
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFumarate production from pyruvate and low concentrations of CO2 with a multi-enzymatic system in the presence of NADH and ATP†
Mika
Takeuchi
a and
Yutaka
Amao
 *ab
*ab
aGraduate School of Science, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: amao@omu.ac.jp
bResearch Centre of Artificial Photosynthesis (ReCAP), Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
First published on 4th September 2024
Abstract
Fumarate is an unsaturated dicarboxylic acid useful as a raw material for unsaturated polyester resins, polybutylene succinate (PBS), poly(propylene fumarate) (PPF), plasticisers, and other products. Biodegradable plastics derived from fumarate are an attractive solution to the serious environmental pollution caused by plastic disposal. A new fumarate production from CO2 and biobased pyruvate using enzymes in aqueous media under ambient temperature and pressure is an environmental approach to overcome plastic pollution and achieve CO2 capture, utilization and storage (CCUS). In this work, fumarate production from pyruvate and low-concentration CO2 below 15% captured from the gas phase using 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-NaOH buffer solution with a multi-enzyme system consisting of pyruvate carboxylase from a bovine liver (PC; EC 6.4.1.1), recombinant malate dehydrogenase from bacteria (MDH; EC 1.1.1.37) and fumarase from a porcine heart (FUM; EC 1.1.1.37) in the presence of adenosine 5′-triphosphate (ATP) and NADH was investigated. It was found that pyruvate can be converted into L-malate in high yields (more than 80%) directly using 15% CO2 equivalent to exhaust gas as a carboxylating agent using a dual-enzyme system consisting of PC and MDH in the presence of ATP and NADH after 5 h incubation. Moreover, fumarate production from 15% CO2 and pyruvate as raw materials was accomplished using a dual-enzyme system consisting of PC and MDH.
Introduction
Plastics are an important and essential material in contemporary life. Plastics have various uses, are lighter than metals and their production costs are relatively low.1 Most plastics in circulation are produced from fossil resources, thereby increasing greenhouse gas emissions through their value chain. Currently, plastics contribute to pollution throughout their life cycle, from production to use and ultimately disposal.2,3 Biodegradable, compostable and biobased plastics are increasingly attracting attention as a potential solution to these issues.4–10 Biodegradable and compostable plastic materials are eventually decomposed by microorganisms into water, CO2, inorganic salts and new biomass.11–15 Biodegradable plastics are classified into two groups based on their synthesis method.16,17 Natural bioplastics are produced from natural resources and synthetic bioplastics from fossil resources.18,19 Natural bioplastics are derived from renewable or biological resources, such as animal, plant, marine or microbial sources, whereas synthetic biodegradable plastics are produced chemically20 from fossil resources, but their versatility can be extended by producing raw materials from biobased materials or CO2.Synthetic biodegradable plastics21 are further classified into three groups: aliphatic polyesters, poly(vinyl alcohol) (PVA)22–25 and poly(vinyl acetate) (PVAC).26–28 Among the synthetic biodegradable plastics based on aliphatic polyesters, poly(L-lactate) (PLA)29–34 and poly(glycolate) (PGA)35–38 are chemically produced from bio-based precursors. In contrast, poly(caprolactone) (PCL)39–43 and poly butylene succinate (PBS)44–46 are chemically produced from synthetic precursors.
For example, PBS is produced via copolymerisation of succinic acid and 1,4-butanediol. PBS precursors succinic acid and 1,4-butanediol can both be synthesised from fumaric acid. Moreover, fumarate is an unsaturated dicarboxylic acid useful as a raw material for unsaturated polyester resins, polybutylene succinate (PBS), poly(propylene fumarate) (PPF), plasticisers, and other products as shown in Fig. 1.47,48 The production of fumarate from biobased materials and CO2 results in a new precursor production method for synthetic biodegradable plastics that is dependent on synthetic precursors derived from fossil resources. Among the biobased materials for biodegradable plastic precursors, pyruvate is a useful material with the following properties. Pyruvate is a product of glucose metabolism, known as glycolysis. One molecule of glucose is used to provide further energy after being broken down into two molecules of pyruvate.49 The two molecules of pyruvate produced by the decomposition of one molecule of glucose are used as a further source of energy. For these reasons, therefore, we focused on fumarate production from the biobased materials pyruvate and CO2 gas. Biocatalytic fumarate production from pyruvate and gaseous CO2 or bicarbonate using malate dehydrogenase decarboxylating type (ME; EC 1.1.1.38)50,51 and fumarase (FUM; EC 4.2.1.2)52–57 in the presence of NADH via an intermediate, L-malate, as shown in Fig. 2(a) has been reported.58,59 Moreover, the visible-light driven fumarate production from pyruvate and bicarbonate or gaseous CO2 with the hybridisation of NAD+ reduction system of triethanol amine as an electron donor, water-soluble zinc porphyrin, zinc tetra(4-sulfonatophenyl) porphyrin tetrasodium salt or zinc tetrakis (N,N,N-aminophenyl) porphyrin as a photosensitizer and Rh coordination complex ([Cp*Rh(bpy)(H2O)]2+; Cp* = pentamethylcyclopentadienyl, bpy = 2,2′-bipyridyl), and a dual-enzyme system consisting of ME and FUM has been developed.60–62
 | ||
| Fig. 1 Current applications of fumaric acid for various plastics and plasticizers. | ||
 | ||
| Fig. 2 Fumarate production from pyruvate and CO2 or bicarbonate with the system using dual-enzyme (ME and FUM) in the presence of NADH (a) and multi-enzyme (PC, MDH and FUM) in the presence of ATP and NADH (b). | ||
ME in Fig. 2(a) catalyses two processes: the carboxylation of pyruvate with bicarbonate or CO2 and the reduction of oxaloacetate to L-malate using NADH as a co-factor. ME is a useful enzyme for L-malate production from pyruvate and CO2; however, ME also catalyses the pyruvate reduction to L-lactate. ME has the disadvantage of catalysing the reduction of pyruvate to L-lactate, especially under conditions of low bicarbonate concentration in solution.63 In other words, one of the major drawbacks is the inability to use low concentration CO2 gas as feedstock in the reaction system using ME as a catalyst. In addition, the Michaelis constant (Km) value of L-malate for FUM-catalysed fumarate production is reported to be 0.65 mM and at least about 2.0 mM of L-malate is required to achieve the maximum rate of fumarate production.64 Another aspect to be improved in the production of biocatalytic fumarate from pyruvate and gaseous CO2 or bicarbonate using ME and FUM in the presence of NADH is the increased rate of ME-catalysed L-malate (intermediate) production. Therefore, improving the production efficiency of the L-malate in the early stages is an important point of improvement for biocatalytic fumarate production viaL-malate from pyruvate and gaseous CO2 or bicarbonate. We therefore devised a way to complement the two catalytic functions of ME with their respective enzymes. One is pyruvate carboxylase (PC; EC 6.4.1.1)65–67 for oxaloacetate production by pyruvate carboxylation with CO2 in the presence of adenosine triphosphate (ATP). The other is malate dehydrogenase (MDH; EC 1.1.1.37)68–70 for oxaloacetate reduction to L-malate in the presence of NADH. By using a multi-enzyme system of PC, MDH and FUM, fumarate production from pyruvate and CO2 in the presence of ATP and NADH can be developed as shown in Fig. 2(b).71 The PC-catalysed pyruvate carboxylation process needs the phosphorylation of bicarbonate by ATP as shown in Fig. 3. As shown in Fig. 3, no direct phosphorylation of CO2 by ATP occurs in PC. For this reason, bicarbonate has been used as a carboxylating agent for pyruvate instead of CO2 gas in PC-catalysed reaction. Furthermore, PC only catalyses the carboxylation of pyruvate, allowing low concentrations of CO2 to be used as a feedstock. This means that the production of L-lactate under conditions of low CO2 concentration, which has been a problem in systems using ME, can be eliminated.
 | ||
| Fig. 3 PC-catalysed pyruvate carboxylation with bicarbonate to produce oxaloacetate in the presence of ATP. | ||
Thus, the direct use of CO2 gas instead of bicarbonate in fumarate production as shown in Fig. 2(b) was devised as a CO2 capture and utilisation (CCU) system. Pure 100% CO2 gas is widely used in many laboratory-scale studies on CCU systems.72 We have also succeeded in producing fumarate by incorporating pure 100% CO2 gas in the gas phase of the reaction vessel into the reaction solution using the system shown in Fig. 2(b).71 One of the challenges for practical application of CCU is to capture 0.04% low concentration of CO2 gas from the atmosphere, and use it for CCU reaction.73 In other words, it would be needless to say that the practical application of CCU technology using the atmospheric CO2 concentration of around 0.04% in the livelihood sphere would require a huge amount of energy and cost. In contrast, the concentration of CO2 in exhaust gas from coal-fired power plants, steel mills and chemical plants ranges from several to 20% of CO2.74 Direct air capture (DAC) technology is gaining attention for capturing such low concentrations of CO2 of around 20%.75–80 More recently, research has also been reported on the linkage of DAC and CCU technologies to convert captured CO2 into useful substances.81,82 One candidate for gaseous CO2 capture technology in aqueous solutions based on DAC is the use of aqueous basic amine solutions. In other words, the use of a weakly basic buffer in the system shown in Fig. 2(b) allows CO2 in the gas phase to be directly captured and converted to bicarbonate in an aqueous medium before being used as a carboxylating agent for pyruvate.
In this study, fumarate production from pyruvate and low-concentration CO2 below 15% captured from the gas phase by 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-NaOH buffer solution with a multi-enzyme system consisting of PC, MDH and FUM in the presence of ATP and NADH was investigated.
Experimental
Materials
MDH recombinant from bacteria (EC 1.1.1.37), NAD+ and NADH were purchased from Oriental Yeast Co., Ltd. ATP disodium hydrate, PC from bovine liver (EC 6.4.1.1) and FUM from porcine heart (EC 1.1.1.37) were purchased from Sigma-Aldrich Co. LLC. Acetyl-CoA trilithium salt, sodium bicarbonate and magnesium chloride hexahydrate were purchased from FUJIFILM Wako Pure Chemical Corporation. ME from Sulfolobus tokodaii was purchased from Thermostable Enzyme Laboratory Co., Ltd (EC 1.1.1.38 MDH-73-01). 4-(2-Hydroxyethyl)-1-piperazine-ethanesulfonic acid (HEPES) was purchased from NACALAI TESQUE, INC.Determination of the capture of CO2 in the gas phase into a sample solution
The amount of capture of CO2 contained in the mixed gas consisting of N2 and CO2 in the gas phase to the sample solution was determined by the following method. The reaction is an isobaric system and the schematic representation of the experimental setup is shown in Fig. 4(a). | ||
| Fig. 4 Outline of the experimental setup for the determination of the amount of CO2 in the gas phase captured into a sample solution (a) and for the isobaric system for direct captured CO2 gas utilisation for a multi-enzyme system (b). | ||
500 mM of HEPES-NaOH buffer (pH 7.8) was used as a sample solution for CO2 capture from the gas phase. The sample solution was deaerated by freeze–pump–thaw cycles repeated 6 times and then introduced in gas phase and syringe (as shown in Fig. 4(a)) with the gas mixture consisting of N2 and CO2 prepared by a gas mixture generator for 10 min. The mixed gas was prepared by controlling the flow rates of N2 and CO2 gases entering the gas mixture generator (KOFLOC PMG-1A-N2-100). The total pressure of the reaction system was kept at 1.01325 × 105 Pa. The volume change in the syringe was measured and the amount of CO2 dissolved in the liquid phase was determined from the change in volume. Here, the CO2 in the gas mixture was considered the real gas and the amount of CO2 (n) in the gas phase was determined using the following eqn (1).
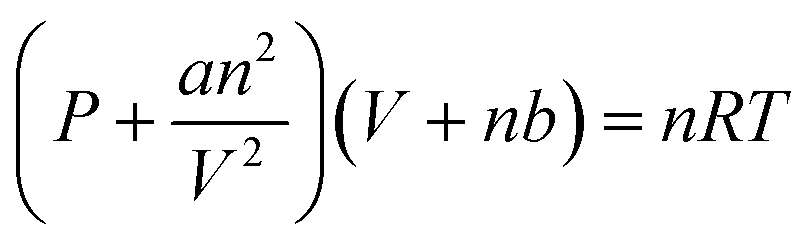 | (1) |
Effect of acetyl-CoA on the L-malate production with PC and MDH in the presence of ATP and NADH
The reaction mixture consisted of sodium pyruvate (2.0 mM), ATP (2.0 mM), sodium bicarbonate (50 mM), manganese chloride (5.0 mM), NADH (2.0 mM), PC (1.0 U), MDH (10 U) and acetyl-CoA in 5.0 mL of 500 mM HEPES buffer-NaOH (pH 7.2). The concentration of acetyl-CoA was varied from 0 and 2.0 mM. The reaction vessel is a clear glass vial, and the reaction is a sealed system. The total volume of the reaction vessel is 14.0 mL. Reactions were carried out in a thermostatic bath (30.5 °C). The concentration of L-malate produced was measured by using an ion chromatograph system with an electrical conductivity detector (Metrohm, Eco IC). Ion chromatographic separation was carried out using an ion exclusion column (Metrosep Organic Acids 250/7.8 Metrohm; column size: 7.8 × 250 mm; composed of 9 μm polystyrene-divinylbenzene copolymer with sulfonic acid groups). Experimental details for L-malate quantification by using an ion chromatograph are explained in the ESI.† The concentration of L-malate was determined using eqn (S1) (ESI†) obtained from a calibration curve (Fig. S1(b), ESI†) based on the chromatogram of the standard sample (Fig. S1(a), ESI†).Determination of kinetic parameters for sodium bicarbonate in the PC and MDH-catalysed L-malate production with ATP and NADH
The reaction mixture consisted of sodium pyruvate (2.0 mM), ATP (2.0 mM), sodium bicarbonate, manganese chloride (5.0 mM), NADH (2.0 mM), PC (1.0 U), MDH (10 U) and acetyl-CoA (1.0 mM) in 5.0 mL of 500 mM HEPES buffer-NaOH (pH 7.2). The concentration of sodium bicarbonate was varied from 0 to 25 mM. The reaction vessel is a clear glass vial, and the reaction is a sealed system. The total volume of the reaction vessel is 14.0 mL. Reactions were carried out in a thermostatic bath (30.5 °C). The concentration of L-malate produced was measured by ion chromatography using the calibration curve based on the chromatogram of a standard sample (Fig. S2 (a) and (b), ESI†) using eqn (S2) (ESI†).Determination of kinetic parameters for NADH in the MDH-catalysed L-malate production
The reaction mixture consisted of sodium oxaloacetate (1.0 mM), NADH and MDH (10 U) in 5.0 mL of 500 mM HEPES buffer-NaOH (pH 7.2). The concentration of NADH was varied from 0 to 1.0 mM. The reaction vessel is a clear glass vial, and the reaction is a sealed system. The total volume of the reaction vessel is 14.0 mL. Reactions were carried out in a thermostatic bath (30.5 °C). Reactions were monitored by the decrease in NADH concentration during the incubation. NADH concentration was monitored by absorption spectra change using UV-visible absorption spectroscopy (SHIMADZU, MaltiSpec-1500) with the molar coefficient of 6220 cm−1 M−1 at 340 nm.83L-Malate production from pyruvate and direct captured CO2 with PC and MDH in the presence of ATP and NADH
The reaction mixture consisted of sodium pyruvate (5.0 mM), ATP (5.0 mM), manganese chloride (5.0 mM), NADH (5.0 mM), PC (1.0 U), MDH (0.5 U) and acetyl-CoA (1.0 mM) in 5.0 mL of 500 mM HEPES buffer-NaOH (pH 7.2). The schematic representation for the experimental setup is also shown in Fig. 4. The sample solution was deaerated by freeze–pump–thaw cycles repeated 6 times to remove dissolved oxygen. After that, the mixture of N2 and CO2 gas was flushed both of gas phase of reaction vessel and connected balloon for 10 min. The shape of the reaction vessel and the method of flushing the mixed gas were as described above. Reactions were carried out in a thermostatic bath (30.5 °C). The concentration of L-malate was detected by ion chromatography and determined from the calibration curve based on the chromatogram of a standard sample (Fig. S2 (a) and (b), ESI†) using eqn (S2) (ESI†). L-Malate production with the system of sodium pyruvate (5.0 mM), magnesium chloride (5.0 mM), NADH (5.0 mM) and ME (0.7 U) in 5.0 mL of 500 mM HEPES buffer-NaOH (pH 7.2) was carried out as a control experiment. The amount of ME enzyme activity in the condition of the control experiment was optimised for L-malate production.64Fumarate production from pyruvate and direct captured CO2 with PC, MDH and FUM in the presence of ATP and NADH
The reaction mixture consisted of sodium pyruvate (5.0 mM), ATP (5.0 mM), manganese chloride (5.0 mM), NADH (5.0 mM), PC (1.0 U), MDH (10 U), FUM (0.5 U) and acetyl-CoA (1.0 mM) in 5.0 mL of 500 mM HEPES buffer-NaOH (pH 7.2). The schematic representation of the experimental setup is also shown in Fig. 4. The sample solution was deaerated by freeze–pump–thaw cycles repeated 6 times to remove dissolved oxygen. After that, the mixture of N2 and CO2 gas was flushed both of gas phase of reaction vessel and connected balloon for 10 min. The shape of the reaction vessel and the method of flushing the mixed gas were as described above. Reactions were carried out in a thermostatic bath (30.5 °C). The concentrations of L-malate and fumarate were detected by ion chromatography and determined from the calibration curve based on the chromatogram of a standard sample (Fig. S3(a) and (b), ESI†) using eqn (S3) (ESI†). Fumarate production with the system of sodium pyruvate (5.0 mM), magnesium chloride (5.0 mM), NADH (5.0 mM), ME (0.7 U) and FUM (0.5 U) in 5.0 mL of 500 mM HEPES buffer-NaOH (pH 7.2) was carried out as a control experiment. The amount of ME or FUM enzyme activity in the control experiment was optimised for fumarate production.64Results and discussion
Determination of the amount of captured CO2 in the gas phase into a sample solution
Fig. 5 shows the time dependence of volume of syringe of the reaction vessel when a gas mixture consisting of 85% N2 and 15% CO2 or 100% CO2 gas61 was introduced as shown in Fig. 4. | ||
| Fig. 5 Time dependence of volume of syringe of the reaction vessel when a gas mixture consisting of 85% N2 and 15% CO2 (blue) and 100% CO2 gas (red)61 was introduced. | ||
As shown in Fig. 5, the volume of syringe of reaction vessel decreased with increasing time. After 5 h of incubation, the volume change of the syringe under a gas mixture consisting of 85% N2 and 15% CO2 or 100% CO2 gas was 6.0 and 17.0 mL,61 respectively. Here, the concentration of CO2 dissolved from the gas phase into the liquid phase was calculated from the volume change of the reaction vessel. The solubility of N2 and CO2 gases in water media at 30 °C has been reported to be 0.016 and 0.162 g kg−1,84 respectively. In this experiment, thus, the dissolution of N2 gas was ignored and the volume change was assumed to be the dissolution of CO2 gas into the water medium. The time dependence of the total CO2 concentration estimated by eqn (1) from the volume change is shown in Fig. 6. As shown in Fig. 6, the estimated CO2 concentration increased with increasing time. After 1 h, 16.2 and 56.6 mM of CO2 under a gas mixture consisting of 85% N2 and 15% CO2 or 100% CO2 gas were dissolved from the gas phase into the liquid phase. We have already reported on the ability of HEPES buffer solution to capture gas-phase CO2 into aqueous solution.85 CO2 in the gas phase is captured by the HEPES buffer solution, as shown in Fig. 7. As shown in Fig. 7, the CO2 captured by the HEPES buffer is converted to bicarbonate. In addition, the proportion of carbonate species present in aqueous solution at pH 7.0 is estimated by the Plummer and Busenberg equation to be 82% bicarbonate and 18% CO2.86 Hence, it is possible that CO2 in the gas phase can be used for PC-catalysed pyruvate carboxylation.
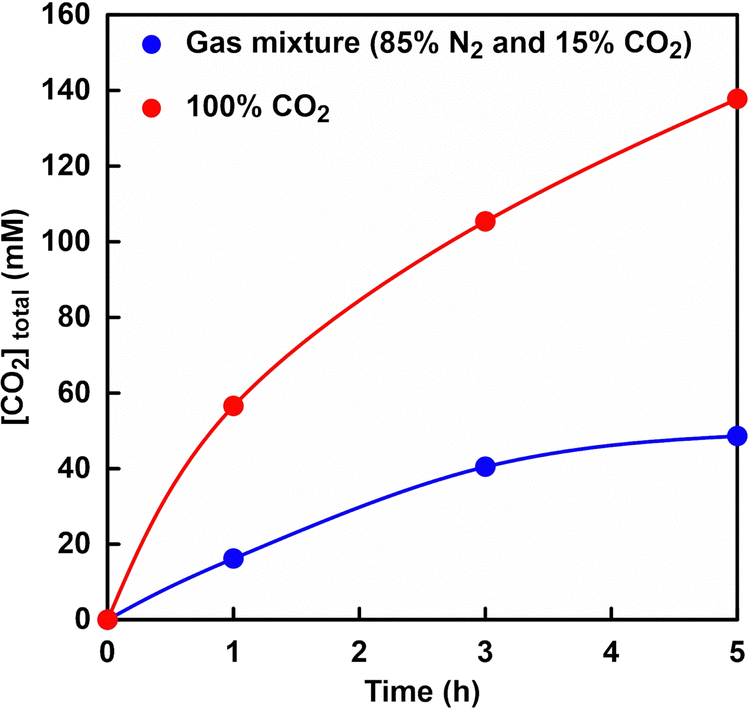 | ||
| Fig. 6 Time dependence of the total CO2 concentration estimated by eqn (1) from the volume change. Gas mixture consisting of 85% N2 and 15% CO2 (blue), and 100% CO2 gas (red). | ||
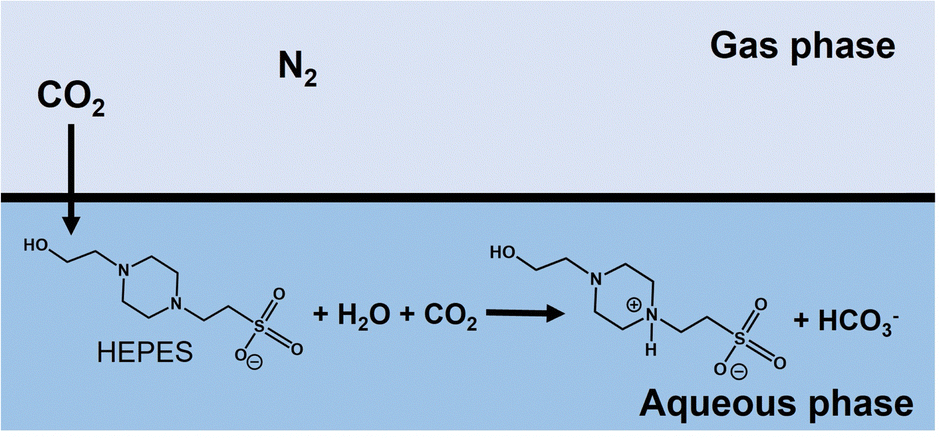 | ||
| Fig. 7 Suggested mechanism for the capture of CO2 in the gas phase using HEPES buffer solution into the aqueous phase. | ||
Effect of acetyl-CoA on the L-malate production with PC and MDH in the presence of ATP and NADH
Fig. 8 shows the relationship between the concentration of acetyl-CoA and L-malate production rate (v0) in the reaction system of sodium pyruvate, ATP, sodium bicarbonate, manganese chloride, NADH, PC, MDH and acetyl-CoA in HEPES buffer-NaOH. The L-malate production rate (v0) was determined from the concentration of L-malate production after 30 min of incubation. Fig. S4 (ESI†) shows an ion chromatography chart of a sample solution after 30 min of incubation. As shown in Fig. 8, the L-malate production rate depends on the concentration of acetyl-CoA. In addition, no L-malate production was observed under conditions without acetyl-CoA. As shown in Fig. 3, acetyl-CoA is not directly involved in the PC-catalysed carboxylation of pyruvate by bicarbonate. However, it has been reported that the allosteric effect of acetyl-CoA on PC promotes pyruvate carboxylation.87 Therefore, acetyl-CoA is also essential in the production of L-malate from pyruvate and bicarbonate using PC and MDH. | ||
| Fig. 8 Relationship between the concentration of acetyl-CoA and the rate of L-malate production (v0) in the reaction system of sodium pyruvate, ATP, sodium bicarbonate, manganese chloride, NADH, PC, MDH and acetyl-CoA in HEPES buffer-NaOH. | ||
Determination of kinetic parameters for sodium bicarbonate in the PC and MDH-catalysed L-malate production with ATP and NADH
The effect of bicarbonate concentration on PC-catalysed pyruvate carboxylation can be investigated to explore the possibility of directly utilising CO2 gas in the gas phase. Fig. 9 shows the relationship between the concentration of sodium bicarbonate and L-malate production rate (v0) in the reaction system of sodium pyruvate, ATP, sodium bicarbonate, manganese chloride, NADH, PC, MDH and acetyl-CoA in HEPES buffer-NaOH.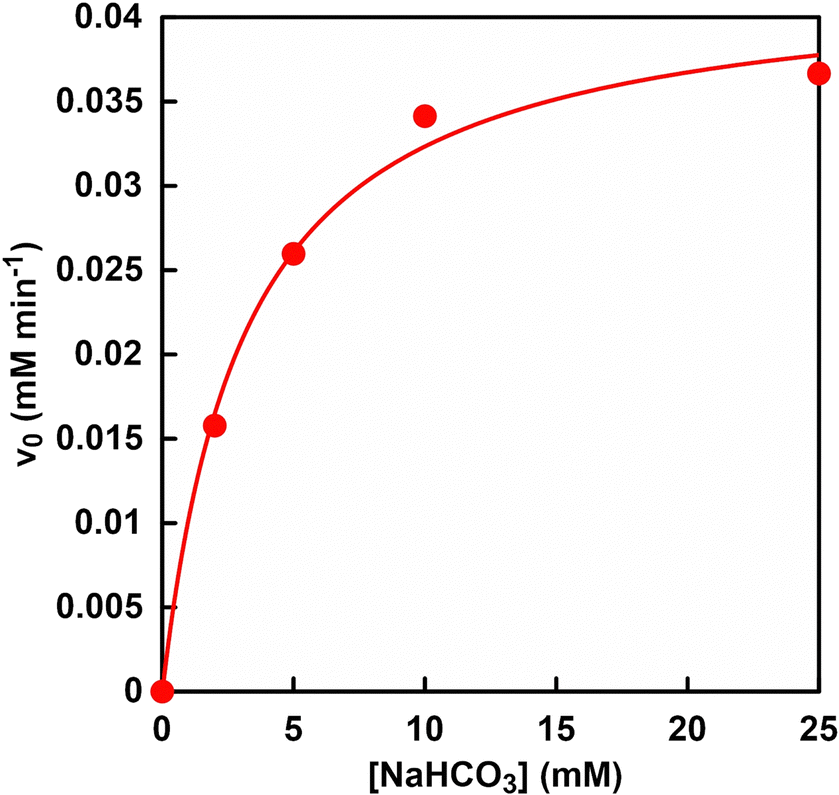 | ||
| Fig. 9 Relationship between the concentration of sodium bicarbonate and the rate of L-malate production (v0) in the reaction system of sodium pyruvate, ATP, sodium bicarbonate, manganese chloride, NADH, PC, MDH and acetyl-CoA in HEPES buffer-NaOH. | ||
The L-malate production rate (v0) was determined from the concentration of L-malate production after 30 min of incubation. Fig. S5 (ESI†) shows an ion chromatography chart of a sample solution after 30 min of incubation. Assuming that PC and MDH are one enzyme, the kinetic parameters of bicarbonate for L-malate production were determined. As shown in Fig. 9, the relationship between bicarbonate concentration and the rate of L-malate production followed a Michaelis–Menten type enzyme reaction. The Michaelis constant Km and maximum rate of L-malate production Vmax of bicarbonate were calculated to be 3.6 mM and 0.04 mM min−1, respectively. On the other hand, Km and Vmax values of bicarbonate for ME-catalysed L-malate production were reported to be 14 mM and 0.03 mM min−1, respectively.61 These results show that the PC and MDH dual-enzyme system can reduce the Km value of bicarbonate by 25% compared to the ME system. Furthermore, a bicarbonate concentration of about three times the Km value is required to produce L-malate at the Vmax using a dual-enzyme system of PC and MDH. As described in the previous section, 16.2 and 56.6 mM of CO2 were dissolved from the gas phase into the liquid phase under a gas mixture consisting of 85% N2 and 15% CO2, and 100% CO2 gas by using the reaction vessel in Fig. 4. From these facts, the bicarbonate concentration captured in aqueous solution from the gas mixture of 85% N2 and 15% CO2 was estimated to be about 4.5 times higher than in Km value. Therefore, a mixed gas of 85% N2 and 15% CO2 was used in subsequent experiments.
Determination of kinetic parameters for NADH in the MDH-catalysed L-malate production
Next, the concentration of NADH for the efficient reduction of oxaloacetate, produced from pyruvate and bicarbonate by PC catalysis, to L-malate by MDH was studied. Fig. 10 shows the relationship between the concentration of NADH and L-malate production rate (v0) in the reaction system of sodium oxaloacetate, ATP, NADH, and MDH in HEPES buffer-NaOH. | ||
| Fig. 10 Relationship between the concentration of NADH and the initial rate (v0) in the reaction system of sodium oxaloacetate, NADH and MDH in HEPES buffer-NaOH. | ||
The initial rate (v0) was determined from the decrease in NADH concentration after 30 min of incubation. As shown in Fig. 10, the relationship between NADH concentration and the initial rate followed a Michaelis–Menten type enzyme reaction. The Michaelis constant Km of NADH and maximum rate Vmax were calculated to be 0.05 mM and 0.07 mM min−1, respectively. On the other hand, Km and Vmax values of NADH for ME-catalysed L-malate production were reported to be 1.5 mM and 0.04 mM min−1, respectively.83 These results show that the MDH system can reduce the Km value of NADH by 3.3% compared to the ME system.
L-Malate production from pyruvate and direct captured CO2 with PC and MDH in the presence of ATP and NADH
Fig. 11 shows the time dependence of L-malate production from pyruvate and CO2 with PC and MDH in HEPES-NaOH buffer including ATP and NADH (The ion chromatograph chart during the incubation is shown in Fig. S6, ESI†). The composition of the gas phase including balloon was adjusted to 85% N2 and 15% CO2 or 100% CO2 gas. The pressure in the reaction vessel was maintained at 1.01325 × 105 Pa. The L-malate concentration increased with increasing incubation time in both cases. After 5 h of incubation, the L-malate concentration was estimated to be ca. 4.0 mM in both cases. The yields for pyruvate to L-malate in both systems after 5 h of incubation were calculated to be ca. 80%. The initial reaction rates under a gas mixture consisting of 85% N2 and 15% CO2, and 100% CO2 gas were estimated to be 0.029 and 0.048 mM min−1, respectively. On the other hand, the maximal rate Vmax of PC and MDH dual-enzyme with bicarbonate was determined to be 0.040 mM min−1. These results indicated that pyruvate is carboxylated using low concentration of gaseous CO2 (15%) as a starting material instead of bicarbonate.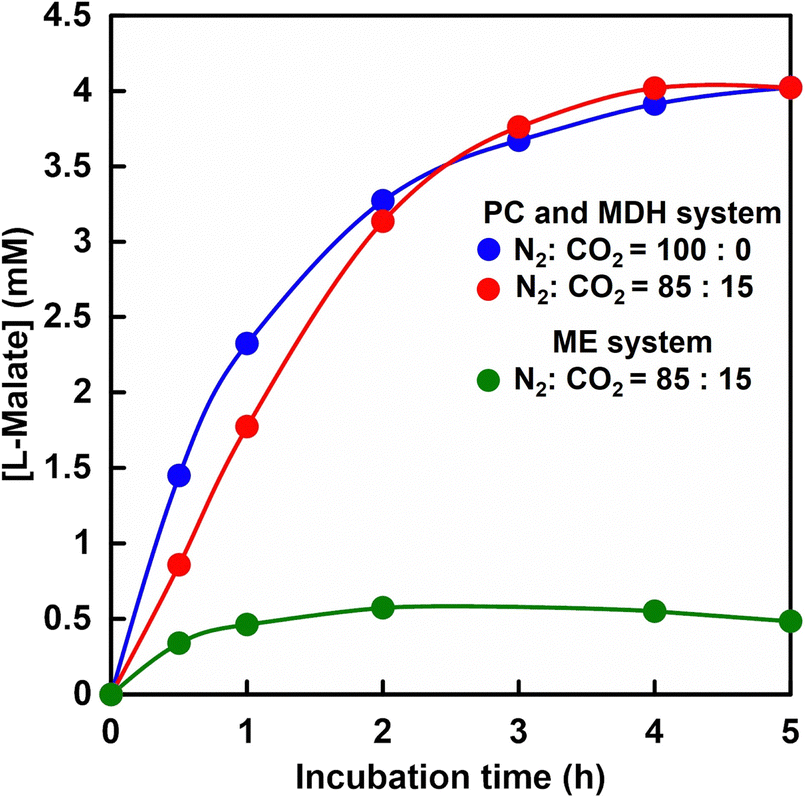 | ||
| Fig. 11 Time dependence of L-malate concentration in the solution of sodium pyruvate, ATP, manganese chloride, acetyl-CoA, NADH, PC and MDH in HEPES-NaOH buffer. The gas phase: the gas mixture of 85% N2 and 15% CO2 (red), and 100% CO2 (blue). Time dependence of L-malate concentration in the solution of sodium pyruvate, magnesium chloride, NADH and ME in HEPES-NaOH buffer. The gas phase: the gas mixture of 85% N2 and 15% CO2 (green). | ||
Time dependence of L-malate production based on ME-catalysed carboxylation of pyruvate under a gas mixture consisting of 85% N2 and 15% CO2 is also shown in Fig. 11. As shown in Fig. 11, the L-malate concentration increased with incubation time but tended to saturate after 1 h of incubation. The L-malate concentration was estimated to be ca. 0.49 mM and the yield for pyruvate to L-malate was calculated to be 9.8% after 5 h of incubation. The yield for pyruvate to L-malate with ME under 100% CO2 gas condition was reported to be 46%.61 The yield of L-malate production based on ME-catalysed carboxylation of pyruvate under a gas mixture consisting of 85% N2 and 15% CO2 or 100% CO2 gas was significantly lower compared with that of the PC and MDH dual-enzyme system.
Let us focus on the products from the ion chromatograph chart. Fig. 12 shows an ion chromatography chart of a sample after 5 h of incubation.
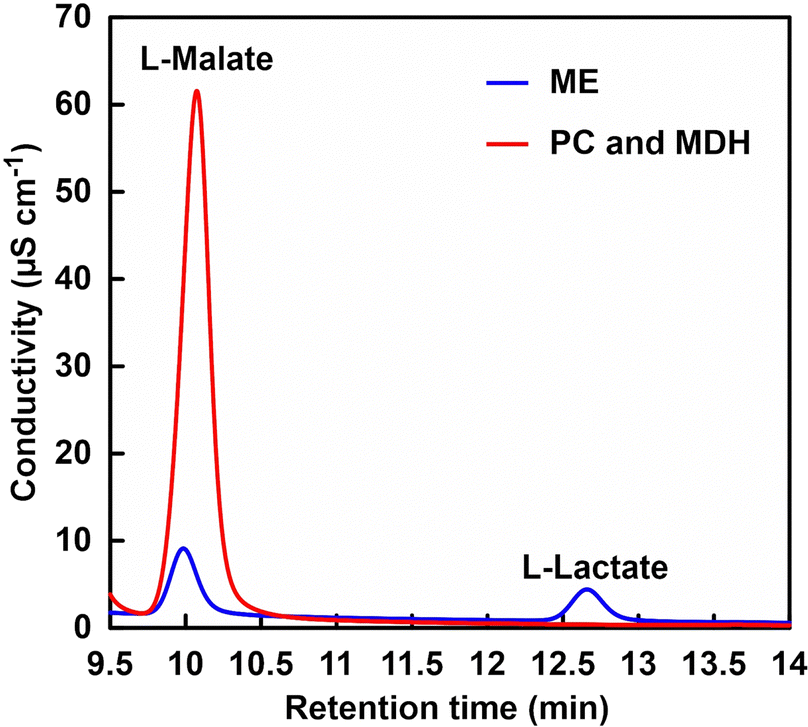 | ||
| Fig. 12 A chart of an ion chromatogram sampled from the reaction solution containing sodium pyruvate (5.0 mM), ATP (5.0 mM), manganese chloride (5.0 mM), NADH (5.0 mM), PC (1.0 U), MDH (10 U) and acetyl-CoA (1.0 mM) in 5.0 mL of 500 mM HEPES buffer-NaOH (pH 7.2) (red). Blue: sodium pyruvate (5.0 mM), magnesium chloride (5.0 mM), NADH (5.0 mM) and ME (0.7 U) in 5.0 mL of 500 mM HEPES buffer-NaOH (pH 7.2). | ||
In the PC and MDH dual-enzyme system, only the peak around 10 min due to the L-malate was observed. In the ME system, on the other hand, in addition to a peak based on L-malate at about 10 min, a peak based on L-lactate was also observed at about 12.7 min. Under the condition of low bicarbonate concentration in ME-catalysed L-malate production, the reduction of pyruvate to L-lactate is preferential over the carboxylation of pyruvate. In other words, L-malate production based on ME-catalysed carboxylation of pyruvate requires high concentration of CO2 in the gas phase as well as bicarbonate in solution. On the other hand, the PC and MDH dual-enzyme system was successfully used to efficiently produce L-malate from pyruvate even under low CO2 concentration condition.
Fumarate production from pyruvate and direct captured CO2 with PC, MDH and FUM in the presence of ATP and NADH
Since efficient pyruvate to L-malate production was successfully achieved under low CO2 concentration condition by using a PC and MDH dual-enzyme system, FUM was added to this system and fumarate production was attempted. Fig. 13 shows the time dependence of L-malate (a) and fumarate (b) concentration with PC and MDH in HEPES-NaOH buffer including ATP and NADH (The ion chromatograph chart during the reaction is shown in Fig. S7, ESI†). The composition of the gas phase including balloon was adjusted to 85% N2 and 15% CO2 or 100% CO2 gas. The pressure in the reaction vessel was maintained at 1.01325 × 105 Pa. L-Malate and fumarate concentrations increased with incubation time under all conditions. After 5 h of incubation, the L-malate concentration was estimated to be ca. 3.2 mM in both cases. On the other hand, ca. 0.8 mM of fumarate was produced in both cases after 5 h of incubation. | ||
| Fig. 13 Time dependence of L-malate (a) and fumarate (b) concentration in the solution of sodium pyruvate, ATP, manganese chloride, acetyl-CoA, NADH, PC, MDH and FUM in HEPES-NaOH buffer. The gas phase: the gas mixture of 85% N2 and 15% CO2 (red) and 100% CO2 (blue). Time dependence of L-malate (a) and fumarate (b) concentration in the solution of sodium pyruvate, magnesium chloride, NADH, ME and FUM in HEPES-NaOH buffer. The gas phase: the gas mixture of 85% N2 and 15% CO2 (green). | ||
The yields for pyruvate to fumarate in both systems after 5 h of incubation were calculated to be ca. 16%. No significant differences in L-malate and fumarate production were observed under conditions of a gas mixture consisting of 85% N2 and 15% CO2 in the PC, MDH and FUM multi-enzyme system. In the PC, MDH and FUM multi-enzyme system, L-malate production tended to saturate to a constant value after 2 h of incubation. In contrast, fumarate tended to produce steadily with incubation time. The initial reaction rates for L-malate production under a gas mixture consisting of 85% N2 and 15% CO2, and 100% CO2 gas were estimated to be 0.03 and 0.05 mM min−1, respectively. On the other hand, the initial reaction rates for fumarate production under a gas mixture consisting of 85% N2 and 15% CO2, and 100% CO2 gas were estimated to be 0.006 and 0.006 mM min−1, respectively. Moreover, the Km value of L-malate for FUM-catalysed fumarate production was estimated to be 0.65 mM. These results suggest that the rate of fumarate production based on L-malate dehydration catalysed by FUM is slower than the rate of L-malate production from pyruvate and CO2 catalysed by PC and MDH. Time dependence of L-malate and fumarate production with ME and FUM under a gas mixture consisting of 85% N2 and 15% CO2 condition is also shown in Fig. 13. As shown in Fig. 13, L-malate and fumarate tended to increase up to 1 h of incubation and then saturate to a constant value. After 5 h of incubation, the L-malate and fumarate concentrations were estimated to be 0.60 and 0.15 mM. The yield for pyruvate to fumarate after 5 h of incubation was calculated to be ca. 3.0%.
The change in the amount of carbonate species in the reaction solution is mentioned here. As an example, changes in the amount of carbonate species during the fumarate production reaction with the PC, MDH and FUM system under the condition of the gas mixture of 85% N2 and 15% CO2 were investigated using ion chromatography.
In the ion chromatography system used in this experiment, it is detected as the sum of carbonate and bicarbonate. By using ion chromatography, the peak around 20.7 min due to the carbonate species was observed as shown in Fig. S8 (ESI†). As shown in Fig. S8 (ESI†), carbonate species in the sample solution increased with incubation time for 3 h. This means that in this reaction system, gaseous CO2 in the gas phase is constantly supplied by the HEPES buffer solution by the mechanism shown in Fig. 7.
Fig. 14 shows an ion chromatography chart of a sample containing sodium pyruvate, ATP, manganese chloride, NADH, PC, MDH, FUM and acetyl-CoA in HEPES buffer-NaOH and containing sodium pyruvate, magnesium chloride, NADH, ME and FUM in HEPES buffer-NaOH under a gas mixture consisting of 85% N2 and 15% CO2 after 2 h of incubation.
 | ||
| Fig. 14 A chart of an ion chromatogram sampled from the reaction solution containing sodium pyruvate (5.0 mM), ATP (5.0 mM), manganese chloride (5.0 mM), NADH (5.0 mM), PC (1.0 U), MDH (10 U), FUM (0.5 U) and acetyl-CoA (1.0 mM) in 5.0 mL of 500 mM HEPES buffer-NaOH (pH 7.2) after 2 h of incubation (red). Blue: sodium pyruvate (5.0 mM), magnesium chloride (5.0 mM), NADH (5.0 mM), ME (0.7 U) and FUM (0.5 U) in 5.0 mL of 500 mM HEPES buffer-NaOH (pH 7.2). The gas phase: the gas mixture of 85% N2 and 15% CO2. | ||
In the PC, MDH and FUM multi-enzyme system, the peaks around 10 min due to the L-malate and around 12.3 min due to the fumarate were observed. In the ME system, on the other hand, in addition to the peaks based on L-malate at about 10 min and fumarate at about 12.3 min, a peak based on L-lactate was also observed at about 12.7 min. Even under low bicarbonate concentration condition in the presence of FUM, the ME-catalysed reduction of pyruvate to L-lactate is preferential over the carboxylation of pyruvate. By using the PC, MDH and FUM multi-enzyme system, utilisation of low concentrations of CO2 as a reagent for the carboxylation of pyruvate and subsequently improving the yield of fumarate production were achieved.
Finally, the effect of CO2 concentration on fumarate production from pyruvate and CO2 using the PC, MDH and FUM multi-enzyme system was investigated. Fig. 15 shows the concentrations of L-malate (a) and fumarate production (b) after 2 h of incubation with the PC, MDH and FUM multi-enzyme system under a gas mixture consisting of 90% N2 and 10% CO2, 85% N2 and 15% CO2, and 100% CO2 (The ion chromatograph chart during the reaction is shown in Fig. S9, ESI†).
 | ||
| Fig. 15 The concentrations of L-malate (a) and fumarate production (b) after 2 h of incubation with the PC, MDH and FUM multi-enzyme system under a gas mixture consisting of 90% N2 and 10% CO2 (blue), 85% N2 and 15% CO2 (red), and 100% CO2 (green). | ||
After 2 h of incubation, 2.6, 2.9 and 3.0 mM of L-malate was produced under the condition of a gas mixture consisting of 90% N2 and 10% CO2, 85% N2 and 15% CO2, and 100% CO2, respectively. 0.52, 0.59 and 0.60 mM of fumarate was produced after 2 h of incubation under the condition of a gas mixture consisting of 90% N2 and 10% CO2, 85% N2 and 15% CO2, and 100% CO2, respectively. The concentrations of L-malate and fumarate production slightly decreased in the PC, MDH and FUM multi-enzyme system under the condition of a gas mixture consisting of 90% N2 and 10% CO2. Under the condition of less than 10% CO2 in the gas mixture, the CO2 concentration in the sample solution is approximately below 5.0 mM, close to the Km value for L-malate production with PC and MDH (3.4 mM), so that L-malate is not produced at Vmax and consequently fumarate production is also expected to be reduced. In contrast, no L-malate and fumarate production was observed in the PC, MDH and FUM multi-enzyme system under the condition of 100% N2 gas. However, by using a multi-enzyme system of PC, MDH and FUM, it is possible to use the equivalent of low pressure (near atmospheric pressure) and low concentration (10–20%) CO2 gas contained in the flue gas of coal-fired power stations and cement plants as a reagent for carboxylation of pyruvate and efficient fumarate production by subsequent dehydration of the produced L-malate could also be achieved.
Conclusions
In conclusion, the improvement of yield for fumarate production from low-concentration CO2 below 15% captured from the gas phase by HEPES-NaOH buffer solution and pyruvate in an aqueous medium using a multi enzyme system consisting of PC, MDH and FUM in the presence of ATP and NADH is achieved. In particular, the dual-enzyme system consisting of PC and MDH drastically improved the yield of L-malate production from pyruvate and low-concentration CO2 to about 80% in the presence of ATP and NADH. It has been shown that pyruvate was converted into L-malate in high yields (approximately 80%) directly using 15% CO2 equivalent to flue gas from coal-fired power plants as a carboxylating agent. Moreover, the development of fumarate production from direct captured low-concentration CO2 and biobased pyruvate as raw materials with a multi-enzyme system containing PC, MDH and FUM in the presence of ATP and NADH has also been successful.Data availability
The authors confirm that the data supporting the findings of this manuscript are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by JSPS KAKENHI 23H05404, 22H01872, 22H01871, 19KK0144, and by Institute for Fermentation, Osaka (IFO) (G-2023-3-050).Notes and references
- G. Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082 CrossRef CAS PubMed.
- T. R. Walker and D. Xanthos, Resour., Conserv. Recycl., 2018, 133, 99 CrossRef.
- J. Hammer, M. H. Kraak and J. R. Parsons, Rev. Environ. Contam. Toxicol., 2012, 220, 1 CAS.
- C. Gioia, G. Giacobazzi, M. Vannini, G. Totaro, L. Sisti, M. Colonna, P. Marchese and A. Celli, ChemSusChem, 2021, 14, 4167 CrossRef CAS PubMed.
- L. Filiciotto and G. Rothenberg, ChemSusChem, 2021, 14, 56 CrossRef CAS PubMed.
- R. Ciriminna and M. Pagliaro, ChemistryOpen, 2020, 9, 8 CrossRef CAS PubMed.
- S. Agarwal, Macromol. Chem. Phys., 2020, 221, 2000017 CrossRef CAS.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50 CrossRef CAS PubMed.
- Handbook of Bioplastics and Biocomposites Engineering Applications, ed. S. Pilla, Wiley-Scrivener, Salem (MA), 2011 Search PubMed.
- Nova-Institute, Bio-based Building Blocks and Polymers, Hurth (Germany): 2019.
- A. S. Al Hosni, J. K. Pittman and G. D. Robso, Waste Manage., 2019, 97, 105 CrossRef CAS PubMed.
- M. Karamanlioglu, A. Houlden and G. D. Robson, Int. Biodeterior. Biodegrad., 2014, 95, 301 CrossRef CAS.
- T. K. Chua, M. Tseng and M. K. Yang, AMB Express, 2013, 3, 8 CrossRef PubMed.
- A. Ammala, Prog. Polym. Sci., 2011, 36, 1015 CrossRef CAS.
- L. Cosgrove, P. L. McGeechan, G. D. Robson and P. S. Handley, Appl. Environ. Microbiol., 2007, 73, 5817 CrossRef CAS PubMed.
- K. Jha, R. Kataria, J. Verma and S. Pradhan, AIMS Mater. Sci., 2019, 6, 119 CAS.
- A. Samir, F. H. Ashour, A. A. A. Hakim and M. Bassyouni, NPJ Mater. Degrad., 2022, 6, 68 CrossRef CAS.
- B. Ghanbarzadeh and A. Hadi, Biodegradable polymers, Biodegradation-life of science, 2013, p. 141 Search PubMed.
- M. P. Ho, H. Wang, J. H. Lee, C. K. Ho, K. T. Lau, J. Leng and D. Hui, Composites, Part B, 2012, 43, 3549 CrossRef CAS.
- A. B. Balaji, H. Pakalapati, M. Khalid, R. Walvekar and H. Siddiqui, “Natural and synthetic biocompatible and biodegradable polymers. Biodegradable and Biocompatible Polymer Composites: Processing, Properties and Applications” 2017.
- S. Mangaraj, A. Yadav, L. M. Bal, S. K. Dash and N. K. Mahanti, Rev. J. Packag. Technol. Res., 2019, 3, 77 Search PubMed.
- Y. Watanabe, N. Hamada, M. Morita and Y. Tsujisaka, Arch. Biochem. Biophys., 1976, 174, 575 CrossRef CAS PubMed.
- S. Matsumura, N. Tomizawa, A. Toki, K. Nishikawa and K. Toshima, Macromolecules, 1999, 32, 7753 CrossRef CAS.
- T. Hatanaka, N. Asahi and M. Tsuji, Biosci., Biotechnol., Biochem., 1995, 59, 1813 CrossRef CAS.
- E. Chiellini, A. Corti, S. D’Antone and R. Solaro, Prog. Polym. Sci., 2003, 28, 963 CrossRef CAS.
- K. Kolter, A. Dashevsky, M. Irfan and R. Bodmeier, Int. J. Pharm., 2013, 457, 470 CrossRef CAS PubMed.
- M. C. Carter, A. Hejl, S. Woodfin, B. Einsla, M. Janco, J. DeFelippis, R. J. Cooper and R. C. Even, ACS Macro Lett., 2021, 10, 591 CrossRef CAS PubMed.
- D. Das, S. Mukherjee, A. Pal, R. Das, S. G. Sahu and S. Pal, RSC Adv., 2016, 6, 9352 RSC.
- G. E. Luckachan and C. Pillai, J. Polym. Environ., 2011, 19, 637 CrossRef CAS.
- R. D. Saini, Int. J. Appl. Chem., 2017, 13, 179 Search PubMed.
- V. Nagarajan, A. K. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2016, 4, 2899 CrossRef CAS.
- O. Martin and L. Avérous, Polymer, 2001, 42, 6209 CrossRef CAS.
- H. R. Kricheldorf and J. M. Jonté, Polym. Bull., 1983, 9, 276 CAS.
- Y. K. Jung, T. Y. Kim and T. Yong, Biotechnol. Bioeng., 2009, 105, 161 CrossRef PubMed.
- P. K. Samantaray, A. Little, D. M. Haddleton, T. McNally, B. Tan, Z. Sun, W. Huang, Y. Ji and C. Wan, Green Chem., 2020, 22, 4055 RSC.
- S. Mohapatra, S. Maity, H. R. Dash, S. Das, S. Pattnaik, C. C. Rath and D. Samantaray, Biochem. Biophys. Rep., 2017, 12, 206 Search PubMed.
- K. Yamane, H. Sato, Y. Ichikawa, K. Sunagawa and Y. Shigaki, Polym. J., 2014, 46, 76 CrossRef.
- E. Göktürk, A. G. Pemba and S. A. Miller, Polym. Chem., 2015, 6, 3918 RSC.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484 RSC.
- J. O. Iroh, in Polymer Data Handbook, ed. J. E. Mark, Oxford University Press, New York, 1999, pp. 361–362 Search PubMed.
- H. Bittiger, R. H. Marchessault and W. D. Niegisch, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 1923 CrossRef CAS.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803 CrossRef CAS.
- Y. Ikada and H. Tsuji, Macromol. Rapid Commun., 2000, 21, 117 CrossRef CAS.
- N. Jacquel, F. Freyermouth, F. Fenouillot, A. Rousseau, J. P. Pascault, P. Fuertes and R. Saint-Loup, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5301 CrossRef CAS.
- D. Minh, M. Besson, C. Pinel, P. Fuertes and C. Petitjean, Top. Catal., 2010, 53, 1270 CrossRef CAS.
- R. M. Deshpande, V. V. Buwa, C. V. Rode, R. V. Chaudhari and P. L. Mills, Catal. Commun., 2002, 3, 269 CrossRef CAS.
- T. Willke and K. D. Vorlop, Microbiol. Biotechnol., 2004, 66, 131 CrossRef CAS PubMed.
- C. A. R. Engel, A. J. J. Straathof, T. W. Zijlmans, W. M. van Gulik and L. A. M. van der Wielen, Appl. Microbiol. Biotechnol., 2008, 78, 379 CrossRef PubMed.
- A. L. Lehninger, D. L. Nelson and M. M. Cox, Principles of Biochemistry, W. H. Freeman and Company, New York, NY, 5th edn, 2008 Search PubMed.
- Y. Liu, J. Song, T. Tan and L. Liu, Appl. Biochem. Biotechnol., 2015, 175, 2823 CrossRef CAS PubMed.
- W. J. Landsperger and B. G. Harris, J. Biol. Chem., 1976, 251, 3599 CrossRef CAS PubMed.
- O. Kolaj-Robin, S. R. OKane, W. Nitschke, C. Leger, F. Baymann and T. Soulimane, Biochim. Biophys. Acta, 2011, 1807, 68 CrossRef CAS PubMed.
- N. Yumoto and M. Tokushige, Biochem. Biophys. Res. Commun., 1988, 153, 1236 CrossRef CAS PubMed.
- S. Beeckmans and L. Kanarek, Eur. J. Biochem., 1977, 78, 437 CrossRef CAS PubMed.
- M. Mescam, K. Vinnakota and D. Beard, J. Biol. Chem., 2011, 286, 21100 CrossRef CAS.
- T. Mizobata, T. Fujioka, F. Yamasaki, M. Hidaka, J. Nagai and Y. Kawata, Arch. Biochem. Biophys., 1998, 355, 49 CrossRef CAS.
- J. S. Keruchenko, I. D. Keruchenko, K. L. Gladilin, V. Zaitsev and N. Y. Chiragadze, Biochim. Biophys. Acta, 1992, 1122, 85 CrossRef CAS PubMed.
- M. Takeuchi and Y. Amao, React. Chem. Eng., 2022, 7, 1931 RSC.
- M. Takeuchi and Y. Amao, RSC Sustainability, 2023, 1, 90 RSC.
- M. Takeuchi and Y. Amao, Sustainable Energy Fuels, 2023, 7, 355 RSC.
- M. Takeuchi and Y. Amao, RSC Sustainability, 2023, 1, 1874 Search PubMed.
- M. Takeuchi and Y. Amao, Dalton Trans., 2024, 53, 418 RSC.
- Y. Morimoto, K. Honda, X. Ye, K. Okano and H. Ohtake, J. Biosci. Bioeng., 2014, 117, 147 CrossRef CAS PubMed.
- M. Takeuchi and Y. Amao, J. Jpn. Petrol. Inst., 2024, 67, 167 CrossRef.
- S. Jitrapakdee, M. St Maurice, I. Rayment, W. W. Cleland, J. C. Wallace and P. V. Attwood, Biochem. J., 2008, 413, 369 CrossRef CAS PubMed.
- G. B. Warren and K. F. Tipton, Biochem. J., 1974, 139, 297 CrossRef CAS PubMed.
- M. C. Scrutton and M. F. Utter, J. Biol. Chem., 1967, 242, 1723 CrossRef CAS.
- Z. D. Wang, B. J. Wang, Y. D. Ge, W. Pan, J. Wang, L. Xu, A. M. Liu and G. P. Zhu, Mol. Biol. Rep., 2011, 38, 1629 CrossRef CAS PubMed.
- T. K. Sundaram, I. P. Wright and A. E. Wilkinson, Biochemistry, 1980, 19, 2017 CrossRef CAS PubMed.
- Y. D. Ge, Y. T. Guo, L. L. Jiang, H. H. Wang, S. L. Hou and F. Z. Su, Protein J., 2023, 42, 14 CrossRef CAS PubMed.
- M. Takeuchi and Y. Amao, RSC Sustainability, 2024, 2, 2491 Search PubMed.
- Y. Yamazaki, M. Miyaji and O. Ishitani, J. Am. Chem. Soc., 2022, 144, 6640 CrossRef CAS PubMed.
- M. R. Smith and S. S. Myers, Nat. Clim. Change, 2018, 8, 834 CrossRef CAS.
- G. V. Last and M. T. Schmick, Environ. Earth Sci., 2015, 74, 1189 CrossRef CAS.
- M. Erans, E. S. Sanz-Pérez, D. P. Hanak, Z. Clulow, D. M. Reiner and G. A. Mutch, Energy Environ. Sci., 2022, 15, 1360 RSC.
- K. S. Lackner, H.-J. Ziock and P. Grimes, Proc. 24th Int. Conf. Coal Util. Fuel Syst., 1999, pp. 885–886.
- F. Zeman and K. S. Lackner, World Res. Rev., 2004, 16, 157 Search PubMed.
- D. W. Keith, Science, 2009, 325, 1654 CrossRef CAS PubMed.
- G. Chichilnisky and P. Eisenberger, Nat. Opin., 2009, 459, 1053 CrossRef CAS PubMed.
- R. A. Pielke Jr., Environ. Sci. Policy, 2009, 12, 216 CrossRef.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87 CrossRef CAS PubMed.
- Z. Zhang, S. Y. Pan, H. Li, J. Cai, A. G. Olabi, E. J. Anthony and V. Manovic, Renewable Sustainable Energy Rev., 2020, 125, 109799 CrossRef CAS.
- R. B. McComb, L. W. Bond, R. W. Burnett, R. C. Keech and G. N. Bowers Jr, Clin. Chem., 1976, 22, 141 CrossRef CAS.
- S. Foltran, M. E. Vosper, N. B. Suleiman, A. Wriglesworth, J. Ke, T. C. Drage, M. Poliakoff and M. W. Georg, Int. J. Greenhouse Gas Control, 2015, 35, 131 CrossRef CAS.
- Y. Kita and Y. Amao, Green Chem., 2023, 25, 2699 RSC.
- L. N. Plummer and E. Busenberg, Geochim. Cosmochim. Acta, 1982, 46, 1011 CrossRef CAS.
- L. E. Westerhold, L. C. Bridges, S. R. Shaikh and T. N. Zeczycki, Biochemistry, 2017, 56, 3492 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj03485f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |