
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamically controlled chemical regeneration of spent battery cathodes using recyclable electron donors under ambient conditions†
Sunghyun
Ko‡
 ab,
Jinkwan
Choi‡
ac,
Jihyun
Hong
*d,
Changsoo
Kim
e,
Uichan
Hwang
af,
Minhyung
Kwon
af,
Gukhyun
Lim
cd,
Seok Su
Sohn
c,
Jinha
Jang
b,
Ung
Lee
egh,
Chan Beum
Park
b and
Minah
Lee
*a
ab,
Jinkwan
Choi‡
ac,
Jihyun
Hong
*d,
Changsoo
Kim
e,
Uichan
Hwang
af,
Minhyung
Kwon
af,
Gukhyun
Lim
cd,
Seok Su
Sohn
c,
Jinha
Jang
b,
Ung
Lee
egh,
Chan Beum
Park
b and
Minah
Lee
*a
aEnergy Storage Research Center, Korea Institute of Science and Technology (KIST), 14 Gil 5 Hwarang-ro, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: minahlee@kist.re.kr
bDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology, 335 Science Road, Daejeon 305-701, Republic of Korea
cDepartment of Materials Science & Engineering, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
dEnergy Materials Research Center, Korea Institute of Science and Technology (KIST), 14 Gil 5 Hwarang-ro, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: jihyunh@kist.re.kr
eClean Energy Research Center, Korea Institute of Science and Technology (KIST), 14 Gil 5 Hwarang-ro, Seongbuk-gu, Seoul 02792, Republic of Korea
fDepartment of Chemical & Biological Engineering, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
gDivision of Energy and Environmental Technology, KIST School, Korea University of Science and Technology (UST), Seoul, 02792, Republic of Korea
hGreen School, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul, 02841, Republic of Korea
First published on 17th April 2024
Abstract
Devising an energy-efficient, profitable, and safe technology to recycle lithium-ion batteries (LIBs) is crucial for their continuous adoption in electric vehicles and grid energy storage. Herein, using recyclable electron donors (REDs) for which the redox potentials range between cathode operation and over-lithiation potentials, we establish thermodynamically controlled Li-coupled electron transfer from REDs to cathodes as a viable route for directly regenerating spent cathodes in dry air at room temperature. Simple soaking of the spent cathode in a regeneration solution enables the complete recovery of the original chemical composition and capacity of the Li-deficient cathode through electron and Li-ion transfer from RED molecules and Li salts, respectively. The RED-based lithiation universally applies to cathode materials with heterogeneous Li loss, allowing the Li quantification and cathode separation processes to be bypassed for recycling. We further demonstrate the practical feasibility of this approach by regenerating spent cathodes from commercial 1-A h pouch cells. In addition, we show that the used regeneration solution can be refreshed by simply mixing with reductants for reuse in lithiation, thereby promising minimal cost and chemical waste for battery recycling. The proposed cathode regeneration method under ambient temperature and pressure will propel the development of facile and scalable LIB recycling technologies.
Broader contextDeveloping an energy-efficient, profitable, and safe technology for recycling lithium-ion batteries (LIBs) is crucial for their continuous integration into electric vehicles and grid energy storage systems. The current LIB recycling technology predominantly involves destructive processes to retrieve valuable metal elements from spent cathodes, necessitating an energy-intensive resynthesis step to produce the original high-value cathode materials. Despite efforts to devise non-destructive methods for regenerating spent cathodes by directly replenishing lost Li in cathodes, the proposed strategies face limitations in industrial viability, as they require impractical lithium quantification to prevent under- and over-lithiation or extreme synthetic conditions such as high pressure/temperature and an inert atmosphere. Herein, we present a viable route for directly regenerating spent cathodes using thermodynamically controlled Li-coupled electron transfer from recyclable electron donors (REDs) to cathodes. The redox potentials of REDs, strategically positioned between cathode operation and over-lithiation potentials, facilitate topotactic lithiation of spent cathodes without inducing detrimental side reactions under ambient conditions. The high chemical stability of REDs enables a closed-loop cathode recycling procedure, minimizing chemical waste. This work introduces an unprecedented chemical regeneration mechanism for spent cathodes, paving the way for the development of scalable and environmentally sustainable LIB recycling technologies. |
Introduction
With the ever-increasing demand for carbon-neutral energy to meet the global targets of reducing greenhouse gas emissions, Li-ion batteries (LIBs) have been extensively applied for medium- and large-scale energy storage such as in electric vehicles (EVs) and grid storage.1–5 The market demand for LIBs is projected to increase by almost 10 fold (2.4 TWh) by 2030,6,7 which would translate into a massive amount of waste batteries given the limited cycle life of LIBs. The global market for spent LIB recycling was worth $6.5 billion in 2022, with a compound annual growth rate of 20.6% over the next decade.8 Among the various LIB components, the primary focus in spent LIB recycling is on the reuse of degraded cathodes. The representative cathodes, layered Li nickel cobalt oxides (LiNixMnyCo1−x−yO2, NMC) consist of expensive metallic elements (Ni, Co, and Li), which are geographically concentrated in specific areas and require harsh environmental and social costs for mining,7,9–12 thereby accounting for more than one-third of the entire cost of LIBs.13,14Despite this urgency, limited options currently exist for bulk LIB recycling, and they mainly involve destructive processes to recover the valuable metal elements from spent cathodes.15,16 Pyrometallurgy methods, the most widely used approach to date, can recover metal (Ni, Co, and Mn) alloys through high-temperature (>1400 °C) smelting and refining.16 However, the high energy consumption during the smelting process poses ecological and economic concerns.17,18 Alternatively, hydrometallurgy methods can dissolve the spent cathodes in acidic leaching solutions at relatively low temperatures to recover valuable metal elements (including Li) in the form of salt precursors (e.g., Li2CO3 and MC2O4·2H2O, M = Ni, Co, Mn) that can be used to synthesize new cathode materials.1,19,20 Nevertheless, the massive amount of wastewater required to neutralize the strong acid solutions used in the process still pose environmental and safety concerns. Furthermore, as the destructive recycling methods merely retrieve low-value materials (such as metal alloys or salt precursors) from the spent cathodes, energy-consuming resynthesis steps are still required to produce the original high-value NMC of ideal stoichiometry and crystal structure.21
In fact, by supplying the lost Li to the spent NMC, the cathode material can be directly reused to build new LIBs. When cycling LIBs, parasitic reactions primarily involving solid–electrolyte interphase formation on the anode surface continuously reduce the amount of active Li in batteries initially stored in NMC cathodes, and the insufficient accessible Li content translates into capacity loss.22–25 Thus, even after fully discharging the aged battery, the spent NMC still possesses a significant number of Li vacancies (VLi) in the crystal structure, which should be refilled through a regeneration process to attain the original capacity.26,27
One of the simplest ways to replenish the lost Li is through solid-state sintering of the spent cathodes with a Li source, typically LiOH or Li2CO3, at high temperatures (>800 °C).23,28–30 This method requires precise control of the Li amount to avoid the undesired formation of impurity phases;31,32 however, the intrinsic compositional heterogeneity of the spent cathodes limits accurate quantification of VLi of NMCs. Another option is to conduct hydrothermal reactions using a concentrated Li solution sealed in a highly pressurized reactor (23 atm) at moderate temperature (∼220 °C).33–39 Unlike solid-state sintering, the hydrothermal method is insensitive to VLi as controlling the temperature and pressure ensures high phase purity even with excess Li. Nevertheless, increasing the size of high-pressure vessels requires extra safety precautions and costs, which necessitates lowering the reaction temperature.27,40,41 Recently, molten salt synthesis has been proposed to regenerate aged cathodes with the advantages of using moderate temperatures and ambient pressure.42 Spent NMCs can be regenerated in a series of molten nitrate, sulfonate, and carbonate salts; however, the corrosive nature of the eutectic molten salts necessitates complex and costly anti-corrosion treatments on reactors.15,43–46 Given the limited industrial viability of the previous direct regeneration methods, developing a simple and scalable strategy based on an innovative lithiation mechanism is crucial to manage the upcoming end-of-life of widely employed LIBs.
Here, we propose a direct cathode regeneration method that uses a spontaneous charge-transfer reaction between electron-donating molecules and NMC at ambient temperature and pressure in dry air. We show that organic electron donors with adequate redox potential between NMC operation and overlithiation potentials, such as 5,10-dimethylphenazine (DMPZ), ferrocene (Fc) and N,N′-diphenyl-p-phenylenediamine (DPPD), facilitate the topotactic lithiation of NMC in an organic solution containing Li salts, such as Li hexafluorophosphate (LiPF6) and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI). The thermodynamically driven lithiation reaction stops when the potential of NMC622 reaches the redox potential of the electron donors, preventing structural degradation caused by overlithiation. As a result, the regenerated NMC622 recovers the original lattice parameters and available capacity, exhibiting electrochemical performance that is comparable to that of the fresh NMC622. Moreover, the charge-transfer reaction can be universally applied to a mixture of cathodes with various Li deficiencies and to other types of cathodes including Ni-rich NMC, LiFePO4 (LFP), LiNi0.5Mn1.5O4 (LNMO), and LiCoO2 (LCO). The simplicity of this solution-based lithiation approach ensures its scalability, as confirmed by the successful regeneration of long-cycled NMC cathodes retrieved from commercial 1-A h-capacity pouch cells. After the regeneration process, the electron donors form stable ion pairs with salt anions in the solution without chemical decomposition, enabling the recycling of the used regeneration solution with general reducing agents (Li2O2, Li2S). The circularity promises minimal cost and chemical waste for the LIB recycling process. This unprecedented practical electron-donor-based cathode regeneration method will pave the way to developing scalable and sustainable LIB recycling technologies.
Results and discussion
Thermodynamics of spontaneous charge transfer from DMPZ to Li-deficient NMC cathode
Unwanted side reactions in batteries, represented by the SEI formation on anodes, steadily and repeatedly occur upon cycling, causing the irreversible loss of active Li within the closed electrochemical cell.22,23 Because of the lack of active Li ions in the given system, even a deep discharging process can only partially lithiate NMC cathodes after long-term operation, leaving vacancies in the Li layers (VLi, Fig. 1(a)).24,33,47 The NMC cathodes with VLi in the crystal structure contain high-valence Ni ions to ensure charge balance and exhibit a higher charging voltage than fresh NMC.40 For example, the charging process of NMC622 with 24.1% VLi initiates at 3.7 V, which is 729-mV higher than the charging voltage of fresh NMC622 containing 7.7% VLi after a formation cycle (Fig. 1(b), Fig. S1 and Table S1, ESI†).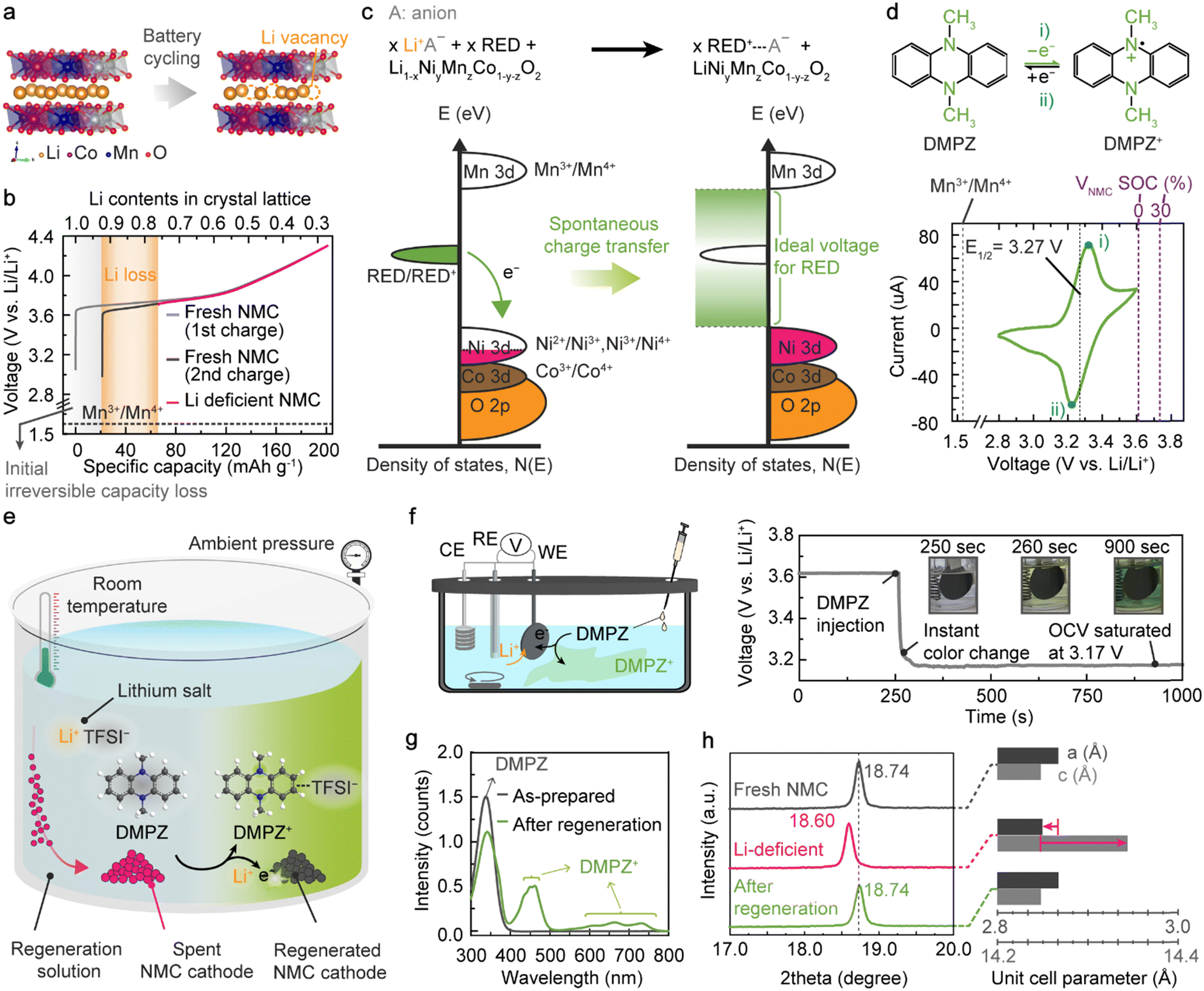 | ||
| Fig. 1 Mechanism and demonstration of RED-based lithiation of spent LIB cathodes. (a) VLi formation in NMC622 cathode during battery cycling. (b) Galvanostatic charge profiles of fresh NMC622 cathode and Li-deficient NMC622 cathode. (c) Ideal energy levels of RED relative to NMC622 density of states for spontaneous lithiation. (d) Redox mechanism of DMPZ molecule and corresponding cyclic voltammogram at a scan rate of 1 mV s−1. (e) Illustration of NMC622 regeneration processes in a solution containing DMPZ and LiTFSI. (f) Changes in OCV of the Li-deficient NMC622 cathode during soaking in the regeneration solution. (g) UV-vis absorption spectra of the regeneration solution before and after the regeneration process. The regeneration solution contained 1.5 times more DMPZ and LiTFSI than the VLi in NMC622. (h) XRD patterns of Fresh (grey), Li-deficient (magenta), and regenerated NMC622 cathode (green), confirming the complete recovery of the Li content through the regeneration process. | ||
To insert Li ions and electrons into the Li-deficient NMC retrieved from the spent batteries, we propose an innovative one-step regeneration strategy exploiting redox-active organic compounds as a recyclable electron donor (RED), which can be used at room temperature in ambient air. The first essential criterion for enabling the spontaneous electron transfer from the neutral RED to NMC cathodes is that the oxidation potential of the RED should be lower than the initial potential of charging NMC (3.7 V), thereby resulting in a negative value of the Gibbs free energy (ΔG = − nFE = − nF(ENMC − ERED/RED+)) for the reaction. When solubilized in a regeneration solution containing Li salts, those REDs can reduce the high-valence Ni to a lower oxidation state through spontaneous electron transfer from the highest occupied molecular orbital (HOMO) of the neutral RED to the Ni 3d orbital of Li-deficient NMC (Fig. 1(c)). To ensure charge neutrality, Li ions are inserted into the crystal lattice of the Li-deficient NMC according to
| Li1−xNiyMnzCo1−y−zO2 + x e− + x Li+ → LiNiyMnzCo1−y−zO2 | (1) |
In parallel, the oxidized REDs (RED+) form ion pairs with the anion remaining from the Li salt through the following reaction:
| x RED + x A− → x RED+-A− + x e− | (2) |
Thus, the overall cathode regeneration reaction can be described by combining eqn (1) and (2):
| Li1−xNiyMnzCo1−y−zO2 + x RED + x A− + x Li+ → x RED+-A− + LiNiyMnzCo1−y−zO2 | (3) |
As the second criterion, the redox potential of the RED should be higher than the potential of Mn3+/Mn4+ redox couples in the layered oxides, approximately 1.6 V vs. Li/Li+, to inhibit the overlithiation forming the Li2TMO2 phase (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 space group), which can cause irreversible structural degradation (Fig. S2, ESI†).47–50 Collectively, the appropriate redox-potential range for the RED is displayed in the density of states (DOS) diagram for the RED/RED+ and NMC-associated transition-metal redox couples (Fig. 1(c)).
m1 space group), which can cause irreversible structural degradation (Fig. S2, ESI†).47–50 Collectively, the appropriate redox-potential range for the RED is displayed in the density of states (DOS) diagram for the RED/RED+ and NMC-associated transition-metal redox couples (Fig. 1(c)).
Among various electron donors, we claim that the organic redox compounds that become stable cations by being oxidized from neutral states under ambient conditions, the so-called p-type redox-active molecules, are ideal for NMC regeneration.51–53 Unlike n-type redox-active compounds, which require a prior reduction process with Li metal to become an electron donor by forming Li-organic complexes,54 the p-type compounds can directly lithiate NMC in the presence of Li salts following eqn (3). More importantly, the redox potentials of most n-type molecules are below 3 V (vs. Li/Li+), limiting their stability in ambient air. Given the high reversibility of the redox reaction, the oxidized p-type molecule after NMC regeneration can be readily reduced when provided with a proper reductant, and thus, the regeneration solution is recyclable for multiple cathode regeneration processes.
To verify the validity of our RED-based cathode regeneration, we first selected DMPZ, for which the redox reaction occurs at 3.27 V (vs. Li/Li+), thereby satisfying our design criterion for an ideal RED. The versatile usage of DMPZ has been demonstrated as an active material in rechargeable organic batteries55 and redox-flow batteries56 and as a soluble redox mediator in Li–O2 or Li–air batteries.57 This widespread adoption of DMPZ in many battery systems indicates its high stability in both neutral and oxidized forms in carbonate- and ether-based electrolytes. In fact, the symmetric current flux measured for both positive and negative scans in the cyclic voltammetry (Fig. 1(d)) confirms the chemical stability of DMPZ upon electrochemical reaction. The reaction between DMPZ (DMPZ/DMPZ+, 3.27 V vs. Li/Li+) and high-valence Ni (Ni2+/Ni3+ or Ni3+/Ni4+, > 3.7 V vs. Li/Li+) is thermodynamically favorable because of the negative Gibbs free energy.40 Thus, upon contact between neutral DMPZ and Li-deficient NMC in a regeneration solution, electrons spontaneously move from the HOMO of DMPZ to the Ni 3d orbitals of NMC, accompanying Li-ion migration into the NMC crystal structure.48,58 In addition, detrimental Mn reduction and overlithiation are thermodynamically unfavorable (ΔG = − nF(EMn3+/Mn4+ − EDMPZ/DMPZ+) > 0), inhibiting the undesirable irreversible phase transition of NMC.
Fig. 1(e) presents a schematic illustration of the one-step process to regenerate spent NMC using a DMPZ and LiTFSI solution. When simply immersing the spent cathode material into the regeneration solution, DMPZ is oxidized and immediately forms a DMPZ+–TFSI− complex, simultaneously promoting a spontaneous Li-coupled electron transfer to the Li-deficient NMC. We monitored the regeneration reaction with a three-electrode system using Li-deficient NMC622 as the working electrode, Pt as the counter electrode, and Li metal as the reference electrode (Fig. 1(f)). The Li-deficient NMC622 was prepared as a model spent cathode with 24.1% Li loss by charging NMC622 to a 30% state-of-charge (SOC) (Fig. S1, ESI†). When the Li-deficient NMC622 was first immersed in DMPZ-free 1,2-dimethoxyethane (DME) solution containing ten times more LiTFSI (71.8 μmol) than VLi in NMC622 (7.1 μmol), the open-circuit voltage (OCV) of the system remained at 3.61 V (vs. Li/Li+), indicating that VLi remained in the NMC622 crystal structure. After injecting DMPZ into the LiTFSI solution, the OCV immediately dropped below 3.2 V (vs. Li/Li+), and the solution turned dark green (Video S1, ESI†). Within 12 min of stirring, the OCV was saturated at 3.17 V (vs. Li/Li+), near the redox potential of DMPZ/DMPZ+. We attribute the immediate OCV drop to spontaneous Li migration from the regeneration solution into VLi in the NMC622 crystal lattice, as described in Fig. 1(c).
We then conducted ex situ characterization of the DMPZ-based regeneration solution and NMC622 cathode to verify the underlying mechanism. The UV-vis spectroscopy analysis revealed new peaks emerging in the range of 420–450 and 600–750 nm for the regeneration solution after treating the Li-deficient NMC622, indicating that the observed dark-green species indeed originated from oxidized DMPZ+ (Fig. 1(g)).55,59 In parallel, the X-ray diffraction (XRD) patterns of the regenerated NMC622 cathodes also confirm their complete reduction (Fig. 1(h), Fig. S3, S4, and Tables S2–S4, ESI†). With 24.1% Li loss in the NMC622, a noticeable down-shift of the (003) peak from its original position was observed, corresponding to an increase in the c lattice parameter resulting from the increased electrostatic repulsion between the adjacent oxygen layers along the c direction due to the VLi.23,60 After the regeneration, we noted that the (003) peak shifted back to higher angle, indicating the replenishment of Li in the cathode crystal structure. The a lattice parameters also significantly increased after regeneration, suggesting the reduction of high-valence Ni to a lower oxidation state with larger ionic radius. In addition, the inductively coupled plasma mass spectrometry (ICP-MS) analysis confirmed the Li replenishment of the NMC622 cathode after the regeneration (Table S1, ESI†). In the case of the Li-deficient double-coated cathode (Fig. S5, ESI†), the most common type of spent cathode encountered in the current battery industries, we also observed an upshift of the (003) peak after the regeneration for both sides. Collectively, these results prove that the DMPZ molecules in the regeneration solution effectively donate electrons and induce Li-ion insertion to the Li-deficient NMC622, restoring the original chemical composition of the NMC622 cathode.
Electrochemical performance of NMC622 cathodes regenerated under practical conditions
To verify the practical feasibility of the RED-based regeneration method, we systematically evaluated the electrochemical performance of NMC622 after regeneration under various conditions mimicking realistic situations (Fig. 2(a)–(c)). To examine the chemical lithiation effect, we compared the initial charge capacity after regeneration with that of the 2nd formation cycle using half cells. We selected the 2nd cycle as a baseline to represent the original charge capacity of the model Li-deficient NMC622 samples because of the high CE. We note that regardless of chemical relithiation, the following discharge capacity of the Li-deficient NMC622 can recover to the original value because of the excessive Li supply from the Li metal anode in the half cell. Still, we used this profile from the half cells to examine if there is any degradation when treating Li-deficient NMC622 with our regeneration solution. First, a partially delithiated NMC622 cathode with 24.1% VLi was galvanostatically cycled in a half cell after immersion in a DMPZ-free LiTFSI solution (22.5 mmol L−1) for 15 min as a baseline condition (Fig. 2(a), top and Fig. S6, ESI†). The initial charge capacity of the baseline sample was 139 mA h g−1, which indicates 23.3% VLi; this result reveals that most of the VLi remained intact and that no Li insertion occurred without REDs. The following discharge capacity was 177 mA h g−1, indicating that the baseline condition barely induces damage to the Li-deficient NMC622.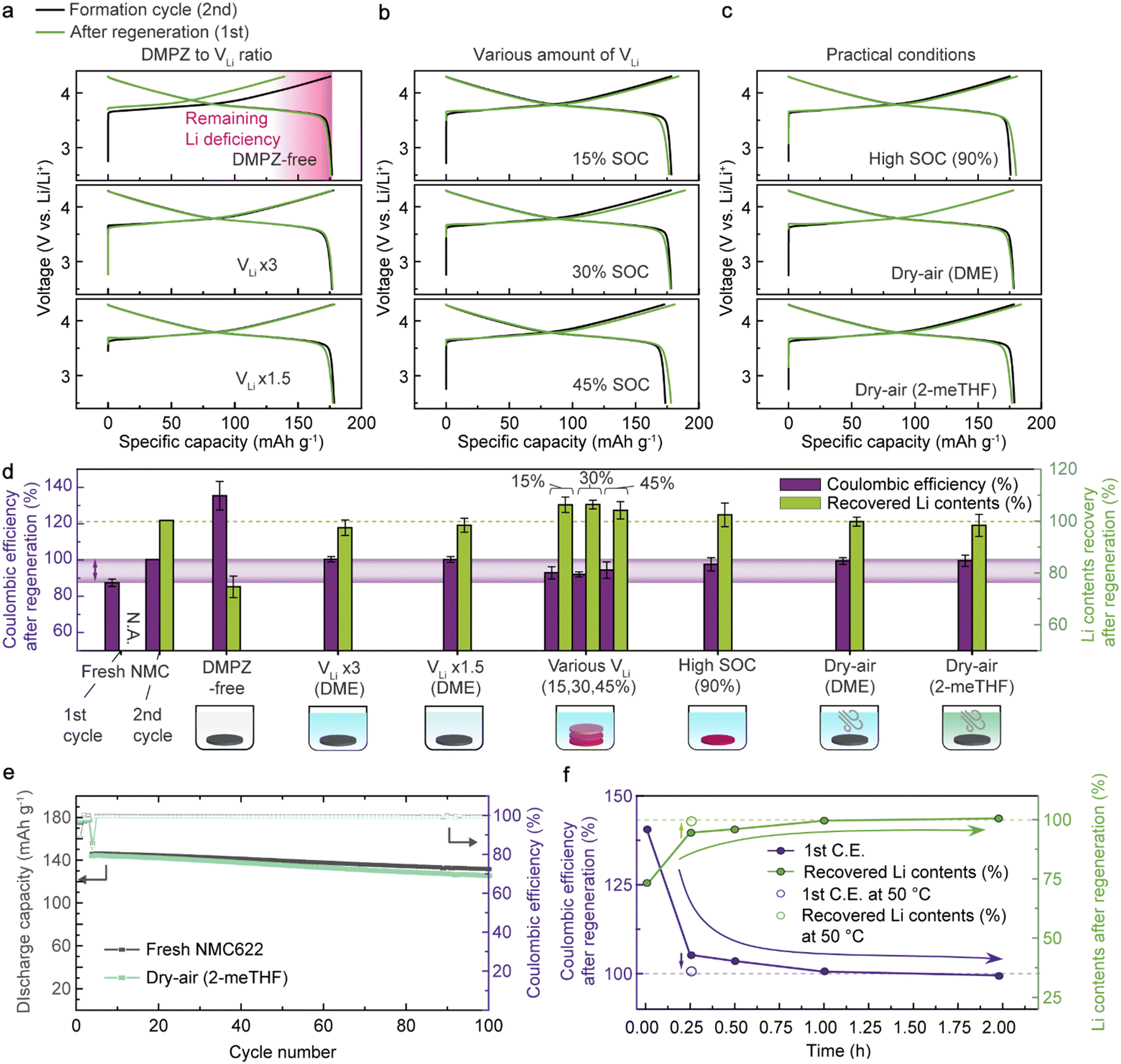 | ||
| Fig. 2 Electrochemical performance of NMC622 cathodes regenerated under practical conditions for battery recycling. (a) The first charge/discharge profiles of NMC622 after regeneration in solutions containing various amounts of DMPZ. The ratio between DMPZ in solution and VLi in NMC was 0, 3, and 1.5. (b) Charge/discharge profiles of NMC of various VLi concentrations regenerated simultaneously in a solution batch. (c) Charge/discharge profiles of NMC622 regenerated under commercially viable conditions for common battery manufacturing facilities; regenerated NMC622 containing high VLi concentration (top), NMC622 regenerated in DME-based DMPZ solution and 2-meTHF-based DMPZ solution in dry air (middle and bottom, respectively). (d) Comparison of CEs (purple bars) and recovered Li contents (green bars) of NMC622 cathodes after regeneration under conditions in (a)–(c). The purple-shaded region indicates the ideal CE range for regenerated NMC622. A CE exceeding 100% means that the initial charge capacity was smaller than the following discharge capacity, caused by incomplete Li recovery. The error bars indicate the standard deviation. (e) Long-term cyclability of fresh NMC622 and NMC622 regenerated under “Dry-air (2-meTHF)” condition. The cathodes were cycled at 3C after the initial three formation cycles at 0.1C. (f) CEs and Li contents of NMC cathodes after regeneration using various processing durations and temperatures. | ||
In contrast, after treating the Li-deficient NMC622 cathode in a regeneration solution containing both DMPZ and LiTFSI for 2 hours, the cathode recovered 100.5% of its charge capacity (177.7 mA h g−1) (Fig. 2(a), middle). Note that the regeneration solution contained three times more DMPZ and LiTFSI than the VLi in NMC622 (denoted as “VLi × 3”). The cathode exhibited a discharge capacity of 176.8 mA h g−1 and a CE of 99.5%. The CE below 100% implies that the amount of Li inserted during the chemical regeneration exceeds the lithiation degree achievable by the conventional electrochemical discharge due to the enhanced charge-transfer kinetics of the chemical lithiation. The voltage profiles barely show an overpotential increase compared to the formation cycle, indicating the stability of the RED-based Li replenishment. Further, the electrode morphology seemed intact, and no residues remained after the chemical regeneration, followed by a brief rinsing of the cathode (Fig. S7 and Table S5, ESI†). We also confirmed the complete lithiation of a thick NMC cathode with a high active loading (17 mg cm−2), verifying the homogeneous Li refill via the effective infiltration of the regeneration solution even within the commercially-viable electrodes (Fig. S8, ESI†). The effect of chemical lithiation can be clearly distinguished in the full-cell demonstration. As shown in Fig. S9 (ESI†), we compared the charge–discharge profiles and capacity retentions of fresh NMC622 (black), Li-deficient NMC622 (red), and regenerated NMC622 (blue) paired with graphite anodes in full cells. Compared to the fresh cell at 0.1C, the full cells of Li-deficient and regenerated NMC622 exhibited 58% and 99% reversible capacities, respectively. At 1C, the full cell with the regenerated cathode recovered 98% of the original energy density while showing comparable cycling stability with the fresh cell. This indicates that RED-based chemical lithiation is an effective way to regenerate the spent cathodes.
We further conducted the lithiation reaction in a dilute regeneration solution having only 1.5 times excess Li ions and RED molecules compared with the VLi in NMC622 (denoted as “VLi × 1.5”). Even with such a dilute solution, the NMC622 cathode recovered 100% of the original charge capacity by chemical Li insertion (Fig. 2(a), bottom). We attribute the high effectiveness of the lithiation using a minimal amount of Li sources to the strong driving force for the charge-transfer reaction caused by the potential difference between NMC622 and DMPZ as opposed to a simple concentration gradient necessitating either high pressure or temprature.61
Next, we assessed the feasibility of the simultaneous regeneration of mixed NMC622 cathodes with multiple Li vacancy concentrations (15%, 30%, and 45% SOCs), imitating the industrial recycling situation where spent LIBs with diverse degradation degrees are collected.27 After simply immersing all the samples together, the recovered charge capacities were 103%, 106%, and 104% for the electrodes with 15%, 30%, and 45% VLi, respectively (Fig. 2(b)). Moreover, we confirmed that the regeneration solution could also refill Li into the highly charged (delithiated) NMC622 with 90% VLi (denoted as “High SOC”) to exhibit 103% of the original charge capacity (Fig. 2(c), top). The results highlight the universal applicability of our regeneration approach for NMC622 cathodes retrieved from spent LIBs suffering a diverse range of Li losses from their operation. This contrasts to solid-state regeneration methods, which require precise control of the Li supply for each condition to prevent irreversible phase transition during the regeneration.29,30
We further tested the regeneration process under the dry-air environments commonly used in industrial battery manufacturing facilities.62 Upon regenerating the Li-deficient NMC622 under dry air (denoted as “Dry-air (DME)”), 100% of the charge capacity was recovered (177.7 mA h g−1), thus resulting in a CE of 100%, demonstrating the compatibility of the process in the dry-air condition (Fig. 2(c), middle). The stability of the oxidized DMPZ+ cations was confirmed by the consistent absorption spectra observed upon one-week exposure to dry air (Fig. S10, ESI†). We attribute the high stability to the high redox potential of DMPZ/DMPZ+ (3.27 V vs. Li/Li+) preventing unwanted side reactions, such as oxygen reduction reactions forming reactive O2˙− radical anions (2.91 V vs. Li/Li+).63,64
In our efforts to identify economically and environmentally sustainable chemicals for our regeneration method, we introduced 2-methyltetrahydrofuran (2-meTHF) as an alternative solvent for DME. 2-meTHF is a representative green ethereal solvent with low toxicity and volatility derived from biomass.65 As shown in the bottom panel of Fig. 2(c), the regenerated NMC622 in 2-meTHF again exhibited 103% charge-capacity recovery and a CE of 96.1%, suggesting that our regeneration strategy will provide a scalable and environmentally friendly solution for LIB recycling.
To aid in comparison, we provide a performance chart in Fig. 2(d) that summarizes the CEs and Li contents of NMC622 cathodes after regeneration under the conditions described above. The Li contents were calculated from the charge capacity and normalized to the second charge capacity of a fresh NMC622 cathode according to
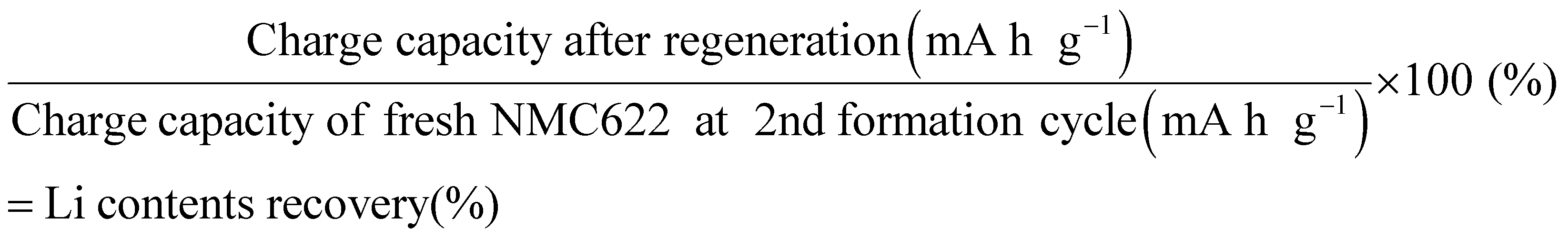
After DMPZ-based regeneration, all the NMC622 cathodes exhibited comparable CEs between 100% and 94%. The CEs below 100% correspond to higher charge capacities than the following discharge capacities, similar to what happens in the initial cycle of the fresh cathode. This finding suggests that the chemical regeneration induces further lithiation than the electrochemical lithiation in batteries, which is attributable to the kinetics of the solution-based charge-transfer reaction through the solid–liquid interface being faster than that of the electrochemical reaction requiring both ion transport and solid-to-solid electron conduction.
We then measured the long-term cycling stability of the NMC622 cathode regenerated with DMPZ and LiTFSI in 2-meTHF in dry air, which is the most viable condition for practical applications (Fig. 2(e)). After 100 cycles, the regenerated NMC622 cathode retained 87.2% of the reversible capacity at 3C, comparable to the fresh NMC622 cycling. Moreover, the voltage profiles showed negligible differences in overpotentials (Fig. S11, ESI†), indicating that no significant evolution occurred in the bulk crystal structure or at the NMC622 surface during regeneration. According to the electrochemical impedance spectroscopy (EIS) measurement and the rate capability test on the fresh and regenerated NMC622 (Fig. S12 and S13, ESI†), the RED-based process has a negligible impact on bulk interfacial electrochemical reaction kinetics.
In addition, we tracked the CE recovery of Li-deficient NMC622 as the regeneration time increased (Fig. 2(f) and Fig. S14, ESI†). At room temperature, using three times more DMPZ and LiTFSI than the VLi in NMC622, the Li replenishment degree was saturated at 100% within 1 hour of regeneration (see Fig. S15 for voltage profiles, ESI†). Notably, a slight increase in the solution temperature to 50 °C could achieve 100% recovery within only 15 min (displayed as empty circles in Fig. 2(f)). We attribute the shortened reaction time at the elevated temperature to the enhanced charge-transfer kinetics. The fast and efficient regeneration using our strategy is desired for industrial applications and can be further optimized by tuning the reaction parameters, which we believe is an essential topic for further study.
Universality of our regeneration strategy
To examine the universality of our regeneration strategy, we adopted other RED molecules and Li salts. Three additional p-type compounds, Fc (E1/2 = 3.49 V vs. Li/Li+), DPPD (E1/2 = 3.46 V vs. Li/Li+) and 1,4-di-tert-butyl-2,5-dimethoxybenzene (DBB, E1/2 = 4.18 V vs. Li/Li+), were tested (Fig. S16, ESI†). The regenerated NMC622 recovered 105.3% and 98% of its original charge capacity in the Fc-LiTFSI and DPPD-LiTFSI solution, whereas the capacity recovery was negligible in the DBB-LiTFSI solution (Fig. S17, ESI†). These results corroborate our design criteria of the RED to achieve thermodynamically driven charge-transfer reactions. In parallel, when LiTFSI was replaced with LiPF6, the most common Li salt for LIB electrolytes, the Li replenishment occurred successfully, showing 98% charge capacity recovery (Fig. S18, ESI†). Given the higher stability of LiTFSI to moisture and light than LiPF6, using LiTFSI would be a solid option for commercialization. However, we believe there is a vast space for exploring various REDs and Li salts to improve both the economic and environmental aspects of our strategy.Additionally, the RED-based strategy was also exploited to regenerate different types of LIB cathodes, including olivine LFP, spinel LNMO, and layered LCO. We prepared Li-deficient LFP, LNMO, and LCO as model spent cathodes by charging each cathode material to a 30% SOC (Fig. S19, ESI†). After immersion in regeneration solutions containing DMPZ and LiTFSI, the Li-deficient LFP, LNMO, and LCO cathodes recovered 102.4%, 117.6%, and 102.1% of their original charge capacities, respectively, while showing negligible differences in overpotentials (Fig. S20, ESI†). We further assembled full-cells using the LFP and LNMO cathodes having Li deficiency (30% SOC) before and after chemical regeneration to show the chemical relithiation effect (Fig. S21, ESI†). Compared with the fresh cells, the regenerated LFP and LNMO recovered 99.2% and 100.9% of the original capacities, respectively, showing a sharp contrast with the full cells without chemical regeneration. These results indicate that our regeneration strategy can be extended to other LIB cathode materials, irrespective of the chemical composition and crystal structure of the cathode.
Circularity in RED-based regeneration process with minimal waste production
A noteworthy advantage of RED-based regeneration is that the used regeneration solution can be readily recycled for multiple cathode-regeneration processes. Because of the high reversibility of the redox reaction between neutral REDs (RED0) and RED+ cations without causing bond breaking, we can make the solution reusable after regeneration by reducing the RED+ cations to RED0 while supplying Li sources (Fig. 3(a)). As a proof-of-concept demonstration, we applied Li peroxide (Li2O2) to the used solution containing DMPZ+ cations, which functions simultaneously as a reducing agent and Li source. The reaction between Li2O2 and DMPZ+ can be described as follows: Li2O2 + 2 DMPZ+-A− → 2 Li+ + 2 DMPZ0 + 2 A− + O2. Because Li2O2 oxidation is known to be an endothermic process in DME,66 the used solution can be recycled without safety risks. As a result, the solution color changed from dark green to bright yellow, indicating the reduction of DMPZ+ to DMPZ0. UV-vis absorption spectroscopy analysis confirmed the high-yield conversion of DMPZ+ to DMPZ0, where the peaks near 450 and 660 nm from DMPZ+ completely disappeared while the peak from DMPZ0 at 338 nm recovered its original intensity after the Li2O2 treatment (Fig. 3(b), see Fig. S22 and Table S6 for the details of quantification, ESI†).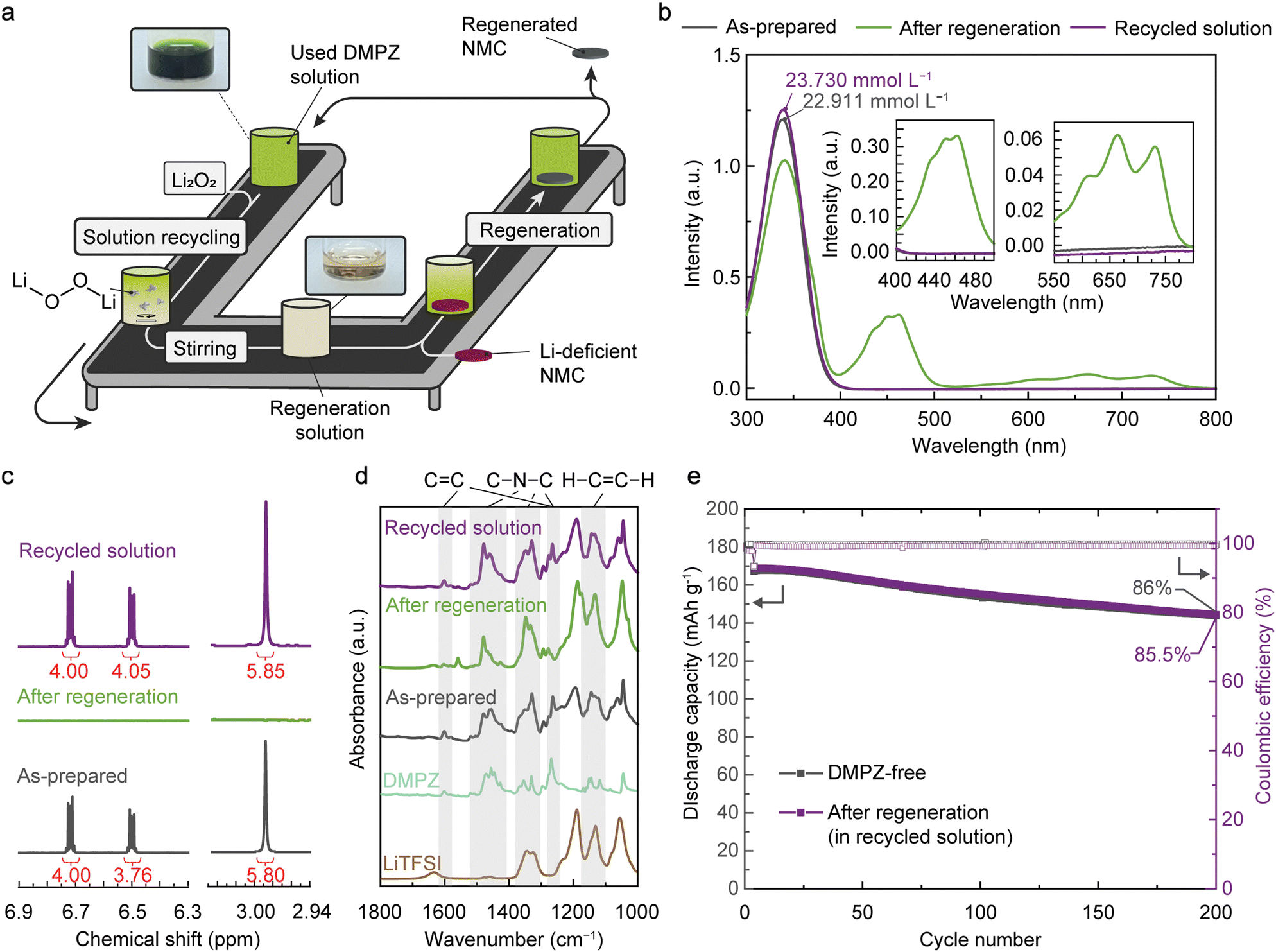 | ||
| Fig. 3 Closed-loop recycling of the used regeneration solution. (a) Schematic illustration of the cathode regeneration-solution recycling processes with circularity. (b) UV/vis absorption spectra, (c) 1H NMR spectra, and (d) FT-IR spectra of DMPZ solution during the closed-loop recycling process. The grey-shaded areas in the FT-IR spectra represent characteristic peaks of DMPZ. (e) Cyclability of NMC622 cathode regenerated using recycled solution compared to that of NMC without regeneration. The cells were cycled at 0.5C after the initial three cycles at 0.1C. | ||
The 1H-nuclear magnetic resonance (NMR) analysis further supports the high stability of the DMPZ+/DMPZ0 redox couple, which barely forms by-products during the cathode regeneration and solution recycling (Fig. 3(c), see Fig. S23–S27 for the whole 1H-NMR spectra, ESI†). During the consecutive NMC622 regeneration and solution recycling processes, the 1H-NMR peaks of DMPZ0 at 6.71, 6.5, and 2.9 ppm disappeared and reappeared reversibly while preserving their intensities (red fonts in Fig. 3(c)), indicating the successful recovery of DMPZ0 after the redox reactions.67,68 The Fourier-transform infrared (FT-IR) spectrum of each regeneration solution also showed the reversible changes of vibrational modes related to stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–N–C bonds in heterocyclic DMPZ molecules, validating the high stability (Fig. 3(d) and Fig. S28, Table S7, ESI†).55,56
C and C–N–C bonds in heterocyclic DMPZ molecules, validating the high stability (Fig. 3(d) and Fig. S28, Table S7, ESI†).55,56
To prove the reusability of the recycled DMPZ solution, we conducted an additional regeneration process on another Li-deficient NMC622 cathode using the recycled solution after the Li2O2 treatment. The NMC622 cathode regenerated for 2 hours in the recycled solution was cycled within a voltage range of 2.5–4.3 V (vs. Li/Li+) in a half cell (Fig. 3(e)). With 102.6% charge capacity recovery and a CE of 97.9% after regeneration (Fig. S29a, ESI†), the regenerated NMC622 cathode retained 85.5% of its reversible capacity (168.7 mA h g−1) after 200 cycles at 0.5C, comparable to that of the control sample (DMPZ-free). The voltage profiles of the regenerated NMC622 were almost identical to those of the control sample during prolonged cycling (Fig. S29b and c, ESI†), indicating that Li replenishment of a Li-deficient NMC622 cathode is feasible in the recycled solution without causing an overpotential increase.
Likewise, Li2S can also function simultaneously as a reducing agent and Li source for solution recycling due to its redox potential (E1/2 = 2.15 V vs. Li/Li+) located below those of REDs (>3.27 V vs. Li/Li+) (Fig. S30, ESI†).53 With the 0.6 molar equivalent of Li2S (Li/RED = 1.2), the oxidized RED was completely reduced, making the solution reusable. Indeed, the recycled solution can regenerate the Li-deficient NMC to recover 111.4% of the original charge capacity with a negligible increase in the overpotential (Fig. S31, ESI†).
Regenerating NMC811 from a commercial pouch cell after extended cycling
Benefiting from the simplicity and scalability of our method, we demonstrated the regeneration of spent cathodes from a commercial 1-Ah battery (Fig. 4(a)). The spent NMC811 cathode was retrieved from the pouch cell after 820 charge/discharge cycles, which resulted in 20% capacity loss (Fig. S32, ESI†). We regenerated 500 mg of the retrieved NMC811 (160 times the mass of NMC622 in the above half-cell demonstrations) having 20% VLi by immersing the cathode powder in 15 mL of DMPZ–LiTFSI solution for 2 hours in a dry room (dew point = 223 K). After regeneration, the resulting used solution could be replaced with fresh regeneration solution for further use by immersing Li2O2. As shown in Fig. 4(b), the regenerated NMC811 exhibited an initial charge capacity of 202.3 mA h g−1, which is 24.2% higher than that of the cycled cathode (162.9 mA h g−1) and comparable to that of the fresh cathode (192.9 mA h g−1). The three cathodes show negligible differences in the discharge profiles at 0.1C, indicating no significant crystal structural deterioration of NMC811 during long-term cycling and regeneration. Instead of using the retrieved powder, we confirmed that directly regenerating the commercial thick electrode is also possible (Fig. S33, ESI†). In addition, the regenerated cathode exhibited a high rate capability (141 mA h g−1 at 3C) and long-term cyclability (82.1% retention at the 150th cycle), similar to the fresh and cycled cathodes (Fig. 4(c), (d) and Fig. S34, ESI†), verifying that the RED-based lithiation barely causes side reactions that deteriorate the structural integrity and interfacial charge-transfer kinetics of the active materials.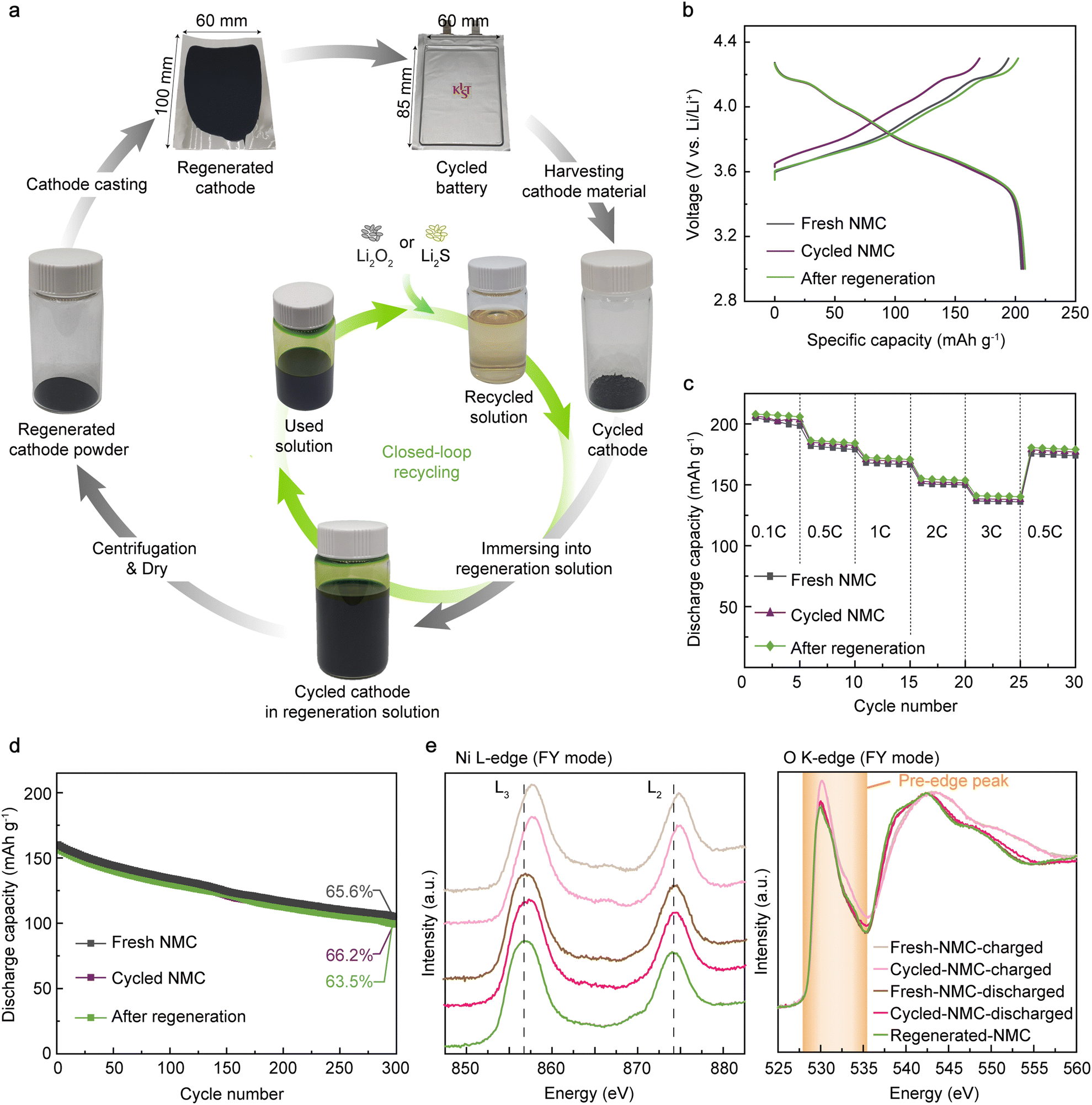 | ||
| Fig. 4 Practical adoption of RED-based lithiation for regenerating NMC811 from commercial-sized battery. (a) Overall regeneration process for cycled NMC811 harvested from commercial 1-A h pouch cells, including solution recycling. (b) Charge/discharge profiles of fresh (after formation cycles), 820 cycled, and regenerated NMC811 after 820 cycles. (c) Rate capability and (d) capacity retention of NMC811 cathodes at 0.5C in (b). (e) NEXAFS Ni L-edge (left) and O K-edge (right) spectra measured in FY mode for NMC811 cathodes at different electrochemical and regeneration states. The Ni L-edge and O K-edge spectra of the regenerated NMC811 resemble those of the fresh NMC811 in the discharged state. | ||
Using ex situ near-edge X-ray absorption fine structure spectroscopy (NEXAFS), we confirmed the successful chemical insertion of Li and the associated reduction of nickel in NMC811 (Fig. 4(e)). To trace the bulk-originated properties, the Ni L-edge and O K-edge NEXAFS spectra were collected in fluorescence yield (FY) mode with a probing depth of 150–200 nm. Compared with the fresh NMC811 in the discharged state (f-NMC-d), the cycled sample in the discharged state after 820 cycles in the pouch cell (c-NMC-d) showed slightly higher Ni oxidation states due to the active Li loss during the prolonged cycling (Fig. S35, ESI†). After regenerating c-NMC-d (r-NMC), the Ni L3 (855–860 eV) and L2 (873–878) peaks shifted toward lower energy, indicating the reduction of nickel ions due to the successful Li replenishment into the NMC811 crystal lattice. More quantitatively, the intensity ratio between the peaks from the t2g (856 eV) and eg (858 eV) levels were 1.1, 1.3, and 1.4 for c-NMC-d, f-NMC-d, and r-NMC, respectively (Fig. S36, ESI†). The r-NMC contained more reduced Ni than f-NMC-d, which is attributed to the sluggish kinetics of the electrochemical reaction at the low SOCs leaving VLi even in the fully discharged state in contrast to the fast chemical lithiation.69,70 Consistently, the O K-edge FY NEXAFS spectra showed decreased intensity of the pre-edge peak at 529–532 eV after the regeneration, indicating the reduced number of holes in the Ni 3d-O 2p hybridized orbitals, e.g., Ni reduction.69,70 The Ni reduction during the regeneration was again observed in the Ni L-edge NEXAFS spectra measured in total electron yield (TEY, probing depth = 5–10 nm) mode (Fig. S37, ESI†), indicating the homogeneous regeneration of NMC811 throughout the surface and subsurface regions. Meanwhile, the Mn L-edge TEY NEXAFS spectra showed negligible differences between the discharged samples (f-NMC-d and c-NMC-d) and r-NMC, thus indicating no formation of undesirable rock-salt phases during the regeneration process (Fig. S38, ESI†).71
We further applied the RED-based regeneration to the spent NMC811 from an even more heavily degraded 1-A h battery with a 60% capacity loss. Fig. S39 (ESI†) shows the voltage profile of the degraded NMC811 in a half cell after retrieving it from the 60% degraded full cell. The reduced initial charge capacity of 125 mA h g−1 indicates the limited Li inventory in the spent cathode. In contrast, the discharge capacity decreased by only 4.8% compared to that of the fresh cathode. Further, the cathode still exhibits an apparent 4.2 V plateau, manifesting the well-preserved original crystal structure and electrochemical activity of the spent NMC811. After the chemical regeneration, on the other hand, the cathode exhibited an initial charge capacity of 222 mA h g−1, reflecting the successful recovery of the original Li inventory. According to the scanning electron microscopy (SEM) images of both NMC811 and graphite from the degraded full cell, while the microstructure of the cathode seemed nearly intact, a significant amount of dendritic SEI covered the entire anode surface, reflecting rigorous Li metal plating and electrolyte consumption during the long-term cycling. The results are consistent with the previous reports that the most significant contributor to the capacity fade in practical full cells is the Li inventory loss at the graphite anode–electrolyte interface,72,73 while the cathode remains relatively intact after cycling.74 As shown in Fig. S40 (ESI†), there was no noticeable morphological evolution of primary and secondary particles during regeneration process, reflecting that our RED-based process successfully recovered the original Li inventory without destroying the morphology of cathode particles.
To investigate the crystal structure evolution in the degraded cathode during chemical regeneration, we performed transmission electron microscopy (TEM) analysis on NMC811 from both 20% and 60% degraded pouch cell. Surprisingly, the additional diffraction spots originating from spinel-like cation-disordered phases in the spent NMC811 disappeared after regeneration in the selected-area electron diffraction (SAED) patterns (Fig. S41 and S42, ESI†), as observed across multiple particles. We also collected scanning transmission electron microscopy (STEM)-high-angle annular dark-field (HAADF) images of the active particles using NMC811 from both 20% and 60% degraded full cells (Fig. S43, ESI†). We note that the thickness of the cation-mixed region in NMC811 from the 60% degraded cell is only slightly higher than that in NMC811 from the 20% degraded cell. The results suggest that the primary cause of capacity loss in full-cell batteries is active Li loss associated with electrolyte decomposition rather than bulk structural disordering in cathodes. Following chemical regeneration, the thickness of the cation-mixed region significantly decreased, which supports the TEM-SAED results where spinel-like phases predominantly disappeared the regeneration.
It is important to note that the degree of surface degradation of the spent cathode can vary with the quality of the electrode, battery assembly/cycling/storage conditions, and sample harvesting methods. In particular, ultrasonication of the cathode in polar solvents has been widely demonstrated in previous literatures,27,33,36,41,75,76 but it's important to recognize that spent cathodes with lithium loss can undergo significant degradation during this separation process. Here, we opted to mechanically separate the spent cathode from the current collector to ideally preserve surface and structure of the cathode, but this process may not be scalable. Nevertheless, we believe that it is crucial to decouple the degradation mechanism between cycling and separation processes to better assess the effectiveness of cathode regeneration strategies. Efforts to advance non-destructive cathode harvesting technology are underway in both academic and industrial sectors, focusing on improving cost-effectiveness, efficiency, and environmental impact.77–80 Moving forward, redesigning battery assemblies for optimal recyclability or developing a direct recycling process for both separation and regeneration purposes, starting with spent electrodes, instead of separated particles, could represent a proactive direction. The commercial viability of the RED-based method relies on proper material collection methods, and thus collaborative efforts between academia, industry, and government entities are essential to advancing non-destructive separation methods.
Fig. 5(a) compares the conventional direct cathode-regeneration methods with our RED-based process. The hydrothermal methods require high-temperature pressurized reactors (>180 °C, >11 bar) to replenish the Li contents in spent cathode materials in most cases, which brings extra safety precautions and costs when expanding the reactor size in large-scale applications (Table S8, ESI†).33–39 Most recently, the hydrothermal temperature could be reduced near 100 °C when combined with reducing agents, and the method is still accompanied by high-temperature annealing for complete cathode regeneration after using strong alkaline solutions.40 Another option is to use eutectic molten salt at above 150 °C, a mixture of more than two Li salts showing a lower melting point than each component, which offers processability at ambient pressure.41,75,76,81 Nevertheless, the highly viscous nature of the molten salts limits the lithiation kinetics due to the low Li-ion mobility, requiring extended time for regeneration. Additionally, the combination of hazardous and corrosive Li salts poses potential environmental concerns for industrial applications.15 In contrast, our RED-based cathode regeneration exploits a redox reaction that occurs under ambient conditions (30 °C, 1 atm, and dry air), thus promising outstanding viability for a large-scale recycling system (Fig. 5(a) and (b)). We highlight that the use of soluble electron-donating p-type molecules with an adequate redox potential (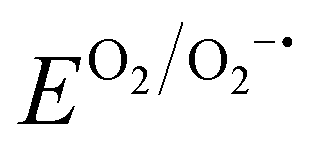 < ERED/RED+ and EHER < ERED/RED+ < EOER) in our study is the key to achieving the high air stability of the regeneration solution, avoiding the reaction with O2 and moisture (2H+ + 2e− → H2; 2H2O → O2 + 4H+ + 4e−). In addition, the redox potential of RED only induces topotactic lithiation of NMC cathodes without causing overlithiation that deteriorates crystal structures. In comparison, Qian et al. recently introduced Li-arene (e.g., aromatic hydrocarbon) complexes (LACs) as reducing agents that enable the chemical lithiation of spent cathodes.54 Unlike the p-type molecules, the redox potentials of most LACs are lower than 1.0 V (vs. Li/Li+), which makes the LACs highly reactive with oxygen and moisture, thereby requiring an inert atmosphere for processing.54,63,64,82,83 More severely, the low redox potential of LAC can lead to over-lithiation followed by irreversible structural degradation of NMC cathodes via a conversion reaction when precise balancing between LAC and VLi is not guaranteed, which requires additional quantification processes.84 Likewise, solid-state sintering, one of the existing direct regeneration methods, also requires accurate Li quantification prior to the process to avoid the formation of undesired impurity phases during the high-temperature sintering.23
< ERED/RED+ and EHER < ERED/RED+ < EOER) in our study is the key to achieving the high air stability of the regeneration solution, avoiding the reaction with O2 and moisture (2H+ + 2e− → H2; 2H2O → O2 + 4H+ + 4e−). In addition, the redox potential of RED only induces topotactic lithiation of NMC cathodes without causing overlithiation that deteriorates crystal structures. In comparison, Qian et al. recently introduced Li-arene (e.g., aromatic hydrocarbon) complexes (LACs) as reducing agents that enable the chemical lithiation of spent cathodes.54 Unlike the p-type molecules, the redox potentials of most LACs are lower than 1.0 V (vs. Li/Li+), which makes the LACs highly reactive with oxygen and moisture, thereby requiring an inert atmosphere for processing.54,63,64,82,83 More severely, the low redox potential of LAC can lead to over-lithiation followed by irreversible structural degradation of NMC cathodes via a conversion reaction when precise balancing between LAC and VLi is not guaranteed, which requires additional quantification processes.84 Likewise, solid-state sintering, one of the existing direct regeneration methods, also requires accurate Li quantification prior to the process to avoid the formation of undesired impurity phases during the high-temperature sintering.23
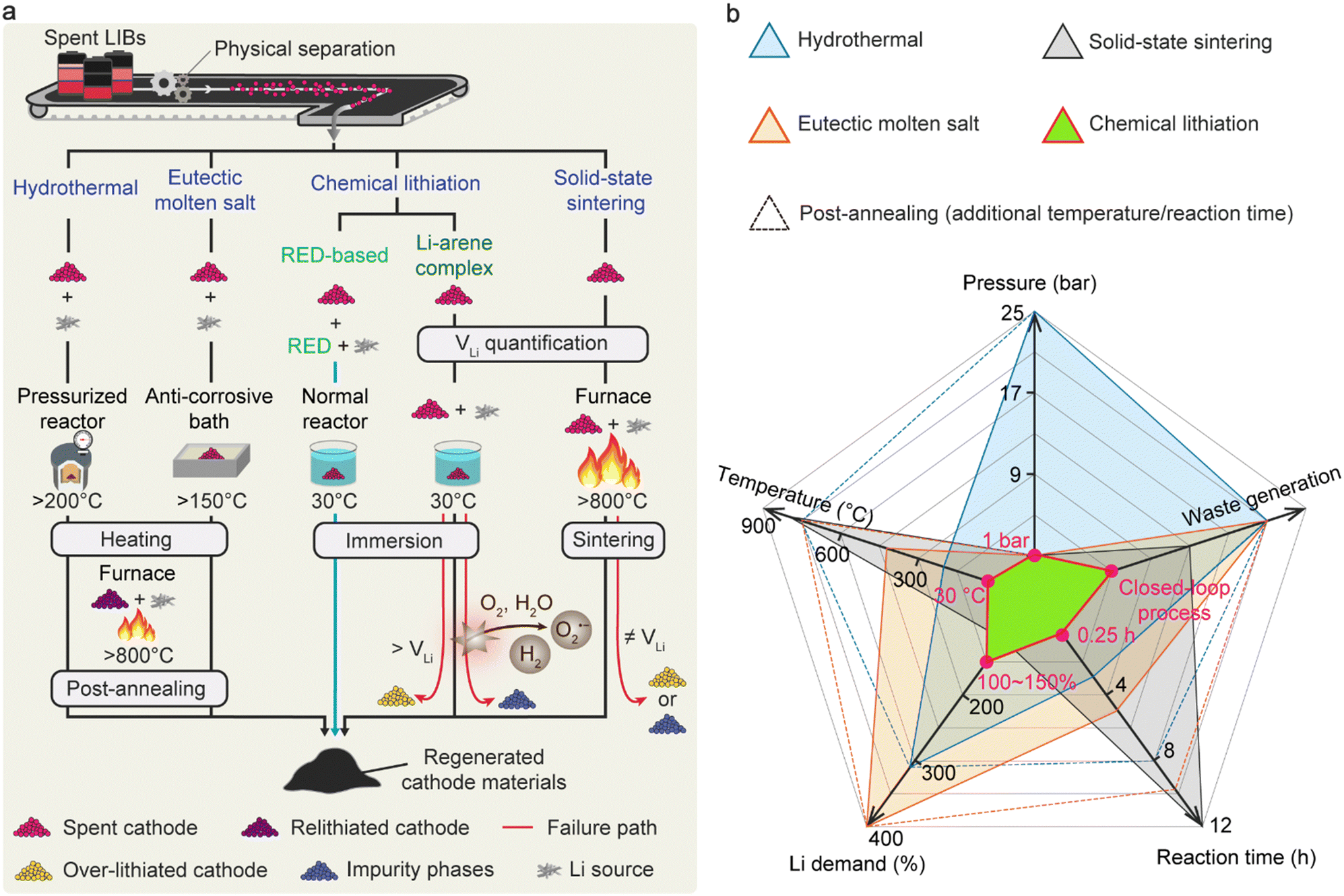 | ||
| Fig. 5 Comparison of procedures and conditions for direct cathode regeneration. (a) Schematic flow chart of representative regeneration methods and our RED-based strategy. (b) Spider diagram manifesting key features of regeneration methods regarding industrial viability. | ||
We further evaluated the environmental and economic impact of our RED-based process using the EverBatt model developed by Argonne National Laboratory and compared it to the reported values for other recycling methods (Fig. S44 and Tables S9–S16, ESI†). The total energy consumption of 2.4 MJ per kg cell and greenhouse gas (GHG) emission of 0.125 kg per kg cell for the RED-based process is significantly lower than those of the conventional pyrometallurgical and hydrometallurgical processes (Fig. S45–S47, ESI†). Also, because the RED-based method can directly produce usable cathode materials without the energy-consuming re-synthesis, a higher profit ($4.23 per kg cell) is potentially expected compared to the destructive processes, which recover metal precursors from the spent cathodes (Fig. S48, ESI†). Among direct regeneration strategies,18,27,37,40,54,85 our RED-based strategy still showed the lowest GHG emission and total energy consumption for recycling 1 kg of spent battery cells (Fig. S49 and S50, ESI†). We note that the calculated costs do not represent a precise economic judgment for immediate industry implementation of the processes, particularly for those necessitating Li quantification and atmosphere control, due to the lack of proper methodology to convert those requirements at a practical scale. Nevertheless, the lowest GHG emissions and energy consumption combined with the competitive recycling cost make our strategy compelling for practical implementation. Even coupling with a post-annealing step to further recover the microstructure and surface crystal structure of cathode materials from spent batteries, given the degradation heterogeneity anticipated in industrial situations, our RED-based regeneration method showed comparatively low greenhouse gas (GHG) emission (0.22 kg) and total energy consumption (3.97 MJ) for recycling 1 kg of spent batteries, with a negligible cost increase ($0.02).
In summary, we designed reusable regeneration solutions composed of soluble electron-donating molecules and Li salts that facilitate thermodynamically controlled cathode recycling under ambient temperature and pressure in dry air. Given the adequate oxidation potential of REDs, the proposed regeneration process allows topotactic lithiation of Li-deficient cathodes while avoiding side reactions with oxygen and detrimental overlithiation causing irreversible cathode degradation. The spontaneous lithiation can be universally applied to revitalize mixed cathodes with various Li and TM stoichiometries, making the chemical quantification process unnecessary. The solution-based process is scalable, as proven by the successful regeneration of a long-cycled cathode from commercially relevant sized 1-A h batteries, indicating the industrial viability of our approach. Finally, given the stable oxidized RED cations after the cathode regeneration, adding a common reductant can refresh the used regeneration solution, thus enabling closed-loop recycling of the spent cathodes with minimal chemical waste. The proposed RED-based cathode regeneration strategy will thrive in the development of profitable, scalable, and environmentally sustainable LIB recycling technologies.
Author contributions
The manuscript was written through contributions of all authors. S. K. and J. C. designed the experiments. S. K., J. C. U. H. and M. K. carried out the chemical lithiation, electrochemical tests, and ex situ analyses. G. L. and J. J. analyzed the experimental results of X-ray diffraction/spectroscopy and NMR spectroscopy, respectively. C. K. and U. L. conducted the life cycle inventory calculation for ferrocene and Li2S and S. K. calculated the economic and environmental impacts of the recycling process using EverBatt. The original idea was conceived by J. H. and M. L., who supervised all the experiments. C. B. P discussed the results and revised the manuscript. All authors discussed the results and gave approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Research Foundation through the Creative Research Initiative Center (grant no. NRF-2015R1A3A2066191). This work was also supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (no. 2021R1A2C2006243, 2021R1C1C1006721). This work was supported by the NRF (grant number: RS-2023-00222078).References
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich, S. Lambert, A. Abbott, K. Ryder, L. Gaines and P. Anderson, Nature, 2019, 575, 75–86 CrossRef CAS PubMed.
- R. E. Ciez and J. F. Whitacre, Nat. Sustain., 2019, 2, 148–156 CrossRef.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- E. C. Evarts, Nature, 2015, 526, S93–S95 CrossRef CAS PubMed.
- E. Fan, L. Li, Z. Wang, J. Lin, Y. Huang, Y. Yao, R. Chen and F. Wu, Chem. Rev., 2020, 120, 7020–7063 CrossRef CAS PubMed.
- M. Chen, X. Ma, B. Chen, R. Arsenault, P. Karlson, N. Simon and Y. Wang, Joule, 2019, 3, 2622–2646 CrossRef CAS.
- H.-H. Ryu, H. H. Sun, S.-T. Myung, C. S. Yoon and Y.-K. Sun, Energy Environ. Sci., 2021, 14, 844–852 RSC.
- L. Narayanan and A. P. Joshi, Lithium-ion Battery Recycling Market, https://www.marketsandmarkets.com/Market-Reports/lithium-ion-battery-recycling-market-153488928.html.
- J. Baars, T. Domenech, R. Bleischwitz, H. E. Melin and O. Heidrich, Nat. Sustain, 2021, 4, 71–79 CrossRef.
- X. Ma, J. Hou, P. Vanaphuti, Z. Yao, J. Fu, L. Azhari, Y. Liu and Y. Wang, Chem, 2022, 8, 1944–1955 CAS.
- Y. Tao, C. D. Rahn, L. A. Archer and F. You, Sci. Adv., 2021, 7, eabi7633 CrossRef CAS PubMed.
- B. Deng, Z. Zhou, W. Wang and D. Wang, ACS Sustainable Chem. Eng., 2020, 8, 14022–14029 CrossRef CAS.
- M. Fan, X. Chang, Y.-J. Guo, W.-P. Chen, Y.-X. Yin, X. Yang, Q. Meng, L.-J. Wan and Y.-G. Guo, Energy Environ. Sci., 2021, 14, 1461–1468 RSC.
- X. Zhang, L. Li, E. Fan, Q. Xue, Y. Bian, F. Wu and R. Chen, Chem. Soc. Rev., 2018, 47, 7239–7302 RSC.
- J. Mao, C. Ye, S. Zhang, F. Xie, R. Zeng, K. Davey, Z. Guo and S. Qiao, Energy Environ. Sci., 2022, 15, 2732–2752 RSC.
- X. Hu, E. Mousa, Y. Tian and G. Ye, J. Power Sources, 2021, 483, 228936 CrossRef CAS.
- J. Wang, K. Jia, J. Ma, Z. Liang, Z. Zhuang, Y. Zhao, B. Li, G. Zhou and H.-M. Cheng, Nat. Sustain., 2023, 6, 797–805 CrossRef.
- G. Ji, J. Wang, Z. Liang, K. Jia, J. Ma, Z. Zhuang, G. Zhou and H.-M. Cheng, Nat. Commun., 2023, 14, 584 CrossRef CAS PubMed.
- X. Zhang, Y. Bian, S. Xu, E. Fan, Q. Xue, Y. Guan, F. Wu, L. Li and R. Chen, ACS Sustainable Chem. Eng., 2018, 6, 5959–5968 CrossRef CAS.
- X. Chen, Y. Chen, T. Zhou, D. Liu, H. Hu and S. Fan, Waste Manage., 2015, 38, 349–356 CrossRef CAS PubMed.
- W. Chu, Y. Zhang, X. Chen, Y. Huang, H. Cui, M. Wang and J. Wang, J. Power Sources, 2020, 449, 227567 CrossRef CAS.
- B.-R. Chen, C. M. Walker, S. Kim, M. R. Kunz, T. R. Tanim and E. J. Dufek, Joule, 2022, 6, 2776–2793 CrossRef CAS.
- Z. Chi, J. Li, L. Wang, T. Li, Y. Wang, Y. Zhang, S. Tao, M. Zhang, Y. Xiao and Y. Chen, Green Chem., 2021, 23, 9099–9108 RSC.
- L. de Biasi, B. Schwarz, T. Brezesinski, P. Hartmann, J. Janek and H. Ehrenberg, Adv. Mater., 2019, 31, 1900985 CrossRef PubMed.
- X.-G. Yang, T. Liu, Y. Gao, S. Ge, Y. Leng, D. Wang and C.-Y. Wang, Joule, 2019, 3, 3002–3019 CrossRef CAS.
- B. Gault and J. D. Poplawsky, Nat. Commun., 2021, 12, 3740 CrossRef CAS PubMed.
- P. Xu, Q. Dai, H. Gao, H. Liu, M. Zhang, M. Li, Y. Chen, K. An, Y. S. Meng, P. Liu, Y. Li, J. S. Spangenberger, L. Gaines, J. Lu and Z. Chen, Joule, 2020, 4, 2609–2626 CrossRef CAS.
- D. Kong, M. Zhang, Y. Xiao, J. Hu, W. Zhao, L. Han and F. Pan, Nano Energy, 2019, 59, 327–335 CrossRef CAS.
- J. Li, L. Hu, H. Zhou, L. Wang, B. Zhai, S. Yang, P. Meng and R. Hu, J. Mater. Sci.: Mater. Electron., 2018, 29, 17661–17669 CrossRef CAS.
- X. Meng, J. Hao, H. Cao, X. Lin, P. Ning, X. Zheng, J. Chang, X. Zhang, B. Wang and Z. Sun, Waste Manage., 2019, 84, 54–63 CrossRef CAS PubMed.
- J. Zhao, W. Zhang, A. Huq, S. T. Misture, B. Zhang, S. Guo, L. Wu, Y. Zhu, Z. Chen, K. Amine, F. Pan, J. Bai and F. Wang, Adv. Energy Mater., 2017, 7, 1601266 CrossRef.
- D. Wang, R. Kou, Y. Ren, C.-J. Sun, H. Zhao, M.-J. Zhang, Y. Li, A. Huq, J. Y. P. Ko, F. Pan, Y.-K. Sun, Y. Yang, K. Amine, J. Bai, Z. Chen and F. Wang, Adv. Mater., 2017, 29, 1606715 CrossRef PubMed.
- Y. Shi, G. Chen, F. Liu, X. Yue and Z. Chen, ACS Energy Lett., 2018, 3, 1683–1692 CrossRef CAS.
- P. Xu, Z. Yang, X. Yu, J. Holoubek, H. Gao, M. Li, G. Cai, I. Bloom, H. Liu, Y. Chen, K. An, K. Z. Pupek, P. Liu and Z. Chen, ACS Sustainable Chem. Eng., 2021, 9, 4543–4553 CrossRef CAS.
- Y. Shi, G. Chen, F. Liu, X. Yue and Z. Chen, ACS Energy Lett., 2018, 3, 1683–1692 CrossRef CAS.
- Y. Guo, X. Liao, P. Huang, P. Lou, Y. Su, X. Hong, Q. Han, R. Yu, Y.-C. Cao and S. Chen, Energy Storage Mater., 2021, 43, 348–357 CrossRef.
- V. Gupta, X. Yu, H. Gao, C. Brooks, W. Li and Z. Chen, Adv. Energy Mater., 2023, 13, 2203093 CrossRef CAS.
- H. Gao, Q. Yan, P. Xu, H. Liu, M. Li, P. Liu, J. Luo and Z. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 51546–51554 CrossRef CAS PubMed.
- Q. Jing, J. Zhang, Y. Liu, W. Zhang, Y. Chen and C. Wang, ACS Sustainable Chem. Eng., 2020, 8, 17622–17628 CrossRef CAS.
- X. Yu, S. Yu, Z. Yang, H. Gao, P. Xu, G. Cai, S. Rose, C. Brooks, P. Liu and Z. Chen, Energy Storage Mater., 2022, 51, 54–62 CrossRef.
- Y. Shi, M. Zhang, Y. S. Meng and Z. Chen, Adv. Energy Mater., 2019, 9, 1900454 CrossRef.
- M. K. Tran, M.-T. F. Rodrigues, K. Kato, G. Babu and P. M. Ajayan, Nat. Energy, 2019, 4, 339–345 CrossRef CAS.
- H. Li, H. Yin, K. Wang, S. Cheng, K. Jiang and D. R. Sadoway, Adv. Energy Mater., 2016, 6, 1600483 CrossRef.
- Y. Wang, B. Goh, P. Nelaturu, T. Duong, N. Hassan, R. David, M. Moorehead, S. Chaudhuri, A. Creuziger, J. Hattrick-Simpers, D. J. Thoma, K. Sridharan and A. Couet, Adv. Sci., 2022, 9, 2200370 CrossRef PubMed.
- D. Di Marino, M. Shalaby, S. Kriescher and M. Wessling, Electrochem. Commun., 2018, 90, 101–105 CrossRef CAS.
- K. Sridharan and T. R. Allen, in Molten Salts Chemistry, ed. F. Lantelme and H. Groult, Elsevier, Oxford, 2013, pp. 241–267 DOI:10.1016/B978-0-12-398538-5.00012-3.
- Y. Sun, H.-W. Lee, Z. W. Seh, N. Liu, J. Sun, Y. Li and Y. Cui, Nat. Energy, 2016, 1, 15008 CrossRef CAS.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- X. Liu, T. Liu, R. Wang, Z. Cai, W. Wang, Y. Yuan, R. Shahbazian-Yassar, X. Li, S. Wang, E. Hu, X.-Q. Yang, Y. Xiao, K. Amine, J. Lu and Y. Sun, ACS Energy Lett., 2021, 6, 320–328 CrossRef CAS.
- G. Sun, F.-D. Yu, L.-F. Que, L. Deng, M.-J. Wang, Y.-S. Jiang, G. Shao and Z.-B. Wang, Nano Energy, 2019, 66, 104102 CrossRef CAS.
- S. Lee, J. Hong and K. Kang, Adv. Energy Mater., 2020, 10, 2001445 CrossRef CAS.
- S. Lee, G. Kwon, K. Ku, K. Yoon, S.-K. Jung, H.-D. Lim and K. Kang, Adv. Mater., 2018, 30, 1704682 CrossRef PubMed.
- Y. Tsao, M. Lee, E. C. Miller, G. Gao, J. Park, S. Chen, T. Katsumata, H. Tran, L.-W. Wang, M. F. Toney, Y. Cui and Z. Bao, Joule, 2019, 3, 872–884 CrossRef CAS.
- C. Wu, J. Hu, L. Ye, Z. Su, X. Fang, X. Zhu, L. Zhuang, X. Ai, H. Yang and J. Qian, ACS Sustainable Chem. Eng., 2021, 9, 16384–16393 CrossRef CAS.
- M. Lee, J. Hong, B. Lee, K. Ku, S. Lee, C. B. Park and K. Kang, Green Chem., 2017, 19, 2980–2985 RSC.
- G. Kwon, S. Lee, J. Hwang, H.-S. Shim, B. Lee, M. H. Lee, Y. Ko, S.-K. Jung, K. Ku, J. Hong and K. Kang, Joule, 2018, 2, 1771–1782 CrossRef CAS.
- S. Ko, Y. Yoo, J. Choi, H.-D. Lim, C. B. Park and M. Lee, J. Mater. Chem. A, 2022, 10, 20464–20472 RSC.
- A. Chakraborty, S. Kunnikuruvan, S. Kumar, B. Markovsky, D. Aurbach, M. Dixit and D. Major, Chem. Mater., 2020, 32, 915–952 CrossRef CAS.
- V. W.-h Lau, I. Moudrakovski, J. Yang, J. Zhang and Y.-M. Kang, Angew. Chem., Int. Ed., 2020, 59, 4023–4034 CrossRef CAS PubMed.
- X. Ma, M. Chen, Z. Zheng, D. Bullen, J. Wang, C. Harrison, E. Gratz, Y. Lin, Z. Yang, Y. Zhang, F. Wang, D. Robertson, S.-B. Son, I. Bloom, J. Wen, M. Ge, X. Xiao, W.-K. Lee, M. Tang, Q. Wang, J. Fu, Y. Zhang, B. C. Sousa, R. Arsenault, P. Karlson, N. Simon and Y. Wang, Joule, 2021, 5, 2955–2970 CrossRef CAS.
- X. Han, M. Ouyang, L. Lu and J. Li, J. Power Sources, 2014, 268, 658–669 CrossRef CAS.
- J. Zhao, Z. Lu, N. Liu, H.-W. Lee, M. T. McDowell and Y. Cui, Nat. Commun., 2014, 5, 5088 CrossRef CAS PubMed.
- S. Renault, S. Gottis, A.-L. Barrès, M. Courty, O. Chauvet, F. Dolhem and P. Poizot, Energy Environ. Sci., 2013, 6, 2124–2133 RSC.
- L. Sieuw, A. E. Lakraychi, D. Rambabu, K. Robeyns, A. Jouhara, G. Borodi, C. Morari, P. Poizot and A. Vlad, Chem. Mater., 2020, 32, 9996–10006 CrossRef CAS.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- H.-K. Lim, H.-D. Lim, K.-Y. Park, D.-H. Seo, H. Gwon, J. Hong, W. A. Goddard, III, H. Kim and K. Kang, J. Am. Chem. Soc., 2013, 135, 9733–9742 CrossRef CAS PubMed.
- W.-J. Kwak, H. Kim, Y. K. Petit, C. Leypold, T. T. Nguyen, N. Mahne, P. Redfern, L. A. Curtiss, H.-G. Jung, S. M. Borisov, S. A. Freunberger and Y.-K. Sun, Nat. Commun., 2019, 10, 1380 CrossRef PubMed.
- W.-J. Kwak, S. A. Freunberger, H. Kim, J. Park, T. T. Nguyen, H.-G. Jung, H. R. Byon and Y.-K. Sun, ACS Catal., 2019, 9, 9914–9922 CrossRef CAS.
- J. P. Singh, J. Y. Park, K. H. Chae, D. Ahn and S. Lee, Nanomaterials, 2020, 10, 759 CrossRef CAS PubMed.
- R. A. House, U. Maitra, L. Jin, J. G. Lozano, J. W. Somerville, N. H. Rees, A. J. Naylor, L. C. Duda, F. Massel, A. V. Chadwick, S. Ramos, D. M. Pickup, D. E. McNally, X. Lu, T. Schmitt, M. R. Roberts and P. G. Bruce, Chem. Mater., 2019, 31, 3293–3300 CrossRef CAS.
- H. Sun, A. Hu, S. Spence, C. Kuai, D. Hou, L. Mu, J. Liu, L. Li, C. Sun, S. Sainio, D. Nordlund, W. Luo, Y. Huang and F. Lin, Adv. Funct. Mater., 2022, 32, 2112279 CrossRef CAS.
- M. Broussely, P. Biensan, F. Bonhomme, P. Blanchard, S. Herreyre, K. Nechev and R. J. Staniewicz, J. Power Sources, 2005, 146, 90–96 CrossRef CAS.
- M. Dubarry, C. Truchot, B. Y. Liaw, K. Gering, S. Sazhin, D. Jamison and C. Michelbacher, J. Power Sources, 2011, 196, 10336–10343 CrossRef CAS.
- J. Kasnatscheew, M. Evertz, B. Streipert, R. Wagner, S. Nowak, I. Cekic Laskovic and M. Winter, J. Phys. Chem. C, 2017, 121, 1521–1529 CrossRef CAS.
- Z. Qin, Z. Wen, Y. Xu, Z. Zheng, M. Bai, N. Zhang, C. Jia, H. B. Wu and G. Chen, Small, 2022, 18, 2106719 CrossRef CAS PubMed.
- J. Yang, W. Wang, H. Yang and D. Wang, Green Chem., 2020, 22, 6489–6496 RSC.
- Y. Bai, N. Muralidharan, J. Li, R. Essehli and I. Belharouak, ChemSusChem, 2020, 13, 5664–5670 CrossRef CAS.
- B. J. Ross, M. LeResche, D. Liu, J. L. Durham, E. U. Dahl and A. L. Lipson, ACS Sustainable Chem. Eng., 2020, 8, 12511–12515 CrossRef CAS.
- Y. Ji, C. T. Jafvert and F. Zhao, Resour., Conserv. Recycl., 2021, 170, 105551 CrossRef CAS.
- H. Kim, S.-K. Kim, J.-S. Sohn, S. Byun, D. Yang and K.-M. Lee, KR Pat., 101999677, 2019 Search PubMed.
- G. Jiang, Y. Zhang, Q. Meng, Y. Zhang, P. Dong, M. Zhang and X. Yang, ACS Sustainable Chem. Eng., 2020, 8, 18138–18147 CrossRef CAS.
- J. Jang, I. Kang, J. Choi, H. Jeong, K.-W. Yi, J. Hong and M. Lee, Angew. Chem., Int. Ed., 2020, 59, 14473–14480 CrossRef CAS PubMed.
- J. Choi, H. Jeong, J. Jang, A. R. Jeon, I. Kang, M. Kwon, J. Hong and M. Lee, J. Am. Chem. Soc., 2021, 143, 9169–9176 CrossRef CAS PubMed.
- X. Liu, Y. Tan, W. Wang, C. Li, Z. W. Seh, L. Wang and Y. Sun, Nano Lett., 2020, 20, 4558–4565 CrossRef CAS PubMed.
- C. Wu, M. Xu, C. Zhang, L. Ye, K. Zhang, H. Cong, L. Zhuang, X. Ai, H. Yang and J. Qian, Energy Storage Mater., 2023, 55, 154–165 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee04528e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |