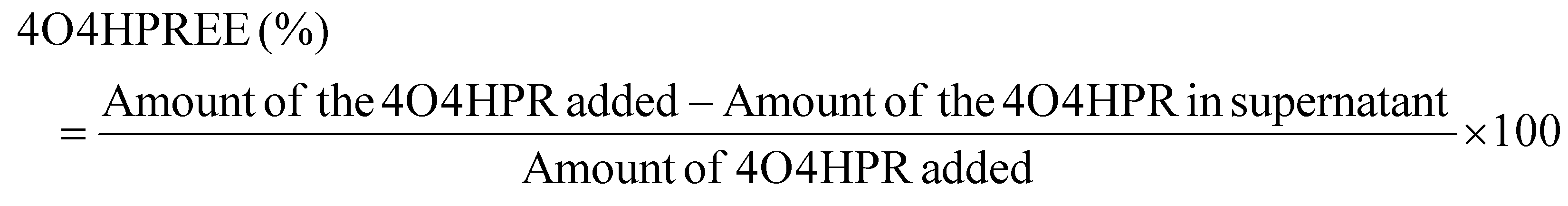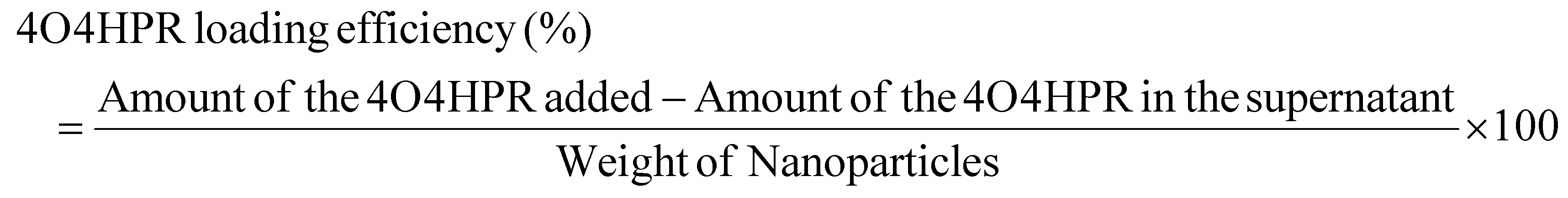DOI:
10.1039/D4NR00642A
(Paper)
Nanoscale, 2024,
16, 16075-16088
Encapsulation of 4-oxo-N-(4-hydroxyphenyl) retinamide in human serum albumin nanoparticles promotes EZH2 degradation in preclinical neuroblastoma models†
Received
14th February 2024
, Accepted 18th July 2024
First published on 1st August 2024
Abstract
Neuroblastoma is the most prevalent and aggressive solid tumor that develops extracranially in children between the ages of 0–14 years, which accounts for 8–10% of all childhood malignancies and ∼15% of pediatric cancer-related mortality. The polycomb repressive complex 2 (PRC2) protein, EZH2, is overexpressed in neuroblastoma and mediates histone H3 methylation at lysine 27 (K27) positions through its methyl transferase activity and is a potential epigenetic silencer of many tumor suppressor genes in cancer. Phosphorylation of EZH2 decreases its stability and leads to proteasomal degradation. The 4-oxo-N-(4-hydroxyphenyl) retinamide (4O4HPR) promotes EZH2 degradation via activation of PKC-δ, but its limited solubility and physiological instability limit its application. In the current study, the encapsulation of 4O4HPR in Human Serum Albumin Nanoparticles (HSANPs) enhanced the solubility and physiological stability of the nanoformulation, leading to improved therapeutic efficacy through G2-M cell cycle arrest, depolarization of mitochondrial membrane potential, generation of reactive oxygen species and caspase 3 mediated apoptosis activation. The molecular mechanistic approach of 4O4HPR loaded HSANPs has activated caspase 3, which further cleaves PKC-δ into two fragments wherein the cleaved fragment of PKC-δ possesses the kinase activity that phosphorylates EZH2 and decreases the protein stability leading to its further ubiquitination in SH-SY5Y cells. Co-immunoprecipitation experiments revealed the direct interaction between PKC-δ and EZH2 phosphorylation, followed by ubiquitination. Moreover, 4O4HPR loaded HSANPs demonstrated improved in vivo biodistribution, greater dispersibility, and biocompatibility and exhibited enhanced protein instability and degradation of EZH2 in the neuroblastoma xenograft mouse model.
Introduction
Neuroblastoma is the most prevalent and fatal tumor of infancy, a cancer of the peripheral sympathetic nervous system arising from the neural crest being the most prevalent extra-cranial solid tumor in infants and children, accounting for 8–10% of all childhood tumors. A wide range of clinical manifestations, from treatment-resistant cancers with poor patient survival to spontaneous regression without any medical intervention, characterizes it. It is responsible for 15% of all cancer-related paediatric fatalities.1,2 Chemotherapy, radiation therapy, and combinatorial therapy are some of the current and traditional treatments for neuroblastoma. These treatments are limited by the non-specificity of the medications, low bioavailability, and numerous side effects.3 Also, the recently developed advanced treatments have their drawbacks in relapse of neuroblastoma after surgical resection.4 Targeted therapies require one or more drugs in combination that target different molecular pathways, which may also lead to recurrence and relapse of the disease.5 Due to substantial treatment resistance and tumor phenotypic heterogeneity, epigenetic therapies have recently come to light as a potential strategy to treat neuroblastoma.6 Recently, epigenetic therapies have been developed targeting DNA methylation and histone modifications such as histone methylation, histone acetylation, non-coding RNA, and mRNA methylation.7 The epigenetic-targeted agents have shown therapeutic efficacy in solid tumors and clinical activity in haematological malignancies.8 The various PcG proteins combine to create the Polycomb repressive complexes 1 and 2 (PRC1 and PRC2, respectively), functionally separate complexes belonging to two major families. The different complexes of each PRC family have unique catalytic activity PRC1 complexes have E3 ligase activity, where monoubiquitinated Histone H2A at lysine 119 position is its most characterised substrate, whereas PRC2 complexes contain the methyltransferase activity that generates the di- or trimethylated form of lysine 27 on histone H3 (H3K27me2 or H3K27me3).9,10 The Polycomb Repressive Complex 2 member, EZH2, in association with its binding partners, SuZ12 (Suppressor of Zeste 12) and EED (Embryonic Ectoderm Development), tri-methylates histone H3 at lysine 27 position (H3K27me3), that serves as an epigenetic hallmark in many tumors.11 EZH2 contributes to crucial biological processes such as cell fate determination, cell cycle control, cell differentiation, senescence, and the onset and spread of cancer.12 It is reported to be highly expressed in neuroblastoma. Moreover, studies have demonstrated that its overexpression is linked to a worse prognosis in neuroblastoma patients.13 A study demonstrated that inhibition of EZH2 using GSK343 in neuroblastoma has significantly decreased proliferation, motility, viability, and tumor growth in vivo.14 EZH2 is often overexpressed in solid tumors, and the increased EZH2 levels correlate with epithelial-mesenchymal transition (EMT).15 Thus, a promising need exists to inhibit EHZ2 directly or indirectly by targeting its upstream regulators. The post-translational modifications of EZH2 play an important role in regulating its downstream functions and silencing target genes. Phosphorylation of EZH2 is so crucial that it controls its stability and biological functions. This study demonstrates that many sites on EZH2 get phosphorylated at serine and threonine residues that inhibit the stability of the protein,16 making it susceptible to polyubiquitination and further degradation. According to a recent study, CDK1-mediated pThr345-EZH2 and pThr487-EZH2 promote ubiquitination and consequent degradation in breast cancer.17 Such phosphorylation is also mediated by S/T kinases, one of which is PKC-δ. A study on lymphoid leukemia demonstrates the translocation of PKC-δ into the nucleus upon treatment of fenretinide (4HPR), thereby inducing apoptosis.18 PKC-δ is known to sensitize the neuroblastoma cells (ACN, GI-MEN, SH-SY-5Y, and SK-N-BE-2C) to L-buthionine sulfoxime and etoposide, thereby inducing the ROS production leading to DNA damage.19 Therefore, we hypothesize that there may be an as-yet-undiscovered interaction between PKC-δ and EZH2 in neuroblastoma. A promising anticancer effect was demonstrated by 4-oxo-N-(4-hydroxyphenyl)retinamide (4-oxo-4-HPR or 4O4HPR), a recently identified20 and an active polar metabolite of the synthetic retinoid N-(4-hydroxyphenyl)retinamide, (4-HPR), with two different independent mechanisms of action21 one causing the mitotic arrest leading to marked G2-M phase arrest of cell cycle in A2780, an ovarian cancer cell line20 and other inducing the apoptosis in cancer cells.22 However, 4O4HPR has poor water solubility23 and physiological instability, preventing it from being used as a therapeutic aid. These limitations can be circumvented by choosing a compatible nano-carrier to encapsulate 4O4HPR. The solubility of hydrophobic drugs increases in the hydrophilic environments when they bind to the hydrophobic patches present in Human Serum Albumin nanoparticles.24 The advantages of human serum albumin (HSA) nanoparticles include covalent alterations with targeted ligands, ease of manufacturing, and biocompatibility. Clinical trials have effectively employed drug delivery strategies based on HSA. For instance, in 2005, the FDA authorised Abraxane, an albumin-nanoformulation of paclitaxel, to treat metastatic breast cancer.25,26 The impact of doxorubicin-loaded human serum albumin (HSA) nanoparticles was investigated in neuroblastoma cells expressing ABCB127 and many other neuroblastoma treatments.
The objective of the current work is to increase the therapeutic effectiveness of 4O4HPR by loading into HSA nanoparticles. The elevated ROS production, depolarised mitochondrial membrane potential, and enhanced apoptosis evident through caspase-3 upregulation demonstrated the neuroblastoma regression. The western blot and quantitative PCR analysis have revealed the significant upregulation of PKC-δ and downregulated EZH2 and its immediate target, trimethyl H3K27. The co-immunoprecipitation studies showed the interaction between PKC-δ and EZH2. The use of MG132, ubiquitin proteasomal pathway inhibitor, followed by western blotting, revealed ubiquitination and degradation of EZH2 via UPP only. PKC-δ siRNA for its knockdown verified the positive correlation between PKC-δ and EZH2. Also, the in vivo biodistribution of 4O4HPR loaded HSANPs in mice has shown better biocompatibility and negligible toxicity. Thus, by upregulating PKC-δ and downregulating the highly expressed epigenetic player EZH2, our nanoformulation has specifically targeted the polycomb-mediated epigenetic mechanism for potential anti-cancer therapy in neuroblastoma preclinical models.
Experimental
Materials
Human serum albumin was procured from Reliance Life Science (A2153), and Ethanol was procured from Merck (1.00983.0511). MTT dye (MB186), Glutaraldehyde (MB222), Propidium Iodide (PI) (TC252) and were purchased from Hi-Media. Fetal Bovine Serum (FBS) (10270106), Penicillin Streptomycin Solution (15140122) from Gibco, and DMEM (12-604F), Trypsin-EDTA (BE02-007E) were purchased from Lonza. Antibodies used were purchased from Affinity Biosciences. Other used chemicals and solvents are AR grade and purchased from local Indian manufacturers.
Synthesis of 4O4HPR loaded HSANPs
The Human Serum Albumin nanoparticles were synthesised using the Desolvation technique in which 20% HSA was diluted to 2% with MilliQ and titrated to 8.0 pH using 0.1 N NaOH. Under constant stirring conditions, @800 rpm, 90% ethanol was added dropwise until HSA was desolvated (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of HSA and ethanol). Then, the desolvated HSA was incubated with 4O4HPR drug dissolved in Ethanol (Merck) for 2 hours in a 1:10 ratio. After two hours, the crosslinking of the nanoparticles was facilitated using 8% glutaraldehyde under constant stirring conditions for 24 hours. Ethanol in the reaction mixture was further separated by vacuum rotary evaporation at 37 °C. After three cycles of centrifugation at 15000 rpm for 30 minutes to remove any remaining impurities, the pellet was disseminated in the original reaction volume with MilliQ, and the nanoparticles were lyophilised.
:2 ratio of HSA and ethanol). Then, the desolvated HSA was incubated with 4O4HPR drug dissolved in Ethanol (Merck) for 2 hours in a 1:10 ratio. After two hours, the crosslinking of the nanoparticles was facilitated using 8% glutaraldehyde under constant stirring conditions for 24 hours. Ethanol in the reaction mixture was further separated by vacuum rotary evaporation at 37 °C. After three cycles of centrifugation at 15000 rpm for 30 minutes to remove any remaining impurities, the pellet was disseminated in the original reaction volume with MilliQ, and the nanoparticles were lyophilised.
Characterization of the nanoformulation
The purified nanoparticles were further characterised for their hydrodynamic size and zeta potential using a Zetasizer Nano ZS (Malvern). The nanoparticles were diluted to 100-fold and drop cast onto a copper grid 300 mesh size coated with carbon (Agar Scientific), and imaging was performed in Transmission Electron Microscope (JOEL, JSM2100) for their morphology. The lyophilised HSA, 4O4HPR, HSANPs, 4O4HPR loaded HSANPs and physical mixture were analysed for their nature in Bruker's D8 X-ray diffractometer. The purified nanoparticles were drop-casted onto the silicon wafer for Field emission scanning electron microscopy. The Fourier Transform Infra-Red (FTIR) spectroscopy of HSA, 4O4HPR, HSANPs, 4O4HPR loaded HSANPs, and physical mixture (10:1) of HSANPs and 4O4HPR were recorded using IR Spectrophotometer. The secondary structure of HSA, HSANPs, and 4O4HPR loaded HSANPs were analysed by circular dichroism spectrophotometer (JASCO J-1500).
Drug loading and encapsulation efficiency, stability, solubility studies
The successful 4O4HPR loading into 4O4HPR loaded HSANPs was estimated indirectly by accurately measuring the drug that remained suspended in the supernatant while processing the drug-loaded nanoparticles. The measurements were recorded in triplicate samples using a UV spectrophotometer. The stability studies were carried out mimicking the different physiological conditions, including 10 mM Phosphate Buffered Saline (pH-7.4), Dulbecco's modified Eagle's Medium (DMEM), DMEM + 2% FBS and DMEM + 10% FBS for HSANPs and 4O4HPR loaded nanoparticles. The zeta potential and hydrodynamic size alterations were analysed using Zetasizer Nano ZS (Malvern). Saturated solutions of 4O4HPR and 4O4HPR loaded HSANPs were prepared in 10 mM PBS (pH 7.4) to evaluate the specific solubility at 37 °C for 24 and 48 h. After the respective time intervals, the samples were centrifuged and further analysed in a UV-visible spectrophotometer.
Drug release studies
In vitro drug (4O4HPR) release studies were performed using HSA as a receiver phase in phosphate-buffered saline (20 mL PBS, pH 7.4) with 2% Polysorbate 80,28 using the dry cellulose acetate membrane method for 1 mg of drug and its equivalent of drug-loaded nanoparticles in triplicates under constant stirring conditions at 180 rpm, at 37 °C. 1 ml of sink buffer was aliquoted at different time points and was replenished with 1 ml fresh buffer. The aliquots in triplicates were analysed using a UV spectrophotometer.
Cellular internalization of nanoparticles
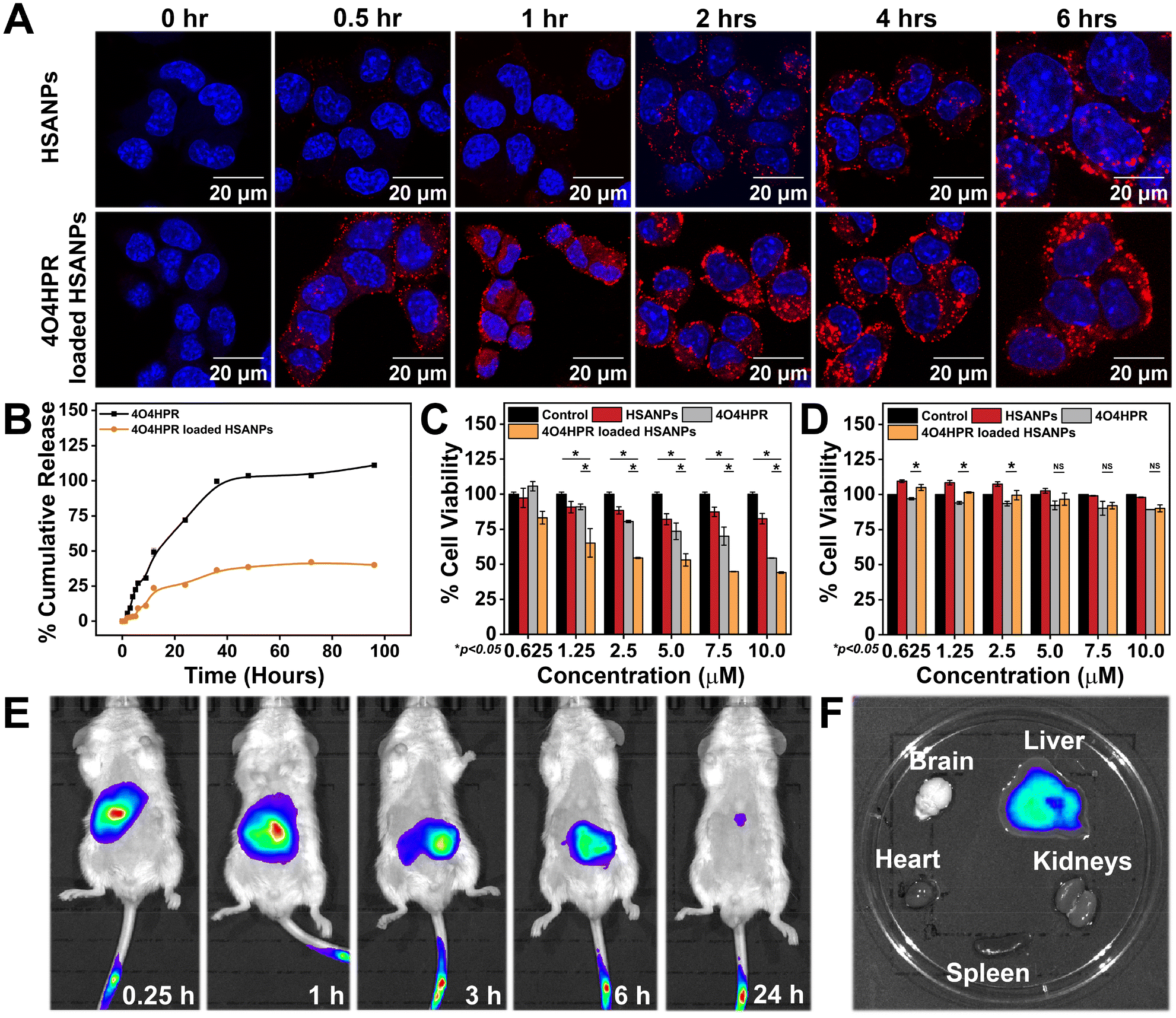
1 × 105 SH-SY5Y cells per well were cultured on coverslips in a 6-well plate with growth media. The next day, the cells were incubated with rhodamine-tagged HSANPs and 4O4HPR loaded HSANPs in a time-dependent manner at time intervals of 0.5 h, 1 h, 2 h, 4 h, and 6 h. After incubating the cells with nanoparticles, they were washed with autoclaved 1× PBS and fixed with 4% formaldehyde solution for 30 min at 4 °C. The cells were further incubated with 5 μL of 1 μg μL−1 DAPI reconstituted in DMSO for 5 min and mounted onto a glass slide using 90% glycerol. Confocal Laser Scanning Microscopic images were acquired with LIECA Confocal Microscope. The cells were seeded directly onto the 6-well plate for flow cytometric analysis, and the Rhodamine-B tagged nanoparticles were treated accordingly. After the incubation, the cells were harvested and analysed live in an Aria Fusion BD FACS flow cytometer.
Cytotoxicity of nanoformulation
Different doses of 4O4HPR and equivalent doses of 4O4HPR loaded HSANPs and HSANPs were used to treat 1 × 104 SH-SY5Y cells for 24 hours. The viability of SH-SY5Y cells after the treatment was assessed by adding the MTT dye. The IC50 dosages of 4O4HPR and 4O4HPR loaded HSANPs were ascertained using non-linear regression.
Cell cycle analysis and ROS estimation
1 × 105 SH-SY5Y cells were synchronized for 48 hours in 2% FBS supplimented DMEM in a 5% CO2 incubator followed by treatment with IC50 doses of 4O4HPR, 4O4HPR loaded HSANPs, and equivalent HSANPs for 24 h. The cells were collected using a scraper, washed with autoclaved 1X PBS and fixed with 70% Merck ethanol at 4 °C followed by propidium iodide (PI) staining for 30 min at room temperature in the dark. The Flow Cytometer (Aria Fusion, BD Biosciences) was used to record the cell cycle distribution based on PI binding to the DNA.29 The reactive Oxygen Species generated were quantified by DCFDA (2′,7′-dichlorofluorescein diacetate) assay.30 Firstly, 1 × 104 SH-SY5Y cells were seeded in a 96-well plate. The following day, the cells were treated with 10 μM DCFDA reconstituted in cell culture grade DMSO and incubated for 45 min in a 5% CO2 incubator. The cells were then washed with PBS and treated with HSANPs, 4O4HPR, and 4O4HPR loaded HSANPs according to the equivalent and IC50 values. As soon as the treatment was given, the cells were evaluated for ROS by setting up a kinetic cycle for 12 hours, then 18 and 24 hours, in an infinite PRO200 plate reader. The fluorescence was determined at 485/527 nm excitation/emission.
JC1 analysis
5 × 105 SH-SY5Y cells were seeded in a 6-well plate and treated with IC50 doses of 4O4HPR, 4O4HPR loaded HSANPs, and equivalent HSANPs for 24 h. The cells were stained with 2 μM JC1 dye and incubated for 45 minutes in a CO2 incubator at 37 °C. After incubation, the cells were collected using a scraper, washed with autoclaved 1× PBS twice, and analysed for J-Aggregates and J-Monomers in a flow cytometer.
RNA isolation and qPCR analysis
1 × 106 SH-SY5Y cells were treated with IC50 doses of 4O4HPR, 4O4HPR loaded HSANPs, and equivalent HSANPs for 24 h. The cells were then collected and washed with 1× autoclaved PBS, and RNA was isolated using the RNA Xpress Reagent (Himedia). High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used to synthesize the cDNA using the isolated RNA, followed by gene expression analysis in Quant Studio 3 (Applied Biosystems) using primers listed in ESI Table 1† and SYBr Green Reagent (iTaq Universal, Bio-Rad Technology). GAPDH was used as an internal control.31
Western blotting – in vitro protein expression study
1 × 106 SH-SY5Y cells were cultured and given treatment with HSANPs, 4O4HPR, and 4O4HPR loaded HSANPs accordingly. Proteins were isolated using RIPA lysis buffer enriched with 1 mM PMSF, protease inhibitor cocktail (PIC), and other additives. During isolation, the cells were washed with autoclaved 1× PBS twice and then incubated with RIPA lysis buffer for 30 min at 4 °C. The cells were collected into MCTs, probe sonicated at 25 amplitudes for 1 min, and centrifuged at 15000 rpm for 15 min. The supernatant was collected and stored at −20 °C. The protein samples were quantified using Bradford Assay. An equal amount of protein was resolved on the Sodium Dodecyl Sulphate-Polyacrylamide (SDS-PAGE) gel. The protein was transferred onto a 0.2 micron-sized PVDF (polyvinylidene difluoride) membrane – Bio-Rad using a semi-dry transfer system (Bio-Rad). The immunoblots were then blocked with blocking buffer for 60 min and then incubated with the primary antibodies β-actin, anti-PKC-δ, anti-EHZ2, and anti-H3K27me3 at 4 °C overnight on a rocking platform. The next day, the blots were washed thrice with TBS-T (0.1% Tween 20) and incubated with the secondary antibodies for 90 min. Clarity western ECL substrate (Bio-Rad) was used to develop the blots, recorded and analysed by the LAS500 gel documentation system.
Interaction studies between PKC-δ and EZH2
The co-immunoprecipitation experiments were conducted to analyse the interaction between PKC-δ and EZH2. 200 μl of total protein lysate was pulled down using anti-PKC-δ (rabbit raised) antibody and A/G beads (sc-2003). Immunoblotting was further executed with anti-EZH2, anti-pSer21-EZH2, and anti-pThr345-EZH2 antibodies (rabbit raised). Anti-EZH2 antibody and A/G beads were used for co-IP, while rabbit-raised anti-PKC-δ and mouse-raised anti-p-Ser, anti-p-Thr, and anti-ubiquitin antibodies were used for immunoblotting.
PKC-δ transient knockdown studies
PKC-δ siRNA was used to transfect 2 × 106 SH-SY5Y cells in incomplete cell culture DMEM according to the protocol provided by the manufacturer. Western blotting was used to evaluate the knockdown mechanism. Control+/siRNA−, 4O4HPR loaded HSANPs+/siRNA−, 4O4HPR loaded HSANPs+/siRNA+, and Control+/siRNA+ groups were all examined using β-actin, anti-Caspase3, anti-PKC-δ, anti-EHZ2 and anti-H3K27me3 antibodies.
Inhibition of EZH2 ubiquitination using MG132
1 × 106 SH-SY5Y cells were given treatment with 1 μM dose of MG132 followed by 4O4HPR loaded HSANPs treatment for 24 hours. The whole cell protein lysate was isolated, and western blot analysis was performed to analyse EZH2 and H3K27me3 levels.
In vivo biodistribution studies
To evaluate tissue distribution and clearance tests in a pre-clinical model, six-week-old BalB/C mice were used to investigate the in vivo whole-body biodistribution studies with the approval of Institutional Animal Ethics Committee (NABI, Mohali). The BalB/C mice were anaesthetised using Isoflurane in the IVIS anaesthesia system. The evacuation pump was operated at a 6 L min−1 flow rate along with an Oxygen supply to the anaesthesia chamber. 2.5% isoflurane was passed into the chamber at a 1.5 L min−1 flow rate. The mice were kept in the anaesthesia chamber. Once anesthetised, they were administered 5 mg ml−1 Indocyanine green (ICG)-tagged HSANPs through the intravenous route, whereas sterile PBS was administered in the control group. The intravenous route of administration of nanoparticles is less cytotoxic due to their reduced extravasation through minute fenestrae present in the capillaries that confer little organ-specific deposition. Biodistribution of 4O4HPR loaded HSANPs was observed in 0.25, 1, 3, 6, and 24-hour intervals; subsequently, the mice were sacrificed. The IVIS Spectrum in vivo imaging system (IVIS Illumina Series-III) was used for both in vivo and ex vivo imaging.
In vivo studies in NBM xenograft
The Institutional Animal Ethics Committee (IISER, Mohali) permitted animal xenograft model studies, and we conducted animal studies by following CPCSEA (Committee for the Purpose of Control and Supervision of Experiments on Animals) guidelines. Our previous published work demonstrated the detailed treatment groups and experimental design.32 Briefly, the groups included were Control, 4O4HPR, and 4O4HPR loaded HSANPs. The doses were 10 mg kg−1 day−1 (Stock prepared in DMSO and diluted in PBS) for 4O4HPR, 125 mg kg−1 day−1 resuspended in PBS for 4O4HPR loaded HSANPs and an equivalent dose of DMSO in the control. Herein, the present study has focused on molecular mechanistic studies exploring molecular pathways by utilizing gifted tumor tissues from our previously performed experimental work.32 The purpose of performing molecular studies by utilizing gifted tumor samples (n = 3) is to minimize the utilization of animals and to get the maximum output from the earlier performed animal studies with similar treatment groups represented in the present study.
Western blotting – in vivo protein expression study
The tumor samples, to remove any blood, were washed multiple times with autoclaved 1× PBS. The tissue samples were then homogenised using a tissue homogenizer and lysed in RIPA lysis buffer enriched with 1 mM PMSF, protease inhibitor cocktail, and other additives. The samples were then probe sonicated at 25 amplitudes for 1 min. The supernatant was then collected and stored at −20 °C after centrifugation at 15000 rpm for 15 min. The concentrations of proteins were determined using Bradford Assay, and an equal amount of protein sample was resolved on Sodium Dodecyl Sulphate-Polyacrylamide (SDS-PAGE) gel. The resolved protein samples were semi-dry transferred onto a PVDF membrane (Bio-Rad) using a semi-dry transfer system (Bio-Rad). The immunoblots were then blocked with blocking buffer for 60 min and then incubated with the primary antibodies β-actin, anti-PKC-δ, anti-EHZ2, and anti-H3K27me3 at 4 °C overnight on a rocking platform. The next day, the blots were washed thrice with TBS-T (0.1% Tween 20) and incubated with the secondary antibodies for 90 min. Clarity western ECL substrate (Bio-Rad) was used to develop the blots, recorded and analysed by the LAS500 gel documentation system.
Immunofluorescence studies
The xenograft tumor tissue samples were subjected to immunofluorescence analysis to determine the expression of PKC-δ and EZH2. After sacrificing the mice, the tumor tissues were collected and fixed in a 4% formalin solution prepared in 10 mM PBS (pH 7.4). The formalin-fixed tumor tissues were subjected to a sucrose solution gradient of 10%, 20%, and 30% for hydration before sectioning on Cryo-microtome (Thermo-Scientific). The tumor tissues were first fixed in Optimum Cutting Temperature (OCT) medium (Shandon, Cryomatrix, Thermo) followed by sectioning in which 20-micron sections were taken onto egg-albumin and glycerol (1:1 ratio) coated glass slides. The glass slides with tissue sections were kept at 95 °C for 10 min in 10 mM sodium citrate buffer for antigen retrieval, and the slides were cooled down to RT. For permeabilization, PBS-Triton-X-100, 0.1% solution was used to wash the slides. 2% goat serum,1% BSA, 0.1% fish gelatin, and 0.05% sodium azide in 1× TBS-T (0.1% Triton-X-100), blocking buffer was used for blocking the sections for 1 hour at RT. Then, the primary antibodies PKC-δ (1:1000) and EZH2 (1:500) were added to the tissue sections at 4 °C for 24 hours, maintaining a humid environment. After the primary antibody incubation, the tissue sections were incubated with secondary antibodies conjugated with a fluorescent dye (anti-mouse FITC and anti-rabbit TRITC) at RT for 1 hour. Washings were performed with TBS-T (0.1% Triton-X-100) for 3 × 10 min each. Slides were counterstained using DAPI and mounted with 90% glycerol to prepare the permanent slides. Confocal Laser Scanning Microscopic images were acquired with LIECA Confocal Microscope.
Statistical analysis
A proper ANOVA was carried out at a significance level of p ≤ 0.05 for each experiment, with a sample size of three (n = 3). The t-test was performed using Tukey and Bonferroni test at a significance level of p ≤ 0.05 using OriginPro8.5.
Results and discussion
Synthesis and characterization of nanoparticles
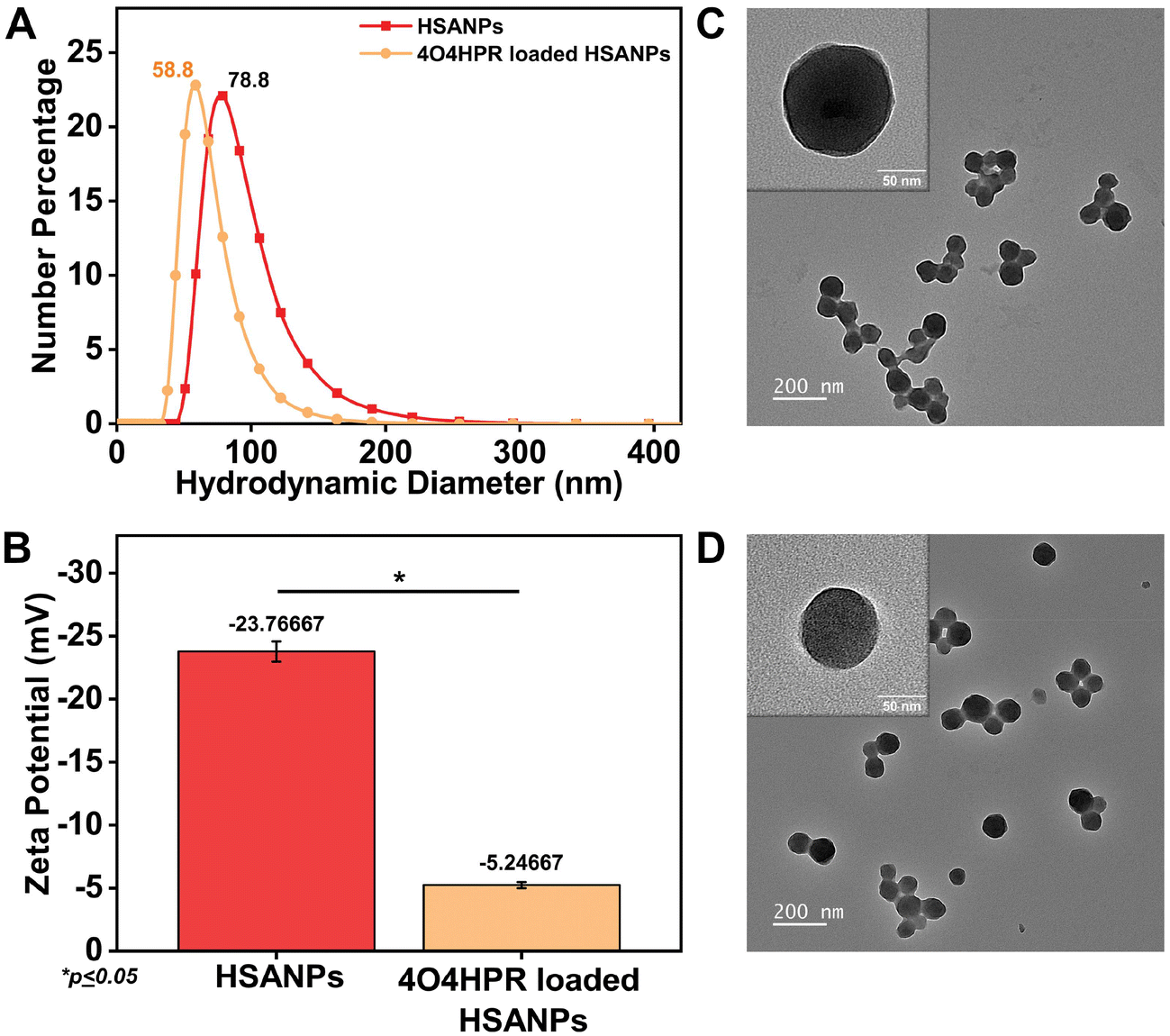
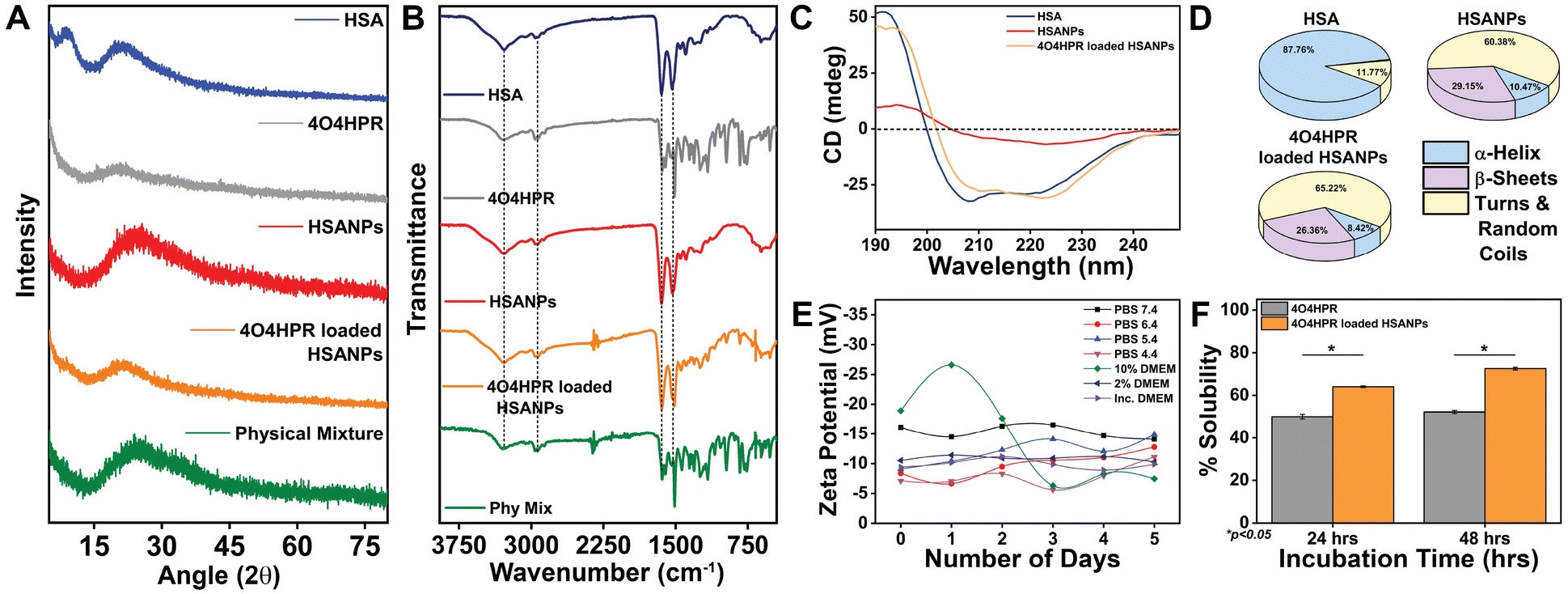
4O4HPR, a polar metabolite of its parent compound 4-HPR, is limited for therapeutics because of its poor water solubility, hydrophobicity, and stability.32 However, these limitations of 4O4HPR can be circumvented by encapsulating the drug into HSA nanoparticles. As reported earlier, the desolvation method was used to synthesise the HSA nanoparticles.21 The mean hydrodynamic diameter of the HSANPs and 4O4HPR loaded HSANPs were 78.8 ± 25 nm and 58.8 ± 30 nm with a polydispersity index of 0.183 and 0.231 respectively (Fig. 1A). This demonstrates that the hydrodynamic size and PDI (Poly Dispersity Index) of the 4O4HPR loaded HSANPs were reduced upon loading, enabling protracted nanoparticle circulation and drug penetration into tumor tissue through the enhanced permeability and retention (EPR) effect.33 A study of different-sized starch nanoparticles has demonstrated how drug loading and encapsulation efficiency decrease with increasing particle size, with speculation that smaller particles have greater specific surface area and adhesion tenacity.34 −23.7 mV and −5.24 mV were the respective zeta potentials of the HSANPs and 4O4HPR loaded HSANPs (Fig. 1B). With a decrease in the hydrodynamic size, polydispersity index, and zeta potential of the 4O4HPR loaded HSANPs, the Zetasizer analysis demonstrates that the nanoformulation is more stable in the liquid medium. The Transmission Electron Microscope (TEM) and Field Emission Scanning Electron Microscope (FESEM) study validated the spherical morphology of the nanoparticles corresponding to the DLS analysis. The TEM analysis (Fig. 1C and D) and FESEM analysis (Fig. S1A–D†) revealed that the HSANPs and 4O4HPR loaded HSANPs have a homogeneous distribution and a spherical morphology. 4O4HPR loaded HSANPs had an estimated 16.03%, drug loading efficiency and 96.2%, encapsulation efficiency. The 4O4HPR loaded HSANPs are appropriate for treating neuroblastoma because the HSA nanoparticle system has excellent encapsulation efficiency with adequate drug loading efficiency. The X-ray powder diffraction analysis HSA, HSANPs, 4O4HPR, 4O4HPR loaded HSANPs, and physical mixture showed the amorphous nature (Fig. 2A). Additionally, the unique peak characteristics of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch, N–O stretch, and CC bend at 1631, 1511, and 966 cm−1 were reflected by the FTIR spectroscopy of 4O4HPR. The HSANPs showed the same infrared peaks for amides I and III as the HSA, while amides II and A & B had shifted to 1526, 3277, and 2957 cm−1, respectively. The peaks of CC, N–O, and CC are visible in the physical mixing (10:1) of HSANPs and 4O4HPR, but these functional groups were obscured in the case of 4O4HPR loaded HSANPs. Similar IR peaks are present in 4O4HPR loaded HSANPs. However, the hydrogen bonding caused a broadening of the amide A peak, suggesting an interaction between the 4O4HPR and HSA (Fig. 2B), which is in accordance with a study involving melatonin loading into HSA and their interaction leading to a change in the secondary structure.35 The circular dichroism studies of HSA revealed the two ultraviolet negative peaks at 208 and 222 nm (Fig. 2C), the characteristic feature of α-helix.36 This showed that HSA nanoparticles had more helix content than HSA and that β-sheets had turned into turns and random coils (Fig. 2D). Our findings are corroborated by another experiment on the α-helical content of HSA after conjugation with picoplatin, as well as secondary structures of HSA that alter when it converts into nanoparticles.37 Further, the time-dependent stability of nanoparticles was analysed. The systemic stability of the 4O4HPR loaded HSANPs was found to be unvarying till 120 h when incubated in Phosphate Buffered Saline with varying pH, incomplete DMEM, DMEM with 10% FBS, and DMEM with 2% FBS with respect to zeta potential (Fig. 2E) and hydrodynamic size (Fig. S2A†) at 37 °C. Nano formulation in Human Serum Albumin enhances the stability of 4O4HPR by the unvaried zeta potential under suitable conditions of incubation in DMEM with 10% and 2% FBS, which are in consonant with a study that included various curcumin nanoformulations.38,39 Furthermore, the encapsulation of 4O4HPR in HSA improves its solubility in a time-dependent fashion (Fig. 2F). HSA interacts hydrophobically with hydrophobic drugs to form bioconjugates and by shielding the molecule from the aqueous environment, it increases the drug's solubility. A study of Cur-HSA nanoparticles has shown that the nanoformulation of hydrophobic drugs increases their stability and, thereby, solubility, supports our findings.39 Thus, the HSA nano formulation overcomes the limitations of 4O4HPR, including the solubility of 4O4HPR, which provides better stability.
C stretch, N–O stretch, and CC bend at 1631, 1511, and 966 cm−1 were reflected by the FTIR spectroscopy of 4O4HPR. The HSANPs showed the same infrared peaks for amides I and III as the HSA, while amides II and A & B had shifted to 1526, 3277, and 2957 cm−1, respectively. The peaks of CC, N–O, and CC are visible in the physical mixing (10:1) of HSANPs and 4O4HPR, but these functional groups were obscured in the case of 4O4HPR loaded HSANPs. Similar IR peaks are present in 4O4HPR loaded HSANPs. However, the hydrogen bonding caused a broadening of the amide A peak, suggesting an interaction between the 4O4HPR and HSA (Fig. 2B), which is in accordance with a study involving melatonin loading into HSA and their interaction leading to a change in the secondary structure.35 The circular dichroism studies of HSA revealed the two ultraviolet negative peaks at 208 and 222 nm (Fig. 2C), the characteristic feature of α-helix.36 This showed that HSA nanoparticles had more helix content than HSA and that β-sheets had turned into turns and random coils (Fig. 2D). Our findings are corroborated by another experiment on the α-helical content of HSA after conjugation with picoplatin, as well as secondary structures of HSA that alter when it converts into nanoparticles.37 Further, the time-dependent stability of nanoparticles was analysed. The systemic stability of the 4O4HPR loaded HSANPs was found to be unvarying till 120 h when incubated in Phosphate Buffered Saline with varying pH, incomplete DMEM, DMEM with 10% FBS, and DMEM with 2% FBS with respect to zeta potential (Fig. 2E) and hydrodynamic size (Fig. S2A†) at 37 °C. Nano formulation in Human Serum Albumin enhances the stability of 4O4HPR by the unvaried zeta potential under suitable conditions of incubation in DMEM with 10% and 2% FBS, which are in consonant with a study that included various curcumin nanoformulations.38,39 Furthermore, the encapsulation of 4O4HPR in HSA improves its solubility in a time-dependent fashion (Fig. 2F). HSA interacts hydrophobically with hydrophobic drugs to form bioconjugates and by shielding the molecule from the aqueous environment, it increases the drug's solubility. A study of Cur-HSA nanoparticles has shown that the nanoformulation of hydrophobic drugs increases their stability and, thereby, solubility, supports our findings.39 Thus, the HSA nano formulation overcomes the limitations of 4O4HPR, including the solubility of 4O4HPR, which provides better stability.
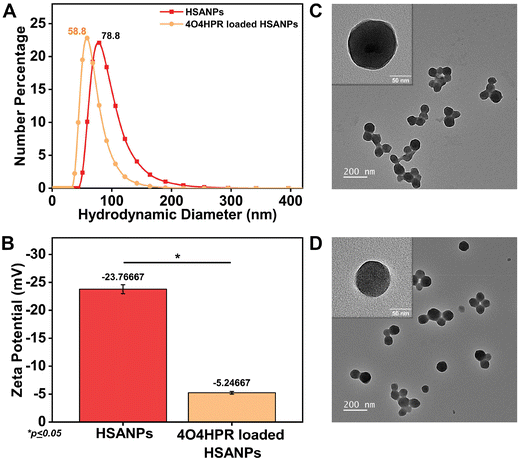 |
| | Fig. 1 (A) The average hydrodynamic diameter of HSANPs and 4O4HPR loaded HSANPs; (B) zeta potential analysis of HSANPs and 4O4HPR loaded HSANPs; morphological characterization of (C) HSANPs and (D) 4O4HPR loaded HSANPs by transmission electron microscope. | |
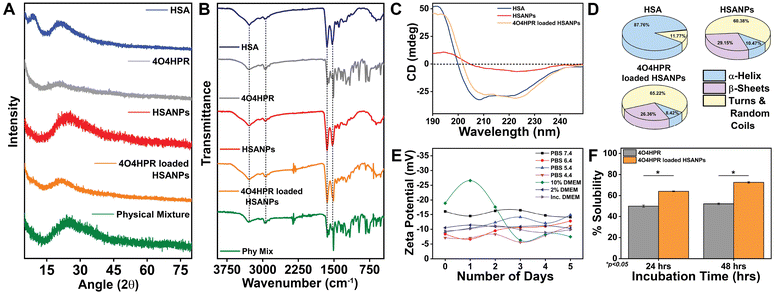 |
| | Fig. 2 (A) The powder X-ray diffraction pattern for HSA, 4O4HPR, HSANPs, 4O4HPR loaded HSANPs, and physical mixture; (B) Fourier-Transform IR spectra of HSA, 4O4HPR, HSANPs, 4O4HPR loaded HSANPs, and physical mixture; (C) circular dichroism spectroscopy depicting the secondary structure of HSA, HSANPs and 4O4HPR loaded HSANPs (D) with quantitative composition of secondary structure conformations (E) stability profile for zeta potential of 4O4HPR loaded HSANPs in PBS with varying pH 7.4, 6.4, 5.4, 4.4, 10% DMEM, 2% DMEM, and Incomplete DMEM; (F) solubility profile of 4O4HPR and 4O4HPR loaded HSANPs; (n = 3), where n is the number of experimental repeats. | |
In vitro efficacy studies
The cumulative drug release profile signified the initial burst release accompanied by sustained release of 4O4HPR and slow but sustained release of 4O4HPR loaded HSANPs (Fig. 3B), which is in accordance with a study of Docetaxel (DTX) loaded NPs that have shown sustained release in comparison to DTX explaining that the encapsulated drugs undergo sustained release.33 Furthermore, the controlled drug release pattern of 4O4HPR and 4O4HPR loaded HSANPs fits into different kinetic models. The fitting of the release patterns of 4O4HPR and 4O4HPR loaded HSANPS in PBS + 2% Tween 80 has predicted R2 values of >0.85 for both the Zero order and Korsmeyer-Peppas model that is supported by the famotidine nanoformulation in gastrointestinal tract study38 (Fig. S4A–D†). The exponents of the release pattern for 4O4HPR for the spherical nanoformulations were determined to be 1.0584 and 1.2982 in both the models, depicting that they follow Super Case II transport mechanism correlating with Candesartan and Cilexetil loaded nanoemulsion.40 The Confocal Laser Scanning Microscope analysis demonstrated the rhodamine B tagged HSANPs and 4O4HPR loaded HSANPs accumulating in the cytoplasm and nuclear periphery of SH-SY5Y cells, respectively (Fig. 3A). Also, the internalization of nanoparticles in SH-SY5Y cells using rhodamine B-tagged HSANPs and 4O4HPR loaded HSANPs was analysed in BD FACS Aria Flow Cytometer (Fig. S3†). According to the research, albumin can particularly bind to the gp60, a 60 kDa glycoprotein receptor, and the SPARC receptor, both of which are overexpressed on the cell membrane of many cancer cell types and are often found on the surface of endothelial cells. This binding facilitates vesicle production, transcytosis, and drug delivery into the sub-endothelial region.41 The cytotoxicity studies carried out in in vitro conditions exhibited the potency of 4O4HPR loaded HSANPs in SH-SY5Y cells when treated with varying doses of 4O4HPR and equivalent doses of 4O4HPR loaded HSANPs and HSANPs, till 24 h. When compared to 4O4HPR alone, 4O4HPR loaded HSANPs significantly killed SH-SY5Y cells, but HSANPs did not exhibit any cytotoxicity (Fig. 3C). The in vitro anti-cancer efficiency of 4O4HPR loaded HSANPs was shown to be superior, with an IC50 of 7.5 ± 0.18 μM, compared to the calculated IC50 dose of 9.35 ± 1.76 μM. Compared to 4O4HPR, the IC50 of the 4O4HPR loaded HSANPs is reduced by ∼1.3 times. The finding is supported by a study that used PRT4165 and PRT4165 loaded HSANPs, where there was a ∼3-fold decrease in IC50 dose of PRT4165 loaded HSANPs compared to PRT4165.42 In addition, the IC50 dose of 4O4HPR loaded HSANPs (7.5 μM) has shown ∼90% viability in the non-cancer derived cell line, HEK293 (Fig. 3D). Our findings are coherent with a similar report on A549 cells, that the chemo-therapeutic drugs are potentiated by the selective death of cancer cells and minimal damage towards normal non-cancerous cells.42 To investigate the degree to which nanoparticles accumulate, persist, and circulate in the in vivo system, the in vivo biodistribution analysis of 4O4HPR loaded HSANPs was carried out in BALB/c mice. By administering the ICG-tagged HSANPs at different time points of 0.25 h, 1 h, 3 h, 6 h, and 24 h, and evaluating the photoluminescent intensity of nanoparticles, the biodistribution of nanoparticles was assessed in BalB/c mice (Fig. 3E). The ex vivo bioimaging of brain, kidneys, spleen, liver, and heart harvested after 24 hours has shown the accumulation of particles in the liver (Fig. 3F) which indicates the clearance by reticuloendothelial system and was quantified using the Living Image 3.1 software. As a result, the in vivo biodistribution of 4O4HPR loaded HSANPs supports a previous study suggesting the nanoformulation is cleared from the body in 24 hours via the reticuloendothelial system.43 The histological evaluation of all the primary body organs after the treatment of 4O4HPR loaded HSANPs was stained by haematoxylin-eosin (H&E) staining and has shown less or no toxicity, thus supporting our findings.32 These results indicate that 4O4HPR loaded HSANPs have good hydrophilic dispersibility and biocompatibility with in vivo systems, and they are therapeutically effective for neuroblastoma treatment.
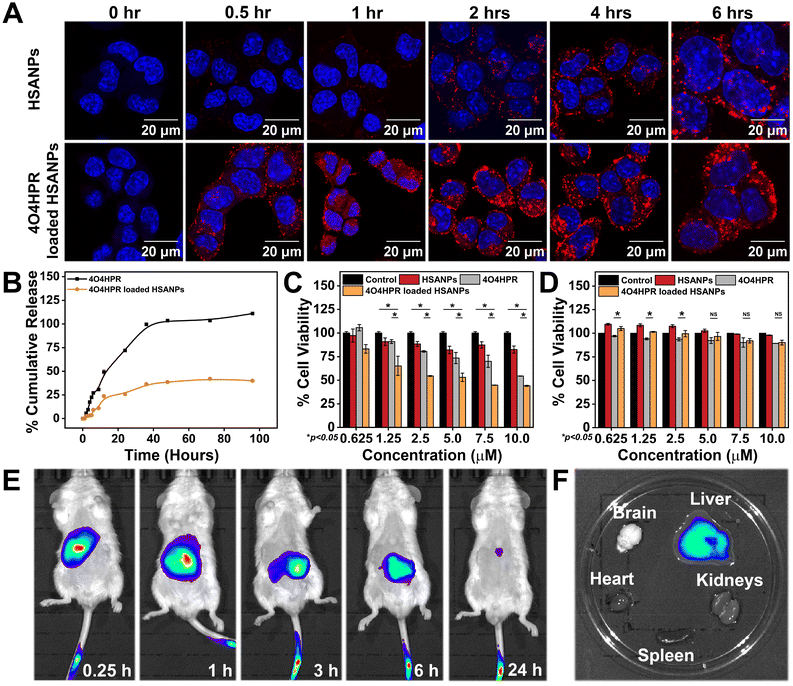 |
| | Fig. 3 (A) The confocal microscope images for cellular internalization of HSANPs and 4O4HPR loaded HSANPs in SH-SY5Y cell line; (B) in vitro cumulative drug release profile of 4O4HPR and 4O4HPR loaded HSANPs; cell viability, MTT assay after treatment with HSANPs, 4O4HPR, and 4O4HPR loaded HSANPs in (C) neuroblastoma SH-SY5Y cell line and (D) normal cell line HEK293; (E) time-dependent in vivo biodistribution of ICG tagged-4O4HPR loaded HSANPs and whole animal imaging 0.25 h, 1 h, 3 h, 6 h, and 24 h, after post intravenous injection; (F) ex vivo imaging of organs like brain, kidneys, spleen, liver, and heart harvested 24 h after injection of nanoparticles. | |
Mechanism of in vitro cytotoxicity
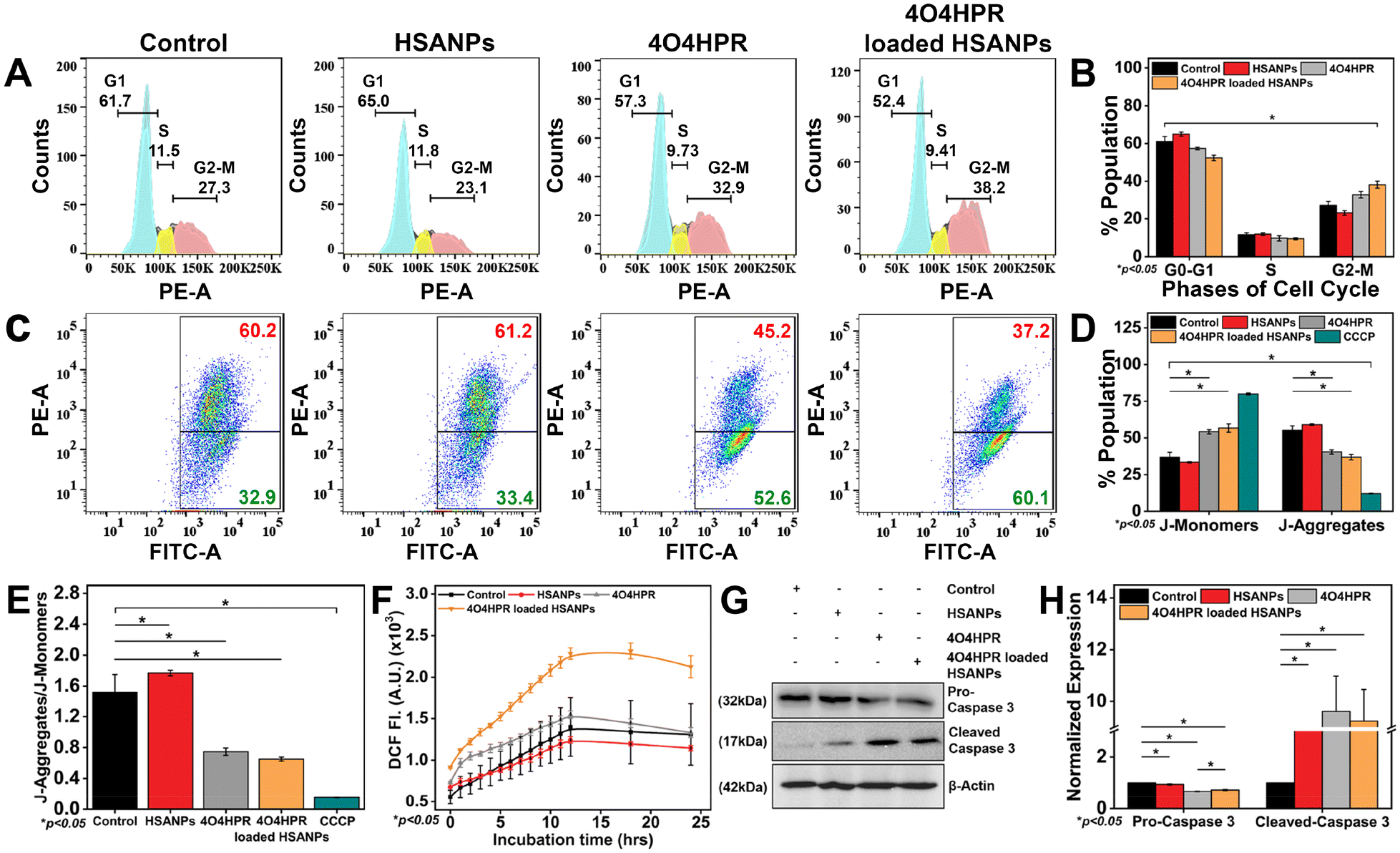
The analysis of the cell cycle using a flow cytometer demonstrated a statistically significant arrest in the G2-M phase of the cell cycle when treated with 4O4HPR loaded HSANPs, which was based on the binding of propidium iodide (PI) to DNA (Fig. 4A and B) and was in accordance with the cell cycle analysis conducted on ovarian A2780 cells and also on SH-SY5Y.22 The antiproliferative effect of 4O4HPR is due to the regulation of cell cycle regulator proteins associated with G2-M cell cycle arrest followed by activation of the caspase cascade, evoking the induction of apoptosis.20,44 During JC-1 staining, the increase in J-monomers or green signal indicated the loss or change in the mitochondrial membrane potential in 4O4HPR and 4O4HPR loaded HSANPs as compared to HSANPs and Control (Fig. 4C & D). Also, 4O4HPR loaded HSANPs induced a significant decrease in the ratio of J-aggregates/J-monomers, demonstrating the depolarization of mitochondrial membrane potential (Fig. 4E), which is the consequence of ROS generation and is evident by DCFHDA staining of SH-SY5Y cells. The time-dependent 2′,7′-dichlorofluorescein (DCF) fluorescence illustrated an increase in the reactive oxygen species (ROS) generation with the treatment of 4O4HPR loaded HSANPs in comparison to other treatment groups (Fig. 4F). According to the reported literature, an increase in the amounts of reactive oxygen species (ROS) in neuroblastoma cells triggers oxidative stress,45 which consecutively serves as the primary mediator of the ROS-dependent, mitochondria-mediated apoptotic cascade supports our findings.46 A study on ovarian cell line A2780 suggested that the 4O4HPR-induced ROS generation was involved in 4O4HPR-induced apoptosis. Also, upon the treatment of 4O4HPR loaded HSANPs, the western blot analysis demonstrated the downregulation of Pro-Caspase-3 (32 kDa) and upregulation of the cleaved caspase-3 (17 kDa) (Fig. 4G & H). The usage of various antioxidants and different kinase inhibitors suggests that 4HPR, the parent compound of 4O4HPR, operates on the mitochondria to promote ROS production and change the mitochondrial membrane potential, which may further lead to induction of apoptosis via the cytochrome c, caspase-3, and caspase-9 pathway in head and neck squamous carcinoma cells.22 These findings demonstrate that 4O4HPR loaded HSANPs induce the SH-SY5Y cell line to undergo apoptosis by generating ROS, increasing the number of unhealthy mitochondria with depolarised mitochondrial membrane potential, and promoting the upregulation of active cleaved caspase-3.
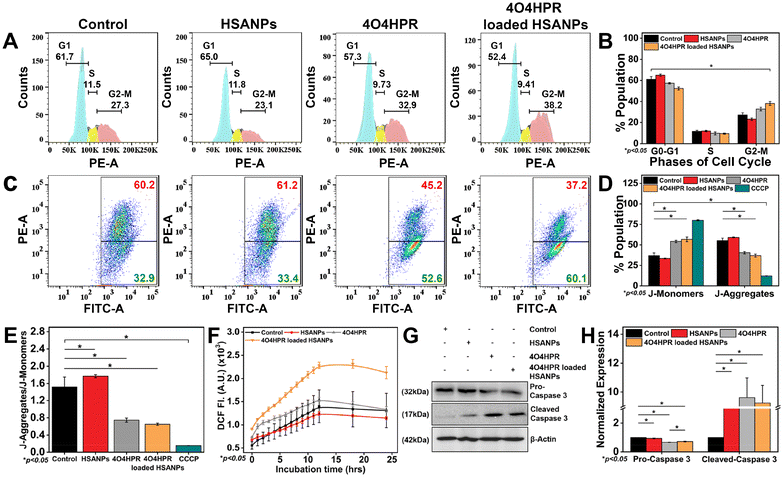 |
| | Fig. 4 (A) Cell cycle analysis in SH-SY5Y cells after treatment with HSANPs, 4O4HPR and 4O4HPR loaded HSANPs; (B) quantification of flow cytometry data of G1, S, and G2-M, phases of cell cycle; (C) analysis of mitochondrial membrane potential using JC-1 dye with the treatment of HSANPs, 4O4HPR, and 4O4HPR loaded HSANPs; (D) quantification of J-monomers and J-Aggregates (E) depolarization of mitochondrial membrane potential depicted by the J-aggregates/J-monomers ratio; (F) time-dependent ROS estimation by DC-FHDA dye after treatment with HSANPs, 4O4HPR and 4O4HPR loaded HSANPs; (G) western blot analysis of pro-caspase 3 and cleaved-caspase 3 expression after treatment with HSANPs, 4O4HPR, and 4O4HPR loaded HSANPs; (H) and their quantification; (n = 3), where n is the number of experimental repeats. | |
Molecular regulation of 4O4HPR loaded HSANPs
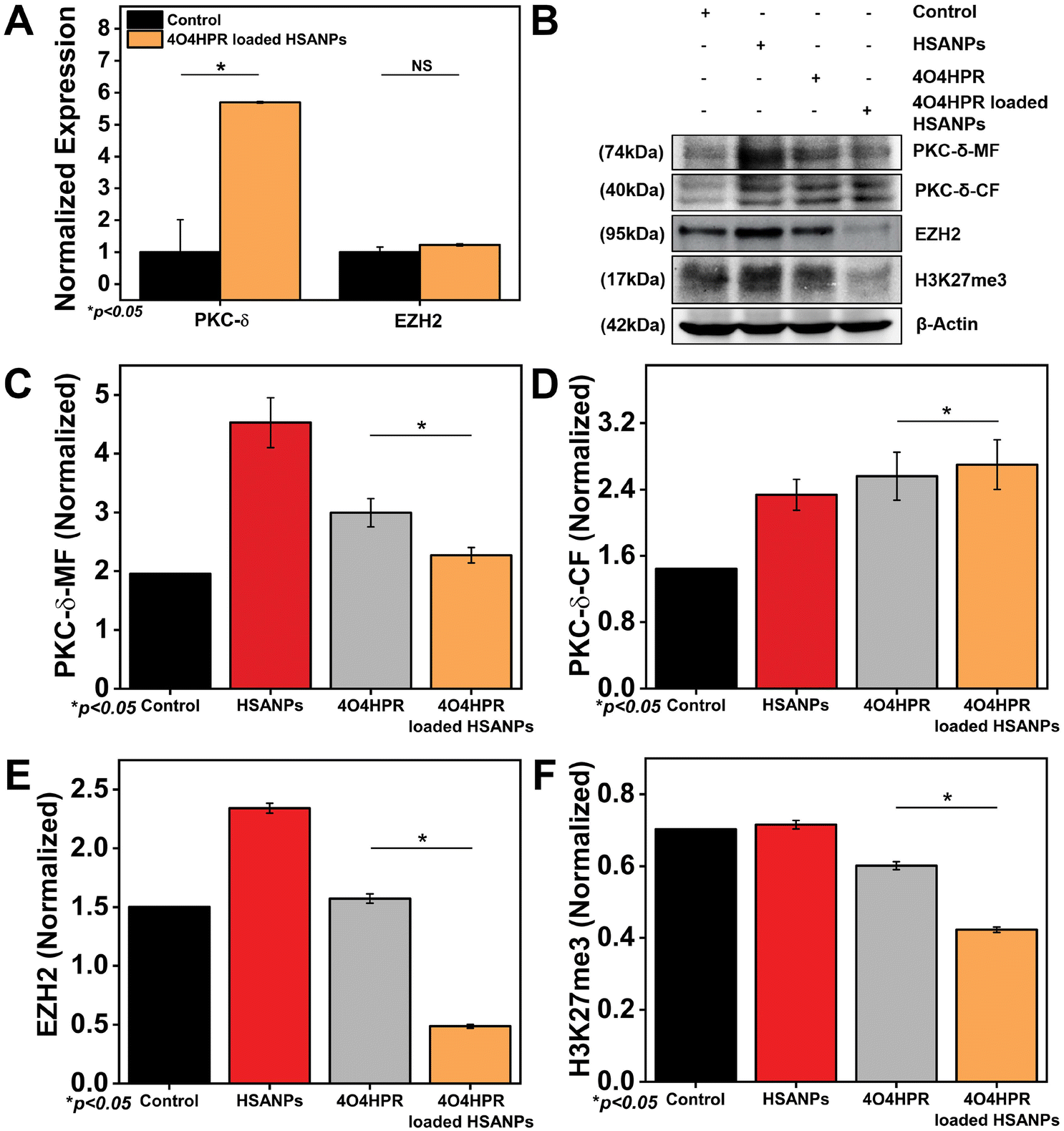
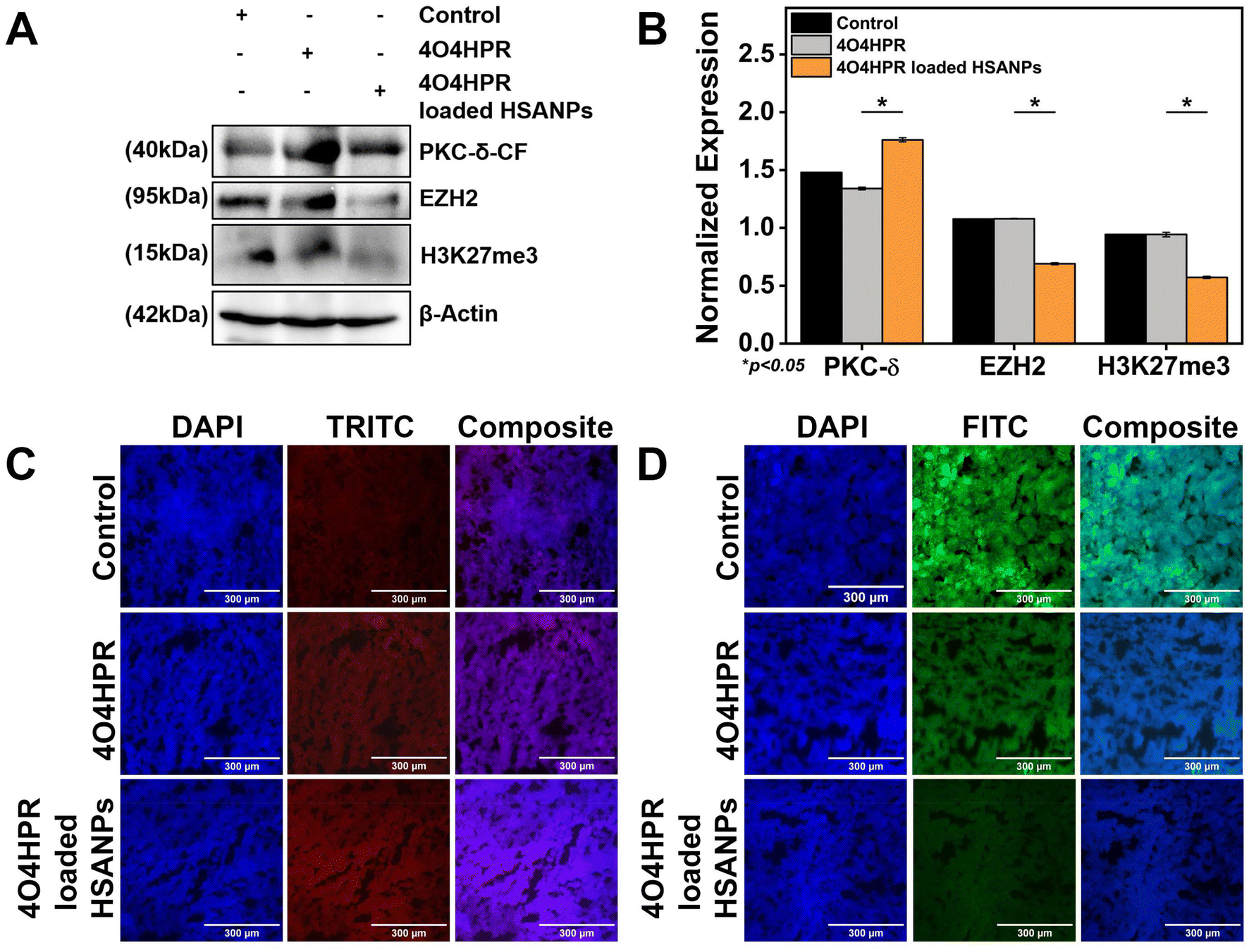
EZH2 is a widely-known methyl transferase that facilitates the tri-methylation of histone H3 at lysine 27 (H3K27me3), linked to the suppression of gene expression and is thought to be a critical epigenetic event throughout the development of tissues and the selection of stem cell fate.47,48 PKC-δ is a novel protein kinase that regulates the expression of many downstream genes by phosphorylating them. 4O4HPR was chosen for its capacity to generate ROS and depolarize mitochondria, thereby leading to the activation of caspase-3, which further acts on mature PKC-δ and cleaves it into two fragments.22,49,50 The C-terminal catalytic fragment possesses the kinase activity, which further phosphorylates EZH2 at several sites, leading to the instability of the protein.15 In several malignancies, EZH2 overexpression has increased carcinogenesis and proliferation of cancer cells and blocked tumor cell differentiation. As a result, research has been going on over the past ten years to create therapies that specifically target EZH2 in neuroblastoma, implicating novel therapeutic approach.51,52 To examine the implications of 4O4HPR-loaded HSANPs, we evaluated the expression of PKC-δ and EZH2 via western blotting and quantitative real-time PCR analysis. The quantitative real-time PCR analysis revealed upregulation of PKC-δ by 5.6 folds and similar expression of EZH2 in the 4O4HPR loaded HSANPs group compared to control, explaining the upregulation of PKC-δ gene expression. At the same time, the EZH2 is regulated at the protein level by post-translational modifications after 4O4HPR loaded HSANPs treatment in the SH-SY5Y cell line (Fig. 5A). Western blot analysis revealed the upregulation of PKC-δ-CF by 0.5 folds (Fig. 5B–D) and downregulation of EZH2 by 3 folds with 4O4HPR loaded HSANPs treatment (Fig. 5B & E). 4O4HPR loaded nanoparticles also downregulated the expression of trimethyl H3K27 by 1.6 folds (Fig. 5F). Nevertheless, HSANPs have not demonstrated any appreciable alterations in gene expression. So, to analyse the PKC-δ and EZH2 interactions and the regulation of EZH2 by PKC-δ, we have performed immunoprecipitation and MG132 inhibitor studies, respectively.
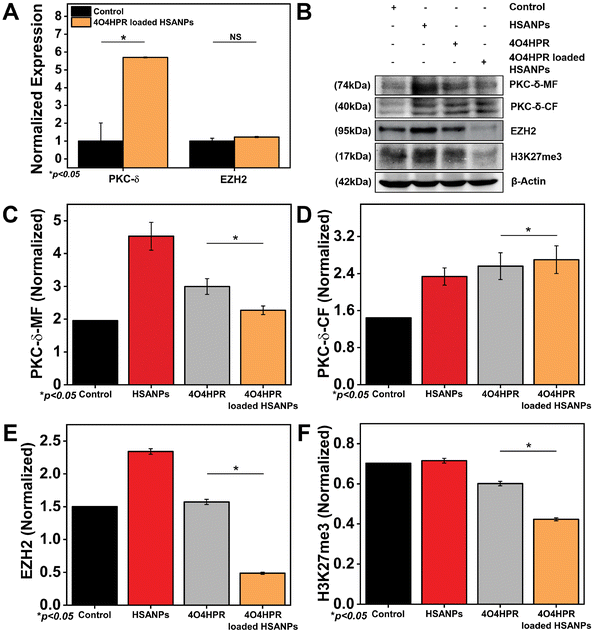 |
| | Fig. 5 (A) qPCR analysis of PKC-δ-CF, EZH2 after treatment with 4O4HPR Loaded HSANPs; (B) western blot analysis of control, HSANPs, 4O4HPR, 4O4HPR loaded HSANPs, for β-actin, PKC-δ-MF, PKC-δ-CF, EZH2, H3K27me3 and their quantification for (C) PKC-δ-MF, (D) PKC-δ-MF, (E) EZH2, (F) H3K27me3. | |
Interaction of PKC-δ and EZH2
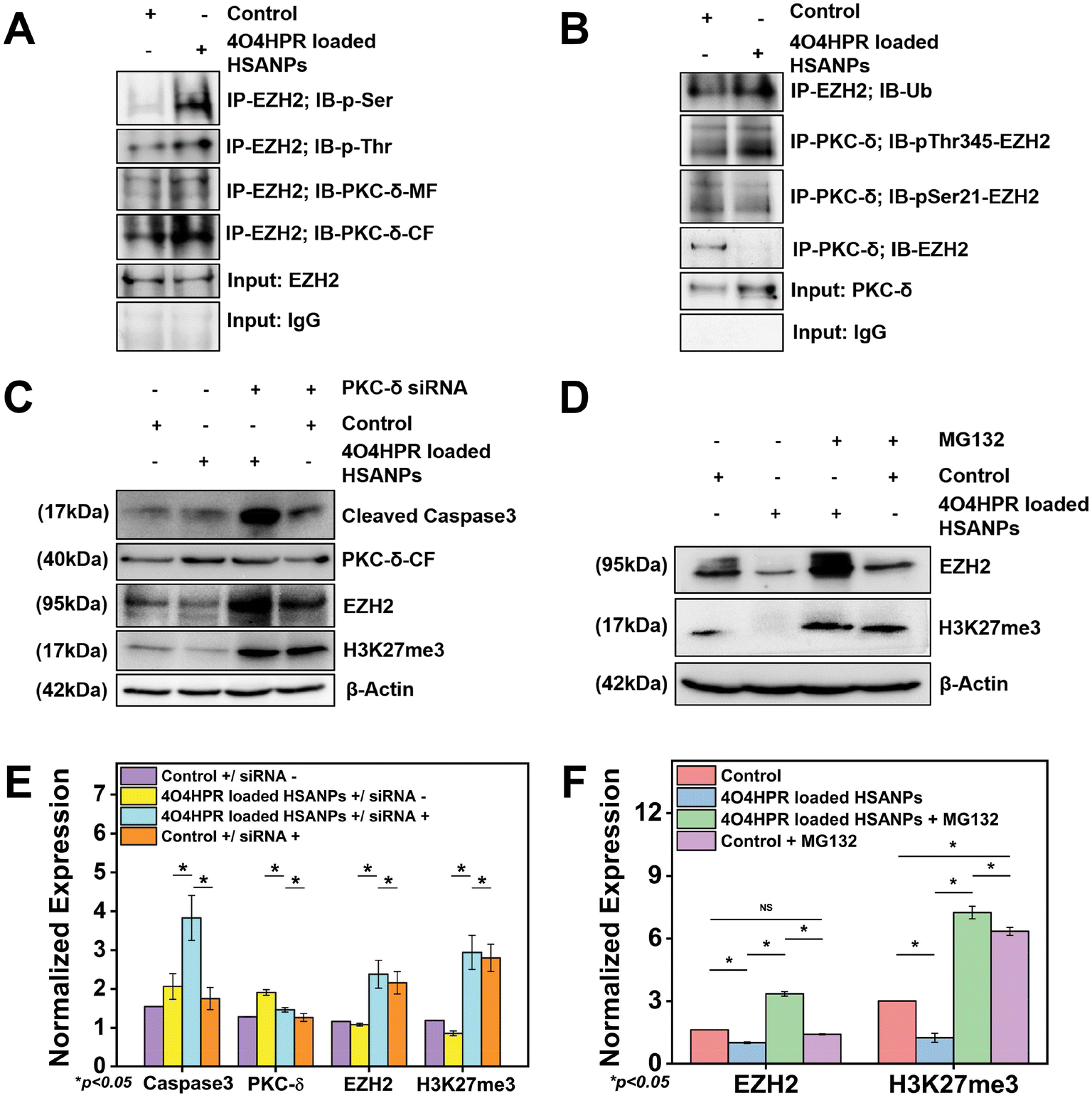
The overexpression of EZH2 mediates the proliferation of neuroblastoma cells, and EZH2 directly controls numerous cascade genes that result in a malignant phenotype in various cancer types; it has been demonstrated that EZH2 knockdown reduces tumorigenesis in neuroblastoma cells.13 Co-immunoprecipitation studies were done to evaluate the interaction between PKC-δ and EZH2. The immunoprecipitation of the whole cell protein with EZH2 and immunoblotting with PKC-δ has shown the interaction between EZH2 and PKC-δ. In contrast, the immunoblotting with Phospho-Serine and Phospho-Threonine has shown increased phosphorylation status after 4O4HPR loaded HSANPs treatment (Fig. 6A). Further pull down of protein with PKC-δ and immunoblotting with EZH2, pSer21-EZH2, pThr345-EZH2, and Ubiquitin has demonstrated the specific phosphorylation of EZH2 by PKC-δ leading to ubiquitination explaining the direct interaction between PKC-δ and EZH2 (Fig. 6B).
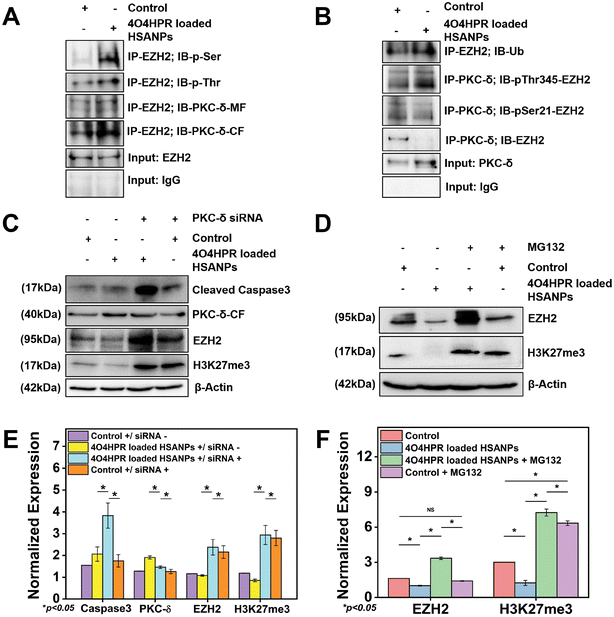 |
| | Fig. 6 (A) Co-immunoprecipitation study to analyse the protein–protein interaction between the PKC-δ and EZH2 while comparing with immunoprecipitated input control, IgG and EZH2 input with immunoblotted PKC-δ, Phospho-Serine, and Phospho-Threonine; (B) the phosphorylation and poly-ubiquitination of EZH2 was measured by co-immunoprecipitation study while comparing immunoprecipitated input control, IgG and EZH2 input with immunoblotted p-Ser21-EZH2, p-Thr345-EZH2, and ubiquitin; (C) western blot analysis after knockdown of PKC-δ with PKC-δ siRNA oligonucleotide followed by treatment with HSANPs, 4O4HPR, 4O4HPR Loaded HSANPs for β-actin, PKC-δ-CF, EZH2, H3K27 and (E) their quantification; (D) for the detection of inhibition of ubiquitin proteasomal pathway SH-SY5Y cells were treated with 1 μM of MG132 followed by treatment with HSANPs, 4O4HPR, 4O4HPR loaded HSANPs and checked for recovery of EZH2 and H3K27me3 and (F) their quantification (n = 3), where n is the number of experimental repeats. | |
PKC-δ transient knockdown study
To scrutinize the specific role of PKC-δ in SH-SY5Y cells, PKC-δ knockdown was carried out by using PKC-δ siRNA oligonucleotide. Western blot analysis demonstrated the significantly downregulated PKC-δ protein when treated with both 4O4HPR loaded HSANPs and siRNA compared to only 4O4HPR loaded HSANPs treatment without siRNA. The siRNA, along with 4O4HPR loaded HSANPs treatment, has increased the expression of caspase3, EZH2, and trimethyl H3K27, explaining the regulation of these proteins by PKC-δ (Fig. 6C & E). A study in Caco2 cells, using PKC-δ siRNA oligonucleotide to knock down PKC-δ, supports our findings.53
Degradation of EZH2 via proteasome function
An important aspect of protein metabolism is the ubiquitin-proteasome pathway (UPP), which controls numerous biological processes like cell cycle regulation, response to DNA damage, apoptosis, etc.54,55 The precise mechanism of EZH2 downregulation has been revealed using MG132, an inhibitor of the ubiquitin-proteasome pathway with a dose optimized by MTT assay (Fig. S5†). In that respect, the SDS-PAGE and western blotting analysis after the treatment of MG132 followed by 4O4HPR loaded HSANPs has shown the recovery of EZH2 by ∼3.3 folds and H3K27me3 by ∼5.7 folds in co-treated groups compared to the 4O4HPR loaded HSANPs treatment only suggesting the ubiquitin-mediated proteasomal degradation of EZH2 after 4O4HPR loaded HSANPs treatment (Fig. 6D & F).
Molecular mechanistic and immunohistochemical analysis of tumor sections
The western blot analysis of protein isolated from the xenograft tumor tissues after the treatment of 4O4HPR and 4O4HPR loaded HSANPs has demonstrated the upregulation of PKC-δ and downregulation of EZH2 and its substrate H3K27me3 (Fig. 7A & B). To assess the expression levels of PKC-δ and EZH2 in neuroblastoma xenograft tumor tissues, immuno-histochemical (IHC) analysis has been carried out. As a result, IHC analysis has shown the upregulation of PKC-δ (Fig. 7C) and downregulation of EZH2 (Fig. 7D) when treated with 4O4HPR loaded HSANPs compared to the control group. 4O4HPR is known to increase ROS levels in ovarian cancer cells,22 which further leads to PKC-δ activation and regulating its downstream signaling molecules.
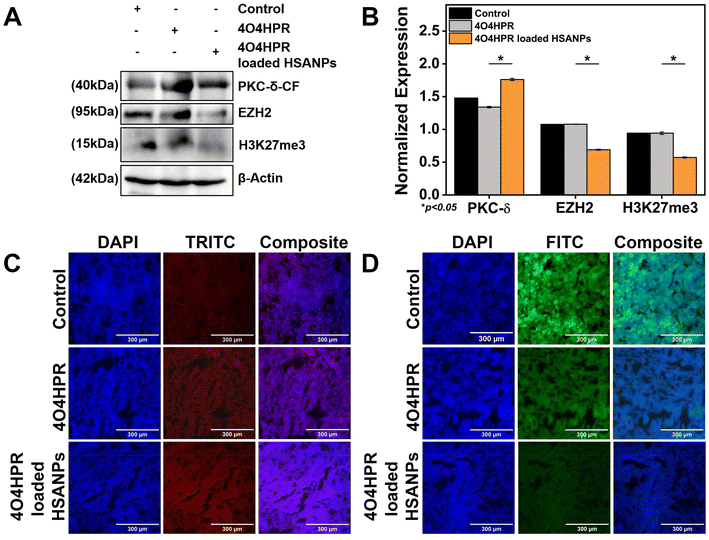 |
| | Fig. 7 (A) Western blot analysis of xenograft tumor tissues after the treatment of 4O4HPR, 4O4HPR loaded HSANPs, for β-actin, PKC-δ-CF, EZH2, H3K27me3 (B) and their quantification; immuno-histochemical expression analysis of neuroblastoma xenograft tumor tissues for (C) PKC-δ and (D) EZH2 demonstrating the upregulation of PKC-δ and downregulation of EZH2 after treatment with 4O4HPR and 4O4HPR loaded HSANPs and was stained using fluorescently conjugated secondary antibodies. | |
Conclusions
The synthesised 4O4HPR loaded HSANPs had overcome the limitations of 4O4HPR corresponding to its limited solubility and stability. Our nanoformulation demonstrated better physiological bio-availability inside the SH-SY5Y cells through SPARC-mediated endocytosis and the Enhanced Permeability and Retention (EPR) effect. The enhanced ROS generation induced increased cytotoxicity, G2-M phase cell cycle arrest, and depolarised mitochondria membrane potential. This led to caspase-dependent apoptosis in SH-SY5Y cells for the anti-cancer effect of 4O4HPR loaded HSANPs. The molecular mechanistic studies revealed the PKC-δ activation that may lead to EZH2 phosphorylation followed by its ubiquitination receding the epigenetic silencing mechanism. Also, the PKC-δ knockdown studies have confirmed the correlation between PKC-δ and EZH2, and immunoprecipitation has validated the direct physical interaction of PKC-δ and EZH2. In addition to this, the anti-cancer effects of 4O4HPR loaded HSANPs studied in mouse neuroblastoma xenograft models exhibited potency in molecular mechanistic studies. In conclusion, 4O4HPR loaded HSANPs nano formulation activated, upregulated PKC-δ, and downregulated EZH2 in vitro and in vivo, inhibiting epigenetic gene silencing in neuroblastoma, contributing to neuroblastoma regression.
Author contributions
BM contributed to the designing of the experiments and manuscript writing. AD performed the animal study. ACK helped in flow cytometry and molecular mechanistic studies. MN has helped in in vitro experiments. SK supervised all the work, including designing the experiments, data analysis, and manuscript writing.
Data availability
All data are incorporated in the article and ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge Indian Council of Medical Research (R11013/09/2021-GIA/HR), Department of Biotechnology, Ministry of Science and Technology, India (BT/PR40647/MED/31/436/2020) and Department of Science and Technology, Ministry of Science and Technology, India (DST/NM/TBI-02/2020) for providing partial funding support. The Facilities of the Indian Institute of Science Education and Research, Mohali (IISER, Mohali), National Agri-Food Biotechnology Institute (NABI, Mohali), and Institute of Nano Science and Technology (INST, Mohali) are duly acknowledged. We are thankful to Dr Nitin Singhal of the National Agri-Food Biotechnology Institute (NABI, Mohali) for providing support to the use of the in vivo imaging facility of NABI, Mohali. Council of Scientific and Industrial Research, India fellowship provided to BM is also acknowledged.
References
- J. R. Park, A. Eggert and H. Caron, Hematol./Oncol. Clin. North Am., 2010, 24, 65–86 CrossRef PubMed.
- J. I. Johnsen, C. Dyberg and M. Wickström, Front. Mol. Neurosci., 2019, 12, 9 CrossRef CAS PubMed.
- E. D. Eisenmann, Z. Talebi, A. Sparreboom and S. D. Baker, Basic Clin. Pharmacol. Toxicol., 2022, 130(Suppl 1), 23–35 CrossRef CAS PubMed.
- F. Li, W. Zhang, H. Hu, Y. Zhang, J. Li and D. Huang, Cancer Manage. Res., 2022, 14, 107–122 CrossRef CAS PubMed.
- X. Zhou, X. Wang, N. Li, Y. Guo, X. Yang and Y. Lei, Front. Pharmacol., 2023, 14 DOI:10.3389/fphar.2023.1114295.
- E. G. Greengard, Children, 2018, 5, 142 CrossRef PubMed.
- L. Jubierre, C. Jiménez, E. Rovira, A. Soriano, C. Sábado, L. Gros, A. Llort, R. Hladun, J. Roma, J. S. d. Toledo, S. Gallego and M. F. Segura, Exp. Mol. Med., 2018, 50, 1–12 CrossRef CAS PubMed.
- N. Jin, T. L. George, G. A. Otterson, C. Verschraegen, H. Wen, D. Carbone, J. Herman, E. M. Bertino and K. He, Clin. Epigenet., 2021, 13, 83 CrossRef CAS PubMed.
- L. Di Croce and K. Helin, Nat. Struct. Mol. Biol., 2013, 20, 1147–1155 CrossRef CAS PubMed.
- S. Aranda, G. Mas and L. Di Croce, Sci. Adv., 2015, 1, e1500737 CrossRef PubMed.
- Y. Shi, X. X. Wang, Y. W. Zhuang, Y. Jiang, K. Melcher and H. E. Xu, Acta Pharmacol. Sin., 2017, 38, 963–976 CrossRef CAS PubMed.
- R. El Baba and G. Herbein, Cells, 2024, 13(6) DOI:10.3390/cells13060541.
- Z. Li, H. Takenobu, A. N. Setyawati, N. Akita, M. Haruta, S. Satoh, Y. Shinno, K. Chikaraishi, K. Mukae, J. Akter, R. P. Sugino, A. Nakazawa, A. Nakagawara, H. Aburatani, M. Ohira and T. Kamijo, Oncogene, 2018, 37, 2714–2727 CrossRef CAS PubMed.
- L. V. Bownes, A. P. Williams, R. Marayati, L. L. Stafman, H. Markert, C. H. Quinn, N. Wadhwani, J. M. Aye, J. E. Stewart, K. J. Yoon, E. Mroczek-Musulman and E. A. Beierle, PLoS One, 2021, 16, e0246244 CrossRef CAS PubMed.
- N. Tiwari, V. K. Tiwari, L. Waldmeier, P. J. Balwierz, P. Arnold, M. Pachkov, N. Meyer-Schaller, D. Schübeler, E. van Nimwegen and G. Christofori, Cancer Cell, 2013, 23, 768–783 CrossRef CAS PubMed.
- Z. Li, M. Li, D. Wang, P. Hou, X. Chen, S. Chu, D. Chai, J. Zheng and J. Bai, Cell Biosci., 2020, 10, 143 CrossRef CAS PubMed.
- Z. Li, P. Hou, D. Fan, M. Dong, M. Ma, H. Li, R. Yao, Y. Li, G. Wang, P. Geng, A. Mihretab, D. Liu, Y. Zhang, B. Huang and J. Lu, Cell Death Differ., 2017, 24, 59–71 CrossRef CAS PubMed.
- V. R. Ruvolo, K. B. Karanjeet, T. F. Schuster, R. Brown, Y. Deng, E. Hinchcliffe and P. P. Ruvolo, J. Signal Transduction, 2010, 2010, 584657 Search PubMed.
- B. Marengo, C. De Ciucis, R. Ricciarelli, M. Passalacqua, M. Nitti, J. M. Zingg, U. M. Marinari, M. A. Pronzato and C. Domenicotti, PLoS One, 2011, 6, e14661 CrossRef CAS PubMed.
- M. G. Villani, V. Appierto, E. Cavadini, A. Bettiga, A. Prinetti, M. Clagett-Dame, R. W. Curley and F. Formelli, Cancer Res., 2006, 66, 3238–3247 CrossRef CAS PubMed.
- P. Tiberio, E. Cavadini, L. Cleris, S. Dallavalle, L. Musso, M. G. Daidone and V. Appierto, Front. Pharmacol., 2017, 8, 226 CrossRef PubMed.
- P. Tiberio, E. Cavadini, G. Abolafio, F. Formelli and V. Appierto, PLoS One, 2010, 5, e13362 CrossRef PubMed.
- L. Musso, P. Tiberio, V. Appierto, R. Cincinelli, E. Cavadini, L. Cleris, M. G. Daidone and S. Dallavalle, Chem. Biol. Drug Des., 2016, 88, 608–614 CrossRef CAS PubMed.
- P. Ma and R. J. Mumper, J. Nanomed. Nanotechnol., 2013, 4, 1000164 Search PubMed.
- E. Miele, G. P. Spinelli, E. Miele, F. Tomao and S. Tomao, Int. J. Nanomed., 2009, 4, 99–105 CrossRef CAS.
- G. Dranitsaris, W. Cottrell, B. Spirovski and S. Hopkins, J. Oncol. Pharm. Pract., 2009, 15, 67–78 CrossRef CAS PubMed.
- H. Onafuye, S. Pieper, D. Mulac, J. Cinatl Jr., M. N. Wass, K. Langer and M. Michaelis, Beilstein J. Nanotechnol., 2019, 10, 1707–1715 CrossRef CAS PubMed.
- K. Nieto, P. Pei, D. Wang, S. R. Mallery and S. P. Schwendeman, Int. J. Pharm., 2018, 538, 48–56 CrossRef CAS PubMed.
- M. J. Cecchini, M. Amiri and F. A. Dick, J. Visualized Exp., 2012, 59 DOI:10.3791/3491.
- H. Wang and J. A. Joseph, Free Radicals Biol. Med., 1999, 27, 612–616 CrossRef CAS PubMed.
- H. J. de Jonge, R. S. Fehrmann, E. S. de Bont, R. M. Hofstra, F. Gerbens, W. A. Kamps, E. G. de Vries, A. G. van der Zee, G. J. te Meerman and A. ter Elst, PLoS One, 2007, 2, e898 CrossRef PubMed.
- A. Dev, M. N. Sardoiwala, M. Boddu, S. J. MohanBhai, S. R. Choudhury and S. Karmakar, ACS Appl. Nano Mater., 2022, 5, 7540–7548 CrossRef CAS.
- C. Weber, J. Kreuter and K. Langer, Int. J. Pharm., 2000, 196, 197–200 CrossRef CAS PubMed.
- N. Qu, Y. Sun, Y. Li, F. Hao, P. Qiu, L. Teng, J. Xie and Y. Gao, Biomed. Eng., 2019, 18, 11 Search PubMed.
- L. Qi, G. Ji, Z. Luo, Z. Xiao and Q. Yang, ACS Sustainable Chem. Eng., 2017, 5, 9517–9526 CrossRef CAS.
- G. Katona, G. T. Balogh, G. Dargó, R. Gáspár, Á. Márki, E. Ducza, A. Sztojkov-Ivanov, F. Tömösi, G. Kecskeméti, T. Janáky, T. Kiss, R. Ambrus, E. Pallagi, P. Szabó-Révész and I. Csóka, Pharmaceutics, 2020, 12(2), 97 CrossRef CAS PubMed.
- Y. Wang, P. Wu, X. Zhou, H. Zhang, L. Qiu and J. Cao, J. Photochem. Photobiol., B, 2016, 162, 611–618 CrossRef CAS PubMed.
- Z. Matloubi and Z. Hassan, Daru, J. Pharm. Sci., 2020, 28, 209–219 CrossRef CAS PubMed.
- K. E. Wong, S. C. Ngai, K. G. Chan, L. H. Lee, B. H. Goh and L. H. Chuah, Front. Pharmacol., 2019, 10, 152 CrossRef CAS PubMed.
- H. H. Ali and A. A. Hussein, AAPS Open, 2017, 3, 4 CrossRef.
- S. M. Vogel, R. D. Minshall, M. Pilipović, C. Tiruppathi and A. B. Malik, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2001, 281, L1512–L1522 CrossRef CAS PubMed.
- A. C. Kushwaha, B. Kaundal, A. Dev, A. K. Srivastava, S. J. Mohanbhai, S. Karmakar and S. R. Choudhury, Appl. Mater. Today, 2020, 21, 100847 CrossRef.
- D. Ding, X. Tang, X. Cao, J. Wu, A. Yuan, Q. Qiao, J. Pan and Y. Hu, AAPS PharmSciTech, 2014, 15, 213–222 CrossRef CAS PubMed.
- A. Spada, J. Emami, J. A. Tuszynski and A. Lavasanifar, Mol. Pharm., 2021, 18, 1862–1894 CrossRef CAS PubMed.
- Z. Pan, X. Zhang, P. Yu, X. Chen, P. Lu, M. Li, X. Liu, Z. Li, F. Wei, K. Wang, Q. Zheng and D. Li, Front. Oncol., 2019, 9, 853 CrossRef PubMed.
- M. Nita and A. Grzybowski, Oxid. Med. Cell. Longevity, 2016, 2016, 3164734 Search PubMed.
- H. J. Kim, N. Chakravarti, N. Oridate, C. Choe, F. X. Claret and R. Lotan, Oncogene, 2006, 25, 2785–2794 CrossRef CAS PubMed.
- J. A. Simon and C. A. Lange, Mutat. Res., 2008, 647, 21–29 CrossRef CAS PubMed.
- Y.-C. Yang, C.-Y. Tsai, c. l. Chen, C.-H. Kuo, C.-W. Hou, S.-Y. Cheng, R. Aneja, C.-Y. Huang and W.-W. Kuo, Aging Dis., 2018, 9, 647–663 CrossRef PubMed.
- X. Yin, S. Yang, M. Zhang and Y. Yue, Biomed. Pharmacother., 2019, 118, 109384 CrossRef CAS PubMed.
- T. A. DeVries, M. C. Neville and M. E. Reyland, EMBO J., 2002, 21, 6050–6060 CrossRef CAS PubMed.
- J. Gao, C. Fosbrook, J. Gibson, T. J. Underwood, J. C. Gray and Z. S. Walters, Cancer Treat. Rev., 2023, 119, 102600 CrossRef CAS PubMed.
- S. R. Cerda, R. Mustafi, H. Little, G. Cohen, S. Khare, C. Moore, P. Majumder and M. Bissonnette, Oncogene, 2006, 25, 3123–3138 CrossRef CAS PubMed.
- Y. Tu, C. Chen, J. Pan, J. Xu, Z. G. Zhou and C. Y. Wang, Int. J. Clin. Exp. Pathol., 2012, 5, 726–738 CAS.
- A. Dev, M. N. Sardoiwala, A. Sharma, S. J. MohanBhai, S. Karmakar and S. R. Choudhury, ACS Med. Chem. Lett., 2022, 13, 1109–1117 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |

 ,
Atul
Dev
,
Avinash Chandra
Kushwaha
,
Mohammed Nadim
Sardoiwala
and
Surajit
Karmakar
,
Atul
Dev
,
Avinash Chandra
Kushwaha
,
Mohammed Nadim
Sardoiwala
and
Surajit
Karmakar