
Protecting Li-metal in O2 atmosphere by a sacrificial polymer additive in Li–O2 batteries†
Xiaohong
Wu‡
*a,
Ben
Niu‡
 b,
Yonglin
Tang
c,
Haiyan
Luo
c,
Zhengang
Li
c,
Xiaoyu
Yu
c,
Xin
Wang
*b,
Chunhai
Jiang
*a,
Yu
Qiao
*cd and
Shi-Gang
Sun
c
b,
Yonglin
Tang
c,
Haiyan
Luo
c,
Zhengang
Li
c,
Xiaoyu
Yu
c,
Xin
Wang
*b,
Chunhai
Jiang
*a,
Yu
Qiao
*cd and
Shi-Gang
Sun
c
aFujian Provincial Key Laboratory of Functional Materials and Applications, Institute of Advanced Energy Materials, School of Materials Science and Engineering, Xiamen University of Technology, Xiamen, 361024, P. R. China. E-mail: xiaohongwu@xmut.edu.cn; chjiang@xmut.edu.cn
bSongshan Lake Materials Laboratory, Dongguan, Guangdong 523808, P. R. China. E-mail: wangxin@sslab.org.cn
cState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: yuqiao@xmu.edu.cn
dFujian Science & Technology Innovation Laboratory for Energy Materials of China (Tan Kah Kee Innovation Laboratory), Xiamen 361005, P. R. China
First published on 20th October 2023
Abstract
Li–O2 batteries (LOBs) with Li-metal as the anode are characterized by their high theoretical energy density of 3500 W h kg−1 and are thus considered next-generation batteries with an unlimited potential. However, upon cycling in a harsh O2 atmosphere, the poor-quality solid electrolyte interphase (SEI) film formed on the surface of the Li-metal anode cannot effectively suppress the shuttle effect from O2, superoxide species, protons, and soluble side products. These issues lead to aggravated Li-metal corrosion and hinder the practical development of LOBs. In this work, a polyacrylamide-co-polymethyl acrylate (PAMMA) copolymer was innovatively introduced in an ether-based electrolyte as a sacrificial additive. PAMMA was found to preferentially decompose and promote the formation of a dense and Li3N-rich SEI film on the Li-metal surface, which could effectively prohibit the shuttle effect from a series of detrimental species in the Li–O2 cell during the discharge/charge process. Using PAMMA, well-protected Li-metal in a harsh O2 atmosphere and significantly enhanced cycling performance of the Li–O2 cell could be achieved. Thus, the use of a sacrificial polymer additive provides a promising strategy for the effective protection of Li-metal in Li–O2 cells in a severe O2 atmosphere during practical applications.
 Yu Qiao | Yu Qiao is currently a professor at the College of Chemistry and Chemical Engineering, Xiamen University. He earned his B.S. degree from the University of Science and Technology of China (USTC) in 2013. He received his PhD from Tsukuba University (Japan) in 2019 and worked as a postdoctoral fellow at the National Institute of Advanced Industrial Science and Technology (AIST, Japan). His research interests are focused on the development of electrochemical energy storage devices, surface/interfacial electrochemistry, and operando spectroscopic characterizations. |
Introduction
Due to its superior theoretical specific capacity (3861 mA h g−1) and ultralow electrochemical potential (−3.04 V vs. standard hydrogen electrode), Li-metal is considered the “Holy Grail” electrode. Furthermore, by coupling the Li-metal anode and O2 as active species in the cathode, Li–O2 batteries (LOBs) exhibit an extra-high energy density of 3500 W h kg−1 and thus have attracted significant attention as secondary battery candidates.1–3 In LOBs, Li-metal provides active Li, which is the only source of Li, for reversible electrochemical cycling. However, the introduction of O2 in LOBs makes the electrochemical environment of Li-metal more complicated than traditional Li-metal batteries (LMBs). Besides the intrinsic growth of dendrites, irreversible Li plating/stripping process, and the corrosion and failure of Li-metal in LOBs in an O2 atmosphere is significantly exacerbated.4–6At present, research is mostly focused on designing specific and functional catalysts and/or redox mediators (RMs) to solve the LOB-related issues, such as sluggish reaction kinetics and high charge overpotential.7–10 However, these modification strategies have missed some important information. The protons derived from solvent hydrogen abstraction (H-abstraction), intermediate superoxide species formed during the oxygen reduction reaction (ORR), H2O moisture from the decomposition of the electrolyte, and O2 can simultaneously shuttle to the anode and rapidly react with the naturally reactive Li-metal to form porous lithium hydroxide (LiOH) or other detrimental byproducts, resulting in dynamic variations (both components and architectures) in the solid electrolyte interphase (SEI) film at the Li-metal/electrolyte interface.11,12 The continuous accumulation of these side products aggravates the roughness of the Li-metal, and more fresh Li is exposed to chemically react with the electrolyte, leading to the further consumption of Li. Based on the premise of inevitable parasitic reactions, the failure of Li-metal will be accelerated and become more serious if there is no effective protection from a high-quality SEI film, resulting in the poor electrochemical performance of LOBs. Therefore, the corrosive issue of Li-metal in LOBs in the harsh O2 atmosphere should not be ignored. Instead, LOBs should be studied as a whole system rather than as an isolated cathode and anode. The shuttle effect of O2, side products, protons, and superoxide species from the cathode side should be considered for Li-metal modification strategies under an O2 atmosphere in LOBs. Especially, for the Li-metal anode, conventional research mostly focused on sealed systems in Li-ion batteries. However, the introduction of O2 in LOBs inevitably creates more complex challenges in the study of the Li-metal anode. Accordingly, it needs to be reconsidered if the modification strategies of Li-metal batteries under an inert Ar condition are feasible in an O2 environment. Therefore, evaluating the stability of the Li-metal anode in an O2 atmosphere is more valuable for the practical development of LOBs. Unlike the popular organic/inorganic materials with a low molecule weight, polymers, which are characterized by tunable functional groups and excellent flexibility, have been rarely reported as effective additives for the modification of the SEI in LOBs to date.13,14
Herein, a polyacrylamide-co-polymethyl acrylate (PAMMA) copolymer with the amide group of –CONH2 was successfully synthesized and solvated in the ether-based electrolyte (1.0 M LiTFSI in TEGDME) of an Li–O2 cell. Due to the localized charge distribution, PAMMA with an electron-deficient –CONH2 group could preferentially decompose more readily than the common electrolyte species (solvent and Li salt) to form an Li3N-rich SEI layer at the Li-metal/electrolyte interface, which significantly protected the Li-metal anode from corrosion by O2, superoxide species, protons and soluble side products. Hence, the stability of the Li-metal in a harsh O2 atmosphere was improved, which promoted the excellent electrochemical performance and long-term cycling lifespan of the Li–O2 cell with PAMMA. The effective polymer additive provides an additional promising option for the protection of the Li-metal anode in Li-O2 cells.
Results and discussion
The process for the synthesis of polyacrylamide-co-polymethyl acrylate (PAMMA) is presented in the Experimental section of the ESI.† Fourier-transform infrared (FTIR) measurement was preferentially conducted to characterize the functional groups and bonds in pure PAMMA in solid form. As exhibited in Fig. 1a, the bands at 3411 and 3181 cm−1 represent the asymmetric and symmetric stretching vibration of the –NH2 groups in PAMMA, respectively. The peaks located at the wavenumbers of 2931 and 1673 cm−1 are assigned to the C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the aliphatic methylene and amide (–CONH2) groups of PAMMA, respectively. The IR absorption band located at around 1432 cm−1 is attributed to the N–H bending mode of the amino group, while the peak at 1031 cm−1 represents the specific vibration modes of the C–O bonds in PAMMA.15 Thus, the FTIR technique effectively confirmed the presence of the characteristic –CONH2 group in PAMMA. Moreover, 5 wt% of PAMMA was fully dissolved in a DMSO-based deuterium-substituted reagent to detect the specific hydrogen environment in PAMMA by 1H NMR spectroscopy. The chemical shifts at 2.5, 0, and 3.3 ppm represent the typical hydrogen environment of DMSO, tetramethylsilane (TMS), and H2O, respectively (Fig. 1b). The peaks in the range of 1.7–1.2 and 2.4–1.9 ppm corresponding to the main-chain groups of –CH2 (marked as a and f in the inset of Fig. 1b) and –CH (b and e) groups in PAMMA, respectively. The chemical shift associated with the –CONH2 group is located at around 7.4–6.6 ppm (c). The peaks at around 3.7–3.4 ppm are assigned to the –CH3 groups (d) in PAMMA.13,16 Thus, the 1H NMR measurement further verified the different hydrogen environments of the functional groups, especially –CONH2 in PAMMA. Gel permeation chromatography (GPC) measurement gave a weight-average molecular weight (Mw) and number-average molecular weight (Mn) of ∼2.2 × 105 and ∼2.5 × 104 g mol−1, respectively, revealing the successful synthesis of the PAMMA polymer, as shown in Fig. S1.†
O stretching vibrations of the aliphatic methylene and amide (–CONH2) groups of PAMMA, respectively. The IR absorption band located at around 1432 cm−1 is attributed to the N–H bending mode of the amino group, while the peak at 1031 cm−1 represents the specific vibration modes of the C–O bonds in PAMMA.15 Thus, the FTIR technique effectively confirmed the presence of the characteristic –CONH2 group in PAMMA. Moreover, 5 wt% of PAMMA was fully dissolved in a DMSO-based deuterium-substituted reagent to detect the specific hydrogen environment in PAMMA by 1H NMR spectroscopy. The chemical shifts at 2.5, 0, and 3.3 ppm represent the typical hydrogen environment of DMSO, tetramethylsilane (TMS), and H2O, respectively (Fig. 1b). The peaks in the range of 1.7–1.2 and 2.4–1.9 ppm corresponding to the main-chain groups of –CH2 (marked as a and f in the inset of Fig. 1b) and –CH (b and e) groups in PAMMA, respectively. The chemical shift associated with the –CONH2 group is located at around 7.4–6.6 ppm (c). The peaks at around 3.7–3.4 ppm are assigned to the –CH3 groups (d) in PAMMA.13,16 Thus, the 1H NMR measurement further verified the different hydrogen environments of the functional groups, especially –CONH2 in PAMMA. Gel permeation chromatography (GPC) measurement gave a weight-average molecular weight (Mw) and number-average molecular weight (Mn) of ∼2.2 × 105 and ∼2.5 × 104 g mol−1, respectively, revealing the successful synthesis of the PAMMA polymer, as shown in Fig. S1.†
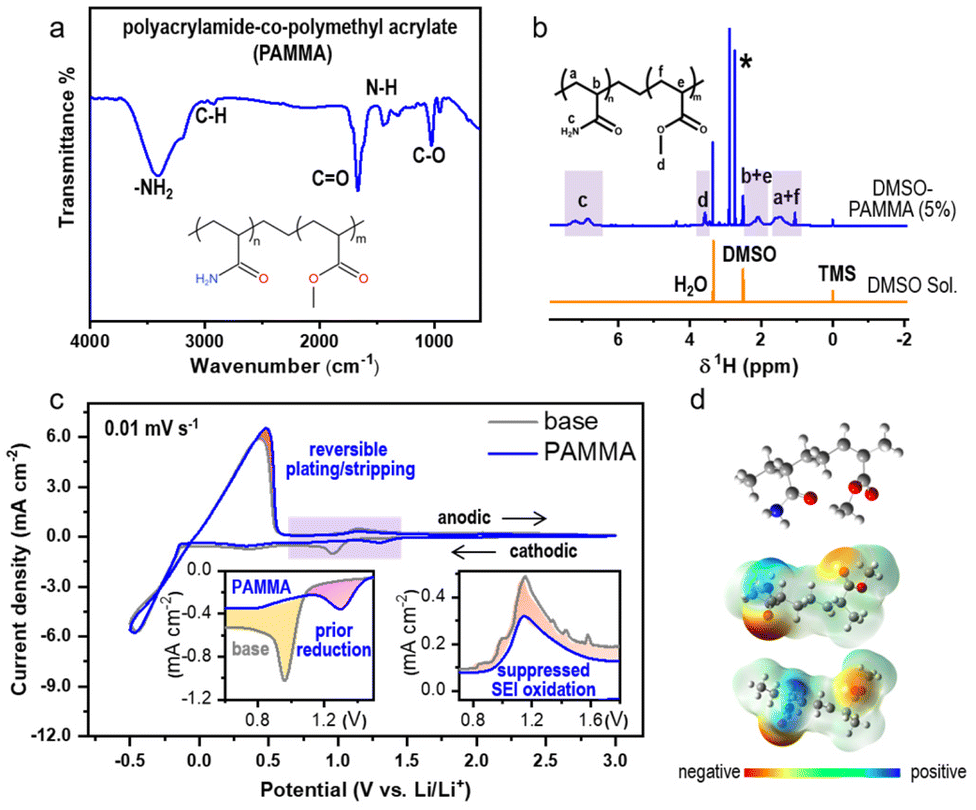 | ||
| Fig. 1 (a) FTIR and (b) 1H-NMR characterization of the PAMMA molecule; structure formulas are exhibited as insets. (c) CV curves in O2 atmosphere without (gray trace, 1.0 M LiTFSI in TEGDME)/with (blue trace) PAMMA addition. Scan rate: 0.01 mV s−1; potential range: −0.5–3.0 V. The insets in the left and right represent partial enlarged cathodic and anodic peaks without and with PAMMA, respectively. (d) ESP calculation of the PAMMA molecule by Gaussian09 with different views. The ball-and-stick model of PAMMA is embedded. | ||
Moreover, the electrochemical performance in ether-based electrolyte (1.0 M LiTFSI in TEGDME) with/without PAMMA under a harsh O2 environment was studied by cyclic voltammetry (CV) measurement with an Li/Cu half-cell. Fig. 1c exhibits a comparison of the CV scans with/without PAMMA on the first cycle in the potential range of −0.5–3.0 V. The ether-based electrolyte without PAMMA is marked as a gray trace, while the blue trace represents the electrolyte with PAMMA. During the cathodic process, only one peak appeared in both the PAMMA-free and PAMMA-containing electrolyte, as shown in the magnified inset on the left (Fig. 1c). The cathodic peak marked as yellow layer in the PAMMA-free electrolyte corresponds to the solvent or Li salt decomposition for the formation of an SEI. Given that only one peak (pink area) was apparently observed in the PAMMA-containing electrolyte and no other peaks overlapped with the yellow area in the PAMMA-free electrolyte, we conclude that PAMMA was preferentially reduced compared to the conventional electrolyte components, involving the formation of an SEI, and the corresponding electrolyte decomposition was significantly prohibited. More importantly, the reduced product of PAMMA was chemically inert given that no other reduction peaks appeared. During the anodic process, the response of current density in PAMMA-present electrolyte is much lower than that in PAMMA-absent electrolyte, as shown in the magnified inset on the right (Fig. 1c), indicating the significantly suppressed oxidative decomposition of the SEI with the addition of PAMMA, and the reconstruction and fragmental accumulation of the SEI film could be avoided during electrochemical cycling. In addition, the obvious difference in the peak-area ratio for the Li plating and stripping process (marked as rose layer) between the PAMMA-free and PAMMA-containing electrolyte further revealed the enhanced reversibility of Li plating/stripping with the addition of PAMMA. Linear scan voltammetry (LSV) measurement was conducted to evaluate the electrochemical stability of PAMMA in an Li/stainless steel cell. In the purple region of the LSV profile (Fig. S2†), the current density response in the PAMMA-containing electrolyte is initially higher than that without PAMMA, corresponding to the preferential oxidation of PAMMA. However, as the voltage increased, the growth rate of the intensity (current density) gradually decreased, and the corresponding current density signal was much lower than that without PAMMA (green region), which is related to the suppressed decomposition of the electrolyte upon the introduction of PAMMA. The variations in both the CV and LSV profiles between the PAMMA-free and PAMMA-containing electrolyte indicate the preferential self-decomposition and restrained electrolyte-related parasitic reactions by PAMMA. The effect of the introduction of PAMMA on the SEI architecture is discussed in detail in the following sections. A ball-and-stick model of the polymer PAMMA is exhibited in the inset in Fig. 1d. Based on the principle of quantum chemistry, the Gaussian09 package was utilized to calculate the surface electrostatic potential map (ESP) of PAMMA. The different enrichment levels of charge density are represented by different colors. Red color usually stands for electron-rich functional groups, while blue color exhibits electron-deficient species.13 As shown in Fig. 1d, only –NH2 in the –CONH2 functional group represents an apparent electron-deficient state, resulting in much easier bond breaking and decomposition than the other groups in PAMMA. This is in good agreement with the PAMMA-containing electrochemical behavior of preferential decomposition in the CV and LSV profiles (Fig. 1c and Fig. S2†).
The FTIR, 1H NMR and GPC measurements all revealed that the PAMMA polymer, which is rich in –CONH2 groups, was successfully synthesized. Also, the electrochemical characterization by CV and LSV and theoretical calculation further confirm that the –CONH2 groups in PAMMA can preferentially decompose and effectively suppress electrolyte-related parasitic reactions.
Before using PAMMA as an effective additive in ether-based electrolyte, the cycling performance of Li/Li symmetrical cells with a series of PAMMA concentrations (0, 0.03, 0.08, and 0.15 wt%) was tested. As shown in Fig. S3,† due to the superior cycling stability of the Li/Li symmetrical cells with 0.08 wt% PAMMA than that with other concentrations, this was determined to be the optimized concentration of PAMMA additive. Unless otherwise indicated, the PAMMA-based electrolyte refers to 0.08 wt% PAMMA additive in 1.0 M LiTFSI-TEGDME. Furthermore, the electrochemical performance of the Li/Li symmetrical cells with/without PAMMA in an O2 atmosphere was studied to simulate the practical working performance of Li-metal in an Li–O2 cell. At the current density of 0.2 mA cm−2 (Fig. 2a and Fig. S4†), the voltage polarization of the Li/Li symmetrical cell without PAMMA (gray trace) started to increase when the cycling time reached 310 h in an O2 atmosphere. However, with the PAMMA additive, the Li/Li symmetrical cell could steadily work for more than 550 h. The longer cycling lifespan of the PAMMA-containing Li/Li symmetrical cell (blue trace) than that of the PAMMA-free cell under an O2 environment indicates the enhanced Li-metal stability with PAMMA additive. Even at a higher current densities of 0.5 mA cm−2 (Fig. 2b), the Li/Li symmetrical cell with PAMMA also exhibited improved cycling stability in an O2 atmosphere. After working for 50 cycles in an O2 atmosphere (0.2 mA cm−2), the surface morphology of the cycled Li-metal without/with PAMMA was characterized by scanning electron microscopy (SEM). Herein, only the Li plating/stripping process was investigated in the Li/Li symmetrical cell without considering the shuttle effect of the byproducts from the cathode. However, the introduction of O2 complicates the electrochemical environment of Li-metal. As shown in Fig. 2c, roughness, cracks and fragments can be obviously observed on the cycled Li-metal surface without PAMMA, which exacerbated the degradation and failure of the cell. Conversely, the uniform and dense morphology in PAMMA-containing cell promoted well-maintained Li-metal (Fig. 2d), which is consistent with the long-term cycling performance (blue trace) of the Li/Li symmetrical cell under an O2 environment, as shown in Fig. 2a and b, respectively. The significant difference in the surface morphology of the cycled Li-metal further highlights the effectiveness of the PAMMA additive.
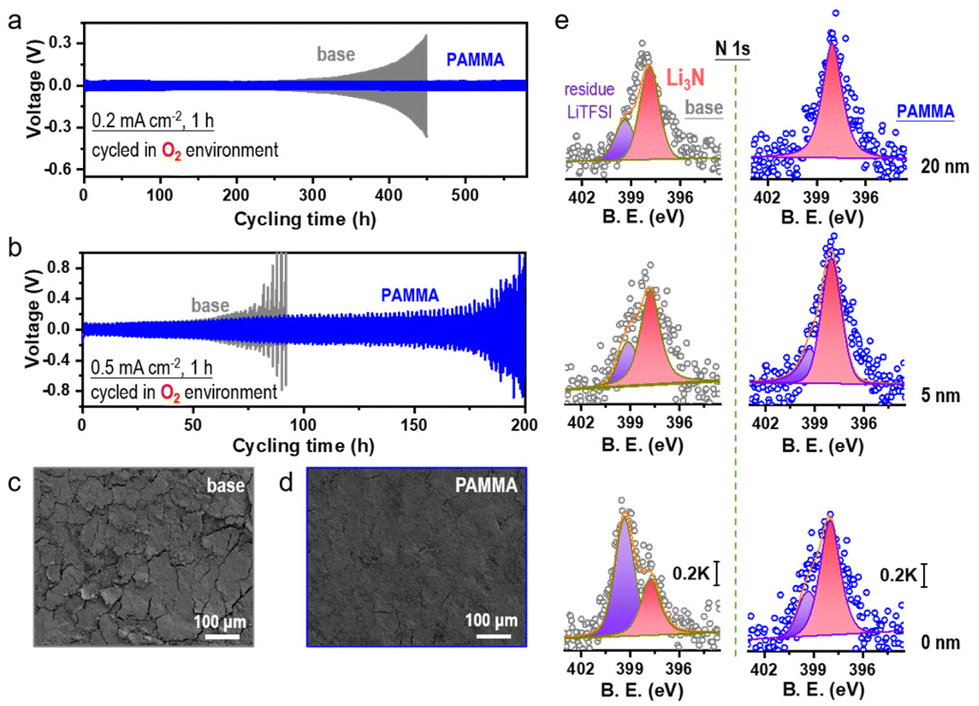 | ||
| Fig. 2 Electrochemical performance of Li/Li symmetrical cells under O2 environment without (gray trace)/with PAMMA (blue trace) at current densities of (a) 0.2 mA cm−2 and (b) 0.5 mA cm−2. Surface morphology characterization of the cycled Li-metal in the Li/Li symmetrical cell under O2 atmosphere (c) without and (d) with PAMMA (after 50 cycles, 0.2 mA cm−2, 1 h). (e) N 1s spectra of the cycled Li-metal surface without/with PAMMA in Li/Li symmetrical cell at different etching depths of 0, 5, and 20 nm (O2 atmosphere, after 50 cycles, 0.2 mA cm−2, 1 h). | ||
Moreover, the specific SEI components on the cycled Li-metal surface without/with PAMMA were measured by X-ray photoelectron spectroscopy (XPS). Fig. 2e and Fig. S5† present the N-related and O-related species in the SEI film on the cycled Li-metal surface without/with PAMMA at different etching depths, respectively. Notably, the original intensity of the XPS spectrum did not excessively change. Hence, the peak intensity could be reasonably utilized to evaluate the relative content of the species.17 As shown in Fig. 2e and Fig. S5,† the scale bar of the N 1s and O 1s spectra under different etching depths are marked as 0.2 K and 10 K, respectively. The N-related species in the PAMMA-containing Li/Li symmetrical cell, specifically Li3N (∼398.3 eV, rose trace), all exhibited a much higher content than that without PAMMA at the etching depth of 0, 5, and 20 nm, respectively, suggesting the formation of an Li3N-rich SEI film on the PAMMA-containing Li-metal surface. Chemically, amide groups can decompose into small molecules in an electrochemical environment under certain conditions.18 Given that PAMMA contains abundant –CONH2 groups and tends to break bonds preferentially than the solvent/salt in the electrolyte (Fig. 1c and Fig. S2†), Li3N was mainly derived from the contribution of PAMMA rather than the decomposition of the electrolyte in the PAMMA-containing cell. Generally, Li3N species, which are characterized by high ionic conductivity, can form a highly uniform conducive network and effectively promote interfacial Li+ migration at the Li-metal/electrolyte interface, realizing homogeneous Li+ plating without the growth of dendrites.19–21 Therefore, the Li3N-rich SEI film generated by PAMMA preferentially decomposed and prohibited electrolyte-related side reactions, promoting the stable electrochemical performance of the PAMMA-containing Li/Li symmetrical cell (Fig. 2a and b, blue trace). The peaks at 399.4 eV, which are assigned to the residual Li salt (LiTFSI, purple trace), presented a much lower content with PAMMA than that in the PAMMA-free cell. This reveals the higher consumption and decomposition of LiTFSI in the PAMMA-free cell and continuous accumulation of the LiTFSI-related byproducts, resulting in the formation of a loose and porous SEI film. Under practical operation conditions, a loose SEI film with porosity can allow the passage of solvent molecules, which makes it easier to for them to directly contact the fresh Li-metal, inducing the formation of a cracked Li-metal surface and accelerating the contact-corrosion of the Li-metal. Hence, the loose and porous SEI film formed in the absence of PAMMA could not facilitate the stable and long-term cycling of the Li-metal (Fig. 2a and b, gray trace). Simultaneously, the issue of Li-metal corrosion resulting from O2 erosion is inevitable and should be considered during cycling in an O2 environment. The loose and porous SEI layer without PAMMA could not prohibit the continuous O2 shuttle. Hence, plentiful LiOH (∼530.2 eV, pink trace) and Li2O (∼528.4 eV, orange trace) species were formed and apparently observed on the Li-metal surface at different etching depths (Fig. S5†).22 In contrast, the Li3N-rich SEI film formed by the decomposition of PAMMA was relatively dense and compact. Simultaneously, the reconstruction and fragmental accumulation of the Li3N-rich SEI film architecture during cycling would be significantly blocked due to the chemical stability of the Li3N species, which could effectively suppress the Li-metal anode corrosion caused by the O2 shuttle. Compared with that without PAMMA, the less LiOH and Li2O at different etching depths indicate a better protected Li-metal anode with PAMMA additive in an O2 environment (Fig. S5†).
An Li-O2 cell with/without PAMMA additive was further assembled to evaluate the ORR performance. As an active reactant, O2 combined with Li+ is reduced to generate Li2O2 product during the ORR process.23,24 The XRD characterization of the discharge product from the Li–O2 cell in Fig. S6† indicates that the discharge mechanism based on Li2O2 was not modified by the PAMMA additive. Fig. 3a and b reveal an obvious difference in the side view of the Li-metal with/without PAMMA, respectively, and the corresponding voltage profiles are presented in Fig. S7.† After discharging for 20 h at a current density of 0.05 mA cm−2, a corrosion layer with a thickness of nearly 45 μm was accumulated on the Li-metal surface in the Li–O2 cell without PAMMA (Fig. 3a). In contrast, a thinner corrosion layer (∼20 μm, Fig. 3b) was formed on the PAMMA-containing Li metal surface. To better understand the specific components and reason for the formation of the corrosion layer on the discharged Li-metal surface, representative XRD measurement was primarily conducted. The standard cards for LiOH (PDF 32-0564, green trace) and Li (PDF 15-0401, pink trace) are exhibited for comparison.22 In Fig. 3c, a peak corresponding to LiOH can be apparently observed on discharged PAMMA-free Li-metal surface (gray trace), indicating that LiOH is one of the main components of the corrosion layer on the Li-metal surface without PAMMA. Alternatively, on the PAMMA-containing Li-metal surface, the XRD peak corresponding to LiOH species is not obvious (blue trace). Generally, the amount of LiOH formed on the Li-metal surface can be employed as a standard index to evaluate the degree of corrosion of the Li-metal.25,26 Moreover, the O 1s XPS spectra of the discharged Li-metal surface was measured, as exhibited in Fig. 3d. The peaks at ∼530.2 eV are attributed to the LiOH species, which are marked as a pink trace. Based on the same scale bar (9 K), it is clear that more LiOH was formed on the PAMMA-free Li-metal surface than that on the Li-metal surface with PAMMA additive. This result reveals that the corrosion of the discharged Li-metal in the severe O2 atmosphere was effectively suppressed by PAMMA.
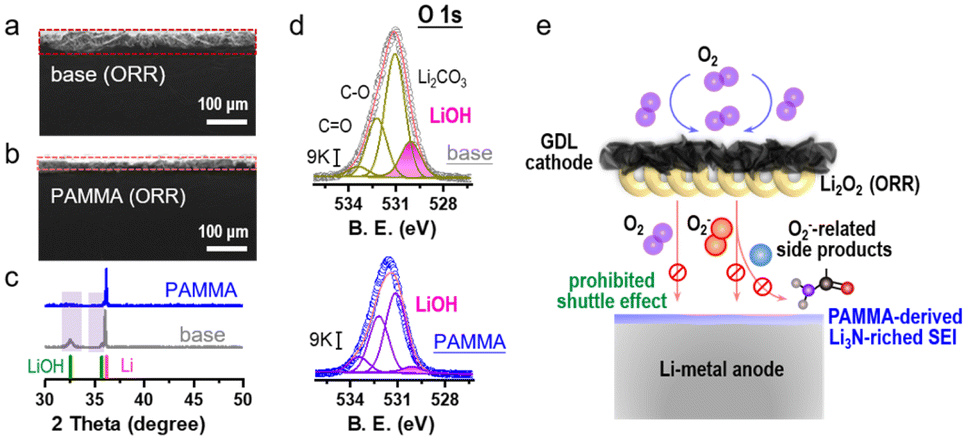 | ||
| Fig. 3 Side views of (a) PAMMA-free and (b) PAMMA-containing discharged Li-metal by SEM measurement (current density: 0.05 mA cm−2; fixed capacity: 1 mA h cm−2). (c) XRD characterization of Li-metal in the Li–O2 cell without/with PAMMA during the ORR process (current density: 0.05 mA cm−2; fixed capacity: 1 mA h cm−2). (d) O 1s spectrum of the Li-metal anode during the ORR process without/with PAMMA. (e) Proposed mechanism related to Li-metal anode protection with the PAMMA additive in the Li–O2 cell. | ||
In Li2O2-based Li–O2 cells, superoxide species are indispensable as intermediates during the ORR process. Acting as a nucleophile, superoxide species are prone to attack the ether solvent and dehydrogenize it, leading to the formation of protons and side products, such as H2O, lithium formate and lithium acetate.17,27,28 O2, superoxide species, protons in the electrolyte and soluble side products can shuttle to the Li-metal anode, resulting in a series of detrimental parasitic reactions. Combined with protons, superoxide species react with Li-metal to generate LiOH. A trace amount of H2O can directly contact with the Li-metal to form LiOH. Also, shuttled O2 can bind with Li+ and H2O, promoting the formation of LiOH.29,30 The corresponding formation mechanism of LiOH (corrosion degree index) at the anode in the Li–O2 cell is illustrated in Fig. S8.† If there is no high-quality protective layer on the Li-metal surface to inhibit the shuttle effect from various hazardous species, the formation of LiOH will be accelerated, leading to an unrecoverable corrosive environment and limited reversibility of the Li-metal. As observed experimentally, because of the poor SEI film, a porous and thick LiOH layer was formed on the Li-metal surface without PAMMA (Fig. 3a). Conversely, the Li3N-riched and dense SEI film derived from PAMMA effectively prevented Li-metal corrosion and the detrimental shuttle effect of O2, H2O, protons and the discharge intermediates, contributing to the formation of a thin LiOH layer (Fig. 3b).31–33 The analysis of the discharge Li-metal in the Li–O2 cell by XPS using the N 1s spectra (Fig. S9†) further verified that richer Li3N species were formed on the PAMMA-containing Li-metal than that on the PAMMA-free Li-metal surface. The proposed mechanism related to the Li-metal anode protection by PAMMA in the harsh O2 environment is shown in Fig. 3e.
The electrochemical performance of the Li–O2 cell was tested to assess the practicability of the PAMMA additive. The typical capacity-voltage profiles without/with PAMMA at a current density of 500 mA g−1 and 1000 mA g−1 are exhibited in Fig. 4a and Fig. S10† (fixed capacity: 1000 mA h g−1), respectively. As shown in Fig. 4b, with the introduction of PAMMA, the cycling lifespan of the Li–O2 cell improved from ∼120 h (without PAMMA, gray trace) to ∼300 h (with PAMMA, blue trace) at a current density of 500 mA g−1 (cut-off voltage: 2.0 V). When the current density increased to 1000 mA g−1, the cycling stability of the Li–O2 cell could still be significantly enhanced by PAMMA, as shown in Fig. 4c. The discharge/charge time with PAMMA was more than 4 times that without PAMMA. As the only lithium source, the stability of the Li-metal in the Li–O2 cell under a harsh O2 atmosphere is critical for the battery performance. Moreover, the morphology of the cycled Li-metal anode in the Li–O2 cell was primarily investigated by SEM measurement. Fragments, cracks and roughness were clearly observed on the PAMMA-free Li-metal surface (Fig. 4d), which are detrimental to long-term cycling and accelerate cell failure. However, the integrity and denseness of the Li-metal anode was well-preserved during continuous discharge/charge process with the introduction of PAMMA, and no cracks appeared (Fig. 4e). The protected Li-metal anode boosted the stable cycling performance of the Li–O2 cell, which is in good agreement with the experimental results in Fig. 4b and c, respectively.
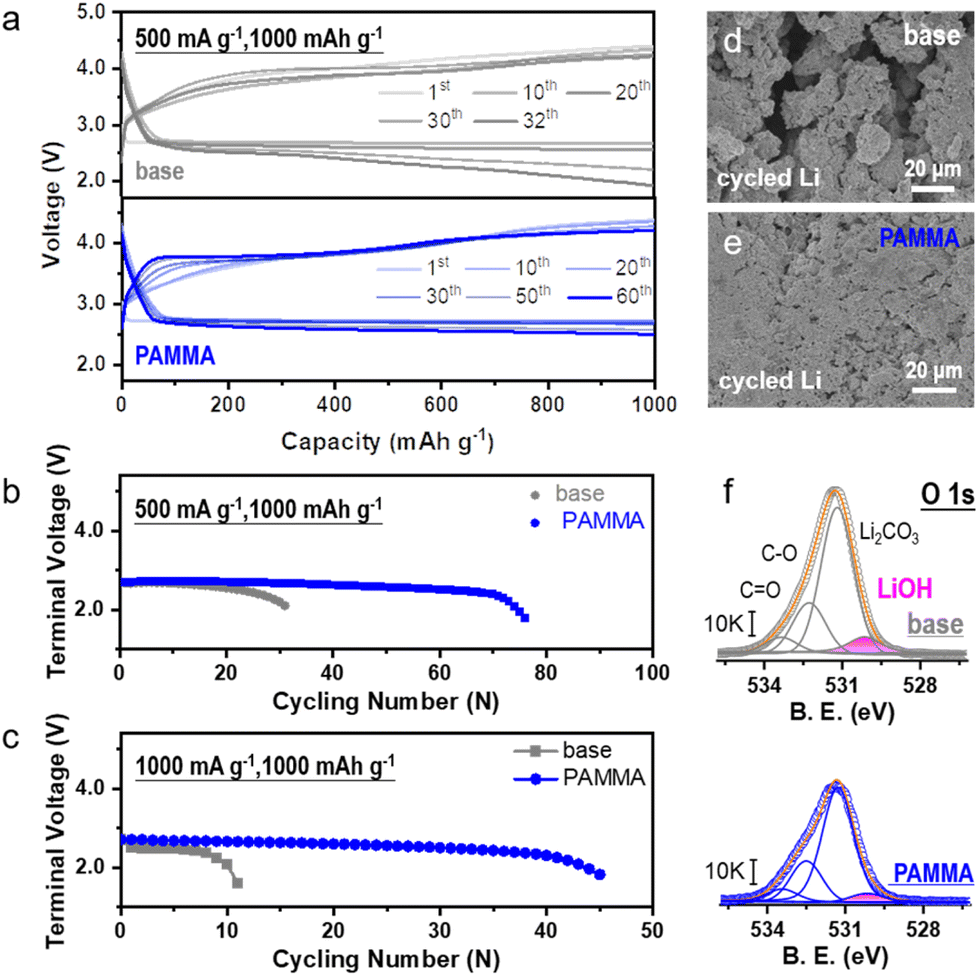 | ||
| Fig. 4 (a) Typical capacity-voltage profiles of Li–O2 cells without (gray trace)/with (blue trace) PAMMA (500 mA g−1, 1000 mA h g−1). Electrochemical performance of Li–O2 cells without and with PAMMA at the current densities of (b) 500 mA g−1 and (c) 1000 mA g−1, respectively. SEM images of Li-metal in the Li–O2 cell after 20 cycles (d) without and (e) with PAMMA. (f) Comparison of O 1s spectra of the cycled Li-metal under O2 atmosphere without/with PAMMA (0.25 mA cm−2, 2 h, after 20 cycles). | ||
To explore the specific components on the cycled Li-metal surface in the Li–O2 cell, XRD and XPS characterization were conducted. As exhibited in Fig. S11,† diffraction peaks at 32.5° and 35.7° corresponding to the Li corrosion product (LiOH) apparently appeared on the PAMMA-free rather than the PAMMA-containing Li-metal surface. The O 1s spectra in Fig. 4f further indicate that a large amount of LiOH, marked as a pink trace, was inevitably formed on the Li-metal surface of the Li–O2 cell without PAMMA during the discharge/charge process, which is consistent with the results obtained by XRD (Fig. S11†). The porous and loose LiOH corrosion layer could not block the shuttle effect from O2, superoxide species, protons and soluble side products. This may have led to a series of serious Li-metal-related parasitic reactions, resulting in the accumulation of LiOH product and further aggravating the degree of corrosion of the Li-metal. Eventually, a thick corrosion layer mainly composed of LiOH was formed the on deteriorated Li-metal surface, shortening the lifespan of the Li–O2 cell without PAMMA. Conversely, the dense and Li3N-rich SEI film derived from the sacrificial PAMMA additive (Fig. 2e and Fig. S12†) effectively isolated the impact from the shuttle effect (O2, superoxide species, protons and soluble side products) on the Li-metal, resulting in the formation of a thin LiOH corrosive layer (Fig. 4f). Furthermore, the D2O-extracted 1H and 19F NMR spectra from the cycled cathode plate and separator in the Li–O2 cell at a current density of 0.25 mA cm−2 were measured to analyze the possible side products formed during cycling. The peaks at 8.3 (Fig. S13†) and 123.2 ppm (Fig. S14†) are assigned to the side products of formate and LiF, respectively, which result from the parasitic reactions related to the decomposition of the electrolyte (both solvent and Li salt) under an O2 environment. Notably, the peak area of the internal standard benzene was normalized before the analysis. The quantitative investigation indicated that the decomposition and degradation of the electrolyte were significantly restrained in the PAMMA-containing Li–O2 cell, which is consistent with the conclusion obtained from the CV and LSV curves (Fig. 1c and Fig. S2†), respectively. Moreover, the soluble byproducts can inevitably cross over to the anode, resulting in the corrosion of the Li-metal anode in the Li–O2 cell. The smaller amount of byproducts (formate, LiF, etc.) exhibited in Fig. S13 and S14† indicates the suppressed shuttle effect in the PAMMA-containing Li–O2 cell. Combined with the PAMMA-derived and shielding SEI film, it effectively protected the Li-metal from passivation and failure under an O2 atmosphere, which is also consistent with the experimental observation in Fig. 4e. Hence, the Li-metal anode was well-protected by the PAMMA-derived SEI film in the Li–O2 cell during long-term cycling. Compared with other related works, the introduction of PAMMA additive has a more prominent effect on the Li–O2 cell at a high current density (Fig. S15†).
Conclusions
In this work, acting as a sacrificial electrolyte additive, a trace amount (0.08 wt%) of PAMMA polymer with the characteristic –CONH2 functional groups could significantly enhance the SEI film quality, which contained Li3N-rich components. Derived by the preferential decomposition of PAMMA, the dense and Li3N-rich SEI film significantly prohibited the hazardous shuttle effect from O2, superoxide species, protons and soluble side products on the Li-metal during cycling, resulting in a well-protected Li-metal anode with a thin LiOH corrosive layer in an O2 environment. Benefiting from these advantages, the Li–O2 cell with PAMMA additive achieved a stable long-term cycling performance, namely, 4 times-higher than that without PAMMA (1000 mA g−1, 1000 mA h g−1). Thus, the rational selection of a polymer with designed functional groups as an effective electrolyte additive provides new insight for Li-metal protection in Li–O2 cells under harsh O2 condition. Different from the tradition electrolyte modification strategies for Li-metal anodes, evaluating and screening suitable electrolyte additives in a severe O2 atmosphere are more feasible for the practical application of Li–O2 cells.Author contributions
X-H. W., B. N., X. W., C. J. and Y. Q. contributed to the design of the research and performed the experimental data analysis. X-H. W. and B. N. contributed equally to this work. X-H. W. conducted the electrochemical characterization and theoretical calculation. B. N. synthesized the polymer molecule and conducted its characterization. Y. T. and H. L. performed the SEM and FTIR characterizations. Z. Li. conducted the XPS characterizations. X. Y. performed XRD test. All authors co-wrote the manuscript. X. W., C. J., Y. Q. and S.-G. S. supervised the work. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Guidance project of The Science and Technology Department of Fujian Province (grant no. 2020H0024) and the Songshan Lake Materials Laboratory (grant no. Y1D1031H311).References
- H. Zhou, Nat. Catal., 2022, 5, 173–174 CrossRef CAS.
- H. Dong, C. Ning, G. Yang, H. Ji and Y. Li, Nanoscale, 2021, 13, 12727–12737 RSC.
- Z. Jiang, Z. Zhu, S. Gao, Q. Lv and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2023, 119, e2202835119 CrossRef PubMed.
- Y. Chen, J. Xu, P. He, Y. Qiao, S. Guo, H. Yang and H. Zhou, Sci. Bull., 2022, 67, 2449–2486 CrossRef CAS PubMed.
- Y.-N. Li, F.-L. Jiang, Z. Sun, O. Yamamoto, N. Imanishi and T. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 16437–16444 CrossRef CAS PubMed.
- X. Chi, M. Li, J. Di, P. Bai, L. Song, X. Wang, F. Li, S. Liang, J. J. Xu and J. Yu, Nature, 2021, 592, 551–557 CrossRef CAS PubMed.
- Y. Huang, J. Geng, Z. Jiang, M. Ren, B. Wen, J. Chen and F. Li, Angew. Chem., Int. Ed., 2023, 62, e202306236 CrossRef CAS PubMed.
- Y. N. Zhang, F. L. Jiang, F. Bai, H. Jiang and T. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 10327–10336 CrossRef CAS PubMed.
- S. Liang, L. C. Zou, L. J. Zheng, F. Li, X. X. Wang, L. N. Song and J. J. Xu, Adv. Energy Mater., 2022, 12, 2103097 CrossRef CAS.
- L. Peng, X. Zhang, Y. Sun and C. Li, Nanoscale, 2021, 13, 16477–16486 RSC.
- F. Qiu, S. Ren, X. Zhang, P. He and H. Zhou, Sci. Bull., 2021, 66, 897–903 CrossRef CAS PubMed.
- Q.-K. Zhang, X.-Q. Zhang, J. Wan, N. Yao, T.-L. Song, J. Xie, L.-P. Hou, M.-Y. Zhou, X. Chen, B.-Q. Li, R. Wen, H.-J. Peng, Q. Zhang and J.-Q. Huang, Nat. Energy, 2023, 8, 725–735 CrossRef CAS.
- X. Wu, B. Niu, H. Zhang, Z. Li, H. Luo, Y. Tang, X. Yu, L. Huang, X. He, X. Wang, Y. Qiao and S. G. Sun, Adv. Energy Mater., 2022, 13, 2203089 CrossRef.
- L. Zhang, Z. Wei, M. Meng, G. Ung and J. He, J. Mater. Chem. A, 2020, 8, 15900–15908 RSC.
- A. S. Singha and A. Guleria, J. Nat. Fibers, 2015, 12, 587–603 CrossRef CAS.
- B. Niu, Z. Li, S. Cai, D. Luo, Y. Qiao, S. Zhou, H. Li, X. He and X. Wang, Chem. Eng. J., 2022, 442, 136217 CrossRef CAS.
- X. Wu, X. Wang, Z. Li, L. Chen, S. Zhou, H. Zhang, Y. Qiao, H. Yue, L. Huang and S. G. Sun, Nano Lett., 2022, 22, 4985–4992 CrossRef CAS PubMed.
- J. Liu, M. Wu, X. Li, D. Wu, H. Wang, J. Huang and J. Ma, Adv. Energy Mater., 2023, 13, 2300084 CrossRef CAS.
- Q. Li, H. Pan, W. Li, Y. Wang, J. Wang, J. Zheng, X. Yu, H. Li and L. Chen, ACS Energy Lett., 2018, 3, 2259–2266 CrossRef CAS.
- Y. K. Z. Yao, M. Hou, J. Huang, J. Zhang, B. Yang, Y. Dai and F. Liang, Adv. Funct. Mater., 2022, 32, 2111919 CrossRef.
- X. Li, R. Zhao, Y. Fu and A. Manthiram, eScience, 2021, 1, 108–123 CrossRef.
- Y. Yu, Y.-B. Yin, J.-L. Ma, Z.-W. Chang, T. Sun, Y.-H. Zhu, X.-Y. Yang, T. Liu and X.-B. Zhang, Energy Storage Mater., 2019, 18, 382–388 CrossRef.
- L. Liu, Y. Liu, C. Wang, X. Peng, W. Fang, Y. Hou, J. Wang, J. Ye and Y. Wu, Small Methods, 2022, 6, 2101280 CrossRef CAS PubMed.
- Z. Lyu, Y. Zhou, W. Dai, X. Cui, M. Lai, L. Wang, F. Huo, W. Huang, Z. Hu and W. Chen, Chem. Soc. Rev., 2017, 46, 6046–6072 RSC.
- Q. C. Liu, J. J. Xu, S. Yuan, Z. W. Chang, D. Xu, Y. B. Yin, L. Li, H. X. Zhong, Y. S. Jiang, J. M. Yan and X. B. Zhang, Adv. Mater., 2015, 27, 5241–5247 CrossRef CAS PubMed.
- J.-L. Shui, J. S. Okasinski, P. Kenesei, H. A. Dobbs, D. Zhao, J. D. Almer and D.-J. Liu, Nat. Commun., 2013, 4, 3255 Search PubMed.
- W.-J. Kwak, R. Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- S. A. Freunberger, Y. Chen, N. E. Drewett, L. J. Hardwick, F. Bardé and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 8609–8613 CrossRef CAS PubMed.
- R. S. Assary, J. Lu, P. Du, X. Luo, X. Zhang, Y. Ren, L. A. Curtiss and K. Amine, ChemSusChem, 2013, 6, 51–55 CrossRef CAS PubMed.
- Y. Qiao, S. Wu, J. Yi, Y. Sun, S. Guo, S. Yang, P. He and H. Zhou, Angew. Chem., Int. Ed., 2017, 129, 5042–5046 CrossRef.
- Z. Huang, J. Ren, W. Zhang, M. Xie, Y. Li, D. Sun, Y. Shen and Y. Huang, Adv. Mater., 2018, 30, e1803270 CrossRef PubMed.
- X. Liu, X. Song, Q. Zhang, X. Zhu, Q. Han, Z. Liu, P. Zhang and Y. Zhao, J. Energy Chem., 2022, 69, 516–523 CrossRef CAS.
- G. Yu, J.-Z. Wang, K. Li, J.-L. Ma and X.-B. Zhang, Adv. Mater., 2020, 32, 2004157 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr04371a |
| ‡ X. Wu and B. Niu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |