
Macrophage-modulating nanomedicine for cancer immunotherapy
Muhammad Muzamil
Khan†
 a,
Yongjiang
Li†
a,
Zhuoming
Zhou†
a,
Abigale
Ni
a,
Qimanguli
Saiding
a,
Duotian
Qin
a,
Wei
Tao
*a and
Wei
Chen
*ab
a,
Yongjiang
Li†
a,
Zhuoming
Zhou†
a,
Abigale
Ni
a,
Qimanguli
Saiding
a,
Duotian
Qin
a,
Wei
Tao
*a and
Wei
Chen
*ab
aCenter for Nanomedicine and Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA. E-mail: wchen123@gate.sinica.edu.tw; wtao@bwh.harvard.edu
bGenomics Research Center, Academia Sinica, Taipei 11529, Taiwan
First published on 5th March 2024
Abstract
Tumor-associated macrophages (TAMs) play crucial roles in the immunosuppressive solid tumor microenvironment (TME). Despite their tumor-promoting functions, TAMs can also be therapeutically modulated to exhibit tumor-killing properties, making them attractive targets for tumor immunotherapy. This review highlights the recent advances in nanomedicine-based strategies centered around macrophages for enhanced cancer immunotherapy. Emerging nanomedicine-based strategies to modulate TAMs in cancer treatment include repolarization of the TAM phenotype, inhibition of monocyte recruitment, depletion of TAMs, and blockage of immune checkpoints. These strategies have shown great promise in significantly improving the efficacy of cancer immunotherapy. Moreover, macrophage-inspired drug delivery systems have demonstrated significant promise in inducing immunotherapeutic effects and enhancing therapeutic efficacy by facilitating evasion from the reticuloendothelial system and promoting accumulation at the tumor site. Finally, we also discuss the challenges and propose future opportunities associated with macrophage-modulating nanomedicine to enhance cancer immunotherapy.
 Wei Chen | Professor Wei Chen received his B.S. and M.S. degrees in Chemistry from National Taiwan University. He then received his Ph.D. degree in Chemistry from the University of California Los Angeles (UCLA) under the supervision of Prof. Jeffrey I. Zink. Following this, he joined Prof. Wei Tao's and Prof. Omid Farokhzad's labs at Brigham and Women's Hospital, Harvard Medical School as a research fellow. He is currently an assistant professor and principal investigator at the Genomics Research Center, Academia Sinica. His research interests include nanomedicine and biomaterials, focusing on their applications in the treatment of cancer, cardiovascular diseases, and other inflammatory diseases. |
1. Introduction
The intricate solid tumor microenvironment (TME) comprises a substantial proportion of stromal cells, including endothelial cells, cancer-associated fibroblasts, tumor-associated macrophages (TAMs), etc.1 The immunosuppressive nature of the TME presents a major obstacle in fully realizing the therapeutic potential of immunotherapies. In addition, a majority of solid tumors are characterized by dense stroma comprising collagen fibers and fibroblasts, which establish a barrier that hinders the intratumoral infiltration of immunostimulatory cells and therapeutic agents and the effective eradication of tumor cells.2 Within the immunosuppressive TME, TAMs play multifaceted roles in tissue homeostasis, contributing to diverse functions such as clearance, phagocytosis, and inflammation regulation; these functions can be harnessed as potential approaches for tumor immunotherapy.3,4 Major TAMs in the TME are pro-tumoral; TAMs play a crucial role in suppressing adaptive immunity and promoting tumor-specific immunosuppression. Moreover, TAMs contribute to tumor progression through diverse mechanisms, including the promotion of metastasis and genetic instability, assistance in the maturation of cancer stem cells, and modulation of adaptive immunity. Furthermore, TAMs play a crucial role in cancer-related inflammation (CRI).3 Consequently, the correlation between poor clinical outcomes resulting from the presence of immunosuppressive TAMs in the TME underscores the critical role of TAMs as potential targets for improving therapeutic outcomes in cancer treatment.Nanomedicine has emerged as a promising therapeutic for cancer treatment5 and various pre-clinical investigations have demonstrated the potential of nanomaterials by modulating TAMs and facilitating cancer immunotherapy. Specifically, the biochemical and physical characteristics of nanoparticles can be rationally designed and modified to enhance the effective delivery of therapeutic payloads, such as nucleic acids, chemotherapeutics, proteins, and peptides.6–9 Importantly, the complicated tumor microenvironment, comprising diverse components such as the extracellular matrix, neovessels, cancer cells, macrophages, and their secreted cytokines, provides an opportunity for leveraging nanomedicine in targeting and modulating TAMs for achieving effective cancer immunotherapy. This review provides a concise overview of the roles of macrophages in the pathophysiology of cancers, emphasizing the application of nanomedicine to modulate these macrophages effectively for cancer immunotherapy. Furthermore, we discuss the current promising nanomedicine-based strategies focused on modulating macrophages to treat cancers. We also present perspectives on the challenges and potential opportunities encountered in the application of nanomedicine to modulate macrophages for cancer immunotherapy. We envision that an extensive understanding of cancer immunology, the fusion of nanotechnology, and accumulated experience in macrophage-modulating nanotherapeutics will drive breakthroughs against cancer through nanomedicine.
2. Role of macrophages in tumor biology
TAMs typically originate from the bone marrow-derived monocytes that are recruited into the TME.1 They are commonly identified by their expression of chemokines categorized as either inducible or inflammatory. Specifically, CC-chemokine ligand 2 (CCL2) interacts with CCR2 and recruits monocytes that differentiate into TAMs. This specific CC-chemokine is the most prevalent chemokine subtype and is presented highly within the TME of diverse tumor types, including gliomas, sarcomas, carcinomas, and melanomas. Furthermore, CCL2 plays a role in prolonging the retention of metastatic-associated macrophages (MAMs) by boosting the secretion of CCL2 at metastatic sites.10,11Macrophages can be polarized by various stimuli into M1- or M2-like phenotypes, capable of secreting various cytokines.12 For example, interferon-γ (IFN-γ) is known to induce M1-like macrophage polarization, characterized by the secretion of pro-inflammatory cytokines such as IL-2, IL-23, and tumor necrosis factor-α (TNF-α), and the production of nitric oxide (NO). M1-like TAMs also express elevated levels of major histocompatibility complex class II (MHC II), CD80, CD86, and inducible nitric oxide synthase (iNOS), contributing to anti-tumor responses. Conversely, IL-4 and colony-stimulating factor 1 (CSF-1) are known inducers of M2-like macrophages, which secrete anti-inflammatory cytokines such as IL-10 and transforming growth factor β (TGF-β), promoting the immunosuppressive TME. This polarization plays a crucial role in shaping the immune microenvironment and influencing the therapeutic outcome. TAMs are major immune cells associated with the tumor microenvironment, playing a “double-edged sword” role in tumor metastasis and immunosuppression. Under the influence of various cytokines, TAMs have the ability to switch their phonotype. Researchers have leveraged this property to design drugs targeting this regulation with a particular focus on the polarization to an anti-tumor phenotype. This strategy holds promise as an immunotherapy approach.13
Moreover, TAMs play a significant role in shaping the immunobiology of tumors, particularly in angiogenesis and immune suppression.14–16 Specifically, tumor-promoting TAMs generate growth factors that can facilitate tumor growth by promoting proliferation, angiogenesis, metastasis, and the dissolution of connective tissues. Through the release of enzymes like matrix metalloproteinases (MMPs), TAMs can degrade components of the extracellular matrix (ECM), thereby facilitating tumor invasion and metastasis. MMPs can create a dynamic environment that supports the remodeling and expansion of the vascular network during angiogenesis in the TME. For example, MMP-9 plays a crucial role in angiogenesis by breaking down the ECM components around blood vessels, facilitating the migration and proliferation of endothelial cells. In addition, MMP-9 promotes the release of growth factors, stimulating the formation of new blood vessels during angiogenesis. While TAMs can facilitate angiogenesis, they can also express anti-angiogenic molecules that inhibit blood vessel growth, such as MMP-12.1,3,4 TAMs can indirectly promote angiogenesis by producing pro-angiogenic growth factors that stimulate tumor growth. This occurs through the accumulation of TAMs in the TME characterized by low oxygen tension due to poor vascularization. Under such conditions, macrophage migration is hindered and their activity is restricted in the avascular and necrotic areas of tumors. This circumstance compels TAMs to collaborate with tumor cells and promote angiogenesis.1 The elevated expression of hypoxia-inducible factor-2a (HIF-2a) has been evidenced in TAMs. HIF-2a as a transcription factor directly induces the production of multiple growth factors, including CXCL8 and vascular endothelial growth factor (VEGF), both of which significantly promote angiogenesis. In addition, pro-tumoral TAMs exhibit reduced expression of positive co-stimulatory molecules compared to anti-tumoral TAMs, resulting in compromised antigen presentation. In contrast, anti-tumoral TAMs can aid in the eradication of early-stage tumors by activating T cells and producing interferons such as interferon-γ secreted by CD8+ T cells, leading to the promotion of M1-like macrophage phenotype that helps tumor eradication.
These TAM-involved processes have been considered compelling therapeutic targets for tumor therapy. However, in TAM-mediated therapy, conventional therapeutics often encounter significant challenges due to limited specificity, high systemic toxicity, and the complex nature of TAM subpopulations within the TME. In recent years, nanoparticles (NPs) have demonstrated superiority in cancer therapy due to their adjustable shape and size, enhancing macrophage phagocytosis for improved targeting and preferential accumulation at tumor sites.7 Currently, nanomedicine-based approaches targeting TAMs for cancer immunotherapy include macrophage polarization from M2 phenotype to M1 to induce tumor inhibitory effects, inhibition of monocyte recruitment by blocking the chemokine signaling pathway and reshaping the TME, depletion of pro-tumoral TAMs by inducing apoptosis, and blocking immune checkpoints to restore phagocytosis of tumor cells (Fig. 1).
 | ||
| Fig. 1 Strategies for macrophage-modulating nanomedicine in cancer immunotherapy include five primary approaches: (i) polarization of macrophages; (ii) inhibition of monocyte recruitment; (iii) depletion of TAMs; (iv) blockage of immune checkpoints; and (v) macrophage-inspired drug delivery system. The figure was created using BioRender. | ||
3. Surface design consideration of the in vivo fate of NPs
The NPs after intravenous administration into the body interact with biomolecules, proteins, and lipids before reaching the tumor site. The NPs interact with various serum proteins, giving rise to a layer termed protein corona (PC),17,18 which impacts various aspects of the in vivo fate of NPs for targeted drug delivery.The PC on the NP surface influences the circulation time of NPs.19 For example, opsonins such as coagulation proteins, immunoglobulins and tissue leakage proteins absorbed on NPs may shorten the circulation time of NPs and facilitate their recognition and phagocytosis by antigen-presenting cells such as dendritic cells and macrophages. For example, immunoglobulin G (IgG) can increase the phagocytosis through recognition of the immunoglobulin Fc portion on PC by Fc receptors on the surface of phagocytic cells. On the other hand, dysopsonins such as apolipoprotein and albumin can help in blocking phagocytosis during circulation to increase the half-life of NPs.20 Therefore, the quantity and composition of opsonins and dysopsonins in PC determine the circulation time of NPs in an interactive manner.
In addition to influencing their circulation time, NP surfaces can be engineered to have a specific PC, enabling more targeted delivery for cancer immunotherapy. For instance, Bai et al. designed multiwalled carbon nanotubes (MWCNTs) and modified the surface with albumin to mimic a PC, facilitating the delivery of ovalbumin (OVA) as an antigen to macrophages.21 The results showed that this strategy led to an increased expression of MHC II on macrophages, which in turn triggered the secretion of pro-inflammatory cytokines such as IL-6 and TNF-α, activating CD8+ T cells and enhancing antitumor effects. The utilization of the PC on NPs shows promise in cancer immunotherapy through the development of NPs with specific materials. In this regard, graphdiyne oxide (GDYO) NPs have shown efficacy.22 They efficiently impede the entry of STAT3 into the cell nucleus by absorbing STAT3 through PC formation. This unique property of GDYO NPs successfully inhibits the expression of M2-like TAMs, ultimately leading to improved immunotherapeutic effects. Collectively, through surface modification, the in vivo destination of NPs and their delivery efficacy could be adjusted to enhance macrophage-mediated anti-tumor therapies.
In addition to the PC, NPs can be coated with the cell membrane or targeting ligands to enhance their accumulation in TAMs for enhanced macrophage-mediated cancer immunotherapy. For example, NPs coated with the cell membrane derived from TAMs enhance the biocompatibility of NPs and avoid clearance by the reticuloendothelial system, ensuring the accumulation of nanoparticles in the tumor microenvironment by the TAM homing effect.23 In another study, PLGA NPs were coated with M2pep, a peptide ligand that selectively targets M2-like macrophages, inducing tumor growth inhibition. Moreover, the M2pep peptide has shown toxicity against M2-like macrophages and extended overall survival in tumor-bearing mice.24 Similarly, glycocalyx-mimicking nanoparticles (GNPs) have proven effective in reprograming TAMs and improving anti-PD-L1-mediated immunotherapy. The GNPs internalize into TAMs via lectin receptors, causing reprogramming of TAMs towards an antitumor phenotype. In vivo studies show that GNPs can significantly enhance the therapeutic potential of αPD-L1 cancer immunotherapy by inhibiting tumor growth.25 These studies highlight the promise of surface-modified NPs for enhancing TAM-mediated cancer immunotherapy.
4. Macrophage-modulating strategies
4.1 Polarization of TAMs
Polarizing TAMs to an antitumoral phenotype is a promising strategy for immunosuppressive tumor treatment. Numerous innovative nanomaterials have been found to induce the polarization of macrophages toward an anti-tumor phenotype. Among them, iron oxide NPs (IONPs) have been discovered to have the ability to shift M2-like macrophages towards an M1-like phenotype, making them attractive candidates for reprogramming the TME. For example, ferumoxytol, an ultrasmall superparamagnetic iron oxide NP, an FDA-approved iron supplement, has shown potential for inhibiting tumor growth and metastasis.26In vitro experiments involving adenocarcinoma cells incubated with ferumoxytol along with macrophages revealed heightened caspase-3 activity, suggesting a potential mechanism through which ferumoxytol could induce apoptosis in these cancer cells. This study demonstrated that the administration of ferumoxytol led to a notable reduction in tumor growth in mice with adenocarcinoma, with an increased population of intra-tumoral M1-like macrophages. The study concluded that the administration of ferumoxytol caused M1-like macrophage polarization, leading to the inhibition of tumor growth. Moreover, it has been recently reported that magnetite-type IONPs have greater efficiency in polarizing macrophages into the M1-like phenotype than hematite-type IONPs.27 The oxidative stress induced by iron ions could be the potential mechanism for inducing M1-like polarization. In addition, magnetite NPs can inhibit arginase-1 (Arg-1) and activate interferon regulatory factor 5 (IRF5), subsequently inhibiting the functions of M2-like TAMs and suppressing tumor growth. Specifically, magnetite NP treatment led to higher levels of M1-like surface markers (CD64, CD80, and CD84) on TAMs than hematite NP treatment. These studies underscore the potential of inorganic NPs in polarizing TAMs and boosting macrophage-mediated anti-tumor immunotherapy.Recently, an innovative “nanodrug-delivering-drug” strategy based on 2D stanene-based nanosheets (STNSP) loaded with β-elemene (ELE) (STNSP@ELE) demonstrated enhanced chemo-immunotherapeutic effects (Fig. 2).28 This study illustrated that both STNSP and ELE can reprogram tumor-supportive macrophages into a tumor-suppressive phenotype through an intracellular reactive oxygen species (ROS)-activating manner. The results from a melanoma mouse model indicated that STNSP@ELE treatment successfully reprogrammed the TME by significantly elevating the ratio of M1/M2-like TAMs and increasing the populations of CD4+ and CD8+ T-lymphocytes and dendritic cells, leading to a robust anti-tumor immunostimulatory response. This study highlights the effectiveness of the STNSP-based nanodrug-delivery approach for treating immunosuppressive tumors and establishes a versatile platform for 2D nanomaterial-driven cancer chemo-immunotherapy.
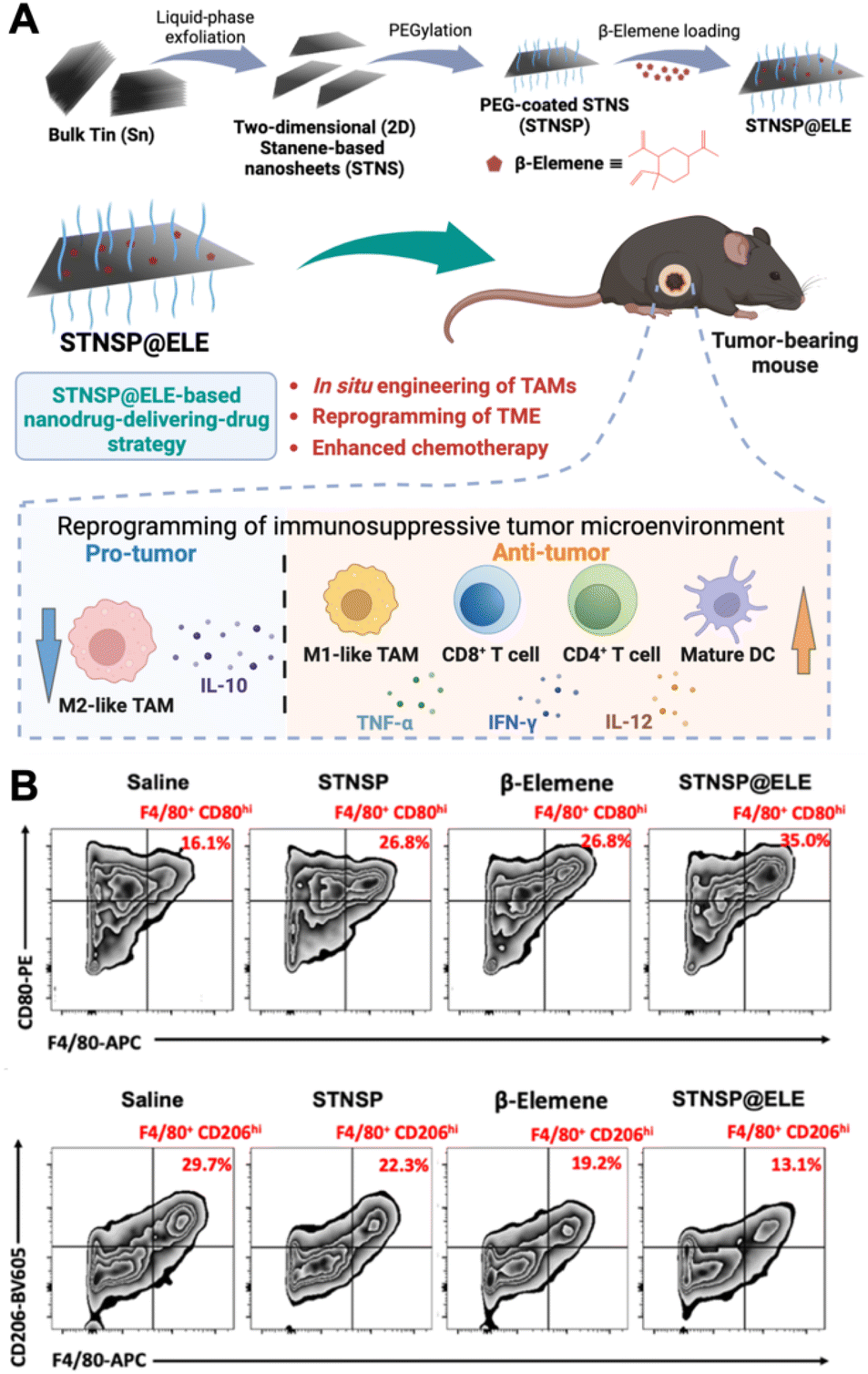 | ||
| Fig. 2 Example of nanomedicine modulating TAM polarization. (A) Schematic illustration of β-elemene-loaded 2D stanene-based nanosheets (STNSP@ELE) for macrophage-mediated cancer chemo-immunotherapy. (B) STNSP@ELE treatment in vivo reversed the immunosuppressive tumor microenvironment by increasing M1-like macrophages and reducing M2-like macrophages within tumors. Reprinted with permission from ref. 28. Copyright 2023 John Wiley & Sons. | ||
Beyond inorganic NPs, organic NPs have shown the ability to carry macrophage-modulating molecules, boosting the macrophage-mediated anti-tumor response. For example, dextran-based NPs exhibit a natural affinity for macrophages, making them promising candidates for macrophage-modulated immunotherapy. In a recent study, β-cyclodextrin NPs (CDNPs) were synthesized through amide bond formation between L-lysin and β-cyclodextrin.29 The delivery of R848 (an agonist of toll-like receptors 7 and 8 (TLR7/8)) and anti-PD1 (an immune checkpoint inhibitor) by CDNPs induced the repolarization of TAMs to an anti-tumoral phenotype, effectively inhibiting tumor growth. Notably, this combination treatment with anti-PD-1 demonstrated improved immunotherapeutic responses, even in mouse models resistant to anti-PD1 therapy.
4.2 Inhibition of monocyte recruitment
Experimental data suggest that the recruitment of monocytes to tumor tissue, their subsequent differentiation into macrophages, and transformation into TAMs constitute a critical process supporting tumor progression. Therefore, disrupting monocyte recruitment holds promise for enhancing immunotherapy responses. A nanomedicine-based approach entails blocking monocyte recruitment to tumor tissue by inhibiting the chemokine signaling pathway through the use of antibodies or small molecule inhibitors. The CCL2/CCR2 signaling pathway is pivotal in circulatory monocyte infiltration into the TME; inhibiting this pathway can significantly boost the antitumor efficacy.30 Studies show that silencing CCL2 using neutralizing antibodies or antagonists inhibits monocyte recruitment, resulting in reduced TAMs and elevated levels of CD8+ and NK cells in the TME.31 Furthermore, cationic NPs composed of PEG–PLA polymer and BHEM-Chol lipid have demonstrated the ability to target circulating monocytes.32 The delivery of siRNA-targeting CCR-2 and DOX successfully inhibited monocyte infiltration into the TME, enhancing chemo-immunotherapeutic effects by disrupting the CCL2–CCR2 signaling axis. Moreover, precise targeted blockage of the CCR2 signaling axis leads to an enhanced immunotherapeutic effect. For example, lipid NPs composed of cationic lipid C-12, DSPC, cholesterol, and DMG-PEG delivered siRNA-targeting CCR-2 to the tumor site. This approach efficiently inhibited CCR2, preventing the accumulation of monocyte recruitment and subsequently reducing the tumor volume.334.3 Depletion of TAMs
The depletion of TAMs is a particularly advantageous strategy to suppress tumor growth achievable through various approaches, including apoptosis and blocking the CSF-1/CSF-1R pathway. Several compounds, such as zoledronate, trabectedin, and clodronate, have demonstrated notable efficacy in inducing TAM apoptosis.34 In addition, bisphosphonates have the ability to eliminate myeloid cells, are readily phagocytized by osteoclasts, and are utilized for preventing bone metastasis.35 For example, clodronate-loaded liposomes have demonstrated the ability to induce apoptosis in TAMs in a dose- and time-dependent manner in a mouse model of hepatocellular carcinoma.36 In a separate study using a KaLwRij mouse model of myeloma, pretreatment with clodronate liposomes effectively eliminated tumors and depleted macrophages within the bone marrow.37 Flow cytometry analysis revealed that liposome pretreatment impaired the retention of myeloid cells in the bone marrow. In addition, in hepatocellular carcinoma xenograft mouse models, the administration of liposomes containing a macrophage depletion agent (clodronate or zoledronic acid) along with a chemotherapeutic agent sorafenib overcame the side effects of sorafenib while increasing the intratumoral infiltration of CD11b+ and F4/80+ cells.38 The study demonstrated that TAM depletion by zoledronic acid or clodronate, along with sorafenib, significantly enhanced the inhibition of tumor growth and metastasis compared to sorafenib or zoledronic acid alone.In another study, immune modulating NPs (IMNPs) coated with mannose were designed for the co-delivery of alendronate (ALN) and bindarit (BIN) to achieve optimal TAM depletion.39 IMNPs were synthesized by conjugating chitosan with ALN, crosslinking it with sodium tripolyphosphate (STPP) to form chitosan NPs, and finally coating these NPs with mannose and a phenylboronic acid-conjugated polymer to prepare IMNPs. These IMNPs effectively suppressed monocyte recruitment and concomitantly released alendronate, enabling TAM depletion. The spatial delivery of BIN and ALN resulted in the effective depletion of TAMs, enhancing the immunotherapeutic effect. Similarly, calcium NPs loaded with zoledronate were coated with lipids and mannose for site-specific delivery of zoledronate (CaZol@pMNPS) to induce apoptosis of TAMs.40 In the S180 tumor mouse model, CaZol@pMNPS significantly reduced TAMs and inhibited angiogenesis, leading to diminished tumor growth. In addition, Li et al. designed a biocompatible alginate-based hydrogel incorporating pexidartinib-encapsulated NPs and platelets conjugated with anti-PD-1 (P-aPD-1).41 The gradual release of pexidartinib-encapsulated NPs at the tumor site enabled continuous blockage of colony-stimulating factor 1 receptors, effectively depleting TAMs. This created a favorable environment to enhance the subsequent delivery of P-aPD-1, inhibiting post-surgery tumor recurrence in mouse models of S180 and B16F10 tumor recurrence (Fig. 3).
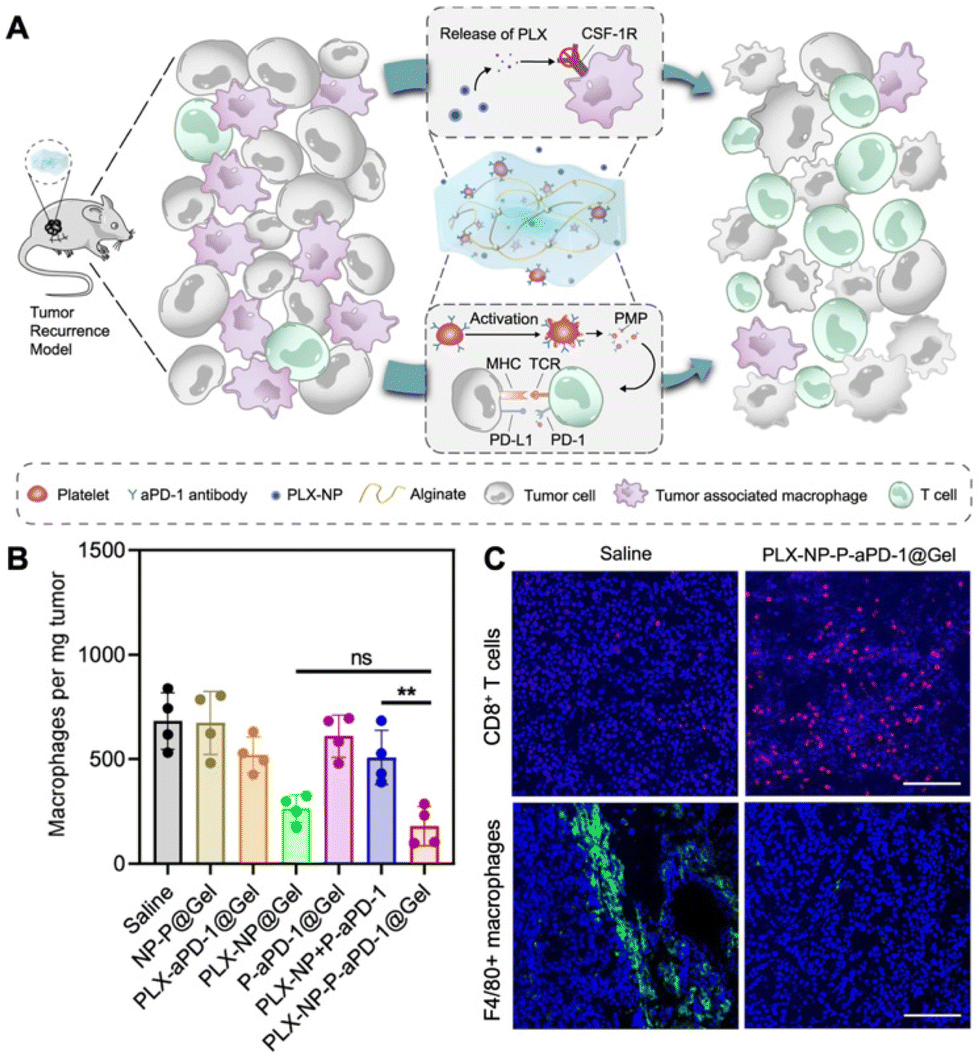 | ||
| Fig. 3 Example of nanomedicine for TAM depletion. (A) Schematic illustration demonstrating modulation of the immunosuppressive tumor microenvironment using pexidartinib (PLX)-encapsulated NPs (PLX-NPs) and anti-PD-1-conjugated platelets (P-αPD-1) loaded into an alginate-based hydrogel in the tumor recurrence model. (B) Quantitative analysis of F4/80+ macrophages per tumor mass after different treatments in recurrent tumor tissues. (C) Confocal microscopy images comparing immune-stained CD8+ T cells and TAMs between the saline group and the PLX-NP-P-αPD-1@Gel group. Reprinted with permission from ref. 41. Copyright 2022 Springer Nature. | ||
4.4 Blockage of immune checkpoints
CD47 serves as an immune checkpoint for macrophages, recognized by signal regulatory protein alpha (SIRPα) abundantly expressed on the surface of myeloid cells, including macrophages, monocytes, and dendritic cells. CD47 is expressed in red blood cells and most cancer cells.42 For example, the use of CD47 antibodies in treatment restores the phagocytic function of macrophages toward tumor cells.43 By blocking the CD47-SIRPα interaction, macrophage-mediated phagocytosis of tumor cells can be reinstated, leading to reduced tumor size, as observed in cholangiocarcinoma. Notably, inhibiting the CD47-SIRPα interaction and restoring phagocytosis remain effective in immune-compromised mice, and this effect is further enhanced when PD-L1 and CD47 are simultaneously blocked.44In another study, calcium carbonate NPs were synthesized and loaded with an anti-CD47 antibody.45 Recognizing the significant challenge of post-surgical tumor recurrence in clinical settings, these NPs were encapsulated in a fibrin gel for in situ application at the surgical wound site, aiming to promote the polarization of TAMs toward an M1-like phenotype. This TAM reprogramming facilitated a robust T cell-mediated antitumor immune response. The results demonstrated that this nanomedicine-based in situ engineering strategy effectively triggered both adaptive and innate immune responses, inhibiting post-surgical tumor recurrence and metastatic spread.
While immune checkpoint blockade therapy represents a promising option for robust immunotherapy, its effectiveness is often limited by a lack of suitable delivery vehicles. In a separate approach, silver NPs coated with sucrose (S-AgNPs) were prepared for the delivery of PD-1 monoclonal antibodies in a melanoma mouse model.46 The findings indicated that S-AgNPs exhibited an enhanced antitumor effect by inducing apoptosis and promoting cytotoxic CD8+ T cell infiltration in the melanoma mouse model. Furthermore, the combination of immune checkpoint blockade inhibitors with chemotherapeutic agents, leading to synergistic antitumor effects, holds great promise in tumor treatment. In an illustrative example, cancer cell biomimetic NPs were engineered for the co-delivery of chemotherapeutic drugs (RA-V) and a PD-1/PD-L1 blockade inhibitor (BMS-202).47 The results demonstrated that BMS/RA@CC-liposomes led to PD-L1 downregulation and induced apoptosis in cancer cells. This system effectively sensitizes hypoxic tumors to the immunotherapeutic effects induced by checkpoint blockade inhibitors.
4.5 Macrophage-inspired delivery systems
The cell membrane coating of NPs presents a promising strategy to improve circulation and achieve targeted delivery to tumors. It has been reported that NPs coated with the TAM membrane (TAMM) exhibit high biocompatibility and enhance immunotherapeutic effects. Specifically, NPs conjugated with a photosensitizer and coated with TAMM (NPR@TAMM) result in an augmented photodynamic immunotherapy effect (Fig. 4).48 This therapeutic strategy induces the polarization of TAMs from an M2-like to an M1-like phenotype, thereby enhancing antitumor immunity. In a separate study, macrophage membrane-camouflaged bismuth selenide NPs encapsulated with quercetin, termed M@BS-QE NPs, were developed for the treatment of breast cancer.49 Engineered M@BS-QE NPs exhibited prolonged circulation life and enhanced tumor tropism compared to their uncoated counterparts, attributed to immune system escape and the CCL-2-mediated recruitment properties of the membrane. This innovative strategy down-regulated the expression of p-Akt and MMP-9 and inhibited metastasis, positioning M@BS-QE NPs as a promising delivery system for breast tumor treatment in a mouse model. | ||
| Fig. 4 Macrophage membrane-coated NPs for tumor therapy. (A) Schematic illustration of TAM membrame (TAMM) coating on upconversion NPs with a conjugated photosensitizer (NPR@TAMMs). (B) In vitro experiments demonstrating the efficient uptake of NPR@TAMMs by tumor cells, inducing cell death after near-infrared (NIR) light irradiation. (C) In vivo anti-tumor therapeutic experiments reveal that NPR@TAMMs significantly suppress tumor aggression and improve survival in the 4T1 tumor-bearing mouse model. Reprinted with permission from ref. 48. Copyright 2021 American Chemical Society. | ||
The effectiveness of membrane-coated NPs can be further amplified by employing membranes derived from two or more cell sources. For example, Gong et al. demonstrated this by preparing pH-responsive poly(lactic-co-glycolic acid) (PLGA) NPs coated with hybrid membranes from macrophages and cancer cells for the co-delivery of metformin and siRNA-targeting fibrinogen-like protein-1 mRNA.50 The hybrid coating strategy facilitated NPs escape from reticuloendothelial system phagocytosis, prolonged circulation time, and enabled effective cytosolic delivery of siRNA and metformin, enhancing tumor immunotherapy. Similarly, PLGA NPs coated with hybrid membranes from RAW264.7 macrophages and breast cancer 4T1 cells were prepared for doxorubicin delivery.51 This approach showcased the advantage of targeting specific tumors and metastases with a propensity to accumulate at inflammation sites. The NPs exhibited excellent anti-metastasis effects and superior therapeutic outcomes against breast cancer both in vitro and in vivo. The innovative use of hybrid membrane-coated NPs holds the potential for targeted and effective breast cancer treatment.
Moreover, macrophage membrane coating nanotechnology extends to various NP types. In another study, HA-modified poly (histidine) polymeric NPs (DHP@M2) were coated with macrophage membranes for doxorubicin delivery.52 This macrophage-camouflaged nanoplatform exhibited specific accumulation at the tumor site, further depleting M2-like macrophages by absorbing CSF-1 and inhibiting the progression of 4T1 tumors in a mouse model through enhanced infiltration of cytotoxic T lymphocytes. In a different application, M1-like macrophage-derived exosomes were employed for miR-192-5p delivery in the treatment of endometrial cancer.53 The results demonstrated that the enhanced delivery of miR-192-5p promoted tumor cell death by inhibiting epithelial–mesenchymal transition through impeding the 1RAK1/NF-κB signaling pathway.
5. Challenges and perspectives
The field of macrophage-modulating nanomedicine for cancer immunotherapy has demonstrated effectiveness across various cancers; nevertheless, several challenges impede its translation into clinical practice. First, a substantial gap exists in tumor biology between murine models and humans, for instance, as evidenced by the lack of success in clinical trials targeting anti-CSF-1R to deplete TAMs. It is imperative to discern whether the anti-CSF-1R strategy inadvertently depletes “beneficial” macrophages. Moreover, the limited understanding of the functionality of antigen-presenting macrophages and immunosuppressive myeloid cells poses another obstacle. Third, the intricate interplay between the TME and the ECM, with numerous biochemical elements involved, adds an extra layer of complexity to the design of suitable nanomaterials. Moreover, the TME in animal models often substantially differs from that in human patients;54 another significant challenge in re-educating TAMs is tumor heterogeneity, marked by epigenetic and phenotypic diversity. The observed heterogeneity in patients is a crucial factor contributing to the limited success in achieving desirable therapeutic outcomes for only a fraction of individuals in clinical trials.55 Recent advances in RNA sequencing hold promise for deciphering the phenotypic description of myeloid cells in the TME,56 consequently providing valuable insights into bridging this gap.There are several technical challenges associated with nanomedicine for macrophage-modulating cancer immunotherapy. For example, standardizing methods for large-scale production and implementing quality control measures are prerequisites for the clinical translation of nanomedicine. Achieving precise control of design and effectiveness at an industrial scale is challenging.6,7,57,58 Moreover, the complexity of NP composition raises concerns about potential toxicity. The metabolic pathway of these NPs may result in their accumulation in specific organs such as the spleen and liver. Undesired stimulation of immune pathways by engineered NPs can lead to toxicity. Importantly, the physicochemical properties of NPs, including size, shape, surface coating, and chemical composition, can influence immunotoxicity.59 Consequently, a systemic investigation of the toxicity of the diverse physicochemical properties of nanoparticles warrants their clinical translation.
Despite these challenges, macrophages stand out as potent therapeutic targets in nanomedicine-mediated tumor immunotherapy.60 Nanomedicine has the potential to transform macrophage-mediated cancer immunotherapies and pre-clinical evidence clearly motivates testing in clinical settings. Importantly, nanomedicine formulations can target cell surface receptors and potentiate the effects of macrophage-mediated immunotherapy for clinical applications. For example, ferumoxytol, an FDA-approved iron supplement consisting of dextran-coated iron oxide NPs, has the potential to repolarize macrophages toward a tumor-inhibitory phenotype. Ferumoxytol NPs inhibit tumor growth by repolarizing TAMs to an M1 phenotype and inducing a pro-inflammatory immune response.26 Continued research into the mechanisms of TAMs holds the key to unlocking innovative strategies in cancer immunotherapy using nanomedicine. Exploring future strategies, especially those centered on macrophage modulation, has the potential to expand and enhance treatment options for cancer patients in clinical settings. Moreover, the exploration of the diversity of TAMs at the single-cell level and the understanding the ontogenetic relationship of macrophages may offer new perspectives for advancing cancer treatment and providing valuable insight into the clinical translation of nanomedicine-based approaches.61
Author contributions
M. M. K., W. T. and W. C. conceived the manuscript. M. M. K., Y. L., Z. Z., A. N., Q. S., D. Q. and W. C. co-wrote the draft. M. M. K., Y. L., Z. Z., W. T., and W. C. refined the draft. All authors discussed and edited the manuscript before its submission.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by an American Heart Association (AHA) Transformational Project Award (No. 23TPA1072337, W. T.), an AHA Collaborative Sciences Award (No. 2018A004190, W. T.), an AHA's Second Century Early Faculty Independence Award (No. 23SCEFIA1151841, W. T.), an American Lung Association (ALA) Cancer Discovery Award (No. LCD1034625, W. T.), an ALA Courtney Cox Cole Lung Cancer Research Award (No. 2022A017206, W. T.), and an Novo Nordisk Validation Award (No. 2023A009607, W. T.). The authors also acknowledge the support and travel grant received from Fulbright organization as a Fulbright Scholar award (M. M. K.). Dr. Wei Chen acknowledges the financial support from the start-up funding and the regular research grant of Genomics Research Center, Academia Sinica (W. C.), as well as the Newly-Appointed Faculty Academic Research Grant, Academia Sinica (W. C.).References
- D. G. DeNardo and B. Ruffell, Nat. Rev. Immunol., 2019, 19, 369–382 CrossRef CAS PubMed
.
- Y. Li, W. Chen, Y. Kang, X. Zhen, Z. Zhou, C. Liu, S. Chen, X. Huang, H.-J. Liu, S. Koo, N. Kong, X. Ji, T. Xie and W. Tao, Nat. Commun., 2023, 14, 6973 CrossRef CAS PubMed
- A. Mantovani, F. Marchesi, A. Malesci, L. Laghi and P. Allavena, Nat. Rev. Clin. Oncol., 2017, 14, 399–416 CrossRef CAS PubMed
- T. A. Wynn, A. Chawla and J. W. Pollard, Nature, 2013, 496, 445–455 CrossRef CAS PubMed
- W. Chen, C.-A. Cheng and J. I. Zink, ACS Nano, 2019, 13, 1292–1308 CAS
- W. Chen, C. A. Glackin, M. A. Horwitz and J. I. Zink, Acc. Chem. Res., 2019, 52, 1531–1542 CrossRef CAS PubMed
- W. Chen, M. Schilperoort, Y. Cao, J. Shi, I. Tabas and W. Tao, Nat. Rev. Cardiol., 2022, 19, 228–249 CrossRef PubMed
- S. Ma, J. H. Kim, W. Chen, L. Li, J. Lee, J. Xue, Y. Liu, G. Chen, B. Tang and W. Tao, Adv. Sci., 2023, 2207768 CrossRef CAS PubMed
- B. E. Ferdows, D. N. Patel, W. Chen, X. Huang, N. Kong and W. Tao, Nanoscale, 2022, 14, 4448–4455 RSC
- B.-Z. Qian, J. Li, H. Zhang, T. Kitamura, J. Zhang, L. R. Campion, E. A. Kaiser, L. A. Snyder and J. W. Pollard, Nature, 2011, 475, 222–225 CrossRef CAS PubMed
- N. Nagarsheth, M. S. Wicha and W. Zou, Nat. Rev. Immunol., 2017, 17, 559–572 CrossRef CAS PubMed
- Y. Zheng, Y. Han, Q. Sun and Z. Li, Exploration, 2022, 2, 20210166 CrossRef PubMed
- S. Xu, Z. Gu, H. Lu, P. Guan and Z. Liu, ACS Appl. Mater. Interfaces, 2023, 15, 27658–27669 CrossRef CAS PubMed
- P. Xiao, Y. Zhang, Y. Zeng, D. Yang, J. Mo, Z. Zheng, J. Wang, Y. Zhang, Z. Zhou, X. Zhong and W. Yan, J. Transl. Med., 2023, 21, 457 CrossRef PubMed
- A. Mantovani, S. Sozzani, M. Locati, P. Allavena and A. Sica, Trends Immunol., 2002, 23, 549–555 CrossRef CAS PubMed
- A. Mantovani, F. Marchesi, A. Malesci, L. Laghi and P. Allavena, Nat. Rev. Clin. Oncol., 2017, 14, 399–416 CrossRef CAS PubMed
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed
- Y. Liu, J. Wang, Q. Xiong, D. Hornburg, W. Tao and O. C. Farokhzad, Acc. Chem. Res., 2021, 54, 291–301 CrossRef CAS PubMed
- I. Rudnik-Jansen and K. A. Howard, J. Controlled Release, 2021, 337, 248–257 CrossRef CAS PubMed
- A. A. Sebak, I. E. O. Gomaa, A. N. ElMeshad, M. H. Farag, U. Breitinger, H.-G. Breitinger and M. H. AbdelKader, Int. J. Nanomed., 2020, 15, 8845–8862 CrossRef CAS PubMed
- W. Bai, A. Raghavendra, R. Podila and J. M. Brown, Int. J. Nanomed., 2016, 11, 4357–4371 CrossRef CAS PubMed
- M. Guo, L. Zhao, J. Liu, X. Wang, H. Yao, X. Chang, Y. Liu, J. Liu, M. You and J. Ren, Nano Lett., 2021, 21, 6005–6013 CrossRef CAS PubMed
- C. Chen, M. Song, Y. Du, Y. Yu, C. Li, Y. Han, F. Yan, Z. Shi and S. Feng, Nano Lett., 2021, 21, 5522–5531 CrossRef CAS PubMed
- L. Pang, Y. Pei, G. Uzunalli, H. Hyun, L. T. Lyle and Y. Yeo, Pharm. Res., 2019, 36, 65 CrossRef PubMed
- Y. Zhang, L. Wu, Z. Li, W. Zhang, F. Luo, Y. Chu and G. Chen, Biomacromolecules, 2018, 19, 2098–2108 CrossRef CAS PubMed
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS PubMed
- Z. Gu, T. Liu, J. Tang, Y. Yang, H. Song, Z. K. Tuong, J. Fu and C. Yu, J. Am. Chem. Soc., 2019, 141, 6122–6126 CrossRef CAS PubMed
- W. Chen, Y. Li, C. Liu, Y. Kang, D. Qin, S. Chen, J. Zhou, H.-J. Liu, B. E. Ferdows, D. N. Patel, X. Huang, S. Koo, N. Kong, X. Ji, Y. Cao, W. Tao and T. Xie, Angew. Chem., Int. Ed., 2023, 62, e202308413 CrossRef CAS PubMed
- C. B. Rodell, S. P. Arlauckas, M. F. Cuccarese, C. S. Garris, R. Li, M. S. Ahmed, R. H. Kohler, M. J. Pittet and R. Weissleder, Nat. Biomed. Eng., 2018, 2, 578–588 CrossRef CAS PubMed
- A. Schmall, H. M. Al-Tamari, S. Herold, M. Kampschulte, A. Weigert, A. Wietelmann, N. Vipotnik, F. Grimminger, W. Seeger and S. S. Pullamsetti, Am. J. Respir. Crit. Care Med., 2015, 191, 437–447 CrossRef CAS PubMed
- X. Li, W. Yao, Y. Yuan, P. Chen, B. Li, J. Li, R. Chu, H. Song, D. Xie and X. Jiang, Gut, 2017, 66, 157–167 CrossRef CAS PubMed
- S. Shen, Y. Zhang, K.-G. Chen, Y.-L. Luo and J. Wang, Mol. Pharm., 2018, 15, 3642–3653 CrossRef CAS PubMed
- F. Leuschner, P. Dutta, R. Gorbatov, T. I. Novobrantseva, J. S. Donahoe, G. Courties, K. M. Lee, J. I. Kim, J. F. Markmann, B. Marinelli, P. Panizzi, W. W. Lee, Y. Iwamoto, S. Milstein, H. Epstein-Barash, W. Cantley, J. Wong, V. Cortez-Retamozo, A. Newton, K. Love, P. Libby, M. J. Pittet, F. K. Swirski, V. Koteliansky, R. Langer, R. Weissleder, D. G. Anderson and M. Nahrendorf, Nat. Biotechnol., 2011, 29, 1005–1010 CrossRef CAS PubMed
- X. Li, R. Liu, X. Su, Y. Pan, X. Han, C. Shao and Y. Shi, Mol. Cancer, 2019, 18, 177 CrossRef PubMed
- H. H. Van Acker, S. Anguille, Y. Willemen, E. L. Smits and V. F. Van Tendeloo, Pharmacol. Ther., 2016, 158, 24–40 CrossRef CAS PubMed
- F. Piaggio, V. Kondylis, F. Pastorino, D. Di Paolo, P. Perri, I. Cossu, F. Schorn, C. Marinaccio, D. Murgia, A. Daga, F. Raggi, M. Loi, L. Emionite, E. Ognio, M. Pasparakis, D. Ribatti, M. Ponzoni and C. Brignole, J. Controlled Release, 2016, 223, 165–177 CrossRef CAS PubMed
- K. S. Opperman, K. Vandyke, K. C. Clark, E. A. Coulter, D. R. Hewett, K. M. Mrozik, N. Schwarz, A. Evdokiou, P. I. Croucher, P. J. Psaltis, J. E. Noll and A. C. W. Zannettino, Neoplasia, 2019, 21, 777–787 CrossRef CAS PubMed
- W. Zhang, X. D. Zhu, H. C. Sun, Y. Q. Xiong, P. Y. Zhuang, H. X. Xu, L. Q. Kong, L. Wang, W. Z. Wu and Z. Y. Tang, Clin. Cancer Res., 2010, 16, 3420–3430 CrossRef CAS PubMed
- C. Zheng, X. Zhao, Y. Wang, Y. Zhao, Y. Zheng, Z. Zhang, Q. Liu, Y. Liu and L. Shi, Chem. Eng. J., 2022, 435, 134779 CrossRef CAS
- X. Zang, X. Zhang, H. Hu, M. Qiao, X. Zhao, Y. Deng and D. Chen, Mol. Pharm., 2019, 16, 2249–2258 CrossRef CAS PubMed
- Z. Li, Y. Ding, J. Liu, J. Wang, F. Mo, Y. Wang, T.-J. Chen-Mayfield, P. M. Sondel, S. Hong and Q. Hu, Nat. Commun., 2022, 13, 1845 CrossRef CAS PubMed
- M. Feng, W. Jiang, B. Y. Kim, C. C. Zhang, Y.-X. Fu and I. L. Weissman, Nat. Rev. Cancer, 2019, 19, 568–586 CrossRef CAS PubMed
- K. Vaeteewoottacharn, R. Kariya, P. Pothipan, S. Fujikawa, C. Pairojkul, S. Waraasawapati, K. Kuwahara, C. Wongkham, S. Wongkham and S. Okada, Transl. Oncol., 2019, 12, 217–225 CrossRef PubMed
- J. T. Sockolosky, M. Dougan, J. R. Ingram, C. C. Ho, M. J. Kauke, S. C. Almo, H. L. Ploegh and K. C. Garcia, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E2646–E2654 CrossRef CAS PubMed
- Q. Chen, C. Wang, X. Zhang, G. Chen, Q. Hu, H. Li, J. Wang, D. Wen, Y. Zhang, Y. Lu, G. Yang, C. Jiang, J. Wang, G. Dotti and Z. Gu, Nat. Nanotechnol., 2019, 14, 89–97 CrossRef CAS PubMed
- X. Kuang, Z. Wang, Z. Luo, Z. He, L. Liang, Q. Gao, Y. Li, K. Xia, Z. Xie, R. Chang, Y. Wang, Y. Liu, S. Zhao, J. Su, Y. Wang, W. Situ, M. Chen, Y. Zhao, X. Chen, H. Xie and H. Liu, J. Colloid Interface Sci., 2022, 616, 189–200 CrossRef CAS PubMed
- Y. Yao, H. Chen and N. Tan, Acta Pharm. Sin. B, 2022, 12, 2103–2119 CrossRef CAS PubMed
- C. Chen, M. Song, Y. Du, Y. Yu, C. Li, Y. Han, F. Yan, Z. Shi and S. Feng, Nano Lett., 2021, 21, 5522–5531 CrossRef CAS PubMed
- H. Zhao, L. Li, J. Zhang, C. Zheng, K. Ding, H. Xiao, L. Wang and Z. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 31124–31135 CrossRef CAS PubMed
- C. Gong, X. Yu, W. Zhang, L. Han, R. Wang, Y. Wang, S. Gao and Y. Yuan, J. Nanobiotechnol., 2021, 19, 58 CrossRef CAS PubMed
- C. Gong, X. Yu, B. You, Y. Wu, R. Wang, L. Han, Y. Wang, S. Gao and Y. Yuan, J. Nanobiotechnol., 2020, 18, 92 CrossRef CAS PubMed
- T. Du, Y. Wang, Z. Luan, C. Zhao and K. Yang, Int. J. Pharm., 2022, 624, 121911 CrossRef CAS PubMed
- Y. Wang, H. Ma, Y. Li and R. Su, Reprod. Sci., 2022, 29, 436–447 CrossRef CAS PubMed
- L. Closset, O. Gultekin, S. Salehi, D. Sarhan, K. Lehti and J. Gonzalez-Molina, Matrix Biol., 2023, 121, 217–228 CrossRef CAS PubMed
- J. Liu, R. Zhang and Z. P. Xu, Small, 2019, 15, 1900262 CrossRef PubMed
- P. S. Hegde and D. S. Chen, Immunity, 2020, 52, 17–35 CrossRef CAS PubMed
- Y.-J. Li, J.-Y. Wu, J. Liu, W. Xu, X. Qiu, S. Huang, X.-B. Hu and D.-X. Xiang, J. Nanobiotechnol., 2021, 19, 242 CrossRef PubMed
- C. Liu, Y. Wang, L. Li, D. He, J. Chi, Q. Li, Y. Wu, Y. Zhao, S. Zhang, L. Wang, Z. Fan and Y. Liao, J. Controlled Release, 2022, 349, 679–698 CrossRef CAS PubMed
- S. Wang, Z. Sun and Y. Hou, Adv. Healthcare Mater., 2021, 10, 2000845 CrossRef CAS PubMed
- D. J. Irvine and E. L. Dane, Nat. Rev. Immunol., 2020, 20, 321–334 CrossRef CAS PubMed
- A. Mantovani, P. Allavena, F. Marchesi and C. Garlanda, Nat. Rev. Drug Discovery, 2022, 21, 799–820 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |