Perfect encapsulation of a guanidinium ion in a helical trinickel(II) metallocryptand for efficient regulation of the helix inversion rate†‡
Shigehisa
Akine
*a,
Masato
Miyashita
b,
Shunjin
Piao
b and
Tatsuya
Nabeshima
*b
aGraduate School of Natural Science and Technology, Kanazawa University, Kakuma-machi, Kanazawa 920-1192, Japan. E-mail: akine@se.kanazawa-u.ac.jp
bFaculty of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8571, Japan. E-mail: nabesima@chem.tsukuba.ac.jp
First published on 20th December 2013
Abstract
Novel tris(salen)-type trinickel(II) metallocryptands, [LNi3] (L = L1, L2), were designed and synthesized. The metallocryptand [L2Ni3] perfectly encapsulated a guanidinium ion in the cavity. The resulting guanidinium complex underwent significantly slow helix inversion compared to the free [L2Ni3]; thus the helix inversion rate was efficiently regulated by guanidinium recognition.
Dynamic helical molecules, which are capable of undergoing reversible helicity inversion, are useful as a basic framework for chiral switching systems.1 There have been many examples of dynamic helical structures such as metal helicates,2 foldamers,3etc. From the viewpoint of chiral information processing, the rate of helix inversion is one of the important features. A switching system based on a dynamic helical structure needs to undergo fast helix inversion enough to rapidly change the helicity when the chiral information is recorded. However, the helix inversion should be suppressed when the information needs to be kept. In this context, it is important to develop dynamic helical systems that can change the helix inversion rate depending on the circumstances. There have been several examples of dynamic helical molecules whose helix inversion rates can be tuned by changing solvent polarity, constituent metal ions of helicates, etc.4 In order to efficiently control the helix inversion rate, combination of such a dynamic helix inversion with host–guest complexation would be effective (Scheme 1).
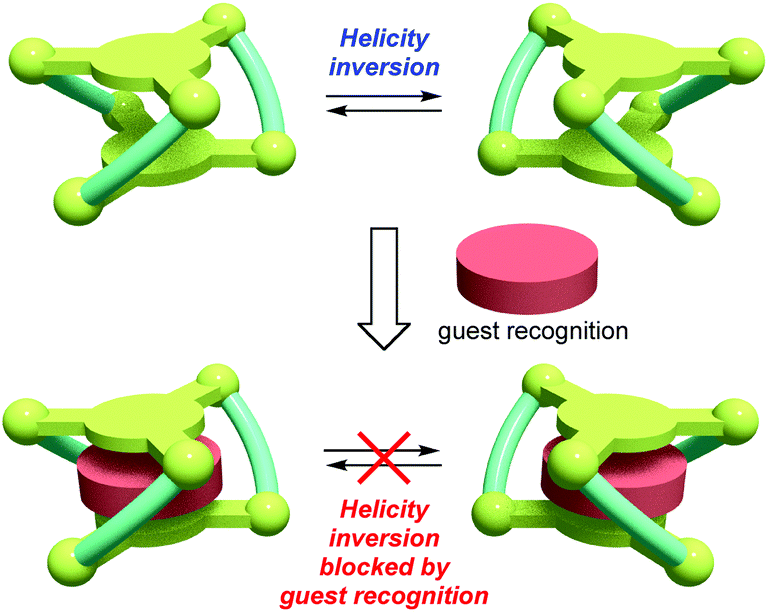 | ||
| Scheme 1 Concept of helicity inversion blocking by guest recognition. | ||
We have already reported that oligosalen-type§ host molecules5–9 having cyclic,7 single-helical,8 and cage-like9 structures showed cation binding affinity due to the negatively polarized phenoxo groups of the salen–metal complex moieties. In this study, we have designed new helical cryptand-like complexes [LNi3] (L = L1, L2; Scheme 2) that have three saloph§ complex moieties connected with two pivotal benzene rings. The complexes have a relatively rigid framework due to the biaryl skeleton and are expected to adopt a triple-helical structure. The helical cage-like cavity surrounded by the three saloph complex moieties would serve as a binding site for a guest molecule, as seen in most cryptands,10 capsular molecules,11etc. In general, cage-like hosts form a stable host–guest complex due to the complete encapsulation of the guest species. In this particular case, strong guest recognition is expected due to the six phenoxo groups in a convergent arrangement as well as the two pivotal benzene rings capable of π-stacking interaction with planar guest species. Here, we found that trinuclear complexes [LNi3], which were prepared from the tris(saloph) cryptand molecules H6L, can form a highly stable host–guest complex with a guanidinium ion. The complete encapsulation of the guanidinium ion significantly retarded the helix inversion rate of the triple helical structure of the [LNi3] scaffold.
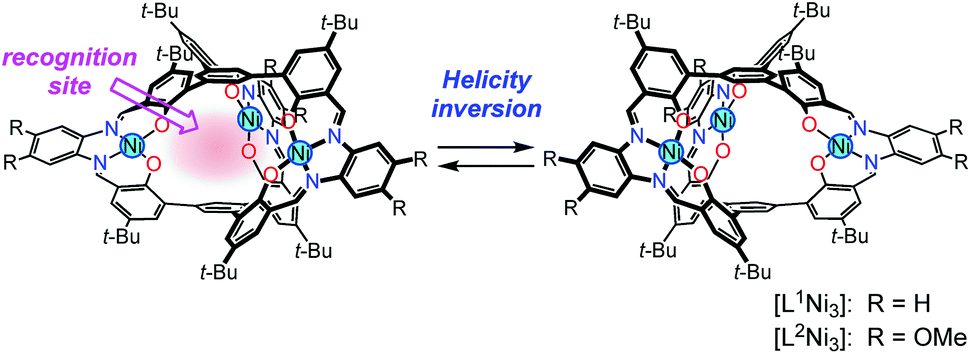 | ||
| Scheme 2 Helical metallocryptand [LNi3] (L = L1, L2) with an inversion rate tunable by guest recognition. | ||
The ligands H6L1 and H6L2 were synthesized from 1,3,5-tribromobenzene (1)12 by a four-step procedure (Scheme 3). The Suzuki-coupling of 1 with 5-tert-butyl-2-methoxyphenylboronic acid (2)13 afforded a triphenylbenzene derivative 3 having three methoxy groups. The lithiation of 3 with n-butyllithium followed by the reaction with N,N-dimethylformamide afforded a trialdehyde 4. The demethylation of this product afforded the precursor 5. The macrocyclization of the trialdehyde 5 with 1,2-phenylenediamine derivatives (6a,b)14 proceeded smoothly to afford the target compounds H6L1 and H6L2. The simple 1H NMR spectral patterns showing one singlet for the imine protons clearly indicated the symmetric structure, and the mass peaks at m/z = 1467.2 for [H6L1 + K]+ and 1632.0 for [H6L2 + Na]+ confirmed the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 stoichiometry of the condensation. The ligands were converted into the corresponding nickel(II) trinuclear complexes [L1Ni3] and [L2Ni3] by the reaction with nickel(II) acetate. The complete conversion was again evidenced by the simple 1H NMR spectra and the mass peaks (m/z = 1621.7 for [L1Ni3 + Na]+; m/z = 1801.6 for [L2Ni3 + Na]+). While the ligand H6L1 and the complex [L1Ni3] without methoxy groups showed low solubility in common organic solvents such as chloroform, the corresponding methoxy derivatives H6L2 and [L2Ni3] showed relatively higher solubility. Consequently, the methoxy derivative [L2Ni3] was used for the binding study.
:3 stoichiometry of the condensation. The ligands were converted into the corresponding nickel(II) trinuclear complexes [L1Ni3] and [L2Ni3] by the reaction with nickel(II) acetate. The complete conversion was again evidenced by the simple 1H NMR spectra and the mass peaks (m/z = 1621.7 for [L1Ni3 + Na]+; m/z = 1801.6 for [L2Ni3 + Na]+). While the ligand H6L1 and the complex [L1Ni3] without methoxy groups showed low solubility in common organic solvents such as chloroform, the corresponding methoxy derivatives H6L2 and [L2Ni3] showed relatively higher solubility. Consequently, the methoxy derivative [L2Ni3] was used for the binding study.
 | ||
| Scheme 3 Synthesis of ligands H6L1, H6L2 and trinuclear complexes [L1Ni3], [L2Ni3]. Reagents and conditions: (a) [Pd(PPh3)4], K2CO3, DMA, EtOH, H2O, 72%; (b) (i) n-BuLi, TMEDA, Et2O, (ii) DMF, (iii) H2O, 99%; (c) (i) BBr3, CH2Cl2, (ii) H2O, 76%; (d) R = H: 6a, CH2Cl2, MeCN, 58%; R = OMe: 6b, CHCl3, MeCN, 58%; (e) R = H: Ni(OAc)2, DMSO, 57%; R = OMe: Ni(OAc)2, CHCl3, DMSO, 72%. | ||
The structures of H6L2 and [L2Ni3] were determined by X-ray crystallography (Fig. 1 and 2).15,16 Both of the compounds had a similar cage-like structure in which the three saloph ligand moieties formed a triple helix. The helix twisting angle17 for [L2Ni3] was about 54°, which was slightly smaller than that for the uncomplexed ligand H6L2 (68°) (Table 1). The introduction of nickel(II) ions probably makes the three saloph moieties more rigid and planar, resulting in the greater dihedral angles of the biaryl moieties and the less twisted triple helical structure. In addition, the three nickel(II) ions significantly shorten the pivot–pivot distance; the distance between the two pivotal benzene rings for [L2Ni3] was 5.523 Å, which was 20% shorter than that of H6L2 (6.866 Å). As a result, the cavity of the cryptand shrank upon the complexation with nickel(II).
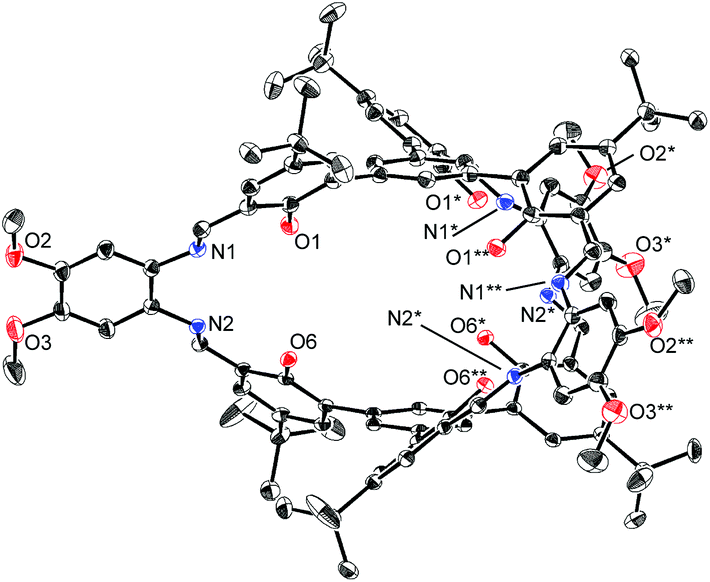 | ||
| Fig. 1 X-ray structure of H6L2 with thermal ellipsoids plotted at the 20% probability level. Hydrogen atoms and less occupied disordered atoms are omitted for clarity. | ||
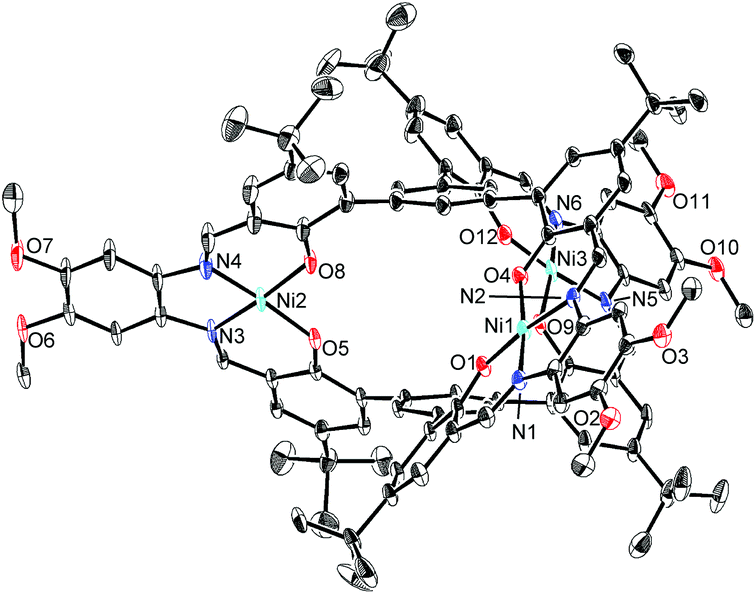 | ||
| Fig. 2 X-ray structure of [L2Ni3] with thermal ellipsoids plotted at the 20% probability level. Hydrogen atoms and less occupied disordered atoms are omitted for clarity. | ||
The phenoxo groups of the salen-type complexes are known to act as a good hydrogen-bond acceptor18 as well as a coordination site for the hard metal ions.5 In addition, the three-fold symmetry of the tris(salen)-type cage [L2Ni3] is expected to be effective for binding to a guest with three-fold symmetry. Thus, we chose the guanidinium ion18a,19 as the guest to be incorporated into the cavity of [L2Ni3].
As expected, the trinuclear metallohost [L2Ni3] showed a high affinity toward the guanidinium ion. In the 1H NMR spectrum of [L2Ni3] in CDCl3–DMSO-d6 (9:1) in the presence of guanidinium chloride, the signal for the bound guanidinium ion appeared as a sharp singlet at 5.06 ppm, which was observed separately from the unbound guanidinium ion (6.98 ppm) (Fig. 3). This indicates that the complexation/decomplexation rate of the guanidinium ion is slow on the NMR timescale. Since 1 equiv. of guanidinium chloride was sufficient to completely convert the [L2Ni3] into the host–guest complex, the binding with guanidinium was significantly strong (log Ka > 8).20 The corresponding metal-free host, H6L2, also formed a host–guest complex with guanidinium (log Ka = 3.61), but the recognition ability of the metallohost [L2Ni3] was much higher (more than 104 times). The signal for the guanidinium ion bound to [L2Ni3] was observed at significantly higher field, probably due to the shielding effect of the two pivotal benzene rings. This indicates the complete encapsulation of the guanidinium ion in the cavity of [L2Ni3]. The formation of the guanidinium complex was further confirmed by the mass peak at m/z = 1838.8, which was assignable to the [L2Ni3·guanidinium]+.
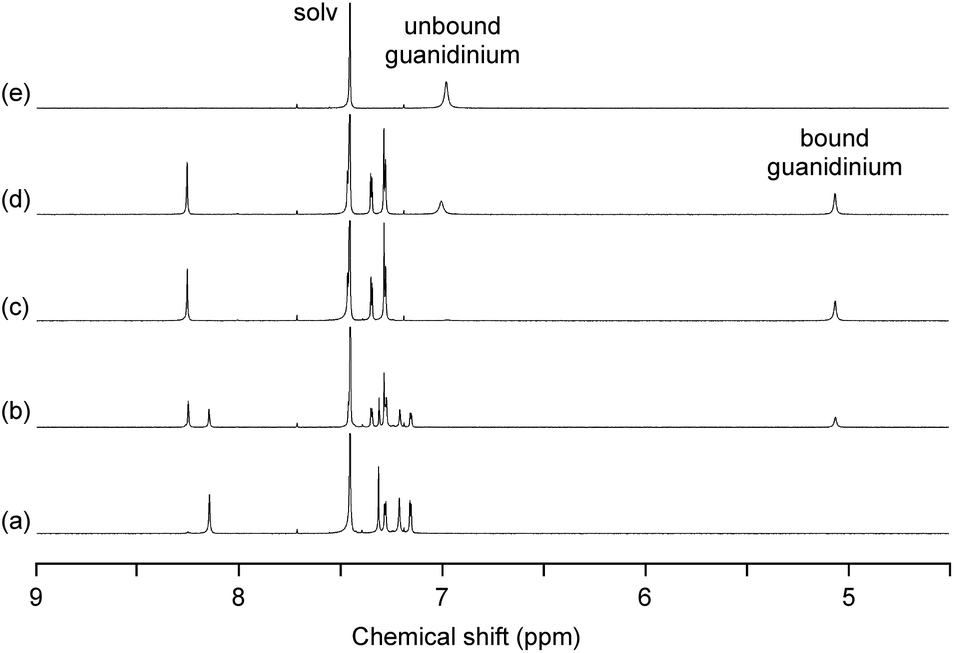 | ||
| Fig. 3
1H NMR spectra of [L2Ni3] (0.5 mM) in the presence of guanidinium chloride (a, 0 equiv.; b, 0.5 equiv.; c, 1.0 equiv.; d, 2.0 equiv.) and (e) guanidinium chloride in CDCl3–DMSO-d6 (9:1) at 400 MHz. | ||
The X-ray crystallographic analysis revealed the structure of the [L2Ni3]–guanidinium complex,21 in which the bound guanidinium ion was completely encapsulated in the cavity of the [L2Ni3] (Fig. 4). The guanidinium ion was located right in the middle of the two pivotal benzene rings in a parallel arrangement. The pivot–guanidinium distances were around 3.03 Å. Such a close contact indicates significant π–π and/or cation–π interactions. In addition, hydrogen bonding also contributed to the guest binding. The six N–O distances between the guanidinium nitrogen atoms and the [Ni(saloph)]-phenoxo oxygen atoms ranged from 2.917 to 2.970 Å, indicating the effective six-fold N–H⋯O hydrogen bonding. It is noteworthy that the geometry of the cage-like framework of [L2Ni3] was only slightly changed by the incorporation of the guanidinium ion. The pivot–pivot distance of [L2Ni3·guanidinium]+ was 6.059 Å, which was only 10% longer than the corresponding distance of the vacant [L2Ni3] (5.523 Å). Thus, the metallohost encapsulated a guanidinium ion via multi-point hydrogen bonding as well as π–π and/or cation–π interactions. Most importantly, the metallohost [L2Ni3] needed to undergo only small structural changes to achieve the multipoint interactions with the guanidinium ion. This shape-complementarity should account for the extremely strong binding with the guanidinium ion.
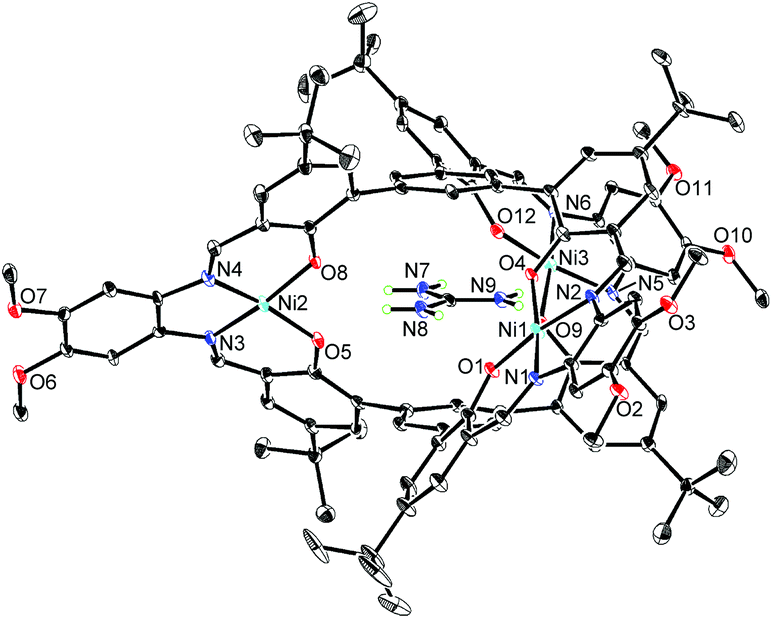 | ||
| Fig. 4 X-ray structure of [L2Ni3·guanidinium]+ with thermal ellipsoids plotted at the 20% probability level. Hydrogen atoms except for those of the guanidinium ion and less occupied disordered atoms are omitted for clarity. | ||
The binding experiments also showed that the metallohost [L2Ni3] interacted with alkylammonium ions. For example, the binding constant for L-phenylalanine methyl ester hydrochloride was determined to be log Ka = 3.0 in CDCl3–DMSO-d6 (9:1), which was smaller than that with the guanidinium ion. Since the cage-like framework of [L2Ni3] adopts a three-fold helical structure, chiral species should interact differently with the right- and left-handed enantiomers. If the two enantiomers are interconvertible, the chiral species might cause a shift of the equilibrium, which could be observable by CD spectroscopy. Indeed, the CD spectrum of [L2Ni3] in the presence of L-phenylalanine methyl ester hydrochloride showed a distinct Cotton effect, indicative of the shift of the equilibrium between the right- and left-handed forms. The CD signals instantly emerged (half-life less than 12 s at 20 °C) after the addition of the chiral ammonium. This quick response indicates that the guest recognition and the interconversion between the right- and left-handed enantiomers of [L2Ni3] occurred within seconds.
From the viewpoint of storing chiral information, it is important to keep the biased right/left ratio even after the chiral source is removed. To achieve the chiral guest removal, a better guest, the guanidinium ion, was added to a mixture of [L2Ni3] and L-phenylalanine methyl ester hydrochloride. As expected, the induced negative CD signals remained essentially intact after the addition of the guanidinium. Only a 6% decrease in the CD intensity was observed after 3 min. It should be noted that the guest exchange by the guanidinium ion immediately completed judging from the 1H NMR spectrum about 3 min after mixing. It is clear that the [L2Ni3] kept the biased ratio even after the guest exchange. The CD signals slowly decayed with a half-life of 2.2 × 103 s at 20 °C (Fig. 5). Therefore, the interconversion rate between the right- and left-handed enantiomers became at least 180-times slower by the complexation with the guanidinium ion.
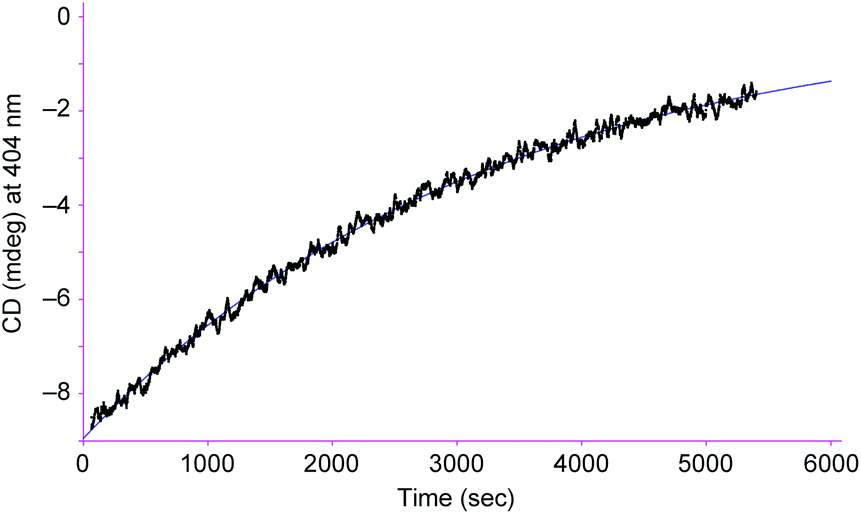 | ||
| Fig. 5 Changes in the CD intensity at 404 nm in chloroform–DMSO (9:1) after 2 equiv. of guanidinium chloride was added to a mixture of [L2Ni3] (0.25 mM) and L-phenylalanine methyl ester hydrochloride (1.25 mM). Path length, 0.5 mm; temperature, 20.0 °C. | ||
In summary, novel helical trinickel(II) metallocryptands [L1Ni3] and [L2Ni3] were synthesized. The metallocryptand [L2Ni3] showed a high affinity toward the guanidinium ion to afford the stable inclusion complex. The encapsulation of the guanidinium significantly retards the helicity inversion between the right- and left-handed forms. Such tuning of the helicity inversion rate by non-covalent interaction would be useful for reversible control of the chiral functions of metal complex moieties.
The research was financially supported by Grants-in-Aid for Scientific Research on Innovative Areas (Coordination Programming Area 2107, no. 22108505 and 24108707 to S.A.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Notes and references
- For recent reviews on dynamic helical structures, see: (a) J. Crassous, Chem. Commun., 2012, 48, 9684–9692 RSC; (b) H. Miyake and H. Tsukube, Chem. Soc. Rev., 2012, 41, 6977–6991 RSC.
- (a) J.-M. Lehn, Supramolecular Chemistry – Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed; (b) C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005–2062 CrossRef CAS PubMed; (c) M. Albrecht, Chem. Rev., 2001, 101, 3457–3497 CrossRef CAS PubMed.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4011 CrossRef CAS PubMed.
- (a) E. Yashima, K. Maeda and Y. Okamoto, Nature, 1999, 399, 449–451 CrossRef CAS; (b) S. Akine, T. Taniguchi, T. Matsumoto and T. Nabeshima, Chem. Commun., 2006, 4961–4963 RSC; (c) J. Gregoliński and J. Lisowski, Angew. Chem., Int. Ed., 2006, 45, 6122–6126 CrossRef PubMed; (d) H. Miyake, H. Kamon, I. Miyahara, H. Sugimoto and H. Tsukube, J. Am. Chem. Soc., 2008, 130, 792–793 CrossRef CAS PubMed; (e) M. Lama, O. Mamula, G. S. Kottas, L. de Cola, H. Stoeckli-Evans and S. Shova, Inorg. Chem., 2008, 47, 8000–8015 CrossRef CAS PubMed; (f) J. Gregoliński, P. Starynowicz, K. T. Hua, J. L. Lunkley, G. Muller and J. Lisowski, J. Am. Chem. Soc., 2008, 130, 17761–17773 CrossRef PubMed.
- (a) S. Akine and T. Nabeshima, Dalton Trans., 2009, 10395–10408 RSC; (b) S. Akine, J. Inclusion Phenom. Macrocyclic Chem., 2012, 72, 25–54 CrossRef CAS.
- For reviews on oligosalen structures, see: (a) M. J. MacLachlan, Pure Appl. Chem., 2006, 78, 873–888 CrossRef CAS; (b) S. J. Wezenberg and A. W. Kleij, Angew. Chem., Int. Ed., 2008, 47, 2354–2364 CrossRef CAS PubMed; (c) A. W. Kleij, Chem.–Eur. J., 2008, 14, 10520–10529 CrossRef CAS PubMed; (d) H. L. C. Feltham and S. Brooker, Coord. Chem. Rev., 2009, 253, 1458–1475 CrossRef CAS.
- (a) S. Akine, T. Taniguchi and T. Nabeshima, Tetrahedron Lett., 2001, 42, 8861–8864 CrossRef CAS; (b) S. Akine, D. Hashimoto, T. Saiki and T. Nabeshima, Tetrahedron Lett., 2004, 45, 4225–4227 CrossRef CAS; (c) T. Nabeshima, H. Miyazaki, A. Iwasaki, S. Akine, T. Saiki, C. Ikeda and S. Sato, Chem. Lett., 2006, 35, 1070–1071 CrossRef CAS; (d) S. Akine, F. Utsuno and T. Nabeshima, Chem. Commun., 2010, 46, 1029–1031 RSC.
- (a) S. Akine, T. Taniguchi and T. Nabeshima, Angew. Chem., Int. Ed., 2002, 41, 4670–4673 CrossRef CAS PubMed; (b) S. Akine, T. Taniguchi, T. Saiki and T. Nabeshima, J. Am. Chem. Soc., 2005, 127, 540–541 CrossRef CAS PubMed; (c) S. Akine, T. Taniguchi and T. Nabeshima, J. Am. Chem. Soc., 2006, 128, 15765–15774 CrossRef CAS PubMed; (d) S. Akine, T. Matsumoto and T. Nabeshima, Chem. Commun., 2008, 4604–4606 RSC; (e) S. Akine, S. Hotate, T. Matsumoto and T. Nabeshima, Chem. Commun., 2011, 47, 2925–2927 RSC; (f) S. Akine, S. Hotate and T. Nabeshima, J. Am. Chem. Soc., 2011, 133, 13868–13871 CrossRef CAS PubMed; (g) S. Akine, S. Sairenji, T. Taniguchi and T. Nabeshima, J. Am. Chem. Soc., 2013, 135, 12948–12951 CrossRef CAS PubMed.
- S. Akine, S. Piao, M. Miyashita and T. Nabeshima, Tetrahedron Lett., 2013, 54, 6541–6544 CrossRef CAS.
- B. Dietrich, J.-M. Lehn and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1973, 15–16 RSC.
- S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS.
- G. H. Coleman and W. F. Talbot, Org. Synth. Coll. Vol. II., 1943, 592–594 Search PubMed.
- J. A. Bryant, R. C. Helgeson, C. B. Knobler, M. P. deGrandpre and D. J. Cram, J. Org. Chem., 1990, 55, 4622–4634 CrossRef CAS.
- X. Ottenwaelder, R. Ruiz-García, G. Blondin, R. Carasco, J. Cano, D. Lexa, Y. Journaux and A. Aukauloo, Chem. Commun., 2004, 504–505 RSC.
- Crystallographic data for H6L2·11DMSO·3H2O (C124H180N6O26S11 = 2523.40): trigonal, R
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , a = 24.1319(10) Å, c = 43.5952(18) Å, V = 21986.3(16) Å3, Z = 6, Dcalcd = 1.143 g cm−3, R1 = 0.0962 (I > 2σ(I)), wR2 = 0.2262 (all data) (CCDC 963501).22.
, a = 24.1319(10) Å, c = 43.5952(18) Å, V = 21986.3(16) Å3, Z = 6, Dcalcd = 1.143 g cm−3, R1 = 0.0962 (I > 2σ(I)), wR2 = 0.2262 (all data) (CCDC 963501).22. - Crystallographic data for [L2Ni3]·3DMSO·H2O·5CH2Cl2·4C6H12 (C137H180Cl10N6Ni3O16S3 = 2793.68): triclinic, P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 17.1680(6) Å, b = 19.0600(7) Å, c = 25.3558(9) Å, α = 67.974(2)°, β = 83.133(2)°, γ = 82.959(2)°, V = 7608.9(5) Å3, Z = 2, Dcalcd = 1.219 g cm−3, R1 = 0.0987 (I > 2σ(I)), wR2 = 0.2444 (all data) (CCDC 963502).22.
, a = 17.1680(6) Å, b = 19.0600(7) Å, c = 25.3558(9) Å, α = 67.974(2)°, β = 83.133(2)°, γ = 82.959(2)°, V = 7608.9(5) Å3, Z = 2, Dcalcd = 1.219 g cm−3, R1 = 0.0987 (I > 2σ(I)), wR2 = 0.2444 (all data) (CCDC 963502).22. - The helix twisting angles were defined as the dihedral angle A1–B1–B2–A2 where A1 and A2 are the tert-butyl quaternary carbon atoms of each saloph moiety and B1 and B2 are the centroids of pivotal benzene rings.
- For complexes of [M(salen)]-type compounds with guanidinium and thiourea, see: (a) A. Giacomelli, C. Floriani and G. Perego, J. Chem. Soc., Chem. Commun., 1982, 650–652 RSC; (b) M. B. Ferrari, G. G. Fava and C. Pelizzi, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 901–908 CrossRef.
- For previous examples of guanidinium receptors, see: (a) E. P. Kyba, R. C. Helgeson, K. Madan, G. W. Gokel, T. L. Tarnowski, S. S. Moore and D. J. Cram, J. Am. Chem. Soc., 1977, 99, 2564–2571 CrossRef CAS; (b) J. A. A. de Boer, J. W. H. M. Uiterwijk, J. Geevers, S. Harkema and D. N. Reinhoudt, J. Org. Chem., 1983, 48, 4821–4830 CrossRef CAS; (c) J. W. H. M. Uiterwijk, C. J. van Staveren, D. N. Reinhoudt, H. J. den Hertog Jr., L. Kruise and S. Harkema, J. Org. Chem., 1986, 51, 1575–1587 CrossRef CAS; (d) T. B. Stolwijk, P. D. J. Grootenhuis, P. D. van der Wal, E. J. R. Sudhölter, D. N. Reinhoudt, S. Harkema, J. W. H. M. Uiterwijk and L. Kruise, J. Org. Chem., 1986, 51, 4891–4898 CrossRef CAS; (e) T. W. Bell, A. B. Khasanov, M. G. B. Drew, A. Filikov and T. L. James, Angew. Chem., Int. Ed., 1999, 38, 2543–2547 CrossRef CAS; (f) A. R. Sanford, L. Yuan, W. Feng, K. Yamato, R. A. Flowers and B. Gong, Chem. Commun., 2005, 4720–4722 RSC.
- The association constant was preliminarily determined by competition binding experiments.
- Crystallographic data for [L2Ni3]·2C(NH2)3Cl·1.5CHCl3·1.8DMSO·6.3EtOAc·4H2O (C134.3H184.7Cl6.5N12Ni3O30.4S1.8 = 2917.90): triclinic, P, a = 17.831(3) Å, b = 21.045(4) Å, c = 24.615(4) Å, α = 106.808(3)°, β = 108.969(3)°, γ = 91.349(3)°, V = 8291(2) Å3, Z = 2, Dcalcd = 1.169 g cm−3, R1 = 0.0859 (I > 2σ(I)), wR2 = 0.2134 (all data) (CCDC 963503).22.
- G. M. Sheldrick, SHELXL 97 program for crystal structure refinement, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Renji Okazaki on the occasion of his 77th birthday, so called Kiju in Japan. |
| ‡ Electronic supplementary information (ESI) available: Synthesis of ligands and metal complexes. CCDC 963501–963503. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qi00067b |
| § H2salen = N,N′-disalicylideneethylenediamine; H2saloph = N,N′-disalicylidene-1,2-phenylenediamine. |
| This journal is © the Partner Organisations 2014 |