
A closer look at molecular mechanisms underlying inhibition of S-adenosyl-L-homocysteine hydrolase by transition metal cations†
Magdalena
Gawel‡
a,
Piotr H.
Malecki‡
a,
Joanna
Sliwiak
a,
Marlena
Stepniewska
a,
Barbara
Imiolczyk
a,
Justyna
Czyrko-Horczak
b,
Dorota
Jakubczyk
a,
Łukasz
Marczak
a,
Marta Eliza
Plonska-Brzezinska
 *c and
Krzysztof
Brzezinski
*a
*c and
Krzysztof
Brzezinski
*a
aInstitute of Bioorganic Chemistry, Polish Academy of Sciences, Noskowskiego 12/14, 61-704 Poznan, Poland. E-mail: kbrzezinski@ibch.poznan.pl
bProvincial Sanitary-Epidemiological Station in Bialystok, Legionowa 8, 15-099, Bialystok, Poland
cMedical University of Bialystok, Department of Organic Chemistry, A. Mickiewicza 2a, 15-222 Bialystok, Poland. E-mail: marta.plonska-brzezinska@umb.edu.pl
First published on 16th August 2024
Abstract
We report biochemical and structural studies on inhibiting bacterial S-adenosyl-L-homocysteine hydrolase by transition metal cations. Our results revealed diverse molecular mechanisms of enzyme inactivation. Depending on the cation, the mechanism is based on arresting the enzyme in its closed, inactive conformation, disulfide bond formation within the active site or oxidation of the intermediate form of a cofactor.
The S-adenosyl-L-methionine (SAM)-dependent methylation reactions are crucial for numerous metabolic processes in all living organisms.1 A by-product of these reactions is S-adenosyl-L-homocysteine (SAH), a potent inhibitor of SAM-dependent methyltransferases. Therefore, SAH must be promptly removed, as its accumulation suppresses all SAM-dependent methylations.2 In numerous organisms, including humans and pathogenic microorganisms, SAH is decomposed to L-homocysteine (Hcy) and adenosine (Ado) only by SAH hydrolase (SAHase). Thus, SAHase is a central regulator of cellular SAM-dependent methylations.3,4 SAHases derived from a variety of organisms have been structurally characterized.5,6 The enzyme's subunit is folded into three domains: the substrate- and NAD+ cofactor binding domains and a smaller C-terminal oligomerization domain. It is worth noting that NAD+-dependent redox steps are required for SAH decomposition. The first two domains are connected by two hinges; one is the alkali metal coordination site (preferably K+, which supports the most efficient catalysis),7 with a bearing on the domain movement essential for the catalytic cycle, during which the enzyme oscillates between open (SAH or Ado and K+ free) and closed (SAH or Ado and K+ bound) states. In addition, the potent inhibitory effect of Zn2+ cations on SAHase dynamics was also revealed.7 However, the biological role of this interaction in a cell metabolism remains unknown. Also, it was suggested that the activity of mammalian SAHase could be abolished by NAD+ cofactor release caused by Cu2+ ions.8
Transition metal cations and their complexes have attracted considerable attention in medicine and drug development as they interact and affect various biological molecules.9,10 They are considered promising therapeutic agents in cancer treatment and infectious diseases.11–13 The inhibition of SAHase by the cations has broad implications for pathogenic microorganisms' vital and virulence processes. Understanding the mechanisms of cation-based SAHase inhibition could accelerate developing novel antimicrobial compounds.14
At first, we analysed the influence of biologically relevant transition metal cations on the activity of SAHase from P. aeruginosa (Fig. S1, ESI†). Finally, we determined that Fe2+, Ni2+, Co2+, Cu+, Cu2+, Zn2+, Cd2+ and Hg2+ affected the enzyme's activity. Of note, we skipped further experiments with Fe2+, as rapid protein precipitation and crystal damaging were observed. The enzyme's inhibition studies revealed that the tested cations undoubtedly affect PaSAHase activity with diverse impacts (Fig. 1A and Fig. S2, Table S1, ESI†). The strongest influence is observed for Cd2+, as shown by an inhibitor constant Ki of 5.00 ± 0.28 nM. Zn2+, Hg2+, Cu2+ and Cu+, were potent or mild inhibitors of PaSAHase with Ki values ranging from 85.00 ± 3.27 nM to 545 ± 7.57 nM. Co2+ and Ni2+ were weak or very weak inhibitors with Ki values 63.64 ± 3.97 μM and 472.78 ± 38.52 μM, respectively.
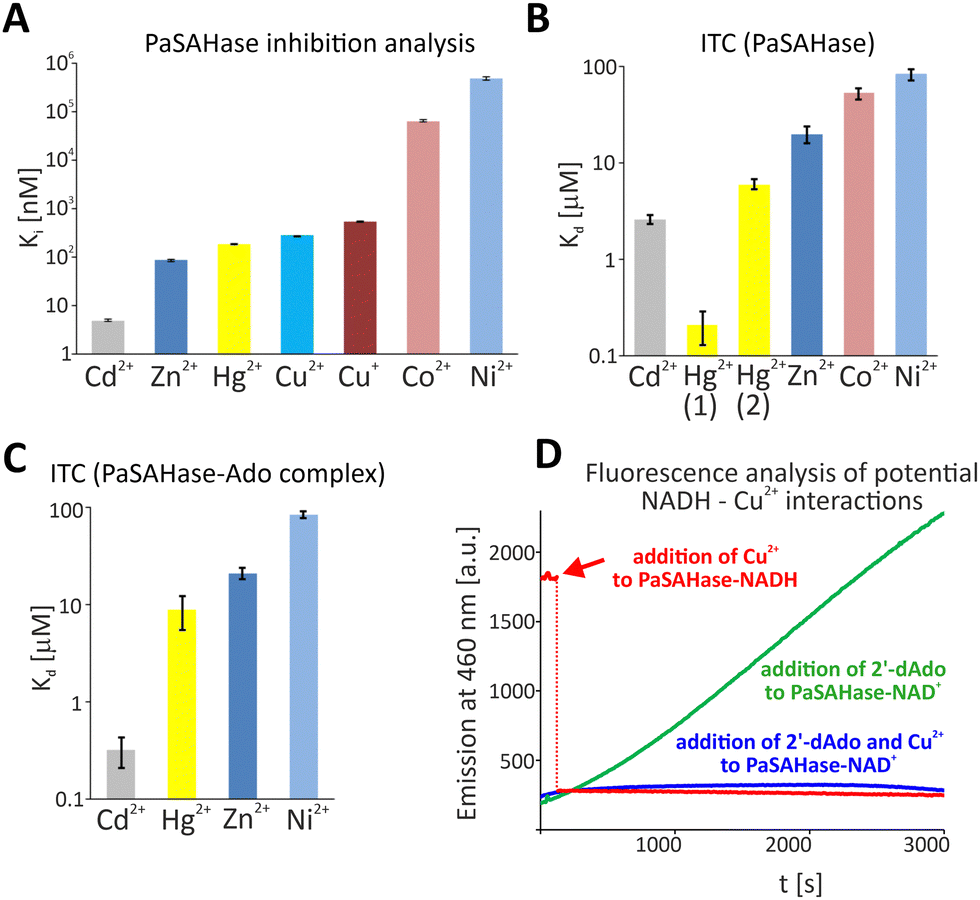 | ||
| Fig. 1 The influence and interactions of transition metal cations with PaSAHase. (A) Enzyme inhibition studies were conducted for the enzyme in the presence of Cd2+, Zn2+, Hg2+, Cu2+, Cu+, Co2+ and Ni2+. The activity was measured at variable concentrations of the cations to determine the IC50 constants, subsequently converted to the presented inhibition constants, Ki, shown on the logarithmic scale. Calorimetric titrations of PaSAHase in its (B) open and (C) closed conformations with the same set of cations, excluding Cu+. The results are presented on a logarithmic scale as dissociation constants, Kd. (D) Changes in the oxidation state of the cofactor initiated by Cu2+ were observed spectrofluorimetrically. For (A) and (B), all measurements were performed at 293 K in duplicates. Bars represent standard errors of the mean values. | ||
Next, we characterized cations-PaSAHase interactions by employing Isothermal Titration Calorimetry (ITC) for the enzyme in closed (PaSAHase-Ado complex) and open (ligand-free enzyme) conformations. However, this excluded Cu+, which is capable of disproportionation to Cu2+ and Cu in solution and may affect the apparatus. Binding parameters obtained from calorimetric titrations of PaSAHase with different divalent cations are summarized in Fig. 1B, C and Fig. S3, Table S2 (ESI†). Surprisingly, although Cu2+ strongly inhibits PaSAHase activity, we did not observe any measurable heat effect for Cu2+ titrations of both open and closed conformations of PaSAHase. The conformation state of the enzyme affected the binding the most in the case of Cd2+, as one could observe that the presence of adenosine strengthened the binding of Cd2+ by ∼8 times (dissociation constants, Kd, equal to 2.6 ± 0.3 or 0.32 ± 0.11 μM for the enzyme in the open or closed conformation, respectively). This tendency could also be observed for the binding of Hg2+, if a model assuming existence of one set of equivalent binding sites was fitted to the obtained raw data. However, in the case of titration of ligand-free PaSAHase with Hg2+, this model did not describe the best-obtained curve, and the model assuming existence of two unequal sets of binding sites could be applied successfully here with a better fit, which indicated that there is one set of Hg2+ binding sites of high affinity (Kd1 ∼ 0.2 μM) and the second set of binding sites of lower affinity (Kd2 ∼ 6 μM). Notably, only the one set of binding sites model could be fitted to data obtained for the titration of PaSAHase-Ado with Hg2+. For the other cations, the Kd values are similar within their errors for both forms of the protein. The obtained binding affinities are ranked as follows: Cd2+ > Hg2+ > Zn2+ > Co2+ > Ni2+.
Binding of a ligand, such as a cation, changes protein thermal stability.15 Thus, we have performed thermal shift assays (TSA) to evaluate the impact of the cations on protein-M2+ stability in both open and closed conformational states (Fig. S4, ESI†). Two ions, Ni2+ and Co2+, did not affect the enzyme's stability in either conformation. On the contrary, other cations usually increased the stability of the open and closed states of protein-metal complexes. However, the extent of the stabilization significantly depends on the protein's conformation. The open enzymes’ thermal stability was increased slightly by 2.5–3.5 °C. In addition, TSA performed in the presence of Cd2+ clearly showed two peaks indicating two effects. As the protein is composed of two principal domains, it indicates the stabilization of one by 3.5 °C and a destabilization by 7.5 °C. Thermal stabilization by the cations is usually more evident for the protein in closed conformation rather than the open, suggesting more rigidifying of the structure. Although some minor destabilization of the protein (by −0.5 °C) is observed in the presence of Hg2+, other cations increased thermal stability from 4.5 to 20.5 °C. For Cu+ and Cu2+, a thermal shift was 4.5 and 6.0 °C, respectively. On the other hand, this effect was spectacular for Cd2+ (increase by 10.5 °C) and Zn2+. Notably, two enzyme domains in the presence of the Zn2+ were thermally more stable by 12.5 and 20.5 °C.
Biochemical and biophysical analyses indicated that transition metal cations affect the function and stability of PaSAHase. However, a detailed mechanism(s) of their action remained elusive. Therefore, we determined the crystal structures of PaSAHase-Ado-M2+ complexes, as crystallization trials of Ado-free PaSAHase failed. Based on X-ray diffraction on protein crystals soaked with the cations, we explained their detailed inhibition mechanism(s). Crystallographic data and refinement statistics are presented in Table S3 (ESI†). From the whole set of investigated ions, Co2+, Zn2+, Hg2+ and Cd2+ were identified in crystal structures (Fig. 2A–L), and two types of M2+ binding sites were found. The first binding site is a specific one located at the interface between substrate- and cofactor-binding domains within the area of the active site entrance. The same site was observed previously for the PaSAHase-Ado-Zn2+ complex, where the low occupancy Zn2+ had been co-purified with the recombinant enzyme.7 Of note, the alkali metal cation coordination site located on the opposite pole of the active site is still occupied by K+ ion. In our crystal structures, each PaSAHase subunit contains one tetrahedrally coordinated Co2+ or Zn2+ ion or one octahedrally coordinated Cd2+ ion. The coordination sphere is formed by three highly conserved amino acid residues located at the entrance to the substrate-binding pocket, which involve one S (C85), one N (H323) and two O (D139) ligand atoms. The octahedral coordination sphere is usually completed by two water molecules. The coordination region is very similar in PaSAHase-Ado-M2+ complexes (Fig. 3A). Therefore, the stability of the complex should mainly depend on the ionic radius of a coordinated cation. In this scenario, lower occupancy of Co2+ in comparison to Zn2+ and Cd2+ most probably results from less effective interaction with the macromolecular environment as a consequence of its lower ionic radius (0.58 Å, coordination number IV) in comparison to Zn2+ (0.60 Å, coordination number IV) and Cd2+ 0.95 Å, coordination number (VI).16 Indeed, our studies indicated a significantly lower influence of Co2+ ions on binding to the enzyme and the enzyme's activity. A different coordination mode is observed for the complex with Hg2+ (ionic radius equals 0.96 Å, coordination number IV), where two possible neighbouring M2+ sites are partially occupied (from 25 to 42%). However, due to a significant disorder that could not be satisfactorily resolved, the geometry of the coordination sphere, where the cations interact with 3–4 ligand atoms, is ambiguous. To conclude, we observed a M2+ binding at the interface between two major domains. The consequence of the binding event is that it arrests the enzyme in its closed conformation, stopping the catalytic cycle. At the same time, it blocks the access to the active site.
 | ||
| Fig. 2 Structural studies of PaSAHase in the closed conformation in a complex with Co2+ (A)–(C), Zn2+ (D)–(F), Hg2+ (G)–(I) and Cd2+ (J)–(L). Each surface representation in two orientations shows particular cation binding sites spread on the protein surface and internal cavities. The black arrows indicate specific coordination sites within the active area. (C), (F), (I) and (L) show close-up views of the M2+ ions (spheres) and the corresponding 2mFo–DFc composite omit electron-density maps (green) contoured at 2σ (Co2+), 12σ (Zn2+), 6σ (Hg2+), 12σ (Cd2+). Possible polar interactions of the cation with macromolecular environments are indicated as black dashed lines. | ||
 | ||
| Fig. 3 Close-up views of the specific coordination site located within the active site area of PaSAHase. Superposition of the specific coordination sites of four PaSAHase crytal structures obtained in the presence of Co2+ (salmon), Zn2+ (pale blue), Cd2+ (yellow), Hg2+ (black) or Cu+ (blue) ions (A). Intramolecular disulfide bond formed in crystals soaked in tetrakis(acetonitrile)copper(I) tetrafluoroborate (B); the 2mFo–DFc composite omit electron density map, which covers disulfide bond formed between C59 and C85 residues is shown in forest green at 1.5σ level. | ||
The second type of M2+ coordination sites, non-specific ones are most probably crystallization artifacts. They coincide with various regions located on a protein surface, where numerous cations: 11 (Co2+), 12 (Zn2+), 16 (Hg2+) and 17 (Cd2+) are bound with partial occupancies and regular or distorted geometries. Despite this fact, the influence of Co2+ on the PaSAHase activity is minimal, supporting the hypothesis that non-specific sites are crystallization artifacts. Details of coordination modes for these specifically bound cations are shown in Table S4 (ESI†).
The remaining three other cations, Ni2+ and redox-active Cu+ and Cu2+ are not observed in the crystal structures of PaSAHase. In the light of our experiments, the lack of specific binding of Ni2+ ions and the modest influence on PaSAHase activity is not surprising and can be explained by the lowest ionic radius in comparison to other cations under investigation (0.55 Å, coordination number IV).16 The geometry of the specific coordination site of PaSAHase lessens its binding potential. Also, Cu+ ions have not been identified in the crystal structure despite the fact that, similarly to Zn2+, the ionic radius of Cu+ equals to 0.60 Å (coordination number IV)16 and tends to form complexes with tetrahedral geometry. However, the striking difference is observed in this crystal structure in the presence of Cu+ when compared to other structures of PaSAHase-Ado-M2+ complexes. In the active site area, two residues, C59 and C85, are oxidized and form an intramolecular disulphide bond within each subunit (Fig. 3B). Notably, the role of Cu+ in this process is supported by the fact that the cation is redox-active and no disulphide bond formation is observed in other structures with non-redox active M2+ ions. The disulfide bridge formation occurs within the substrate-binding domain, far from the interface between two major domains involved in enzyme dynamics. Thus, we speculate that the mechanism does not involve arresting the enzyme in its closed conformation. This finding is consistent with thermal shift assays, which indicated only a slight increase in thermal stability in the presence of Cu+. It is known that any modification of residue C85 located in the active site area might affect substrate binding. Our speculation is supported by a study on rat SAHase that had indicated that modifications (substitutions or disulfide bridge formation with other cysteine residues within the subunit) of C85 counterpart in the rat enzyme had affected its catalytic activity.17
A puzzling situation was noted in the case of Cu2+. TSA and ITC analyses revealed no specific binding of Cu2+. Also, no binding site for Cu2+ was identified in the crystal structure. Nevertheless, Cu2+ strongly inhibits PaSAHase activity. A possible explanation is that the lower ionic radius of the Cu2+, equal to 0.57 Å (coordination number IV) or 0.73 (coordination number VI)16 might be too small to form a stable complex with tetrahedral coordination geometry. Moreover, Cu2+ preferably forms complexes with square planar geometry instead of tetrahedral. Therefore, coordination of Cu2+ in the specific rigid binding site with four ligands is less probable. Also, Cu2+ is not complexed with octahedral coordination geometry, as observed for the PaSAHase-Ado-Cd2+ complex. The lower ionic radious of Cu2+ compared to Cd2+ (0.73 versus 0.78 Å, coordination number VI)16 might be one of the reasons for the absence of Cu2+ in the crystal structure. To elucidate a mechanism of inhibition, we performed additional experiments to explore if the redox properties of Cu2+ are related to the inhibition of PaSAHase. Firstly, we monitored spectrofluorimetrically time course inactivation of PaSAHase by 2′-deoxyadenosine (2′-dA), a SAHase inhibitor that deactivates the enzyme through the reduction of NAD+ to NADH (Fig. 1D).7 However, after the simultaneous addition of 2′-dA and Cu2+, no emission increase at 460 nm was detected in the course of time, suggesting that NADH is not formed under this condition. Secondly, we prepared the inactive variant of PaSAHase containing the reduced form of the cofactor, NADH, instead of NAD+, and analysed it spectrofluorimetrically. A strong emission at 460 nm was observed for such a sample. However, after adding Cu2+, the emission significantly dropped (Fig. 1D), indicating the oxidation of NADH. These two experiments revealed that the reduced form of the cofactor bound to the enzyme could be oxidized in a solution in the presence of Cu2+. In fact, NADH is formed at one of the initial steps of the catalytic reaction when NAD+ attracts the C3′–H hydride anion from the SAH to form 3′-keto-SAH.5 NAD+ is regenerated at the final step of the reaction during a transfer of hydride anion from NADH to 3′-keto-Ado to produce Ado. Therefore, any interaction of Cu2+ with NADH formed during the catalytic cycle could affect the enzyme's activity. Based on this assumption, we analysed the reaction products of SAH hydrolysis catalysed by PaSAHase in the absence or presence of Cu2+ through mass spectrometry (MS, Fig. S5, ESI†). Notably, SAH was detected as a major compound in the reaction performed in the presence of Cu2+. This observation also might suggest that the intermediate product, 3′-keto-SAH, is transformed back to SAH (in the Cu2+-dependent, e.g., reduction of the 3′-keto group or Cu2+-independent way), rather than eliminate of Hcy, as Hcy, Ado and other adenosine-based intermediates were detected in only trace amounts, with MS. To conclude, we demonstrated that transition metal cations disturb the function of PaSAHase with diverse mechanisms. These include arresting the enzyme in one conformation through the cation coordination on the interface between two major domains (Cd2+, Zn2+, Hg2+ and Co2+), limiting the access to the active site through the formation of interdomain disulfide bridge (Cu+) and interrupting the catalytic cycle through the oxidation of intermediate form of the cofactor, NADH (Cu2+). Understanding distinct mechanisms of PaSAHase inhibition is essential for developing novel, metal-cation-based antibacterial compounds.
We gratefully acknowledge the financial support of the National Science Centre, Poland, grants #2013/09/B/NZ1/01880 and #2018/30/E/NZ1/00729 to KB, and the Medical University of Bialystok, grant #B.SUB.24.314 to MEP-B. The synchrotron data was collected at beamline P13 operated by EMBL Hamburg at the PETRA III storage ring (DESY, Hamburg, Germany). We would like to thank Saravanan Panneerselvam for his assistance in using the beamline.
Data availability
The data supporting this article have been included as part of the ESI.† The atomic coordinates and structure factors can be accessed at PDB. Raw diffraction images have been deposited in the MX-RD Repository.Conflicts of interest
There are no conflicts to declare.References
- T. C. Petrossian and S. G. Clarke, Mol. Cell. Proteomics, 2011, 10, M110.000976 CrossRef PubMed.
- P. K. Chiang, Pharmacol. Ther., 1998, 77, 115–134 CrossRef CAS PubMed.
- O. Tehlivets, et al. , Biochim. Biophys. Acta, 2013, 1832, 204–215 CrossRef CAS PubMed.
- P. Vizán, et al. , Front. Cell Dev. Biol., 2021, 9, 654344 CrossRef PubMed.
- M. A. Turner, et al. , Nat. Struct. Biol., 1998, 5, 369–376 CrossRef CAS PubMed.
- K. Brzezinski, Biomolecules, 2020, 10, 1682 CrossRef CAS PubMed.
- J. Czyrko, et al. , Sci. Rep., 2018, 8, 11334 CrossRef PubMed.
- M. Li, et al. , Biochemistry, 2007, 46, 11451–11458 CrossRef CAS PubMed.
- P. C. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed.
- L. D. Palmer and E. P. Skaar, Annu. Rev. Genet., 2016, 50, 67–91 CrossRef CAS PubMed.
- M. A. Malik, et al. , MedChemComm, 2018, 9, 409–436 RSC.
- C. Imberti, et al. , Angew. Chem., 2020, 132, 61–73 CrossRef.
- S. Nayak and S. L. Gaonkar, ChemMedChem, 2021, 16, 1360–1390 CrossRef CAS.
- S. K. Nandanwar and H. J. Kim, ChemistrySelect, 2019, 4, 1706–1721 CrossRef CAS.
- M. W. Pantoliano, et al. , SLAS Discov., 2001, 6, 429–440 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr. Sect. A, 1976, 32, 751–767 CrossRef.
- R. R. Aksamit, et al. , J. Biol. Chem., 1994, 269, 4084–4091 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03143a |
| ‡ Magdalena Gawel and Piotr H. Malecki contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |