
New tricopper(II) cores self-assembled from aminoalcohol biobuffers and homophthalic acid: synthesis, structural and topological features, magnetic properties and mild catalytic oxidation of cyclic and linear C5–C8 alkanes†
Sara S. P.
Dias
a,
Marina V.
Kirillova
a,
Vânia
André
a,
Julia
Kłak
b and
Alexander M.
Kirillov
*a
aCentro de Química Estrutural, Complexo I, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001, Lisbon, Portugal. E-mail: kirillov@ist.utl.pt
bFaculty of Chemistry, University of Wrocław, ul. F. Joliot-Curie 14, 50-383 Wroclaw, Poland
First published on 30th March 2015
Abstract
Two new crystalline materials [Cu3(μ2-H3bis-tris)2(μ2-Hhpa)2]·H2O (1) and [Cu3(μ2-H2tea)2(μ2-hpa)(μ3-hpa)]n (2) bearing distinct tricopper(II) cores were easily generated by the aqueous medium self-assembly method from copper(II) nitrate, bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane (H5bis-tris) or triethanolamine (H3tea) aminoalcohol biobuffers and homophthalic acid (H2hpa). The obtained products were characterised by IR, UV-vis and EPR spectroscopy, ESI-MS(±), thermogravimetric, elemental and single crystal X-ray diffraction analysis. Apart from possessing different dimensionality, the crystal structures of the discrete 0D trimer 1 and the zigzag 1D coordination polymer 2 show distinct symmetric [Cu3(μ-O)4(μ-COO)2] and asymmetric [Cu3(μ-O)3(μ-COO)2] tricopper(II) cores, respectively. An intense pattern of intermolecular O–H⋯O hydrogen bonds provides a 0D → 3D (1) or 1D → 2D (2) extension of the structures into intricate topologically unique H-bonded nets. After additional simplification, these were classified as a uninodal 6-connected 3D framework with the snk topology in 1 and a binodal 3,5-connected 2D layer with the 3,5L50 topology in 2. Variable-temperature magnetic susceptibility studies indicate a predominant ferromagnetic coupling [J = 39.1(1) and 29.5(1) cm−1 for 1 and 2, respectively] within the mixed-bridged tricopper(II) cores. Both compounds 1 and 2 were also applied as rather efficient bio-inspired pre-catalysts for the mild homogeneous oxidation, by aqueous H2O2 at 50 °C in acidic MeCN–H2O medium, of cyclic (cyclopentane, cyclohexane, cycloheptane and cyclooctane) and linear (n-pentane, n-hexane, n-heptane and n-octane) alkanes to the corresponding alcohols and ketones with overall yields up to 26% based on the alkane. The effects of different reaction parameters (type of pre-catalyst and acid promoter, reaction time and substrate scope) and various selectivity features were investigated and discussed, supporting a free-radical mechanism in the present alkane oxidations.
Introduction
Within the vast development of crystal engineering research in recent years,1 the design of novel molecular materials bearing diverse multicopper(II) cores with promising functional properties has attracted an increased attention in view of the versatile redox, magnetic, biological, catalytic and coordination behaviour of copper ions.2,3 In many cases, rather unusual multicopper(II) aggregates of diverse nuclearity can be constructed via simple self-assembly protocols employing a variety of organic building blocks.4,5 Thus, further exploration of this research direction can consist of finding attractive organic molecules capable of self-assembling with copper ions and generating multinuclear cores with desired structural and functional characteristics.Aiming to search for such organic molecules, we have focused our attention on a pair of aminoalcohols as main building blocks, namely bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane (H5bis-tris) and triethanolamine (H3tea), which are commonly applied as buffers in biochemistry and molecular biology.6 As a potential linker, we have chosen homophthalic acid (H2hpa) that combines both aromatic and aliphatic carboxylate functionalities (Scheme 1). However, in spite of being commercially available and featuring aqueous solubility and coordination versatility, the application of such aminoalcohols and carboxylic acid as potentially multidentate building blocks in crystal engineering of mixed-ligand multicopper materials has remained somewhat underexplored,7 although some notable examples of polynuclear Cu compounds derived from H3tea and carboxylic acids have been documented.5,8 Thus, the first aim of the current study consisted of probing the combination of the above building blocks for the self-assembly generation of novel multicopper(II) molecular materials, whereas the second aim concerned the investigation of their intrinsic structural, magnetic and catalytic properties.
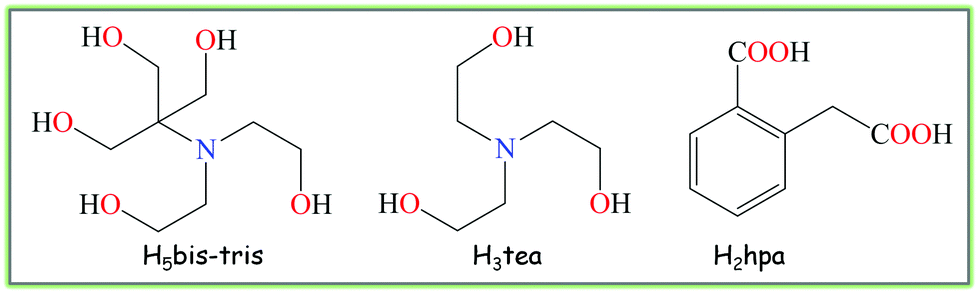 | ||
| Scheme 1 Structural formulae of organic building blocks. | ||
Hence, we report herein the facile aqueous medium self-assembly preparation, spectral features, crystal structures, topological analysis, thermal and magnetic properties as well as catalytic application of the two novel crystalline materials [Cu3(μ2-H3bis-tris)2(μ2-Hhpa)2]·H2O (1) and [Cu3(μ2-H2tea)2(μ2-hpa)(μ3-hpa)]n (2) that bear distinct linear tricopper(II) cores. The type of the aminoalcohol biobuffer ligand plays a key structure-driven role in defining the dimensionality and kind of the tricopper(II) core in these coordination compounds as well as the topology of extended H-bonded networks. In addition, by acting as homogeneous pre-catalysts for the mild oxidation of cyclic and linear C5–C8 alkanes and possessing the alkoxo-carboxylato tricopper(II) cores, both compounds 1 and 2 can be considered as bio-inspired catalytic materials4,9 with some relevance to particulate methane monooxygenase (pMMO), a unique copper enzyme that bears an active site based on a tricopper cluster with an N,O-environment capable of hydroxylating alkanes.10
Experimental section
Materials and methods
All synthetic work was performed in air and at room temperature (r.t., ∼25 °C). All chemicals were obtained from commercial sources and used as received. C, H and N elemental analyses were carried out by the Microanalytical Service of the Instituto Superior Técnico. Infrared spectra (4000–400 cm−1) were recorded on a JASCO FT/IR-4100 instrument in KBr pellets (abbreviations: vs – very strong, s – strong, m – medium, w – weak, br. – broad, sh. – shoulder). ESI-MS(±) spectra were run on a 500-MS LC Ion Trap instrument (Varian Inc, Alto Palo, CA, USA) equipped with an electrospray (ESI) ion source, using ∼10−3 M solutions of 1 and 2 in MeCN–H2O and MeOH, respectively. The UV-vis spectra were recorded in MeCN–H2O or MeCN solutions on the Agilent Cary 60 UV-Vis Spectrophotometer with a fiber optic dip probe. The thermogravimetric analyses (TGA) were carried out on a Setaram Setsys TG-DTA 16 instrument by heating the crystalline samples (8–10 mg) of 1 and 2 under N2 at the 10 °C min−1 rate in the 30–750 °C temperature range. EPR spectra of powdered samples 1 and 2 were recorded at 293 and 77 K on a Bruker ESP 300 spectrometer operating at X-band equipped with an ER 035M Bruker NMR gaussmeter and HP 5350B Hewlett-Packard microwave frequency counter. Gas chromatographic (GC) analyses were run on an Agilent Technologies 7820A series gas chromatograph (He as carrier gas) equipped with a FID detector and BP20/SGE (30 m × 0.22 mm × 0.25 μm) capillary column. For simplicity, the term “aminoalcohol” was used in this work to denote a general kind of the ligand in 1 and 2, understanding that the derived H3bis-tris and H2tea ligands are in fact partially deprotonated aminoalcoholate(ato).General synthetic procedure and characterisation for 1 and 2
To an aqueous 0.1 M solution of Cu(NO3)2·3H2O (10 mL, 1 mmol) was added an aqueous 1 M solution (1 mL, 1 mmol) of aminoalcohol [bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane (H5bis-tris) for 1 and triethanolamine (H3tea) for 2] with continuous stirring at r.t. Then, homophthalic acid (H2hpa; 180.2 mg, 1 mmol) and an aqueous 1 M solution of NaOH (3 mL, 3 mmol; up to pH ∼ 8) were added to the reaction mixture. This was stirred for 1 day and then filtered off, and the filtrate was left to evaporate in a beaker at r.t. Greenish blue (1) or blue (2) crystals, including those suitable for single crystal X-ray diffraction analysis, were formed in 2–3 weeks, then collected and dried in air to give the compounds 1 and 2 in ∼50% yield, based on copper(II) nitrate.X-ray crystallography
Crystals of 1 and 2 suitable for X-ray diffraction study were mounted with Fomblin® in a cryoloop. Data were collected on a Bruker AXS-KAPPA APEX II diffractometer with graphite-monochromated radiation (Mo Kα, λ = 0.17073 Å) at 150 K. The X-ray generator was operated at 50 kV and 30 mA and the X-ray data collection was monitored by the APEX2 program.11 All data were corrected for Lorentzian, polarisation and absorption effects using SAINT and SADABS programs.11 SIR9712 and SHELXS-9713 were used for structure solution, and SHELXL-9713 was applied for full matrix least-squares refinement on F2. These three programs are included in the package of programs WINGX-Version 1.80.05.14 Non-hydrogen atoms were refined anisotropically. A full-matrix least-squares refinement was used for the non-hydrogen atoms with anisotropic thermal parameters. All the hydrogen atoms were inserted in idealised positions and allowed to refine in the parent carbon or oxygen atom, except for the OH hydrogen atoms in the aminoalcohol ligands that were located from the electron density map. For 1, it was not possible to locate the water hydrogen atoms and the H atoms of HO groups in the H3bis-tris ligand are most likely disordered precluding a perfect refinement. TOPOS 4.015 and PLATON16 were used for topological analysis and hydrogen bond interactions, respectively. Crystal data and details of data collection for 1 and 2 are reported in Table 1.| 1 | 2 | |
|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2. | ||
| Formula | C17H24Cu1.5NO9.25 | C30H40Cu3N2O14 |
| Fw | 485.68 | 843.26 |
| Crystal form, colour | Block, blue | Plate, blue |
| Crystal size (mm) | 0.16 × 0.14 × 0.06 | 0.20 × 0.08 × 0.04 |
| Crystal syst. | Tetragonal | Monoclinic |
| Space group | I41/a | P21/c |
| a/Å | 29.809(4) | 18.2009(7) |
| b/Å | 29.809(4) | 13.5600(4) |
| c/Å | 8.4384(15) | 13.3152(5) |
| α/° | 90.00 | 90.00 |
| β/° | 90.00 | 99.471(2) |
| γ/° | 90.00 | 90.00 |
| Z | 16 | 4 |
| V/Å3 | 7498(2) | 3241.5(2) |
| T/K | 150(2) | 150(2) |
| D c/g cm−3 | 1.721 | 1.728 |
| μ(Mo Kα)/mm−1 | 1.771 | 2.024 |
| θ range/° | 2.51–26.49 | 2.16–26.37 |
| Refl. collected | 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 761 761 |
29395 |
| Independent refl. | 3835 | 6609 |
| R int | 0.0947 | 0.0513 |
|
R
1a, wR2b [I ≥ 2σ(I)] |
0.0735, 0.1707 | 0.0302, 0.0697 |
| GOF on F2 | 1.014 | 1.066 |
Magnetic studies
The magnetisation of powdered samples 1 and 2 was measured over the 1.8–300 K temperature range using a Quantum Design SQUID-based MPMSXL-5-type magnetometer. The superconducting magnet was generally operated at a field strength ranging from 0 to 5 T. Sample measurements were made at magnetic field 0.5 T. The SQUID magnetometer was calibrated with the palladium rod sample. Corrections are based on subtracting the sample-holder signal and the χD contribution was estimated from the Pascal's constants.17Catalytic studies
The alkane oxidations were carried out in an air atmosphere in round bottom flasks equipped with a condenser, under vigorous stirring at 50 °C and using MeCN as the solvent (up to 5.0 mL total volume). In a typical experiment, a solid pre-catalyst 1 or 2 (0.01 mmol) and GC internal standard (MeNO2, 50 μL) were introduced into the reaction mixture, followed by the addition of an acid co-catalyst (0.1 mmol) used as a stock solution in MeCN. The alkane substrate (1 mmol) was then introduced, and the reaction started upon addition of hydrogen peroxide (50% in H2O, 5 mmol) in one portion. The reactions were monitored by withdrawing small aliquots after different periods of time, which were treated with PPh3 (following a method developed by Shul'pin)18 for reduction of remaining H2O2 and alkyl hydroperoxides that are typically formed as major primary products in alkane oxidations. The samples were analysed by gas chromatography using nitromethane as an internal standard. Attribution of peaks was made by comparison with chromatograms of authentic samples.Various control tests were also performed (see Tables S2–S4 in ESI†) using different combinations of Cu(NO3)2, H5bis-tris, H3tea, H2hpa and trifluoroacetic acid as stock solutions in MeCN. In a typical experiment, acetonitrile solutions of Cu(NO3)2 (0.01 mmol), H5bis-tris or H3tea (0.05 mmol; optional), H2hpa (0.05 mmol; optional), trifluoroacetic acid (0.1 mmol; optional) and GC internal standard (MeNO2, 50 μL) were introduced into the reaction mixture. The alkane substrate (1 mmol) was then added, and the reaction started upon addition of hydrogen peroxide (50% in H2O, 5 mmol) in one portion. Small aliquots were then taken, treated with PPh3 and analysed by GC as described above. The performed tests confirmed that alkane oxidations do not proceed in the absence of any copper pre-catalyst (less than 0.2% overall yields, Table S4†), whereas the control Cu(NO3)2/TFA/H2O2 system leads to significantly inferior overall yields (2–5% in C5–C8 alkane oxidation, Table S2†) than those in the 1/TFA/H2O2 system (10–26%, Table 2). The model systems comprising a mixture of copper(II) nitrate, aminoalcohol, homophthalic and trifluoroacetic acid (optional) are also active in alkane oxidations, although resulting in lower product yields in comparison with those obtained when using the pre-catalysts 1 or 2 and TFA (see Table S4 in ESI†).
| Alkane | Yieldb/% | ||
|---|---|---|---|
| Alcohol(s)c | Ketone(s)c | Total | |
| a Reaction conditions: pre-catalyst 1 (0.01 mmol), TFA (0.1 mmol), alkane (1.0 mmol), H2O2 (50% aq., 5.0 mmol), MeCN (up to 5 mL total volume), 50 °C, 3 h. b Based on alkane substrate, calculated from GC analysis after treatment of the reaction mixture with PPh3. c In the case of n-alkanes, the indicated total product yields correspond to the sum of yields of various isomeric alcohols and ketones (aldehydes), see Table S3 for details. | |||
| Cyclopentane | 12.0 | 4.0 | 16.0 |
| Cyclohexane | 15.9 | 4.0 | 19.9 |
| Cycloheptane | 16.8 | 9.0 | 25.8 |
| Cyclooctane | 5.5 | 12.1 | 17.6 |
| n-Pentane | 6.5 | 3.0 | 9.5 |
| n-Hexane | 6.9 | 3.1 | 10.0 |
| n-Heptane | 9.6 | 4.4 | 14.0 |
| n-Octane | 10.3 | 4.5 | 14.8 |
Results and discussion
Synthesis and characterisation
To explore the application of bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane (H5bis-tris) and triethanolamine (H3tea) aminoalcohol building blocks along with the homophthalic acid (H2hpa) linker towards the design of mixed-ligand multicopper(II) compounds, we followed a versatile aqueous medium self-assembly protocol.5,19 Thus, an aqueous solution of copper(II) nitrate was simply combined at ∼25 °C in air with an aminoalcohol [H5bis-tris for 1 or H3tea for 2] as a main building block, homophthalic acid as a linker and sodium hydroxide as a pH-regulator. As a result, two new compounds composed of tricopper(II) cores, namely a discrete 0D derivative [Cu3(μ2-H3bis-tris)2(μ2-Hhpa)2]·H2O (1) and a 1D coordination polymer [Cu3(μ2-H2tea)2(μ2-hpa)(μ3-hpa)]n (2) were self-assembled and isolated as air-stable microcrystalline materials (Scheme 2). They were characterised by IR and EPR spectroscopy, thermogravimetric, elemental and single crystal X-ray diffraction analysis.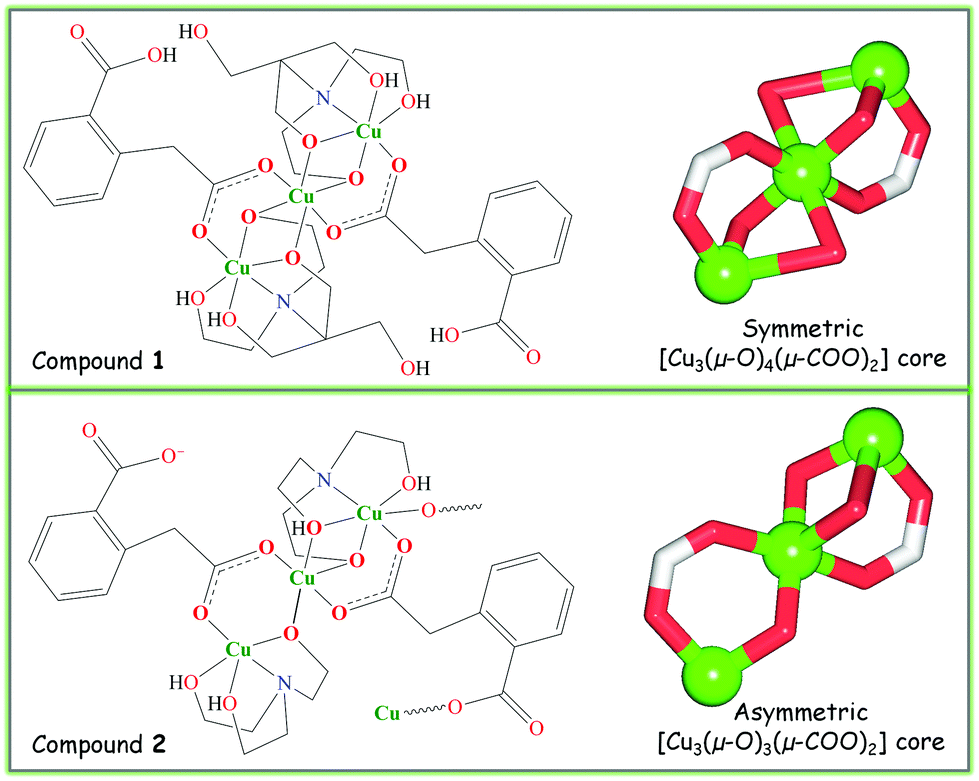 | ||
| Scheme 2 Structural formulae of 1 and 2 (left) with ball-and-stick representation of their tricopper(II) cores (right). | ||
The IR spectra of 1 and 2 reveal a number of characteristic vibrations owing to the presence of aminoalcohol (H3bis-tris or H2tea) and aromatic carboxylate (Hhpa or hpa) ligands, as well as a crystallisation H2O molecule (in 1).19,20 Thus, two ν(OH/H2O) bands were observed in 1 at 3445 and 3176 cm−1, whereas the ν(OH) vibration in 2 has a maximum at 3330 cm−1. The broad nature of these bands in both compounds suggests intense H-bonding interactions.20 The νas(CH) and νs(CH) vibrations are also present as one or several weak intensity bands in the 3015–2835 cm−1 interval. The most characteristic vibrations include the very strong and broad νas(COO) and νs(COO) major bands with maxima at 1562 and 1388 cm−1 (Δ = 174 cm−1) in 1 and 1569 and 1396 cm−1 (Δ = 173 cm−1) in 2. The observed frequency difference Δ below 200 cm−1 is indicative of the syn–syn-μ2–η1:η1 coordination mode21 of the principal μ2-carboxylate group in the bridging hpa moieties, whereas the presence of several inferior intensity shoulders at the major νas(COO) and νs(COO) bands is related to the η0:η0 (in 1) or η1:η0 (in 2) modes of the second COOH/COO group of homophthalate ligands. The UV-vis spectra of 1 and 2 in MeCN–H2O exhibit broad absorptions with maxima at 673 and 678 nm, respectively, which are related to d → d transitions involving Cu(II) ions. There are also two intense bands [305 and 244 nm in 1; 278 and 227 nm in 2] of ligand-based transitions. The ESI-MS(−) spectrum of 1 reveals a molecular [Cu3(H3bis-tris)2(Hhpa)(hpa)]− (m/z = 962) peak and various tri- and dicopper fragments, namely [Cu3(H3bis-tris)2(Hhpa)(H2O)]− (m/z = 803), [Cu2(H3bis-tris)2(Hhpa)(H2O)]− (m/z = 739) and [Cu2(H2bis-tris)(Hhpa)2]− (m/z = 692), corresponding to the elimination of one Hhpa ligand, Cu(Hhpa) or Cu(H3bis-tris) moieties, respectively. Similar type of tricopper fragments, [Cu3(tea)2(Hhpa)]− (m/z = 663) and [Cu3(H2tea)2(hpa) + H2O + H]+ (m/z = 685), are also detected in negative and positive mode mass spectra of 2, respectively.
To evaluate the thermal behaviour of 1 and 2, we have run their thermogravimetric analyses under a N2 atmosphere in the 30–750 °C interval (for TG–DTA plots, see Fig. S1 and S2†). In 1, the elimination of one crystallisation H2O molecule (mass loss: Δm = 2.1% exp., 1.8% calcd) is observed during the endothermic effect in the 50–140 °C temperature range, with the dehydrated sample remaining stable up to 215 °C. A sharp endothermic effect in the 215–240 °C interval can be associated with the removal of one H3bis-tris moiety (Δm = 18.1% exp., 18.8% calcd), whereas the second H3bis-tris fragment and two Hhpa ligands almost fully decompose in the 240–450 °C range. Similarly, the decomposition of 2 begins at 215 °C and the endothermic effect (215–270 °C) corresponds to the elimination of two H2tea moieties (Δm = 29.5% exp., 29.4% calcd). An almost complete removal of two hpa fragments is observed in the 270–450 °C interval, although the sample continues to lose weight up to 750 °C attaining a total weight loss of 71.4% that is very close to the calculated value of 71.7% assuming the formation of CuO residue.
Description of crystal structures
The discrete 0D structure of 1 consists of the neutral tricopper(II) [Cu3(μ2-H3bis-tris)2(μ2-Hhpa)2] unit (Fig. 1) and one partially occupied water molecule in the asymmetric unit. Within the Cu3 unit, the “central” six-coordinate Cu2 atom exhibits an ideal octahedral {CuO6} environment, the equatorial sites of which are occupied by two pairs of the symmetry equivalent O6 and O2 atoms from the μ2-H3bis-tris and μ2-Hhpa moieties [Cu2–O6 1.967(5), Cu2–O2 2.022(5) Å], respectively, while the axial positions are taken by a pair of O7 atoms [Cu2–O7 2.391(6) Å] from the μ2-H3bis-tris ligand. The two “outer” Cu1 centres are also six-coordinate and exhibit a distorted octahedral {CuNO5} geometry filled by the N1, O6 and O8 atoms of the μ2-H3bis-tris and the O1 atom of the μ2-Hhpa ligands in equatorial positions [Cu1–N1 2.001(5), Cu1–O1 1.903(5), Cu1–O6 1.956(5), Cu1–O8 2.0500(10) Å], whereas the apical sites are occupied by the remaining O5 and O7 atoms of the μ2-H3bis-tris moiety with the Cu1–O5 and Cu1–O7 distances of 2.3361(10) and 2.609(6) Å, respectively. Although the latter bond length is rather long, it is well below the sum of the van der Waals radii of Cu and O atoms [∼2.92 Å].22 The Cu2 and Cu1 centres are triply interconnected by means of the bridging O6 and O7 atoms of the pentadentate μ2-H3bis-tris ligands and via the O1–C2–O2 carboxylate groups of μ2-Hhpa that adopt a syn–syn-μ2–η1:η1 coordination mode, thus furnishing a symmetric linear [Cu3(μ-O)4(μ-COO)2] core with the Cu1⋯Cu2 and Cu1⋯Cu1i separations of 3.0594(9) and 6.119(1) Å, respectively. The second O3–C10–O4 carboxylic group of μ2-Hhpa and one of the hydroxymethyl fragments (O9) of μ2-H3bis-tris remain uncoordinated. In general, the bonding parameters in 1 are comparable to those of other very rare copper compounds7 derived from H5bis-tris23 or H2hpa.24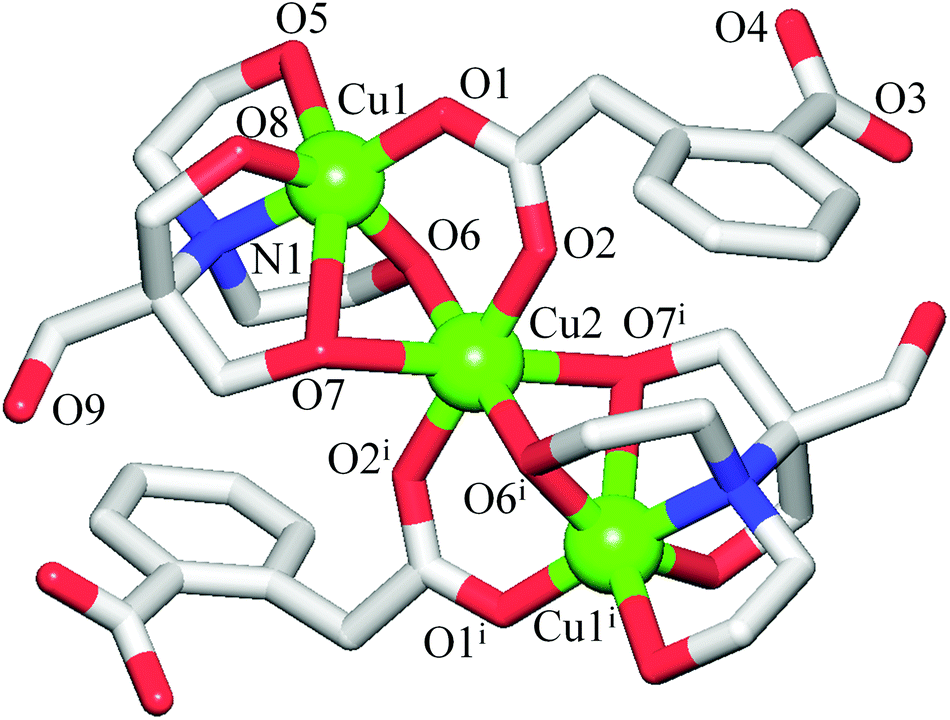 | ||
| Fig. 1 Structure of 1 showing tricopper(II) unit with atom numbering scheme. H atoms and crystallisation H2O molecules are omitted for clarity. Colour codes: Cu (green), O (red), N (blue), and C (grey). Selected distances (Å): Cu1–N1 2.001(5), Cu1–O1 1.903(5), Cu1–O5 2.3361(10), Cu1–O6 1.956(5), Cu1–O7 2.609(6), Cu1–O8 2.0500(10), Cu2–O2 2.022(5), Cu2–O6 1.967(5), Cu2–O7 2.391(6), Cu1⋯Cu2 3.0594(9). Symmetry code: (i) −x, −y + 1, −z. | ||
In contrast to 1, the crystal structure of 2 discloses a zigzag 1D coordination polymer assembled from the repeating tricopper(II) [Cu3(μ2-H2tea)2(μ2-hpa)(μ3-hpa)] units (Fig. 2). Although these resemble those Cu3 blocks in 1, the main differences of 2 consist of (i) the presence of one μ3-hpa spacer possessing the syn–syn-μ2–η1:η1 and η1:η0 coordination modes of carboxylic groups, (ii) the existence of three symmetry non-equivalent copper atoms with two of them (Cu2, Cu3) adopting different five-coordinate environments and one (Cu1) having a six-coordinate geometry, and (iii) the distinct linkage of the “central” Cu2 atom with the “outer” Cu1 and Cu3 atoms. These differences give rise to the generation of an asymmetric [Cu3(μ-O)3(μ-COO)2] core in 2, which slightly deviates from the linearity [Cu1⋯Cu2⋯Cu3 angle of 177.04(1)°] and possesses the distinct Cu1⋯Cu2 [3.0961(4) Å] and Cu2⋯Cu3 [3.3016(4) Å] separations. The “central” Cu2 atom adopts a distorted {CuO5} square-pyramidal environment (τ5 = 0.10 in 2; τ5 = 0 for an idealised square-pyramidal geometry),25 occupied by the two triethanolaminate (O2, O14) and two homophthalate (O5, O8) oxygen atoms in equatorial sites [Cu2–O 1.9211(15)–1.9746(15) Å], while an axial position is filled by the O3 atom of the μ2-H2tea moiety [Cu2–O3 2.452(2) Å]. The {CuNO4} coordination geometry of the “outer” Cu3 atom is better described as a highly distorted trigonal-bipyramid (τ5 = 0.66 in 2; τ5 = 1 for idealised trigonal-bipyramidal geometry),25 filled by the triethanolaminate N12 and O12 and carboxylate O9 atoms in equatorial positions [Cu3–N2 2.0130(19), Cu3–O12 2.0950(16), Cu3–O9 1.9038(17) Å], and by the O13 and O14 atoms of the μ2-H2tea ligand in apical sites [Cu3–O13 2.1359(18), Cu3–O14 1.9296(15) Å]. In contrast to Cu3, Cu1 atom exhibits a distorted {CuNO5} octahedral environment with equatorial sites filled by the triethanolaminate N1 and O2 and μ3-homophthalate O4 and O6i atoms [Cu1–N1 2.0406(18), Cu1–O2 1.9397(15), Cu1–O4 1.9549(15), Cu1–O6i 1.9651(15) Å], while the axial positions are taken by the remaining O1 and O3 atoms of μ2-H2tea [Cu1–O1 2.3966(17), Cu1–O3 2.638(2) Å]. Thus, a notable feature of 2 consists of the observation of distinct coordination geometries in all three copper(II) centres. It should also be highlighted that the obtained compounds represent the first examples of trimetallic cores supported by homophthalate ligands,7 thus opening up the application of H2hpa as a useful linker for the design of polynuclear coordination compounds.
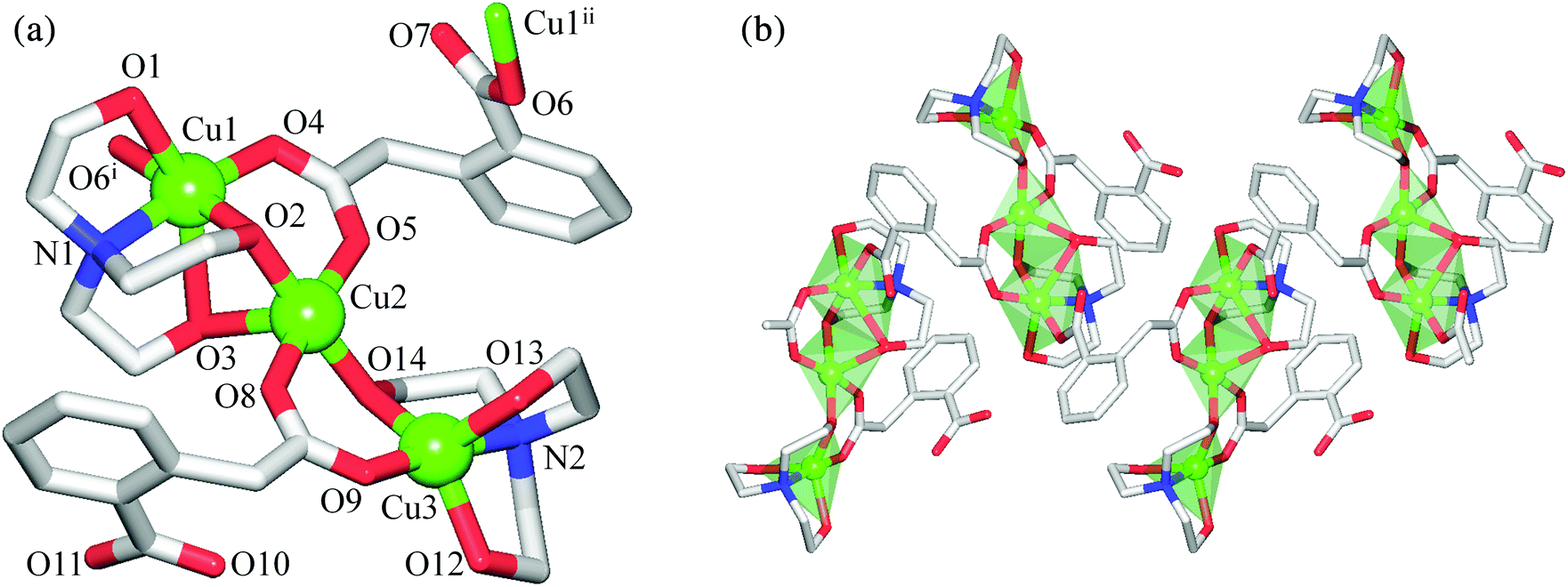 | ||
| Fig. 2 Structural fragments of 2 showing: (a) tricopper(II) unit with atom numbering scheme and (b) zigzag 1D metal–organic chain with polyhedral representation of the coordination environments around Cu atoms. H atoms are omitted for clarity. Colour codes: Cu (green), O (red), N (blue), and C (grey). Selected distances (Å): Cu1–N1 2.0406(18), Cu1–O1 2.3966(17), Cu1–O2 1.9397(15), Cu1–O3 2.638(2), Cu1–O4 1.9549(15), Cu1–O6i 1.9651(15), Cu2–O2 1.9280(14), Cu2–O3 2.452(2), Cu2–O5 1.9746(15), Cu2–O8 1.9652(15), Cu2–O14 1.9211(15), Cu3–N2 2.0130(19), Cu3–O9 1.9038(17), Cu3–O12 2.0950(16), Cu3–O13 2.1359(18), Cu3–O14 1.9296(15), Cu1⋯Cu2 3.0961(4), Cu2⋯Cu3 3.3016(4), Cu1⋯Cu1ii 6.6787(5). Symmetry codes: (i) x, −y + 1.5, z + 0.5; (ii) x, −y + 1.5, z − 0.5. | ||
Topological analysis of H-bonded networks
In recent years, various topological analysis methods have become quite important in crystal engineering research, contributing to the identification, classification and prediction of topological motifs in a variety of coordination and supramolecular compounds.15,26,27 Although a multitude of topological types (>75000) have been determined or theoretically predicted, the identification of novel topologies is a constantly growing research direction, as attested by an increasing number of publications on this topic, including the state-of-the-art reviews and perspective articles.15,16,27 Some of these studies are entirely focused on the topological analysis of hydrogen-bonded networks27 which, due to their high complexity, represent a considerable potential not only to disclose new topological types but allow an identification of the theoretically predicted topological patterns. Moreover, the topological analysis of H-bonded nets can contribute to the rationalisation and prediction of supramolecular motifs, thus facilitating the design of desired structures.15a
The structures of 1 and 2 reveal a rather intense pattern of the intermolecular O–H⋯O hydrogen bonds between aminoalcohol and homophthalate moieties (Table S1†), which are responsible for the extension of discrete 0D units in 1 and infinite 1D chains in 2 to furnish intricate 3D and 2D supramolecular networks, respectively. To gain further insight into these complex H-bonded nets, we have run their topological analysis using the concept of the simplified underlying net.15,26
To simplify the 3D H-bonded framework of 1 we have applied a specific methodology developed for the topological analysis of H-bonded nets built from the discrete molecular units.15,27 Hence, the molecular [Cu3(μ2-H3bis-tris)2(μ2-Hhpa)2] blocks have been reduced to their centroids which, along with the centroids of crystallisation H2O molecules, give rise to a very complex binodal 4,8-connected net with a unique topology (Fig. S3†). Further simplification of this net by omitting H2O nodes resulted in an alternative framework composed of the 6-connected [Cu3(μ2-H3bis-tris)2(μ2-Hhpa)2] nodes (Fig. 3a). This uninodal 6-connected net features the snk topology defined by the (410·52·63) point symbol. Although this topological type has been theoretically predicted,15 to our knowledge, compound 1 represents the first example wherein such a topology has been experimentally identified.
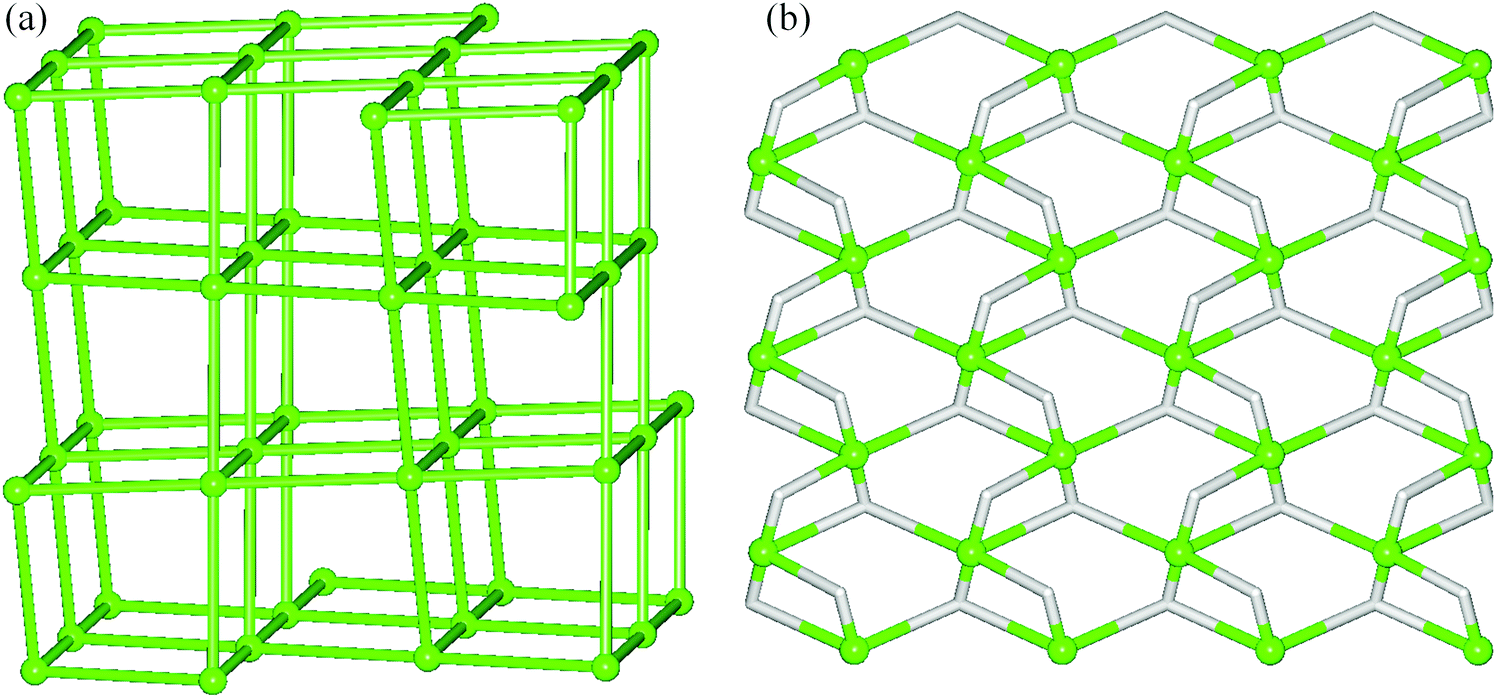 | ||
| Fig. 3 Topological representations of the underlying H-bonded networks in 1 (a) and 2 (b). Further details: (a) a uninodal 6-connected 3D net in 1 with the snk topology and the point symbol of (410·52·63); green balls correspond to centroids of 6-connected [Cu3(μ2-H3bis-tris)2(μ2-Hhpa)2] molecular nodes; view along the c-axis. (b) A binodal 3,5-connected 2D net in 2 with the 3,5L50 topology and the point symbol of (3·52)(32·53·64·7); centroids of 5-connected [Cu3(μ2-H2tea)2]4+ nodes (green balls), centroids of 3- and 2-connected hpa nodes and linkers (grey); view along the a-axis. | ||
For the coordination polymer 2, a different network simplification procedure has been applied.15,26 Thus, an underlying 2D H-bonded layer of 2 has been obtained by reducing all the ligands to their centroids, generating a very complex hexanodal 3,4-connected net with a unique topology. This net has been further simplified by treating the [Cu3(μ2-H2tea)2]4+ fragments as cluster Cu3 nodes, leading to a derived net (Fig. 3b) assembled from the 5-connected [Cu3(μ2-H2tea)2]4+ and 3-connected hpa nodes, as well as 2-connected hpa linkers. Its topological analysis discloses a binodal 3,5-connected layer with a 3,5L50 topology and a point symbol of (3·52)(32·53·64·7), wherein the (3·52) and (32·53·64·7) notations are those of the hpa and [Cu3(μ2-H2tea)2]4+ nodes, respectively.
Magnetic properties
The magnetic behaviour of 1 and 2 was studied over the 1.8–300 K temperature range. Plots of magnetic susceptibility χmT product vs. T (χm is the molar magnetic susceptibility for three CuII ions) are shown in Fig. 4a. The experimental χmT values at room temperature (1.44 and 1.28 cm3 mol−1 K for 1 and 2, respectively) are somewhat larger than those expected for the three magnetically uncoupled copper(II) ions [χmT = 3(Nβ2g2/3k)S(S + 1) = 1.23 cm3 mol−1 K, wherein g = 2.1 is the spectroscopic splitting factor, N – the Avogadro's number, β – the Bohr magneton, k – the Boltzmann's constant, and S = 1/2].28 Upon cooling the sample, the χmT increases and attains a maximum of 1.94 or 1.75 cm3 mol−1 K at 16 or 10 K for 1 and 2, respectively. This behaviour is indicative of a strong ferromagnetic coupling between the adjacent Cu(II) atoms within the tricopper blocks. Below the maximum values, the χmT decreases rapidly down to 1.39 (1) and 1.34 (2) cm3 mol−1 K, what can be attributed either to a zero-field splitting (ZFS) within the quartet ground state and/or to small antiferromagnetic intermolecular interactions within the compounds. The values of Curie and Weiss constants determined from the χm−1 = f(T) relationship above 16 and 10 K are equal to 1.41 cm3 mol−1 K and 9.9 K for 1 and 1.26 cm3 mol−1 K and 8.7 K for 2. Positive Weiss constants confirm also the occurrence of ferromagnetic interactions between the metal centres in the Cu3 cores.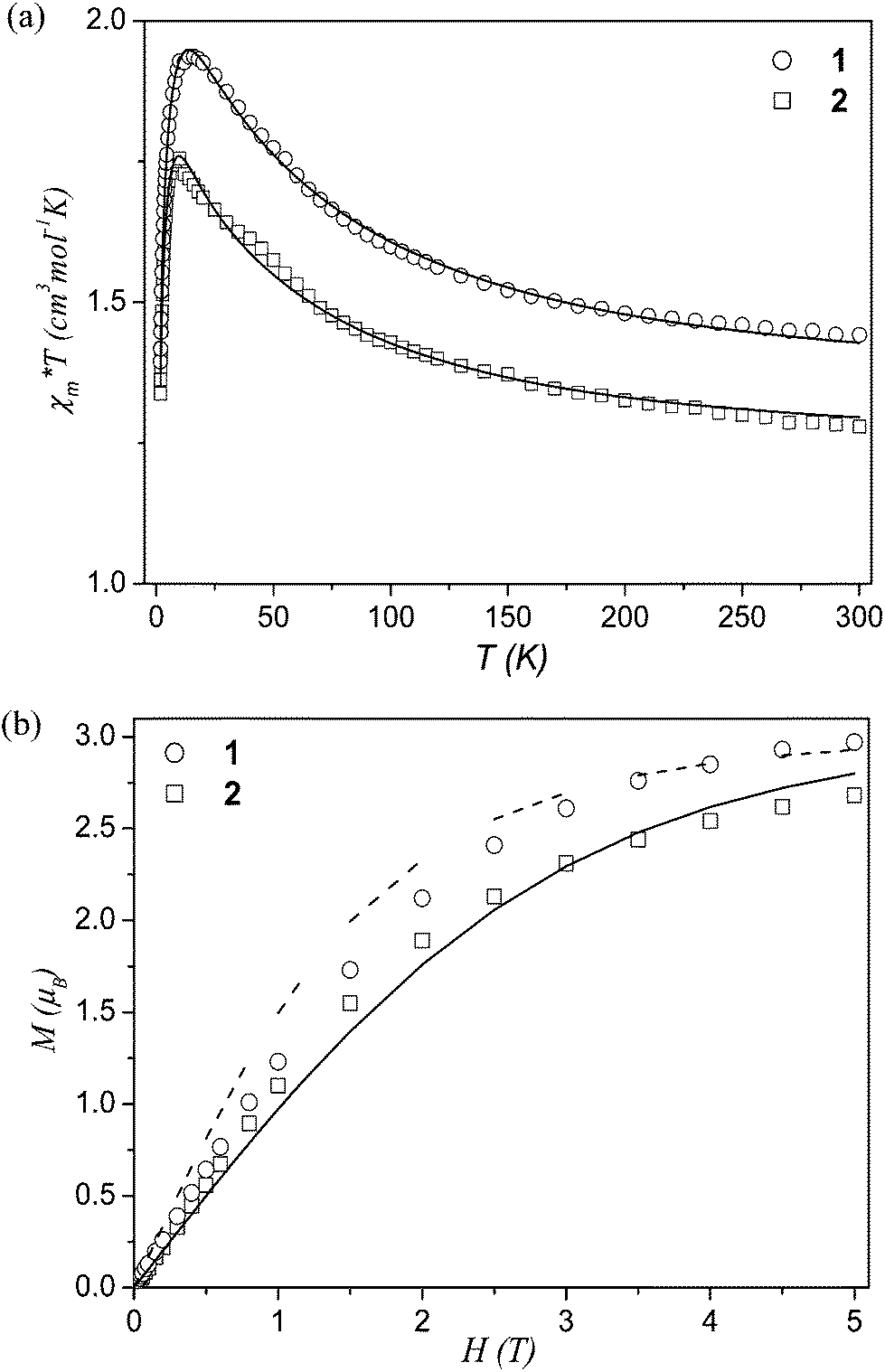 | ||
| Fig. 4 (a) Temperature dependence of experimental χmT (χm per 3 CuII atoms) for 1 and 2. The solid lines are the calculated curves derived from eqn (2). (b) Field dependence of the magnetisation (M per Cu3 entities) for 1 and 2. The solid line is the Brillouin function curve for the system of three uncoupled spins with S = 1/2 and g = 2.0; the dashed line is the Brillouin function curve for a S = 3/2 state of the Cu3 unit. | ||
To elucidate the nature of the ground state in 1 and 2, we also investigated the variation of the magnetisation (M) with respect to the field (H), at 2 K. The results are given in Fig. 4b, where the molar magnetisation M (per Cu3 entities) is expressed in μB units. The magnetisation increases linearly at low applied fields up to ∼0.8 T and then progressively tends to saturation with values of 2.9 (1) and 2.7 (2) μB at 5 T. The magnetisation curves were reproduced by the equation M = gβSNBs(x)(S = 3·SCu), where Bs(x) is the Brillouin function and x = gβH/kT.28 The experimental magnetisation of both compounds is greater than that of three non-interacting S = 1/2 systems, being very close to the Brillouin function of S = 3/2 state. This behaviour can be attributed to incomplete population of the S = 3/2 ground state.
From the structural point of view, the compounds 1 and 2 bear tricopper(II) entities, which are further assembled into a zigzag 1D chain in the case of 2. It is expected that the coupling between the adjacent Cu3 blocks is much weaker than the intratrimer coupling. The spin Hamiltonian appropriate to describe the exchange interaction in two linear trimers has the following form (eqn (1)):29
| H = −2J12(Ŝ1Ŝ2 + Ŝ2Ŝ3) − 2J13Ŝ1Ŝ3 | (1) |
The exchange constant J12 refers to the interaction between the “central” and “outer” copper(II) centres, while the J13 parameter is the interaction between the two “outer” Cu(II) atoms. The energies of the spin states are given in terms of E(ST, S+) with total spin ST = S1 + S2 + S3 and S+ = S1 + S3. For three interacting S = 1/2 centres, a quartet state, E(3/2, 1) = −J12 − ½J13 and two doublet states, E(1/2, 0) = 3/2J13 and E(1/2, 1) = 2J13 − ½J13, are obtained. Using the van Vleck equation,28,29 the following theoretical expression for the temperature dependence of χm is obtained (eqn (2)):
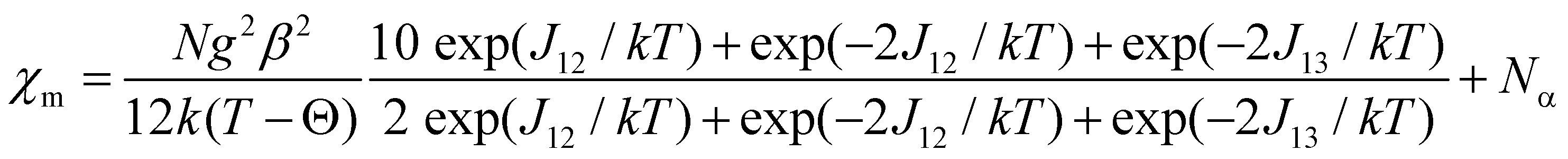 | (2) |
Weiss constant θ was introduced to take into account the decrease of the magnetic moments at low temperatures. The parameters J12, J13, g and θ were evaluated by fitting eqn (2) to the experimental susceptibilities (Na = 60 × 10−6 cm3 mol−1). A least-squares fitting of the experimental data led to the following values: J12 = 39.1(1) cm−1, J13 = −6.1(2) cm−1, g = 2.15(1), θ = −0.9(1) K (R = 1.99 × 10−5) for 1 and J12 = 29.5(1) cm−1, J13 = −6.7(2) cm−1, g = 2.11(2), θ = −0.8(2) K (R = 5.98 × 10−5) for 2. The criterion for the determination of the best fit was based on the minimisation of the sum of squares of the deviation, R = ∑(χexpT − χcalcT)2/∑(χexpT)2. The calculated curves very well reproduce the magnetic behaviour in the whole temperature range (Fig. 4a). The obtained results indicate that the predominant ferromagnetic coupling in 1 and 2 is due to the interaction between the neighbouring Cu(II) centres. A smaller antiferromagnetic contribution results from the interaction between the “outer” copper(II) atoms in a Cu3 unit. Although the J13 value is expected to be close to zero from the studies of other linear copper(II) trimers29 and relatively large distances between the terminal centres, a considerably worse agreement between the experimental and calculated susceptibilities using J13 = 0 implies an interaction which is far from negligible.
Ferromagnetic exchange interaction observed in 1 and 2 is in agreement with the coupling constants reported for a series of mixed-bridged hydroxo and carboxylato trinuclear copper(II) derivatives with the J12 values in the 30–90 cm−1 range.29,30 Such ferromagnetic coupling appears to be unexpected because of the syn–syn carboxylato and alkoxo bridges, since Oalkoxo–Cu–Oalkoxo angles larger than 97.5° are known to cause antiferromagnetic coupling.31 However, when the bridging ligands are different, the two bridges may either add or counterbalance their effects. This problem has already been addressed by Nishida et al.32 and McKee et al.33 who identified these phenomena as orbital complementarity and countercomplementarity, respectively. In the case of 1 and 2, the antiferromagnetic contributions of each bridge almost cancel each other out (i.e., these bridges exhibit an orbital countercomplementarity) and the ferromagnetic term dominates (JT = JAF + JF, with |JAF| being smaller than |JF|). However, some differences in the exchange coupling constants between adjacent copper(II) centres in the Cu3 units of 1 and 2 are consistent with their structural features and may arise from the presence in 2 of three symmetry non-equivalent copper(II) atoms and slight deviation from linearity of the asymmetric tricopper(II) core. In addition, a zigzag 1D chain structure of 2 may also cause some distinction of its magnetic behaviour in comparison with 1.
The EPR spectra of powdered samples 1 and 2 (Fig. S5†) provide evidence for the presence of the spin-quartet ground state with a zero-field splitting.34 There is one transition in low field part of the spectra [at 1500–2000 G (forbidden transition, ΔMS = ±2)], confirming the exchange interaction between the copper(II) centres in the Cu3 units, as detected by the direct magnetic measurements (Fig. 4a). Compared with the spectrum at 293 K, the signals at 77 K are much sharper and stronger.
Mild catalytic oxidation of cyclic and linear C5–C8 alkanes
In pursuit of our interest in the development of multicopper(II) catalytic materials for different oxidative transformations of saturated hydrocarbons,5,35,36 the compounds 1 and 2 have been applied as pre-catalysts for the mild homogeneous oxidation of alkanes by aqueous H2O2 in acetonitrile solution and in the presence of an acid promoter (co-catalyst), resulting in the formation of corresponding alcohols and ketones. Cyclohexane has been used as a model substrate while testing the influence of various reaction parameters. Control tests have shown that the oxidation of C6H12 does not proceed without a Cu pre-catalyst, either in the absence or in the presence of an acid promoter (Table S4†). Due to rather close structural characteristics, both compounds 1 and 2 appear to exhibit practically equal catalytic activities (Fig. 5a) in the oxidation of C6H12 to a mixture of cyclohexanol and cyclohexanone that proceeds in the presence of a small amount of the trifluoroacetic acid (TFA) co-catalyst. In fact, the C6H12 oxidations catalysed by the 1/TFA and 2/TFA systems proceed with the same reaction rate, achieving the maximum value of the total product yield (20% based on substrate) after 75 min time. Kinetic curves of the product accumulation in the presence of 1 (Fig. 5b) show that cyclohexanol is formed predominantly compared to cyclohexanone, the latter is however present since the beginning of the reaction. Since a slight difference in the product composition (alcohol-to-ketone molar ratio) has been observed before and after the treatment of the reaction solution with PPh3 (Shul'pin's method),18 the cyclohexane oxidation proceeds via the formation of cyclohexyl hydroperoxide as an intermediate product, which however rapidly decomposes (most likely via a Cu-catalysed process) to give alcohol and ketone as final products.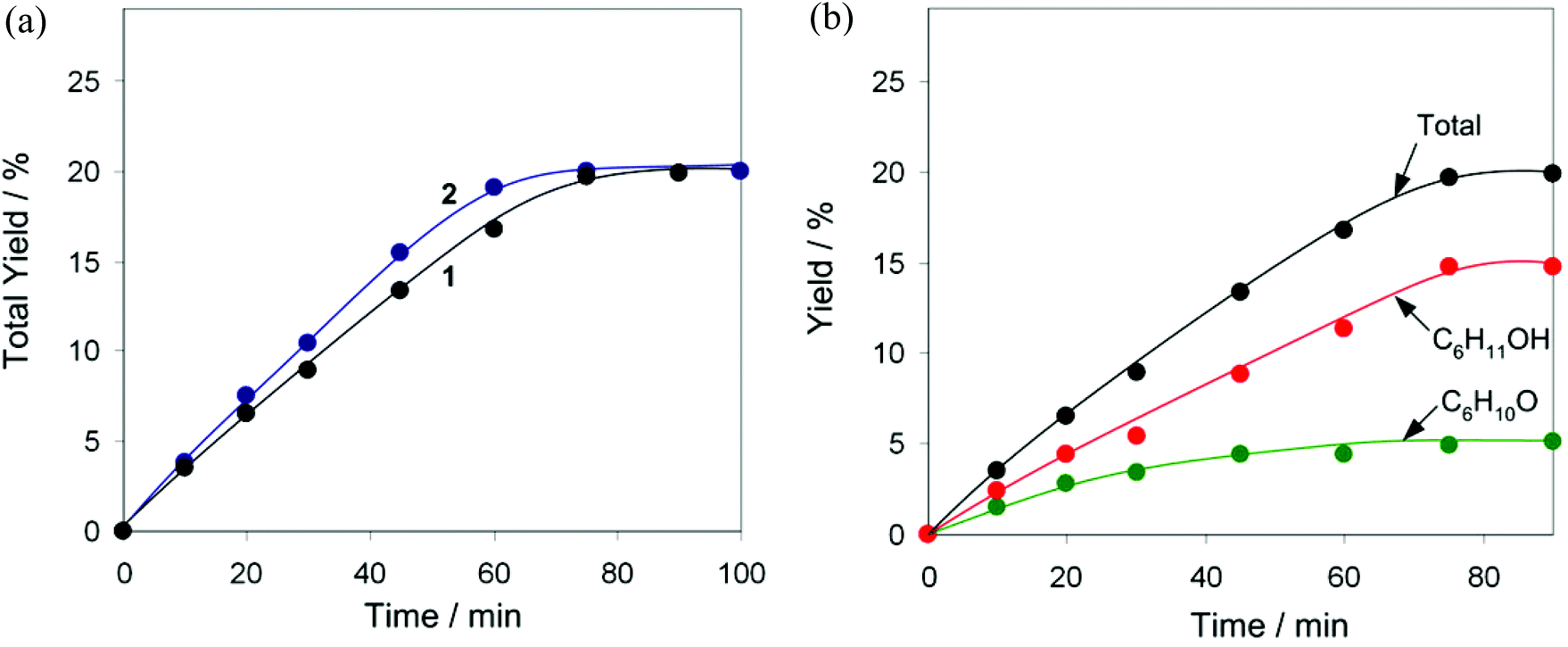 | ||
| Fig. 5 Oxidation of cyclohexane to cyclohexanol and cyclohexanone by H2O2. (a) Evolution of the total product yield with time in the reactions using pre-catalysts 1 or 2 and TFA. (b) Accumulation of cyclohexanol and cyclohexanone with time in the reaction with pre-catalyst 1 and TFA. General conditions: C6H12 (2 mmol), H2O2 (50% aq., 10 mmol), pre-catalyst 1 or 2 (0.01 mmol), TFA (0.1 mmol), 50 °C, MeCN (up to 5 mL). | ||
Both pre-catalysts 1 and 2 are almost inactive unless a small amount of an acid co-catalyst is added (see Fig. 6a for behaviour of 1 in the absence of an acid promoter and Table S4† for additional control tests). Aiming to verify whether the type of acid co-catalyst has an influence on the efficiency of catalytic system, we tested different acids (TFA, HCl, H2SO4 and HNO3) in the oxidation of C6H12 using 1 as a pre-catalyst (Fig. 6a). Although addition of TFA leads to the maximum total yield of 20% after 75 min (TOF = 32 h−1), the oxidation is exceptionally quicker in the presence of HCl resulting in a comparable total yield (18%) reached in 5 min reaction time (TOF = 430 h−1). The oxidation of C6H12 also proceeds with H2SO4 as a co-catalyst (18% total yield after 150 min), albeit is slower than in the 1/TFA system. In contrast, HNO3 shows a weaker promoting behaviour (13% total yield after 90 min).
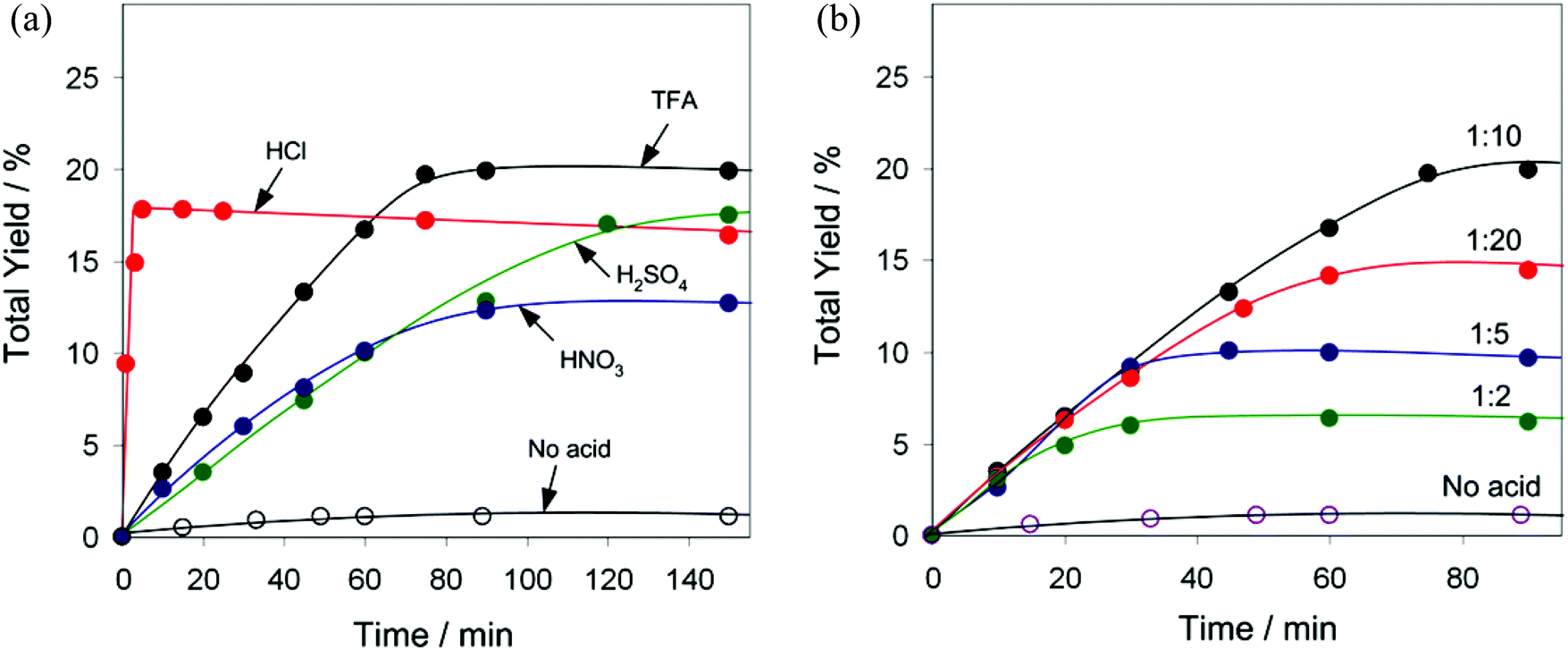 | ||
| Fig. 6 Oxidation of cyclohexane to cyclohexanol and cyclohexanone by H2O2 in the presence of pre-catalyst 1. (a) Evolution of the total product yield with time in the absence and in the presence of different acid co-catalysts (HNO3, H2SO4, HCl, TFA; 0.1 mmol). (b) Effect of TFA amount on the evolution of the total product total yield with time (1:TFA molar ratios: 1:0, 1:2, 1:5, 1:10, 1:20). General conditions: C6H12 (2 mmol), H2O2 (50% aq., 10 mmol), pre-catalyst 1 (0.01 mmol), 50 °C, MeCN (up to 5 mL). | ||
Besides, we studied the effect of TFA amount on the total yield and reaction rate of cyclohexane oxidation in the presence of 1 (Fig. 6b). It appears that the maximum initial reaction rate does not depend on the TFA concentration, although this mainly affects the oxidation time and the total yield. Such an observation suggests that TFA is not directly involved in the rate-determining step of the reaction, but is necessary to activate the pre-catalyst, presumably via additional protonation and partial decoordination of aminoalcohol and carboxylate moieties.5,37 The optimum TFA amount corresponds to a 1:TFA molar ratio of 1:10, whereas at lower co-catalyst loading (1:TFA = 1:2 or 1:5) the reaction is complete after 20 or 30 min, affording only 5 or 10% total product yields, respectively. A higher TFA concentration (1:TFA = 1:20) shows an unfavourable effect with the total product yield dropping to 15%. The importance and possible role of various acid promoters in metal-complex-catalysed alkane oxidations have been summarised in recent reviews.5,37a In brief, the function of an acid promoter consists in its involvement in proton transfer steps, activation of a pre-catalyst by ligand protonation and unsaturation of the metal centres, and enhancement of the oxidative properties of hydrogen peroxide and prevention of its decomposition (i.e., catalase activity).5,37a
To investigate the substrate versatility of the 1/TFA/H2O2 system, we have tested the oxidation of various cyclic and linear C5–C8 alkanes (Table 2). Under similar reaction conditions, the oxidation of cyclopentane and cyclooctane is less efficient in comparison with cyclohexane (16 and 18% vs. 20% total product yield for C5H10 and C8H16vs. C6H12, respectively), whereas a higher total yield (26%) is achieved in the case of cycloheptane substrate. In contrast to cycloalkanes, linear C5–C8 alkanes are less reactive substrates showing the total yields of oxidation products (isomeric alcohols and ketones) ranging from ∼10% for n-pentane and n-hexane oxidation to ∼14–15% for n-heptane and n-octane oxidation. Under the same reaction conditions, the control Cu(NO3)2/TFA/H2O2 system composed of simple copper(II) salt reveals much lower activity (∼2–5% overall yields, Table S2, ESI†) in comparison with the 1/TFA/H2O2 system (10–26% overall yields, Table 2) in the oxidation of both cyclic and linear C5–C8 alkanes. Besides, the alkane oxidations with the Cu(NO3)2 pre-catalyst appear to not depend on the presence of TFA, as attested by similar product yields obtained in the reactions using cyclohexane (∼3% with and without TFA) and n-hexane (∼4% with and without TFA) substrates (Table S2†).
Aiming to get some additional information on the reaction mechanism, we have investigated the oxidation of n-heptane, n-octane, methylcyclohexane, adamantane, cis-1,2-dimethylcyclohexane, trans-1,2-dimethylcyclohexane, cyclohexane and cyclohexane-d12 by the 1/H2O2/TFA and 2/H2O2/TFA systems and measured various selectivity parameters (Table 3), which appeared to be rather similar in both catalytic systems. The oxidation of linear chain alkanes, n-heptane and n-octane, proceeds without specific preference to any secondary carbon atom of the hydrocarbon chain, as attested by modest regioselectivity parameters C(1):C(2):C(3):(C4) of 1:(4–5):(4–6):(4–5).37,38 In the oxidation of methylcyclohexane, the normalised bond selectivity parameters 1°:2°:3° of 1:6:(12–15) suggest that the tertiary C atom is oxidised with some preference over the secondary C atoms (3°/2° ratio of 2–2.5). As expected, this behaviour is also observed in the adamantane oxidation that shows the 3°/2° parameter in the 3.5–3.8 range. The oxidation reactions with cis- or trans-1,2-dimethylcyclohexane as substrates generally proceed in a non-stereoselective manner, as confirmed by the trans/cis ratios of 0.7–0.9 between the generated isomeric tertiary alcohols with the mutual trans and cis orientation of the methyl groups. A partial inversion of the configuration has also been detected, with the cis isomers being predominant products in both cis- and trans-1,2-dimethylcyclohexane oxidations. Such a type of partial inversion of the configuration was previously described in various catalytic systems based on a tetracopper(II) triethanolamine derived complex [Cu4(μ4-O)(tea)4(BOH)4][BF4]2,36c as well as in other copper and vanadium based systems.37,38 Besides, a competitive oxidation of cyclohexane and cyclohexane-d12 revealed a low kinetic isotope effect (KIE = 1.1), which is indicative of a powerful and rather indiscriminate oxidizing species.39 By analogy with other Cu containing catalysts for alkane oxidation,5,36 all the above selectivity parameters (regioselectivity, bond selectivity, stereoselectivity, KIE; Table 3) are close to those previously reported for the catalytic systems operating with hydroxyl radicals, thus pointing out their involvement as principal oxidizing species in the present 1/TFA/H2O2 and 2/TFA/H2O2 systems.
| Selectivity parameter (alkane substrate) | 1 | 2 |
|---|---|---|
|
a Reaction conditions: pre-catalyst (0.01 mmol), TFA (0.1 mmol), alkane (2 mmol), H2O2 (50% aq., 10 mmol), MeCN up to 5 mL total volume, 50 °C. All parameters were measured after reduction of the reaction mixtures with PPh3 before GC analysis and calculated based on the ratios of isomeric alcohols. The calculated parameters were normalised, i.e., recalculated taking into account the number of H atoms at each carbon atom.
b Parameters C(1):C(2):C(3):C(4) are relative reactivities of H atoms at carbons 1, 2, 3 and 4 of the n-heptane or n-octane chain.
c Parameters 1°:2°:3° are relative normalised reactivities of the H atoms at primary, secondary and tertiary carbons of methylcyclohexane.
d Parameters 3°/2° are relative normalised reactivities of the H atoms at tertiary and secondary carbons of adamantane, determined as the ratio of the formed tertiary and secondary alcohol isomers.
e Parameter trans/cis is determined as the ratio of the formed tertiary alcohol isomers with mutual trans and cis orientation of the methyl groups.
f The kinetic isotope effect (KIE) was calculated as a ratio between the reaction rates of C6H12 and C6D12 oxidations.
|
||
| Regioselectivity | ||
| C(1):C(2):C(3):C(4) (n-C7H16)b |
1:5:6:5 |
1:4:5:5 |
| C(1):C(2):C(3):C(4) (n-C8H18)b |
1:5:5:5 |
1:4:4:4 |
| Bond selectivity | ||
| 1°:2°:3° (methylcyclohexane)c |
1:6:12 |
1:6:15 |
| 3°/2° (adamantane)d | 3.5 | 3.8 |
| Stereoselectivity | ||
| trans/cis (cis-1,2-Dimethylcyclohexane)e | 0.8 | 0.9 |
| trans/cis (trans-1,2-Dimethylcyclohexane)e | 0.8 | 0.7 |
| Kinetic isotope effect (KIE)f | 1.1 | 1.1 |
It should also be noted that 1 and 2 are not intact in the course of catalytic experiments and, upon treatment with an acid co-catalyst and oxidant, generate catalytically active species, the exact nature of which is still to be established. In fact, the UV-vis studies of compounds 1 and 2 indicate that their parent bands with a maximum at 673–678 nm (Fig. S6a,b†) are shifted to 755–756 nm upon addition of trifluoroacetic acid (Fig. S6c,d†), whereas further introduction of hydrogen peroxide leads to the disappearance of these bands due to the generation of copper(I) species (Fig. S6e,f†). Although a somewhat similar trend (i.e., slight shift of the d–d transition band from 746 to 762 nm upon the addition of TFA and its disappearance when introducing H2O2, Fig. S7a–d†) is observed in the UV-vis spectra of Cu(NO3)2 as well as various model solutions comprising ligands (Fig. S7e–j†), one should mention that the spectra of the latter are not equal to those derived from the parent samples 1 and 2 (Fig. S6†). These observations might explain a different activity of the pre-catalysts 1 and 2 in comparison with the corresponding model solutions.
In fact, various control tests have disclosed that alkane oxidations can also proceed in model systems comprising a mixture of copper(II) salt, aminoalcohol and homophthalic acid, either with or without TFA (Table S4†). The oxidation of cyclohexane by the Cu(NO3)2/H3tea/H2hpa/TFA/H2O2 system (Cu:H3tea:H2hpa molar ratio of 1.5:1:1) that models the behaviour of 2 results in an overall yield of ∼11%, whereas the use of higher amounts of ligands (Cu:H3tea:H2hpa molar ratio of 1:5:5) increases the yield up to ∼16% (Table S4†). A similar behaviour is observed when applying the Cu(NO3)2/H5bis-tris/H2hpa/TFA/H2O2 system as a model of 1, which leads to a maximum yield of ∼13%. Due to the presence of homophthalic acid, both the above-mentioned model systems are also active in the absence of TFA, although resulting in slightly inferior yields (∼12–14%, Table S4†). These results indicate that the parent pre-catalysts 1 or 2 with TFA are more efficient in the cyclohexane oxidation (∼19% overall yields) in comparison with different model systems, either solely based on copper(II) nitrate (∼3% overall yields) or comprising a combination of aminoalcohol, homophthalic and trifluoroacetic acids (∼8–16% overall yields, Table S4†). Hence, these facts confirm the influence of ligands and their structural arrangement in 1 and 2 on the observed catalytic behaviour. It should also be mentioned that the 3-fold lower loadings of 1 and 2 (0.0033 mmol) relative to those (0.01 mmol) typically applied in the present work do not lead to essentially different product yields, but instead affect the rate of the oxidation reaction. As depicted in Fig. S8,† the cyclohexane oxidation by the 1/TFA/H2O2 system shows only a low difference in total product yields (∼20 vs. 19% for 0.01 and 0.0033 mmol loading of 1, respectively), although different reaction times (75 vs. 180 min, respectively) are needed to achieve these yields. A similar behaviour is observed when applying the 2/TFA/H2O2 system (Table S4†).
Although a detailed investigation of the mechanism and characterisation of catalytic intermediates was out of the scope of the present work, we have attempted an ESI-MS study of the pre-catalyst 1 after its treatment with the TFA co-catalyst and hydrogen peroxide in MeCN–H2O medium and under the conditions similar to those of catalytic experiments. Although the obtained ESI-MS(−) pattern reveals similar tri- and dicopper fragments detected in the spectrum of the parent compound 1, their relative intensity is decreased. In addition, two new rather intense fragments emerge, namely [Cu3(H2bis-tris)2]− (m/z = 603) and [Cu(H3bis-tris)(H2O)]− (m/z = 289), which can potentially correspond to catalytically active species. This can be indirectly confirmed by the presence of a highly intense [Hhpa]− (m/z = 179) peak in 1 or [Hhpa + H2O]− (m/z = 197) fragment in 1/TFA/H2O2 and the absence of any signal corresponding to a free aminoalcohol ligand in both samples, thus suggesting that H3bis-tris moieties are strongly bound to copper centres and are important for the observed catalysis. This observation can also explain a significantly higher catalytic activity of model solutions composed of a copper(II) salt and an aminoalcohol in comparison with systems comprising only a copper(II) salt (Table S4†).
Based on the above-mentioned data and literature background5,36,37 we can propose that the general reaction mechanism involves the following steps. An acid promoter interacts with copper pre-catalyst causing the formation of labile sites (via an additional protonation and partial decoordination of aminoalcohol and/or elimination of carboxylate ligands, L) and generation of LCuII species. These participate in the formation of HO˙ radicals from H2O2, possibly via Fenton-like steps [2LCuII + H2O2 → 2LCuI + 2H+ + O2; LCuI + H2O2 → LCuII + HO˙ + HO−].36c,37a Then, the hydroxyl radicals abstract H atoms from an alkane forming the alkyl radicals R˙ [RH + HO˙ → R˙ + H2O], which further react with O2 (e.g., from air) resulting in the ROO˙ radicals [R˙ + O2 → ROO˙] that are transformed to alkyl hydroperoxides ROOH as primary intermediate products [ROO˙ + CuI → ROO− + CuII; ROO− + H+ → ROOH].36c However, alkyl hydroperoxides rapidly decompose (conceivably by Cu-catalysed processes) to furnish the corresponding alcohols and ketones as final products.5,36–38
Conclusions
In the present work, we have further widened a versatile aqueous medium self-assembly synthetic method to a different combination of aminoalcohol and aromatic dicarboxylate building blocks, allowing the design of two novel crystalline materials 1 and 2 that feature distinct tricopper(II) cores with various coordination geometries of copper(II) centres. The obtained products not only extend the copper coordination chemistry of aminoalcohol biobuffers that still remains unexplored for H5bis-tris, but also provide very rare examples of Cu compounds derived from homophthalic acid.7 Besides, both compounds reveal interesting and rather complex 3D (1) or 2D (2) supramolecular networks assembled via multiple intermolecular H-bonds. These supramolecular nets have been analysed from the topological viewpoint, allowing an identification of the uninodal 6-connected 3D framework in 1 and binodal 3,5-connected 2D layer in 2 with the snk and 3,5L50 topology, respectively. Thus, the current work also contributes towards the topological classification of supramolecular networks in metal–organic materials.15,26,27 In fact, in spite of being theoretically predicted, the snk topological net had not been experimentally identified before the present study. Besides, the magnetic properties of the obtained compounds have been investigated indicating a predominant ferromagnetic coupling between the “central” and “outer” copper(II) atoms in the Cu3 cores, while a weak antiferromagnetic interaction has been detected between the two “outer” copper(II) centres. Although symmetric and asymmetric tricopper(II) cores in 1 and 2 reveal a number of structural differences, both compounds show essentially similar magnetic behaviour that is also in good agreement with other mixed-bridged alkoxo and carboxylato trinuclear copper(II) derivatives.Furthermore, we have shown that 1 and 2 act as versatile and rather efficient bio-inspired pre-catalysts towards the homogeneous oxidation, by H2O2 under mild conditions (50 °C, atmospheric pressure) in acidic MeCN–H2O medium, of diverse cyclic and linear C5–C8 alkanes to give the corresponding alcohols and ketones. Given the fact that both pre-catalysts represent a comparable level of activity, the catalytic action of 1 has been studied in more detail. The influence of various acid promoters (TFA, HCl, H2SO4 and HNO3) has been investigated, disclosing the highest activity of trifluoroacetic acid, although the cyclohexane oxidation has been exceptionally quicker (TOF up to 430 h−1) when using HCl as a co-catalyst. The substrate versatility of the 1/TFA/H2O2 system has been studied, showing that cycloalkanes are generally more reactive substrates than the corresponding linear hydrocarbons. The bond, regio- and stereoselectivity parameters as well as kinetic isotope effect have also been determined for both 1/TFA and 2/TFA catalytic systems, testifying an involvement of hydroxyl radicals as main oxidizing species in the present alkane oxidations.
The results obtained herein indicate that common biological buffers such as bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane (H5bis-tris) and triethanolamine (H3tea) can act as versatile multidentate N,O-building blocks towards crystal engineering, by facile aqueous medium self-assembly method, of novel multicopper(II) materials that not only disclose a number of interesting structural, topological and magnetic features, but also furnish bio-inspired pre-catalysts for the mild oxidation of alkanes. Further research on widening the types of self-assembled multicopper(II) cores derived from various aminoalcohol biobuffers and search for their application in molecular magnetism and biomimetic oxidation catalysis is underway.
Acknowledgements
This work was supported by the Foundation for Science and Technology (FCT) (projects PTDC/QUI-QUI/121526/2010, RECI/QEQ-QIN/0189/2012, PEst-OE/QUI/UI0100/2013, IF/01395/2013 and SFRH/BPD/78854/2011), Portugal. We also thank Dr. M. C. Oliveira and Ms. A. Dias for ESI-MS (IST-node of the RNEM/FCT).References
- (a) G. R. Desiraju, J. J. Vittal and A. Ramanan, Crystal Engineering: A Textbook, World Scientific, 2011 Search PubMed; (b) Engineering of Crystalline Materials Properties: State of the Art in Modeling, Design and Applications, ed. J. J. Novoa, D. Braga and L. Addadi, Springer, 2008 Search PubMed; (c) D. Braga and F. Grepioni, Making Crystals by Design: Methods, Techniques and Applications, Wiley, 2007 Search PubMed.
- (a) R. Mukherjee in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty, T. J. Meyer and D. E. Fenton, Elsevier, Dordrecht, 2nd edn, 2003, vol. 6, chap. 6.6, pp. 747–910 Search PubMed; (b) R. E. P. Winpenny in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty, T. J. Meyer, M. Fujita, A. Powell and C. Creutz, Elsevier, Dordrecht, 2nd edn, 2003, vol. 7, chap. 7.3, pp. 125–175 Search PubMed.
- (a) C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815 CrossRef CAS PubMed; (b) S. Mukherjee and P. S. Mukherjee, Acc. Chem. Res., 2013, 46, 2556 CrossRef CAS PubMed; (c) C. Adhikary and S. Koner, Coord. Chem. Rev., 2010, 254, 2933 CrossRef CAS PubMed.
- (a) S. R. Batten, D. R. Turner and S. M. Neville, Coordination Polymers: Design, Analysis and Application, Royal Society of Chemistry, London, 2009 Search PubMed; (b) A. J. Blake, N. R. Champness, P. Hubberstey, W. S. Li, M. A. Withersby and M. Schroder, Coord. Chem. Rev., 1999, 138, 117 CrossRef.
- A. M. Kirillov, M. V. Kirillova and A. J. L. Pombeiro, Coord. Chem. Rev., 2012, 256, 2741 CrossRef CAS PubMed.
- (a) H. Eagle, Science, 1971, 174, 500 CrossRef CAS; (b) K. H. Scheller, T. H. Abel, P. E. Polanyi, P. K. Wenk, B. E. Fischer and H. Sigel, Eur. J. Biochem., 1980, 107, 455 CrossRef CAS PubMed; (c) R. Beynon and J. Easterby, Buffer Solutions Basics, Oxford Univ. Press Inc., New York, 1st edn, 1996 Search PubMed.
- See the Cambridge Structural Database (CSD, version 5.35, 2014): F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed.
- (a) A. M. Kirillov, Y. Y. Karabach, M. V. Kirillova, M. Haukka and A. J. L. Pombeiro, Cryst. Growth Des., 2012, 12, 1069 CrossRef CAS; (b) A. M. Kirillov, Y. Y. Karabach, M. Haukka, M. F. C. G. da Silva, J. Sanchiz, M. N. Kopylovich and A. J. L. Pombeiro, Inorg. Chem., 2008, 47, 162 CrossRef CAS PubMed; (c) G. Xu, X. He, J. Lv, Z. Zhou, Z. Du and Y. Xie, Cryst. Growth Des., 2012, 12, 3619 CrossRef CAS; (d) Z. Boulsourani, V. Tangoulis, C. P. Raptopoulou, V. Psycharis and C. Dendrinou-Samara, Dalton Trans., 2011, 40, 7946 RSC.
- (a) R. S. Brissos, S. Garcia, A. Presa and P. Gamez, Comments Inorg. Chem., 2011, 32, 219 CrossRef CAS; (b) P. Gamez, P. G. Aubel, W. L. Driessen and J. Reedijk, Chem. Soc. Rev., 2001, 30, 376 RSC.
- (a) R. A. Himes and K. D. Karlin, Curr. Opin. Chem. Biol., 2009, 13, 119 CrossRef CAS PubMed; (b) R. L. Lieberman and A. C. Rosenzweig, Nature, 2005, 434, 177 CrossRef CAS PubMed.
- Bruker Analytical Systems Madison, WI, 2005.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- (a) V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CrossRef CAS; (b) V. A. Blatov, IUCr CompComm Newslett., 2006, 7, 4 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- (a) G. B. Shul'pin, C. R. Chim., 2003, 6, 163 CrossRef; (b) G. B. Shul'pin, J. Mol. Catal. A: Chem., 2002, 189, 39 CrossRef; (c) G. B. Shul'pin, Mini-Rev. Org. Chem., 2009, 6, 95 CrossRef.
- (a) S. S. P. Dias, V. André, J. Kłak, M. T. Duarte and A. M. Kirillov, Cryst. Growth Des., 2014, 14, 3398 CrossRef CAS; (b) M. V. Kirillova, A. M. Kirillov, A. N. C. Martins, C. Graiff, A. Tiripicchio and A. J. L. Pombeiro, Inorg. Chem., 2012, 51, 5224 CrossRef CAS PubMed; (c) A. M. Kirillov, J. A. S. Coelho, M. V. Kirillova, M. F. C. G. da Silva, D. S. Nesterov, K. R. Gruenwald, M. Haukka and A. J. L. Pombeiro, Inorg. Chem., 2010, 49, 6390 CrossRef CAS PubMed; (d) P. J. Figiel, A. M. Kirillov, M. F. C. G. da Silva, J. Lasri and A. J. L. Pombeiro, Dalton Trans., 2010, 39, 9879 RSC; (e) Y. Y. Karabach, A. M. Kirillov, M. Haukka, J. Sanchiz, M. N. Kopylovich and A. J. L. Pombeiro, Cryst. Growth Des., 2008, 8, 4100 CrossRef CAS; (f) Ł. Jaremko, A. M. Kirillov, P. Smoleński and A. J. L. Pombeiro, Cryst. Growth Des., 2009, 9, 3006 CrossRef.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 5th edn, Wiley, New York, 1997 Search PubMed.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- (a) A. M. Kirillov, Y. Y. Karabach, M. V. Kirillova, M. Haukka and A. J. L. Pombeiro, Dalton Trans., 2011, 40, 6378 RSC; (b) Y. Inomata, Y. Gochou, M. Nogami, F. S. Howell and T. Takeuchi, J. Mol. Struct., 2004, 702, 61 CrossRef CAS PubMed.
- (a) G.-Z. Liu, J. Zhang and L.-Y. Wang, Polyhedron, 2011, 30, 1487 CrossRef CAS PubMed; (b) A. M. Atria, G. Corsini, M. T. Garland and R. Baggio, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2011, 67, m342 CAS.
- (a) L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC; (b) A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- (a) M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675 CrossRef PubMed; (b) The Reticular Chemistry Structure Resource (RCSR) Database; M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 30, 1782 CrossRef PubMed; (c) M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343 CrossRef CAS PubMed.
- (a) P. N. Zolotarev, M. N. Arshad, A. M. Asiri, Z. M. Al-amshany and V. A. Blatov, Cryst. Growth Des., 2014, 14, 1938 CrossRef CAS; (b) I. A. Baburin, V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2008, 10, 1822 RSC.
- O. Kahn, Molecular Magnetism, VCH, New York, Weinheim and Cambridge, 1993 Search PubMed.
- (a) W. Haase and S. Gehring, J. Chem. Soc., Dalton Trans., 1985, 2609 RSC; (b) S. Gehring, P. Fleischhauer, H. Paulus and W. Haase, Inorg. Chem., 1993, 32, 54 CrossRef CAS.
- (a) S. Gehring, H. Astheimer and W. Haase, J. Chem. Soc., Faraday Trans. 2, 1987, 83, 347 RSC; (b) G. A. van Albada, I. Mutikainen, O. Roubeau, U. Turpeinen and J. Reedijk, Inorg. Chim. Acta, 2002, 331, 208 CrossRef CAS.
- (a) A. Rodríguez-Fortea, P. Alemany, S. Alvarez and E. Ruiz, Chem. – Eur. J., 2001, 7, 627 CrossRef; (b) E. Ruiz, P. Alemany, S. Alvarez and J. Cano, J. Am. Chem. Soc., 1997, 119, 1297 CrossRef CAS; (c) Y. Song, P. Gamez, O. Roubeau, M. Lutz, A. L. Spek and J. Reedijk, Eur. J. Inorg. Chem., 2003, 2924 CrossRef CAS PubMed; (d) G. A. van Albada, P. J. van Koningsbruggen, I. Mutikainen, U. Turpeinen and J. Reedijk, Eur. J. Inorg. Chem., 1999, 2269 CrossRef CAS; (e) A. Roth, J. Becher, C. Herrmann, H. Görls, G. Vaughan, M. Reiher, D. Klemm and W. Plass, Inorg. Chem., 2006, 45, 10066 CrossRef CAS PubMed; (f) T. C. Higgs, K. Spartalian, C. J. O'Connor, B. F. Matzanke and C. J. Carrano, Inorg. Chem., 1998, 37, 2263 CrossRef CAS PubMed; (g) M. H. W. Lam, Y. Y. Tang, K. M. Fung, X. Z. You and W. T. Wong, Chem. Commun., 1997, 957 RSC.
- (a) Y. Nishida and S. Kida, J. Chem. Soc., Dalton Trans., 1986, 2633 RSC; (b) Y. Nishida, M. Takeuchi, K. Takahashi and S. Kida, Chem. Lett., 1985, 631 CrossRef CAS.
- (a) V. McKee, M. Zvagulis and C. A. Reed, Inorg. Chem., 1985, 24, 2914 CrossRef CAS; (b) V. McKee, M. Zvagulis, J. V. Dagdigian, M. G. Patch and C. A. Reed, J. Am. Chem. Soc., 1984, 106, 4765 CrossRef CAS.
- (a) R. Boca, Coord. Chem. Rev., 2004, 248, 757 CrossRef CAS PubMed; (b) F. E. Mabbs and D. Collison, Electron Paramagnetic Resonance of d Transition Metal Compounds, Elsevier Science, Amsterdam, 1992 Search PubMed.
- (a) M. V. Kirillova, A. M. Kirillov and A. J. L. Pombeiro, Chem. – Eur. J., 2010, 16, 9485 CrossRef CAS PubMed; (b) M. V. Kirillova, A. M. Kirillov and A. J. L. Pombeiro, Adv. Synth. Catal., 2009, 351, 2936 CrossRef CAS PubMed.
- (a) A. M. Kirillov, M. V. Kirillova, L. S. Shul'pina, P. J. Figiel, K. R. Gruenwald, M. F. C. G. da Silva, M. Haukka, A. J. L. Pombeiro and G. B. Shul'pin, J. Mol. Catal. A: Chem., 2011, 350, 26 CrossRef CAS PubMed; (b) M. V. Kirillova, A. M. Kirillov, D. Mandelli, W. A. Carvalho, A. J. L. Pombeiro and G. B. Shul'pin, J. Catal., 2010, 272, 9 CrossRef CAS PubMed; (c) M. V. Kirillova, Y. N. Kozlov, L. S. Shul'pina, O. Y. Lyakin, A. M. Kirillov, E. P. Talsi, A. J. L. Pombeiro and G. B. Shul'pin, J. Catal., 2009, 268, 26 CrossRef CAS PubMed.
- (a) A. M. Kirillov and G. B. Shul'pin, Coord. Chem. Rev., 2013, 257, 732 CrossRef CAS PubMed; (b) A. M. Kirillov, M. V. Kirillova and A. J. L. Pombeiro, Adv. Inorg. Chem., 2013, 65, 1 CrossRef CAS.
- (a) G. B. Shul'pin, Dalton Trans., 2013, 42, 12794 RSC; (b) G. B. Shul'pin, Org. Biomol. Chem., 2010, 8, 4217 RSC.
- M. Costas, K. Chen and L. Que Jr., Coord. Chem. Rev., 2000, 200–202, 517 CrossRef CAS.
Footnote |
| † TG-DTA plots (Fig. S1, S2), additional topological representations (Fig. S3, S4), EPR spectra (Fig. S5), UV-vis spectra (Fig. S6, S7), hydrogen bonding (Table S1) and catalytic (Tables S2–S4) details for 1 and 2. CCDC 1014687 and 1014688. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00220b |
| This journal is © the Partner Organisations 2015 |