
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiplexing strategy for simultaneous detection of redox-, phospho- and total proteome – understanding TOR regulating pathways in Chlamydomonas reinhardtii†
Silas P.
Rodrigues‡
a,
Sophie
Alvarez‡
b,
Emily G.
Werth
a,
William O.
Slade
a,
Brian
Gau
b,
Edgar B.
Cahoon
c and
Leslie M.
Hicks
*a
aDepartment of Chemistry, University of North Carolina at Chapel Hill, 125 South Road, CB#3290, Chapel Hill, NC 27599, USA. E-mail: lmhicks@unc.edu; Fax: +1-919-962-2388; Tel: +1-919-843-6903
bDonald Danforth Plant Science Center, 975 N. Warson Road, St. Louis, MO 63132, USA
cCenter for Plant Science Innovation and Department of Biochemistry, University of Nebraska-Lincoln, Lincoln, NE 68588, USA
First published on 29th May 2015
Abstract
New methods for studying the complexity of multiple PTMs in functional proteomics are required to understand cell signaling processes. In this study, a multiplexing 2DE-based approach is introduced for parallel analysis of the redox-, phospho-, and total-proteome. This triplexing approach uses spectrally distinct fluorophores, is not matrix-specific and requires relatively low sample amounts with applicability to any cell/tissue type. This methodology was applied for the study of Target of Rapamycin (TOR) regulating pathways in Chlamydomonas reinhardtii. With emerging research demonstrating a complex yet unclear relationship between TOR kinase, autophagy, and lipid metabolism, rapamycin treatment was used to induce TOR inhibition in C. reinhardtii and redox-, phospho- and total proteome changes were assessed using the triplexing approach. We identified a total of 68 spot abundance changes in response to TOR inhibition which provide a basis for understanding this highly conserved, master regulator in algae.
Introduction
Over the past decade, research focused on identification and characterization of post-translational modifications (PTMs) has expanded with the advancement of techniques enabling large-scale proteomics studies. While PTMs are largely investigated independently, examination into how different PTMs interact will reveal a more complete understanding of the underlying biology.1 For example, a co-enrichment strategy for the investigation of ubiquitylation/phosphorylation cross-talk with regard to protein degradation identified putative novel regulatory mechanisms of kinases via proteasome-independent ubiquitylation.1 Further, identifying multiple PTM changes will reveal the co-regulation and interdependence of PTM signaling cascades on a number of cellular processes.2 However, approaches for co-enrichment/detection of PTMs are underdeveloped. Thus, there is an immediate need for robust methods to assess PTM cross regulation in large-scale proteomics research.Herein, we report a multiplexing 2DE-based approach for parallel analysis of the redox-, phospho-, and total-proteome in a single sample. The redox environment of the cell affects the biological activity of proteins. Redox PTMs, which consist of disulfide bonds and oxidation of cysteine thiol groups by reactive oxygen (ROS) and nitrogen (RNS) species, are involved in numerous cellular processes dependent on thiol-based mechanisms.3 Previously thought to be deleterious agents, ROS are now considered signaling molecules involved in programmed cell death and serve as defense signaling components in tissues.4 With tightly regulated intracellular concentrations, ROS/RNS play important roles in relaying downstream intracellular signals promoting conformational changes through the formation of disulfide bonds in response to the changing cellular redox environment.3,5 Similarly, protein phosphorylation is an essential PTM in eukaryotic organisms involved in regulation of cellular processes critical for cell metabolism, signaling, proliferation and cell survival.6 Due to its reversible nature, phosphorylation acts as a molecular switch turning on or off activity resulting in downstream effects based on environmental stimuli. While phosphorylation events are common in cellular processes and signaling, a challenge in studying phosphoproteomics comes from the substoichiometric levels of phosphorylation modification, requiring utilization of phosphopeptide enrichment strategies.1
Currently, 2DE approaches are used for proteome-wide redox or phosphorylation status monitoring.7–9 Fluorescent reagents are relatively inexpensive, offer excellent sensitivity (low ng), and allow good linearity (103–104).10,11 Further, as fluorescent reagents can be spectrally distinct, the opportunity for multiplexing exists. For example, 5-iodoacetamidofluorescein (IAF) labels cysteine thiols in proteins prior to 2DE separation to analyze the redox proteome,12 and we previously demonstrated compatibility with SYPRO Ruby total protein staining in a 2-plex experiment.13 Pro-Q Diamond stain, a phosphosensor fluorophore capable of ultrasensitive global detection and quantitation of phosphorylated amino acid residues in proteins,14 has demonstrated multiplexing compatibility with SYPRO.15,16 These approaches have been applied in photosynthetic organisms to reveal phospho-17–19 and redox-proteomes,13,20 respectively.
Here, we demonstrate triplexing capability through the combination of cysteine IAF-labeling, Pro-Q, and SYPRO staining in a single 2DE of a single sample. This method was applied to Chlamydomonas reinhardtii to allow for the parallel analysis of the redox-, phospho-, and total proteomes changes in response to exogeneous rapamycin to investigate pathways regulated by Target of Rapamycin (TOR). TOR is a conserved Ser/Thr kinase that controls many cellular processes, including energy homeostasis and autophagy.21 In turn, autophagy is regulated by lipid metabolism,22 suggesting these processes share regulatory mechanisms. In yeast and plants, downregulation of TOR induces autophagy and lipid metabolism, resulting in the production of autophagosomes and the accumulation of TAGs,23,24 respectively. Both processes are modulated by reactive oxygen species (ROS), which induce autophagy24 and lipid metabolism.25 Thus, there is a complex and unclear dynamic among TOR, autophagy, and lipid metabolism, all of which are regulated by intracellular energy status. In Chlamydomonas reinhardtii, exogenous rapamycin inhibits TOR via a mechanism that requires TOR complex 1,26 suggesting C. reinhardtii is a photosynthetic organism that can be used as a model for understanding TOR function.
Results and discussion
Evaluation of the triplexing labeling 2DE method
A reverse-labeling strategy is implemented through protein extraction buffer containing N-ethylmaleimide (NEM) that readily reacts with cysteine thiols to form non-reversible thioether bonds (Fig. 1).27 Proteins are then reduced using 1,4-dithio-DL-threitol (DTT) to expose originally oxidized thiols,28 which can now react with IAF to form stable thioether bonds.29 Proteins are separated using 2DE and the occurrence of IAF-labeled proteins is revealed by imaging using appropriate excitation lasers and emission filters. The gels are further stained/imaged for phosphoproteins using Pro-Q followed by a final staining/imaging for total proteins using SYPRO.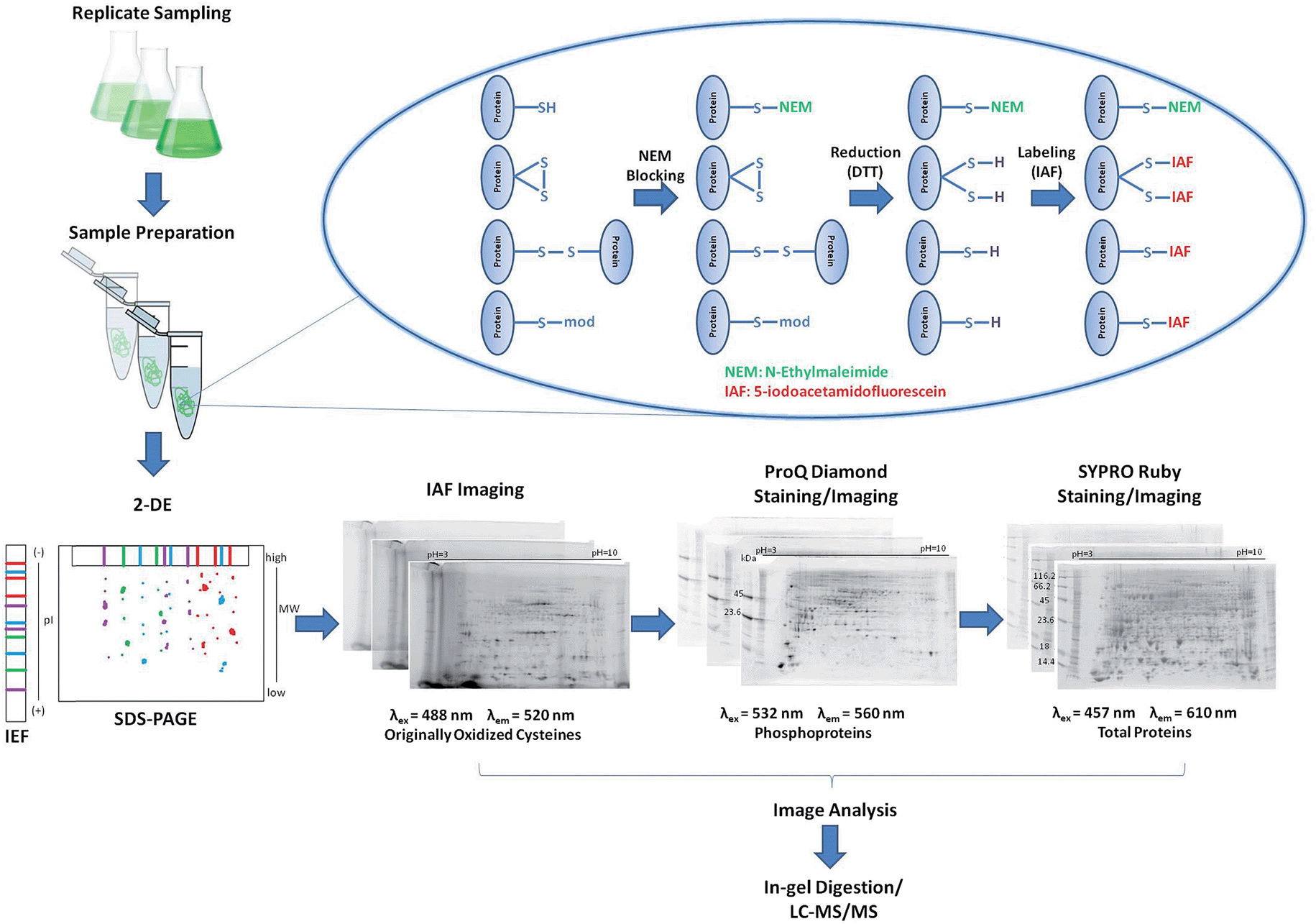 | ||
| Fig. 1 Schematic of the multiplexing 2DE approach. A reverse-labeling strategy is employed to extracted proteins to label originally oxidized cysteines with IAF. Samples are separated by 2DE and imaged for IAF revealing the redox proteome profile. Gels are subsequently stained using Pro-Q Diamond and the phosphoproteome profile obtained. Total proteome abundances are obtained after SYPRO Ruby staining and imaging. | ||
Reagents used for protein labeling and gel staining were evaluated for sequential gel-imaging compatibility. Protein lysates were prepared and labeled using IAF as described in the methods. Negative controls were labeled using IAM. Protein lysates were separated using 1D SDS-PAGE followed by imaging (Fig. 2). As expected, using λex = 488 nm and λem = 520 nm, IAF labeled proteins were observed as a complex band profile not observed for IAM labeled controls (Fig. 2(A)). The gels were scanned immediately after IAF imaging using Pro-Q settings (λex = 532 nm and λem = 560 nm) revealing negligible background (Fig. 2(B)). Imaging after Pro-Q staining is shown in Fig. 2(C), followed by imaging using SYPRO settings (λex = 457 nm and λem = 610 nm, Fig. 2(D)). Fig. 2(E) then shows the image for SYPRO settings after staining and imaging. In Fig. 2(B) and (D), the low background observed is probably associated with intrinsic protein light absorption properties in the specified wavelengths since the same result was observed for both IAF and IAM labeled samples.
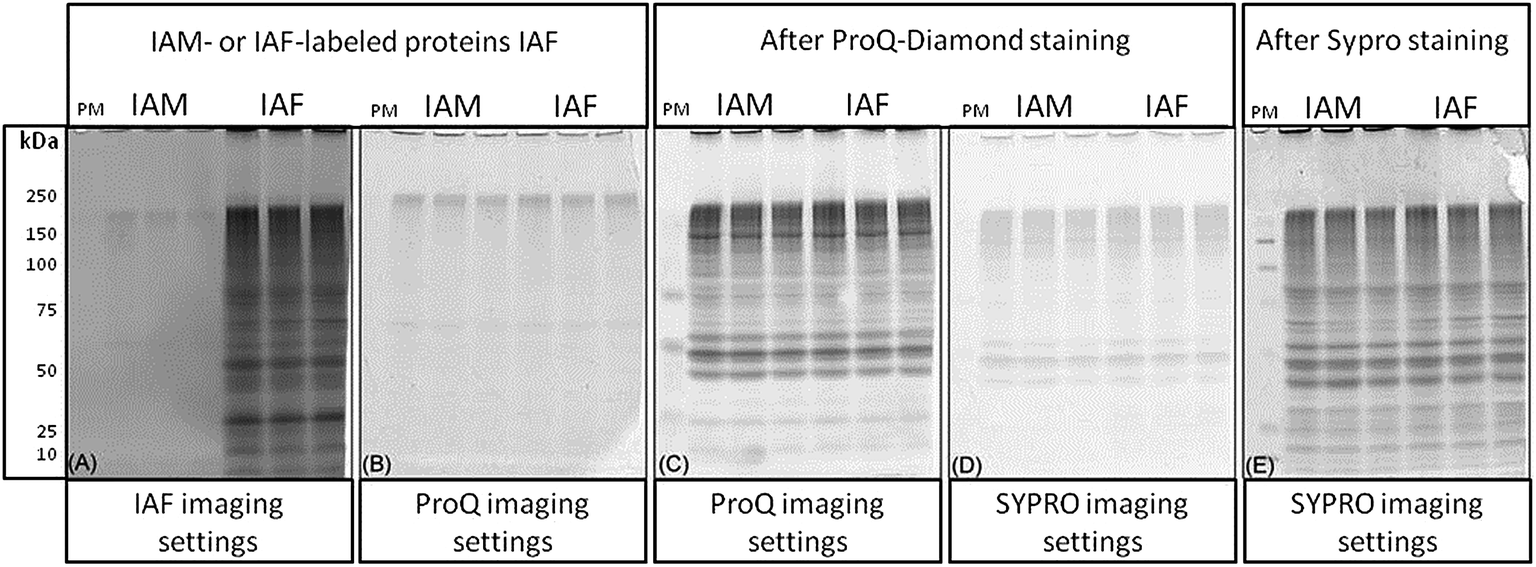 | ||
| Fig. 2 Evaluation of reagent compatibility for multiplexing. C. reinhardtii proteins were extracted, blocked, reduced and originally oxidized cysteine thiols were labeled with IAF. Negative controls were labeled using IAM. Proteins were separated using 1D SDS-PAGE and imaged as follows. (A) Proteins were imaged using IAF settings. (B) The gel was subsequently scanned using Pro-Q settings. (C) After Pro-Q gel staining, the phosphoprotein profile was obtained. (D) Pro-Q stained gels were imaged using SYPRO settings. (E) Total proteins profiles were obtained after SYPRO staining and imaging. PM = phosphoprotein marker. | ||
To test that IAF labeling of the proteins does not interfere with the 2D-gel separation, protein lysates were prepared and labeled using IAM (negative control) or IAF as described in the methods. The gels were then scanned using the IAF settings. Fig. 3 confirms that proteins modified with IAM do not appear in these settings (Fig. 3(A and B)). The gels were then stained with SYPRO and imaged using the SYPRO settings (Fig. 3(C and D)). The alignment of the two SYPRO-stained gels from IAM and IAF labeling show perfect alignment and therefore demonstrates that overall protein migration is not affected by the IAF labeling (Fig. 3(E)). The alignment of the gels from the IAF labeled proteins and from the SYPRO stained proteins show that the spots intensities from the IAF-labeled proteins do not correlate necessarily with protein abundance (Fig. 3(F)). Here we are able to detect very low abundance Cys-containing proteins labeled with IAF, making this approach a very sensitive method for detection of redox changes.
 | ||
| Fig. 3 Evaluation of the IAF labeling on 2D-gels. C. reinhardtii proteins were extracted, blocked, reduced and originally oxidized cysteine thiols were labeled with IAF, then with SYPRO. Negative controls were labeled using IAM. Proteins were separated using 2D SDS-PAGE and imaged as follows: IAM- (A) and IAF- (B) labeled proteins imaged using IAF settings. IAM- (C) and IAF- (D) labeled proteins imaged using SYPRO settings. 2D-gels were aligned using SameSpots and overlapping images displayed as follows: (E) SYPRO stained gels from the IAM- (green) and IAF- (pink) (F) IAF- (green) and SYPRO- (pink) labeled proteins from the IAF labeled sample. | ||
This method was then used to explore the effects of TOR inhibition at the protein level using this tri-plexing labeling approach for parallel analysis of the C. reinhardtii redox-, phospho-, and total proteome in response to exogenous rapamycin treatment.
Redox, phospho and total differential proteomes in response to TOR inhibition in Chlamydomonas
2-DE profiles (IAF, Pro-Q and SYPRO) of 3 biological replicates of rapamycin-treated and untreated Chlamydomonas cells were obtained for each gel and used for image analysis. Representative images for each data channel are shown in Fig. 4. Spot volumes were used to quantify the PTM and total protein levels. In total, 68 differential spots were found (p ≤ 0.05): 6 (3 Up- and 3 Down-regulated) for redox, 28 (10 U and 18 D) for phosphorylation and 34 (17 U and 17 D) for total (Fig. 5). It is interesting to note that 3 out of the 6 spots from the proteins showing a redox change were also showing a change in protein abundance in the same direction which may indicate that the change in redox is the consequence of the protein expression change. Similarly, 10 spots showing a phosphorylation change overlapped with changes in protein abundance. All of them except one showed a similar change. While these 9 quantitative phosphorylation changes may also be the result of the protein expression, the remaining 19 clearly seem to be the consequence of differential phosphatase and kinase protein activities in response to the treatment.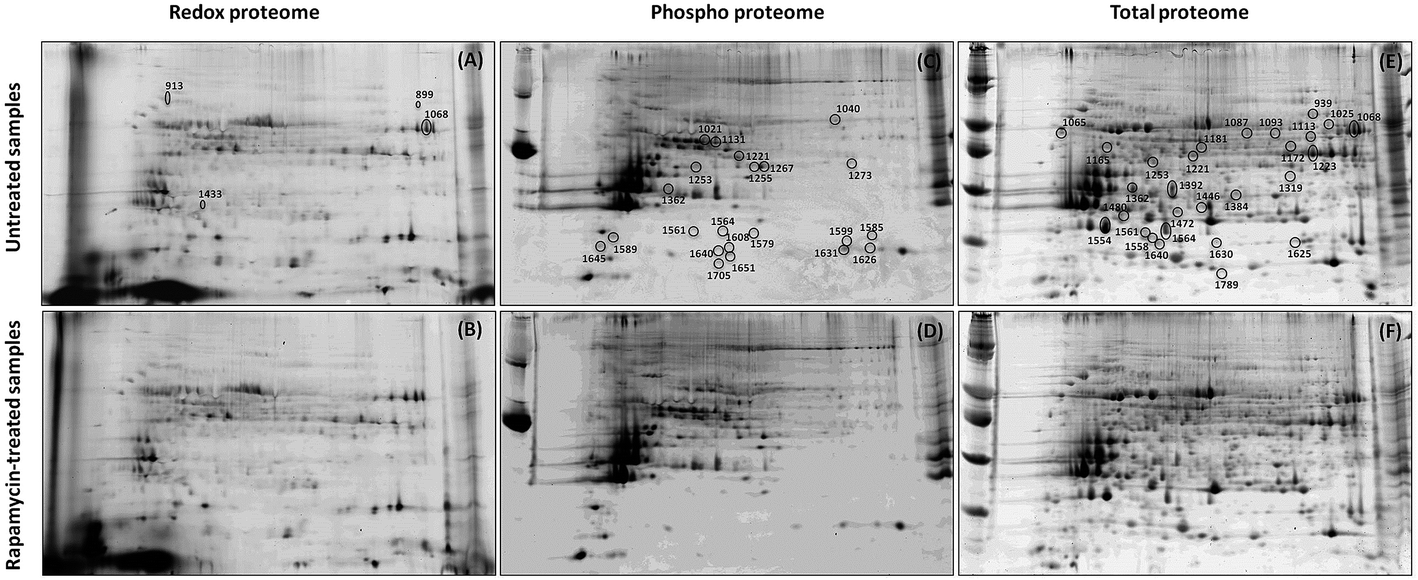 | ||
| Fig. 4 Redox (A and B), phospho (C and D) and total (E and F) proteomes from untreated (A, C and E) and rapamycin-treated (B, D and F) samples. Spots showing an intensity change are indicated on the gel. | ||
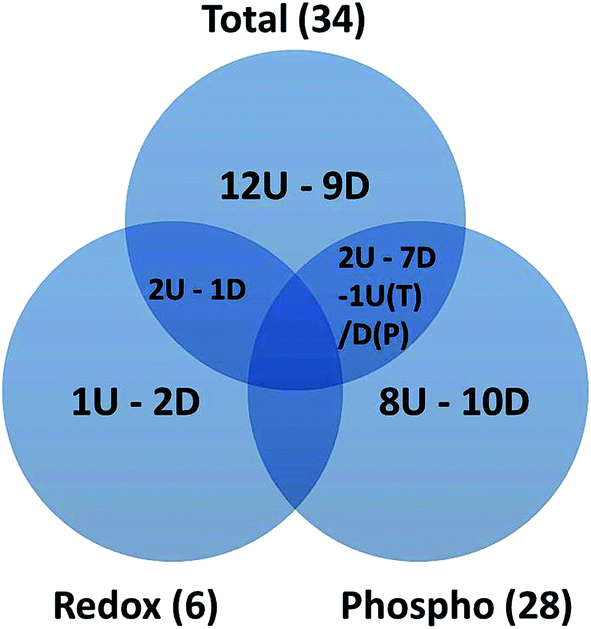 | ||
| Fig. 5 Venn diagram showing the overlapping between the redox, phospho and total spots identified with a significant change in spot abundance. The number of spots up- and down regulated are marked as U and D, respectively. For the overlap between total and phospho-proteins, one spot shows an opposite change specific to each indicated by T and P. | ||
The proteins from the spots were identified using a bottom-up approach. Because more than one protein was identified per spot, in order to narrow down the possible candidates, only the top proteins determined by the emPAI were used for further analysis. A total of 153 unique proteins were identified with at least 2 peptides and a False Discovery Rate (FDR) ≤ 1%. As a note, as protein descriptions/annotations in this algal model organism are still being refined, careful attention must be made to data analysis and interpretation of large-scale omics studies. Amongst those identified, 38 proteins were identified in multiple spots within the same group or across the redox, phospho and total proteins showing a spot intensity change. Fourteen proteins (5 U, 9 D) for redox (Table S1†), 115 (42 U and 73 D) for phosphorylation (Table S2†) and 121 (48 U and 73 D) for total (Table S3†) were identified. Proteins modulated by rapamycin treatment included those putatively related to the TOR pathway involved in nutrient and energy production to promote cell growth and redox homeostasis.30,31 Although known TOR targets, such as S6 Kinase 1,32 protein phosphatase 2A (PP2A),33 and AMP-activated protein kinase,34 were not detected – reflecting the dependence of proteome profiles on sensitivity/specificity of technology and protocol – we did identify several proteins representing processes involved in the response to rapamycin such as amino acid metabolism, citric acid cycle and carbon fixation (Table S2 and S3†). This observation was also recently shown at the transcriptomic level in a very similar experiment using rapamycin treatment in a cell wall-deficient strain of Chlamydomonas.35 This observation suggests that our treatment to inhibit TOR mimics those of other systems.
TOR-dependent redox homeostasis
The redox status of a cell regulates the synthesis, degradation, and recycling of cellular components. In C. reinhardtii, it occurs in part through the interplay between ROS and autophagy which are induced successively by inhibition of TOR.21,36,37 The burst of ROS causes oxidative damage to proteins, lipids, and small molecules, while also contributing to controlled cell degradation. In turn, autophagy limits the damage caused by ROS.38 Enzymes involved in maintaining cell redox homeostasis through the main antioxidant route, ascorbate/glutathione cycle, were not identified in the study. However amongst the other ROS detoxification systems known in plants, the thioredoxin (TRX), the peroxiredoxin (PRX) and the glutathione peroxidase (GPX) antioxidant pathways were identified with possible changes in phosphorylation level and protein abundance (Tables S2 and S3†). Thus, these results suggest that changes in ROS accumulation mediated by TOR inhibition are regulated by multiple antioxidant routes but may not include the main ascorbate/glutathione cycle as suggested in ref. 35 where it was observed that the metabolism for cysteine, one of the glutathione precursors, is blocked in response to rapamycin treatment in Chlamydomonas. In addition to redox homeostasis, it is important to note that the antioxidant systems also act as regulatory switches through redox regulation, and in some cases through cross-talk with other PTM regulation. For example, in mammalians, peroxiredoxin type I was shown to be the target of phosphorylation and this event would inactivate the peroxiredoxin activity leading to the accumulation of ROS around the membrane to act in redox signaling.39An additional antioxidant route, glutaredoxin (GRX), was identified in response to rapamycin through the IAF labeling. Here, the treatment decreased the oxidation (spot 1433, Table S1†) of a glutaredoxin CGFS-type (GRX) (Table S1†), which could be the result of an increase in oxidized glutathione levels indicative of an oxidative stress. This CGFS-type GRX is a monothiol glutaredoxin which has been shown to bind iron–sulfur clusters and to be involved with iron homeostasis40 and iron trafficking.41 In C. reinhardtii, it was demonstrated that CGFS-type GRX3 had, among other types of GRX, the unique property to control deglutathionylation of proteins42 and that these proteins reacted specifically with CFGS type GRX, allowing a specific response and regulation of cell metabolism in response to oxidative stress.43
TOR-dependent ATP balance
TOR coordinates environmental and intracellular signals to regulate an organism's metabolic state, which is partly defined by the relationship among adenylate levels in the cell (e.g. ATP, ADP, and AMP). This balance functions as an intracellular indicator of energy status and is regulated by the production of ATP, which is controlled by the activity of the mitochondrial F0–F1 ATP-synthase complex. Inhibition of ATP-synthase using α-ketoglutarate extends life span in C. elegans and downregulates TOR pathway activity, suggesting ATP-synthase functions upstream of TOR.44 Here, inhibition of TOR modulated the abundance and phosphorylation of several ATP synthase chains from various organelles (Table S2 and S3†). While the chloroplastic ATP synthase subunit delta and gamma decreased in phosphorylation level and also abundance for the delta subunit (spots 1561 and 1651), mitochondrial and vacuolar ATP synthase increased in level of phosphorylation and/or abundance (spots 1253 and 1319). These results suggest that TOR modulates an organism's metabolic state in part by a feedback loop that regulates ATPase activity through the control of its level of abundance and phosphorylation.Lipid accumulation induced by TOR inhibition
Although it is known in yeast and plants that TOR plays a role in lipid metabolism, only one protein related to the lipid pathway was identified in this study. The reasons could be because of the low level of abundance of proteins involved in lipid metabolism or the level of regulation of their activity. Here, the dihydrolipoyl dehydrogenase (DLD) was found with an increase in protein abundance (Table S3†). DLD is one member of the multi-enzyme pyruvate dehydrogenase complex (PDC) which consists of a four step conversion of pyruvate to acetyl-CoA which is fed to the citric acid cycle and fatty acid biosynthesis.To further confirm and characterize the effect of TOR inhibition in lipid biosynthesis and accumulation in C. reinhardtii, cells were grown in liquid culture to early log phase and treated with rapamycin (Fig. 6(A)). As lipid droplet (LD) accumulation was detected at 5 μM rapamycin and cell viability in these conditions was 95% (Fig. 6(B)), a time course from 2, 4, 6, 8, and 24 h was assayed and neutral lipids (Fig. 6(C)) and total triacylglycerols (TAGs) were determined as previously described (Fig. 6(D)).45 Rapamycin treatment induced total TAGs to accumulate 1.2-, 1.2-, and 3.2-fold (two-tailed unequal variance T-test) at 6, 8 and 24 h, respectively. Of the TAG species we were able to analyze and quantify via fatty-acid methyl ester analysis, rapamycin induced accumulation of several 16- and 18-carbon derivatives after 6 and 8 h (Fig. S1†). LD accumulation was evident after 6 h of treatment which may also explain for not identifying more proteins involved in lipid metabolism in the proteomics study which was performed at 4 h rapamycin treatment. The protein levels and/or protein PTM regulation may occur at a later stage between 4 and 6 h after rapamycin treatment.
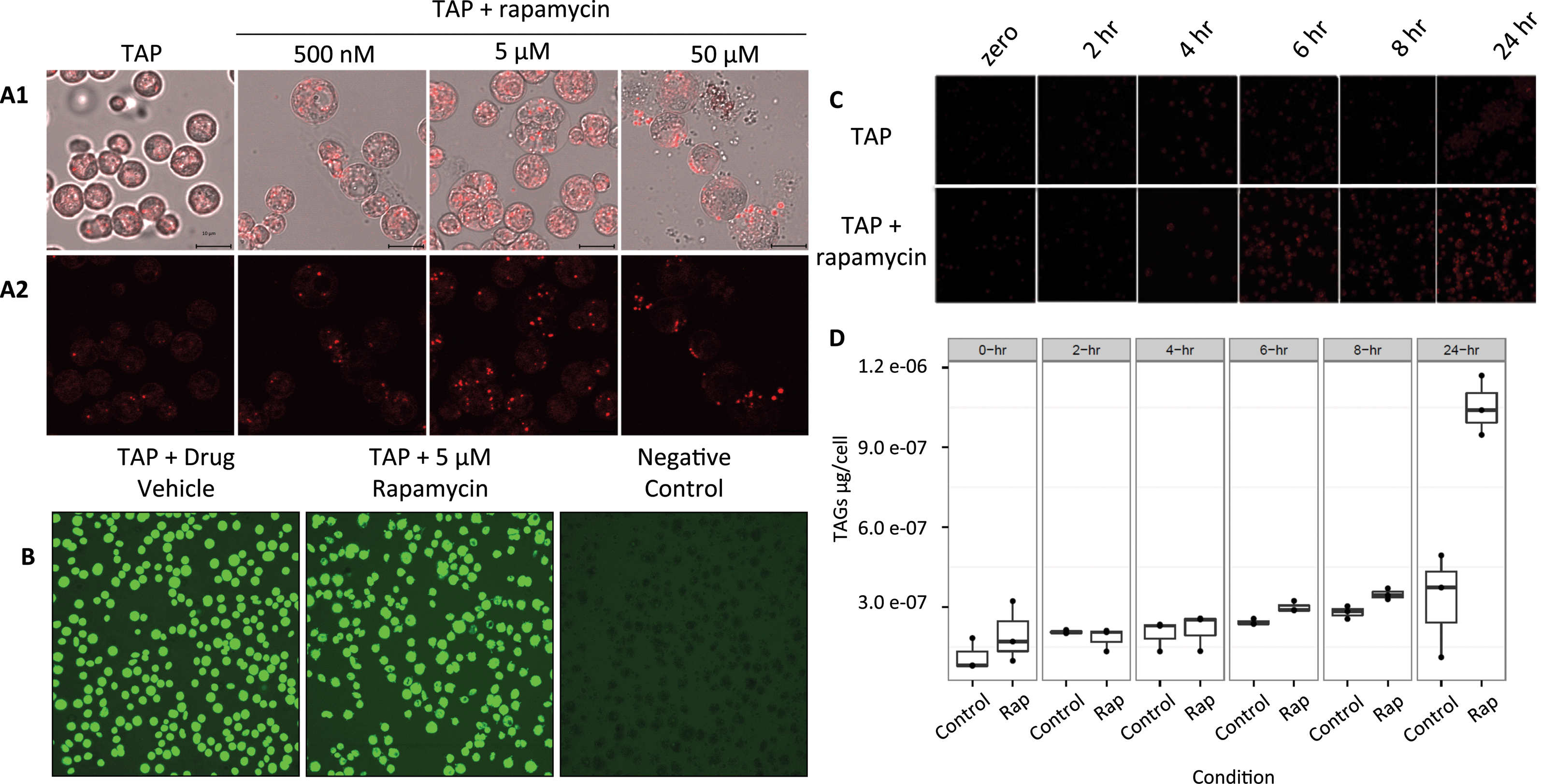 | ||
| Fig. 6 Effects of rapamycin treatment on C. reinhardtii cells grown in TAP medium. (A1) Mid-log phase C. reinhardtii cells were treated for 24 h with 500 nM, 5 μM, or 50 μM of rapamycin in TAP medium with cells grown in TAP without rapamycin (TAP) as negative control. (A2) Nile red dye was used for neutral lipid staining with red fluorescence indicating the presence of lipid droplets. Briefly, cells were stained using Nile Red, pelleted using centrifugation (5 min, 600 × g), transferred to glass slides, and observed using laser scanning confocal microscopy (Zeiss LSM 510, λex = 543 nm and λem = 565–615 nm). Bars = 10 μm. (B) Cell viability at 24 h of 5 μM rapamycin treatment was confirmed using 50 μg mL−1 fluorescein diacetate (FDA) for 10 min at RT, followed by cell pelletting (5 min at 600 × g at RT), and imaging by confocal microscopy (λex = 488 nm and λem = 500–530 nm). Negative control cells were frozen at −80 °C for 10 min prior to FDA exposure. Green fluorescence is the indicator of living cells. (C) 5 μM rapamycin-treated cells were then subjected to a time course with 2, 4, 6, 8 and 24 h of treatment and stained with Nile red for detection of lipid droplets. (D) Total triacylglycerols (TAGs) were extracted and quantified across the time course using GC-MS. | ||
Conclusion
We present here a gel-based multiplexing strategy for the differential and parallel analysis of the redox-, phospho-, and total-proteome in a single sample. Because our method uses relatively low amounts of protein and inexpensive reagents, this technique is an attractive alternative to MS-based quantitative methods that require expensive labeling and/or enrichment. Along with cost, the major advantage of multiplexing is targeted detection and a reduction in LC-MS/MS analysis time necessary for each experiment. As our method is not matrix-specific, we expect facile adaptation to any cell/tissue type. Proof-of-principle for the gel-based multiplexing approach was demonstrated for the parallel analysis of the C. reinhardtii redox-, phospho-, and total proteome in response to inhibition of TOR. We not only have identified specific redox homeostasis and energy production pathways regulated by TOR, but also shown for the first time the lipid accumulation in response to rapamycin treatment in C. reinhardtii. The data reported here serves as a basis for understanding the interplay between TOR, autophagy, and lipid metabolism in photosynthetic organisms.Experimental procedures
C. reinhardtii growth and drug treatment
C. reinhardtii cw15 mt-strain (CC-4349) was obtained from Professor Ursula Goodenough (Washington University in Saint Louis, MO). C. reinhardtii was cultivated in Tris–acetate–phosphate (TAP) medium (Harris, 1989) using 250 mL flasks incubated at 21 °C under 100 μmol m−2 s−1 and continuous illumination (Osram fluora) on an orbital shaker (110 rpm; INFORS HT Multitron). The cells were taken in mid-log phase by centrifugation (600 × g, RT, 5 min) and washed once in fresh TAP medium. For confocal microscopy, the cell suspension (1.0 × 107 cells per mL) was treated with rapamycin (LC Laboratories, Worburn, MA) dissolved in 90% ethanol and 10% Tween 20. For protein extraction, cells (2.0 x 107 cells mL−1) treated with 5 μM rapamycin during 4 h were collected by centrifugation and immediately frozen in liquid nitrogen for 2 min. Three biological replicates composed of cell populations growing in three different flasks were used.Protein extraction and IAF labeling
Protein extraction proceeded as described previously13 with minor modifications. Frozen C. reinhardtii cells (∼200 mg) were homogenized in 600 μL of extraction buffer 0.1 M Tris–HCl, pH 8.8, 10 mM EDTA, 0.9 M sucrose, 10 mM N-ethylmaleimide (NEM, Sigma, St. Louis, MO), 1× PhosSTOP (Roche, Indianapolis, IN), 1× complete EDTA-free (Roche) and 600 μL of Tris-pH 8.0 buffered phenol (Sigma). Samples were vortexed for 30 min at 4 °C, centrifuged (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, 20 min, 4 °C) and the phenolic phase was collected. The remaining aqueous phase was back extracted using 600 μL phenol solution. The combined phenolic phases were mixed with 0.1 M ammonium acetate in 100% methanol and incubated at −20 °C overnight. The pellet obtained by centrifugation (20000 × g, 20 min, 4 °C) was washed once using 0.1 M ammonium acetate in 100% methanol, twice using 80% acetone and once with 70% methanol. The pellet was resuspended in buffer (7 M urea, 2 M thiourea, 1× PhosSTOP (Roche), 50 mM Tris pH 8.0), and the protein concentration was estimated using a CBX protein assay (G-Biosciences, St. Louis, MO).
000 × g, 20 min, 4 °C) and the phenolic phase was collected. The remaining aqueous phase was back extracted using 600 μL phenol solution. The combined phenolic phases were mixed with 0.1 M ammonium acetate in 100% methanol and incubated at −20 °C overnight. The pellet obtained by centrifugation (20000 × g, 20 min, 4 °C) was washed once using 0.1 M ammonium acetate in 100% methanol, twice using 80% acetone and once with 70% methanol. The pellet was resuspended in buffer (7 M urea, 2 M thiourea, 1× PhosSTOP (Roche), 50 mM Tris pH 8.0), and the protein concentration was estimated using a CBX protein assay (G-Biosciences, St. Louis, MO).
Proteins (1.3 mg mL−1) were reduced in resupension buffer containing 50 mM DTT for 15 min at room temperature in order to reduce any cysteine residues oxidized in vivo. After DTT removal by precipitation using 100% methanol for 30 min at −20 °C, proteins (1.3 mg mL−1) were labeled using 200 mM 5-(iodoacetamido)fluorescein (IAF, Sigma) dissolved in labeling buffer (40 mM HEPES, pH 7.5, 50 mM NaCl), containing 7 M urea, 2 M thiourea and 1× PhosSTOP (Roche). After 15 min of incubation in the dark, proteins were precipitated using 100% cold methanol. The pellet was washed using 100% methanol before resuspension in DeStreak rehydration solution (GE Healthcare, Waukesha, WI), containing 1% IPG buffer, pH 3–10 NL (GE Healthcare). Cysteine thiols in control samples were alkylated using 200 mM iodoacetamide (IAM, Sigma) dissolved in labeling buffer. In contrast to,13 proteins were precipitated once after DTT reduction and IAF labeling to shorten sample processing steps.
2DE procedure and imaging
IAF-labeled proteins (250 μg) were loaded onto immobilized pH strips 3–10 NL (11 cm, Bio-Rad, Hercules, CA) and 2DE performed as described previously,13 with minor modifications. The second-dimension was conducted immediately after IEF, eliminating the gel strip equilibration steps. SDS-PAGE standards (Bio-Rad) and PeppermintStick phosphoprotein standards (Invitrogen, Eugene, OR) were loaded in the positive and negative pH strip edges, respectively. After the second-dimension separation on 8–16% Tris–HCl Criterion gels (Bio-Rad), imaging was immediately performed using a Typhoon 9410 (GE Healthcare) to detect IAF-labeled proteins (λex = 488 nm and λem = 520 nm, 700 PMT). The gels were fixed for 30 min using 50% methanol and 10% acetic acid (v/v), then overnight using the same solution. The gels were stained using Pro-Q Diamond Phosphoprotein Gel Stain (Invitrogen) and the phosphoproteins were imaged (λex = 532 nm and λem = 560 nm, 540 PMT). Then, the gels were stained using SYPRO Ruby Protein Gel Stain (Invitrogen), and the total proteins imaged (λex = 457 nm and λem = 610 nm, 600 PMT).Gel image analysis
Gel alignment, spot averaging and normalization were employed using the ‘Multiple Stain’ experimental setup in SameSpots (Nonlinear Dynamics, Newcastle, UK). Normalized spot volumes were exported to Microsoft Excel. The average ratios obtained from three biological replicates (1–3) from control and treated samples were compared using a two sample T-test assuming equal variances. Differential spots (p ≤ 0.05) were excised and identified using a bottom-up proteomics approach.Protein identification using LC-MS/MS
Briefly, spots were excised and digested using trypsin and then analyzed using nano-LC-MS/MS. In-gel trypsin digestion was conducted as previously described.13 The resulting peptides were diluted in 10 μL of mobile phase A (2% (v/v) acetonitrile and 0.1% (v/v) formic acid), and 5 μL was injected into a trap column (350 μm × 0.5 mm, Chrom XP C18, 3 μm, 120 Å, Eksigent, Monmouth Junction, NJ). Nano-LC separation (nano-LC 2D, Eksigent) was performed using a reverse phase capillary column (150 × 0.075 mm, 3C18-CL-120, 3 μm, 120 Å, Eksigent). The elution was carried out in a linear gradient from 2 to 35% solvent B (acetonitrile and 0.1% (v/v) formic acid) over 60 min at 300 nL min−1. The eluted peptides were directly introduced into a nanospray III ionization source coupled to a Triple TOF 5600 mass spectrometer (AB Sciex, Framingham, MA). Full scan mass spectrometric analysis and product ion MS/MS analysis were performed using Information Dependent Acquisition (IDA) experiments, in high resolution mode (>30000). A cycle of one full scan (MS) (350–1600 m/z) followed by multiple tandem mass spectra (MS/MS) was applied using a rolling collision energy (RCE) and a RCE spread relative to the m/z and charge state of the precursor ion. Data was acquired for 59 min with a cycle time of 2.0 s (total of 1768 cycles). The maximum number of candidate ions monitored per cycle was 20 and the ion tolerance was 100 ppm. The switch criteria were set to exclude former target ions for 10 s and to include isotopes within 2 Da. The raw data files (.wiff) were converted to mascot generic files (.mgf) before to be used for protein identification.
Data analysis
Protein identifications were obtained utilizing Mascot Server v.2.5.1 (Matrix Science, London, UK) search engine using the Chlamydomonas reinhardtii database (version 5.5, 19526 sequences total) from Phytozome V10 (ref. 46) (http://www.phytozome.net). Searches of MS/MS data used a trypsin protease specificity and the possibility of two missed cleavages, peptide/fragment mass tolerances of 10 ppm/0.08 Da, variable modifications of fluorescein and N-ethylmaleimide at cysteine, oxidation at methionine, acetylation at the protein N-terminal, deamidation of Gln and Asn, and phosphorylation of Ser, Thr, and Tyr. Results from Mascot were then processed using Scaffold (version Scaffold 4.4.1.1, Proteome Software Inc., Portland, OR) which was used to validate MS/MS based peptide and protein identifications. Only peptide identifications with 100% probability established by the Peptide Prophet algorithm47 with Scaffold delta-mass correction were further used. Additionally, peptide identifications were also filtered based on Mascot thresholds. Mascot identifications required ion scores to be greater than both the associated identity scores and ion score of 30, 40 and 50 for doubly, triply and quadruply charged peptides. Protein identifications were accepted if they could be established at greater than 99.0% probability assigned by the Protein Prophet algorithm48 and contained at least 2 identified peptides. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters. Because more than one protein per spot was identified for most of the spots, the emPAI (exponentially modified protein abundance index) as described previously49 was used to identify the top proteins for each spot. Proteins for which the emPAI was >5% of the total emPAI for each spot were kept for further analysis. Proteins with no Cys residues identified from the spots showing a redox change were removed from the analysis. The protein annotation was obtained from blasting the sequences from Phytozome to NCBInr.
Acknowledgements
This work was supported by CABS, an EFRC funded by the U.S. DOE, Office of Science, BES Award #DE-SC0001295 (LMH) and NSF EPSCoR Research Infrastructure Improvement Grant Track 1: Nanohybrid Materials and Algal Biology under EPS-1004094 (EBC) for lipidomic analyses.References
- D. L. Swaney, P. Beltrao, L. Starita, A. Guo, J. Rush, S. Fields, N. J. Krogan and J. Villén, Nat. Methods, 2013, 10, 676–682 CrossRef CAS PubMed.
- P. Minguez, L. Parca, F. Diella, D. R. Mende, R. Kumar, M. Helmer-Citterich, A. C. Gavin, V. van Noort and P. Bork, Mol. Syst. Biol., 2012, 8, 599 CrossRef PubMed.
- J. Couturier, K. Chibani, J. P. Jacquot and N. Rouhier, Front. Plant Sci., 2013, 4, 105 Search PubMed.
- S. Kangasjarvi, J. Neukermans, S. Li, E.-M. Aro and G. Noctor, J. Exp. Bot., 2012, 63, 1619–1636 CrossRef PubMed.
- L. M. Hicks, R. E. Cahoon, E. R. Bonner, R. S. Rivard, J. Sheffield and J. M. Jez, Plant Cell, 2007, 19, 2653–2661 CrossRef CAS PubMed.
- T. E. Thingholm, O. N. Jensen and M. R. Larsen, Proteomics, 2009, 9, 1451–1468 CrossRef CAS PubMed.
- N. Navrot, C. Finnie, B. Svensson and P. Hagglund, J. Proteomics, 2011, 74, 1450–1462 CrossRef CAS PubMed.
- T. Xing and A. Laroche, Plant Signaling Behav., 2011, 6, 1469–1474 CrossRef CAS PubMed.
- N. V. Bykova and C. Rampitsch, Proteomics, 2013, 13, 579–596 CrossRef CAS PubMed.
- J. X. Yan, R. A. Harry, C. Spibey and M. J. Dunn, Electrophoresis, 2000, 21, 3657–3665 CrossRef CAS.
- T. H. Steinberg, B. J. Agnew, K. R. Gee, W. Y. Leung, T. Goodman, B. Schulenberg, J. Hendrickson, J. M. Beechem, R. P. Haugland and W. F. Patton, Proteomics, 2003, 3, 1128–1144 CrossRef CAS PubMed.
- J. W. Baty, M. B. Hampton and C. C. Winterbourn, Proteomics, 2002, 2, 1261–1266 CrossRef CAS.
- S. Alvarez, A. Galant, J. M. Jez and L. M. Hicks, Proteomics, 2011, 11, 1346–1350 CrossRef CAS PubMed.
- B. Schulenberg, T. N. Goodman, R. Aggeler, R. A. Capaldi and W. F. Patton, Electrophoresis, 2004, 25, 2526–2532 CrossRef CAS PubMed.
- C. Marondedze, K. Lilley and L. Thomas, Methods Mol. Biol., 2013, 1016, 139–154 CAS.
- J. Wu, N. J. Lenchik, M. J. Pabst, S. S. Solomon, J. Shull and I. C. Gerling, Electrophoresis, 2005, 26, 225–237 CrossRef CAS PubMed.
- A. J. Howden, M. Salek, L. Miguet, M. Pullen, B. Thomas, M. R. Knight and L. J. Sweetlove, New Phytol., 2011, 190, 49–56 CrossRef CAS PubMed.
- J. Lin, D. Tritschler, K. Song, C. F. Barber, J. S. Cobb, M. E. Porter and D. Nicastro, J. Biol. Chem., 2011, 286, 29175–29191 CrossRef CAS PubMed.
- Z. B. Yang, D. Eticha, H. Fuhrs, D. Heintz, D. Ayoub, A. Van Dorsselaer, B. Schlingmann, I. M. Rao, H. P. Braun and W. J. Horst, J. Exp. Bot., 2013, 64, 5569–5586 CrossRef CAS PubMed.
- H. Wang, S. Wang, Y. Lu, S. Alvarez, L. M. Hicks, X. Ge and Y. Xia, J. Proteome Res., 2012, 11, 412–424 CrossRef CAS PubMed.
- S. Diaz-Troya, F. J. Florencio and J. L. Crespo, Eukaryotic Cell, 2008, 7, 212–222 CrossRef CAS PubMed.
- R. Singh, S. Kaushik, Y. Wang, Y. Xiang, I. Novak, M. Komatsu, K. Tanaka, A. M. Cuervo and M. J. Czaja, Nature, 2009, 458, 1131–1135 CrossRef CAS PubMed.
- Y. Zhang, S. Persson and P. Giavalisco, Mol. Plant, 2013, 6, 1731–1733 CrossRef CAS PubMed.
- S. Diaz-Troya, M. E. Perez-Perez, F. J. Florencio and J. L. Crespo, Autophagy, 2008, 4, 851–865 CrossRef CAS.
- O. Blokhina and K. V. Fagerstedt, Plant Physiol. Biochem., 2010, 48, 359–373 CrossRef CAS PubMed.
- J. L. Crespo, S. Diaz-Troya and F. J. Florencio, Plant Physiol., 2005, 139, 1736–1749 CrossRef CAS PubMed.
- D. G. Smyth, O. O. Blumenfeld and W. Konigsberg, Biochem. J., 1964, 91, 589–595 CAS.
- W. L. Zahler and W. W. Cleland, J. Biol. Chem., 1968, 243, 716–719 CAS.
- J. J. Gorman, Anal. Biochem., 1987, 160, 376–387 CrossRef CAS.
- Y. Xiong and J. Sheen, Plant Physiol., 2014, 164, 499–512 CrossRef CAS PubMed.
- F. John, S. Roffler, T. Wicker and C. Ringli, Plant Signaling Behav., 2011, 6, 1700–1705 CrossRef CAS PubMed.
- H. Nojima, C. Tokunaga, S. Eguchi, N. Oshiro, S. Hidayat, K. Yoshino, K. Hara, N. Tanaka, J. Avruch and K. Yonezawa, J. Biol. Chem., 2003, 278, 15461–15464 CrossRef CAS PubMed.
- E. Jacinto and M. N. Hall, Mol. Cell. Biol., 2003, 4, 117–126 CAS.
- N. Kimura, C. Tokunaga, S. Dalal, C. Richardson, K. Yoshino, K. Hara, B. E. Kemp, L. A. Witters, O. Mimura and K. Yonezawa, Genes Cells, 2003, 8, 65–79 CrossRef CAS.
- S. Kleessen, S. Irgang, S. Klie, P. Giavalisco and Z. Nikoloski, Plant J., 2015, 81, 822–835 CrossRef CAS PubMed.
- R. Scherz-Shouval, E. Shvets, E. Fass, H. Shorer, L. Gil and Z. Elazar, EMBO J., 2007, 26, 1749–1760 CrossRef CAS PubMed.
- Y. Chen and S. B. Gibson, Autophagy, 2008, 4, 246–248 CrossRef CAS.
- R. Scherz-Shouval and Z. Elazar, Trends Biochem. Sci., 2011, 36, 30–38 CrossRef CAS PubMed.
- S. G. Rhee, H. A. Woo, I. S. Kil and S. H. Bae, J. Biol. Chem., 2012, 287, 4403–4410 CrossRef CAS PubMed.
- A. Picciocchi, C. Saguez, A. Boussac, C. Cassier-Chauvat and F. Chauvat, Biochemistry, 2007, 46, 15018–15026 CrossRef CAS PubMed.
- E. Herrero and M. A. de la Torre-Ruiz, Cell. Mol. Life Sci., 2007, 64, 1518–1530 CrossRef CAS PubMed.
- M. Zaffagnini, L. Michelet, V. Massot, P. Trost and S. D. Lemaire, J. Biol. Chem., 2008, 283, 8868–8876 CrossRef CAS PubMed.
- X. H. Gao, M. Zaffagnini, M. Bedhomme, L. Michelet, C. Cassier-Chauvat, P. Decottignies and S. D. Lemaire, FEBS Lett., 2010, 584, 2242–2248 CrossRef CAS PubMed.
- R. M. Chin, X. Fu, M. Y. Pai, L. Vergnes, H. Hwang, G. Deng, S. Diep, B. Lomenick, V. S. Meli, G. C. Monsalve, E. Hu, S. A. Whelan, J. X. Wang, G. Jung, G. M. Solis, F. Fazlollahi, C. Kaweeteerawat, A. Quach, M. Nili, A. S. Krall, H. A. Godwin, H. R. Chang, K. F. Faull, F. Guo, M. Jiang, S. A. Trauger, A. Saghatelian, D. Braas, H. R. Christofk, C. F. Clarke, M. A. Teitell, M. Petrascheck, K. Reue, M. E. Jung, A. R. Frand and J. Huang, Nature, 2014, 510, 397–401 CAS.
- J. Msanne, D. Xu, A. R. Konda, J. A. Casas-Mollano, T. Awada, E. B. Cahoon and H. Cerutti, Phytochemistry, 2012, 75, 50–59 CrossRef CAS PubMed.
- S. S. Merchant, S. E. Prochnik, O. Vallon, E. H. Harris, S. J. Karpowicz, G. B. Witman, A. Terry, A. Salamov, L. K. Fritz-Laylin, L. Maréchal-Drouard, W. F. Marshall, L. H. Qu, D. R. Nelson, A. A. Sanderfoot, M. H. Spalding, V. V. Kapitonov, Q. Ren, P. Ferris, E. Lindquist, H. Shapiro, S. M. Lucas, J. Grimwood, J. Schmutz, P. Cardol, H. Cerutti, G. Chanfreau, C. L. Chen, V. Cognat, M. T. Croft, R. Dent, S. Dutcher, E. Fernández, H. Fukuzawa, D. González-Ballester, D. González-Halphen, A. Hallmann, M. Hanikenne, M. Hippler, W. Inwood, K. Jabbari, M. Kalanon, R. Kuras, P. A. Lefebvre, S. D. Lemaire, A. V. Lobanov, M. Lohr, A. Manuell, I. Meier, L. Mets, M. Mittag, T. Mittelmeier, J. V. Moroney, J. Moseley, C. Napoli, A. M. Nedelcu, K. Niyogi, S. V. Novoselov, I. T. Paulsen, G. Pazour, S. Purton, J. P. Ral, D. M. Riaño-Pachón, W. Riekhof, L. Rymarquis, M. Schroda, D. Stern, J. Umen, R. Willows, N. Wilson, S. L. Zimmer, J. Allmer, J. Balk, K. Bisova, C. J. Chen, M. Elias, K. Gendler, C. Hauser, M. R. Lamb, H. Ledford, J. C. Long, J. Minagawa, M. D. Page, J. Pan, W. Pootakham, S. Roje, A. Rose, E. Stahlberg, A. M. Terauchi, P. Yang, S. Ball, C. Bowler, C. L. Dieckmann, V. N. Gladyshev, P. Green, R. Jorgensen, S. Mayfield, B. Mueller-Roeber, S. Rajamani, R. T. Sayre, P. Brokstein, I. Dubchak, D. Goodstein, L. Hornick, Y. W. Huang, J. Jhaveri, Y. Luo, D. Martínez, W. C. Ngau, B. Otillar, A. Poliakov, A. Porter, L. Szajkowski, G. Werner, K. Zhou, I. V. Grigoriev, D. S. Rokhsar and A. R. Grossman, Science, 2007, 318, 245–250 CrossRef CAS PubMed.
- A. Keller, A. I. Nesvizhskii, E. Kolker and R. Aebersold, Anal. Chem., 2002, 74, 5383–5392 CrossRef CAS.
- A. I. Nesvizhskii, A. Keller, E. Kolker and R. Aebersold, Anal. Chem., 2003, 75, 4646–4658 CrossRef CAS.
- Y. Ishihama, Y. Oda, T. Tabata, T. Sato, T. Nagasu, J. Rappsilber and M. Mann, Mol. Cell. Proteomics, 2005, 4, 1265–1272 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ay00521c |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |