
Versatile electrooxidative amino- and oxyselenation of alkenes†
Renjie
Wang‡
a,
Nana
Zhang‡
a,
Yonghong
Zhang
 a,
Bin
Wang
a,
Yu
Xia
a,
Kai
Sun
b,
Weiwei
Jin
*ac,
Xinyong
Li
*d and
Chenjiang
Liu
*a
a,
Bin
Wang
a,
Yu
Xia
a,
Kai
Sun
b,
Weiwei
Jin
*ac,
Xinyong
Li
*d and
Chenjiang
Liu
*a
aUrumqi Key Laboratory of Green Catalysis and Synthesis Technology, Key Laboratory of Oil and Gas Fine Chemicals, Ministry of Education & Xinjiang Uygur Autonomous Region, State Key Laboratory of Chemistry and Utilization of Carbon Based Energy Resources, College of Chemistry, Xinjiang University, Urumqi 830017, P. R. China. E-mail: wwjin0722@163.com; pxylcj@126.com
bCollege of Chemistry and Chemical Engineering, Yantai University, Yantai 264005, P. R. China
cKey Laboratory of Specialty Agri-Product Quality and Hazard Controlling Technology of Zhejiang Province, College of Life Sciences, China Jiliang University, Hangzhou 310018, P. R. China
dAsymchem Life Science (Tianjin) Co., Ltd, No. 71, 7th Avenue, TEDA, Tianjin, 300457, P. R. China. E-mail: lixinyong@asymchem.com.cn
First published on 12th April 2023
Abstract
Herein, we describe a general and eco-friendly electrochemical methodology for amino- and oxyselenation of alkenes under transition-metal catalyst- and additional-oxidant-free conditions. This electrocatalytic difunctionalisation reaction exhibits excellent chemoselectivity, ample substrate scope, and high functional group tolerance. To our delight, the selenation products (118 examples, up to 99% yield) were constructed from various alkenes including the challenging 1-aryl-1,3-dienes, unactivated aliphatic alkenes, and various N- or O-centered nucleophiles. Preliminary mechanistic studies were conducted. The practical utility of this protocol is highlighted by the gram-scale synthesis and late-stage modification of bioactive molecules.
Introduction
Alkenes are important bulk chemicals which can be produced from the traditional petrochemical industry and emerging coal chemical industry. The difunctionalisation of accessible alkene feedstocks into varied highly value-added fine chemicals with increased molecular complexity is a cutting-edge research hotspot in synthetic chemistry.1,2 During the past few years, a wide spectrum of elegant alkene oxidative difunctionalisation reactions,3 which can concurrently introduce two vicinal C–C/C–C, C–hetero/C–hetero, or C–C/C–hetero bonds to multiple bonds, have been well established with the strategies of transition-metal catalysis,4–6 visible-light catalysis,7 and organocatalysis.8 Meanwhile, the asymmetric version of the alkene difunctionalisation reactions as a much more challenging topic has also achieved rapid progress.9,10Organoselenium compounds are widespread in the areas of the pharmaceutical industry,11,12 polymer chemistry,13 and advanced organic functional materials14 due to their abundant biological activities and specific photophysical and photochemistry properties. They are also exploited as small molecule catalysts15,16 and polydentate ligands17 in organic synthesis. The direct selenofunctionalisation of alkene skeletons with the formation of adjacent C–Se and C–X (X = carbon and heteroatoms) bonds has been proved to be a straightforward and powerful protocols for synthesising organic selenium-containing products.18–30 However, the use of transition-metal catalysts, excessive chemical oxidants, various reaction additives, and harsh reaction conditions of some of the methods mentioned above usually result in unwanted byproducts and large amounts of waste, thus increasing the difficulties of the workup procedure and bringing the risk of environmental pollution.
In recent years, electrochemical organic synthesis31,32 and the merging of electrosynthesis with transition-metal catalysis33 or visible-light photoredox catalysis34,35 have achieved remarkable advances in the functionalisation of olefins. Among them, tandem electrochemical oxidative inter- or intramolecular carbonselenation,36–38 aminoselenation,39 oxyselenation,39–50 selenosulfonylation,51 and fluoroselenation52 of alkene derivatives with C–X (X = C, N, O, S, F) and C–Se bonds formation have been successfully reported. Lei's group described an efficient electrooxidative amino- and oxyselenation of styrenes with benzotriazoles, carboxylic acids, and alcohols as the nucleophiles.39 However, only 35% of the target product is obtained when saccharin is used as the nitrogen source. In contrast, other electrochemical oxyselenation processes are mainly confined to the intramolecular annulation reactions of unactivated alkenes, such as olefinic carbonyl, olefinic alcohols, alkenoic acids, allylphenol, β,γ-unsaturated amides, and olefinic amides. Notably, some electrooxidative carbonselenation,53,54 oxyselenation,55,56 and diselenylation57 of alkynes have also been realised.
Compared with the extensively studied difunctionalisation of activated alkenes, the conversion of inactive aliphatic olefins has rarely been researched and the general and green protocol for the difunctionalisation of activated alkenes58–63 and challenging 1-aryl-1,3-dienes64,65 is far from established. Based on our previous work on the electrooxidative trifunctionalisation of styrenes,66 we herein present a universal methodology for the regioselective aminoselenation reaction of alkenes including activated arylalkenes, challenging 1-aryl-1,3-dienes, and non-activated aliphatic alkenes, and excitingly the oxyselenation of inert alkenes was also successful from diversified carboxylic acids (Scheme 1).
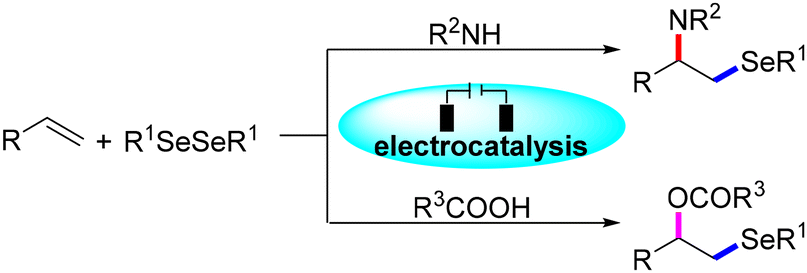 | ||
| Scheme 1 Electrooxidative amino- and oxyselenation of alkenes. | ||
Results and discussion
After the systematic screening of reaction parameters (see Tables S1–S3 in the ESI† for details), the reaction conditions for the aminoselenation of activated alkenes were optimised as follows: in an undivided cell with C(+)/Ni(−) electrodes, constant current electrolysis was performed at 10 mA with Et4BrN as the electrolyte in CH3CN at room temperature for 2 h. With optimized reaction conditions in hand, the scope for the aminoselenation of activated alkenes 1 was primarily studied (Scheme 2). Generally, under standard conditions, a wide variety of substituted styrenes with varied electron-donating and -withdrawing groups can be conveniently transformed into their corresponding products 4a–4p in moderate to excellent yields (53–93%). Moreover, 2-vinylnaphthalene, 2-vinylthiophene, and vinyl ferrocene also delivered their respective aminoselenation products 4q–4s in 94%, 84%, and 45% yields. Even substrates containing steric hindrance, such as 1H-indene, α-methlystyrene, and β-methlystyrene, smoothly produce the target products 4t–4v with good yields.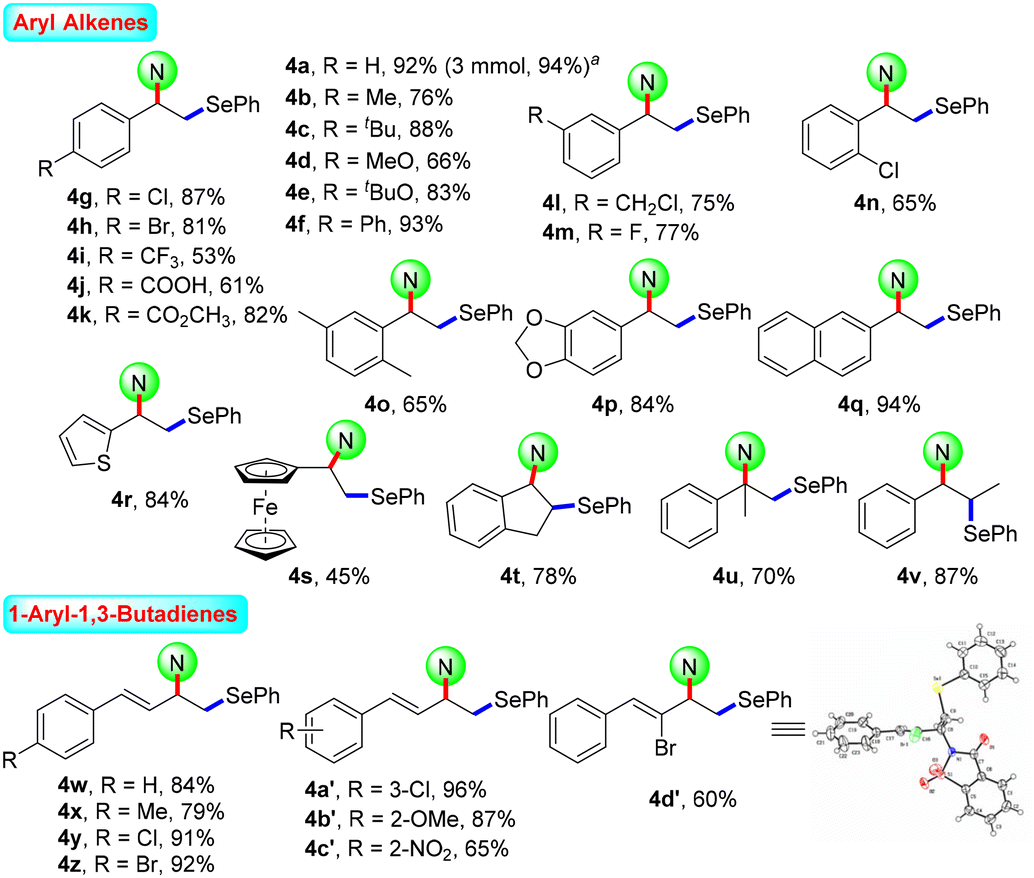 | ||
Scheme 2 Substrate scope for the aminoselenation of activated alkenes. Reaction conditions: 1 (0.3 mmol), 2a (0.2 mmol), 3a (0.15 mmol), Et4NBr (0.2 mmol), CH3CN (4.0 mL), C anode, Ni cathode, 10 mA, 2 h, r.t., undivided cell. Isolated yields. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Gram-scale reaction. Gram-scale reaction. | ||
1,3-Dienes often function as versatile feedstocks to assemble structurally diverse, complex molecular architectures by way of transition-metal- or visible-light photoredox-catalyzed alkene-functionalization reactions. In light of 1,3-dienes containing multiple reaction sites, the regioselective alkene functionalisation of 1,3-dienes is a challenging research topic. Delightfully, this methodology was successfully expanded to 1-aryl-1,3-dienes under standard electrolytic conditions with exclusive 1,2-regio- and stereoselectivity. The substituent effects of the aromatic ring had little influence on the reaction efficiency. A series of functional groups, including –Me, –MeO, –Cl, –Br, and even strongly deactivated –NO2, were well tolerated, forming the products 4w–4c′ in good to excellent yields. Notably, the reaction of (Z)-(2-bromobuta-1,3-dien-1-yl)benzene with saccharin (2a) and diphenyl diselenide (3a) proceeded well to result in the target product 4d′ in 60% yield. The molecular structure of 4d′ was determined by single-crystal X-ray diffractometry.
Aliphatic alkenes usually have lower reaction activity and are difficult to activate. The functionalisation of this kind of skeleton is a promising and cutting-edge research field, and has attracted much attention from synthetic chemists all over the world. Various non-activated aliphatic olefins were selected to probe the generality and limitations of this catalytic system. As shown in Scheme 3, a wide range of substituted allylbenzenes were selected as substrates, furnishing the corresponding products 5a–5g in 44–79% yields. Using homoallylbenzene as the starting material, the desired aminoselenation product 5h was also generated in moderate yield. In the same fashion, unactivated propylenes with attached useful synthetic functional groups such as phenol oxygen, ester, carbonate, amide, and phthalimide were suitable substrates, producing the difunctionalisation products 5i–5o in acceptable yields. Moreover, diselenide substrate scope was also investigated using allylbenzene as the olefin source. Under optimal electrolytic conditions, substituted diaryl- and dibenzyl diselenides were subjected to this reaction, showing moderate reaction efficiencies for the generation of the corresponding products 5p–5x.
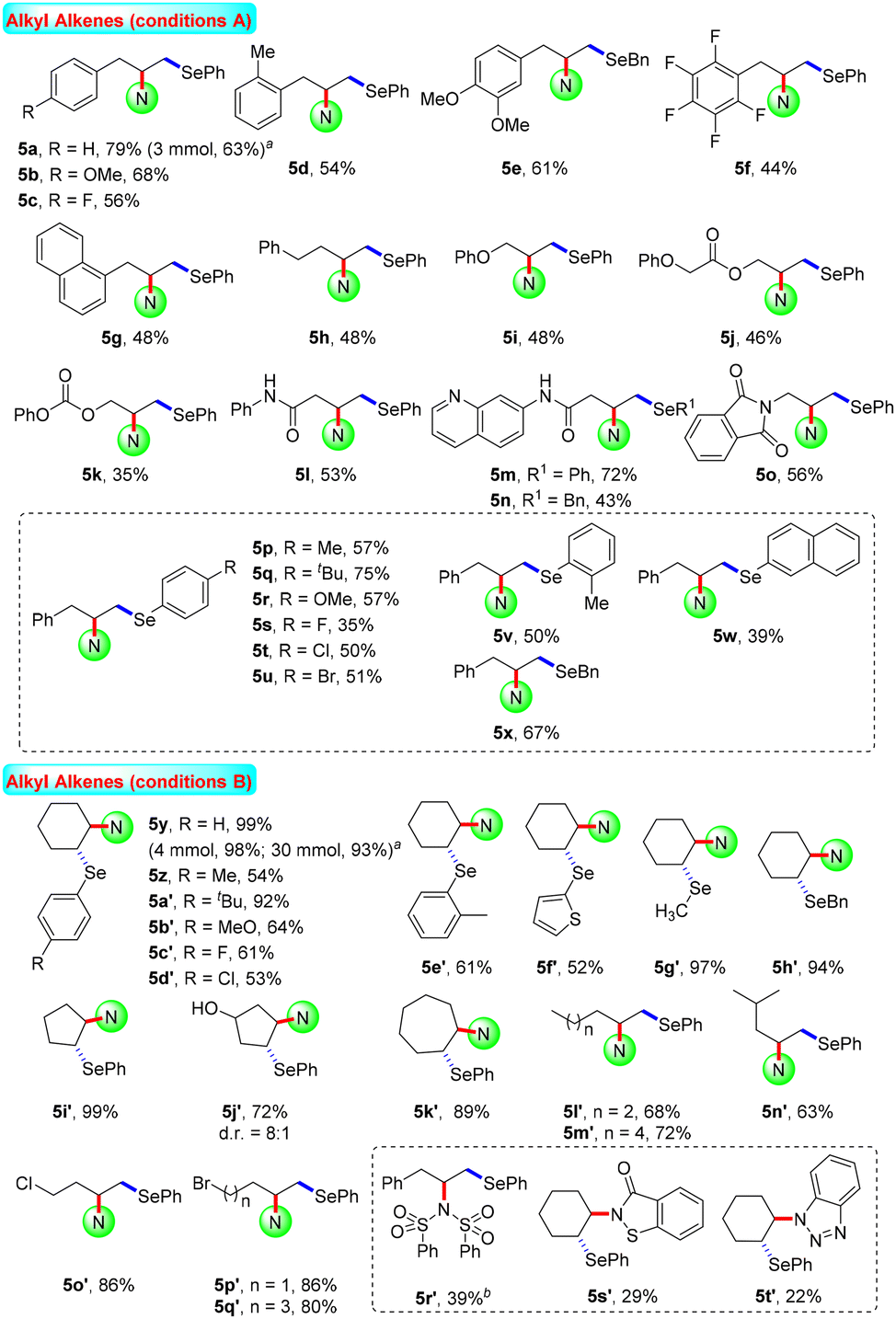 | ||
| Scheme 3 Substrate scope for the aminoselenation of unactivated alkenes. Reaction conditions: 1 (0.3 mmol), 2a (0.2 mmol), 3 (0.15 mmol), r.t., undivided cell. Conditions A: Et4NBr (0.2 mmol), CH3CN (4.0 mL), C anode, Ni cathode, 10 mA, 3 h; conditions B: VBImBr (0.2 mmol), CH3CN/TFE (4 mL, v:v = 1:3), C anode, C cathode, 5 mA, 2 h. Isolated yields. aGram-scale reaction. bUnder conditions A. VBImBr = 1-butyl-3-vinylimidazolium bromide. TFE = 2,2,2-trifluoroethanol. | ||
For cyclohexene, a moderate isolated yield (66%) of the target product 5y was obtained under the current standard conditions A, as shown in Scheme 3. Under modified electrolysis conditions (conditions B in Scheme 3, see Tables S4–S11† for details), the desired product 5y was formed with up to a quantitative yield. Furthermore, a suite of symmetric diselenides containing diaryl, diheteroaryl, and dialkyl diselenides were well involved in this electrochemical reaction system to assemble the expected disubstituted products 5z–5h′ in moderate to excellent yields (52–97%). In addition, 5-/7-membered cyclic and long chain aliphatic alkenes were proved to be good starting materials and led to the desired aminoselenation products 5i′–5q′ in satisfactory yields. Unfortunately, due to the relatively poor nucleophilic ability, other amine sources as nucleophiles only presented lower reaction activities (5r′–5t′). The possible reason may be the smaller reduction current of saccharin than other N-centered nucleophiles; this gives it a higher nucleophilicity by smoothly losing the protons (see Fig. S4 in the ESI† for details).
Inspired by the good performance of the electrooxidative aminoselenation of various alkenes, we envisioned that this protocol may be qualified to realise the oxyselenation of inert olefins with similar electrolytic processes. Pleasingly, under the slightly altered reaction conditions of Scheme 3, B (see Tables S12–S15† for details), the electrochemical oxyselenation of unactivated alkenes proved to be feasible and the desired product 7a was produced in 90% yield (Scheme 4). Benzoic acids decorated with electron-rich (–Me, –Et, –tBu, –MeO, –MeS, and –PPh2) and -deficient groups (–F, –Cl, –Br, –I, –CF3, –CN, and –NO2) on the phenyl ring were compatible to give rise to 7b–7p in 71–97% yields. Other analogues of carboxylic acid, such as condensed, polyhalogenated, and aryl alkynyl carboxylic acid reacted well with saccharin and diphenyl diselenide to generate the corresponding products 7q–7t in moderate to good yields. Importantly, the treatment of aliphatic carboxylic acids formed the oxyselenation products 7u–7y in good yields. In the cases of 5-, 7-, 8-membered cyclic alkenes and the linear aliphatic alkenes, the desired products 7z–7d′ were obtained with up to 99% yield. It should be pointed out that these oxyselenation products substituted with plenty of easily modified function groups can be transformed for the further production of synthetically useful organic molecules. The structures of products 5y and 7m were finally determined by X-ray single crystal analysis (Fig. 1). Unfortunately, no desired difunctionalization products were formed when aliphatic alcohols such as methanol and ethanol were used as nucleophiles.
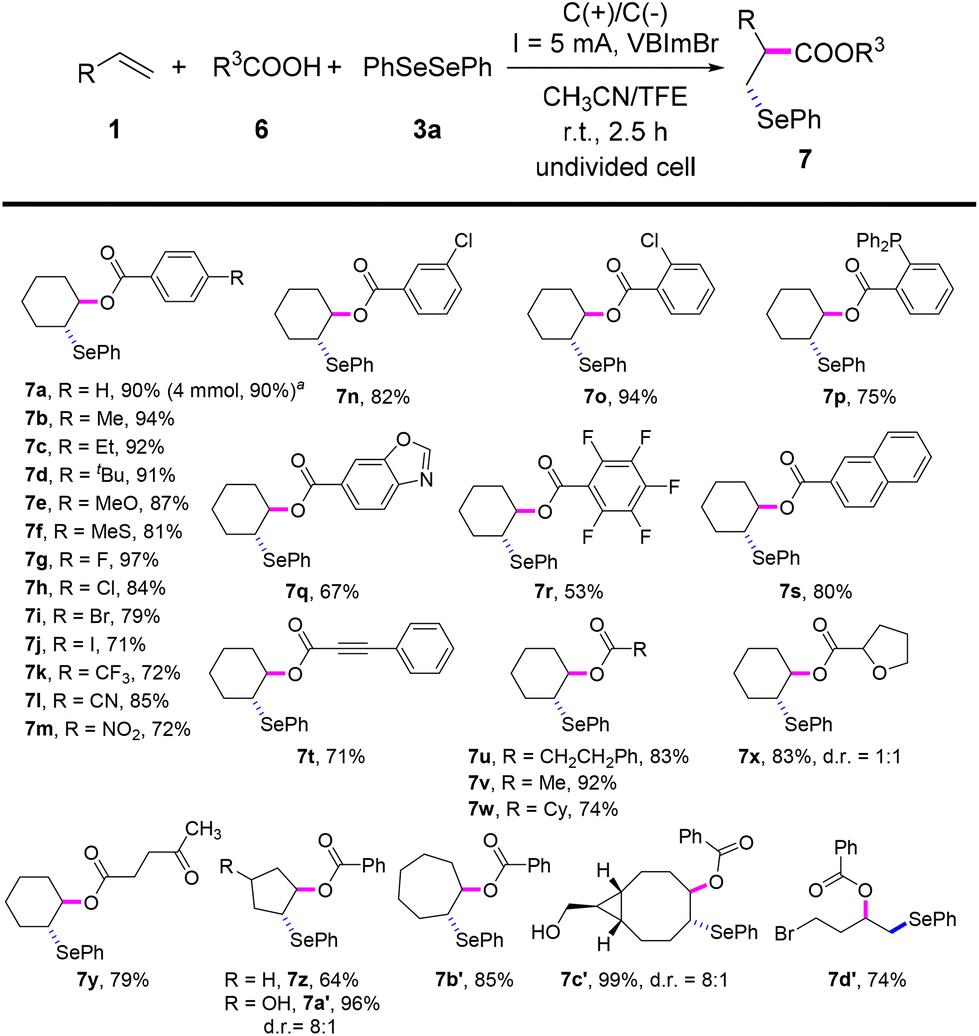 | ||
| Scheme 4 Substrate scope for the oxyselenation of unactivated alkenes. Reaction conditions: 1 (0.2 mmol), 6 (0.4 mmol), 3a (0.15 mmol), VBImBr (0.2 mmol), CH3CN/TFE (4 mL, v:v = 2:2), C anode, C cathode, 5 mA, 2.5 h, r.t., undivided cell. Isolated yields. aGram-scale reaction. | ||
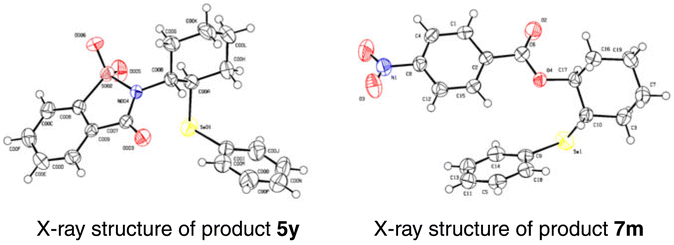 | ||
| Fig. 1 Crystal structures of compounds 5y and 7m. | ||
The practicability of this protocol was confirmed by the late-stage modification of bioactive molecules (Scheme 5). Estrone derivatives were smoothly disubstituted to give selenating products 8a and8b in 75% and 91% yields, respectively. Aryl-, cinnamyl-, and heteroarylcarboxylic acid had moderate to good reaction efficiencies (8c–8h). With regard to the aliphatic carboxylic acid, the expected oxyselenation products (8i–8l) were obtained in satisfactory yields. Moreover, this electrosynthetic technology can be easily scaled up to gram scale for activated and unactivated alkenes (4a, 5a, 5y, and 7a).
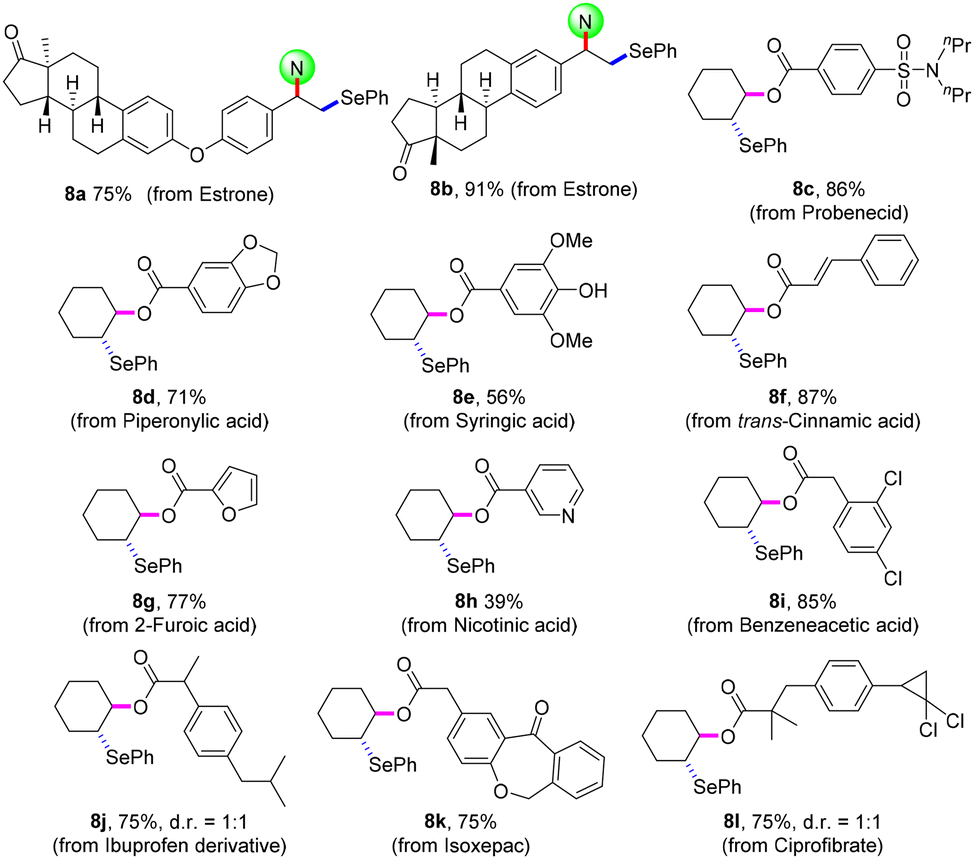 | ||
| Scheme 5 Late-stage modification of bioactive molecules. Reaction conditions: for 8a and8b, conditions in Scheme 1; for 8c–8l, conditions in Scheme 4. Isolated yields. | ||
According to our mechanistic probe experiments (see the ESI† for details) and literature reports, a plausible mechanism for this electrochemical amino- and oxyselenation procedure is proposed. As shown in Scheme 6 path I, diselenide 3 is first oxidized at the anode to give the cationic radical intermediate A, which is dissociated into selenium cation B and selenium radical C. Cation B then reacts with alkene 1 to form the cyclic selenonium ion intermediate D. Finally, the desired product is obtained following the nucleophilic attack of intermediate D by the N- or O-based nucleophile, as well as deprotonation. Another possible free radical mechanism cannot be completely excluded (path II). In this process, carbon radical H is generated by the addition of selenium radical C to the carbon–carbon double bond of alkene 1. H is then converted to its cation I by anodic oxidation. Subsequently, the desired product is produced by concessive nucleophilic attack and a deprotonation sequence.
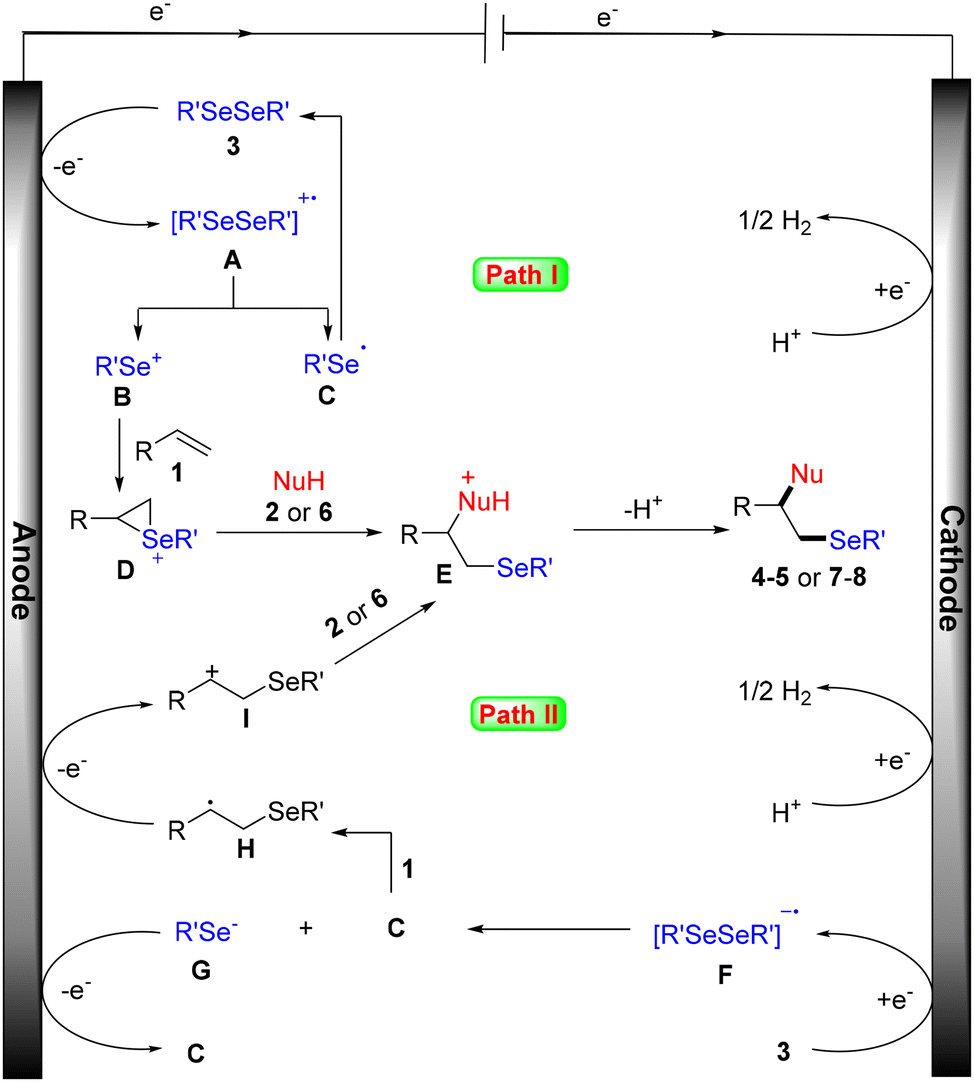 | ||
| Scheme 6 Plausible mechanism. | ||
Conclusions
In conclusion, we developed an environmentally friendly synthetic methodology for the sustainable amino- and oxyselenation of varied olefins with clean electric energy. A wide spectrum of readily available alkenes including arylalkenes, 1-aryl-1,3-dienes, and non-activated aliphatic alkenes are involved in this protocol. The successful implementation of scale-up experiments and the late-stage modification of bioactive molecules prove the potential application for producing selenium-containing drugs. Preliminary mechanistic studies suggest that the reaction was induced by both the free radical mechanism and the ionic mechanism. This strategy provides a universal and modular route to organoselenium compounds from easily available starting materials by the electrooxidative difunctionalisation of carbon–carbon double bonds.Author contributions
Renjie Wang and Nana Zhang carried out the experiments and data analyses and prepared the original manuscript. Yonghong Zhang, Bin Wang, Yu Xia, and Kai Sun provided experimental assistance and constructive suggestions. Weiwei Jin rewrote and revised the manuscript. Weiwei Jin, Xinyong Li, and Chenjiang Liu directed the project and supervised the whole experiment. All the authors discussed and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 21702175, 21961037, and 22161044), the Program for Tianshan Innovative Research Team of the Xinjiang Uygur Autonomous Region (2021D14011), and the Natural Science Foundation of the Xinjiang Uygur Autonomous Region (2020D01C077) for supporting this research.Notes and references
- X.-W. Lan, N.-X. Wang and Y. Xing, Eur. J. Org. Chem., 2017, 5821–5851 CrossRef CAS.
- X. Fu and W. Zhao, Chin. J. Org. Chem., 2019, 39, 625–647 CrossRef CAS.
- J. Lin, R.-J. Song, M. Hu and J.-H. Li, Chem. Rec., 2019, 19, 440–451 CrossRef CAS PubMed.
- J. Liu, X. Xiao, Y. Lai and Z. Zhang, Org. Chem. Front., 2022, 9, 2256–2279 RSC.
- Y. Li, D. Wu, H.-G. Cheng and G. Yin, Angew. Chem., Int. Ed., 2020, 59, 7990–8003 CrossRef CAS PubMed.
- R. K. Dhungana, R. R. Sapkota, D. Niroula and R. Giri, Chem. Sci., 2020, 11, 9757–9774 RSC.
- X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed.
- R. M. Romero, T. H. Wöste and K. Muñiz, Chem. – Asian J., 2014, 9, 972–983 CrossRef CAS PubMed.
- Z.-L. Li, G.-C. Fang, Q.-S. Gu and X.-Y. Liu, Chem. Soc. Rev., 2020, 49, 32–48 RSC.
- F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS PubMed.
- C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255–6285 CrossRef CAS PubMed.
- G. Mugesh, W.-W. du Mont and H. Sies, Chem. Rev., 2001, 101, 2125–2179 CrossRef CAS PubMed.
- S. Ye, V. Lotocki, H. Xu and D. S. Seferos, Chem. Soc. Rev., 2022, 51, 6442–6474 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- L. Liao and X. Zhao, Acc. Chem. Res., 2022, 55, 2439–2453 CrossRef CAS PubMed.
- Y. Li, Y. Wang, T. Yang, Z. Lin and X. Jiang, Green Chem., 2021, 23, 2986–2991 RSC.
- A. Kumar, G. K. Rao, F. Saleem and A. K. Singh, Dalton Trans., 2012, 41, 11949–11977 RSC.
- G. Perin, E. J. Lenardão, R. G. Jacob and R. B. Panatieri, Chem. Rev., 2009, 109, 1277–1301 CrossRef CAS PubMed.
- Y.-Q. Jiang, Y.-H. Wang, C.-F. Zhou, Y.-Q. Zhang, Y. Ling, Y. Zhao and G.-Q. Liu, J. Org. Chem., 2022, 87, 14609–14622 CrossRef CAS PubMed.
- K. Sun, X. Wang, C. Zhang, S. Zhang, Y. Chen, H. Jiao and W. Du, Chem. – Asian J., 2017, 12, 713–717 CrossRef CAS PubMed.
- Z. Zhang, S. Wang, P. Tan, X. Gu, W. Sun, C. Liu, J. Chen, J. Li and K. Sun, Org. Lett., 2022, 24, 2288–2293 CrossRef CAS PubMed.
- K. Sun, X. Wang, C. Li, H. Wang and L. Li, Org. Chem. Front., 2020, 71, 3100–3119 RSC.
- E. Tang, W. Wang, Y. Zhao, M. Zhang and X. Dai, Org. Lett., 2016, 18, 176–179 CrossRef CAS PubMed.
- K. Sun, X. Wang, Y. Lv, G. Li, H. Jiao, C. Dai, Y. Li, C. Zhang and L. Liu, Chem. Commun., 2016, 52, 8471–8474 RSC.
- L.-L. Zhu, L. Tian, B. Cai, G. Liu, H. Zhang and Y. Wang, Chem. Commun., 2020, 56, 2979–2982 RSC.
- X. Li, J. Huang, L. Xu, P. Lin and Y. Wei, J. Org. Chem., 2022, 87, 10684–10697 CrossRef CAS PubMed.
- H. Guan, H. Wang, D. Huang and Y. Shi, Tetrahedron, 2012, 68, 2728–2735 CrossRef CAS.
- X. Li, P. He, H.-B. Zhou and C. Dong, Org. Biomol. Chem., 2018, 16, 2150–2155 RSC.
- M. Chaitanya and P. Anbarasan, Org. Lett., 2018, 20, 1183–1186 CrossRef CAS PubMed.
- E. Tang, Y. Zhao, W. Li, W. Wang, M. Zhang and X. Dai, Org. Lett., 2016, 18, 912–915 CrossRef CAS PubMed.
- J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed.
- N. Chen and H.-C. Xu, Green Synth. Catal., 2021, 2, 165–178 CrossRef.
- C. A. Malapit, M. B. Prater, J. R. Cabrera-Pardo, M. Li, T. D. Pham, T. P. McFadden, S. Blank and S. D. Minteer, Chem. Rev., 2022, 122, 3180–3218 CrossRef CAS PubMed.
- M.-J. Luo, Q. Xiao and J.-H. Li, Chem. Soc. Rev., 2022, 51, 7206–7237 RSC.
- G.-Q. Liu, C.-F. Zhou, Y.-Q. Zhang, W. Yi, P.-F. Wang, J. Liu and Y. Ling, Green Chem., 2021, 23, 9968–9973 RSC.
- Y. J. Kim and D. Y. Kim, Org. Lett., 2019, 21, 1021–1025 CrossRef CAS PubMed.
- X.-Y. Wang, Y.-F. Zhong, Z.-Y. Mo, S.-H. Wu, Y.-L. Xu, H.-T. Tang and Y.-M. Pan, Adv. Synth. Catal., 2021, 363, 208–214 CrossRef CAS.
- L. Sun, L. Wang, H. Alhumade, H. Yi, H. Cai and A. Lei, Org. Lett., 2021, 23, 7724–7729 CrossRef CAS PubMed.
- L. Sun, Y. Yuan, M. Yao, H. Wang, D. Wang, M. Gao, Y.-H. Chen and A. Lei, Org. Lett., 2019, 21, 1297–1300 CrossRef CAS PubMed.
- J. Chen, L. Mei, H. Wang, L. Hu, X. Sun, J. Shi and Q. Li, ChemistryOpen, 2019, 8, 1230–1234 CrossRef CAS PubMed.
- Z. Guan, Y. Wang, H. Wang, Y. Huang, S. Wang, H. Tang, H. Zhang and A. Lei, Green Chem., 2019, 21, 4976–4980 RSC.
- X.-J. Meng, P.-F. Zhong, Y.-M. Wang, H.-S. Wang, H.-T. Tang and Y.-M. Pan, Adv. Synth. Catal., 2020, 362, 506–511 CrossRef CAS.
- Y. Kim, J. Jang and D. Y. Kim, Asian J. Org. Chem., 2021, 10, 3271–3274 CrossRef CAS.
- S. Mallick, M. Baidya, K. Mahanty, D. Maiti and S. De Sarkar, Adv. Synth. Catal., 2020, 362, 1046–1052 CrossRef CAS.
- M. R. Scheide, A. R. Schneider, G. A. M. Jardim, G. M. Martins, D. C. Durigon, S. Saba, J. Rafique and A. L. Braga, Org. Biomol. Chem., 2020, 18, 4916–4921 RSC.
- A. Kharma, C. Jacob, Í. A. O. Bozzi, G. A. M. Jardim, A. L. Braga, K. Salomão, C. C. Gatto, M. F. S. Silva, C. Pessoa, M. Stangier, L. Ackermann and E. N. da Silva Júnior, Eur. J. Org. Chem., 2020, 4474–4486 CrossRef CAS.
- X. Cheng, B. Hasimujiang, Z. Xu, H. Cai, G. Chen, G. Mo and Z. Ruan, J. Org. Chem., 2021, 86, 16045–16058 CrossRef CAS PubMed.
- H. Li, F. Lu, J. Xu, J. Hu, H. Alhumade, L. Lu and A. Lei, Org. Chem. Front., 2022, 9, 2786–2791 RSC.
- K. Lin, J. Lan and T. Zhu, Adv. Synth. Catal., 2022, 364, 3466–3471 CrossRef CAS.
- F. Lu, J. Xu, H. Li, K. Wang, D. Ouyang, L. Sun, M. Huang, J. Jiang, J. Hu, H. Alhumade, L. Lu and A. Lei, Green Chem., 2021, 23, 7982–7986 RSC.
- X. Kong, K. Yu, Q. Chen and B. Xu, Asian J. Org. Chem., 2020, 9, 1760–1764 CrossRef CAS.
- S.-F. Wu, Y. Yu, Y. Yuan, Z. Li and K.-Y. Ye, Eur. J. Org. Chem., 2022 DOI:10.1002/ejoc.202201032.
- J. Hua, Z. Fang, M. Bian, T. Ma, M. Yang, J. Xu, C. Liu, W. He, N. Zhu, Z. Yang and K. Guo, ChemSusChem, 2020, 13, 2053–2059 CrossRef CAS PubMed.
- Z.-W. Hou, L. Li and L. Wang, Org. Chem. Front., 2022, 9, 2815–2820 RSC.
- X. Lin, Z. Fang, C. Zeng, C. Zhu, X. Pang, C. Liu, W. He, J. Duan, N. Qin and K. Guo, Chem. – Eur. J., 2020, 26, 13738–13742 CrossRef CAS PubMed.
- D. Maiti, A. Halder, A. S. Pillai and S. De Sarkar, J. Org. Chem., 2021, 86, 16084–16094 CrossRef CAS PubMed.
- P. Zhou, H. Jiao, K. Niu, H. Song, Y. Liu and Q. Wang, ACS Sustainable Chem. Eng., 2023, 11, 2607–2612 CrossRef CAS.
- H.-Y. Tu, S. Zhu, F.-L. Qing and L. Chu, Synthesis, 2020, 1346–1356 CAS.
- Y. Li, Y. Li, H. Shi, H. Wei, H. Li, I. Funes-Ardoiz and G. Yin, Science, 2022, 376, 749–753 CrossRef CAS PubMed.
- Y. Lei, J. Yang, Y. Wang, H. Wang, Y. Zhan, X. Jiang and Z. Xu, Green Chem., 2020, 22, 4878–4883 RSC.
- L. Song, S. Luo and J.-P. Cheng, Org. Chem. Front., 2016, 3, 447–452 RSC.
- F.-H. Cui, Y. Hua, Y.-M. Lin, J. Fei, L.-H. Gao, X. Zhao and H. Xia, J. Am. Chem. Soc., 2022, 144, 2301–2310 CrossRef CAS PubMed.
- P. Zhou, K. Niu, H. Song, Y. Liu and Q. Wang, Green Chem., 2022, 24, 5760–5763 RSC.
- D.-K. Wang, L. Li, Q. Xu, J. Zhang, H. Zheng and W.-T. Wei, Org. Chem. Front., 2021, 8, 7037–7049 RSC.
- P.-Z. Wang, W.-J. Xiao and J.-R. Chen, Chin. J. Catal., 2022, 43, 548–557 CrossRef CAS.
- G. Qin, R. Wang, Z. Cheng, Y. Zhang, B. Wang, Y. Xia, W. Jin and C. Liu, Green Synth. Catal., 2022 DOI:10.1016/j.gresc.2022.10.008 , in press.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2233790, 2191956 and 2227321. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc00837a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |