
Improvement of analytical performance in inductively coupled plasma optical emission spectrometry without compromising robustness using an infrared-heated sample introduction system with a pneumatic nebulizer
Yoseif
Makonnen
a,
John
Burgener
b and
Diane
Beauchemin
*a
aDepartment of Chemistry, Queen's University, Kingston, Ontario K7L 3N6, Canada. E-mail: diane.beauchemin@chem.queensu.ca; Fax: +1-613-533-6669; Tel: +1-613-533-2619
bBurgener Research Inc, 1680 Lakeshore Road West, Mississauga, Ontario L5J 1J5, Canada
First published on 12th September 2014
Abstract
A simple enhanced sample introduction system, using pneumatic nebulization (PN), was developed for inductively coupled plasma optical emission spectrometry (ICP OES). The aerosol generated by a Burgener parallel-flow nebulizer, coupled to a single-pass flip chamber (FC), was heated to 230 °C using a ceramic infrared (IR) heater. Multivariate optimizations were conducted to find operating conditions that maximized analyte sensitivity while maintaining plasma robustness, as measured by the Mg II/Mg I intensity ratio. Other spray chamber designs, such as a single-pass (SP) and a custom Scott-type double-pass (DPE), were also tested in order to find the ideal combination with the Burgener nebulizer. Under optimum conditions and compared to conventional pneumatic nebulization at room temperature, a 6 fold improvement in sensitivity and a 4–7 fold improvement in detection limit was obtained for 38 elements using FC(IR), SP(IR) and DPE(IR). The improvement was more significant for ionic rather than atomic emission lines. Plasma robustness also increased significantly over room temperature PN. Moreover, the improvements in analytical performance are observed for FC(IR), SP(IR) and DPE(IR), despite using a sample introduction rate over 10 times lower than room temperature PN.
Introduction
Inductively coupled plasma (ICP) optical emission spectrometry (OES) and mass spectrometry (MS) are widely used high throughput, multi-elemental trace and ultra-trace detection techniques with a wide linear dynamic range for the analysis of various geological/environmental samples.1 On the basis of sensitivity, ICP OES is typically used for the determination of major, minor and trace elements, while ICP-MS is usually preferable for ultra-trace analysis. On the other hand, ICP OES, which passively measures the emitted light, is inherently more robust than ICP-MS, in which the analyte ions must be physically extracted from the plasma. The continuous nebulization of solutions with dissolved solid content ≥0.2% m/v in ICP-MS leads to clogging of the sampler and/or skimmer cones, whereas no such problem exists in ICP OES.2 Hence, a much greater degree of dissolved solid content can be tolerated in ICP OES, up to several % m/v in some cases. This translates into less sample dilution or pre-treatment and thus a higher sample throughput if the dilution or pre-treatment is not done on-line. Clearly then, improving the detection limits of ICP OES, without sacrificing robustness, would be very advantageous.In ICP OES, the plasma can be viewed laterally (side-on), at 90° to the central channel, or axially (end-on) to improve detection limits.3,4 However, when samples with complex matrices are analysed and robust plasma conditions are required, the lateral (side-on) view configuration offers the best performance in terms of analytical figures of merit.4,5 From the ICP literature,6–10 high radio frequency (R.F.) power, low nebulizer gas flow rate and low sample uptake rate, and/or a wider bore injector produce robust plasma conditions, under which the matrix composition of the sample has little effect on analyte intensity. Such conditions provide improved sample desolvation, vaporization, atomization and ionization through greater energy transfer from the bulk plasma to the central channel,6,7 and can be characterized by monitoring the Mg II (280.270 nm)/Mg I (285.213 nm), ionic to atomic, line emission ratio. In general, a Mg II/Mg I ratio > 10 constitutes robust plasma conditions.8–10
Because the detection limit is inversely proportional to analyte sensitivity (slope of the calibration curve) and directly proportional to the background noise level (standard deviation of the blank), it can be improved via an increase in sample introduction efficiency and/or a reduction of noise sources. In ICP-based spectrometry, aqueous sample solutions are typically introduced into the plasma via a pneumatic nebulization (PN) system comprised of a pneumatic nebulizer and a spray chamber,11 where at least 95% of the sample typically goes down to the drain. The polydisperse nature of the sample aerosol reaching the plasma is a significant source of noise. Large droplets that undergo desolvation and vaporization cool the plasma in the vicinity of smaller droplets, which have reached the atomization, ionization and/or excitation stage, thereby reducing the number of excited atoms and ions.2 Thus, conditions that lead to smaller, or even vaporized, sample aerosol droplets will drastically reduce this source of noise.
An alternative sample introduction system that increases sample introduction efficiency, while reducing noise from variable droplet size, is the ultrasonic nebulizer (USN) equipped with a desolvation system. Typically, a heater/condenser (HC) is used to vaporize and condense, respectively, a large majority of the solvent, thereby reducing the average aerosol droplet size. Further reduction of the solvent load can be accomplished using a membrane desolvator (MD), especially for organic solvents.12 The increased sample introduction efficiency (30% for USN-HC-MD vs. 5% for PN) and the effective pre-concentration of the analyte, via desolvation, leads to significant improvements in sensitivity and detection limit over conventional PN.13,14 However, since the matrix is pre-concentrated along with the sample, matrix effects are exacerbated for USN systems11,13 compared to conventional PN3,15 or hydride generation systems.16 There may be some analytes lost during desolvation (i.e. Hg) and the desolvation system itself can lead to memory effects and blockages in the MD due to salt depositions.17,18 The removal of water via desolvation also has a detrimental effect on the excitation and ionization capability of the plasma,16 because water is the main source of hydrogen and oxygen in the plasma (which facilitate energy transfer between the bulk plasma and the central channel) and it acts as a load buffer (which minimizes matrix effects).19,20 All of these factors make the analysis of environmental/geological samples (complex matrices) with USN systems quite difficult.21–23
On the other hand, the sample aerosol can be vaporized, without removing water, by inserting a heated (∼400 °C) pre-evaporation tube (PET) between the spray chamber and the torch, leading to improvements in both sensitivity and detection limit in ICP-MS.24–26 For most of the analytes, the improvement in detection limit surpasses that seen for sensitivity, which would suggest a reduction in noise arising from droplet desolvation, as it decreases the average size of sample aerosol droplets entering the plasma.24,26 Similarly, replacing the whole desolvation system of USN-HC-MD with a PET improved sensitivity, detection limit and plasma robustness in ICP OES.2,27 The significant improvements were attributed to the water vapor that was preserved by the PET,27 in contrast to introducing water aerosol, which greatly increases the background noise level and decreases both the ionization temperature and the electron number density of the ICP.19 Significant improvement in sensitivity and detection limit was also demonstrated using a low sample consumption system with a spray chamber heated to 105 °C.28 Coupling a multi-mode sample introduction system (MSIS) with a USN-PET improved sensitivity and detection limits, in particular for hydride-forming elements.29 Replacing the heating tape with an infrared (IR) heater in a USN-PET system allowed vastly increased sample uptake rates because IR heating is more uniform and efficient than convective heating.2 The efficacy of IR heating over conduction-convection based heating had also been demonstrated in earlier studies with the Mistral desolvation system for ICP OES.30–32
Another enhanced PN sample introduction system is the torch-integrated sample introduction system (TISIS) being developed by Todolí and Mermet, which uses a micro-nebulizer coupled to a heated single-pass spray chamber to minimize washout time and memory effects without degrading sensitivity, stability and detection limits.33 Such a system can achieve 100% sample introduction efficiency when operated at very low sample delivery rates (i.e. less than 20 μL min−1).34 It was successfully applied to the analysis of petroleum derivatives, with no matrix effects.35 It was also successfully coupled to ICP-MS, for the analysis of representative certified reference samples (spinach leaves, marine plankton, bone tissue, human blood).36 However, the very small sample uptake rate limits sensitivity and the speed of analysis.
The goal of this work was to develop a simple enhanced PN sample introduction system involving direct coupling of a Burgener nebulizer to an IR-heated conventional spray chamber. This is in contrast to the TISIS, which is limited to <20 μL min−1 by the small size of the spray chamber. By achieving complete pre-evaporation, IR heating of a conventional size spray chamber should allow for a much greater sample uptake rate while enabling aerosol formation without the aerosol striking the chamber's wall and going down the drain. In theory, it should thus be possible to obtain similar improvements as reported for the USN-PET-(IR) system, but using a Burgener nebulizer instead of an expensive USN. The ultimate aim was to find conditions providing essentially 100% sample introduction efficiency at relatively high sample uptake rates. To this end, fundamental studies on IR heating of the aerosol produced by a Burgener nebulizer, coupled to different spray chamber designs, were conducted to determine the ideal combination to improve both detection limits and plasma robustness. A multivariate experimental design was used to optimize plasma and sample introduction parameters so as to maximize sensitivity, while also maintaining robustness, as measured by the Mg II/Mg I ratio. The analytical performance was then compared to a conventional PN sample introduction system at room temperature, as well as to previously published results for the TISIS, PN-MSIS-PET and USN-PET-(IR) systems.
Experimental
Instrumentation
Research was conducted on a lateral view (side-on) ARCOS ICP OES instrument (SPECTRO Analytical Instruments, Kleve, Germany) fitted with a parallel-flow HP Ari Mist Burgener nebulizer (Burgener Research Inc., Mississauga, Canada), different spray chambers, and a torch with an integrated sheathing device (SPECTRO Analytical Instruments, Kleve, Germany). A conventional torch without a sheathing device (SCP Science, Baie d’Urfé, Quebec, Canada) was also used for comparative purposes.A single-pass flip chamber (FC) (Burgener Research Inc., Mississauga, Canada), standard Scott double-pass spray chamber (DP) (SPECTRO Analytical Instruments, Kleve, Germany and Precision Glass Blowing, Colorado, USA), a custom Scott double-pass spray chamber with a glass extension nebulizer adaptor (DPE) (Precision Glass Blowing, Colorado, USA) and a single-pass spray chamber (SP) (Burgener Research Inc., Mississauga, Canada) were used, whose dimensions are given in Fig. 1. The optimal plasma operating conditions and measurement parameters for the different spray chamber designs are summarised in Table 1.
 | ||
| Fig. 1 Spray chambers used and their dimensions: top, custom Scott double-pass spray chamber with a glass extension nebulizer adaptor (DPE) (the regular Scott double-pass spray chamber (DP) had the same dimensions as the DPE); middle, single-pass spray chamber (SP); bottom, single-pass flip chamber (FC). | ||
| Parameter | Range | DP(RT)a | FC(IR)b | SP(IR)c | DPE(IR)d |
|---|---|---|---|---|---|
| a Room temperature Scott double-pass spray chamber. b Infrared-heated single-pass flip chamber. c Infrared-heated single-pass spray chamber. d Infrared-heated Scott double-pass spray chamber with integrated glass extension instead of a Teflon adaptor to couple to the nebulizer. | |||||
| Plasma gas flow rate (L min−1) | 12.0–15.0 | 12.0 | 14.5 | 14.5 | 14.5 |
| Auxiliary gas flow rate (L min−1) | 0.6–3.0 | 1.0 | 0.8 | 0.8 | 0.9 |
| R.F. power (kW) | 1.4–1.7 | 1.4 | 1.7 | 1.7 | 1.7 |
| Sheath gas flow rate (L min−1) | 0–0.7 | 0 | 0.55 | 0.60 | 0.50 |
| Nebulizer gas flow rate (L min−1) | 0.6–1.2 | 1.0 | 1.0 | 1.0 | 1.0 |
| Plasma observation height (mm) | 10–14 | 10 | 10 | 10 | 10 |
| Sample uptake rate (mL min−1) | 0.05–2.0 | 1 | 0.08 | 0.0925 | 0.09 |
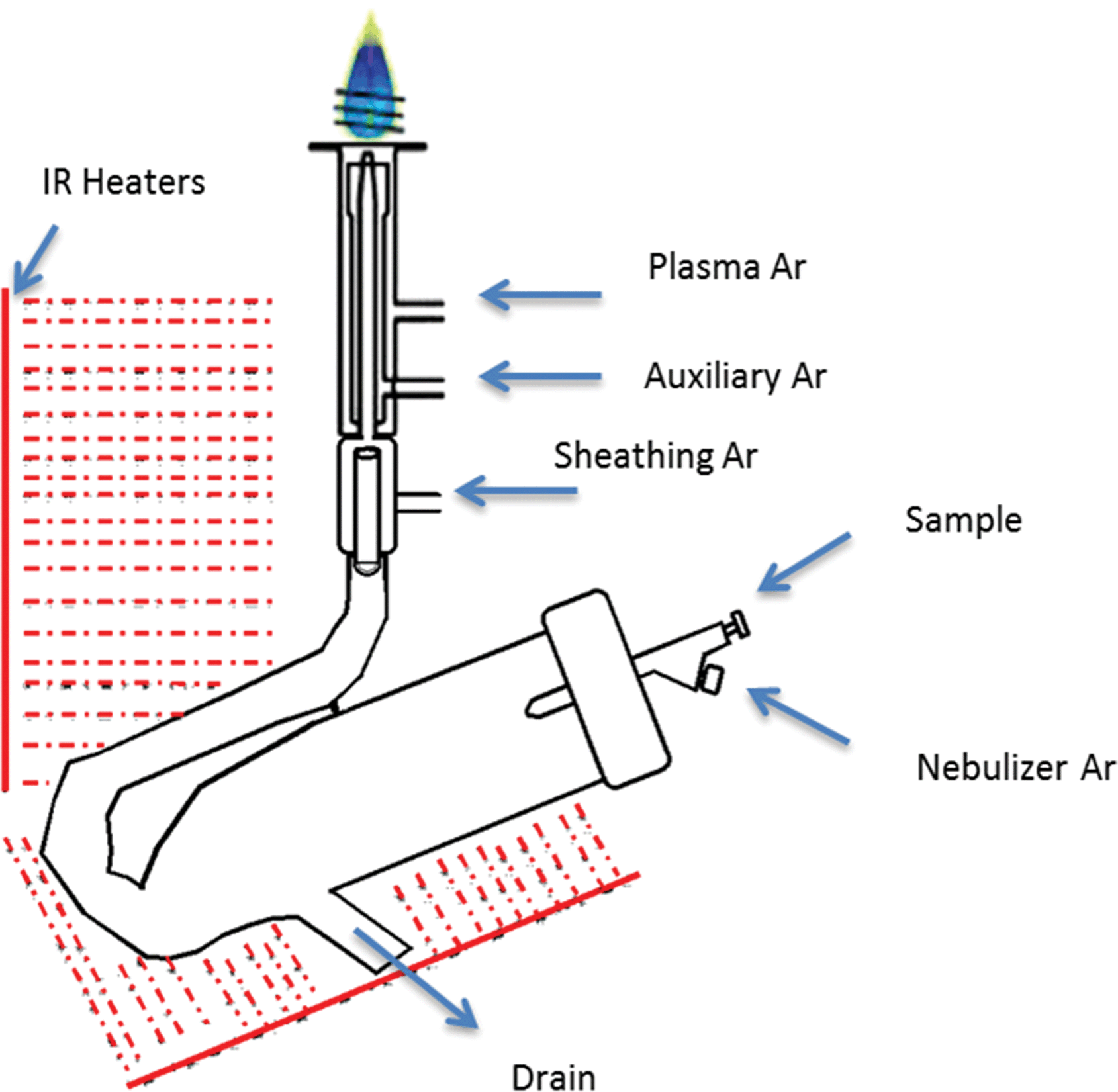 | ||
| Fig. 2 Graphical depiction of the IR-heated spray chamber setup for SPECTRO ARCOS ICP OES. | ||
Reagents
A 7.5 mg L−1 multi-elemental standard solution, containing 38 elements in 2% (v/v) HNO3, was prepared daily from a stock 100 mg L−1 multi-elemental standard solution, which was made from commercially available 1000 mg L−1 single element standard solutions (SCP Science, Baie d’Urfé, Québec, Canada) and doubly deionized water (DDW) (Arium Pro UV/DI System, Sartorius Stedim Biotech, Goettingen, Germany). A corresponding blank was also prepared and these solutions were used for optimization and validation experiments. A blank and five multi-elemental external calibration standards over the 0.1–10 mg L−1 range were also prepared in 2% v/v HNO3, for the determination of detection limits. All HNO3 (ACS grade; Fisher Scientific, Ottawa, Canada) was purified prior to use using a DST-1000 sub-boiling distillation system (Savillex, Minnetonka, USA). Certified reference drinking water EP-L-3 and waste water EU-L-3 (SCP Science, Baie d’Urfé, Québec, Canada) were used for method validation.Optimization
For the multivariate optimization experiments, under a given set of operating conditions, a 7.5 mg L−1 multi-element standard solution in 2% v/v HNO3 and the corresponding blank were nebulised in order of increasing analyte concentration, after rinsing the sample introduction system for 2 min with DDW. A central composite response surface experimental design was used for the multivariate optimization of the IR temperature applied to face of the heater, the Ar nebulizer gas flow rate and the Ar sheath gas flow rate (Table 2) to maximize analyte sensitivity, as well as the Mg II/Mg I ratio, in order to maintain plasma robustness. A progressive factorial approach was then used to optimize the auxiliary gas flow rate, nebulizer gas flow rate, sheath gas flow rate, observation height and sample uptake rate, while keeping other parameters at their optimal values. This was again done with the goal of maximizing analyte sensitivity and the Mg II/Mg I ratio, with a value of 10 signifying minimal matrix effects. To compare the performance of the IR-heated sample introduction system with that of a conventional sample introduction system at room temperature, an HP Ari Mist Burgener nebulizer coupled to a Scott double-pass spray chamber (sample uptake rate of 1.0 mL min−1) was used with no sheathing device and a standard torch. Experiments were repeated over ten months to ensure the reproducibility of the results.| Set # | IR heater temperature (°C) | Ar nebulizer gas flow rate (L min−1) | Ar sheath gas flow rate (L min−1) |
|---|---|---|---|
| 1 | 0 | 0.6 | 0.3 |
| 2 | 230 | 0.6 | 0.3 |
| 3 | 0 | 1.1 | 0.3 |
| 4 | 230 | 1.1 | 0.3 |
| 5 | 0 | 0.6 | 1 |
| 6 | 230 | 0.6 | 1 |
| 7 | 0 | 1.1 | 1 |
| 8 | 230 | 1.1 | 1 |
| 9 | 0 | 0.85 | 0.65 |
| 10 | 230 | 0.85 | 0.65 |
| 11 | 115 | 0.6 | 0.65 |
| 12 | 115 | 1.1 | 0.65 |
| 13 | 115 | 0.85 | 0.3 |
| 14 | 115 | 0.85 | 1 |
| 15 | 115 | 0.85 | 0.65 |
Data processing
Sensitive ionic and atomic emission lines were selected that were free from potential spectroscopic interference (Smart Analyzer Vision software, SPECTRO Analytical Instruments, Kleve, Germany). Data obtained from the experiments were processed with Minitab 17 and Microsoft Office Excel 2010. The signal intensity of the blank was subtracted from that of its corresponding 7.5 mg L−1 multi-element standard solution to give the net signal intensity for the standard solution. Detection limits for analytes in 2% HNO3 were calculated as 3 times the standard deviation of the average signal intensity of at least seven consecutive blanks divided by the slope of the calibration curve (i.e., the sensitivity).Results and discussion
Selection of compromised optimum parameters
The IR-heated sample introduction setup described in Fig. 2, using a single-pass flip chamber and an HP Ari Mist Burgener nebulizer, will be used as representative model for this discussion. Table 1 shows that the optimal experimental parameters for the different spray chamber designs are very similar. The Mn II 257.611 nm emission line was selected as a representative element line for this discussion. Because an increase in R.F. power was required to sustain the plasma upon IR heating of the sample aerosol, the multivariate optimizations and performance validations were conducted at 1.7 kW. As expected from the literature,6,9 the higher R.F. power increased robustness, with the Mg II/Mg I ratio rising from 10 at 1.4 kW to 14 at 1.7 kW.Contour plots (of averages from 3 replicates) of the sensitivity and the Mg II/Mg I ratio as a function of the nebulizer gas flow rate, the sheath gas flow rate and the IR heater temperature are shown in Fig. 3. While increasing the temperature of the IR heater significantly increased both analyte sensitivity and plasma robustness, it had to be limited to 230 °C to avoid deforming the HP Ari Mist Burgener nebulizer body or inner capillary, which are made from PEEK (polyether ether ketone) and Teflon, respectively. At this temperature, 1.0 L min−1 nebulizer gas flow rate and 0.55 L min−1 sheath gas flow rate provided the best compromise with respect to sensitivity.
 | ||
| Fig. 3 Results of the multivariate optimization of the IR heated flip chamber on SPECTRO ARCOS ICP OES with sample introduction system (PN-FC(IR) in this case) while varying the nebulizer gas flow rate, the sheath gas flow rate and the IR heater temperature. Left: effect on the blank-subtracted signal for 7.5 mg L−1 Mn II 257.611 nm. Right: effect on the blank-subtracted ratio of Mg II 280.270 nm/Mg I 285.213 nm. | ||
Increasing the nebulizer gas flow rate from 0.6 to 1.1 L min−1 made the Mg II/Mg I ratio go through a maximum at 0.9–1.0 L min−1, as reported previously,16,27,37 which indicates that a minimum amount of solvent is necessary to improve thermal conductivity in the plasma whereas too high a nebulizer gas flow rate shortens the analyte residence time too much. The similarity in contour plots (at a set IR temperature) for different elements and the Mg II/Mg I ratio as a function of nebulizer gas flow rate is also in agreement with a previous study on the effect of pre-evaporation on ion distributions in ICP-MS, where a single axial sampling depth provided optimal sensitivity for nearly all elements.26 On the other hand, further optimization of plasma robustness was needed, given that the Mg II/Mg I ratio was below 10 (Fig. 3).
The auxiliary gas flow rate, the sample uptake rate and the observation height were thus optimized in progressive succession (Fig. 4) to find that 0.8 L min−1 auxiliary gas flow rate, 0.08 mL min−1 sample uptake rate and 10 mm observation height provided a large increase in not only plasma robustness (Mg II/Mg I ∼ 14), but also in sensitivity with the heated flip chamber. The selection of the lower observation height above the load coil increased both the Mg II/Mg I emission ratio and the Mn II emission intensity, in agreement with previous studies.17,27 The observation height was measured as the height above the load coil and the center of the plasma observation window, which is itself 11 mm in diameter. The optimum sample uptake rate (Table 1) was slightly higher with the other spray chambers, whose larger volumes presumably allowed for a greater degree of pre-evaporation. The maximum sample uptake rate before extinguishing the plasma was 0.150 mL min−1 for all IR-heated spray chambers tested in this work.
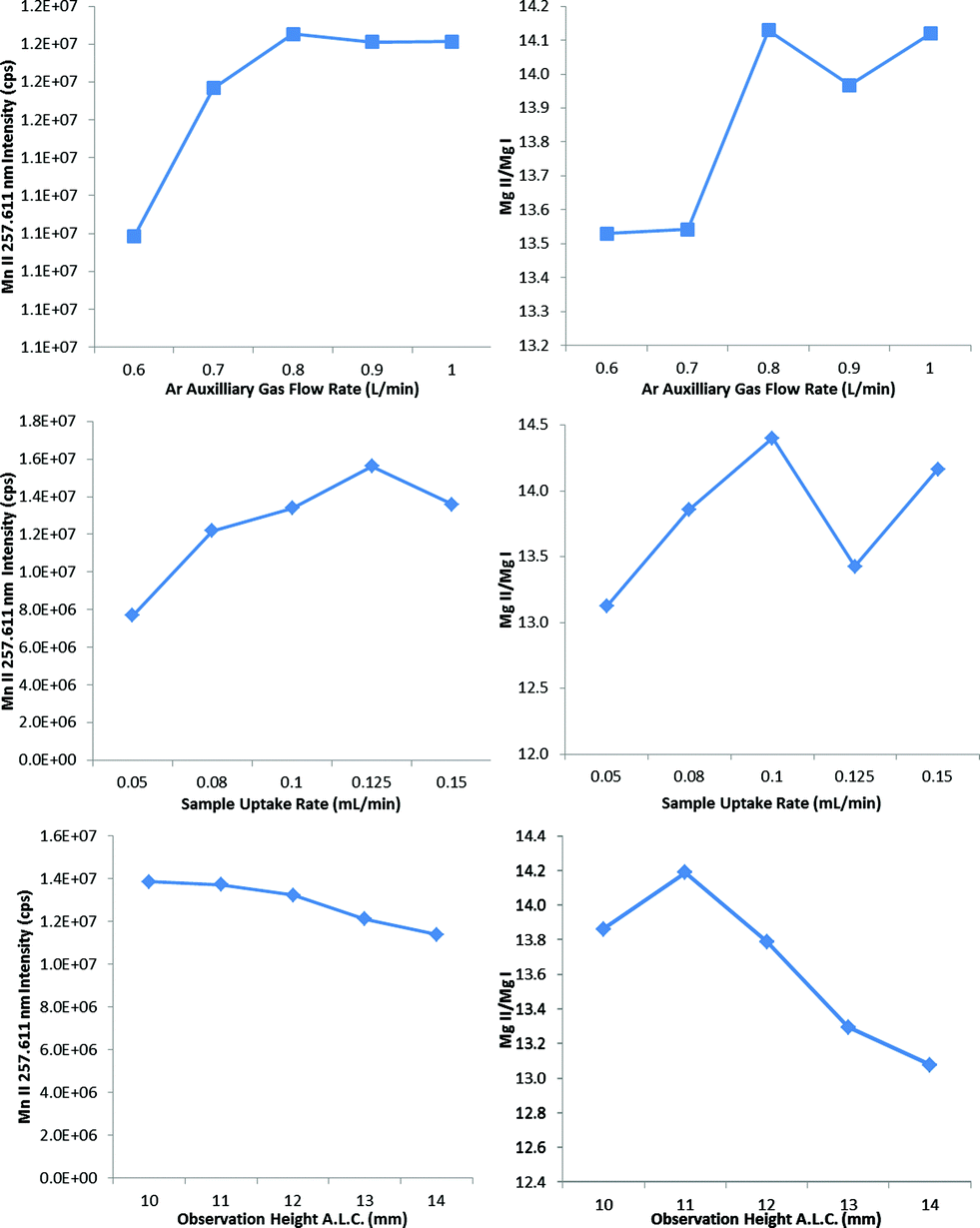 | ||
| Fig. 4 Results of the multivariate optimization of the PN-FC(IR) while varying the auxiliary gas flow rate, the sample uptake rate and the observation height. Left: effect on the blank-subtracted signal for 7.5 mg L Mn II 257.611 nm. Right: effect on the blank-subtracted ratio of Mg II 280.270 nm/Mg I 285.213 nm. | ||
The IR heater temperature of 230 °C is considerably lower than the 350 °C of the TISIS with petroleum products35 and the 400 °C of the USN-PET(IR)2,27 and PN-MSIS-PET29 systems, but higher than the 100–150 °C of the low consumption TISIS system for aqueous samples.28,36 However, in the latter case, the spray chamber wall temperature was reported whereas the temperature at the face of the IR heater was measured here. In any case, the optimized PN-FC(IR) system was able to tolerate nearly double the sample loading compared to the working range of the TISIS (0.020–0.100 mL min−1),28,35 likely as a result of the higher power used (1.7 kW) than with the TISIS (∼1.3 kW). The apparent higher sample loading with the USN-PET(IR) system (1.5 mL min−1) is commensurate with the fact that only the aerosol exiting the spray chamber was pre-evaporated, not that in the spray chamber.
Sensitivities, detection limits and precision
Tables 3 and 4 show the factors of improvement in sensitivity and detection limit that were obtained upon moving from room temperature PN, at a conventional sample uptake rate of 1 mL min−1, to the optimized IR-heated setups in the operating conditions of Table 1. The average sensitivity improvement for all analytes was 6.1 ± 1.1, 6.5 ± 3.6 and 6.4 ± 4.0 for the PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups, respectively, while the average improvement in detection limit was 4.7 ± 3.1, 7 ± 14 and 4.6 ± 3.4 for the PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups, respectively. The slightly higher improvement in detection limits for the PN-SP(IR) setup likely arises from the fact that it is essentially a straight tapered cylinder, without a central tube or kinks, which allowed the aerosol to follow a straight path through the spray chamber, thereby reducing noise and thus improving detection limits further.| Analyte/nm | FC(IR) | SP(IR) | DPE(IR) | USN-PET(IR)2 | PN-MSIS-PET29 |
|---|---|---|---|---|---|
| Al II 167.078 | 7.3 | 20.0 | 24.2 | 23.3 | |
| As I 189.042 | 5.5 | 6.0 | 5.0 | 16.7 | 35.0 |
| Au I 242.795 | 5.5 | 6.7 | 5.1 | ||
| Be II 313.107 | 6.5 | 7.8 | 10.3 | 9.9 | 0.9 |
| Bi II 190.241 | 3.4 | 4.0 | 5.4 | 8.0 | 36.7 |
| Ca II 396.847 | 4.0 | 3.9 | 4.9 | 20.0 | |
| Cd II 226.502 | 7.3 | 6.8 | 7.1 | 28.0 | 2.1 |
| Ce II 413.765 | 5.7 | 4.9 | 4.3 | ||
| Co II 228.616 | 7.1 | 6.3 | 6.9 | 16.6 | 1.5 |
| Cr II 205.618 | 7.0 | 6.4 | 6.6 | 14.0 | 1.5 |
| Cu II 224.700 | 5.2 | 6.6 | 4.2 | 7.6 | 1.6 |
| Eu II 381.967 | 5.1 | 4.4 | 4.0 | ||
| Fe II 238.204 | 7.3 | 6.1 | 6.6 | 16.0 | 1.2 |
| Ga I 287.424 | 4.8 | 4.9 | 4.1 | ||
| Ge II 164.919 | 5.2 | 21.9 | 19.7 | 10.0 | 67.5 |
| Hg I 184.950 | 6.9 | 6.7 | 1.5 | 7.5 | 74.0 |
| In II 230.606 | 5.6 | 6.4 | 5.4 | ||
| K I 766.491 | 6.8 | 4.5 | 3.3 | 8.0 | |
| La II 333.749 | 6.2 | 4.9 | 4.9 | ||
| Li I 670.780 | 5.9 | 4.2 | 4.9 | 6.3 | |
| Mg II 280.270 | 7.1 | 6.1 | 6.8 | 13.9 | |
| Mn II 257.611 | 6.7 | 5.8 | 6.0 | 13.4 | 1.2 |
| Mo II 202.095 | 7.0 | 5.9 | 5.8 | 15.0 | 1.3 |
| Na I 589.592 | 4.6 | 4.3 | 3.7 | 12.0 | |
| Ni II 231.604 | 5.4 | 5.4 | 7.2 | 16.7 | 1.8 |
| Pb II 220.353 | 6.3 | 6.3 | 6.3 | 20.0 | 1.6 |
| Pd I 324.270 | 6.6 | 6.8 | 5.4 | ||
| Pt II 214.423 | 7.0 | 6.8 | 7.2 | ||
| S I 182.034 | 5.1 | 6.9 | 5.7 | 18.0 | |
| Sb I 206.833 | 4.2 | 5.0 | 4.2 | 12.0 | 82.9 |
| Sc II 335.373 | 7.2 | 5.3 | 5.8 | 11.2 | |
| Se I 204.050 | 6.3 | 6.6 | 5.8 | 25.0 | |
| Sr II 421.552 | 4.9 | 4.4 | 4.3 | 8.4 | |
| Ti II 334.941 | 6.9 | 5.4 | 5.6 | 11.4 | |
| V II 292.402 | 7.0 | 5.5 | 6.1 | 11.7 | 1.7 |
| Y II 324.228 | 6.6 | 5.2 | 4.9 | 10.4 | |
| Zn II 206.200 | 7.9 | 7.6 | 8.2 | 27.4 | 2.0 |
| Zr II 339.198 | 6.8 | 5.1 | 5.1 | 11.0 |
| Analyte/nm | FC(IR) | SP(IR) | DPE(IR) | USN-PET(IR)2 | PN-MSIS-PET29 |
|---|---|---|---|---|---|
| Al II 167.078 | 5.5 | 36.9 | 20.1 | 29.0 | |
| As I 189.042 | 5.6 | 4.1 | 1.6 | 10.0 | 25.0 |
| Au I 242.795 | 1.2 | 3.7 | 3.1 | ||
| Be II 313.107 | 10.2 | 5.2 | 4.1 | 10.0 | 1.0 |
| Bi II 190.241 | 2.7 | 1.0 | 1.4 | 7.5 | 40.0 |
| Ca II 396.847 | 2.1 | 1.3 | 0.9 | 20.0 | |
| Cd II 226.502 | 3.3 | 4.1 | 4.0 | 20.0 | 1.6 |
| Ce II 413.765 | 2.5 | 1.3 | 6.0 | ||
| Co II 228.616 | 8.9 | 3.7 | 7.8 | 10.0 | 1.4 |
| Cr II 205.618 | 4.2 | 2.0 | 5.8 | 13.0 | 1.0 |
| Cu II 224.700 | 5.4 | 3.0 | 4.1 | 15.0 | 1.3 |
| Eu II 381.967 | 2.2 | 0.9 | 4.7 | ||
| Fe II 238.204 | 3.5 | 3.5 | 6.5 | 5.0 | 0.1 |
| Ga I 287.424 | 6.8 | 2.6 | 3.7 | ||
| Ge II 164.919 | 1.1 | 31.0 | 11.0 | 15.0 | 50.0 |
| Hg I 184.950 | 2.6 | 6.0 | 0.3 | 17.0 | 50.0 |
| In II 230.606 | 2.6 | 2.8 | 2.7 | ||
| K I 766.491 | 2.5 | 1.4 | 2.9 | 13.0 | |
| La II 333.749 | 12.9 | 2.1 | 2.6 | ||
| Li I 670.780 | 5.5 | 1.3 | 2.6 | 5.0 | |
| Mg II 280.270 | 4.0 | 6.7 | 3.8 | 33.0 | |
| Mn II 257.611 | 4.0 | 3.5 | 5.0 | 18.0 | 0.7 |
| Mo II 202.095 | 3.9 | 2.1 | 3.6 | 13.0 | 1.0 |
| Na I 589.592 | 1.6 | 3.7 | 2.2 | 5.0 | |
| Ni II 231.604 | 2.3 | 2.4 | 4.3 | 25.0 | 0.7 |
| Pb II 220.353 | 2.1 | 3.1 | 3.8 | 10.0 | 1.3 |
| Pd I 324.270 | 10.2 | 2.0 | 4.2 | ||
| Pt II 214.423 | 2.4 | 4.1 | 1.6 | ||
| S I 182.034 | 2.3 | 6.1 | 3.2 | 20.0 | |
| Sb I 206.833 | 2.3 | 1.8 | 2.2 | 20.0 | 20.0 |
| Sc II 335.373 | 4.8 | 2.1 | 5.7 | 20.0 | |
| Se I 204.050 | 4.1 | 2.0 | 4.4 | 20.0 | |
| Sr II 421.552 | 1.7 | 0.5 | 6.2 | 20.0 | |
| Ti II 334.941 | 7.1 | 3.5 | 5.3 | 10.0 | |
| V II 292.402 | 5.4 | 2.7 | 8.4 | 8.0 | 2.0 |
| Y II 324.228 | 10.2 | 2.0 | 6.7 | 10.0 | |
| Zn II 206.200 | 9.9 | 80.6 | 3.5 | 17.0 | 1.7 |
| Zr II 339.198 | 10.4 | 1.9 | 5.0 | 10.0 |
The enhancements in sensitivity are in agreement with those reported for the TISIS (8–15 fold),28 PN-MSIS-PET (1–50 fold)29 and USN-PET(IR) (10–25 fold)2 systems, and are not due to a sole increase in R.F. power. Indeed, the average improvement in sensitivity with room temperature PN-DP at 1 mL min−1 sample uptake rate upon increasing power from 1.4 to 1.7 kW was 1.62 ± 0.42. Furthermore, the PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups improved sensitivity and detection limit for 38 elements, whereas the PN-MSIS-PET system only offered enhancements for 14 elements.29 Moreover, the factors of improvement for the TISIS and USN-PET(IR) systems are reported relative to conventional PN at a similar sample uptake rate, whereas those for PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups (at 0.080–0.0925 mL min−1 sample uptake rate) are reported relative to conventional PN operating at over 10 times the sample uptake rate (1 mL min−1). Compared to room temperature PN-DP operated at 0.080 mL min−1, the average improvement in sensitivity with the PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups were 14 ± 6, 19 ± 15 and 16 ± 14, respectively. This is a clear demonstration of the efficacy of the new PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups, which result in sensitivity enhancements despite aspirating over 10 times less sample volume.
The conversion of the aerosol into vapor upon IR heating appeared to be visibly complete within the integrated sheathing device of the ICP torch. Compared to the previous USN-PET(IR) system, the pre-evaporation region in the new PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups is much shorter, with condensation more likely to occur at the base of the torch, in turn leading to noise and plasma instability, unless uniform thermal insulation is present from the spray chamber to the torch base. Increasing the length of the pre-evaporation region was attempted but abandoned because it drastically increased sample washout time. With better insulation, similar or larger improvements in detection limit than in sensitivity should be obtained for all elements instead of a few. This will be the subject of future work. Nonetheless, the larger improvement in sensitivity than in detection limit is in agreement with what was reported when the sample introduction efficiency was increased in other systems.38
Generally, the improvement in sensitivity and detection limit was greater for ionic than atomic lines (Tables 3 and 4). For example, the improvement factor in sensitivity when using ionic lines for Be, Mg and Zn was, on average, 1.43 ± 0.12, 1.29 ± 0.12 and 1.56 ± 0.18 fold that achieved using atomic lines with PN-FC(IR), PN-SP(IR) and PN-DPE(IR), respectively, versus PN. This improvement is directly related to the total excitation potential (TEP), which is the excitation potential for atomic lines, and the sum of ionization and excitation potentials for ionic lines.39,40
A similar improvement was observed when using Be II 313.042 nm and Zn II 206.200 nm (TEP = 13.3 and 15.5 eV, respectively) over Be I 234.861 nm and Zn I 213.856 nm lines (TEP = 5.3 and 5.8 eV, respectively). This is in agreement with the literature, where a larger improvement was seen with a higher TEP, regardless of whether or not vapor formation had occurred.29 Lines with a higher TEP are also more easily affected by changes in plasma excitation conditions. For example, the intensities for emission lines with a TEP > 13 eV were depressed to a greater extent with USN than with PN in the presence of a Ca matrix.3 Larger improvements in sensitivity and detection limit were observed with ionic lines versus atomic lines when moving from PN to PN-FC(IR) or PN-SP(IR) or PN-DPE(IR) as a result of the pre-evaporated aerosol providing improved plasma excitation conditions.
The instrumental precision, expressed as relative standard deviation (RSD), obtained for various sample introduction systems is summarised in Table 5. The average %RSD was 1.4 ± 0.9, 1.2 ± 0.5 and 0.5 ± 0.5 for the PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups, respectively. The %RSD values for the IR-heated PN setups are comparable amongst one another and with the %RSD for conventional PN at room temperature (0.6 ± 0.2), within error. Further improvement in %RSD should be possible for the IR-heated setups with improved thermal insulation ensuring uniform heat distribution within the sample introduction system. The %RSD values are also comparable with the previous USN-PET(IR) and PN-MSIS-PET systems. However, the precision with the new PN-SP(IR) and PN-DPE(IR) setups is better than that obtained with an earlier USN-PET using heating tape instead of IR heating (where %RSD was 2.6 ± 0.7).27
| Analyte/nm | PN | FC(IR) | SP(IR) | DPE(IR) | USN-PET(IR)2 | PN-MSIS-PET29 |
|---|---|---|---|---|---|---|
| Al II 167.078 | 0.5 | 0.7 | 0.5 | 0.6 | 1.4 | |
| As I 189.042 | 0.5 | 0.3 | 1.4 | 0.9 | 1.4 | 4.1 |
| Au I 242.795 | 0.8 | 2.2 | 1.1 | 0.5 | ||
| Be II 313.107 | 0.5 | 1.4 | 0.8 | 0.8 | 0.7 | 1.5 |
| Bi II 190.241 | 0.5 | 0.5 | 1.5 | 1.1 | 1.6 | 1.4 |
| Ca II 396.847 | 0.4 | 0.1 | 2.6 | 0.9 | 1.9 | |
| Cd II 226.502 | 0.9 | 1.2 | 1.0 | 0.5 | 0.3 | 1.4 |
| Ce II 413.765 | 0.5 | 1.8 | 1.7 | 0.3 | ||
| Co II 228.616 | 0.7 | 1.7 | 1.0 | 0.4 | 2.4 | 2.3 |
| Cr II 205.618 | 0.6 | 1.6 | 1.1 | 0.6 | 1.7 | 2 |
| Cu II 224.700 | 0.9 | 1.9 | 0.7 | 0.2 | 2 | 1.9 |
| Eu II 381.967 | 0.5 | 2.2 | 1.9 | 0.2 | ||
| Fe II 238.204 | 0.5 | 1.8 | 0.9 | 0.3 | 2.1 | 1.6 |
| Ga I 287.424 | 0.6 | 0.5 | 1.0 | 0.3 | ||
| Ge II 164.919 | 0.8 | 0.5 | 0.4 | 0.8 | 1.3 | 2.9 |
| Hg I 184.950 | 1.3 | 0.7 | 1.1 | 3.0 | 1.7 | 1.3 |
| In II 230.606 | 0.6 | 0.7 | 0.9 | 0.4 | ||
| K I 766.491 | 0.5 | 0.5 | 1.5 | 0.3 | 1.9 | |
| La II 333.749 | 0.3 | 2.1 | 0.9 | 0.3 | ||
| Li I 670.780 | 0.5 | 2.1 | 1.6 | 0.4 | 1.6 | |
| Mg II 280.270 | 0.8 | 2.7 | 1.4 | 1.0 | 0.2 | |
| Mn II 257.611 | 0.6 | 2.3 | 0.7 | 0.4 | 0.7 | 1.4 |
| Mo II 202.095 | 0.5 | 1.8 | 1.2 | 0.8 | 1.8 | 1.6 |
| Na I 589.592 | 0.4 | 1.7 | 1.8 | 0.3 | 2 | |
| Ni II 231.604 | 0.7 | 0.7 | 0.8 | 0.3 | 1.8 | 1.1 |
| Pb II 220.353 | 0.9 | 0.2 | 0.9 | 0.3 | 1.3 | 3.7 |
| Pd I 324.270 | 0.4 | 2.7 | 0.9 | 0.3 | ||
| Pt II 214.423 | 0.8 | 1.2 | 1.3 | 0.7 | ||
| S I 182.034 | 1.0 | 0.2 | 0.5 | 0.4 | 1.8 | |
| Sb I 206.833 | 0.7 | 1.0 | 1.1 | 0.3 | 1.4 | 4.1 |
| Sc II 335.373 | 0.5 | 2.6 | 1.0 | 0.2 | 0.3 | |
| Se I 204.050 | 0.5 | 0.2 | 1.3 | 0.3 | 2.1 | |
| Sr II 421.552 | 0.4 | 0.2 | 3.0 | 0.4 | 0.2 | |
| Ti II 334.941 | 0.4 | 2.7 | 0.9 | 0.2 | 1 | |
| V II 292.402 | 0.6 | 1.9 | 1.0 | 0.2 | 1.6 | 2.8 |
| Y II 324.228 | 0.4 | 2.7 | 0.9 | 0.3 | 0.9 | |
| Zn II 206.200 | 0.6 | 1.0 | 0.9 | 0.7 | 0.8 | 1.4 |
| Zr II 339.198 | 0.4 | 2.0 | 1.0 | 0.3 | 0.2 |
Plasma robustness
The Mg II 280.270 nm/Mg I 285.213 nm emission intensity ratio was used to evaluate plasma excitation conditions when using the different sample introduction systems,3,16,37 as shown in Fig. 5. The PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups all clearly improved plasma robustness compared to that achieved with conventional PN and the previous PN-MSIS-PET system, as well as compared to the previous TISIS system operated at 100 °C (Mg II/Mg I ∼ 12, not shown).28 Even when operating at sample uptake rates about 20 times lower than the USN-PET(IR) systems, the PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups all provide similar Mg II/Mg I emission ratios (Fig. 5), suggesting similar sample introduction efficiency, as the amount of water vapor entering the plasma is known to improve plasma excitation conditions.19,28 The lowest Mg II/Mg I emission ratio was indeed seen for conventional PN at room temperature (9.9 ± 0.5). The significantly higher Mg II/Mg I emission ratios obtained for the PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups are better than previously reported values for PN at room temperature (7–12) in axial and lateral view ICP OES,3,37 even under robust conditions.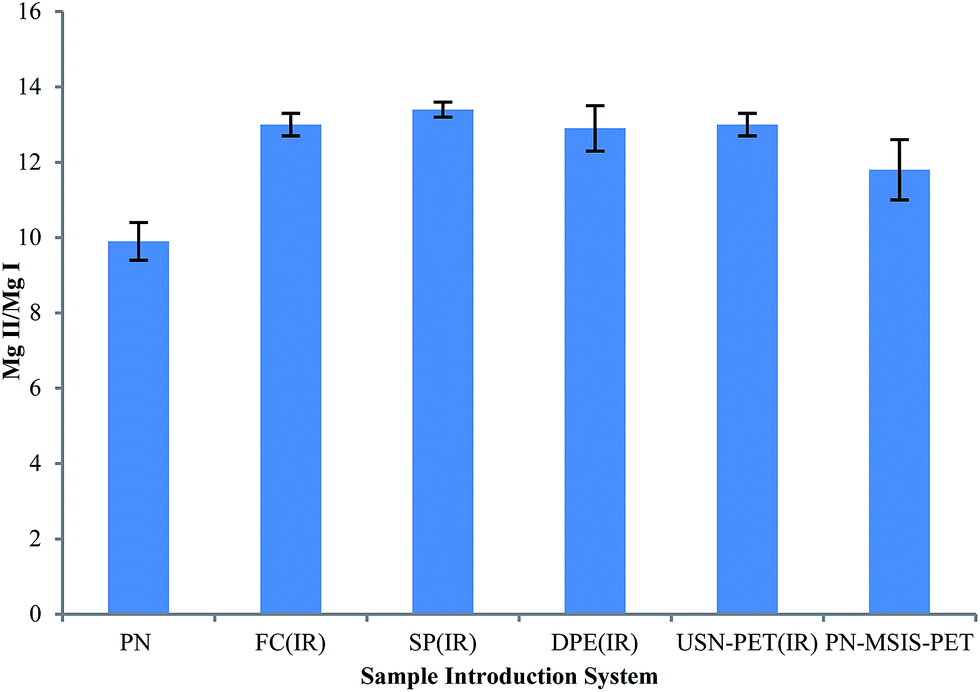 | ||
| Fig. 5 Average Mg II 280.270 nm/Mg I 285.213 nm line intensity ratio (Mg II/Mg I), with 95% confidence limit (n = 5), for different sample introduction systems for the SPECTRO ARCOS instrument. | ||
The performance of the PN-FC(IR) setup was demonstrated in Table 6 by the direct analysis of two certified reference materials, without internal standardization or matrix matching. As the expiration date had passed for these two materials, close agreement with the certified values was not expected. Nonetheless, the results are within the tolerance limits and are very similar to those obtained with room temperature PN-DP.
| Drinking water EP-L-3 | |||
|---|---|---|---|
| Analyte | Certified ± tolerance limits | PN-DP (at 1 mL min−1) | PN-FC(IR) (at 0.08 mL min−1) |
| Al | 100 ± 10 | 92.3 ± 1.2 | 90.87 ± 0.79 |
| As | 10.6 ± 1.8 | 9.76 ± 0.15 | 9.43 ± 0.10 |
| Be | 1.98 ± 0.12 | 1.97 ± 0.02 | 1.96 ± 0.02 |
| Cd | 1.97 ± 0.23 | 1.60 ± 0.04 | 1.71 ± 0.04 |
| Co | 9.8 ± 1.2 | 8.95 ± 0.20 | 9.17 ± 0.07 |
| Cr | 12.7 ± 1.0 | 11.56 ± 0.24 | 11.83 ± 0.10 |
| Cu | 15.6 ± 2.0 | 15.07 ± 0.26 | 14.61 ± 0.10 |
| Fe | 27.9 ± 3.8 | 25.74 ± 0.36 | 25.62 ± 0.19 |
| K | 404 ± 42 | 419.7 ± 8.0 | 381.24 ± 3.6 |
| Mn | 5.85 ± 0.58 | 5.39 ± 0.11 | 5.37 ± 0.04 |
| Mo | 22.6 ± 2.8 | 21.10 ± 0.42 | 21.40 ± 0.17 |
| Ni | 19.9 ± 2.0 | 17.94 ± 0.43 | 18.26 ± 0.16 |
| Pb | 4.00 ± 0.35 | 3.78 ± 0.07 | 3.74 ± 0.03 |
| V | 13.6 ± 1.1 | 12.50 ± 0.23 | 12.65 ± 0.10 |
| Waste water EU-L-3 | |||
|---|---|---|---|
| Analyte | Certified ± tolerance limits | DP (at 1 mL min−1) | FC(IR) (at 0.08 mL min−1) |
| Al | 6.3 ± 1.5 | 6.11 ± 0.05 | 5.61 ± 0.11 |
| Be | 1.23 ± 0.15 | 1.17 ± 0.02 | 1.06 ± 0.02 |
| Cd | 2.28 ± 0.42 | 2.02 ± 0.01 | 1.83 ± 0.03 |
| Cr | 6.3 ± 1.4 | 5.54 ± 0.04 | 5.20 ± 0.07 |
| Cu | 10.6 ± 1.9 | 10.27 ± 0.06 | 9.36 ± 0.12 |
| K | 207 ± 40 | 232.4 ± 6.4 | 186.2 ± 2.4 |
| Mo | 3.97 ± 0.70 | 3.64 ± 0.04 | 3.37 ± 0.05 |
| Sr | 14.0 ± 3.8 | 13.23 ± 0.39 | 12.58 ± 0.28 |
| V | 4.95 ± 0.62 | 4.53 ± 0.03 | 4.30 ± 0.06 |
| Zn | 3.1 ± 1.8 | 2.61 ± 0.02 | 2.33 ± 0.03 |
In addition to the above robustness, no significant buildup of condensed products was observed either inside the sample introduction system or the torch injector after several days of continuous operation. The sample introduction components did not need to be cleaned any more frequently than with PN at room temperature, unlike the previous PN-MSIS-PET system, which had to be cleaned after only two days.29 The washout time was about 3–4 s longer than that with the standard system at room temperature, despite the fact that the sample pumping rate was over 10 times smaller. Operating at 1.7 kW, i.e. under very robust conditions, which was required to handle the increased sample load, did not have any noticeable effect on the lifetime of the torch.
Conclusions
Applying IR heating to a conventional PN sample introduction system is a simple way to significantly improve the analytical performance (sensitivity, detection limit and plasma robustness) of ICP OES. The newly optimized PN-FC(IR), PN-SP(IR) and PN-DPE(IR) setups all managed to improve sensitivity by a factor of 6, on average, despite the sample uptake rate being over an order of magnitude smaller than that used at room temperature. This indicates an increase in sample introduction efficiency. Future work will include the optimization of spray chamber design, a study of sample washout times and the analysis of a variety of certified reference materials. Perhaps the use of an Ar–N2–H2 mixed-gas plasma in order to provide further plasma power density and increase sample loading will also be tested. This system will also be coupled to ICP-MS to see if it will provide similar improvements in analytical performance.Acknowledgements
The authors gratefully acknowledge the Natural Sciences and Engineering Research Council of Canada and Anglo American Pty for the SPECTRO ARCOS instrument, and funding from the Mitacs Accelerate Industrial Internship Program, as well as the financial support of Telegistics Inc.References
- J. D. Winefordner, I. B. Gornushkin, T. Correll, E. Gibb, B. W. Smith and N. Omenetto, J. Anal. At. Spectrom., 2004, 19, 1061–1083 RSC.
- A. Asfaw, W. R. MacFarlane and D. Beauchemin, J. Anal. At. Spectrom., 2012, 27, 1254–1263 RSC.
- I. B. Brenner, M. Zischka, B. Maichin and G. Knapp, J. Anal. At. Spectrom., 1998, 13, 1257–1264 RSC.
- F. V. Silva, L. C. Trevizan, C. S. Silva, A. R. A. Nogueira and J. A. Nobrega, Spectrochim. Acta, Part B, 2002, 57, 1905–1913 CrossRef.
- J. M. Mermet and E. Poussel, Appl. Spectrosc., 1995, 49, 12–18 CrossRef.
- X. Romero, E. Poussel and J. M. Mermet, Spectrochim. Acta, Part B, 1997, 52, 487–493 CrossRef.
- J. W. Tromp, M. Pomares, M. Alvarez-Prieto, A. Cole, H. Ying and E. D. Salin, Spectrochim. Acta, Part B, 2003, 58, 1927–1944 CrossRef.
- M. Murillo and J. M. Mermet, Spectrochim. Acta, Part B, 1989, 44, 359–366 CrossRef.
- J. M. Mermet, Anal. Chim. Acta, 1991, 250, 85–94 CrossRef CAS.
- J. M. Mermet, Spectrochim. Acta, Part B, 1989, 44, 1109–1116 CrossRef.
- J. Mora, S. Maestre, V. Hernandis and J. L. Todoli, TrAC, Trends Anal. Chem., 2003, 22, 123–132 CrossRef CAS.
- R. I. Botto and J. J. Zhu, J. Anal. At. Spectrom., 1994, 9, 905–912 RSC.
- J. Borkowska- Burnecka, A. Lesniewicz and W. Zymicki, Spectrochim. Acta, Part B, 2006, 61, 579–587 CrossRef PubMed.
- K. V. Desboeufs, R. Losno and J. L. Colin, Anal. Bioanal. Chem., 2003, 375, 567–573 CAS.
- Y. C. Sun, S. H. Wu and C. C. Lee, J. Anal. At. Spectrom., 2003, 18, 1163–1170 RSC.
- M. Grotti, C. Lagomarsino and J. M. Mermet, J. Anal. At. Spectrom., 2006, 21, 963–969 RSC.
- I. Novotny, J. C. Farinas, J. L. Wan, E. Poussel and J. M. Mermet, Spectrochim. Acta, Part B, 1996, 51, 1517–1526 CrossRef.
- M. Bensimon, J. Bourquin and A. Parriaux, J. Anal. At. Spectrom., 2000, 15, 731–734 RSC.
- S. E. Long and R. F. Browner, Spectrochim. Acta, Part B, 1988, 43, 1461–1471 CrossRef.
- P. E. Walters and C. A. Barnardt, Spectrochim. Acta, Part B, 1988, 43, 325–337 CrossRef.
- M. Hoenig, H. Docekalova and H. Baeten, J. Anal. At. Spectrom., 1998, 13, 195–199 RSC.
- B. Budic, Fresenius' J. Anal. Chem., 2000, 368, 371–377 CrossRef CAS.
- E. Vassileva and M. Hoenig, Spectrochim. Acta, Part B, 2001, 56, 223–232 CrossRef.
- G. R. Peters and D. Beauchemin, Anal. Chem., 1993, 65, 97–103 CrossRef CAS.
- S. Liu and D. Beauchemin, Spectrochim. Acta, Part B, 2006, 61, 965–970 CrossRef PubMed.
- S. Liu and D. Beauchemin, Spectrochim. Acta, Part B, 2006, 61, 157–163 CrossRef PubMed.
- A. Asfaw and D. Beauchemin, Spectrochim. Acta, Part B, 2010, 65, 376–384 CrossRef PubMed.
- E. Paredes, M. Grotti, J. M. Mermet and J. L. Todoli, J. Anal. At. Spectrom., 2009, 24, 903–910 RSC.
- A. Asfaw and D. Beauchemin, J. Anal. At. Spectrom., 2012, 27, 80–91 RSC.
- A. R. Eastgate, R. C. Fry and G. H. Gower, J. Anal. At. Spectrom., 1993, 8, 305–308 RSC.
- C. Gueguen, J. Dominik, M. Pardos, C. Benninghoff and R. L. Thomas, Lakes Reservoirs, 2000, 5, 59–66 CrossRef.
- W. Schron and U. Muller, Fresenius' J. Anal. Chem., 1997, 357, 22–26 CrossRef.
- J. L. Todolí and J. M. Mermet, Can. J. Anal. Sci. Spectrosc., 2002, 47, 164–170 Search PubMed.
- J. M. Mermet and J. L. Todoli, Anal. Bioanal. Chem., 2004, 378, 57–59 CrossRef CAS PubMed.
- R. Sanchez, J. L. Todoli, C. P. Lienemann and J. M. Mermet, J. Anal. At. Spectrom., 2012, 27, 937–945 RSC.
- M. Grotti, F. Ardini and J. L. Todoli, Anal. Chim. Acta, 2013, 767, 14–20 CrossRef CAS PubMed.
- I. B. Brenner, A. Zander, M. Cole and A. Wiseman, J. Anal. At. Spectrom., 1997, 12, 897–906 RSC.
- A. Asfaw and G. Wibetoe, J. Anal. At. Spectrom., 2006, 21, 1027–1035 RSC.
- I. B. Brenner and A. T. Zander, Spectrochim. Acta, Part B, 2000, 55, 1195–1240 CrossRef.
- Y. Ralchenko, A. Kramida and J. Reader, NIST Atomic Spectra Database (ver. 5.1), http://physics.nist.gov/asd Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |