
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTurning down the heat: catalyst-free, low-temperature chemical degradation of thermoplastic polyurethanes
Adrià
Roig
 ,
Hao
Wang
and
Filip E.
Du Prez
*
,
Hao
Wang
and
Filip E.
Du Prez
*
Polymer Chemistry Research Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, Krijgslaan 291 S4, 9000 Ghent, Belgium. E-mail: Filip.DuPrez@UGent.be
First published on 22nd October 2025
Abstract
Thermoplastic polyurethanes (TPUs) constitute one of the most industrially relevant polymers because of their versatile and tunable properties, which makes them very interesting for a wide range of applications. However, their chemical degradability and recyclability is only achievable through hydrolysis or glycolysis of the urethane moieties, which typically occurs when using catalysts and elevated temperatures, thus resulting in a poorly cost- and energy-efficient process. Herein, we report a novel strategy where variable amounts of chemically degradable β-amino ester (BAE) moieties are incorporated into the main backbone of the thermoplastic urethanes. The amino group in the β-position of the ester was thought to act as an internal, covalently bonded catalyst that not only enables the preparation of materials without the use of external and potentially leachable toxic catalysts, but also the chemical degradation of the resulting TPUs under relatively mild conditions and shorter times via a straightforward transesterification process. The addition of different amounts of BAE groups has been investigated for both aliphatic and aromatic polyurethane thermoplastic backbones. Their chemical, structural and thermal properties have been investigated in-depth, and kinetic studies have been additionally performed with low molar mass model compounds to find the optimal conditions for their chemical degradation. The final aim of this bottom-up molecular design was not only to enable a catalyst-free polyurethane synthesis but also to open new avenues for developing TPUs with an enhanced, cost-effective and energy-efficient degradation process.
Introduction
Over the years, polyurethane (PU) materials have emerged as a crucial class of polymers because of their wide range of household applications such as packaging, furniture, mattresses, sport goods or automotive parts. Their global market size was estimated at more than USD 78 billion in 2023 and is projected to reach USD 105 billion by 2030, representing ca. 4.5% annual growth.1 Among different types of PUs, thermoplastic polyurethanes (TPUs) – mostly linear and rubber-like copolymers usually consisting of alternating soft and hard chain segments – represent a large part of the total PU market (more than USD 2.8 billion).2 Their popularity originates from the high flexibility and versatility of the applied monomers (diols, diisocyanates and chain extenders), which enable an easy fine-tuning of their properties including tensile strength, elasticity, corrosion resistance, ductility and (re)processability according to the desired end-use.3,4 For this reason, TPUs can find applications in a wide array of applications including adhesives, textiles, films, electronic devices, cable jacketing, or even construction and automobiles.5,6However, despite their large-scale production and widespread applications, TPUs usually require the use of toxic metal catalysts (e.g., dibutyltin dilaurate (DBTDL), tin octanoate) for their preparation, and their chemical degradation/recycling is mainly achieved by glycolysis or hydrolysis.7 This process typically requires large amounts of solvents, elevated temperatures, and the addition of external catalysts or harsh pressure conditions, and suffers from poor yields and limited purity of the recovered polyols,8 posing significant environmental concerns due to the generation of chemical waste and energy-intensive conditions. Therefore, in the past two decades, researchers have focused their attention on the investigation of the chemical degradability of TPUs. For instance, in 2011 Wang et al. reported an in-depth study of the potential chemical degradation of TPUs using ethylene glycol (EG) and ethanolamine (EA).9 After a first screening of different conditions, the authors demonstrated an efficient degradation of the TPUs at 170 °C using an EG/EA ratio of 9/2 in the presence of 1 wt% of lithium acetate as a catalyst. Additionally, they could also recover the original diol in relatively high purity after an extraction with benzene, followed by a harsh vacuum distillation process. Recently, the attention has shifted toward the incorporation of other functional groups – either as side chains or in the main backbone – aimed at enhancing the (bio)degradability of the resulting TPUs. These modifications not only slightly improve the recovery of the original monomers but also can eliminate the use of external catalysts, thereby increasing the sustainability of the materials. In this context, Hillmyer and coworkers reported the preparation of chemically recyclable biobased polyurethanes containing ester groups.10 For that, they used in-house synthesized hydroxy te-lechelic poly(β-methyl-δ-valerolactone) (MVL) diols of different molecular weights as polyols for the preparation of different PUs (schematically shown in Fig. 1, top).
 | ||
| Fig. 1 Overview of previous work describing the strategy of introducing labile ester moieties into the backbone of TPUs and this work incorporating β-amino esters (BAE) units with a highlight of their advantages and disadvantages. | ||
Their materials showcased similar properties to those of petrol-derived TPUs. In addition, they could recover the initial MVL in high yields and purity through urethane dissociation and subsequent depolymerization at elevated temperatures and pressure (200–250 °C and 100 mTorr). This concept was further developed by Yan et al. in 2022 who substitute the MVL by δ-caprolactone (CL).8 Their strategy proved to be more effective since the recovery of the CL after thermolysis could be performed at lower temperatures (180 °C) and using lower loadings of catalyst (0.05 mol%). Moreover, higher yields and purities were obtained. More recently, in the frame of covalent adaptable networks, Agarwal and coworkers reported the preparation PU-networks using different amounts of a self-made tertiary amine-containing diol.11 This tertiary amine as a pending chain could act as a covalently-embedded catalyst for the preparation of the materials as well as for the further bond exchange reactions, thereby avoiding the use of toxic metal catalysts and enhancing the sustainability of the resulting materials.
Additionally, the incorporation of ester groups in the main backbone of TPUs also has opened new avenues for the potential biodegradability of these materials.12–15 For example, Gunatillake and coworkers prepared an ester-containing chain extender based on a diol derived from lactide.16 They demonstrated that the biodegradability of the resulting TPUs was enhanced from 0% in 365 days for TPUs without lactide-derived diol to 100% in 180 days for TPUs containing only this diol. In a more recent study, Cao et al. used a polylactic acid diol and polycaprolactone diol to prepare biodegradable TPU-based pressure sensitive adhesives (TPU-PSA).17 Apart from demonstrating excellent PSA characteristics, the materials could be degraded up to 60% already in 60 days, which undoubtedly highlights their potential for renewable and recyclable PSA.
Despite the efficient chemical degradation reports previously mentioned, there is an urgent need to develop TPUs capable of being chemically degraded and recycled at milder conditions in order to establish more simple and cost-effective protocols that allow them to be industrially viable. To address this, we propose here a straightforward synthetic strategy to incorporate chemically degradable β-amino ester (BAE) groups into the main backbone of both aliphatic and aromatic TPUs, using only a bisamine and 2-hydroxyethyl acrylate (HEA) as bulk chemicals on top of the usual chemicals to make conventional TPUs (Fig. 1, bottom). The approach is first evaluated via model studies to explore the potential chemical degradation via a simple transesterification reaction with MeOH. Subsequently, the incorporation of different amounts of BAE groups is thoroughly investigated and the properties of the resulting TPUs explored by means of NMR, FTIR, differential scanning calorimetry (DSC), thermogravimetric analysis (TGA) and size-exclusion chromatography (SEC). Finally, the chemical degradation of all polymers in MeOH at 70 °C is evaluated in detail.
Results and discussion
Chemical degradation study using model compounds
First, a low molecular weight model compound study was conducted to evaluate how the incorporation of β-amino ester (BAE) moieties into aliphatic and aromatic urethane matrices affect their degradability. To this end, two model compounds containing BAE groups and either an aromatic- (M1) or an aliphatic urethane (M2) were synthesized via a straightforward, two-step synthetic protocol with quantitative yield and only making use of bulk chemicals (see SI for experimental details and Fig. 2 for chemical structures) and their purity was subsequently checked by NMR (Fig. S1–S8). In order to use relatively mild conditions for their chemical degradation, M1 or M2 were dissolved in MeOH in the absence of catalyst and heated at 70 °C. To determine the degradation kinetics, aliquots were taken at different times and analyzed using 1H NMR (Fig. 2A for M1 and Fig. S18 for M2). After 150 min, the NMR-spectra revealed the complete disappearance of the singlet at 4.34 ppm, corresponding to the protons of the methylene groups of M1 (blue dot), while two signals at 4.25 and 3.83 ppm corresponding to the ethylene unit of the formed hydroxy-urethane compound (green dots) could be observed. Additionally, the presence of an intense singlet at 3.67 ppm, attributed to the formed methyl ester (red dot), further confirmed the transesterification reaction and thus the successful conversion of M1. Similar results were obtained for the aliphatic urethane M2 (see Fig. S18), yet, complete degradation was only achieved after 6 hours (see also Fig. 2B). To test the effect of the covalently-bonded tertiary amine, a model compound with an ester group in the absence of an amino group (E2) was synthesized (see Fig. S13 and S14). When testing the potential transesterification in the same conditions, only 2% of the expected methyl ester formed was observed after 3 h (Fig. 2B, black squares and Fig. S19), highlighting the strong catalytic effect of the embedded tertiary amine. Notably, in all the performed tests, no NMR-signals corresponding to a methyl urethane, which would be generated from the potential nucleophilic attack of the MeOH to the urethane, were observed, suggesting an excellent selectivity of the alcohol groups towards the transesterification reaction rather than the transcarbamoylation. This selectivity is attributed to the amine group in the β-position of the ester, which can form a hydrogen-bonded intermediate transition state between the ester and the alcohol, thereby favoring the transesterification reaction (Scheme 1).18,19 On the other hand, if the amine would be in close proximity to the urethane, then transcarbamoylation reaction would be much more favored resulting in a much lower selectivity.19 | ||
| Fig. 2 (A) Stacked 1H NMR spectra in CDCl3 of the degradation of M1 with boiling MeOH at different reaction times. (B) Fraction of the initial reagent as a function of time for M1, M2, R1, R2, and E2 calculated from 1H NMR spectra. | ||
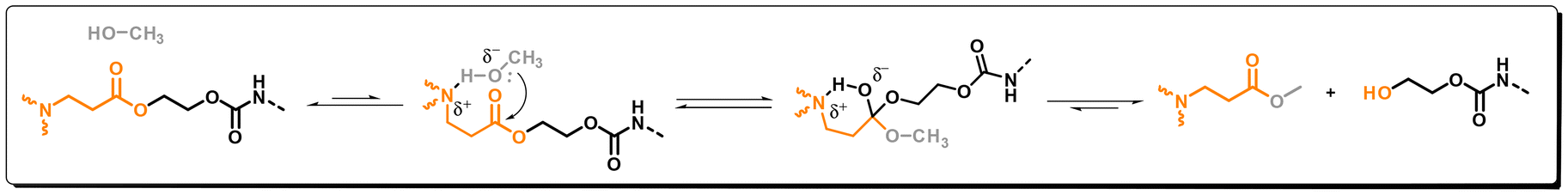 | ||
| Scheme 1 Proposed mechanism of the transesterification reaction taking place with the BAE-containing compound in the presence of methanol. | ||
To further prove that the transcarbamoylation is not taking place at the used conditions, two reference aromatic (R1) and aliphatic (R2) low molecular weight urethane model compounds without the BAE functionality were synthesized (Fig. 2 and S9–S12) and subjected to the same tests with the addition of an organic base (5 mol% of triethylamine (NEt3)) in order to create a similar basic environment as in the case of M1 and M2 (Fig. S20 and S21). The 1H NMR spectra revealed no changes even after 6 hours in both cases, confirming that the transcarbamoylation reaction is not taking place at 70 °C in the presence of a base. This result is expected as NEt3 is reported to be a weak catalyst for this type of reaction.20,21
These results are a first indication that the incorporation of BAE moieties into a urethane matrix could become a promising strategy of novel chemically degradable TPUs.
Design and characterization of the BAE-containing thermoplastic urethanes
For the design of the thermoplastic polyurethanes (TPU) materials, a BAE-containing diol (BAE-OH) was first synthesized through a fast, quantitative yielding aza-Michael addition between bulk N,N-dimethyl-1,6-hexanediamine and HEA (Fig. 3A), which high purity was confirmed by 1H and 13C NMR (Fig. S15 and S16). Then, different amounts of BAE-OH (1–100 mol%) were combined in a solvent-free way with tetraethylene glycol (TEG) and reacted with readily available 1,6-hexamethylene diisocyanate (HDI) or tolylene-2,4-diisocyanate (TDI) in a conventional oven at 80 °C for 24 h to obtain a library of TPU materials containing BAE groups in the main backbone (Fig. 3 and Table S1, see SI for experimental details). Those polymers are further referred to as HDI-x%BAE. | ||
| Fig. 3 Schematic overview of the preparation of the catalyst-free TPU materials highlighting the most relevant functional groups. Mixtures of BAE-OH and TEG reacted with HDI or TDI were put in a conventional oven at 80 °C for 24 h. | ||
Importantly, this reaction proceeded without adding an external catalyst, as the tertiary amine moiety present in BAE-OH efficiently catalyzes the polyaddition of alcohols and isocyanates. Indeed, even 1 mol% of covalently incorporated BAE was sufficient to promote the reaction, which is consistent with the fact that tertiary amines such as NEt3 or pyridine are effective catalysts for polyurethane synthesis.11,22 Notably, the absence of external (and potentially toxic) catalysts such as DBTDL or triazabicyclodecene (TBD), together with the solvent-free synthesis protocol, further underscores the sustainability advantages of the strategy reported herein for the manufacturing of industrially relevant TPUs.
To evaluate the completeness of the reaction and prove that the covalently-bonded amine is an effective internal catalyst for the polyaddition reaction, FTIR spectra of the different TPUs were recorded (Fig. 4A and Fig. S22, S23).
 | ||
| Fig. 4 (A) Overlayed FTIR spectra of BAE-OH and all the HDI-derived TPUs prepared. (B) Stacked 1H NMR spectra in DMF-d7 of HDI-derived TPUs containing 0%- (black), 10%- (orange) and 100% (blue) of BAE-OH. | ||
The complete vanishment of the band corresponding to the free isocyanate group at around 2150 cm−1 together with the appearance of the bands at 1681 and 1531 cm−1, attributed to the stretching and bending vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H of the urethane group, indeed confirmed the successful synthesis of the TPU materials. Additionally, the band corresponding to the ester group at around 1732 cm−1 gradually decreased with a lower BAE content. Similar results were obtained for aromatic TDI-derived samples, thereby confirming the incorporation of BAE moieties into both aliphatic and aromatic TPU backbones.
O and N–H of the urethane group, indeed confirmed the successful synthesis of the TPU materials. Additionally, the band corresponding to the ester group at around 1732 cm−1 gradually decreased with a lower BAE content. Similar results were obtained for aromatic TDI-derived samples, thereby confirming the incorporation of BAE moieties into both aliphatic and aromatic TPU backbones.
1H NMR spectroscopy was then used to characterize the structure of all TPU polymers (see Fig. S24 and S25). For comparison purposes, Fig. 4B displays the 1H NMR spectra of HDI-derived TPUs with a 100%, 25% and 0% (reference) content of BAE-OH. As can be seen in this figure, the appearance of the peak between 6.9–7.1 ppm corresponding to the –NH– proton suggested the successful synthesis of polyurethanes. As expected, the peaks at around 2.60–2.25 ppm, assigned to the α- and β-protons of the ester, respectively showed decreased intensity with lower BAE content.
In the case of TDI-derived samples, a similar trend can be observed. However, when increasing the amount of BAE-OH, the content of the tertiary amines also increases which, together with the inherent high reactivity of TDI, generated a large exotherm during the mixing. This reaction possibly altered the stoichiometry and caused the occurrence of multiple side reactions, such as the formation of isocyanurates, ureas or allophanates, which explain the complex 1H NMR spectra of TDI-25%BAE and TDI-100%BAE (Fig. S25). In order to avoid this exotherm, we considered the use of a solvent during the preparation for the dilution of the reaction mixture. Therefore, TDI-100%BAE was prepared using a similar protocol but adding 50 mL of THF as solvent at 50 °C. As can be seen in the corresponding 1H NMR analysis (Fig. S26), the absence of acrylate signals confirms that the use of solvent could indeed be an alternative but industrially less attractive strategy to avoid such side reactions.
The thermal properties of the TPU materials were analyzed by DSC and TGA. First, DSC analysis was performed to evaluate the thermal transitions of both TPU series (HDI-derived and TDI-derived). The second heating curves as a function of temperature for HDI-derived and TDI-derived materials are shown in Fig. 5A and D, respectively whereas the main data extracted is reported in Tables 1 and 2. These analyses show that increasing the content of BAE-OH in the TPUs does not have a significant effect on the glass transition temperature (Tg) of the HDI-derived polymers (Tgs of −15 to −13 °C). Only when more than 25 mol% BAE is used (HDI-100%BAE), the Tg shows a slight decrease, probably due to the longer aliphatic chain of BAE-OH compared to TEG as well as to the lower molecular weight (Mw) achieved in these polymers (vide infra).
 | ||
| Fig. 5 (A) Thermograms (second heating) obtained from DSC analysis of all HDI-derived TPU-like polymers. (B) TGA plots under N2 atmosphere of all HDI-derived TPU materials with a temperature ramp from 30 to 800 °C and a heating rate of 10 °C min−1. (C) SEC traces of all HDI-derived TPU-like polymers using DMAc as a solvent. (D) Thermograms (second heating) obtained from DSC analysis of all TDI-derived TPU-like polymers. (E) TGA plots under N2 atmosphere of all TDI-derived TPU-like polymers with a temperature ramp from 30 to 800 °C and a heating rate of 10 °C min−1. (F) SEC traces of all TDI-derived TPU-like polymers using DMAc as a solvent. | ||
| Sample | T g (°C) | T m (°C) | ΔHc (J g−1) | T 5% (°C) | M n (kDa) | M w (kDa) | Đ |
|---|---|---|---|---|---|---|---|
| a T g determined from DSC analysis performed at a heating rate of 10 °C min−1. b T m determined from DSC analysis at a heating rate of 10 °C min−1. c Enthalpy determined from the integration of the endothermic peaks on the DSC thermograms. d TGA onset temperatures after 5% weight loss. e Number average molecular weight determined from SEC using DMAc as a solvent. f Weight average molecular weight determined from SEC using DMAc as a solvent. g Dispersity obtained using SEC. | |||||||
| HDI-ref | −15.3 | 82.8 | 35.5 | 302 | 25.8 | 52.1 | 2.13 |
| HDI-1%BAE | −14.4 | 85.1 | 34.7 | 314 | 40.3 | 90.6 | 2.25 |
| HDI-2.5%BAE | −13.6 | 86.6 | 9.5 | 308 | 37.1 | 82.6 | 2.23 |
| HDI-5%BAE | −13.9 | 85.0 | 9.0 | 297 | 39.0 | 87.9 | 2.25 |
| HDI-10%BAE | −13.3 | 83.5 | 1.9 | 295 | 39.3 | 93.8 | 2.39 |
| HDI-25%BAE | −13.3 | — | — | 267 | 34.9 | 74.2 | 2.13 |
| HDI-100%BAE | −15.1 | — | — | 236 | 19.7 | 50.5 | 2.56 |
| Sample | T g (°C) | T m (°C) | ΔHc (J g−1) | T 5% (°C) | M n (kDa) | M w (kDa) | Đ |
|---|---|---|---|---|---|---|---|
| a T g determined from DSC analysis performed at a heating rate of 10 °C min−1. b T m determined from DSC analysis at a heating rate of 10 °C min−1. c Enthalpy determined from the integration of the endothermic peaks of the DSC thermograms. d TGA onset temperatures after 5% weight loss. e Number average molecular weight determined from SEC using DMAc as a solvent. f Weight average molecular weight determined from SEC using DMAc as a solvent. g Dispersity obtained using SEC. | |||||||
| TDI-ref | 41.8 | — | — | 278 | 27.8 | 50.5 | 1.82 |
| TDI-1%BAE | 41.5 | — | — | 280 | 25.1 | 48.9 | 1.95 |
| TDI-2.5%BAE | 41.2 | — | — | 284 | 16.6 | 32.7 | 1.97 |
| TDI-5%BAE | 39.9 | — | — | 280 | 16.0 | 31.9 | 1.99 |
| TDI-10%BAE | 39.1 | — | — | 272 | 13.8 | 26.4 | 1.92 |
| TDI-25%BAE | 30.0 | — | — | 240 | 8.0 | 15.7 | 1.97 |
| TDI-100%BAE | 11.6 | — | — | 200 | 4.0 | 8.7 | 2.17 |
Interestingly, varying the content of BAE groups does have a clear influence on the crystallization of the HDI-derived TPU polymers. While the reference polymer (HDI-ref) shows a clear semi-crystalline behavior with a melting peak (Tm) at around 83 °C and an enthalpy (ΔH) of 36 J g−1, the use of only 2.5 mol% of BAE-OH maintains the same Tm but drastically decreases the enthalpy to 10 J g−1. Further increasing the BAE content resulted in a decrease of ΔH up to a certain threshold (between 10 and 25% of BAE-OH content), for which the materials become completely amorphous due to the relatively high content of the long chain of BAE-OH and therefore the lower content of hard segments.
Notably, all semi-crystalline HDI-derived materials displayed a cold crystallization effect, which is also observed in ether-containing TPUs.23 This effect diminishes with increasing BAE-OH content, which can be attributed to the longer aliphatic chain of BAE-OH (relative to TEG) and the presence of pendant methyl groups on the tertiary amine functionality. These structural features are expected to hinder the hard-phase formation in the PU materials, thereby affecting crystallization and leading to lower melting enthalpies.24,25
In the case of TDI-derived samples (Fig. 5D and Table 2), increasing the BAE content from 1 to 10 mol% did not have a significant impact on the Tg-value of around 40 °C. However, when 25 mol% and 100 mol% of BAE-OH were used, the Tg dramatically decreased to 30 and 12 °C, respectively, which can be attributed to the lower Mw achieved (vide infra). The huge drop of Tg can be also be ascribed to the occurrence of side reactions, as previously stated. It is also worth mentioning that all TDI-derived polymers displayed an amorphous behavior (even with 1 mol% of BAE-OH), which can be mainly attributed to the use of an isomeric mixture of 2,4 and 2,6-TDI for the preparation of the TPUs.
TGA analyses were then performed to evaluate the thermal stability of all the TPU materials (see Fig. 5B and Table 1 for HDI-derived samples and Fig. 5E and Table 2 for TDI-derived polymers). Both for aliphatic and aromatic TPUs, an increase of BAE groups resulted in a gradual decrease of thermal stability. Notably, a significant decrease in the thermal stability was only observed for samples with BAE contents higher than 10 mol%.
When comparing HDI- and TDI-derived TPUs, the former samples showed T5% values higher than the latter ones, due to the higher thermal stability of HDI compared to TDI.26 Generally, all TPUs showed a main degradation step in DTGA curves (Fig. S27), which consists in the thermal decomposition of the urethane linkage. Additionally, a small peak at around 460 °C appears, which is attributed to the dissociation of C–C bonds. However, samples with 100 mol% of BAE-OH (HDI-100%BAE and TDI-100%BAE) showcased a different and more complex degradation pattern, probably due to the occurrence of the side reaction previously mentioned. However, it is worth mentioning that TPUs with less than 10 mol% of BAE moieties showcased remarkable thermal stability (up to 310 °C), suggesting that the incorporation of small amounts of such labile groups do not alter the thermal properties of the polymers.
Finally, the Mw of the prepared TPUs was determined by SEC (Fig. 5C, F and Tables 1, 2). As observed, HDI-derived TPUs displayed monomodal SEC traces with Mw values ranging from 50 to 94 kDa and dispersities (Đ) always below 2.6. Particularly, HDI-derived TPUs containing 1 to 10 mol% of BAE groups displayed the highest values. These results demonstrate that, despite of the solvent-free synthesis and the absence of an external catalyst, incorporating small amounts of labile BAE moieties still enables to obtain TPUs with relatively high Mw. However, increasing the content of BAE groups above 25 mol% resulted in a decrease of molecular weight. This can be attributed to the high concentration of tertiary amines, which catalyze the isocyanate–alcohol reaction, producing a pronounced exotherm that promotes side reactions such as allophanate formation.27,28 In addition, the elevated temperature can induce dissociation of β-amino ester groups, yielding free amines that subsequently react to form urea linkages. This could also be confirmed with the results obtained from TDI-derived materials. In this case, despite the almost monomodal SEC curves, the obtained Mws were significantly lower than those of their HDI-counterparts. These materials showed the same trend as for the HDI samples but in a much clearer way. Specifically, when the BAE content is too high (TDI-100%BAE), Mw drops to 8 kDa, due to the occurrence of stoichiometric imbalances and side reactions caused by the large exotherm during mixing, which leads to the dissociation of BAE moieties. For this, we refer to acrylate-related signals between 6.5 and 5.7 ppm (Fig. S25). These findings were also well in accordance with the NMR, DSC and TGA results previously obtained. For this reason, TPUs containing more than 25 mol% of BAE were not further investigated.
Chemical degradation of the TPUs
As reported in the literature, the use of BAE bonds can enable the occurrence of transesterification reactions at high temperatures.29,30 Therefore, the incorporation of these groups in TPUs was expected to allow their potential degradation under milder conditions than the ones currently used to degrade this type of polymers.9 For this purpose, all the TPU materials were subjected to the same degradation conditions as the model compound study (vide supra), which consisted in using MeOH at 70 °C for a certain period of time. Then, SEC analyses were performed on the degraded samples and compared to the original TPUs (Fig. 6, Fig. S28, S29 and Table S2). | ||
| Fig. 6 (A) SEC traces of the original TDI-1%BAE (black) and the sample degraded for 5 h (red) and 24 h (dashed orange). (B) SEC traces of the original HDI-2.5%BAE (black) and the sample degraded for 5 h (red) and 24 h (dashed orange). | ||
Notably, all TDI-derived TPUs could be degraded within 5 h under the reported mild conditions. It should be noted that the introduction of only 1 mol% of BAE groups was sufficient to chemically degrade the TPUs, as the Mw of this material shifted significantly towards lower molecular weights (Fig. 6A). Nevertheless, keeping the samples in MeOH at 70 °C for longer times (24 h) did not further decrease the Mw of the polymers.
In contrast, for HDI-derived materials, a BAE content of at least 2.5 mol% was needed to be able to degrade the TPUs to approximately half of their Mw. Again, extending the degradation time to 24 h did not show any differences in the Mw values. It is worth mentioning that aliphatic TPU materials showed higher Mw compared to the aromatic ones, which undoubtedly can affect the potential degradation.
However, it could be concluded that even TPUs containing only 2.5 mol% of BAE moieties and Mw up to 94 kDa could be easily (partially) degraded by using mild conditions without the use of any external, toxic catalyst.
To confirm that the degradation takes place through the transesterification reaction and not through a transcarbamoylation pathway, HDI-ref and TDI-ref (without BAE groups) were subjected to the same protocol (see Fig. S28A and S29A). Even after 24 h, these polymers did not degrade at all, indicating that the presence of BAE groups is indeed essential to initiate degradation of these TPUs at these conditions.
Finally, all TPUs were kept in MeOH at room temperature for 24 h to demonstrate that no degradation occurs at ambient conditions, as the transesterification reaction should not take place at service temperatures. As can be seen in Fig. S30, only TPUs with more than 25 mol% of BAE groups exhibited a slight degradation, probably caused by the their lower Mw values.
Conclusions
In this study, we demonstrated a straightforward protocol to incorporate chemically degradable β-amino ester moieties into the main backbone of thermoplastic polyurethanes. First, the chemical degradation of urethanes with BAE groups in MeOH at 70 °C was assessed through model compounds. This demonstrated that the presence of BAE moieties could indeed induce the transesterification reaction at 70 °C without the use of external catalysts, fully degrading the compounds in less than 150 min for BAE-containing aromatic urethanes and in less than 360 min for BAE-containing aliphatic urethanes. Subsequently, a straightforward and scalable synthetic protocol using two bulk chemicals was used to obtain a BAE-containing diol (BAE-OH) in high yield. Then, different amounts (0–100 mol%) of BAE-OH were successfully incorporated into the main backbone of TPUs through a solvent- and catalyst-free preparation protocol. We then demonstrated that the use of low contents of BAE groups (<10 mol%) did not alter the Tg values either of aliphatic-derived TPUs (Tgs ≈ −13 °C) or aromatic-derived TPUs (Tgs ≈ 40 °C) but did have an influence on the crystallinity behavior of the aliphatic TPUs, since high contents of BAE groups resulted in fully amorphous materials. All TPUs showed good thermal stability (T5% > 270 °C), regardless of the type of urethane and concentration of BAE. This strategy also allowed us to obtain TPUs with relatively high Mw (up to 93 kDa for aliphatic and up to 50 kDa for aromatic TPUs), particularly when less than 10 mol% of BAEOH was used.Finally, the incorporation of more than 2.5 mol% of BAE groups into the main backbone of TPUs allowed for a successful chemical degradation of all polymers in MeOH at 70 °C, which highlights the potential use of this strategy on an industrial scale, in order to have a cost-effective and energy-efficient degradability process for TPUs without modifying their thermal and structural properties.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: details about instrumentation, synthesis and experimental procedures, NMR-, FTIR-, SEC-, ESI-MS- spectra, as well as DSC and TGA thermograms. See DOI: https://doi.org/10.1039/d5py00881f.Acknowledgements
F. D. P. acknowledges the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme 101021081 (ERC-AdG-2020, CiMaC-project). A. R. acknowledges the Bijzonder Onderzoeksfonds (BOF) of Ghent University for his postdoctoral fellowship (01P02924). The NMR expertise center (Ghent University) is also acknowledged for providing support and access to NMR infrastructure funded by a Research Foundation Flanders (FWO I006920N) and the Bijzonder Onderzoeksfonds (BOF.BAS.2022.0023.01). The authors thank Bernhard De Meyer for technical support as well as Dr Nezha Badi, Dr Aitor Hernández, Loc Tan Nguyen and Jens Van Hoorde for the fruitful discussions held during the duration of the study.References
- Grand View Research, Polyurethane Market Size, Share & Growth Report 2023, Grand View Research, San Francisco, 2023 Search PubMed.
- Thermoplastic Polyurethane Global Market Report, Thermoplastic Polyurethane Global Market Report 2023, New York, 2023 Search PubMed.
- R. H. Aguirresarobe, S. Nevejans, B. Reck, L. Irusta, H. Sardon, J. M. Asua and N. Ballard, Prog. Polym. Sci., 2021, 114, 101362 CrossRef CAS.
- J. Zhao, J. Zhu, J. Zhang, Z. Huang and D. Qi, React. Funct. Polym., 2024, 199, 105886 CrossRef CAS.
- A. Mouren and L. Avérous, Chem. Soc. Rev., 2023, 52, 277–317 RSC.
- A. Pierrad, S. F. Melo, C. Oury, C. Detreumbleur and C. Jérôme, Eur. Polym. J., 2025, 233, 113985 CrossRef.
- D. Simón, A. M. Borreguero, A. de Lucas and J. F. Rodríguez, Waste Manage., 2018, 76, 147–171 CrossRef PubMed.
- Q. Yan, C. Li, T. Yan, Y. Shen and Z. Li, Macromolecules, 2022, 55, 3860–3868 CrossRef CAS.
- X. Wang, H. Chen, C. Chen and H. Li, Fibers Polym., 2011, 12, 857–863 CrossRef CAS.
- D. K. Schneiderman, M. E. Vanderlaan, A. M. Mannion, T. R. Panthani, D. C. Batiste, J. Z. Wang, F. S. Bates, C. W. Macosko and M. A. Hillmyer, ACS Macro Lett., 2016, 5, 515–518 CrossRef CAS PubMed.
- L. Schwarzer and S. Agarwal, Macromol. Chem. Phys., 2024, 225, 2400072 CrossRef CAS.
- G. T. Howard, Int. Biodeterior. Biodegrad., 2002, 49, 245–252 CrossRef CAS.
- B. F. Pierce, A. H. Brown and V. V. Sheares, Macromolecules, 2008, 41, 3866–3873 CrossRef CAS.
- H.-Y. Mi, X. Jing, B. S. Hagerty, G. Chen, A. Huang and L.-S. Turng, Mater. Des., 2017, 127, 106–114 CrossRef CAS.
- A. Jaisingh, M. W. Halloran, B. S. Rajput and M. D. Burkart, ACS Appl. Polym. Mater., 2024, 6, 9724–9734 CrossRef CAS.
- L. Tatai, T. G. Moore, R. Ahikari, F. Malherbe, R. Jayasekara, I. Griffiths and P. A. Gunatillake, Biomaterials, 2007, 28, 5407–5417 CrossRef CAS PubMed.
- Y. Cao, X. Wang, L. Sun, J. Zhang, S. Hu and H. Yu, J. Appl. Polym. Sci., 2025, 142, e57008 CrossRef CAS.
- M. Delahaye, F. Tanini, J. O. Holloway, J. M. Winne and F. E. Du Prez, Polym. Chem., 2020, 11, 5207–5215 RSC.
- A. Hernández, H. A. Houck, F. Elizalde, M. Guerre, H. Sardon and F. E. Du Prez, Eur. Polym. J., 2022, 168, 111100 CrossRef.
- B. Rhone and V. Semetey, Synlett, 2017, 28, 2004–2007 CrossRef CAS.
- C. Bakkali-Hassani, D. Berne, V. Ladmiral and S. Caillol, Macromolecules, 2022, 55, 7974–7991 CrossRef CAS.
- L. Ruiduan, L. Ling, L. Yanjie, W. Ben, Y. J. Jun and Z. Jibo, Top. Chem. Mater. Eng., 2018, 1, 54–57 Search PubMed.
- Y. Deng, S. Li, J. Zhao, Z. Zhang, J. Zhang and W. Yang, RSC Adv., 2014, 4, 43406 RSC.
- K. Kojo, S. Nakamura and M. Furukawa, Polymer, 2004, 45, 8147–8152 CrossRef.
- K. Kojio, M. Furukawa, Y. Nonaka and S. Nakamura, Materials, 2010, 3, 5097–5110 CrossRef CAS PubMed.
- J. C. Q. Amado, J. Res. Updates Polym. Sci., 2019, 8, 85–93 Search PubMed.
- A. Lapprand, F. Boisson, F. Delolme, F. Méchin and J.-P. Pascault, Polym. Degrad. Stab., 2005, 90, 363–373 CrossRef CAS.
- M. Spirkova, M. Kubín and K. Dusek, J. Macromol. Sci., Chem., 1987, 10, 1151–1166 Search PubMed.
- C. Taplan, M. Guerre and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 9140–9150 CrossRef CAS PubMed.
- H. Kassem, E. H. Samat, L. Imbernon and F. E. Du Prez, Adv. Funct. Mater., 2024, 34, 2409263 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |