
Aluminum intercalation behaviours of {[Fe(Tp)(CN)3]2[M(H2O)2]} cyanido-bridged chain compounds in an ionic liquid electrolyte†
Na
Li
ab,
Yanling
Li
 a,
Hans Jurgen
von Bardeleben
d,
Damien
Dambournet
*ac and
Rodrigue
Lescouëzec
*b
a,
Hans Jurgen
von Bardeleben
d,
Damien
Dambournet
*ac and
Rodrigue
Lescouëzec
*b
aSorbonne Université, CNRS, Physico-chimie des Électrolytes et Nano-Systèmes Interfaciaux, PHENIX, F-75005 Paris, France. E-mail: damien.dambournet@sorbonne-universite.fr
bSorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire, ERMMES, F-75005 Paris, France. E-mail: rodrigue.lescouezec@sorbonne-universite.fr
cRéseau sur le Stockage Electrochimique de l'Energie (RS2E), FR CNRS 3459, 80039 Amiens, France
dSorbonne Université, CNRS, Institut des Nanosciences de Paris, UMR 7588, F-75005 Paris, France
First published on 19th June 2024
Abstract
As the development of aluminum-ion batteries is still in its infancy, researchers are still dedicated to exploring suitable host materials and investigating their aluminum intercalation behaviours. Here, a series of cyanido-bridged chain compounds with the formula {[FeIII(Tp)(CN)3]2[MII(H2O)2]}n (M = Ni, Co, Mn, Zn, Cu) are studied as cathode electrodes for aluminum-ion batteries with [EMIm]Cl–AlCl3 (1-ethyl-3-methylimidazolium chloride–AlCl3) ionic liquid as the electrolyte. The electrochemical properties suggested Fe3+/Fe2+ to be the redox-active couple during the aluminum intercalation and deintercalation processes of these compounds, and the observed maximum specific capacity obtained by the Fe–Co compound is 200 mA h g−1 despite the rapid specific capacity fading. To gain a deeper understanding of the capacity decay suffered by these compounds, further investigation was conducted to explore the evolution of compounds during the electrochemical measurements. It has been attributed to the following reasons: 1. thermodynamic instability results in the transformation/damage of two of the chain structures (for the Fe–Ni and Fe–Co compounds) during heat treatment on electrodes, a crucial step in electrode preparation; 2. the acidic nature of the electrolyte triggers the destruction of the chain structure, with the appearance of partial reduction of Fe3+ to Fe2+, and a new interaction of the cyano group with aluminum; 3. the high charge density of inserted Al ions makes the chain structure suffer from structural damage during both the charging and discharging processes. The progressive accumulation of trapped intercalated ions hampers their involvement in the reaction, consequently decreasing electrochemical reversibility.
1. Introduction
The contemporary increasing demand for energy has prompted extensive research into energy storage/conversion technologies, and the continuous search for new electroactive materials is amongst the solutions. Transition metal cyanides because of the structural robustness and electronic tuning of polycyanido-metallate [M(CN)n]m− building blocks (M: transition metal) have been recognized as promising candidates as electrode materials.1 Prussian Blue (PB) and its analogues (PBAs), of general formula AxM′[M(CN)6]z·nH2O (M and M′ = divalent or trivalent transition metal ions and A = alkali metal ions), are among the best-known examples that have been continuously reported as electrode materials for secondary batteries. A lot of effort is currently focused on the study of heterometallic PBAs and the influence of their structuration in ion batteries.2–6 Since these cyanide-based compounds have led to appealing results, we and other groups started a few years ago to explore the potentialities of related cyanido-based molecular materials with more complex but more versatile and tuneable electronic properties.7 We more specifically focused on the use of the triscyanido, fac-[FeIII(Tp)(CN)3]− complex (Tp: hydrotris(pyrazolyl)borate),8 which has proven effective at leading discrete and polymeric frameworks of cyanide materials.9 The scorpionate Tp− ligand acts as an ancillary ligand to facilitate the preparation of low-dimensional materials. Interestingly, it can also be functionalized to adjust the electronic properties of the Fe complex. When the cyanometallate complex fac-[FeIII(Tp)(CN)3]− is reacted with metal cations, polymeric frameworks, generally heterometallic chain structures, can be formed. In a seminal work,10 we reported the lithium-ion storage capabilities of the iron–nickel bimetallic one-dimensional (1D) coordination polymer, {[FeIII(Tp)(CN)3]2[NiII(H2O)2]}n. The result first confirmed the reversible Li+ (de)intercalation in the 1D cyanide-bridged molecular material. This result aroused our interest in further exploring the possible insertion of aluminium ions into such 1D coordination polymers. Aluminium-ion batteries (AIBs) are recognized as a promising future post-Li battery technology, particularly in terms of safety, low cost and high theoretical capacity (Al metal: 2978 mA h g−1 and 8034 mA h L−1).11 However, the development of AIBs is full of challenges. A major difficulty is finding suitable cathode materials able to reversibly intercalate such high-valent cations whose diffusion suffers from poorer kinetics due to the higher electrostatic interactions when compared to monovalent Li ions. Another difficulty lies in the choice of an appropriate electrolyte. Despite the availability of molten salt electrolytes, aqueous electrolytes and ionic liquid electrolytes have prevailed in recent years in AIBs. Although the research on AIBs dates back to the early 1970s,12 cathode electrode materials reported in the literature mainly focus on carbon,13–17 sulphur,18–20 vanadium-containing compounds,21–23 metal oxides, metal sulphides,24–27etc., while fewer studies have been undertaken toward the application of molecular materials in AIBs. In particular, the reports on cyanido-metallate compounds are limited.28–32PBAs appear to be the only transition metal cyanide-based materials that have been investigated in AIBs. Pang et al.28 constructed aqueous AIBs with potassium cobalt hexacyanoferrate (K2Co[Fe(CN)6]) as the cathode in the electrolyte of 1 M Al(NO3)3. A reversible discharge capacity of 50 mA h g−1 at 0.1 A g−1 was obtained in the potential window of 0–1.2 V with an excellent capacity retention (76% after 1600 cycles). Aqueous electrolytes of Al2(SO4)3 and AlCl3 were also applied in this work for comparison, and it was shown that they exhibit less favorable results than the Al(NO3)3 electrolyte in terms of charge/discharge capacity and impedance. So far, most research studies with molecular materials as electrode materials for AIBs have been conducted using aqueous electrolytes, while the utilization of ionic liquids (ILs) in AIBs with molecular materials remains scarce. For example, copper hexacyanoferrate has been widely applied in aluminium batteries with aqueous electrolytes, while the only work with a non-aqueous electrolyte displays an undesirable electrochemical performance and a deep understanding of the energy storage mechanism and capacitance decay needs to be gained. In an organic electrolyte Al(OTf)3 in diglyme, cubic particles of Cu1.5[Fe(CN)6]·6H2O delivered a very low reversible capacity (between 5 and 14 mA h g−1) within an electrochemical window between 0.1 and 0.7 V despite an initial discharge capacity of 60 mA h g−1.29 In an aqueous Al2(SO4)3 electrolyte, Gao et al.30 investigated KCu[Fe(CN)6]·8H2O nanoparticles and observed a specific capacity of 62.9 mA h g−1 at 50 mA g−1 in the voltage range of 0.2 to 1.2 V, with capacity retention of 54.9% after 1000 cycles. Moreover, Wills et al.31 and Chiang et al.32 reported separately aqueous aluminium ion intercalation with copper hexacyanoferrate as the cathode. Although aqueous AIBs are assembled with different anode materials and electrolytes, they all present a specific capacity of approximately 50–60 mA h g−1. It is worth mentioning that the discharge voltage in Wills's work is as high as 1.5 V, which surpasses many reported operating voltages.
Compared with aqueous electrolytes being limited by the stability window of water, ILs featuring high conductivity, nonflammability, nonvolatility, and high thermal stability could be operated within a relatively wide electrochemical window.33 In particular, 1-methyl-3-methylimidazolium chloride ([EMIm]Cl) outperforms most non-aqueous electrolytes in terms of electrochemical window width, liquidus temperature and aluminium reactivity and maintains the conductivity, viscosity, Lewis acidity range and solvating power of other salts.34 Therefore, we selected [EMIm]Cl–AlCl3 ionic liquid as the electrolyte and investigated a series of one-dimensional (1D) materials with the formula {[FeIII(Tp)(CN)3]2[MII(H2O)2]}n (M = Ni, Co, Mn, Zn, Cu) as intercalation cathode materials. We expect that the lower dimensionality and open framework of these compounds could permit easier ion (de)intercalation and a better Al-ion host capability. We can also hypothesize that the presence of an organic shell (Tp ligands) in the chains could favor weaker electrostatic interactions between the inserted multivalent cation and the framework, and thus a better diffusion. Furthermore, comparisons between compounds bridged by different divalent metals, including inactive zinc, are intended to help understand the multifaceted effects of bridged metals on intercalation performances.
2. Materials and methods
2.1. Materials synthesis
The precursor (NBu4)[Fe(Tp)(CN)3] was prepared following the procedure previously reported by Lescouëzec et al., but substituting PPh4Cl salt by NBu4Cl.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. A solution of Ni(NO3)2·6H2O (145 mg, 0.5 mmol) in 20 mL of the same solvent was added dropwise to the former solution. The resulting solution was stirred for 18 h and filtered. The orange-red solid product was obtained after drying in air at room temperature.
:3. A solution of Ni(NO3)2·6H2O (145 mg, 0.5 mmol) in 20 mL of the same solvent was added dropwise to the former solution. The resulting solution was stirred for 18 h and filtered. The orange-red solid product was obtained after drying in air at room temperature.
2.2. Characterization
Elemental analyses for C, H, and N were performed on a PerkinElmer 240C analyser.The powder X-ray diffraction (PXRD) data were collected on a D8 Advance diffractometer equipped with a linear LynEye detector and an X-ray tube with a copper anode, working with the Ka1 and Ka2 doublet of copper.
FT-IR spectra were recorded for all samples in the 400–4000 cm−1 range at room temperature under an ambient atmosphere on a Vertex 70 Bruker instrument using the attenuated total reflection (ATR) technique (with a 4 cm−1 resolution).
Thermogravimetric analysis (TGA) was performed at a rate of 2 °C min−1 under a nitrogen or air flow up to 600 °C on a PerkinElmer STA-Q600 simultaneous thermal analyser.
SEM micrographs and EDS (energy dispersive spectroscopy) analyses were recorded with a FEI Magellan 400 scanning electron microscope with an Oxford EDS probe.
Magnetic measurements were carried out on a MPMS3 magnetometer in the temperature range of 2–300 K. The applied fields of 500 Oe and of 5000 Oe were used respectively for low temperature (2–30 K) and high temperature (20–300 K) measurements. The sample packed in a tin capsule was introduced under a helium flow at 200 K to avoid solvent evaporation. All data were corrected by estimating the diamagnetic contribution of the sample and sample holder.
Electron Paramagnetic Resonance (EPR) spectra were recorded in quartz tubes with a Bruker ER200 instrument and a CW X-band (9.33 GHz) spectrometer, equipped with a liquid helium cryostat.
Electrochemical measurements were conducted on electrochemical workstation with three-electrode Swagelok-type cells. The cell assembling was completed in a glovebox using an Al sheet as the cathode and Al wire as the reference electrode. The electrolyte was [EMIm]Cl–AlCl3 (1-ethyl-3-methylimidazolium chloride–AlCl3 mixture, molar ratio 1:1.1) ionic liquid. The working electrode was composed of 80 wt% active material, 10 wt% conductive carbon and 10 wt% binder (PTFE) and molybdenum sheets were used as the current collector. The working electrode was air-dried overnight, and vacuum heated at 100 °C for 4 hours prior to assembly.
3. Results and discussion
3.1. Materials synthesis and characterization
This series of materials, denoted as Fe–M, with the formula {[FeIII(Tp)(CN)3]2[MII(H2O)2]}n (M = Ni, Co, Mn, Zn, Cu) were obtained by the direct precipitation method. The metal Fe/M ratios of different samples were monitored through energy dispersive spectroscopy. The analysis of two different randomly selected regions of the samples, (Fig. S1–S5†) confirmed that the metal Fe/M ratio is consistent with the molecular formula, almost all of which is 2:1. Their crystal structures were verified by comparing their powder X-ray diffraction patterns with those known from bibliographical data collected in the Cambridge Structural Database (see below). The experimental formulas were determined by combining elemental analysis and thermogravimetry. Table S1† lists the exact formulas and the concordance between theoretical and experimental content for the materials studied here. The experimental XRD patterns corresponding to the different products and the calculated powder X-ray diffraction patterns are presented in Fig. 1 and the enlarged fragments of patterns are presented in Fig. S6.†
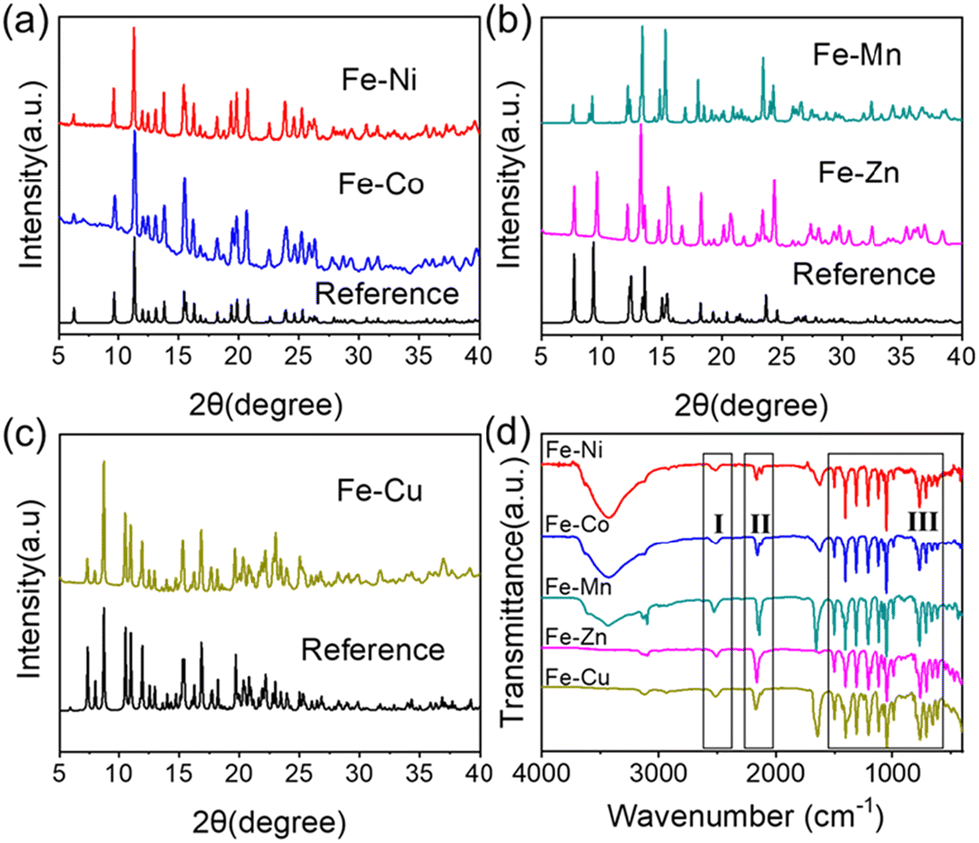 | ||
| Fig. 1 Comparison between the experimental powder X-ray diffraction patterns of the synthesized compounds and calculated powder X-ray diffraction patterns from single-crystal structures (a) Fe–Ni and Fe–Co, (b) Fe–Mn and Fe–Zn, (c) Fe–Cu; and (d) infrared spectra of the compounds studied in this work. | ||
The XRD patterns of Fe–Ni and Fe–Co compounds shown in Fig. 1a are almost identical, and they match well with the calculated powder XRD pattern obtained from previously reported single crystal data.10 Note that the broad background shown on the XRD pattern of the Fe–Co compound is due to the fluorescence phenomenon. Therefore, Fe–Ni and Fe–Co compounds are isomorphic, belonging to the C2/c space group, and possess a chain structure. This chain structure is composed of Fe2IIINi2II squares (Fe2IIICo2II squares) where the cyanide bridges are almost linear, as shown in Scheme 1. Within each square, the [Fe(Tp)(CN)3]− unit binds two NiII (CoII) ions through two of its three cyanide groups, the third one remaining terminal. The NiII (CoII) are shared between two squares and their coordination is thus made of four N atoms from four cyanide groups. Two water molecules in the trans position complete their coordination sphere. The Fe–Mn and Fe–Zn compounds, which have not been previously reported, exhibit similar XRD patterns as depicted in Fig. 1b. Notably, the XRD peaks of both Fe–Mn and Fe–Zn display similarities to the diffractogram of the {FeIII(Tp)(CN)3}4{FeII(H2O)2FeII} compound (abbreviated as Fe–Fe) which exhibits a chain structure in the C2 space group, as reported by Sato et al.35 Small shifts and deviations (Fig. 1b and Fig. S6†) are, however, observed, which might be attributed to the different divalent metals and more importantly to the differences in reaction solvents. The reference compound Fe–Fe is synthesized in a mixed solution of water and acetonitrile, while Fe–Mn in DMF, and Fe–Zn in a mixed solution of water and methanol. Within the chain (Scheme 1), each [Fe(Tp)(CN)3]− entity acts as a bidentate ligand alternately linking octahedrally coordinated MnII (ZnII) and tetrahedrally coordinated MnII (ZnII), as shown in the yellow frame in Scheme 1. Although prepared in the DMF solvent, two cis positions of octahedral MnII are occupied by two water molecules, similarly to the Fe–Zn compound obtained in the mixed water/methanol solvent. The Fe–Cu product, which crystallizes in the monoclinic space group P21/c, exhibits a different chain structure, which is in complete agreement with that reported in the previous literature (Fig. 1c).36 Unlike the above-mentioned compounds, in this chain, divalent copper ions are coordinated by two bulkier DMF molecules, which induce an increase of the spacing between neighbouring chains in the crystal lattice.
 | ||
| Scheme 1 Schematic illustration for the synthesis of the chain compounds. The images on the right are perspective views of the (Fe2M)n chain crystal structures. | ||
Here, Fe–Ni and Fe–Co compounds were obtained in the mixed solvent of methanol and water applying a previously reported protocol,10,36 and Fe–Cu was synthesized in DMF following You et al.'s work.36 The newly constructed chain structures of Fe–Zn and Fe–Cu in our work used the procedure of synthesizing Fe–Ni/Fe–Co and Fe–Cu, respectively. Despite the difference in the reaction solvent, both of them present the same structure as a reported chain structure connected by FeIII and FeII.35
Fig. 1d exhibits the collection of IR spectra of the chain complexes studied herein. Despite the change of the metal ion coordinated to the FeIII ion, the IR spectra are very similar. In Fig. 1d, all spectra show characteristic peaks of the anionic Tp ligand in region III ranging from 900 to 1550 cm−1. The presence of a DMF molecule in compounds Fe–Mn and Fe–Cu is revealed by a characteristic peak at ca. 1650 cm−1 ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration. The broad absorption in region I corresponds to the B–H stretching vibration from the Tp ligand in the series of complexes, and the values are collected in Table 1. The region II emphasizes the characteristic cyanide stretching vibrations including the characteristic bridging FeIII–CN–MII (M refers to different metals) at higher vibrations and the terminal FeIII–CN cyanide at lower vibrations, which are locally magnified and summarized (Table 1).
O stretching vibration. The broad absorption in region I corresponds to the B–H stretching vibration from the Tp ligand in the series of complexes, and the values are collected in Table 1. The region II emphasizes the characteristic cyanide stretching vibrations including the characteristic bridging FeIII–CN–MII (M refers to different metals) at higher vibrations and the terminal FeIII–CN cyanide at lower vibrations, which are locally magnified and summarized (Table 1).
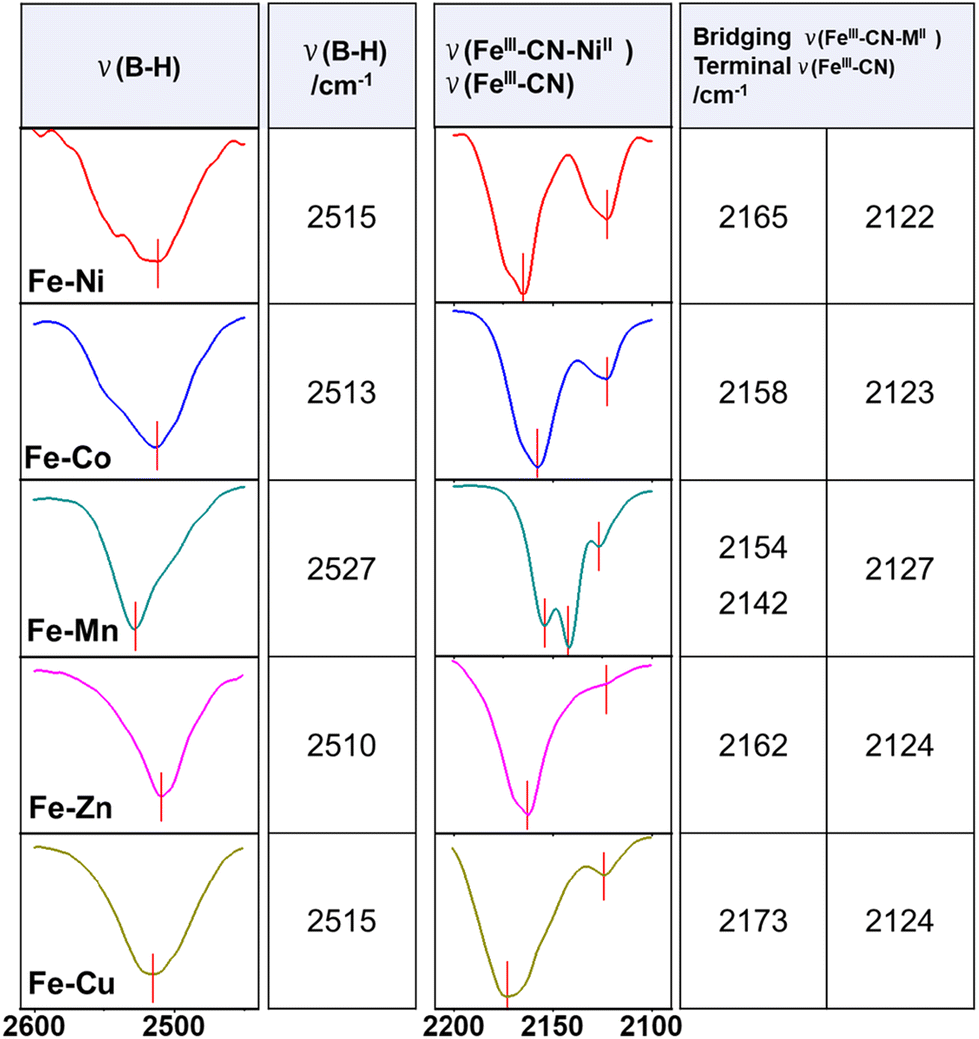
|
Thermogravimetric analysis (TGA) was used to investigate the thermal stability of the compounds under both air and nitrogen atmospheres. Almost coincident curves exhibiting three major mass losses are observed in air and N2 for Fe–Ni before reaching 200 °C (Fig. 2). In contrast, Fe–Co shows one weight loss at lower temperature in air, suggesting the tighter bond of solvent molecules to nickel than cobalt. This fits nicely with the Irving Williams series where Ni presents stronger binding force than Co.37 Besides, the percentages of weight loss (12.2% and 11.9%) of Fe–Ni and Fe–Co samples at low temperature are consistent with the calculated ones (12.6% and 12.0%) for the crystallization solvents and two coordinated water molecules, respectively. Crystallization solvent molecules of Fe–Ni and Fe–Co products are thus removed completely below 200 °C.
 | ||
| Fig. 2 (a–e) Thermogravimetric analysis of the compounds in nitrogen and air atmospheres; (f) calculated percentage of solvent to the total weight of the molecule. | ||
In the case of Fe–Mn, the substantially identical thermogravimetric profiles in both air and nitrogen present two mass reductions before reaching 200 °C, corresponding to a weight loss percentage of 15.6% (Fig. 2c). This value is exactly the expected one, considering the crystallization solvents (water and DMF) and coordinated water molecules. In Fig. 2d, whether in air or nitrogen, Fe–Zn exhibits little mass loss (2.4%) at low temperature, which matches with the calculated weight loss percentage of 2.3%, corresponding to the removal of free solvent molecules and the coordinated water molecule in the Fe–Zn sample. The thermal behaviour observed for Fe–Cu is quite different, as Fe–Cu shows little mass loss (1.8%) when heated up to 140 °C, which is close to the expected percentage for the loss of free solvents (1.6%). Then, a noticeable loss of mass is observed between 140 and 200 °C, which cannot be ascribed to the removal of the only bound DMF molecule and thus suggests a partial decomposition. In air, the Fe–Cu compound shows a bump between 200 and 300 °C which is not seen under N2 and thus probably a result of an oxidation reaction in air.
Finally, for all the Fe2M chain compounds, a large weight loss is observed above 200 °C, which accounts for their thermal decomposition. As all these degradation temperatures are notably higher than 100 °C at which we treated electrodes containing these compounds in a tube furnace, all these compounds are suitable for subsequent electrochemical studies from a thermal stability perspective.
As mentioned in the Experimental section, all the materials were dried in a vacuum at 100 °C for 4 h before the assembly of the electrochemical cells. Subsequently, XRD patterns and partial FT-IR spectra were recorded, and they are shown in Fig. 3 and the full FT-IR spectra are exhibited in Fig. S7.† After the thermal treatment under vacuum, Fe–Zn and Fe–Cu compounds retain the original IR spectra and their XRD patterns are unchanged (Fe–Zn and Fe–Cu in Fig. 3 and Fig. S7†), while the other compounds are modified to some degree. In Fig. 3, Fe–Ni after vacuum heating presents a completely different XRD pattern from the previous one, and heated Fe–Co becomes amorphous. For the Fe–Co compound, we observed that upon heating it is prone to amorphization (Fig. 3). While we do not have a definitive explanation for this observation, it is possible that the labile character of the Co–N bond together with the ability of the CoII to easily change from octahedral to tetrahedral surrounding upon desolvation, favours the occurence of defects in the structure that ultimately lead to amorphization. Considering their IR spectra, Fe–Ni before and after vacuum heating show similar features in both the fingerprint region of the spectra and the cyanide stretching vibration region (Fig. S7† and partial IR spectra on the right, Fig. 3). In contrast, Fe–Co cyanide vibrations display changes: a broadening of the peaks occurs (2124 cm−1 appears as a shoulder) and more importantly, the appearance of a weak peak at 2067 cm−1, which could accounts for a partial reduction of Fe(III) to Fe(II).38,39 Further insight into this compound may require PDF (pair diffusion function) analysis or XAS (X-ray absorption spectroscopy), which are suitable techniques for investigating local structures. A broadening of the CN stretching vibration is also observed in the IR spectrum of Fe–Mn under drying which could account for a higher structural disorder. Actually, the XRD patterns of Fe–Mn in Fig. 3 reflect a relatively lower crystallinity compared to the fresh compound. Therefore, we hypothesize that Fe–Ni may have lost part of the solvent molecules while maintaining the chain structure, and Fe–Co may have suffered from some degree of crystal structure collapse, which is present to a lower extent in Fe–Mn. Fe–Cu and Fe–Zn exhibit relatively good thermal stability probably because they are on the extremity of the Irving Williams series and are expected to show the strongest bonding with the N cyanide. To further probe this hypothesis, we conducted magnetic measurements on Fe–Ni and Fe–Co compounds before and after vacuum heating.
 | ||
| Fig. 3 Powder X-ray diffraction patterns and partial IR spectra of compounds before and after vacuum heating (denoted as VH) at 100 °C. | ||
The temperature dependence of the χMT product in the temperature range of 300–1.9 K is shown in Fig. 4 (where χM is the magnetic molar susceptibility per FeIII2MII unit). At 300 K, the χMT of fresh and vacuum heated Fe–Ni chain (Fig. 4a) are almost the same, 2.70 and 2.66 cm3 mol−1 K, respectively, and they both agree with the expected value for one octahedral Ni(II) ion and two low-spin Fe(III) ions with orbital contribution.10,40 Thus, the drying process does not seem to affect the metal ions’ redox state. For both compounds, upon decreasing temperature, the χMT product first gradually increases until 50 K and then suddenly increases reaching maximum values of 110 cm3 mol−1 K and 37 cm3 mol−1 K at 5 K, followed by an abrupt decrease until 2 K. The continuous increase upon lowering T accounts for the occurrence of ferromagnetic interactions between the Fe(III) and Ni(II) ions through the cyanide bridge. This is coherent with the Kahn's model that predicts ferromagnetic exchange coupling between the orthogonal magnetic orbitals: t2g on the Fe(III) and eg on the Ni(II) ions.41 The sudden increase of χMT to high values below 50 K is coherent with the one-dimensional character of the compound as the correlation length of the ferromagnetic domains increases with decreasing temperature.40,42 Interestingly, the increase is not so pronounced in the dried sample, which could be due to the occurrence of defects or distortion in the chain structure upon desolvation.
 | ||
| Fig. 4 Thermal dependence of the χMT product of Fe–Ni (a) and Fe–Co (b) in the temperature range of 1.9–300 K. | ||
The fresh Fe–Co sample exhibits similar properties to the Fe–Ni ones. The χMT values at 300 K, 4.13 cm3 mol−1 K are in the range of the expected theoretical values for magnetically independent two low-spin Fe(III) and one high-spin Co(II), ca.4.0–4.8 cm3 mol−1 K (both of them having a significant magnetic orbital contribution). Upon decreasing the temperature, a first gradual then abrupt (below 50 K) increase of the χMT value is observed, which accounts for the occurrence of ferromagnetic exchange coupling propagating along the 1D compound (as previously observed in similar compounds).43 The dried Fe–Co sample shows differences that can be observed using the chemical analyses, FT-IR and TGA. First, the χMT value at 300 K (3.52 cm3 mol−1 K) is slightly smaller than that of the fresh sample. This can be associated with a partial reduction of the Fe(III) ion into the diamagnetic Fe(II) center which is coherent with the new characteristic cyanide stretching vibrations (Fig. 3). This hypothesis is in fact coherent with the magnetic measurement at lower temperature. The χMT value only smoothly increases reaching only 6.43 cm3 mol−1 K at 5 K. This is coherent with the loss of long-range ordering along the 1 D compound.44,45 Moreover, as the compound loses its crystallinity, the occurrence of structural defects cannot be discarded. Finally, TGA indicated an almost complete loss of solvent and coordinated water molecule which could indicate the occurrence of tetrahedral Co(II) centers which would also account for a reduction of the χMT product (Fig. S8†).
In summary, the process of vacuum heating for electrode manufacture does not significantly alter the crystal structure of the Fe–Mn, Fe–Zn and Fe–Cu compounds, while it causes noticeable modification of the structure of Fe–Ni and Fe–Co compounds. However, the magnetic data of Fe–Ni remains coherent with the preservation of a one-dimensional character. Greater damage is observed in the Fe–Co compound which also suffers from partial reduction of the Fe(III) center, resulting in the amorphous state with a short-range order.
3.2. Electrochemical characterization
Since Fe–Ni has been confirmed to be capable of lithium storage in a previous study,10 we are interested in exploring the possibility of employing it as an aluminum storage host and comparing this material with similar Fe–M chain compounds. Swagelok cells were assembled to characterize the electrochemical behavior of these materials, with the as-prepared samples as the working electrode and aluminum metal as counter and reference electrodes. The ionic liquid 1-ethyl-3-methyl-imidazolium chloride (EMImCl) mixed with AlCl3 in the ratio 1:1.1 for our studies was selected as the electrolyte due to a few attractive features such as very low vapor pressure, relatively high electrical conductivity and a suitable electrochemical potential window.34
In such an electrolyte, the molar ratio of [EMIm]Cl/AlCl3 controls the acidity of the electrolyte and determines the predominant anion species in the electrolyte. The mixture of more than 50 mol-% AlCl3 is considered as Lewis acidic and the aluminium species in the form of Al2Cl7− appears, while those with a 1:1 (50 mol-% EMImCl to 50 mol-% AlCl3) composition are Lewis neutral characterised by AlCl4− and less than 50 mol-% AlCl3 are Lewis basic, mainly consisting of Cl−.46 However, the organic cations in EMImCl–AlCl3 ionic liquid are reduced at a higher potential than the dominant AlCl4− anions, indicating the aluminium deposition from AlCl4− in a Lewis neutral ionic liquid is not possible. Therefore, aluminium deposition, which ensures aluminum-ion battery operation, only occurs in acidic electrolytes according to the following reaction, in which the Al2Cl7− anions are reduced (−0.2 V vs. Al|Al(III)):46
| 4Al2Cl7− + 3e− → Al + 7AlCl− |
To provide more insight into the Al3+ storage within this series of one-dimensional compounds, the galvanostatic charge–discharge (GCD) cycling was performed on the five compounds at a current of 10 mA g−1 in selected voltage windows (Fig. 5). Firstly, the assembled cells with different metal compound cathodes were discharged from 1.8 to 0 V. In the case of Fe–Ni and Fe–Co compounds, the cut-off voltage had to be increased to 0.4 V to avoid the cathode damaging observed beyond this value. The corresponding GCD curves are shown in Fig. 5(a) and (c), while those of Fe–Mn, Fe–Zn and Fe–Cu compounds are shown in Fig. 5(e), (g) and (i).
 | ||
| Fig. 5 Galvanostatic discharge–charge profiles of the compounds Fe–Ni (a–b), Fe–Co (c–d), Fe–Mn (e–f), Fe–Zn (g–h), Fe–Cu (i–j) in the voltage range of 0–1.8 V/0.4–1.8 V and 1.0–1.8 V. | ||
The GCD curve of Fe–Ni in the voltage range of 0.4–1.8 V only shows a discharge plateau at 1.4 V, and its specific capacity is only 29 mA h g−1. This is the lowest first discharge specific capacity of this series of compounds, and the specific capacity drops to 6 mA h g−1 by the second cycle. In Fig. 5(c), Fe–Co exhibits two plateaus at 1.4 V and 0.4 V, respectively, and its first discharge process presents a similar behavior to that of Fe–Zn with two plateaus at 1.4 V and 0.2 V of its first discharge curve. The second discharge plateaus are both longer, making their first discharge specific capacity as high as 200 mA h g−1 for Fe–Co and 196 mA h g−1 for Fe–Zn, which are relatively high compared with the specific capacity reported in the literature for this class of materials in aluminum ion batteries.47,48 It could be suggested that long discharge process damages or destroys the chain structure, which results in a great capacity fading, with the specific capacity of the second cycle for Fe–Co and Fe–Zn falling to 85 mA h g−1 and 15 mA h g−1, respectively. Although their initial discharge specific capacity is high, the specific capacity deteriorates deeply, showing an irreversible electrochemical process. This deterioration of specific capacity also occurs on the cathodes composed of Fe–Mn and Fe–Cu compounds. The GCD curve of Fe–Mn presents three discharge plateaus at ca. at 1.4 V, 1.1 V and 0.5 V (Fig. 5(e)), and the capacity in the first discharge process is 44 mA h g−1, which is more than twice that of the second circle (21 mA h g−1). Similarly, the first-discharge capacity of the electrode based on the Fe–Cu compound could reach a high specific capacity of 116 mA h g−1, although it decreases to 24 mA h g−1 for the second discharge process. The GCD curve of Fe–Cu in Fig. 5(g) exhibits two clear plateaus at 1.4 V and 0.2 V.
Preliminary electrochemical charge–discharge tests of these five compounds in the selected potential window (0–1.8 V and 0.4–1.8 V) show an irreversible electrochemical process after the first long discharge process. We believe that the high capacity of the first discharge process derives mainly from the large number of cavities in the one-dimensional structure. After the initial discharge, a large amount of aluminum ions could be stuck in cavities and not completely deintercalated during the subsequent charging process. Considering the high charge density of aluminum ions, both intercalation and deintercalation may cause structural damage. As the number of charges and discharges increases, this phenomenon intensifies, reflected in a sharp decrease in specific capacity. Then, compared with the other three compounds, the chain structures of the Fe–Ni and Fe–Co compounds are even more unstable, as they cannot even withstand the insertion of large amounts of aluminum ions when discharging at the potential window of 0–1.8 V. So, the potential window between 0.4–1.8 V with increased cutoff voltage was applied on Fe–Ni and Fe–Co compounds to limit structural damage or evolution of the cathode materials.
It is worth noting that the first discharge plateau on the GCD curves of all compounds appears at approximately 1.4 V, which may correspond to the redox reaction of Fe3+ that is common to all the compounds. The variability in the subsequent discharge curves is due to the different nature of the compounds. The excellent first-cycle discharge specific capacities obtained, accompanied by a large discharge plateau near 1.4 V, especially for Fe–Co and Fe–Zn compounds suggests a favourable aluminum ion intercalation in this process. Nonetheless, the severe specific capacity deterioration of the second cycle suggests that this process is a one-time event, as poor cycling stability resulting from structural damage or collapse occurs. To limit the material damage, the cut-off voltage was reduced, aiming to increase the reversibility of electrochemical intercalation and deintercalation at the expense of the first-cycle discharge specific capacity, thereby improving cycling stability.
The galvanostatic charge–discharge curves of these compounds in the voltage range of 1–1.8 V at a current of 10 mAg−1 are shown in the right column of Fig. 5. The discharge curves for Fe–Ni and Fe–Mn show a consistent discharge plateau at 1.4 V, aligning with previous tests. The first-cycle discharge capacity of Fe–Ni does not change significantly because there is no obvious redox reaction of Fe–Ni within the potential window of 0.4 V–1 V. For Fe–Mn electrode, as the cut-off voltage increases to 1 V, the specific capacity drops to 18 mA h g−1, and the capacity retention rate referring to that of the second cycle increases from 48% to 61%. In Fig. 5(d) and (h), Fe–Co and Fe–Zn exhibit similar discharge behavior with two plateaus at 1.4 V and 1.2 V, respectively. Due to the increase in cut-off voltage, both Fe–Co and Fe–Zn lose the long structure-damaging redox process at lower voltages, meanwhile their specific capacities decrease from as high as about 200 mA h g−1 to 25 mA h g−1 and 13 mA h g−1 respectively. The discharge curve in the 1–1.8 V voltage range of the Fe–Cu electrode shown in Fig. 5(j) also displays two discharge plateaus as in the previous test. Although its first-cycle specific capacity is reduced to 49 mA h g−1, its second-cycle specific capacity is close to the previous one, which means that the capacity retention rate was improved.
The first-cycle and second-cycle specific capacities in different voltage windows and the corresponding capacity retention of all compounds are listed in Table 2. It is obvious that increasing cut-off voltage reduces the specific capacities of these compounds, while the capacity retention rates are improved (except for the Fe–Co). In addition, in the voltage window of 1.0–1.8 V, Fe–Cu displayed the maximum capacity of 49 mA h g−1 among these compounds whereas the capacity of Fe–Co is as high as 200 mA h g−1 in the previous situation. Considering their capacity retentions, Fe–Zn exhibited the most stable structure while the stability of Fe–Co was the worst. Yet, even so, the relatively high irreversible capacities in the first discharge process and the much lower reversible capacities in the following electrochemical cycles make them not impressive for application in reversible aluminum storage when compared with cathode materials for AIBs with good performance reported so far. For example, Guo et al. investigated a 2D copper-based MOF and optimized it by in situ coating reduction graphene oxide (rGO). Then, the Al–MOF battery based on [EMIm]Cl–AlCl3 electrolyte is constructed with this optimized MOF as the cathode, delivering a high reversible capacity (184 mA h g−1 at 50 mA g−1), and excellent cycling stability (beyond 1000 cycles).49 In addition, before further investigating the Al3+ ions trapped in cavities causing capacity decay, we conducted the “Compounds Stability Investigation” section and found issues with Al3+ ions being trapped within structures of compounds and interactions with the electrolyte leading to their dissolution. This structural deterioration during their electrochemical reactions complicates quantitative analysis of the trapped Al3+ ions, as some may return to the electrolyte.
| Q Theoretical (Fe3+/Fe2+) | 0–1.8 V/0.4–1.8 V | 1.0–1.8 V | |||||
|---|---|---|---|---|---|---|---|
| 1st cycle (mA h g−1) | 2nd cycle (mA h g−1) | Capacity retention (%) | 1st cycle (mA h g−1) | 2nd cycle (mA h g−1) | Capacity retention (%) | ||
| {Fe(Tp)(CN)3}2{Ni(H2O)2} | 31.13 | 29 | 6 | 21 | 30 | 8 | 27 |
| {Fe(Tp)(CN)3}2{Co(H2O)2} | 31.32 | 200 | 85 | 43 | 25 | 6 | 24 |
| {Fe(Tp)(CN)3}4 {Mn(H2O)2Mn} | 30.21 | 44 | 21 | 48 | 18 | 11 | 61 |
| {Fe(Tp)(CN)3}4 {Zn(H2O)2Zn} | 29.20 | 196 | 15 | 8 | 13 | 9 | 69 |
| {Fe(Tp)(CN)3}2{Cu(DMF)} | 34.48 | 116 | 24 | 21 | 49 | 22 | 45 |
3.3. Compounds stability investigation
So far, the substantial performance degradation of compounds. during the electrochemical charge and discharge process has led us to realize that the damage and collapse of the structure may be caused by the insertion and deintercalation of ions with high charge density. However, the role played by the Lewis acidic ionic liquid electrolyte remains to be investigated. Therefore, open circuit voltage (OCV) was monitored over time to study the effect of the electrolyte on compounds and to anticipate the active materials’ behavior and their possible evolution when inserted in a battery device. Cyclic voltammetry (CV) curves recorded after some delays were also measured and compared to those described above and obtained straight after battery preparation in the voltage window of 1–1.8 V.The OCV curve of the cell assembled with the Fe–Co compound as the electrode material exhibits the worst stability in the GCD test (1–1.8 V) as presented in Fig. 6. It displays a downward trend over time, falling sharply from 1.5 V down to 0 V within five hours. The initial CV curve (in red) exhibits clear redox peaks between 1.4 V and 1.5 V, which match well with the position of the first plateau in the GCD curve (Fig. 5c). After 24 h, the open circuit voltage of the assembled cell experienced a sharp decline (blue dot in Fig. 6), and the voltage after 72 h was close to 0 V. When CV curves collected at different time delays are compared, the obvious decrease of the peak area and loss of redox peaks are revealed. This is consistent with the performance of the Fe–Co electrode in the galvanostatic charge–discharge test which shows a strong capacity decrease and an evolution of the material when applied to the battery device.
 | ||
| Fig. 6 Fe–Co product: Open circuit voltage (OCV) and aggregate curves of CVs with different delay times. | ||
The Fe–Zn compound which shows the most stable performance in galvanostatic charge–discharge test in 1–1.8 V also shows a time dependent OCV indicating an evolution of the materials once assembled in battery devices (Fig. 7). Actually, its open circuit voltage first decreases before rising to a maximum value near 24 h, and then it decreases and stabilizes at around 1.53 V. Although there is an upward and downward trend throughout the process, the open circuit voltage is controlled between 1.5–1.6 V. All CV curves collected with delay times of 0 h, 24 h, and 72 h have obvious redox peaks, indicating that this compound retains good redox ability after 72 hours, which is in contrast to the CV curve of Fe–Co after 72 hours in Fig. 6. However, the covering area of the CV curve in yellow is much smaller than the red one, demonstrating that even the Fe–Zn electrode delaying after 72 h is capable of redox reaction, it has been changed when in contact with the electrolyte for 72 h.
 | ||
| Fig. 7 Fe–Zn product: Open circuit voltage (OCV) and aggregate curves of CVs with different delay times. | ||
Other compounds including Fe–Ni, Fe–Mn and Fe–Cu suffer from the same evolution when in contact with the electrolyte for a period, as shown in Fig. S9, S10 and S11,† respectively. The OCV curves of Fe–Ni and Fe–Cu electrodes have experienced the same process of falling, rising and falling again like that of the Fe–Zn electrode. The Fe–Ni retains a downward trend even after 146 hours while Fe–Cu, similar to Fe–Zn, soon stabilizes between 1.5 and 1.6 V. Regarding the CV curve of Fe–Ni in 1.0–1.8 V, it has almost lost redox ability after 24 hours of soaking with the electrolyte, as there are no redox peaks in corresponding CV curve. The CV curves of the Fe–Cu electrode delayed for 24 h and 72 h only reserved narrow areas. Therefore, we believe that a large number of aluminum ions were trapped in the structural framework of the Fe–Cu compound, resulting in the loss of redox ability. In Fig. S10,† the open circuit voltage of Fe–Mn keeps rising until 168 h, but the CV curve collected at 72 hours of the rising process no longer has a redox peak. Without exception, Fe–Mn changed within 72 hours of interacting with the electrolyte, showing very unstable electrochemical properties.
Overall, not only do the intercalation and deintercalation of ions lead to structural damage and collapse, but these compounds also suffer from the interaction of the ionic liquid electrolyte over a very short period of time. Firstly, among them, Fe–Ni and Fe–Co are relatively more reactive, which is reflected in the inability to discharge to 0 V in the galvanostatic charge–discharge test, and the loss of the redox peaks in the 24 h-delayed curves in the cyclic voltammetry test. Then, in the voltage range of 1.0–1.8 V, Fe–Cu delivered the highest first-cycle discharge capacity. This might be correlated to the presence of bulkier coordinated DMF on the divalent ion in the Fe–Cu compound, which leads to an increase of the spacing between neighboring chains.37 This could lead to a better Al-ion insertion and a higher specific capacity. The capacity retention of Fe–Zn in the voltage range of 1.0–1.8 V indicates its better stability compared to other compounds, although the specific capacity is low. It may result from the relatively stable valence state of Zn2+. In addition, it is worth mentioning that although these compounds are coordinated with different divalent metals, the redox peaks of their initial state CV curves in the 1.0–1.8 V potential window are all located near 1.4 V. Therefore, we believe that the process of storing ions in these compounds mainly relies on the Fe3+/Fe2+ redox reaction. The shift of the redox peak positions and the difference of specific capacities between these compounds result from the influence of another metal in each compound.
In order to explore the stability of the one-dimensional compounds in the ionic liquid electrolyte, we dissolved selected compounds in the electrolyte, and then we performed EPR and FT-IR measurements on both dissolved and bulk compounds for comparison.
For EPR, we used a diluted mixture compared with the concentration used in batteries (where 100 uL of the electrolyte is used for ca. 1 mg of active material) to avoid dipolar interaction between molecules, and the Fe–Zn and Fe–Cu compounds were selected to facilitate the interpretation of the EPR spectra. On the one hand, the Zn2+ ion is EPR-silent in the Fe–Zn compound and we thus expected to observe recognizable signals of the anisotropic low-spin Fe(III) ions. On the other hand, the EPR spectra of Cu2+ compounds are well known in the literature and easily trackable.
Fig. 8 shows the EPR spectra of powder (a) and dissolved Fe–Zn (b) and powder (c) and dissolved Fe–Cu (d) at 4 K. The powder spectrum of Fe–Zn (Fig. 8a) is typically that of low-spin Fe(III), exhibiting a rhombic electronic structure with g1 = 3.03, g2 = 1.98 and g3 = 1.55.50 In contrast, the spectrum of dissolved 1D Fe–Zn (Fig. 8b) presents a narrow peak with g = 2.00 and several weak peaks with g ![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 4.29. This clearly accounts for a decomposition of the [FeIII(Tp)(CN)3] subunits According to the literature, the high g value (g 4.29) could account for the occurrence of rhombic high spin Fe(III) ions, while the signal g = 2 could be attributed to the presence of iron coordination clusters containing coupled Fe(III) ions.51–56 The powder spectrum of the pristine Fe–Cu compound (Fig. 8c) shows a typical signal of an integer-spin species resulting from the magnetic exchange interaction between low-spin Fe(III) (S = ½) and Cu(II) (S = ½) integrated into the chain.57 In contrast, the spectrum of the dissolved Fe–Cu material (Fig. 8d) seems to be the superposition of two signals: (i) a signal at g = 2.00 similar to that observed in Fe–Zn suggests again a decomposition of the Fe(III) subunit and (ii) a signal with g1 = 2.06, g2 = 2.04 and g3 = 2.02 typical of rhombic Cu(II) ions which is also coherent with a material dissociation. In particular, the hyperfine interactions of the Cu(II) ions are clearly visible on the gX component of the spectrum. Overall, the EPR spectra indicate that both chains are dissociated in the electrolyte into individual metal complexes that can then evolve.
4.29. This clearly accounts for a decomposition of the [FeIII(Tp)(CN)3] subunits According to the literature, the high g value (g 4.29) could account for the occurrence of rhombic high spin Fe(III) ions, while the signal g = 2 could be attributed to the presence of iron coordination clusters containing coupled Fe(III) ions.51–56 The powder spectrum of the pristine Fe–Cu compound (Fig. 8c) shows a typical signal of an integer-spin species resulting from the magnetic exchange interaction between low-spin Fe(III) (S = ½) and Cu(II) (S = ½) integrated into the chain.57 In contrast, the spectrum of the dissolved Fe–Cu material (Fig. 8d) seems to be the superposition of two signals: (i) a signal at g = 2.00 similar to that observed in Fe–Zn suggests again a decomposition of the Fe(III) subunit and (ii) a signal with g1 = 2.06, g2 = 2.04 and g3 = 2.02 typical of rhombic Cu(II) ions which is also coherent with a material dissociation. In particular, the hyperfine interactions of the Cu(II) ions are clearly visible on the gX component of the spectrum. Overall, the EPR spectra indicate that both chains are dissociated in the electrolyte into individual metal complexes that can then evolve.
 | ||
| Fig. 8 EPR spectrum of samples of the pristine compounds Fe–Zn (a) and Fe–Cu (c) and their mixture with the-electrolyte (respectively (b) and (d)) at 4 K. | ||
IR spectroscopy provides another view of the instability of these compounds in the electrolyte. In this case, the powder sample of the vacuum heated Fe–Ni compound was immersed in the electrolyte and the amount of active material is much higher in this experiment than in the battery set-up. After 24 hours, the compounds seemed to dissolve, and then a mixture of the dissolved sample and electrolyte in the slurry state was obtained and probed by FT-IR spectroscopy. Surprisingly, almost all traces of organic component disappeared, and only weak cyanide vibrations could be observed as shown in Fig. 9. Three weak stretching vibrations were thus observed at 2193, 2134 and 2090 cm−1 in the Fe–Ni sample (which do not appear in the spectroscopy results for the pure electrolyte). The peaks at 2134 and 2090 cm−1 clearly point to a partial reduction of the Fe(III) to Fe(II) in the material, while the one at a higher wavenumber could be tentatively assigned to the formation of the FeIII–CN–AlIII bridge, as shown in the zoomed-in images on the right side of Fig. 9(b) and (c).
 | ||
| Fig. 9 Infrared spectra of pure electrolyte, sample after vacuum heating (noted as VH) at 100 °C and mixture of electrolyte and samples after vacuum heating: (a) electrolyte; (b and c) Fe–Ni; and the (d and e) (NBu4)[Fe(Tp)(CN)3] precursor. | ||
Furthermore, using the same protocol, a large amount of precursor (NBu4)[Fe(Tp)(CN)3] was mixed with the electrolyte and then the FT-IR spectrum was recorded (Fig. 9(e) and (f)). Although the fingerprint region corresponding to the electrolyte and precursor mixture is consistent with that of the electrolyte, a new stretching vibration appears at 2190 cm−1. As mentioned above, this vibration probably accounts for the occurrence of new FeIII–CN–AlIII bridging units. Actually, when the terminal cyano-group is metal-coordinated, its v(CN) stretching frequency will shift to higher wavenumber.58 Note that similar stretching vibrations for the FeIII–CN–AlIII unit were also reported by Bertrán et al. at ca. 2185 cm−1.59 In summary, the chain compounds when interacting with the electrolyte probably suffered from competitive coordination of the cyanide with Al3+ cations, partial reduction of Fe(III) to Fe(II), and some decomposition.
4. Conclusion
In this work, we investigated the Al intercalation properties in a series of related cyanido-bridged chain compounds coordinated with different divalent metals (Ni, Co, Mn, Zn, Cu). We showed that these low dimensional counterparts of the well-known Prussian blue analogues have the capability of accommodating aluminium ions when using an ionic liquid electrolyte and Al metal as the counter electrode. Interestingly, these compounds exhibited identical voltage discharge plateaus in GCD curves and consistent redox peak positions in CV curves, suggesting a unified redox-active site at 1.4 V, attributed to the redox couple Fe3+/Fe2+. Besides, a maximum first-cycle capacity of about 200 mA h g−1 was observed for the Fe–Co compound in the potential window of 0–1.8 V. However, this capacity falls quickly in the following cycles. The thermal and electrochemical stabilities of all these compounds were investigated and compared, for better understanding the differences in structural and electrochemical behaviours caused by different bridging divalent metals. Although the Fe–Zn compound appears relatively stable thermally and electrochemically among these compounds, the study shows that a high first-cycle discharge specific capacity obtained by a large potential window (0–1.8 V) and a good capacity retention obtained by increasing the cut-off voltage (1–1.8 V) are incompatible. Furthermore, we studied the impact of ionic liquid electrolytes on the compounds and revealed that the anchorage of intercalated aluminum ions to cyanido groups could occur. Thus, the formation of the new cyanide bridge (FeIII–CN–AlIII) may create some resistance to the deintercalation of aluminum ions. This gives us some inspiration for exploring possibly more suitable Al-ion host candidates such as higher dimensional materials (2D and 3D) with more stable organic ligands, due to the higher stability of materials with higher dimensions and the consideration of avoiding potential overly tight binding of Al3+ to donor sites.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support provided by the Centre National de la Recherche Scientifique (CNRS), Sorbonne University, and the China Scholarship Council (CSC).References
- N. Imanishi, T. Morikawa, J. Kondo, Y. Takeda, O. Yamamoto, N. Kinugasa and T. Yamagishi, J. Power Sources, 1999, 79, 215–219 Search PubMed.
- Y. Xu, S. Zheng, H. Tang, X. Guo, H. Xue and H. Pang, Energy Storage Mater., 2017, 9, 11–30 Search PubMed.
- Q. Wang, J. Li, H. Jin, S. Xin and H. Gao, InfoMat, 2022, 4, e12311 Search PubMed.
- B. Singh and A. Indra, Mater. Today Energy, 2020, 16, 100404 Search PubMed.
- J. Qian, C. Wu, Y. Cao, Z. Ma, Y. Huang, X. Ai and H. Yang, Adv. Energy Mater., 2018, 8, 1702619 Search PubMed.
- J. Long, D. Asakura, M. Okubo, A. Yamada, Y. Guari and J. Larionova, Inorg. Chem., 2016, 55, 7637–7646 Search PubMed.
- M. Okubo, J. Long, D. R. Talham and R. Lescouëzec, C. R. Chim., 2019, 22, 483–489 Search PubMed.
- R. Lescouëzec, J. Vaissermann, F. Lloret, M. Julve and M. Verdaguer, Inorg. Chem., 2002, 41, 5943–5945 Search PubMed.
- T. Shiga, N. Mihara and M. Nihei, Coord. Chem. Rev., 2022, 472, 214763 Search PubMed.
- J.-R. Jiménez, A. Sugahara, M. Okubo, A. Yamada, L.-M. Chamoreau, L. Lisnard and R. Lescouëzec, Chem. Commun., 2018, 54, 5189–5192 Search PubMed.
- P. Wang, H. Chen, N. Li, X. Zhang, S. Jiao, W.-L. Song and D. Fang, Energy Storage Mater., 2018, 13, 103–111 Search PubMed.
- G. L. Holleck, J. Electrochem. Soc., 1972, 119, 1158 Search PubMed.
- D.-Y. Wang, C.-Y. Wei, M.-C. Lin, C.-J. Pan, H.-L. Chou, H.-A. Chen, M. Gong, Y. Wu, C. Yuan, M. Angell, Y.-J. Hsieh, Y.-H. Chen, C.-Y. Wen, C.-W. Chen, B.-J. Hwang, C.-C. Chen and H. Dai, Nat. Commun., 2017, 8, 14283 Search PubMed.
- N. P. Stadie, S. Wang, K. V. Kravchyk and M. V. Kovalenko, ACS Nano, 2017, 11, 1911–1919 Search PubMed.
- Z. Liu, J. Wang, H. Ding, S. Chen, X. Yu and B. Lu, ACS Nano, 2018, 12, 8456–8466 Search PubMed.
- K. V. Kravchyk, S. Wang, L. Piveteau and M. V. Kovalenko, Chem. Mater., 2017, 29, 4484–4492 Search PubMed.
- G. A. Elia, I. Hasa, G. Greco, T. Diemant, K. Marquardt, K. Hoeppner, R. J. Behm, A. Hoell, S. Passerini and R. Hahn, J. Mater. Chem. A, 2017, 5, 9682–9690 Search PubMed.
- X. Yu and A. Manthiram, Adv. Energy Mater., 2017, 7, 1700561 Search PubMed.
- T. Gao, X. Li, X. Wang, J. Hu, F. Han, X. Fan, L. Suo, A. J. Pearse, S. B. Lee, G. W. Rubloff, K. J. Gaskell, M. Noked and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 9898–9901 Search PubMed.
- G. Cohn, L. Ma and L. A. Archer, J. Power Sources, 2015, 283, 416–422 Search PubMed.
- H. Wang, Y. Bai, S. Chen, X. Luo, C. Wu, F. Wu, J. Lu and K. Amine, ACS Appl. Mater. Interfaces, 2015, 7, 80–84 Search PubMed.
- N. Jayaprakash, S. K. Das and L. A. Archer, Chem. Commun., 2011, 47, 12610 Search PubMed.
- M. Chiku, H. Takeda, S. Matsumura, E. Higuchi and H. Inoue, ACS Appl. Mater. Interfaces, 2015, 7, 24385–24389 Search PubMed.
- Y. Liu, S. Sang, Q. Wu, Z. Lu, K. Liu and H. Liu, Electrochim. Acta, 2014, 143, 340–346 Search PubMed.
- S. Liu, J. J. Hu, N. F. Yan, G. L. Pan, G. R. Li and X. P. Gao, Energy Environ. Sci., 2012, 5, 9743 Search PubMed.
- Z. Li, B. Niu, J. Liu, J. Li and F. Kang, ACS Appl. Mater. Interfaces, 2018, 10, 9451–9459 Search PubMed.
- L. Geng, J. Scheifers, C. Fu, J. Zhang, B. P. T. Fokwa and J. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 21251–21257 Search PubMed.
- Y. Ru, S. Zheng, H. Xue and H. Pang, Chem. Eng. J., 2020, 382, 122853 Search PubMed.
- L. D. Reed, S. N. Ortiz, M. Xiong and E. J. Menke, Chem. Commun., 2015, 51, 14397–14400 Search PubMed.
- S. Liu, G. L. Pan, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2015, 3, 959–962 Search PubMed.
- A. Holland, R. D. Mckerracher, A. Cruden and R. G. A. Wills, J. Appl. Electrochem., 2018, 48, 243–250 Search PubMed.
- Z. Li, K. Xiang, W. Xing, W. C. Carter and Y. Chiang, Adv. Energy Mater., 2015, 5, 1401410 Search PubMed.
- W. Zhou, M. Zhang, X. Kong, W. Huang and Q. Zhang, Adv. Sci., 2021, 8, 2004490 Search PubMed.
- B. Craig, T. Schoetz, A. Cruden and C. Ponce De Leon, Renewable Sustainable Energy Rev., 2020, 133, 110100 Search PubMed.
- Y.-J. Zhang, T. Liu, S. Kanegawa and O. Sato, J. Am. Chem. Soc., 2009, 131, 7942–7943 Search PubMed.
- H.-R. Wen, C.-F. Wang, Y. Song, S. Gao, J.-L. Zuo and X.-Z. You, Inorg. Chem., 2006, 45, 8942–8949 Search PubMed.
- J. Montero, D. Arenas-Esteban, D. Ávila-Brande, E. Castillo-Martínez, S. Licoccia and J. Carretero-González, Electrochim. Acta, 2020, 341, 136063 Search PubMed.
- H. Niwa, T. Moriya, T. Shibata, Y. Fukuzumi and Y. Moritomo, Sci. Rep., 2021, 11, 4119 Search PubMed.
- A. Mondal, Y. Li, M. Seuleiman, M. Julve, L. Toupet, M. Buron-Le Cointe and R. Lescouëzec, J. Am. Chem. Soc., 2013, 135, 1653–1656 Search PubMed.
- V. Costa, R. Lescouëzec, J. Vaissermann, P. Herson, Y. Journaux, M. H. Araujo, J. M. Clemente-Juan, F. Lloret and M. Julve, Inorg. Chim. Acta, 2008, 361, 3912–3918 Search PubMed.
- O. Kahn, MOLECULAR MAGNETISM, VCH Publishers, Weinheim, 1993 Search PubMed.
- D.-P. Dong, Y.-J. Zhang, H. Zheng, P.-F. Zhuang, L. Zhao, Y. Xu, J. Hu, T. Liu and C.-Y. Duan, Dalton Trans., 2013, 42, 7693 Search PubMed.
- L. M. Toma, C. Ruiz-Pérez, J. Pasán, W. Wernsdorfer, F. Lloret and M. Julve, J. Am. Chem. Soc., 2012, 134, 15265–15268 Search PubMed.
- C. Mathonière, D. Mitcov, E. Koumousi, D. Amorin-Rosario, P. Dechambenoit, S. Fatima Jafri, P. Sainctavit, C. Cartier Dit Moulin, L. Toupet, E. Trzop, E. Collet, M.-A. Arrio, A. Rogalev, F. Wilhelm and R. Clérac, Chem. Commun., 2022, 58, 12098–12101 Search PubMed.
- C. Mathonière, Eur. J. Inorg. Chem., 2018, 2018, 248–258 Search PubMed.
- T. Schoetz, O. Leung, C. P. De Leon, C. Zaleski and I. Efimov, J. Electrochem. Soc., 2020, 167, 040516 Search PubMed.
- F. Wu, H. Yang, Y. Bai and C. Wu, Adv. Mater., 2019, 31, 1806510 Search PubMed.
- C. Li, C.-C. Hou, L. Chen, S. Kaskel and Q. Xu, EnergyChem, 2021, 3, 100049 Search PubMed.
- Y. Guo, W. Wang, H. Lei, M. Wang and S. Jiao, Adv. Mater., 2022, 34, 2110109 Search PubMed.
- I. Rutkowska, K. Dzilinski, T. Kaczmarzyk and J. Stanek, Nukleonika, 2013, 58, 419–423 Search PubMed.
- P. Fejes, I. Kiricsi, K. Lázár, I. Marsi, A. Rockenbauer and L. Korecz, Appl. Catal., A, 2003, 252, 75–90 Search PubMed.
- T. Balić, Z. Jagličić, E. Sadrollah, F. Jochen Litterst, M. Počkaj, D. Baabe, E. Kovač-Andrić, J. Bijelić, D. Gašo-Sokač and I. Djerdj, Inorg. Chim. Acta, 2021, 520, 120292 Search PubMed.
- R. Naik, S. C. Prashantha, H. Nagabhushana and K. M. Girish, J. Lumin., 2018, 197, 233–241 Search PubMed.
- V. Vercamer, G. Lelong, H. Hijiya, Y. Kondo, L. Galoisy and G. Calas, J. Non-Cryst. Solids, 2015, 428, 138–145 Search PubMed.
- P. Fejes, Appl. Catal., A, 2003, 252, 75–90 Search PubMed.
- S. S. Mal, M. H. Dickman, U. Kortz, A. M. Todea, A. Merca, H. Bögge, T. Glaser, A. Müller, S. Nellutla, N. Kaur, J. van Tol, N. S. Dalal, B. Keita and L. Nadjo, Chem. - Eur. J., 2008, 14, 1186–1195 Search PubMed.
- E. Garribba, G. Micera, D. Sanna and L. Strinna-Erre, Inorg. Chim. Acta, 2000, 299, 253–261 Search PubMed.
- D. Karaağaç, Bull. Chem. Soc. Ethiop., 2020, 34, 365–376 Search PubMed.
- J. F. Bertrán, J. B. Pascual and E. R. Ruiz, Spectrochim. Acta, Part A, 1990, 46, 685–689 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01316f |
| This journal is © The Royal Society of Chemistry 2024 |