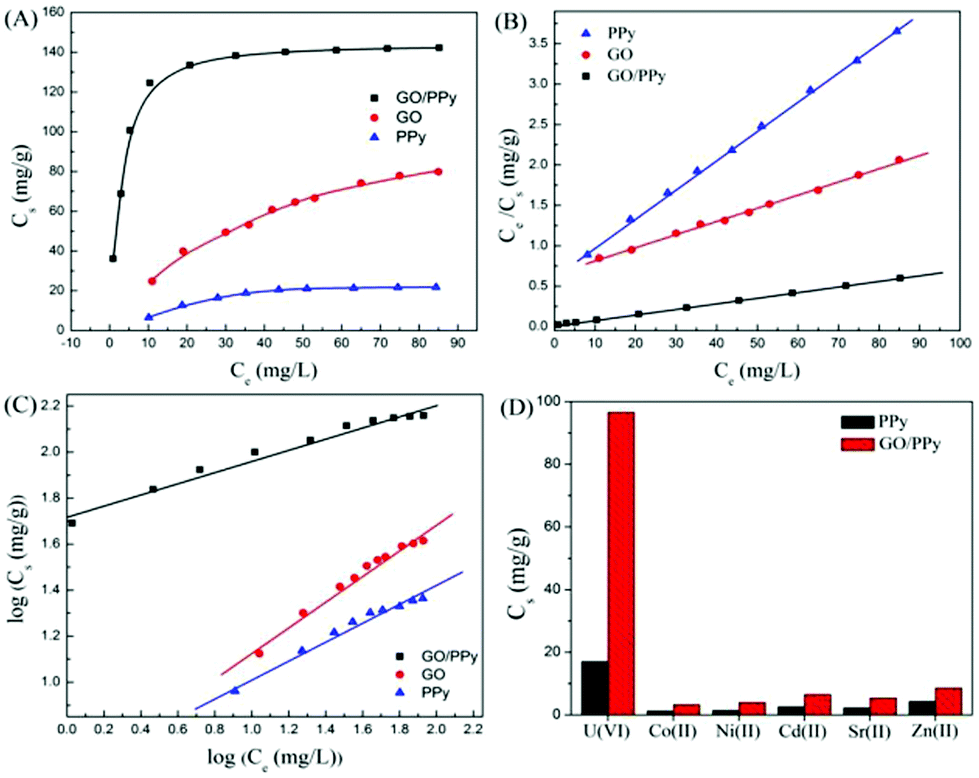 | ||
| Fig. 10 (A) Sorption isotherms, (B) Langmuir isotherms and (C) Freundlich isotherms of U(VI) ions on GO, PPy and GO/PPy composites; (D) selective sorption capacities of coexistent ions on PPy and GO/PPy composites at Cinitial = 1.5 × 10−4 mol L−1 for all cations. T = 298 ± 2 K, pH = 5.0 ± 0.1, m/V = 0.3 g L−1, I = 0.01 mol L−1 NaClO4.144 | ||
In recent years, the magnetic separation technique has been considered as an effective and quick method for separating magnetic particles. In general, magnetic nanomaterials usually have a high sorption efficiency, sorption capacity, and selectivity for radionuclides. More importantly, the intrinsic magnetic properties may make solid–liquid separation easy and effective under an applied magnetic field without centrifugation or filtration. Zong and co-workers prepared Fe3O4/GO from natural flake graphite and employed it as an adsorbent to remove U(VI) ions.162 The maximum sorption capacity of U(VI) on Fe3O4/GO was about 69.49 mg g−1. Amidoximated magnetite/GO (AOMGO) composites were synthesized and applied to U(VI) sorption, and had a maximum sorption capacity of 284.92 mg g−1.158 The temperature-dependent sorption isotherms (Fig. 11) suggested that the sorption of U(VI) on AOMGO composites was an endothermic and spontaneous process. MnO2–Fe3O4–RGO was successfully prepared by Tan et al. and used for the removal of U(VI).163 The maximum sorption capacity was 108.7 mg g−1. In addition, U(VI)-loaded MnO2–Fe3O4–RGO was efficiently renewed.
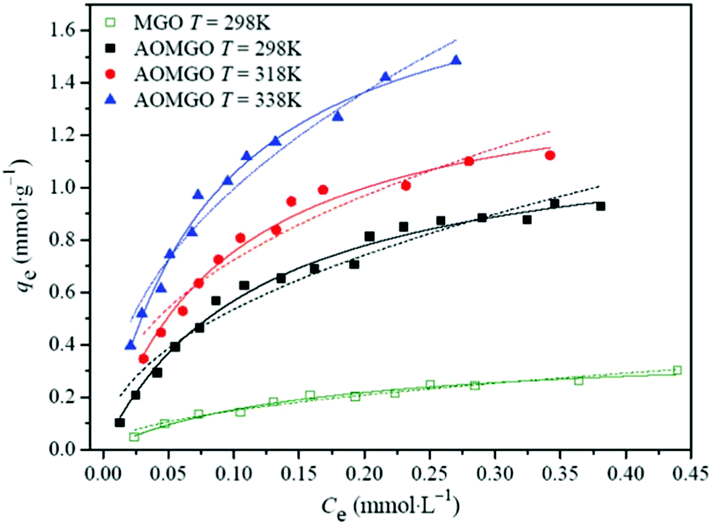 | ||
| Fig. 11 Sorption isotherms of U(VI) on MGO and AOMGO composites. pH = 5.0 ± 0.1, I = 0.01 mol L−1 NaClO4, m/V = 0.2 g L−1. The scattered points represent experimental data, the solid lines represent the Langmuir model and the dashed lines represent the Freundlich model.158 | ||
3.2 Eu(III) sorption
As an important goal in radioactive waste disposal, efficient immobilization of radionuclides in wastewater attracts intensive research interest. Because europium (Eu) and actinide radionuclides are chemical homologues, the investigation of Eu(III) sorption behavior at the solid–liquid interface can be helpful in assessing the risks of radioactive exposure to trivalent actinides.179,180 However, reports on the application of GO-based materials for Eu(III) treatment in nuclear waste management are still scarce.GO nanosheets were synthesized and used for the sorption of Eu(III).4,45 The maximum sorption capacity of Eu(III) on GO was 175.44 mg g−1 at pH 6.0 and 28.70 mg g−1 at pH 4.0. Ding et al. found that the removal rate of Eu(III) by GO was connected with the relative distribution of Eu(III) species in solution and the surface properties of the GO.45 At low pH values, Eu3+ was the main species, while the hydrolyzed mononuclear and multinuclear species (i.e., Eu(OH)2+, Eu2(OH)24+) increased with increasing pH. The precipitated Eu(OH)3 appeared at pH 8.0. The Eu(III) sorption increased between pH 2.0 to 6.0 due to electrostatic attraction, and then retained this level at pH 6.0–9.0 due to surface complexation.45
There are still some drawbacks of GO when used in the treatment of large-scale wastewater. Firstly, the biggest obstacle of using GO for practical water treatment is its high cost. Secondly, GO agglomerates easily after drying, which not only reduces its specific surface area, but also reduces its hydrophilicity, thus affecting the sorption efficacy. To overcome these problems, Li et al. presented titanium phosphate (TiP) modified GO (GTiP) composites.171 GTiP exhibited excellent sorption characteristics for Eu(III). It took less than 1.5 min to reach half-equilibrium, while its sorption capacity was as high as 64.33 mg g−1. The excellent sorption capability of GTiP was due to the synergistic effect of the materials, where GO increased the specific surface area and sorption capacity, and TiP enhanced the hydrophilicity and mechanical properties of GTiP.
GO-supported polyaniline (PANI@GO) composites were used for the removal of Eu(III) and the maximum sorption capacity was 250.74 mg g−1.142 Compared to granular activated carbon (AC), PANI, and GO (Fig. 12), the enhanced sorption of Eu(III) on the PANI@GO composites was due to the occurrence of a large number of nitrogen- and oxygen-containing functional groups, which form strong complexes with Eu(III). Song et al. synthesized polyacrylamide (PAM) grafted GO (PAM/GO) using the plasma-induced polymerization technique and applied it to the removal of Eu(III).174 The maximum sorption capacity of Eu(III) on PAM/GO was 189.20 mg g−1 at pH = 5.0 and T = 295 K. Magnetite-decorated graphene oxide (MGO) was fabricated for the highly efficient removal of Eu(III) from aqueous systems.160 The combination of the excellent sorption capacity of GO and the magnetic properties of magnetite can give a promising candidate for the removal of Eu(III) and related radionuclides in nuclear waste management.
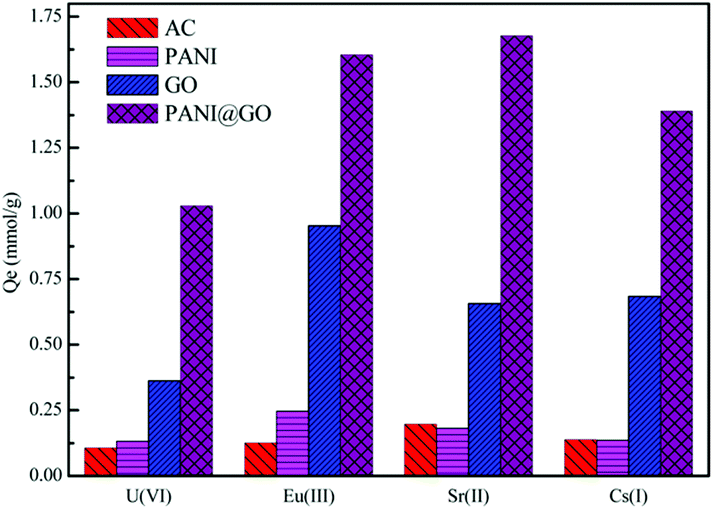 | ||
| Fig. 12 Comparison of the sorption capacity of PANI@GO composite with other adsorbents for the uptake of radionuclides; pH = 3.0, T = 298 K, m/V = 0.25 g L−1.142 | ||
3.3 Sr(II) sorption
Strontium (Sr) is a radionuclide that is incorporated into the bone structure of the human body because it cannot be distinguished from Ca2+. Such incorporated Sr(II) cannot be removed from an organism and, having a half-life of 28.8 years, causes cancer.181 Given the large release of Sr(II) into the environment during the last two major nuclear power plant accidents, i.e., Chernobyl (Sr released into the atmosphere) and Fukushima (Sr released into seawater), there is an urgent need for efficient remediation.The interaction of GO with Sr(II) was studied by Romanchuk et al. and they found that the maximum sorption capacity of GO for Sr(II) was evaluated to be 23.83 mg g−1.155 Sorption capacity is controlled by the functional groups on the surface. The modification of graphene with suitable functional groups or an anticoagulant could reduce the self-aggregation and enhance the dispersion in aqueous systems, resulting in significant improvement of its sorption capacity.182 3-dimensional hierarchical flower-like GO–hydroxyapatite (GO–HAp) nanocomposites were synthesized using a simple biomimetic method in a modified simulated body fluid.3 A maximum sorption capacity of 702.18 mg g−1 was achieved on GO–HAp, almost two fold higher than that of bare HAp and nine fold higher than that of GO (Fig. 13). Chen and co-workers prepared poly(amidoxime) grafted graphene (PAO-g-RGO) composites, which exhibited improved hydrophilicity and a maximum sorption capacity for Sr(II) of 99.4 mg g−1.145 In a similar manner, Sun et al. synthesized GO-supported polyaniline (PANI@GO) composites for Sr(II) removal from aqueous systems.142 The sorption of Sr(II) onto magnetite/GO (M/GO) was investigated by Chen et al. and they found that M/GO can be separated by magnetic separation from an aqueous system on a large scale.161
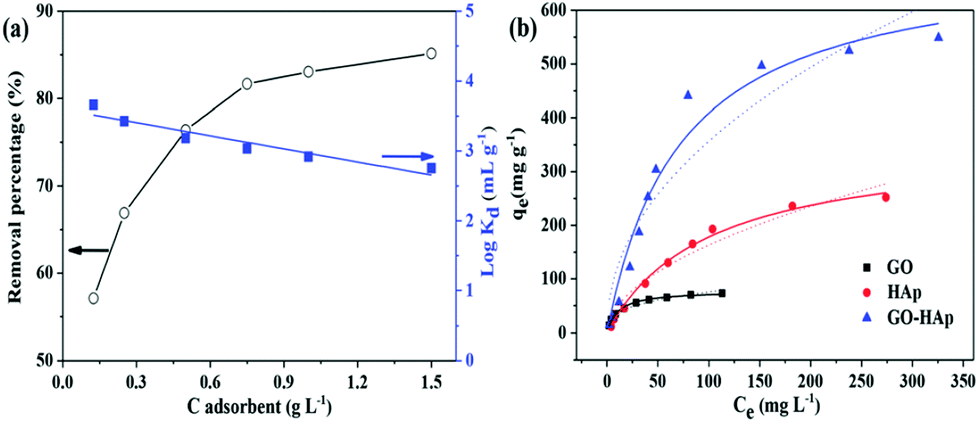 | ||
| Fig. 13 (a) Removal of Sr(II) from solution onto GO–HAp as a function of GO–HAp content, pH = 7.0 ± 0.1, C[Sr(II)]initial = 20 mg L−1, and I = 0.01 mol L−1 NaNO3. (b) Sorption isotherms of Sr(II) on GO, HAp and GO–HAp samples. Symbols denote experimental data, the solid lines represent the Langmuir model simulation, and the dashed lines represent the Freundlich model. All sorption isotherms were measured at pH = 7.0 ± 0.1, m/V = 0.5 g L−1, and I = 0.01 mol L−1 NaNO3.3 | ||
Molecular imprinting as a new molecular/ion recognition technology has developeds rapidly in recent years, and is a straightforward and versatile method for the preparation of molecular/ion imprinting polymers (MIPs/IIPs) with remarkable recognition properties for templates. A novel hydrophilic ion-imprinted polymer based on GO was synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization with the surface imprinting technique.172 Compared with traditional free radical polymerization (TRP-IIP) and non-imprinted polymers (NIPs), RAFT polymerization (RAFT-IIP) resulted in higher sorption capacity (145.77 mg g−1) and excellent selectivity for the rebinding of Sr(II).172
3.4 Other radionuclides
There are also other radionuclides that are removed efficiently by GO-based materials. Bai et al. reported that GO had a high sorption capacity (214.6 mg g−1) for Th(IV) because of the sufficient exposure of active sites of abundant oxygen-containing functional groups on the GO surface and the huge surface area of GO.60 RGO was prepared by the reduction of GO using thiourea dioxide (TUD) for the sorption of Th(IV), and the maximum sorption capacity was 0.21 mmol g−1.157Magnetic prussian blue (PB/Fe3O4) and PB/Fe3O4/GO nanocomposites were developed by Yang and co-workers for the removal of Cs(I) from water.164 The maximum sorption capacity of PB/Fe3O4/GO was 55.56 mg g−1, which was higher than that of PB/Fe3O4 (46.30 mg g−1). This can be explained by in situ anchoring of the magnetic prussian blue (PB) and PB onto the large surface area of GO, which efficiently increased the PB/Fe3O4/GO dispersion. They further synthesized PB/Fe3O4/GO nanocomposites encapsulated in calcium alginate microbeads (PFGM) for the elimination of Cs(I).173 The microbeads were stable in natural water, seawater, and solutions with pH values ranging from 4.0 to 10.0, without collapsing and leaching PB. Most importantly, these microbeads could be separated effectively from the aqueous system (or soil suspensions) using an external magnetic field, which was convenient for large-scale treatment of cesium-contaminated water.
4. Characterization of sorption
For the long-term performance assessment of nuclear waste repositories, knowledge of the interactions of radionuclides with GO-based materials is imperative. The fate of released radionuclides is strongly dependent on the sorption and desorption process occurring at GO-based material–water interface. Therefore, it is necessary to characterize the surface species formed and to elucidate the reaction mechanisms involved. The sorption of radionuclides depends on a variety of factors, such as temperature, pH, redox conditions and the concentration of complexing inorganic and organic ligands.180,183 The definition of sorption includes all adsorption, absorption, surface precipitation, and coprecipitation processes resulting in the removal of adsorbate from solution by the adsorbent.149 Herein, the sorption between radionuclides and GO-based materials mainly means a physical and chemical process that occurs when radionuclides accumulate on the surface or access the interior of GO-based materials by hydrogen bonding, intermolecular forces or chemical bond forces. Specific cases of sorption are covered by the following: surface complexation, ion exchange, precipitation, etc.74 The interactions between radionuclides and GO-based materials are mainly inner sphere sorption. There are a wide variety of available analytical techniques for understanding the speciation of radionuclides on surfaces and the reaction mechanisms of sorption, such as kinetic analysis, thermodynamic analysis, surface complexation models, spectroscopic techniques and theoretical calculations.4.1 Kinetic analysis
For the purpose of investigating the mechanisms of sorption and its potential rate-controlling steps, which include mass transport and the chemical reaction process, kinetic models have been exploited to test the experimental data.184 From kinetic analysis, the solute uptake rate, which determines the residence time required for completion of the sorption reaction, may be established. Also, one can know the scale of a sorption apparatus based on the kinetic information.185 Generally, sorption reactions take place rapidly in the initial stages and gradually slow down upon reaching the equilibrium state. The time to reach equilibrium varies with the adsorbates, adsorbents, initial concentration and the condition of the solution. Numerous kinetic models have been applied to fit the sorption process, including pseudo-first-order, pseudo-second-order, Lagergren, Zeldowitsch and Elovich kinetic models.186 Generally, the sorption of radionuclides follows the pseudo-second order model.The Eu(III) and U(VI) sorption by GO was evaluated, and the kinetic data were appropriately fitted with pseudo-second-order kinetics.45 Song et al. concluded that the kinetic sorption process included two main stages (Fig. 14): (I) initial rapid sorption (amount of 85–90%) extended over the first 2 h of contact time; and (II) slow approach to equilibrium covering about 6 h.174 The kinetics of U(VI) sorption on GO/PPy composites followed the pseudo-second order model, suggesting that strong chemical sorption or surface complexation was the main mechanism for the sorption of U(VI) ions on GO/PPy composites.144 The removal of Eu(III) by GTiP occurred within 20 min, and the strong interaction between Eu(III) and GTiP was due to the carboxyl and hydroxyl groups on GO as well as the phosphate groups on TiP.171
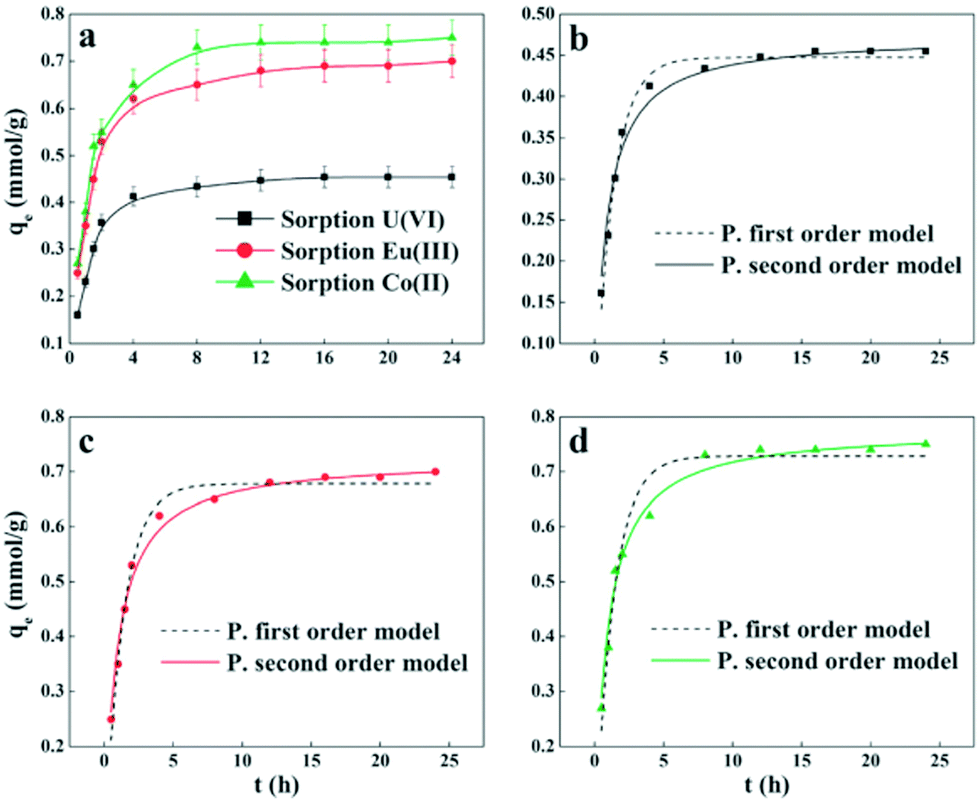 | ||
| Fig. 14 Effect of contact time on radionuclide sorption on PAM/GO (a); comparison of the two kinetic models of U(VI) (b); Eu(III) (c) and Co(II) (d) sorption on PAM/GO (pH = 5.0 ± 0.1, T = 295 K, m/V = 0.2 g L−1, Cinitial = 0.2 mmol L−1, I = 0.01 mol L−1 NaCl).174 | ||
4.2 Thermodynamic analysis
In general, a sorption isotherm is an invaluable curve describing the phenomenon governing the retention (or release) or mobility of a substance from the aqueous porous media or aquatic environments to a solid-phase at a constant temperature and pH.187 Its physicochemical parameters, together with the underlying thermodynamic assumptions, provide an insight into the sorption mechanism and surface properties as well as the degree of affinity of the adsorbents.188 Over the years, a wide variety of equilibrium isotherm models (i.e., Langmuir, Freundlich, Brunauer–Emmett–Teller, Redlich–Peterson, Dubinin–Radushkevich, Temkin, Toth, Koble–Corrigan, Sips, Flory–Huggins and Radke–Prausnitz isotherms), have been applied to describe the experimental data of sorption isotherms.189 The range of different models have been described in the references,45,59,153,158 so won't be covered again herein.Thermodynamic parameters are the result of the sorption process, and give subsequent important information on chemical engineering technologies and ultimate uptake of adsorbate onto the adsorbents. They also provide very useful information on the sorption mechanisms. The thermodynamic parameters, such as the Gibbs free energy change (ΔG), the enthalpy change (ΔH) and the entropy change (ΔS), calculated from temperature-dependent sorption data, were crucial to understanding the interaction behavior of radionuclides with GO-based materials. The thermodynamics of radionuclide sorption have been investigated extensively.145,154,158,163,167,170 Generally, the sorption of radionuclides on GO-based materials is an endothermic and spontaneous process.
The thermodynamic parameters for the sorption of U(VI) on GO and GO-supported chitosan (GO-Ch) were calculated to predict the nature of the sorption process.153 The removal of U(VI) increased with increasing temperature. Negative values of ΔG indicate the feasibility of the process and spontaneous nature of the sorption process. The positive value of ΔH (25.20 kJ mol−1) indicates the endothermic nature of the process. The positive value of ΔS (117.14 J mol−1 K−1) reflects the increased randomness at the solid–liquid interface, which also reveals that U(VI) sorption on GO-Ch and GO was a spontaneous process.153 In the studies on the capability and behavior of Eu(III) sorption on GO, the ΔH and ΔG values of Eu(III) on GO showed an endothermic and spontaneous process.45 Chen et al. calculated the ΔG, ΔH and ΔS values of Sr(II) sorption on M/GO to be −17.89 kJ mol−1, 9.18 kJ mol−1 and 89.34 J mol−1 K−1 at 303 K, respectively.161 Song et al. reported the ΔG, ΔH and ΔS values of Eu(III) sorption on PAM/GO to be −8.71 kJ mol−1, 3.96 kJ mol−1 and 29.53 J mol−1 K−1 at 295 K, respectively.174 The sorption of different radionuclides on different adsorbents is quite different, and the thermodynamic parameters of radionuclide sorption are dominated by the nature of radionuclides, nature of adsorbents, solution conditions, ionic strength, experimental conditions, etc.
4.3 Surface complexation models
Over the past forty years, surface complexation models (SCMs) have emerged as a powerful tool for describing sorption processes at solid–water interfaces.190,191 SCMs have enabled predictions of the impact of solution chemistry (e.g., pH and ionic strength) on the binding of inorganic aqueous solutes to solid surfaces with a single set of model parameters.192,193 The three widely-used forms of SCMs are the diffuse-layer model (DLM), constant-capacitance model (CCM), and triple-layer model (TLM).74 Among them, the DLM is the simplest and has been widely used to model the sorption behavior of radionuclides in GO-based materials.With the aid of FITEQL 3.2, Sr(II) sorption on M/GO was fitted using the DLM.161 The modeling results explained the changes in sorption morphology at different pH values and ionic strengths in detail (Fig. 15). The results showed that at pH < 4.5, ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SOHSr2+ was the dominant surface species; in a pH range from 4.5 to 9.0, SOSr+ was the dominant component among the surface species, and WOSr+ became the dominant surface species at pH > 9.0.
SOHSr2+ was the dominant surface species; in a pH range from 4.5 to 9.0, SOSr+ was the dominant component among the surface species, and WOSr+ became the dominant surface species at pH > 9.0.
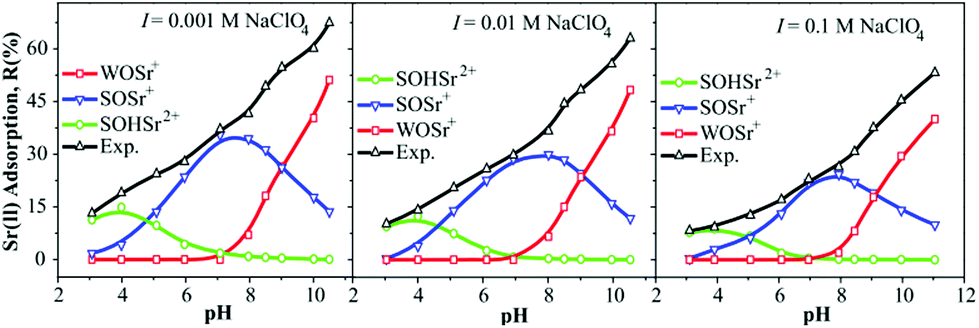 | ||
| Fig. 15 The modeling results of Sr(II) sorption onto the M/GO composite as a function of pH at different ionic strengths; m/V = 0.4 g L−1, T = 303 K, CSr(II) initial = 6.0 mg L−1, I = 0.01 mol L−1 NaClO4.161 | ||
Sun et al. simulated the sorption of Eu(III) and U(VI) on GO nanosheets using a diffuse double-layer model (DDLM) with the aid of FITEQL v4.0 code (Fig. 16),4,45 and they found that the logK values of U(VI) and Eu(III) were 4.62 and 4.23, respectively. The higher logK values substantiated the higher sorption capacity of U(VI) on GO nanosheets compared to Eu(III). They also found that the main species were SOM(n−1)+ and SOMOH(n−2)+ under low and high pH conditions, respectively. The main sorption reactions can be expressed by:
| SOH + Mn+ → SO M(n−1)+ + H+ | (1) |
| SOH + Mn+ + H2O → SO MOH(n−2)+ + 2H+ | (2) |
SOH represents the amphoteric hydroxyl groups on the surface of the GO nanosheets. Mn+ refers to Eu(III) or U(VI) cations.
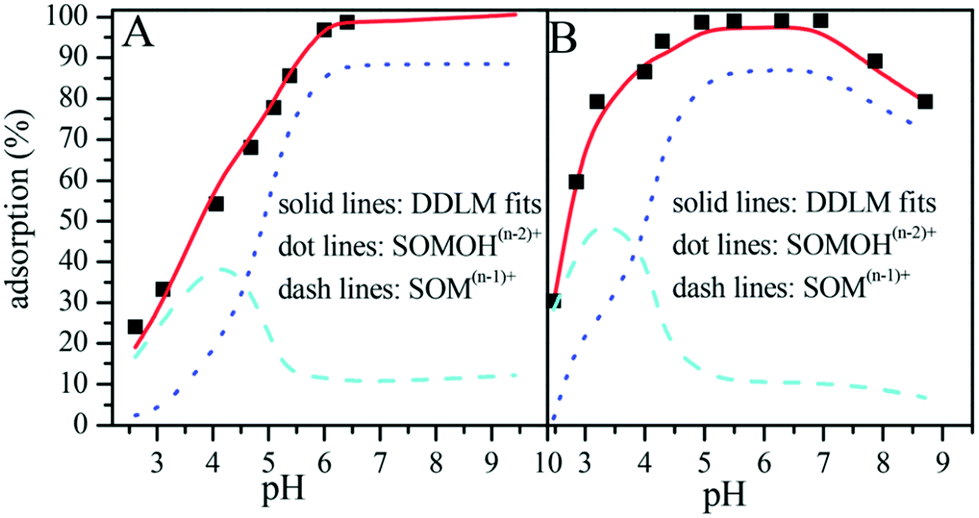 | ||
| Fig. 16 Simulation of sorption with a diffuse double-layer model (DDLM); A: Eu(III), B: U(VI); C0 = 10.0 mg L−1, m/V = 0.20 g L−1, I = 0.01 mol L−1 NaClO4, T = 303 K.45 | ||
The surface complexation model assumptions with regard to sorption mechanisms and surface speciation cannot be directly derived from wet chemistry data. Equilibrium-based models simply describe the macroscopic sorption data and do not definitively prove a reaction mechanism.194,195 Spectroscopic analysis can help to increase the reliability of model predictions by examining sorption mechanisms, surface speciation and local atomic structures at a molecular level. To confirm the main sorption sites, it is essential to identify binding sites using spectroscopic techniques.196
4.4 Spectroscopic techniques
Surface spectroscopic techniques can directly probe the chemical identity and the structure of the surface complexes formed between radionuclide ions and surface sites. With growing challenges for radionuclide sorption investigations, spectroscopic techniques experienced a boom over the past decade.197,198 Extended X-ray absorption fine structure (EXAFS) spectroscopy, X-ray absorption near-edge structure (XANES) spectroscopy, X-ray photoelectron spectroscopy (XPS), Fourier transform infrared (FTIR) spectroscopy and Raman spectroscopy have been employed to provide information for the analysis of radionuclide sorption mechanisms at a molecular level.X-ray absorption fine structure (XAFS), consisting of XANES spectroscopy and EXAFS spectroscopy, has proved to be a powerful tool in characterization of radionuclide structures on solid surfaces. The sorption behaviors and interaction mechanisms of radionuclides with various traditional environment-associated materials, such as clay minerals199–202 and oxides203–205 have been extensively studied. To investigate the local atomic structure of U(VI) adsorbed on GO, Li et al. applied EXAFS spectroscopy to analyze the interaction between U(VI) and GO (Fig. 17).59 They found that the sorption mechanism of the surface complexation with U(VI) and the coordination number of U(VI) was estimated to be ∼5 in the equatorial plane of U(VI). Bai et al. investigated the interaction mechanism of Th(IV) and GO by EXAFS spectroscopy,60 and Th(IV) was found to bind to ∼8 or 9 O atoms and the average bond length of Th–O was estimated to be ∼2.45 Å in the first coordination shell. The abundant oxygen-containing functional groups on the surfaces of GO therefore have the capacity to form stable inner-sphere complexes with Th4+ rather than simple physical sorption.
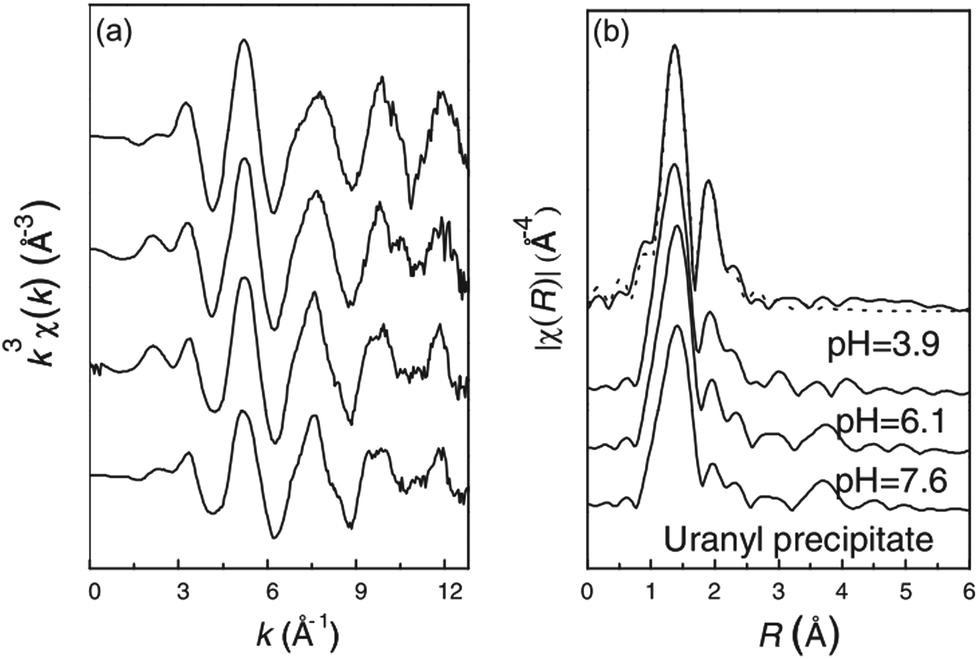 | ||
| Fig. 17 (a) Raw U LIII-edge k3-weighted χ(k) EXAFS spectra and (b) their corresponding Fourier transforms of U(VI) adsorbed on GO sheets at pH 3.9, 6.1 and 7.6. C0(U) = 0.5 mmol L−1, CGO = 0.4 g L−1, and contact time = 24 h. The bottom-most spectrum is from uranyl hydroxide precipitate prepared as a solubility-limiting phase.59 | ||
The interaction mechanism between Eu(III) and GO nanosheets (GONS) was determined, from mononuclear monodentate complexes to binuclear bidentate surface complexes with increasing pH, by using EXAFS spectroscopy.4 The results of the EXAFS spectra analysis indicated that Eu(III) bonded to ∼6–7 O atoms at a bond distance of ∼2.44 Å in the first coordination shell. The sorption of Eu(III) on mesoporous Al2O3/expanded graphite (EG) composites was observed by EXAFS (Fig. 18).165 The sorption mechanism between Eu(III) and mesoporous Al2O3/EG composites shifts from outer-sphere surface complexation to inner-sphere surface complexation with increasing pH values. Sun et al. studied the U(VI)-reacted nZVI/RGO composites using XANES and EXAFS.46 The XANES spectra indicated that the extent of U(VI) reduction to U(IV) significantly increased with increasing reaction time. EXAFS spectra analysis showed the formation of inner-sphere surface complexes on the nZVI/RGO composites.
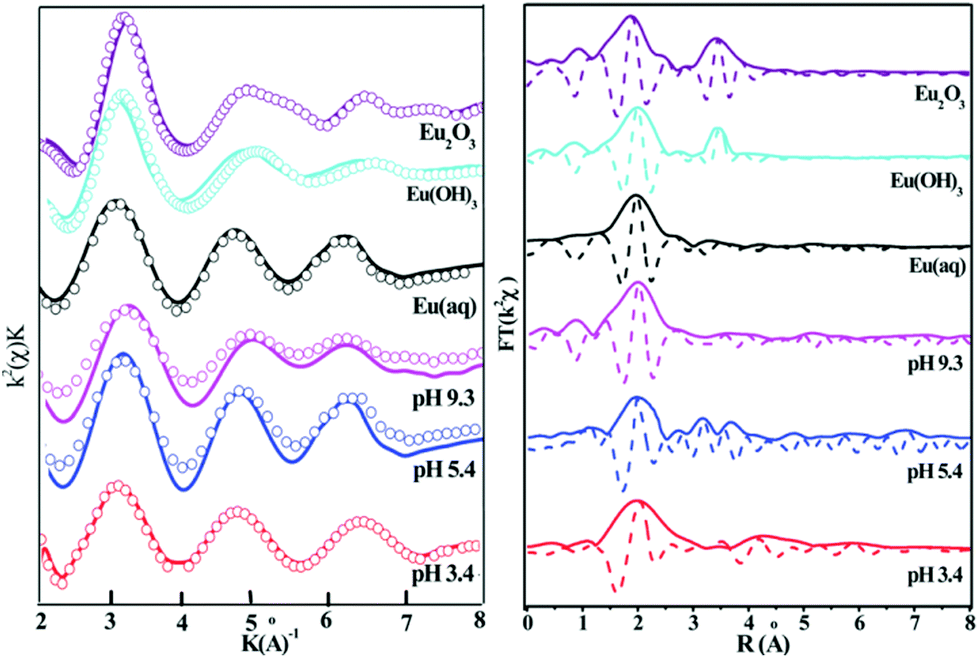 | ||
| Fig. 18 The k2-weighted Eu LIII-edge EXAFS spectra (A) and the corresponding Fourier transforms (B) of the reference samples and selected sorption samples at different pH values; C (Eu(III)) = 10 mg L−1, m/V = 1.2 g L−1, I = 0.01 mol L−1 NaClO4, T = 293 K.165 | ||
The XPS spectroscopy technique has been used to determine the interaction mechanism between radionuclides and GO-based materials.3,45,46,166 According to the XPS analysis, the irreversible sorption of U(VI) on GO nanosheets at variable radionuclide concentrations was attributed to the oxygen-containing functional groups (e.g., hydroxyl and carboxyl groups).45 Curve-fitting analysis of the XPS spectra demonstrated that the significant improvement of U(VI) removal by the nZVI/RGO composites was attributed to the enrichment of Fe2+ on the RGO surface and the large number of chemisorbed OH− groups on RGO.46 The interaction mechanism between Sr(II) and the GO–HAp composite was investigated by XPS.3 There were two mechanisms of Sr(II) sorption: ion-exchange and the formation of complex compounds with HAp active surface sites. The main sorption reactions can be described as:
| HAp-Ca2+ + Sr2+ → HAp-O-Sr2+ + Ca2+ | (3) |
| HAp-OH + Sr2+ → HAp-O-Sr2+ + H2+ | (4) |
| 2HAp-OH + Sr2+ → (HAp-O)2Sr2+ + 2H+ | (5) |
The adsorbed U(VI) on the PANI/GO composite surface was confirmed using FTIR and XPS spectroscopy techniques (Fig. 19).166 They suggested that the –COOH group was an effective active site for the sorption of U(VI) ions. Moreover, amino groups can also coordinate with U(VI) ions through electrostatic interactions and hydrogen bonds. The sorption reaction equations of U(VI) on the surface of the PANI/GO composite can be described as follows:
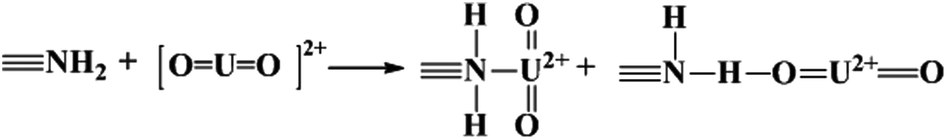 | (6) |
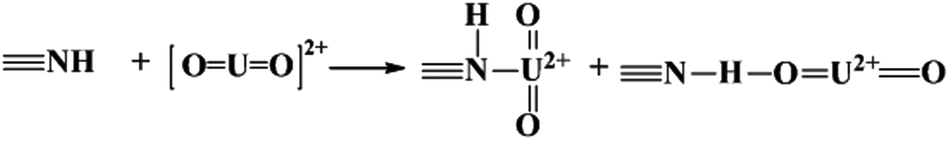 | (7) |
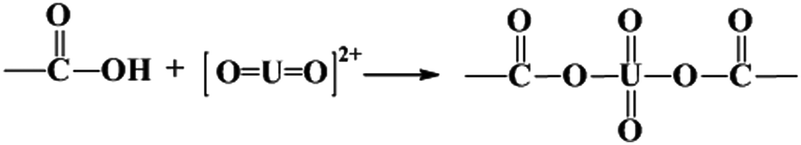 | (8) |
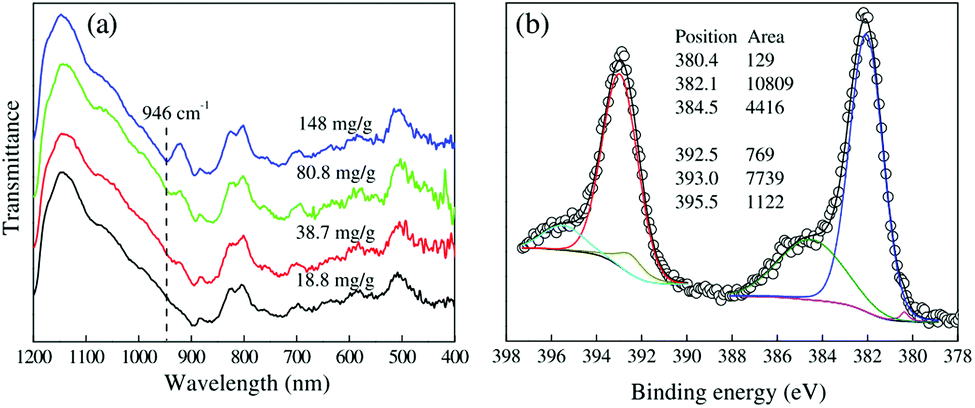 | ||
| Fig. 19 Characterization of U(VI)-loaded PANI/GO. (a) FTIR spectra of PANI/GO after the sorption of U(VI). (b) XPS U4f5/2 and U4f7/2 peaks of PANI/GO after the sorption of U(VI).166 | ||
4.5 Theoretical calculation
In addition to the increasing importance and remarkable improvement of the experimental surface methods, theoretical calculation is also becoming routinely used as an indispensable tool in chemical research. As one of the most useful theoretical tools, density functional theory (DFT) arises mainly because of its ability of accounting for the correlation energy in a very efficient way, which is usually applied to describe the sorption of molecules on a surface as a localized interaction.206 DFT is adopted to give an insight into the sorption process at an atomic level and to interpret experimental results from XPS, TRLFS, EXAFS, etc.207–209 DFT calculations yield structural, force, and total energy information, as well as the system charge density. These properties enable comparison and connection with experimental results and identification of underlying mechanisms through analysis of the electronic structure.196Shi et al. investigated the interaction mechanism between Th(IV) and GO using DFT calculations.60 Seven structures with Th(IV) coordinated to carboxyl or hydroxyl oxygen atoms on the edge and middle sites of GO were optimized (Fig. 20).60 They found that three of the nitrate anions acted as bidentate ligands and the other nitrate anions coordinated in a monodentate fashion to Th4+ for structures A to C, whereas all the nitrate anions were coordinated as bidentate ligands for D to G. All the Th–O bond lengths for the hydroxyl oxygen atoms were longer than those for the carboxyl oxygen atoms, which suggested that the carboxyl oxygen atoms had a stronger coordination ability with Th(IV) than the hydroxyl oxygen atoms.
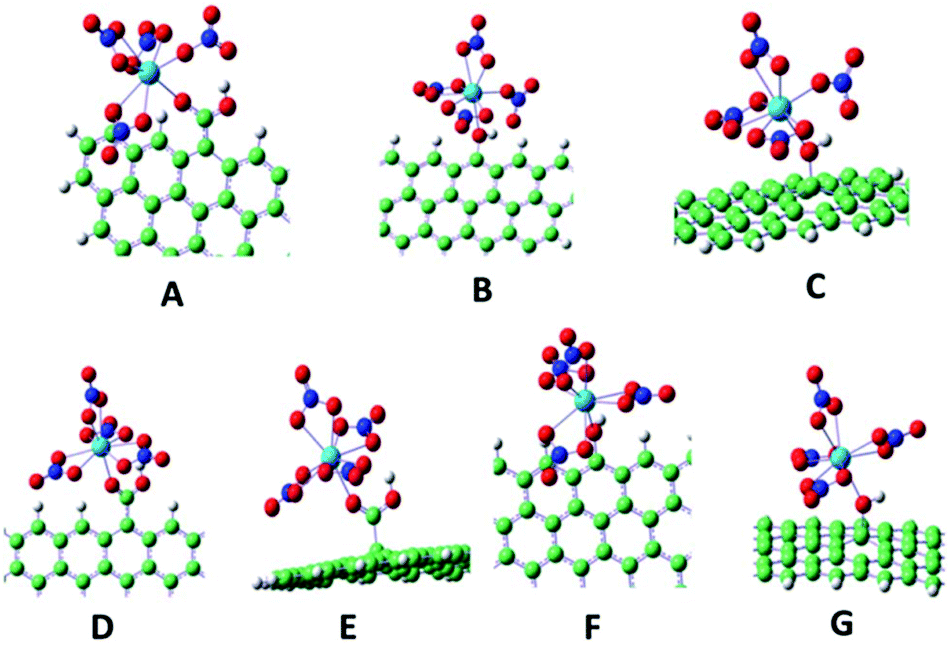 | ||
| Fig. 20 Optimized binding geometries for the coordination of Th(IV) with GO. Red, white, blue, green and light blue spheres represent O, H, N, C and Th, respectively.60 | ||
The bond orders and atomic charges between U(VI) and GO were investigated and the calculated Wiberg bond indices (WBIs) of the U–O bonds and Mulliken atomic charges on the U and O atoms of GO for the uranyl/GO complexes were reported.61 They suggested that electrostatic interactions dominated the U–O bonds for neutral GO/uranyl complexes. They also gave a hint that the U–OG bonds had more covalent character than the U–NG bonds. Furthermore, the Mulliken atomic charges on the nitrogen atoms were larger than those of the oxygen atoms, which also supported that the U–OG bonds had more covalent character.
To analyze the interaction modes between radionuclides and GO, Yang et al. investigated individual and competitive sorption of Pb(II), Ni(II) and Sr(II) on GOs using DFT calculations.210 The results (Fig. 21) indicated that Pb(II) and Ni(II) preferred to interact with COH groups while Sr(II) interacted with COH and COC comparably. Furthermore, Pb(II) tended to absorb OH from GOs because of the small energy barrier and very stable Pb(OH)⋯GO product, which was different from Ni(II) and Sr(II). The DFT calculation results further supported the experimental ones, i.e., the interaction of Pb(II) with GO was stronger than that of Ni(II) and Sr(II) with GO.
 | ||
| Fig. 21 The DFT-optimized geometry of the complexes composed of Pb(II), Ni(II), Sr(II) and GO.210 | ||
Using the first-principle density-functional theory, the sorption and desorption of four kinds of main radioactive products (Cs, I, Sr and Ag) on a graphite surface in high temperature gas cooled reactors (HTGRs) were studied by Luo et al.211 It turned out that the sorption of Cs, I and Sr atoms was chemisorption while the sorption of Ag was a pure physisorption (Fig. 22). When a vacancy was introduced in the graphite surface, nuclide adatoms were trapped by the vacancy and formed chemical bonds with three nearest neighbor carbon atoms, leading to a significant increase in the sorption energy, especially for Ag, whose sorption changed from physisorption to chemisorption.
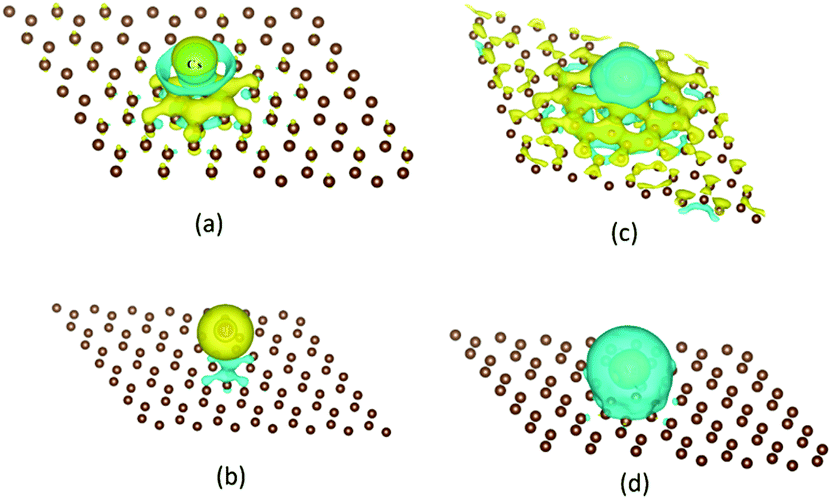 | ||
| Fig. 22 The sketch of charge density difference to show electron transfer between an adatom and graphite surface, calculated with 6 × 6 primitive cells. (a) Cs, (b) I, (c) Sr, (d) Ag. Yellow represents positive electronic clouds that accept from others and cyan represents negative electronic clouds that donate to others.211 | ||
The structures, bonding nature, and binding energies of Np(V) and Pu(IV, VI) complexes with GO were investigated systematically using scalar-relativistic DFT.47 The highest occupied molecular orbit (HOMO) and the lowest unoccupied molecular orbit (LUMO) diagrams of four types of GO are presented in Fig. 23. It was found that Np(V) and Pu(VI) complexes with GO were five-coordinate at the equatorial plane, while Pu(IV) complexes preferred eight-coordination. The bond between Pu(IV) ions and GO possessed more covalency, whereas electrostatic interaction dominated the Np/Pu–OG bond. Koh et al. studied the sorption and vibrations of 235UF6 and 238UF6 on graphene-based materials, including single and double layered graphene, graphane (hydrogenated graphene) and fluorographene using generalized gradient approximation (GGA) DFT as well as dispersion corrected DFT (DFT-D).212 Different graphene-based materials showed a range of sorption strengths, from physisorption on fluorographene to chemisorption on graphane and (single or double layer) graphene.
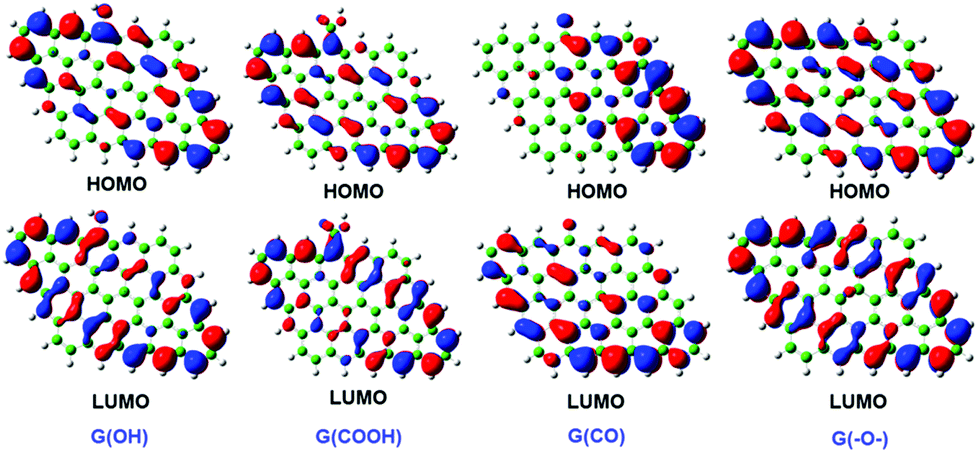 | ||
| Fig. 23 HOMO and LUMO diagrams of G(OH), G(COOH), G(CO), and G(–O–).47 | ||
5. Conclusions and future work
Graphene oxide has unique morphology, chemical structure and sorption properties. With respect to its sorption properties and applications, the main advantages of GO-based materials include their high surface area and functionalized surfaces. This paper reviews the applications of GO-based materials in the sorption of radionuclides from aqueous systems. The sorption mechanisms can be characterized by the application of kinetic analysis, thermodynamic analysis, surface complexation models, spectroscopic techniques and theoretical calculations to scrutinize and quantify radionuclide speciation and their interface reactions.Much work remains to be done in facilitating the practical applications of GO-based materials and broadening the sorption character of radionuclides in the future. Specific attention should be focused on the synthesis and functionalization of GO-based materials. The large scale production of GO by current methods is limited by the concentrated acids required and the variability in sheet size, as well as surface functionality. Furthermore, we should also pay attention to the complete determination of the structure of GO-based materials at the molecular level, further exploration of the sorption properties of GO-based materials and a better understanding of the effect of defects on the sorption properties of GO-based materials. Despite the number of remaining challenges, it is clear that GO-based materials offer many advantages for promising applications in the removal of radionuclides, and researchers are now making rapid progress in this area. Undoubtedly, the future of GO-based materials remains very bright and exciting.
In the context of safety evaluations of nuclear waste disposal, thorough analysis is needed for the understanding of sorption mechanisms of radionuclides at solid–water interfaces. The combination of batch techniques, theoretical modelling and spectroscopy analysis at the molecular level is necessary for understanding the sorption behavior of radionuclides on GO-based materials. The regeneration of the adsorbent is likely to be a key factor for improving wastewater process economics. Furthermore, the renewable radionuclides may be used for nuclear energy. By studying the dissolution and desorption behavior of radionuclides at solid–water interfaces, we believe that we can deeply understand the recycling properties of adsorbents. As a final word of recommendation we would like to point out that GO-based materials are promising materials in the application of nuclear waste management or radioactive contamination treatment.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21377132, 21225730 and 91326202), the Jiangsu Provincial Key Laboratory of Radiation Medicine and Protection and the Priority Academic Program Development of Jiangsu Higher Education Institutions are acknowledged.References
- N. Chapman and A. Hooper, Proc. Geol. Assoc., 2011, 123, 46–63 CrossRef PubMed.
- K. Buesseler, M. Aoyama and M. Fukasawa, Environ. Sci. Technol., 2011, 45, 9931–9935 CrossRef CAS PubMed.
- T. Wen, X. L. Wu, M. C. Liu, Z. H. Xing, X. K. Wang and A. W. Xu, Dalton Trans., 2014, 43, 7464–7472 RSC.
- Y. B. Sun, Q. Wang, C. L. Chen, X. L. Tan and X. K. Wang, Environ. Sci. Technol., 2012, 46, 6020–6027 CrossRef CAS PubMed.
- Y. Z. Tang and R. J. Reeder, Geochim. Cosmochim. Acta, 2009, 73, 2727–2743 CrossRef CAS PubMed.
- X. X. Wang, S. W. Zhang, J. X. Li, J. Z. Xu and X. K. Wang, Inorg. Chem. Front., 2014, 1, 641–648 RSC.
- J. R. Bargar, R. Reitmeyer, J. J. Lenhart and J. A. Davis, Geochim. Cosmochim. Acta, 2000, 64, 2737–2749 CrossRef CAS.
- Y. B. Sun, S. T. Yang, G. D. Sheng, Z. Q. Guo, X. L. Tan, J. Z. Xu and X. K. Wang, Sep. Purif. Technol., 2011, 83, 196–203 CrossRef PubMed.
- D. D. Shao, Z. Q. Jiang, X. K. Wang, J. X. Li and Y. D. Meng, J. Phys. Chem. B, 2009, 113, 860–864 CrossRef CAS PubMed.
- Q. H. Fan, M. Tanaka, K. Tanaka, A. Sakaguchi and Y. Takahashi, Geochim. Cosmochim. Acta, 2014, 135, 49–65 CrossRef CAS PubMed.
- W. Yang and A. Zaoui, J. Hazard. Mater., 2013, 261, 224–234 CrossRef CAS PubMed.
- P. K. Verma, P. N. Pathak, P. K. Mohapatra, S. V. Godbole, R. M. Kadam, A. A. Veligzhanin, Y. V. Zubavichus and S. N. Kalmykov, Environ. Sci.: Processes Impacts, 2014, 16, 904–915 CAS.
- S. Kerisit and C. X. Liu, Environ. Sci. Technol., 2014, 48, 3899–3907 CrossRef CAS PubMed.
- S. Choung, M. Kim, J. S. Yang, M. G. Kim and W. Um, Environ. Sci. Technol., 2014, 48, 9684–9691 CrossRef CAS PubMed.
- Z. M. Wang, J. M. Zachara, J. Y. Shang, C. Jeon, J. Liu and C. X. Liu, Environ. Sci. Technol., 2014, 48, 7766–7773 CrossRef CAS PubMed.
- X. L. Li, W. J. Mu, X. Xie, B. J. Liu, H. Tang, G. H. Zhou, H. Y. Wei, Y. Jian and S. Z. Luo, J. Hazard. Mater., 2014, 264, 386–394 CrossRef CAS PubMed.
- W. C. Song, M. C. Liu, R. Hu, X. L. Tan and J. X. Li, Chem. Eng. J., 2014, 246, 268–276 CrossRef CAS PubMed.
- Y. G. Zhao, J. X. Li, L. P. Zhao, S. W. Zhang, Y. S. Huang, X. L. Wu and X. K. Wang, Chem. Eng. J., 2014, 235, 275–283 CrossRef CAS PubMed.
- S. T. Yang, P. F. Zong, J. Hu, G. D. Sheng, Q. Wang and X. K. Wang, Chem. Eng. J., 2013, 214, 376–385 CrossRef CAS PubMed.
- S. T. Yang, G. D. Sheng, G. Montavon, Z. Q. Guo, X. L. Tan, B. Grambow and X. K. Wang, Geochim. Cosmochim. Acta, 2013, 121, 84–104 CrossRef CAS PubMed.
- M. S. Mauter and M. Elimelech, Environ. Sci. Technol., 2008, 42, 5843–5859 CrossRef CAS.
- X. Y. Zhang, J. L. Yin, C. Peng, W. Q. Hu, Z. Y. Zhu, W. X. Li, C. H. Fan and Q. Huang, Carbon, 2011, 49, 986–995 CrossRef CAS PubMed.
- Y. Chang, S. T. Yang, J. H. Liu, E. Donga, Y. Wang, A. Cao, Y. Liu and H. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef CAS PubMed.
- O. Akhavan and E. Ghaderi, ACS Nano, 2010, 4, 5731–5736 CrossRef CAS PubMed.
- E. C. Salas, Z. Sun, A. Luttge and J. M. Tour, ACS Nano, 2010, 4, 4852–4856 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS PubMed.
- Y. K. Zhang, T. Yan, L. G. Yan, X. Y. Guo, L. M. Cui, Q. Wei and B. Du, J. Mol. Liq., 2014, 198, 381–387 CrossRef CAS PubMed.
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS PubMed.
- S. F. Pei and H. M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS PubMed.
- L. Liu, Y. R. Zhang, Y. J. He, Y. F. Xie, L. H. Huang, S. Z. Tan and X. Cai, RSC Adv., 2015, 5, 3965–3973 RSC.
- N. Sui, L. N. Wang, X. H. Wu, X. H. Li, J. Sui, H. L. Xiao, M. H. Liu, J. Wan and W. W. Yu, RSC Adv., 2015, 5, 746–752 RSC.
- L. Y. Chai, T. Wang, L. Y. Zhang, H. Y. Wang, W. C. Yang, S. Dai, Y. Meng and X. R. Li, Carbon, 2015, 81, 748–757 CrossRef CAS PubMed.
- X. Zhou, W. Y. Huang, J. Shi, Z. X. Zhao, Q. B. Xia, Y. W. Li, H. H. Wang and Z. Li, J. Mater. Chem. A, 2014, 2, 4722–4730 CAS.
- R. Hu, S. Y. Dai, D. D. Shao, A. Alsaedi, B. Ahmad and X. K. Wang, J. Mol. Liq., 2015, 203, 80–89 CrossRef CAS PubMed.
- Y. Shen, Q. L. Fang and B. L. Chen, Environ. Sci. Technol., 2015, 49, 67–84 CrossRef CAS PubMed.
- A. H. Hung, R. J. Holbrook, M. W. Rotz, C. J. Glasscock, N. D. Mansukhani, K. W. MacRenaris, L. M. Manus, M. C. Duch, K. T. Dam, M. C. Hersam and T. J. Meade, ACS Nano, 2014, 8, 10168–10177 CrossRef CAS PubMed.
- L. Kong, A. Enders, T. S. Rahman and P. A. Dowben, J. Phys.: Condens. Matter, 2014, 26, 443001–443028 CrossRef PubMed.
- X. Y. Liu and J. M. Zhang, Appl. Surf. Sci., 2014, 293, 216–219 CrossRef CAS PubMed.
- J. P. Sun, Q. L. Liang, Q. Han, X. Q. Zhang and M. Y. Ding, Talanta, 2015, 132, 557–563 CrossRef CAS PubMed.
- L. M. Cui, X. Y. Guo, Q. Wei, Y. G. Wang, L. Gao, L. G. Yan, T. Yan and B. Du, J. Colloid Interface Sci., 2015, 439, 112–120 CrossRef CAS PubMed.
- S. Yang, L. Y. Li, Z. G. Pei, C. M. Li, X. Q. Shan, B. Wen, S. Z. Zhang, L. R. Zheng, J. Zhang, Y. N. Xie and R. X. Huang, Carbon, 2014, 75, 227–235 CrossRef CAS PubMed.
- C. C. Ding, W. C. Cheng, Y. B. Sun and X. K. Wang, Dalton Trans., 2014, 43, 3888–3896 RSC.
- Y. B. Sun, C. C. Ding, W. C. Cheng and X. K. Wang, J. Hazard. Mater., 2014, 280, 399–408 CrossRef CAS PubMed.
- Q. Y. Wu, J. H. Lan, C. Z. Wang, Y. L. Zhao, Z. F. Chai and W. Q. Shi, J. Phys. Chem. A, 2014, 118, 10273–10280 CrossRef CAS PubMed.
- C. Lattao, X. Y. Cao, J. D. Mao, K. Schmidt-Rohr and J. J. Pignatello, Environ. Sci. Technol., 2014, 48, 4790–4798 CrossRef CAS PubMed.
- X. B. Wang, S. S. Huang, L. H. Zhu, X. L. Tian, S. H. Li and H. Q. Tang, Carbon, 2014, 69, 101–112 CrossRef CAS PubMed.
- X. Y. Guo, Q. Wei, B. Du, Y. K. Zhang and X. D. Xin, Appl. Surf. Sci., 2013, 284, 862–869 CrossRef CAS PubMed.
- G. X. Zhao, D. D. Shao, C. L. Chen and X. K. Wang, Appl. Phys. Lett., 2011, 98, 183114 CrossRef PubMed.
- Y. B. Sun, S. B. Yang, G. X. Zhao, Q. Wang and X. K. Wang, Chem. – Asian J., 2013, 8, 2755–2761 CrossRef CAS PubMed.
- G. X. Zhao, X. M. Ren, X. Gao, X. L. Tan, J. X. Li, C. L. Chen, Y. Y. Huang and X. K. Wang, Dalton Trans., 2011, 40, 10945–10952 RSC.
- G. X. Zhao, J. X. Li, X. M. Ren, C. L. Chen and X. K. Wang, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS PubMed.
- G. X. Zhao, J. X. Li and X. K. Wang, Chem. Eng. J., 2011, 173, 185–190 CrossRef CAS PubMed.
- T. Wen, X. L. Wu, X. L. Tan, X. K. Wang and A. W. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 3304–3311 CAS.
- X. L. Wu, L. Wang, C. L. Chen, A. W. Xu and X. K. Wang, J. Mater. Chem., 2011, 21, 17353–17359 RSC.
- M. C. Liu, C. L. Chen, J. Hu, X. L. Wu and X. K. Wang, J. Phys. Chem. C, 2011, 115, 25234–25240 CAS.
- Z. J. Li, F. Chen, L. Y. Yuan, Y. L. Liu, Y. L. Zhao, Z. F. Chai and W. Q. Shi, Chem. Eng. J., 2012, 210, 539–546 CrossRef CAS PubMed.
- Z. Q. Bai, Z. J. Li, C. Z. Wang, L. Y. Yuan, Z. R. Liu, J. Zhang, L. R. Zheng, Y. L. Zhao, Z. F. Chai and W. Q. Shi, RSC Adv., 2014, 4, 3340–3347 RSC.
- Q. Y. Wu, J. H. Lan, C. Z. Wang, C. L. Xiao, Y. L. Zhao, Y. Z. Wei, Z. F. Chai and W. Q. Shi, J. Phys. Chem. A, 2014, 118, 2149–2158 CrossRef CAS PubMed.
- J. M. Tour, A. Slesarev, D. Kosynkin, A. Y. Romanchuk and S. N. Kalmykov, PCT Int. Appl, WO 2012170086 A1, 2012 Search PubMed.
- A. G. Gallastegui, M. Shaffer, A. O. Alyoubi and S. Basahel, PCT Int. Appl, WO 2013132259 A1, 2013 Search PubMed.
- H. P. Cong, J. F. Chen and S. H. Yu, Chem. Soc. Rev., 2014, 43, 7295–7325 RSC.
- X. Wang, B. Liu, Q. P. Lu and Q. S. Qu, J. Chromatogr. A, 2014, 1362, 1–15 CrossRef CAS PubMed.
- K. C. Kemp, H. Seema, M. Saleh, N. H. Le, K. Mahesh, V. Chandra and K. S. Kim, Nanoscale, 2013, 5, 3149–3171 RSC.
- S. B. Wang, H. Q. Sun, H. M. Ang and M. O. Tadé, Chem. Eng. J., 2013, 226, 336–347 CrossRef CAS PubMed.
- Q. Wang, X. K. Wang, Z. F. Chai and W. P. Hu, Chem. Soc. Rev., 2013, 42, 8821–8834 RSC.
- F. Kepák, Isotopenpraxis, 1988, 24, 1–5 Search PubMed.
- F. Macášek, NATO ASI Ser., Ser. E, 1999, 362, 109–135 Search PubMed.
- D. Li and D. I. Kaplan, J. Hazard. Mater., 2012, 243, 1–18 CrossRef CAS PubMed.
- N. Das, Clean: Soil, Air, Water, 2012, 40, 16–23 CrossRef CAS PubMed.
- A. Stockdale and N. D. Bryan, Earth-Sci. Rev., 2013, 121, 1–17 CrossRef CAS PubMed.
- X. L. Tan, X. M. Ren, C. L. Chen and X. K. Wang, Trends Anal. Chem., 2014, 61, 107–132 CrossRef CAS PubMed.
- A. Dahal and M. Batzill, Nanoscale, 2014, 6, 2548–2562 RSC.
- V. Chabot, D. Higgins, A. P. Yu, X. C. Xiao, Z. W. Chen and J. J. Zhang, Energy Environ. Sci., 2014, 7, 1564–1596 CAS.
- J. Zhao, Z. Y. Wang, J. C. White and B. S. Xing, Environ. Sci. Technol., 2014, 48, 9995–10009 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- H. Y. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- J. Rafiee, M. A. Rafiee, Z. Z. Yu and N. Koratkar, Adv. Mater., 2010, 22, 2151–2154 CrossRef CAS PubMed.
- X. Q. Zhang, S. H. Wan, J. B. Pu, L. P. Wang and X. Q. Liu, J. Mater. Chem., 2011, 21, 12251–12258 RSC.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Y. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- A. Dato, V. Radmilovic, Z. Lee, J. Phillips and M. Frenklach, Nano Lett., 2008, 8, 2012–2016 CrossRef CAS PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- B. C. Brodie, Philos. Trans. R. Soc. London, Ser. A, 1859, 14, 249–259 CrossRef.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481–1487 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- Z. H. Tang, H. L. Kang, Z. L. Shen, B. C. Guo, L. Q. Zhang and D. M. Jia, Macromolecules, 2012, 45, 3444–3451 CrossRef CAS.
- L. M. Veca, F. S. Lu, M. J. Meziani, L. Cao, P. Y. Zhang, G. Qi, L. W. Qu, M. Shrestha and Y. P. Sun, Chem. Commun., 2009, 2565–2567 RSC.
- Y. S. Liu, J. Y. Zhou, X. L. Zhang, Z. B. Liu, X. J. Wan, J. G. Tian, T. Wang and Y. S. Chen, Carbon, 2009, 47, 3113–3121 CrossRef CAS PubMed.
- H. F. Yang, C. S. Shan, F. H. Li, D. X. Han, Q. X. Zhang and L. Niu, Chem. Commun., 2009, 3880–3882 RSC.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabó, A. Szeri and I. Dékány, Langmuir, 2003, 19, 6050–6055 CrossRef CAS.
- R. Salvio, S. Krabbenborg, W. J. M. Naber, A. H. Velders, D. N. Reinhoudt and W. G. Wiel, Chem. – Eur. J., 2009, 15, 8235–8240 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS PubMed.
- C. Xu, X. D. Wu, J. W. Zhu and X. Wang, Carbon, 2008, 46, 386–389 CrossRef CAS PubMed.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W. F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS PubMed.
- Y. C. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- G. X. Zhao, L. Jiang, Y. D. He, J. X. Li, H. L. Dong, X. K. Wang and W. P. Hu, Adv. Mater., 2011, 23, 3959–3963 CrossRef CAS PubMed.
- Q. Wang, J. X. Li, Y. Song and X. K. Wang, Chem. – Asian J., 2013, 8, 225–231 CrossRef CAS PubMed.
- A. Dato, V. Radmilovic, Z. Lee, J. Phillips and M. Frenklach, Nano Lett., 2008, 8, 2012–2016 CrossRef CAS PubMed.
- Z. Y. Ji, X. P. Shen, J. L. Yang, G. X. Zhu and K. M. Chen, Appl. Catal., B, 2014, 144, 454–461 CrossRef CAS PubMed.
- X. M. Wen, P. Yu, Y. R. Toh, Y. C. Lee, K. Y. Huang, S. J. Huang, S. Shrestha, G. Conibeer and J. Tang, J. Mater. Chem. C, 2014, 2, 3826–3834 RSC.
- Y. K. Zhang, L. G. Yan, W. Y. Xu, X. Y. Guo, L. M. Cui, L. Gao, Q. Wei and B. Du, J. Mol. Liq., 2014, 191, 177–182 CrossRef CAS PubMed.
- S. Wang, B. M. Goh, K. K. Manga, Q. Bao, P. Yang and K. P. Loh, ACS Nano, 2010, 4, 6180–6186 CrossRef CAS PubMed.
- T. H. Han, W. J. Lee, D. H. Lee, J. E. Kim, E. Y. Choi and S. O. Kim, Adv. Mater., 2010, 22, 2060–2064 CrossRef CAS PubMed.
- N. Ruecha, R. Rangkupan, N. Rodthongkum and O. Chailapakul, Biosens. Bioelectron., 2014, 52, 13–19 CrossRef CAS PubMed.
- U. Rana, P. M. G. Nambissan, S. Malik and K. Chakrabarti, Phys. Chem. Chem. Phys., 2014, 16, 3292–3298 RSC.
- N. B. Trung, T. V. Tam, H. R. Kim, S. H. Hur, E. J. Kim and W. M. Choi, Chem. Eng. J., 2014, 255, 89–96 CrossRef CAS PubMed.
- C. X. Li, C. G. Hu, Y. Zhao, L. Song, J. Zhang, R. Huang and L. T. Qu, Carbon, 2014, 78, 231–242 CrossRef CAS PubMed.
- Q. Li, H. Y. Pan, D. Higgins, R. G. Cao, G. Q. Zhang, H. F. Lv, K. B. Wu, J. Cho and G. Wu, Small, 2015, 11, 1443–1452 CrossRef CAS PubMed.
- J. W. Liu, Y. Zhang, X. W. Chen and J. H. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 10196–10204 CAS.
- C. H. Lu, H. H. Yang, C. L. Zhu, X. Chen and G. N. Chen, Angew. Chem., Int. Ed., 2009, 121, 4879–4881 CrossRef PubMed.
- H. Chang, L. Tang, Y. Wang, J. Jiang and J. Li, Anal. Chem., 2010, 82, 2341–2346 CrossRef CAS PubMed.
- Y. Wang, Z. Li, D. Hu, C. T. Lin, J. Li and Y. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef CAS PubMed.
- Z. J. Yang, S. F. Luo, J. Li, J. Shen, S. H. Yu, X. Y. Hu and D. D. Dionysiou, Anal. Chim. Acta, 2014, 839, 67–73 CrossRef CAS PubMed.
- K. W. Kim, W. Song, M. W. Jung, M. A. Kang, S. Y. Kwon, S. Myung, J. Lim, S. S. Lee and K. S. An, Carbon, 2015, 82, 96–102 CrossRef CAS PubMed.
- V. K. Gupta, N. Atar, M. L. Yola, Z. Üstündağ and L. Uzun, Water Res., 2014, 48, 210–217 CrossRef CAS PubMed.
- Y. Wei, X. Zuo, X. Q. Li, S. S. Song, L. W. Chen, J. Shen, Y. Y. Meng, Y. Zhao and S. D. Fang, Mater. Res. Bull., 2014, 53, 145–150 CrossRef CAS PubMed.
- J. Liu, S. Fu, B. Yuan, Y. Li and Z. Deng, J. Am. Chem. Soc., 2010, 132, 7279–7281 CrossRef CAS PubMed.
- J. F. Shen, M. Shi, N. Li, B. Yan, H. W. Ma, Y. Z. Hu and M. X. Ye, Nano Res., 2010, 3, 339–349 CrossRef CAS PubMed.
- Q. Wang, M. M. Song, C. L. Chen, Y. Wei, X. Zuo and X. K. Wang, Appl. Phys. Lett., 2012, 101, 033103 CrossRef PubMed.
- Q. Wang, M. M. Song, C. L. Chen, W. P. Hu and X. K. Wang, ChemPlusChem, 2012, 77, 432–436 CrossRef CAS PubMed.
- L. N. Gao, W. B. Yue, S. S. Tao and L. Z. Fan, Langmuir, 2013, 29, 957–964 CrossRef CAS PubMed.
- Y. Zhao, D. L. Zhao, C. L. Chen and X. K. Wang, J. Colloid Interface Sci., 2013, 405, 211–217 CrossRef CAS PubMed.
- D. L. Zhao, G. D. Sheng, C. L. Chen and X. K. Wang, Appl. Catal., B, 2012, 111–112, 303–308 CrossRef CAS PubMed.
- C. Chen, W. M. Cai, M. C. Long, B. X. Zhou, Y. H. Wu, D. Y. Wu and Y. J. Feng, ACS Nano, 2010, 4, 6425–6432 CrossRef CAS PubMed.
- K. Dai, L. H. Lu, C. H. Liang, J. M. Dai, Q. Z. Liu, Y. X. Zhang, G. P. Zhu and Z. L. Liu, Electrochim. Acta, 2014, 116, 111–117 CrossRef CAS PubMed.
- S. Chen, J. W. Zhu, X. D. Wu, Q. F. Han and X. Wang, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS PubMed.
- H. J. Huang and X. Wang, Nanoscale, 2011, 3, 3185–3191 RSC.
- G. D. Sheng, Y. M. Li, X. Yang, X. M. Ren, S. T. Yang, J. Hu and X. K. Wang, RSC Adv., 2012, 2, 12400–12407 RSC.
- J. Li, S. W. Zhang, C. L. Chen, G. X. Zhao, X. Yang, J. X. Li and X. K. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 4991–5000 CAS.
- M. C. Liu, T. Wen, X. L. Wu, C. L. Chen, J. Hu, J. Li and X. K. Wang, Dalton Trans., 2013, 42, 14710–14717 RSC.
- J. Li, Z. Y. Shao, C. L. Chen and X. K. Wang, RSC Adv., 2014, 4, 38192–38198 RSC.
- A. H. Ye, W. Q. Fan, Q. H. Zhang, W. P. Deng and Y. Wang, Catal. Sci. Technol., 2012, 2, 969–978 CAS.
- P. Gao, J. C. Liu, D. D. Sun and W. Ng, J. Hazard. Mater., 2013, 250–251, 412–420 CrossRef CAS PubMed.
- A. N. Cao, Z. Liu, S. S. Chu, M. H. Wu, Z. M. Ye, Z. W. Cai, Y. L. Chang, S. F. Wang, Q. H. Gong and Y. F. Liu, Adv. Mater., 2010, 22, 103–106 CrossRef CAS PubMed.
- J. Chen, F. Xu, J. Wu, K. Qasim, Y. D. Zhou, W. Lei, L. T. Sun and Y. Zhang, Nanoscale, 2012, 4, 441–443 RSC.
- X. Yang, C. L. Chen, J. X. Li, G. X. Zhao, X. M. Ren and X. K. Wang, RSC Adv., 2012, 2, 8821–8826 RSC.
- Y. B. Sun, D. D. Shao, C. L. Chen, S. B. Yang and X. K. Wang, Environ. Sci. Technol., 2013, 47, 9904–9910 CrossRef CAS PubMed.
- S. W. Zhang, M. Y. Zeng, W. Q. Xu, J. X. Li, J. Li, J. Z. Xu and X. K. Wang, Dalton Trans., 2013, 42, 7854–7858 RSC.
- R. Hu, D. D. Shao and X. K. Wang, Polym. Chem., 2014, 5, 6207–6215 RSC.
- H. Chen, D. D. Shao, J. X. Li and X. K. Wang, Chem. Eng. J., 2014, 254, 623–634 CrossRef CAS PubMed.
- C. Petit and T. J. Bandosz, Adv. Mater., 2009, 21, 4753–4757 CAS.
- W. Z. Bao, Z. A. Zhang, Y. H. Qu, C. K. Zhou, X. W. Wang and J. Li, J. Alloys Compd., 2014, 582, 334–340 CrossRef CAS PubMed.
- S. J. Yu, H. Y. Mei, X. Chen, X. L. Tan, B. Ahmad, A. Alsaedi, T. Hayat and X. K. Wang, J. Mol. Liq., 2015, 203, 39–46 CrossRef CAS PubMed.
- K. A. Rod, W. Um and M. Flury, Environ. Sci. Technol., 2010, 44, 8089–8094 CrossRef CAS PubMed.
- S. İnan and Y. Altaş, Chem. Eng. J., 2011, 168, 1263–1271 CrossRef PubMed.
- R. A. Crane, M. Dickinson and T. B. Scott, Chem. Eng. J., 2015, 262, 319–325 CrossRef CAS PubMed.
- S. Handley-Sidhu, J. A. Hriljac, M. O. Cuthbert, J. C. Renshaw, R. A. D. Pattrick, J. M. Charnock, B. Stolpe, J. R. Lead, S. Baker and L. E. Macaskie, Environ. Sci. Technol., 2014, 48, 6891–6898 CrossRef CAS PubMed.
- W. C. Cheng, M. L. Wang, Z. G. Yang, Y. B. Sun and C. C. Ding, RSC Adv., 2014, 4, 61919–61926 RSC.
- G. X. Zhao, T. Wen, X. Yang, S. B. Yang, J. L. Liao, J. Hu, D. D. Shao and X. K. Wang, Dalton Trans., 2012, 41, 6182–6188 RSC.
- A. Y. Romanchuk, A. S. Slesarev, S. N. Kalmykov, D. V. Kosynkin and J. M. Tour, Phys. Chem. Chem. Phys., 2013, 15, 2321–2327 RSC.
- N. Pan, J. G. Deng, D. B. Guan, Y. D. Jin and C. Q. Xia, Appl. Surf. Sci., 2013, 287, 478–483 CrossRef CAS PubMed.
- S. P. Chen, J. X. Hong, H. X. Yang and J. Z. Yang, J. Environ. Radioact., 2013, 126, 253–258 CrossRef CAS PubMed.
- Y. G. Zhao, J. X. Li, S. W. Zhang, H. Chen and D. D. Shao, RSC Adv., 2013, 3, 18952–18959 RSC.
- H. X. Cheng, K. F. Zeng and J. T. Yu, J. Radioanal. Nucl. Chem., 2013, 298, 599–603 CrossRef CAS PubMed.
- Y. Li, G. D. Sheng and J. Sheng, J. Mol. Liq., 2014, 199, 474–480 CrossRef CAS PubMed.
- H. Chen, J. X. Li, S. W. Zhang, X. M. Ren, Y. B. Sun, T. Wen and X. K. Wang, Radiochim. Acta, 2013, 101, 785–794 CrossRef CAS.
- P. F. Zong, S. F. Wang, Y. L. Zhao, H. Wang, H. Pan and C. H. He, Chem. Eng. J., 2013, 220, 45–52 CrossRef CAS PubMed.
- L. C. Tan, J. Wang, Q. Liu, Y. B. Sun, X. Y. Jing, L. H. Liu, J. Y. Liu and D. L. Song, New J. Chem., 2015, 39, 868–876 RSC.
- H. J. Yang, L. Sun, J. L. Zhai, H. Y. Li, Y. Zhao and H. W. Yu, J. Mater. Chem. A, 2014, 2, 326–332 CAS.
- Y. B. Sun, C. L. Chen, X. L. Tan, D. D. Shao, J. X. Li, G. X. Zhao, S. B. Yang, Q. Wang and X. K. Wang, Dalton Trans., 2012, 41, 13388–13394 RSC.
- D. D. Shao, G. S. Hou, J. X. Li, T. Wen, X. M. Ren and X. K. Wang, Chem. Eng. J., 2014, 255, 604–612 CrossRef CAS PubMed.
- D. D. Shao, J. X. Li and X. K. Wang, Sci. China: Chem., 2014, 57, 1449–1458 CrossRef CAS.
- W. C. Song, D. D. Shao, S. S. Lu and X. K. Wang, Sci. China: Chem., 2014, 57, 1291–1299 CrossRef CAS.
- S. P. Dubey, A. D. Dwivedi, I. C. Kim, M. Sillanpaa, Y. N. Kwon and C. Lee, Chem. Eng. J., 2014, 244, 160–167 CrossRef CAS PubMed.
- L. C. Tan, Y. L. Wang, Q. Liu, J. Wang, X. Y. Jing, L. H. Liu, J. Y. Liu and D. L. Song, Chem. Eng. J., 2015, 259, 752–760 CrossRef CAS PubMed.
- C. R. Li, Y. Huang and Z. Lin, J. Mater. Chem. A, 2014, 2, 14979–14985 CAS.
- Y. Liu, X. G. Meng, M. Luo, J. Qiu, Z. Y. Hu, F. F. Liu, G. X. Zhong, Z. C. Liu, L. Ni and Y. S. Yan, J. Mater. Chem. A, 2015, 3, 1287–1297 CAS.
- H. J. Yang, H. Y. Li, J. L. Zhai, L. Sun, Y. Zhao and H. W. Yu, Chem. Eng. J., 2014, 246, 10–19 CrossRef CAS PubMed.
- W. C. Song, X. X. Wang, Q. Wang, D. D. Shao and X. K. Wang, Phys. Chem. Chem. Phys., 2015, 17, 398–406 RSC.
- B. Benedict, T. H. Pigford and H. W. Levi, Nuclear Chemical Engineering, McGrawHill, New York, 1981 Search PubMed.
- S. B. Xie, J. Yang, C. Chen, X. J. Zhang, Q. L. Wang and C. Zhang, J. Environ. Radioact., 2008, 99, 126–133 CrossRef CAS PubMed.
- U.S. EPA, EPA Integrated Risk Information System (IRIS) Electronic Database, U.S. Environmental Protection Agency, Washington, DC, 1996 Search PubMed.
- C. Li, H. Bai and G. Shi, Chem. Soc. Rev., 2009, 38, 2397–2409 RSC.
- T. Rabung, H. Geckeis, J. I. Kim and H. P. Beck, Radiochim. Acta, 1998, 82, 243–248 CAS.
- Q. H. Fan, X. L. Tan, J. X. Li, X. K. Wang, W. S. Wu and G. Montavon, Environ. Sci. Technol., 2009, 43, 5776–5782 CrossRef CAS.
- Z. Sofer, L. Wang, K. Klímová and M. Pumera, RSC Adv., 2014, 4, 26673–26676 RSC.
- Y. S. Huang, L. Taylor, X. Chen and N. Ayres, J. Polym. Sci., Part A: Pol. Chem., 2013, 24, 5230–5238 CrossRef PubMed.
- G. D. Sheng, R. P. Shen, H. P. Dong and Y. M. Li, Environ. Sci. Pollut. Res., 2013, 20, 3708–3717 CrossRef CAS PubMed.
- G. X. Zhao, X. L. Wu, X. L. Tan and X. K. Wang, Open Colloid Sci. J., 2011, 4, 19–31 CrossRef.
- F. C. Wu, R. L. Tseng, S. C. Huang and R. S. Juang, Chem. Eng. J., 2009, 151, 1–9 CrossRef CAS PubMed.
- Y. S. Ho, J. Hazard. Mater., 2006, 136, 681–689 CrossRef CAS PubMed.
- K. Y. Foo and B. H. Hameed, Chem. Eng. J., 2010, 156, 2–10 CrossRef CAS PubMed.
- E. Bulut, M. Ozacar and I. A. Sengil, Microporous Mesoporous Mater., 2008, 115, 234–246 CrossRef CAS PubMed.
- R. Hu, X. K. Wang, S. Y. Dai, D. D. Shao, T. Hayat and A. Alsaedi, Chem. Eng. J., 2015, 260, 469–477 CrossRef CAS PubMed.
- T. Hiemstra and W. H. V. Riemsdijk, J. Colloid Interface Sci., 1996, 179, 488–508 CrossRef CAS.
- Z. M. Wang and D. E. Giammar, Environ. Sci. Technol., 2013, 47, 3982–3996 CrossRef CAS PubMed.
- P. L. Brezonik and W. A. Arnold, Environ. Sci. Technol., 2012, 46, 5650–5657 CrossRef CAS PubMed.
- C. Koretsky, J. Hydrol., 2000, 230, 127–171 CrossRef CAS.
- R. Dähn, A. M. Scheidegger, A. Manceau, M. L. Schlegel, B. Baeyens, M. H. Bradbury and D. Chateigner, Geochim. Cosmochim. Acta, 2003, 67, 1–15 CrossRef.
- P. J. Pretorius and P. W. Linder, Appl. Geochem., 2001, 16, 1067–1082 CrossRef CAS.
- X. L. Tan, M. Fang and X. K. Wang, Molecules, 2010, 15, 8431–8468 CrossRef CAS PubMed.
- X. L. Tan, X. K. Wang, H. Geckeis and T. Rabung, Environ. Sci. Technol., 2008, 42, 6532–6537 CrossRef CAS.
- H. Nitsche, J. Alloys Compd., 1995, 223, 274–279 CrossRef CAS.
- X. M. Ren, S. T. Yang, F. C. Hu, B. He, J. Z. Xu, X. L. Tan and X. K. Wang, J. Hazard. Mater., 2013, 252–253, 2–10 CrossRef CAS PubMed.
- J. Hu, X. L. Tan, X. M. Ren and X. K. Wang, Dalton Trans., 2012, 41, 10803–10810 RSC.
- X. L. Tan, Q. H. Fan, X. K. Wang and B. Grambow, Environ. Sci. Technol., 2009, 43, 3115–3121 CrossRef CAS.
- J. Hu, Z. Xie, B. Hu, G. D. Sheng, C. L. Chen, J. X. Li, Y. X. Chen and X. K. Wang, Sci. China: Chem., 2010, 53, 1420–1428 CrossRef CAS.
- C. L. Chen, X. Yang, J. Wei, X. L. Tan and X. K. Wang, J. Colloid Interface Sci., 2013, 393, 249–256 CrossRef CAS PubMed.
- X. L. Tan, X. M. Ren, J. X. Li and X. K. Wang, RSC Adv., 2013, 3, 19551–19559 RSC.
- X. Gao, G. D. Sheng and Y. Y. Huang, Sci. China: Chem., 2013, 56, 1658–1666 CrossRef CAS.
- A. C. Q. Ladeira, V. S. T. Ciminelli, H. A. Duarte, M. C. M. Alves and A. Y. Ramos, Geochim. Cosmochim. Acta, 2001, 65, 1211–1217 CrossRef CAS.
- H. Perron, J. Roques, C. Domain, R. Drot, E. Simoni and H. Catalette, Inorg. Chem., 2008, 47, 10991–10997 CrossRef CAS PubMed.
- R. Drot, J. Roques and É. Simoni, C. R. Chim., 2007, 10, 1078–1091 CrossRef CAS PubMed.
- T. Hattori, T. Saito, K. Ishida, A. C. Scheinost, T. Tsuneda, S. Nagasaki and S. Tanaka, Geochim. Cosmochim. Acta, 2009, 73, 5975–5988 CrossRef CAS PubMed.
- S. B. Yang, Y. Chen, C. L. Chen, D. Q. Wang and X. K. Wang, ChemPlusChem., 2015, 80, 480–484 CrossRef CAS PubMed.
- X. F. Luo, C. Fang, X. Li, W. S. Lai, L. F. Sun and T. X. Liang, Appl. Surf. Sci., 2013, 285, 278–286 CrossRef CAS PubMed.
- Y. W. Koh, K. Westerman and S. Manzhos, Carbon, 2015, 81, 800–806 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2015 |