
Electrochemical dual α,β-C(sp3)–H functionalization of cyclic N-aryl amines†
Tian
Feng
a,
Zile
Zhu
a,
Dongmei
Zhang
b,
Siyi
Wang
a,
Ruopu
Li
a,
Zhaolin
Zhu
a,
Xinxing
Zhang
 *b and
Youai
Qiu
*a
*b and
Youai
Qiu
*a
aState Key Laboratory and Institute of Elemento-Organic Chemistry, Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai, University, 94 Weijin Road, Tianjin, 300071, China. E-mail: qiuyouai@nankai.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Renewable Energy Conversion and Storage Center (RECAST), Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai University, 94 Weijin Road, Tianjin, 300071, China. E-mail: Zhangxx@nankai.edu.cn
First published on 9th March 2023
Abstract
Herein, a straightforward and efficient route for the construction of dual α,β-C(sp3)–H functionalized cyclic N-aryl amines using a combination of electrocatalysis and iron catalysis is disclosed. This method is achieved by multiple single electron oxidations and is carried out in an operationally simple manner with high chemo- and site-selectivity, setting the stage for the challenging multiple site selective C–H functionalization. The mechanism is supported by a variety of experimental probes and theoretical investigations, including cyclic voltammetry tests, a bipolar ultramicroelectrode (BUME) methodology combined with nano-electrospray ionization mass spectrometry, and density functional theory calculations.
Introduction
Electrocatalysis has gained popularity over the past few years as an alternative and environmentally-friendly method for sustainable transformations in modern organic synthesis. It benefits from bypassing the use of stoichiometric oxidants or reductants through efficient electron transfer1 and has been applied to C–H activation,2 alkene functionalization,3 C–H oxidation,4 cross-coupling reactions,5 and others.6 Among which, electro-oxidative C(sp3)–H bond functionalization attracts much attention because the C(sp3)–H bond is one of the most ubiquitous motifs in organic molecules, including the benzylic C(sp3)–H bond,4e,7 the allylic C(sp3)–H bond,2g,4b,8 the C(sp3)–H bond adjacent to an X (X = N, O, S) atom6f,9 represented by Shono oxidation,10 and even the inert C(sp3)–H bond (Fig. 1a)2c,4c,11 Despite the significant progress in this field, the vast majority of such methods have mainly focused on the transformation of the single C(sp3)–H bond, whereas many synthetic targets require the transformation of multiple C–H bonds which have been far elusive. One of the major challenges of such reactions is that the installation of one functional group is inclined to deactivate the surrounding C–H bonds. Hence, the development of simultaneous electrochemical methodologies for direct, efficient, and selective multiple C–H functionalization in a greener, more general, and economic way, facilitating potential practical applications, is still scarce and highly desirable yet challenging.12 Notably, Lambert and co-workers reported an elegant electrophotocatalytic diamination of vicinal C(sp3)–H bonds by successive Ritter-type aminations,13 using a combination of light and electrical energy,14 and this strategy was also further successfully applied to the oxygenation of multiple adjacent C(sp3)–H bonds,15 which are elegant examples of synthesizing multi-functionalized compounds by electrophotocatalysis with the potential for further applications.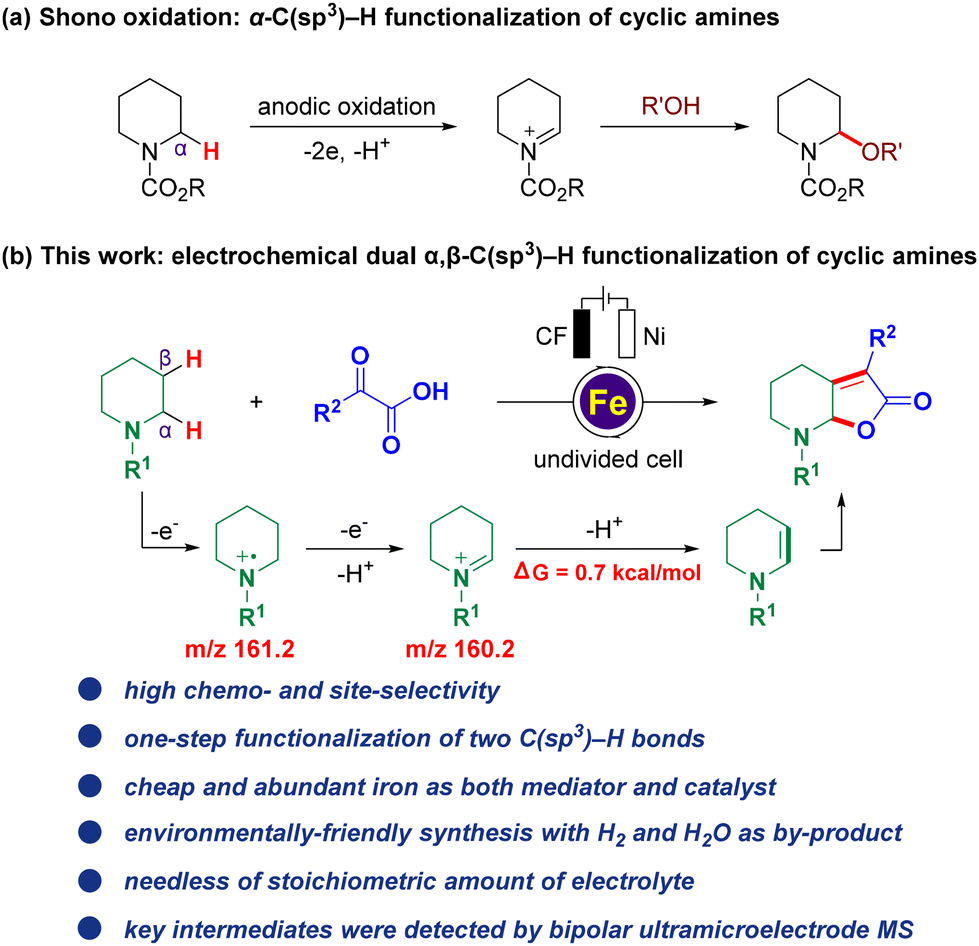 | ||
| Fig. 1 Background and current work. (a) Shono oxidation: α-C(sp3)–H functionalization of cyclic amines. (b) Electrochemical dual α,β-C(sp3)–H functionalization (this work). | ||
With our continuous interest in the electrochemical C–H functionalization of cyclic amines16 and sustainable electrochemical transformation,17 herein, we report the first iron-catalyzed electrochemical dual α,β-C(sp3)–H functionalization of cyclic amines (Fig. 1b). Notable features include: (a) high chemo- and site-selectivity, (b) one-step functionalization of vicinal C(sp3)–H bonds, (c) an inexpensive and Earth-abundant iron complex used as both a mediator and catalyst, (d) environmentally-friendly synthesis with H2 and H2O as by-products and (e) non-requirement of stoichiometric amounts of the electrolyte.
Results and discussion
Optimization of electrochemical dual C(sp3)–H functionalization
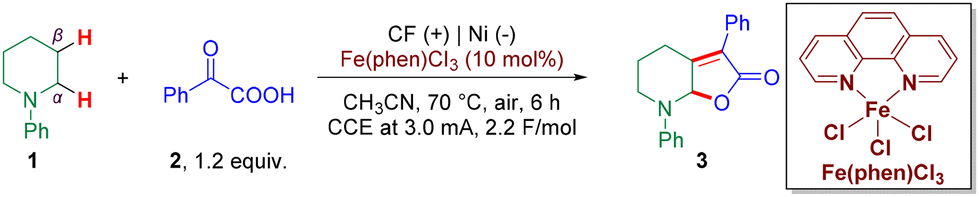
We began our investigation by employing N-phenyl piperidine 1 and 2-oxo-2-phenylacetic acid 2 as model substrates for the initial optimization of the envisioned transformation. To our delight, the desired dual C(sp3)–H functionalized product 3 was obtained in 80% yield with constant current electrolysis selectively performed in the presence of a catalytic amount of the iron catalyst (Fe(phen)Cl3 (10 mol%)) with cheap carbon felt (CF, price of CF: 25 dollars per m2, and the CF could also be reused.) being employed as the anode and a nickel plate as the cathode (Table 1, entry 1). To further optimize this reaction, several reaction parameters were examined. Firstly, control experiments showed that only a trace amount of the dual functionalized product was obtained in the absence of Fe(phen)Cl3, which demonstrated the crucial role of the iron catalyst (Table 1, entry 2). Interestingly, 7% of 3 was obtained without electricity, and it was considered that the Fe(III) complex could undergo a single electron transfer (SET) with 1 (Table 1, entry 3). Using FeCl3 (10 mol%) and 1,10-phenanthroline (10 mol%) in place of Fe(phen)Cl3 resulted in a lower yield of 3 (59%) (Table 1, entry 4), and in the absence of 1,10-phenanthroline, the yield of the product harshly decreased to 44% with a considerable amount of the starting material remaining, which demonstrated the significant role of the ligand (Table 1, entry 5). Different valences of iron catalysts were also investigated, such as Fe(phen)Cl2, which gave the product in a lower yield (58%) (Table 1, entry 6). Next, considering that the iron catalyst could also act as a Lewis acid in this transformation, we explored other commonly used Lewis acids such as ZnCl2, CuCl2, and B(C6F5)3, and the results showed a much lower efficiency (Table 1, entry 7) or even complete impediment of the reaction (Table 1, entry 8). Using a graphite felt anode instead of carbon felt afforded the desired product in 56% (Table 1, entry 9). Replacing the Ni plate with a Pt plate as the cathode furnished the product with a slight decrease in the yield (Table 1, entry 10), and when a larger specific surface area Ni foam was also used as the cathode, it resulted in only 38% yield of 3 (Table 1, entry 11). Next, we investigated the influence of the constant current and temperature, and the results showed that a higher current led to a lower yield of 3 (54%) possibly due to overoxidation (Table 1, entry 12). The reaction worked well at a lower temperature (50 °C) although in a slightly lower yield (66%, Table 1, entry 13), and 35% yield of 3 was obtained at room temperature (Table 1, entry 14).|
|
||
|---|---|---|
| Entry | Variation | Yieldb/% |
| a Reaction conditions: undivided cell, carbon felt (CF) anode, nickel plate cathode, constant current = 3.0 mA, N-phenyl piperidine 1 (0.3 mmol), 2-oxo-2-phenylacetic acid 2 (0.36 mmol, 1.2 equiv.), Fe(phen)Cl3 (10 mol%), CH3CN (5.0 mL), 70 °C, 6 h. b Yield of the isolated product. GF = graphite felt. RT = room temperature. | ||
| 1 | None | 80 |
| 2 | No Fe(phen)Cl3 | Trace |
| 3 | No electricity | 7 |
| 4 | FeCl3 (10 mol%) + 1,10-phen (10 mol%) | 59 |
| 5 | w/o 1,10-phen | 44 |
| 6 | Fe(phen)Cl2 | 58 |
| 7 | ZnCl2 or CuCl2 | 26 or 33 |
| 8 | B(C6F5)3 | — |
| 9 | GF ( + ) | 56 |
| 10 | Pt (−) | 73 |
| 11 | Ni foam (−) | 38 |
| 12 | 5.0 mA | 54 |
| 13 | 50 °C | 66 |
| 14 | RT | 35 |
Substrate scope
Encouraged by the successful optimization of the reaction conditions, we explored the generality of this electrochemical dual C(sp3)–H functionalization on various cyclic N-aryl amines and aryl keto acids (Fig. 2). Firstly, a diverse range of N-aryl piperidines bearing different electron donating groups (EDGs) or electron withdrawing groups (EWGs) were screened, which all worked well to afford the corresponding products in moderate to good yields (4–13), and no obvious electronic affect was observed. Other aryl motifs, including naphthalene (Np) and dihydrobenzofuran, were all well tolerated and gave the corresponding products in moderate yields (14, 15). Substrates bearing different substituents on the piperidine motif such as 2-Me, 3-Me, 4-Me, or even a potentially sensitive ester group at the 4-position all worked smoothly and generated the corresponding products with a diastereomeric ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1.67:1 for 16 and 17, respectively. Remarkably, 4-Me and 4-CO2Et substituted piperidine substrates could furnish the corresponding products in high levels of stereoselectivity (18, 19), and the configuration of 19 was verified by X-ray analysis. To our delight, substrates with different ring sizes of cyclic N-phenyl amines, also successfully led to the formation of the corresponding seven-membered product with good efficiency (for example, 20). Next, we focused on the scope of the α-aryl keto acid component. Both EDGs and EWGs at the ortho-, meta- or para-position of the benzene ring could furnish the corresponding desired products in moderate to good yields (21–30), and no obvious electronic effect was observed. In addition, this electrochemical C(sp3)–H dual functionalization was also compatible with other aromatic motifs such as 2-Np and MDB (1,2-methylenedioxybenzene) substituted keto acids, furnishing 31 and 32 in good yields.
:1 and 1.67:1 for 16 and 17, respectively. Remarkably, 4-Me and 4-CO2Et substituted piperidine substrates could furnish the corresponding products in high levels of stereoselectivity (18, 19), and the configuration of 19 was verified by X-ray analysis. To our delight, substrates with different ring sizes of cyclic N-phenyl amines, also successfully led to the formation of the corresponding seven-membered product with good efficiency (for example, 20). Next, we focused on the scope of the α-aryl keto acid component. Both EDGs and EWGs at the ortho-, meta- or para-position of the benzene ring could furnish the corresponding desired products in moderate to good yields (21–30), and no obvious electronic effect was observed. In addition, this electrochemical C(sp3)–H dual functionalization was also compatible with other aromatic motifs such as 2-Np and MDB (1,2-methylenedioxybenzene) substituted keto acids, furnishing 31 and 32 in good yields.
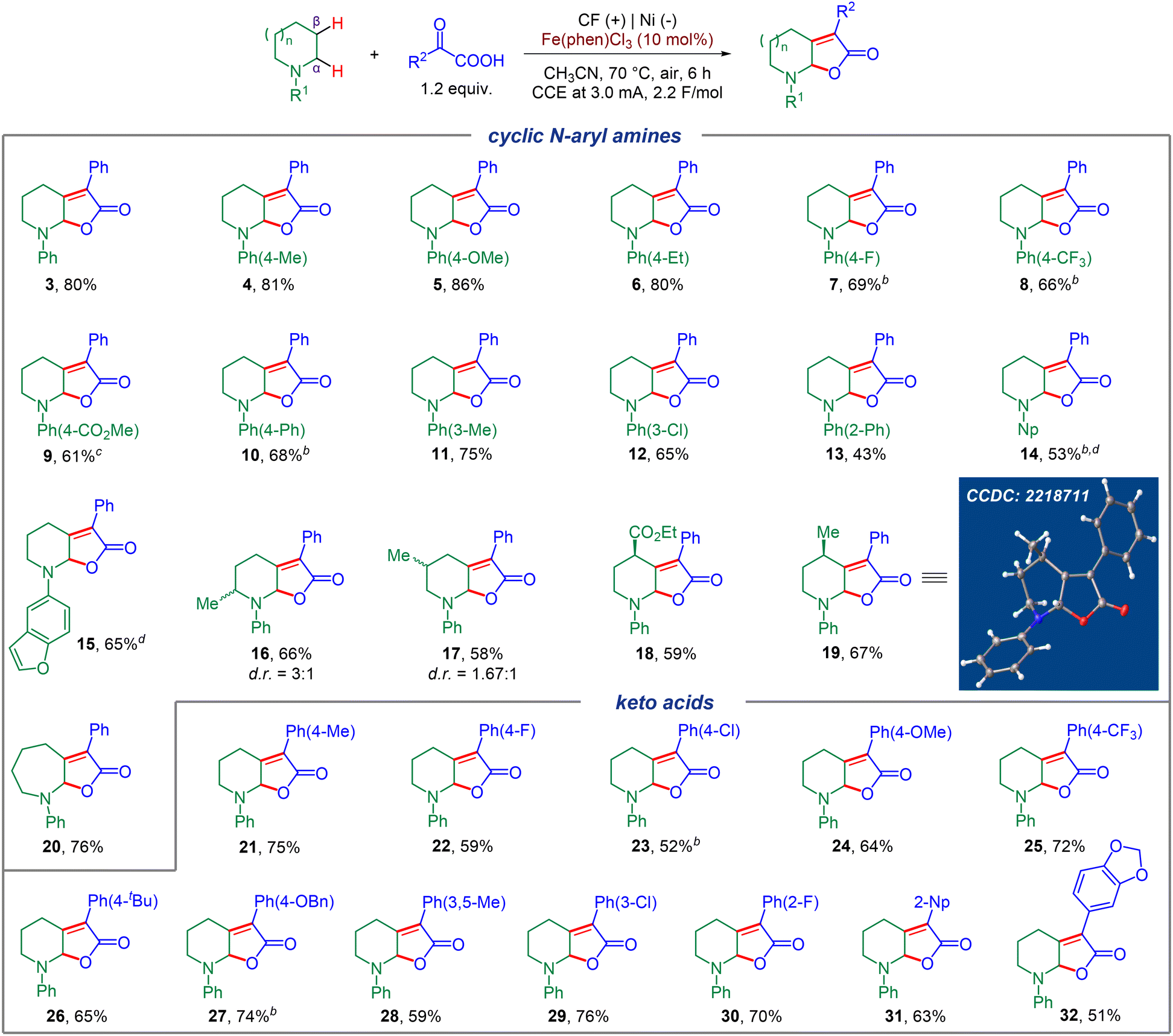 | ||
| Fig. 2 Scope of the electrochemical dual C(sp3)–H functionalization. Reaction conditions: carbon felt anode, Ni plate cathode, cyclic N-aryl amines (0.3 mmol), aryl keto acid (0.36 mmol, 1.2 equiv.), Fe(phen)Cl3 (10 mol%), CH3CN (5.0 mL), constant current = 3.0 mA, 70 °C, air, 6 h. a5 mA. b7 mA. c24 h. | ||
We next investigated the application of this electrochemical dual C(sp3)–H functionalization for late-stage modification (Fig. 3). To our delight, a variety of complex compounds were suitable substrates. Carbohydrates such as D-Galactose (33) could furnish the corresponding dual functionalization product in 58% yield with a diastereomeric ratio of 1:1 (see the ESI for details†). Moreover, different configurations of D-mannofuranose reacted smoothly to generate products 34 and 35 in 53% and 61% yields, respectively. Furthermore, the use of natural product derivatives such as DL-isoborneol and DL-menthol furnished the desired products with good efficiency (36, 37, and 38, respectively). Notably, a potentially sensitive ester group was well tolerated and gave the corresponding product 38 in 68% yield (dr = 1:1). The unique ability of this dual functionalization strategy was also highlighted by the modification of derivatives of α-amino acids. To this end, natural amino acid derivatives of L-phenylalanine bearing a piperidine motif were investigated and the corresponding products with α-phenyl (4-H, 4-OMe) keto acids were obtained in good yields (39, 40).
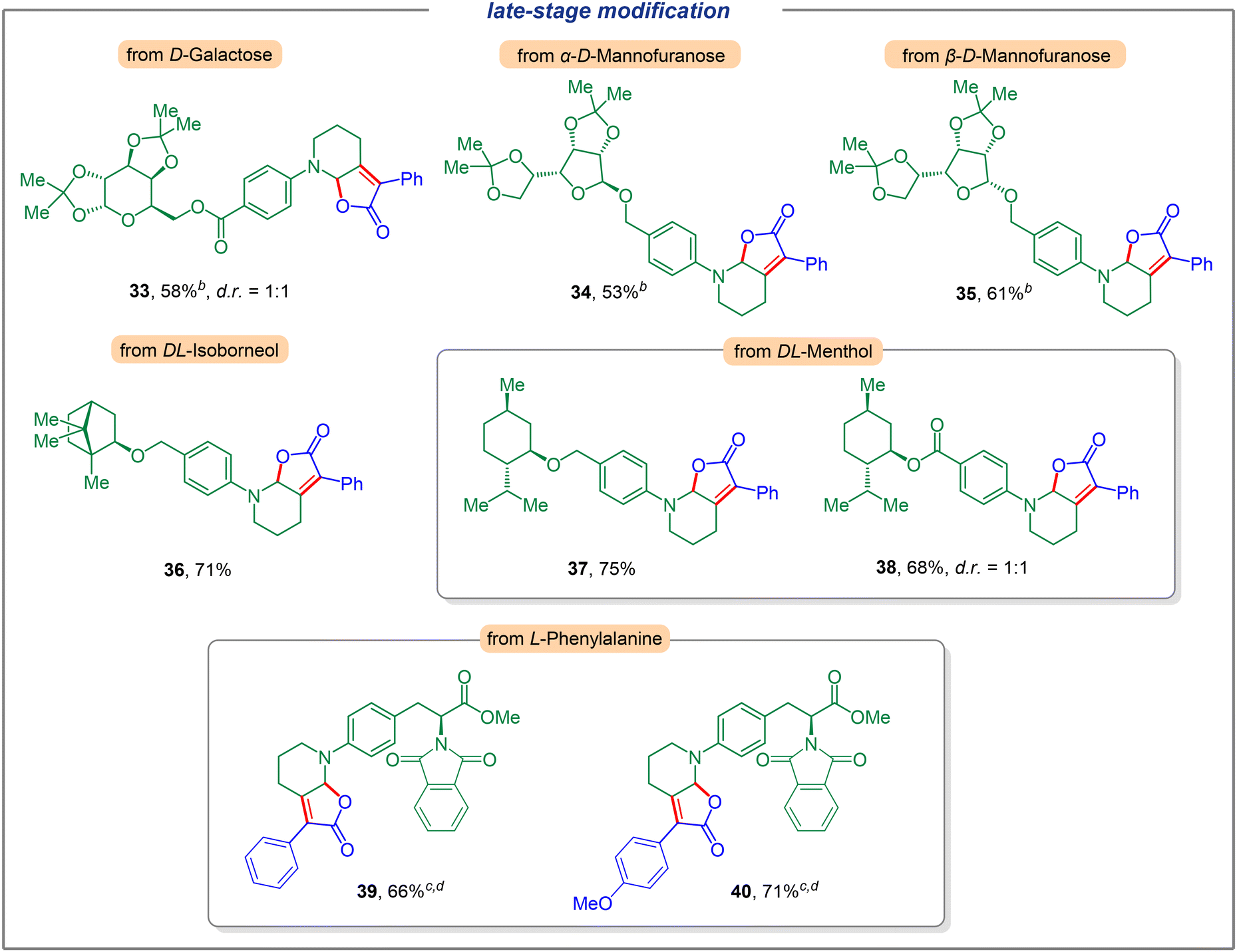 | ||
| Fig. 3 Late-stage modification. Reaction conditions: carbon felt anode, Ni plate cathode, N-aryl piperidine (0.3 mmol), aryl keto acid (0.36 mmol, 1.2 equiv.), Fe(phen)Cl3 (10 mol%), CH3CN (5.0 mL), constant current = 3.0 mA, 70 °C, air, 6 h. a5 mA. b7 mA. c24 h. | ||
Mechanistic studies
To further illustrate the synthetic utility of this method, we carried out a gram-scale preparation of the product. As a result, the reaction of N-phenyl piperidine 1 (10 mmol, 1.61 g) with 2-oxo-2-phenylacetic acid 2 (12 mmol, 1.2 equiv.) under a constant current at 50 mA, furnished product 3 in 61% isolated yield (1.78 g) after 12 hours (Fig. 4A). Notably, the catalyst loading could be further reduced to 3 mol%.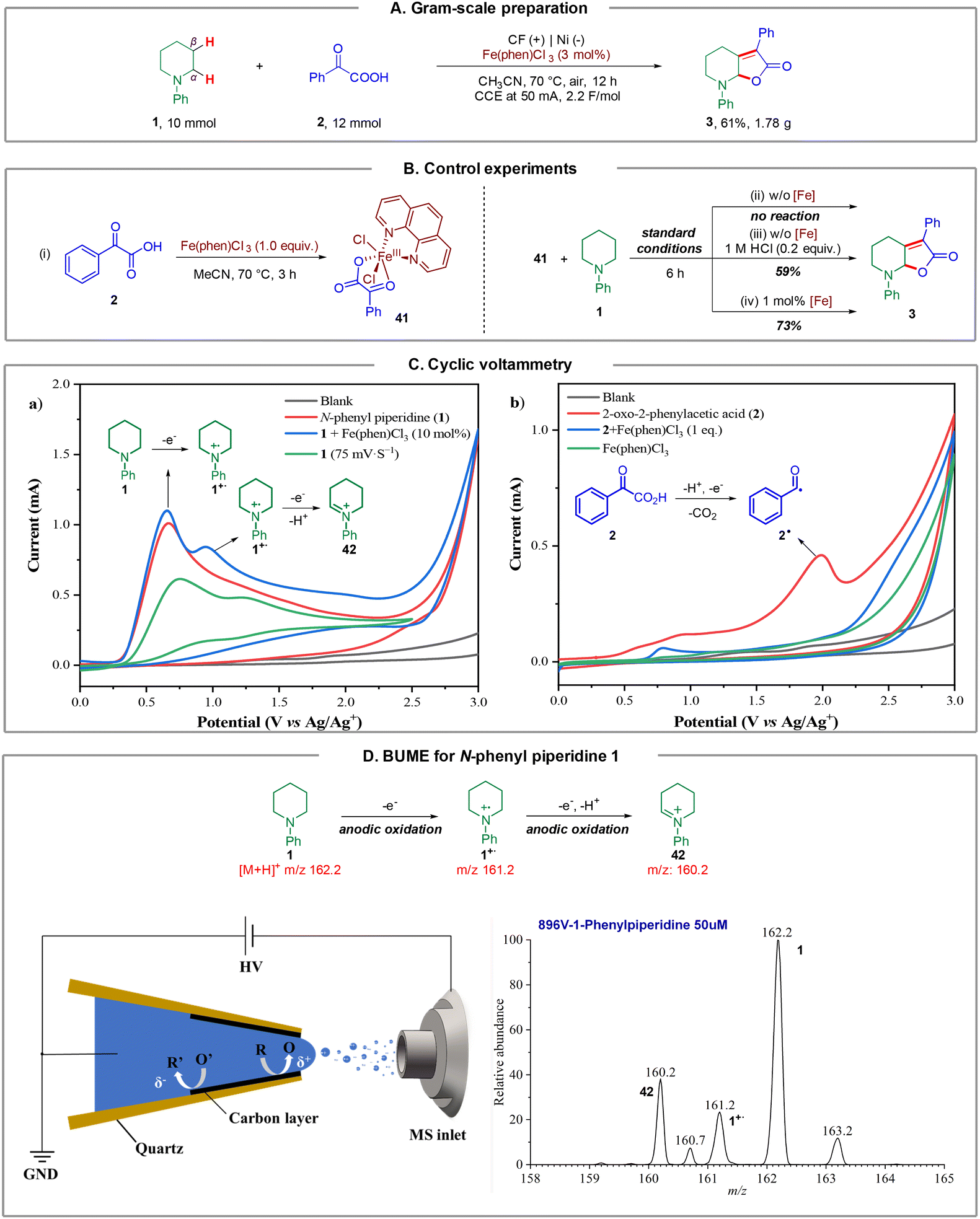 | ||
| Fig. 4 Mechanistic studies. (A) Gram-scale preparation. (B) Control experiments. (C) Cyclic voltammetry, using glass carbon as the working electrode, Pt plate as the counter electrode and Ag/Ag+ as the reference electrode. (CH3CN, 1 (10.0 mM), 2 (10.0 mM), Fe(phen)Cl3 (1.0 mM), 0.1 M nBu4NBF4, 100 mVs−1). (D) BUME of the twice single electron oxidation. | ||
Next, we sought to demonstrate the role of the iron catalyst. Firstly, we synthesized and isolated Fe complex 41 by treating 2-oxo-2-phenylacetic acid 2 with Fe(phen)Cl3 in CH3CN at 70 °C for 3 h, according to the method of a previous report (Fig. 4B, (i)),18 and we hypothesized that this Fe complex could furnish the transformation directly. However, it turned out that 41 could not furnish product 3 under the standard conditions (Fig. 4B, (ii)). This result revealed that Fe(phen)Cl3 could not dissociate and participate in the catalytic cycle under standard conditions. The dissociation of the iron catalyst involves a chlorine anion to generate Fe(phen)Cl3 from 41, as a consequence, a catalytic amount of HCl (1.0 M, 0.2 equiv.) was added to the reaction, and the desired product 3 could be obtained in 59% yield, which supported the abovementioned hypothesis (Fig. 4B, (iii)). Next, the reaction of N-phenyl piperidine 1 and 41 could furnish the desired product 3 in 73% yield on introducing 1 mol% Fe(phen)Cl3 (Fig. 4B, (iv)). These results support that Fe(phen)Cl3 could act as both a catalyst and an electro-mediator.
In order to further understand the reaction mechanism of this electrochemical dual C(sp3)–H functionalization, several cyclic voltammetry (CV) tests were performed. As depicted in Fig. 4C, an irreversible oxidation peak of N-phenyl piperidine 1 at E = 0.66 V (vs. Ag/Ag+) was observed at a scan rate of 100 mV s−1 (Fig. 4C (a), red line). Intriguingly, another oxidation peak of 1 at E = 1.17 V (vs. Ag/Ag+) was observed (Fig. 4C (a), green line) when the CV test was performed at a different scan rate (75 mV s−1). We speculated that this oxidation peak could be the second oxidation of N-phenyl piperidine 1 according to previous reports.19 Surprisingly, an obvious catalytic current appeared and the second oxidation peak increased distinctly from 0.49 mA to 0.75 mA in the presence of Fe(phen)Cl3 (Fig. 4C (a), blue line), which demonstrated that the Fe catalyst likely participates in the second electron transfer process from the N radical cation to the iminium ion as an oxidative mediator. In addition, 2-oxo-2-phenylacetic acid 2 exhibited an oxidation peak at E = 1.99 V (vs. Ag/Ag+) as shown in Fig. 4C (b) (red line), representing the electrochemical decarboxylation process. No peaks of Fe(phen)Cl3 could be observed (Fig. 4C (b), green line) and, in sharp contrast, the oxidation peak of 2 surprisingly disappeared in the presence of 1.0 equivalent of Fe(phen)Cl3 (Fig. 4C (b), blue line), which supports that the presence of Fe(phen)Cl3 clearly restrained the oxidation of the keto acid and hence we consequently speculated that Fe(phen)Cl3 could also prevent the keto acid from decarboxylation.
To capture and identify short-lived reactive intermediates in electrochemical reactions, a bipolar ultramicroelectrode (BUME) methodology combined with nano-electrospray ionization mass spectrometry was also developed. The fabrication of the bipolar ultramicroelectrode has been described in details previously.20 With this method, the electrospray ionization and faradaic reaction occurred simultaneously, permitting a rapid transfer of the electrochemically generated short-lived intermediates into an LTQ-XL mass spectrometer (Thermo-Fisher, Waltham, MA) for analysis. When a positive high voltage is applied, half-reactions occur on the far end of the carbon film that is closer to the spray tip and the mass spectrometer inlet, facilitating the mass analysis of the oxidation products. In this experiment, a positive voltage of 896 V was applied to the Cu wire to induce a redox reaction from a BUME nanopipette filled with 50 μM N-phenyl piperidine 1 in acetonitrile. As a result, the N-phenyl piperidine radical cation 1˙+ (m/z 161.2) was immediately captured, and subsequently, the radical cation underwent another single electron oxidation, followed by deprotonation to generate iminium 42 (m/z 160.2), which was also observed by mass spectrometry (Fig. 4D), and suggested that the radical played a key role in initiating the reaction.
Then, density functional theory (DFT) calculations ((Fig. 5, for details, please see the ESI†) were conducted. Firstly, we explored the formation of the enamine intermediate 43. According to DFT calculations, the iminium species 42 would undergo a proton transfer with N-phenyl piperidine 1 to afford enamine 43 and ammonium salt 1′ which provided support for this electrolyte-free electrochemical transformation. Next, Fe(phen)Cl3 was proposed as the starting species, which can be coordinated with keto acid 2via ligand exchange to afford N-phenyl piperidinium chloride and Fe complex Int1 located at −2.0 kcal mol−1. Simultaneously, multiple electron oxidations of N-phenyl piperidine 1, along with deprotonation generated the enamine intermediate 43. Besides, the nitrogen radical cation 1˙+ could also be oxidized to form 42via single electron transfer by FeIII(phen)Cl3 according to the CV tests, and the subsequently generated FeII(phen)Cl2 could be oxidized by the anode to regenerate FeIII(phen)Cl3. Then, an intermolecular nucleophilic addition occurred to form intermediate Int3 with 2.7 kcal mol−1via the transition state TS1 located at 15.6 kcal mol−1, followed by intermolecular hydrogen transfers to generate the enamine intermediate Int5 with 1.9 kcal mol−1 (path A) by traversing TS2 located at 17.3 kcal mol−1, and Int5 then needed a barrier of 14.8 kcal mol−1 for C–O bond breakage to form the intermediate Int6 with 13.2 kcal mol−1. Then, a hydrogen transfer followed by ligand exchange and the subsequent dehydration assisted by 1·HCl formed Int9 with −0.4 kcal mol−1 of energy. Finally, the dissociation of Fe(phen)Cl3 and intramolecular nucleophilic addition gave the target product 3 by traversing the transition state TS4 located at 1.9 kcal mol−1. For the overall reaction, the energy span (δE) of 19.3 kcal mol−1 indicates an effective FeIII-catalytic process, with TS2 as the TDTS (TOF-determining transition state), and Int1 as the TDI (TOF-determining intermediate). In order to test the sequence of the dissociation and the dehydration process, we also conceived another plausible reaction pathway, in which the dissociation of Fe(phen)Cl3 occurred before the dehydration step (path B), and it was found that the energy barrier of this process was much higher than that of path A (via transition state TS3′ located at 43.7 kcal mol−1 and Int9′ with 31.0 kcal mol−1). Therefore, DFT calculations supported path A as the reaction mechanism for this electrochemical dual functionalization reaction.
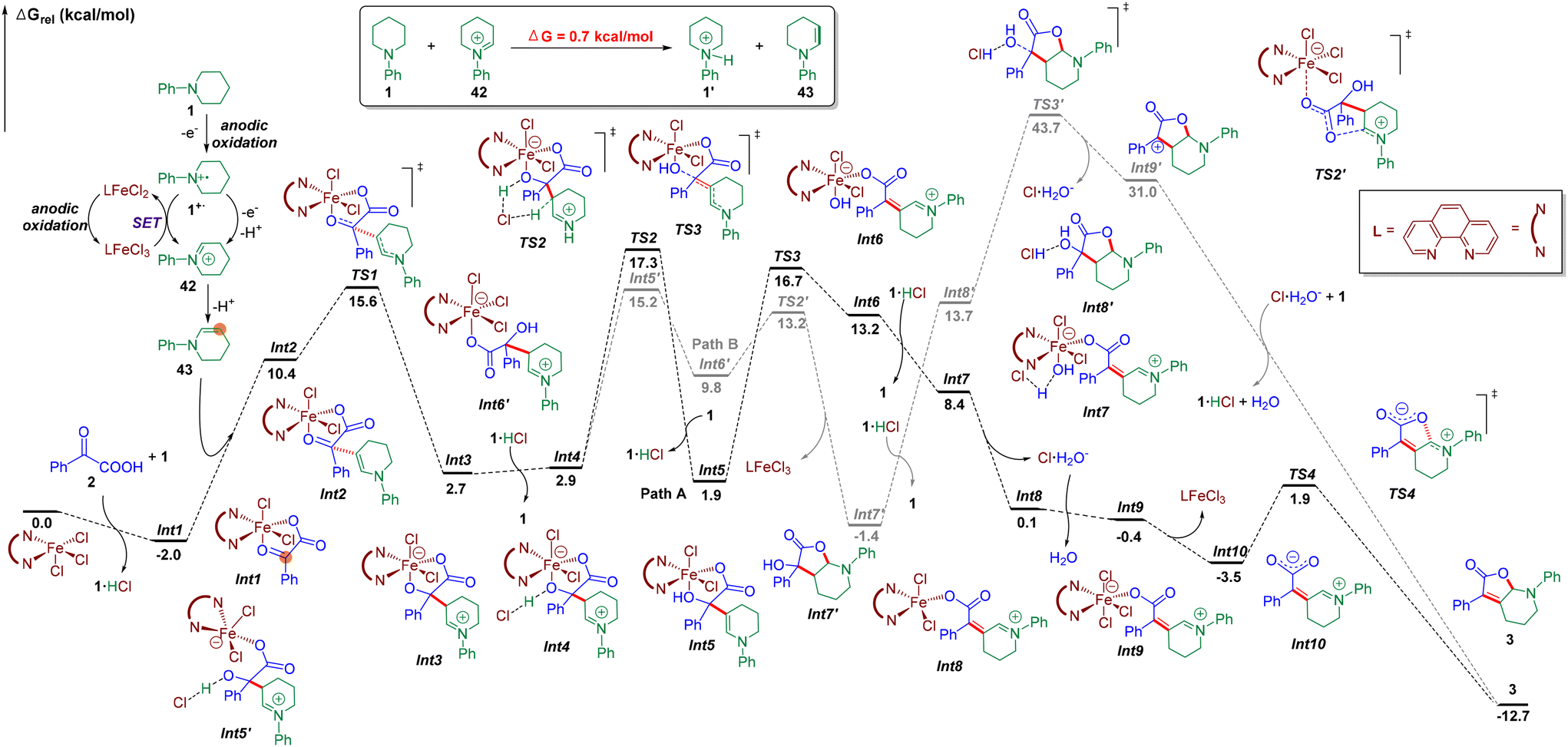 | ||
| Fig. 5 DFT Calculations. | ||
Based on the mechanistic studies and DFT calculations, a plausible mechanism was proposed (Fig. 6). The reaction was initiated by multiple electron and proton transfer of N-phenyl piperidine 1 on the anode to afford enamine 43. Besides, the nitrogen radical cation 1˙+ could also be oxidized to form 42via single electron transfer by FeIII(phen)Cl3 according to the CV tests, and the subsequently generated FeII(phen)Cl2 could be oxidized by the anode to regenerate FeIII(phen)Cl3. Next, Fe(phen)Cl3 could coordinate with keto acid 2via ligand exchange to afford N-phenyl piperidinium chloride and Fe complex I. Then, an intermolecular nucleophilic addition occurred, followed by intermolecular hydrogen transfers to generate the enamine intermediate II. Then, a hydrogen transfer followed by ligand exchange and the subsequent dehydration assisted by hydrochloric acid formed III. Finally, the dissociation of Fe(phen)Cl3 formed intermediate IV and intramolecular nucleophilic addition occurred to furnish the target product 3.
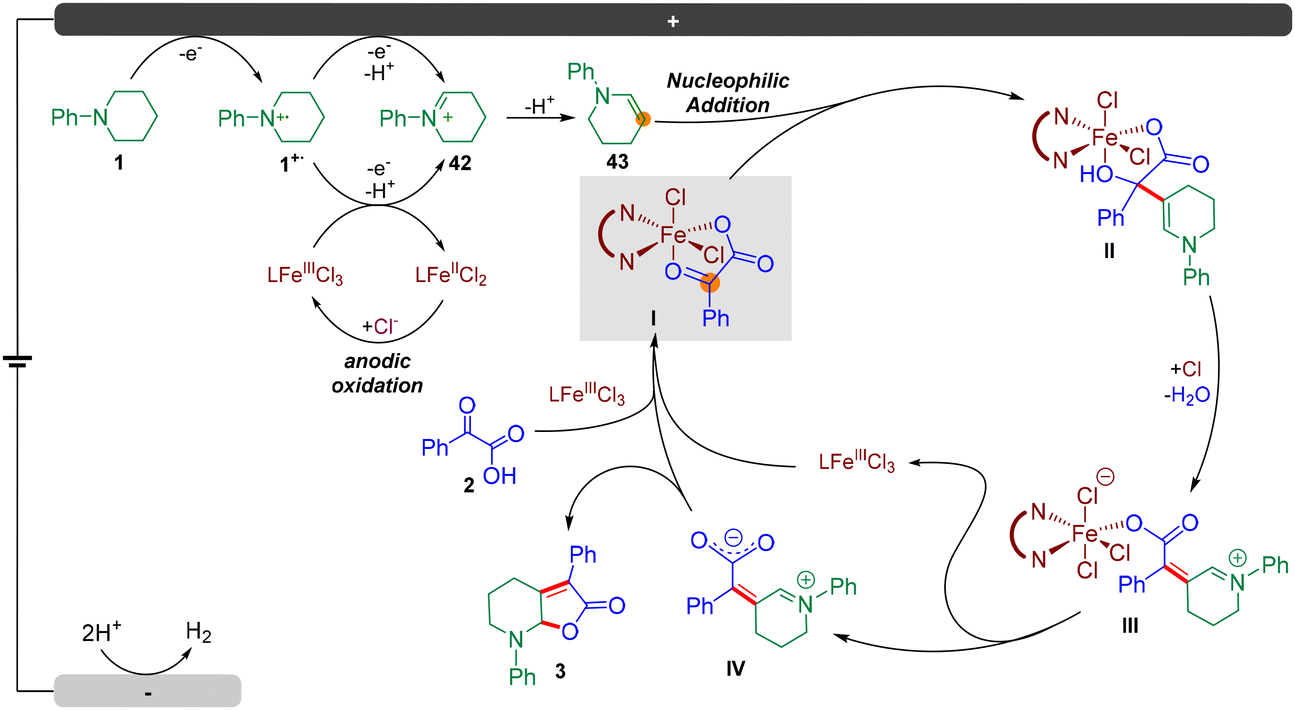 | ||
| Fig. 6 The proposed catalytic cycle. | ||
Conclusions
In conclusion, we developed a facile, selective, and straightforward dual α,β-C(sp3)–H functionalization of cyclic amines with keto acids by merging electrochemistry and iron catalysis. This transformation relied on the in situ generation of an enamine intermediate, and the multiple single electron oxidations and deprotonations of amines, as well as the coordination between the iron catalyst and the aryl keto acid, resulted in the desired products with high chemo- and site-selectivity. The reaction proceeded smoothly in the absence of stoichiometric amounts of the electrolyte under mild conditions, possibly due to the in situ generated ammonium salt acting as an electrolyte. In addition, cheap carbon felt was used as the anode and Earth-abundant metal nickel was employed as the cathode. The gram-scale preparation and late-stage modification of natural product derivatives showed the potential application of the method. Specific mechanistic studies using CV tests, BUME, and DFT calculations showed consistency and support of the proposed reaction pathway. We envisioned that the potent power of this functionalization of dual sp3 C–H bonds by electrocatalysis would give a potentially new perspective on the functionalization of cyclic amines and even unactivated alkyl C(sp3)–H bonds. Further electrochemical transformations of cyclic amines are currently underway in our laboratory.Author contributions
Y. Qiu, X. Zhang, and T. Feng conceived and designed the study and wrote the manuscript. T. Feng, Z. Zhu, D. Zhang, S. Wang, R. Li, Z. Zhu, X. Zhang, and Y. Qiu performed the experiments and mechanistic studies and revised the manuscript. All authors contributed to the analysis and interpretation of the data.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support from the National Key R&D Program of China (2022YFA1503200), the Fundamental Research Funds for the Central Universities (No. 63223007), the Natural Science Foundation of China (Grant No. 22188101), the Frontiers Science Center for New Organic Matter, Nankai University (Grant No. 63181206) is gratefully acknowledged. Nankai University is gratefully acknowledged and we sincerely thank Professor Qi-Lin Zhou for his generosity and key support.References
- (a) J. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2018, 108, 2265–2299 CrossRef PubMed; (b) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (c) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (d) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed; (e) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed; (f) C. W. Anson and S. S. Stahl, Chem. Rev., 2020, 120, 3749–3786 CrossRef CAS PubMed; (g) L. F. T. Novaes, J.-J. Liu, Y.-F. Shen, L.-X. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (h) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed; (i) X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS; (j) C. A. Malapit, M. B. Prater, J. R. Cabrera-Pardo, M. Li, T. D. Pham, T. P. McFadden, S. Blank and S. D. Minteer, Chem. Rev., 2022, 122, 3180–3218 CrossRef CAS PubMed.
- (a) Y. Qiu, C. Zhu, M. Stangier, J. Struwe and L. Ackermann, CCS Chem., 2020, 2, 1529–1552 Search PubMed; (b) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS PubMed; (c) Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293–3298 CrossRef CAS PubMed; (d) N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef CAS PubMed; (e) Y. Qiu, C. Tian, L. Massignan, T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5818–5822 CrossRef CAS PubMed; (f) Q. Wang, X. Zhang, P. Wang, X. Gao, H. Zhang and A. Lei, Chin. J. Chem., 2021, 39, 143–148 CrossRef CAS; (g) M. Chen, Z.-J. Wu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2022, 61, e202115954 CAS.
- (a) M.-J. Luo, Q. Xiao and J.-H. Li, Chem. Soc. Rev., 2022, 51, 7206–7237 RSC; (b) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed; (c) P. Xiong, H. Long, J. Song, Y. Wang, J.-F. Li and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 16387–16391 CrossRef CAS PubMed; (d) L. Song, N. Fu, B. G. Ernst, W. H. Lee, M. O. Frederick, R. A. DiStasio Jr. and S. Lin, Nat. Chem., 2020, 12, 747–754 CrossRef CAS PubMed; (e) J. Ke, W. Liu, X. Zhu, X. Tan and C. He, Angew. Chem., Int. Ed., 2021, 60, 8744–8749 CrossRef CAS PubMed; (f) D. S. Chung, S. H. Park, S.-G. Lee and H. Kim, Chem. Sci., 2021, 12, 5892–5897 RSC; (g) H. Huang and T. H. Lambert, J. Am. Chem. Soc., 2021, 143, 7247–7252 CrossRef CAS PubMed; (h) X. Wu, C. N. Gannett, J. Liu, R. Zeng, L. F. T. Novaes, H. Wang, H. D. Abruña and S. Lin, J. Am. Chem. Soc., 2022, 144, 17783–17791 CrossRef CAS PubMed; (i) S. H. Park, J. Jiang, K. Shin and H. Kim, ACS Catal., 2022, 12, 10572–10580 CrossRef CAS.
- (a) J.-E. Bäckvall and A. Gogoll, J. Chem. Soc., Chem. Commun., 1987, 1236–1238 RSC; (b) E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef CAS PubMed; (c) Y. Kawamata, M. Yan, Z.-Q. Liu, D.-H. Bao, J.-S. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS PubMed; (d) Y. Liang, S.-H. Shi, R. Jin, X. Qiu, J. Wei, H. Tan, X. Jiang, X. Shi, S. Song and N. Jiao, Nat. Catal., 2021, 4, 116–123 CrossRef CAS; (e) M. A. Hoque, J. Twilton, J. Zhu, M. D. Graaf, K. C. Harper, E. Tuca, G. A. DiLabio and S. S. Stahl, J. Am. Chem. Soc., 2022, 144, 15295–15302 CrossRef CAS PubMed.
- (a) H. Wang, X. Gao, Z. T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed; (b) J. L. Röckl, D. Pollok, R. Franke and S. R. Waldvogel, Acc. Chem. Res., 2020, 53, 45–61 CrossRef PubMed; (c) A. Kirste, G. Schnakenburg, F. Stecker, A. Fischer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2010, 49, 971–975 CrossRef CAS PubMed; (d) P. Wang, S. Tang, P. Huang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009–3013 CrossRef CAS PubMed; (e) Z.-J. Wu and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 4734–4738 CrossRef CAS PubMed; (f) T. J. DeLano and S. E. Reisman, ACS Catal., 2019, 9, 6751–6754 CrossRef CAS PubMed; (g) H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS; (h) G.-Q. Sun, W. Zhang, L.-L. Liao, L. Li, Z.-H. Nie, J.-G. Wu, Z. Zhang and D.-G. Yu, Nat. Commun., 2021, 12, 7086 CrossRef CAS; (i) W. Zhang, L. Lu, W. Zhang, S. D. Ware, J. Mondragon, J. Rein, N. Strotman, D. Lehnherr, K. A. See and S. Lin, Nature, 2022, 604, 292–297 CrossRef CAS PubMed.
- (a) F. Kakiuchi, T. Kochi, H. Mutsutani, N. Kobayashi, S. Urano, M. Sato, S. Nishiyama and T. Tanabe, J. Am. Chem. Soc., 2009, 131, 11310–11311 CrossRef CAS PubMed; (b) A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS PubMed; (c) J. Xiang, M. Shang, Y. Kawamata, H. Lundberg, S. H. Reisberg, M. Chen, P. Mykhailiuk, G. Beutner, M. R. Collins, A. Davies, M. Del Bel, G. M. Gallego, J. E. Spangler, J. Starr, S. Yang, D. G. Blackmond and P. S. Baran, Nature, 2019, 573, 398–402 CrossRef CAS PubMed; (d) T.-J. He, Z.-R. Ye, Z.-F. Ke and J.-M. Huang, Nat. Commun., 2019, 10, 833 CrossRef PubMed; (e) Y. Qiu, A. Scheremetjew and L. Ackermann, J. Am. Chem. Soc., 2019, 141, 2731–2738 CrossRef CAS PubMed; (f) L. F. T. Novaes, J. S. K. Ho, K. Mao, K. Liu, M. Tanwar, M. Neurock, E. Villemure, J. A. Terrett and S. Lin, J. Am. Chem. Soc., 2022, 144, 1187–1197 CrossRef CAS PubMed; (g) C. Huang, W. Ma, X. Zheng, M. Xu, X. Qi and Q. Lu, J. Am. Chem. Soc., 2022, 144, 1389–1395 CrossRef CAS PubMed; (h) J. Zhang, B. Das, O. Verho and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2022, 61, e202212131 CAS; (i) L.-H. Jie, B. Guo, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2022, 144, 2343–2350 CrossRef CAS PubMed; (j) L.-L. Liao, Z.-H. Wang, K.-G. Cao, G.-Q. Sun, W. Zhang, C.-K. Ran, Y. Li, L. Chen, G.-M. Cao and D.-G. Yu, J. Am. Chem. Soc., 2022, 144, 2062–2068 CrossRef CAS PubMed.
- (a) M. Rafiee, F. Wang, D. P. Hruszkewycz and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 22–25 CrossRef CAS PubMed; (b) Z.-W. Hou, D.-J. Liu, P. Xiong, X.-L. Lai, J.-S. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2021, 60, 2943–2947 CrossRef CAS PubMed; (c) Z. Ruan, Z. Huang, Z. Xu, S. Zeng, P. Feng and P.-H. Sun, Sci. China: Chem., 2021, 64, 800–807 CrossRef CAS; (d) M. Stangier, A. Scheremetjew and L. Ackermann, Chem. – Eur. J., 2022, 28, e202201654 CrossRef CAS PubMed.
- S. R. Waldvogel and M. Selt, Angew. Chem., Int. Ed., 2016, 55, 12578–12580 CrossRef CAS PubMed.
- (a) F. Wang, M. Rafiee and S. S. Stahl, Angew. Chem., Int. Ed., 2018, 57, 6686–6690 CrossRef CAS PubMed; (b) Z.-H. Wang, P.-S. Gao, X. Wang, J.-Q. Gao, X.-T. Xu, Z. He, C. Ma and T.-S. Mei, J. Am. Chem. Soc., 2021, 143, 15599–15605 CrossRef CAS PubMed; (c) H. Wang, M. He, Y. Li, H. Zhang, D. Yang, M. Nagasaka, Z. Lv, Z. Guan, Y. Cao, F. Gong, Z. Zhou, J. Zhu, S. Samanta, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2021, 143, 3628–3637 CrossRef CAS PubMed.
- T. Shono, H. Hamaguchi and Y. Matsumura, J. Am. Chem. Soc., 1975, 97, 4264–4268 CrossRef CAS.
- (a) Y. Takahira, M. Chen, Y. Kawamata, P. Mykhailiuk, H. Nakamura, B. K. Peters, S. H. Reisberg, C. Li, L. Chen, T. Hoshikawa, T. Shibuguchi and P. S. Baran, Synlett, 2019, 1178–1182 CAS; (b) Y. Liu, B. Shi, Z. Liu, R. Gao, C. Huang, H. Alhumade, S. Wang, X. Qi and A. Lei, J. Am. Chem. Soc., 2021, 143, 20863–20872 CrossRef CAS PubMed; (c) L. Zhang, Y. Fu, Y. Shen, C. Liu, M. Sun, R. Cheng, W. Zhu, X. Qian, Y. Ma and J. Ye, Nat. Commun., 2022, 13, 4138 CrossRef PubMed.
- (a) G.-Q. Xu, J.-T. Xu, Z.-T. Feng, H. Liang, Z.-Y. Wang, Y. Qin and P.-F. Xu, Angew. Chem., Int. Ed., 2018, 57, 5110–5114 CrossRef CAS PubMed; (b) X. Shi, Y. He, X. Zhang and X. Fan, Adv. Synth. Catal., 2018, 360, 261–266 CrossRef CAS; (c) A. F. Prusinowski, R. K. Twumasi, E. A. Wappes and D. A. Nagib, J. Am. Chem. Soc., 2020, 142, 5429–5438 CrossRef CAS PubMed; (d) W. Chen, A. Paul, K. A. Abboud and D. Seidel, Nat. Chem., 2020, 12, 545–550 CrossRef CAS PubMed; (e) D. A. Valles, S. Dutta, A. Paul, K. A. Abboud, I. Ghiviriga and D. Seidel, Org. Lett., 2021, 23, 6367–6371 CrossRef CAS PubMed; (f) X. Liu, Z. Wang, Q. Wang and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202205493 CAS.
- T. Shen and T. H. Lambert, Science, 2021, 371, 620–626 CrossRef CAS PubMed.
- (a) L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed; (b) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed; (c) S. Wu, J. Kaur, T. A. Karl, X. Tian and J. P. Barham, Angew. Chem., Int. Ed., 2022, 61, e202114672 Search PubMed.
- T. Shen, Y.-L. Li, K.-Y. Ye and T. H. Lambert, Nature, 2023, 614, 275–280 CrossRef CAS PubMed.
- (a) T. Feng, S. Wang, Y. Liu, S. Liu and Y. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202115178 CAS; (b) T. Feng, S. Wang and Y. Qiu, Synlett, 2022, 1582–1588 CAS.
- (a) P. Li, C. Guo, S. Wang, D. Ma, T. Feng, Y. Wang and Y. Qiu, Nat. Commun., 2022, 13, 3774 CrossRef CAS PubMed; (b) Y. Wang, S. Tang, G. Yang, S. Wang, D. Ma and Y. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202207746 CAS; (c) Y. Wang, Z. Zhao, D. Pan, S. Wang, K. Jia, D. Ma, G. Yang, X.-S. Xue and Y. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202210201 CAS; (d) Z. Zhao, Y. Liu, S. Wang, S. Tang, D. Ma, Z. Zhu, C. Guo and Y. Qiu, Angew. Chem., Int. Ed., 2023, 62, e202214710 CAS; (e) S. Wang, T. Feng, Y. Wang and Y. Qiu, Chem. - Asian J., 2022, 17, e202200543 CAS.
- R. D. Jana, D. Sheet, S. Chatterjee and T. K. Paine, Inorg. Chem., 2018, 57, 8769–8777 CrossRef CAS PubMed.
- D. Gunasekera, J. P. Mahajan, Y. Wanzi, S. Rodrigo, W. Liu, T. Tan and L. Luo, J. Am. Chem. Soc., 2022, 144, 9874–9882 CrossRef CAS PubMed.
- (a) U. Karst, Angew. Chem., Int. Ed., 2004, 43, 2476–2478 CrossRef CAS PubMed; (b) M. Lu, Y. Liu, R. Helmy, G. E. Martin, H. D. Dewald and H. Chen, J. Am. Soc. Mass Spectrom., 2015, 26, 1676–1685 CrossRef CAS PubMed; (c) C. Mu, W. Wang, J. Wang, C. Gong, D. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 21515–21519 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2218711. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc00344b |
| This journal is © The Royal Society of Chemistry 2023 |