
Control of molecular conductance by pH
Bangyao
Wu
,
Weiyi
Guo
,
Jianming
An
 and
Haixing
Li
*
and
Haixing
Li
*
Department of Physics, City University of Hong Kong, Kowloon 999077, Hong Kong, P. R. China. E-mail: haixinli@cityu.edu.hk
First published on 6th June 2022
Abstract
pH plays a fundamental role in regulating various processes occurring in ecosystems, biological organisms, and chemistry laboratories, and lately, has been observed to drastically impact material electronic properties at the single molecule level. The use of pH represents one route both to investigate the fundamental charge transport processes at nanoscale junctions and to create new electronic device functions. In this review, we describe how pH affects and controls the conductance of various single molecule junctions, their underlying mechanisms and technological implications, and how such rules of pH–property relationship need to be further developed so we can ultimately use pH as a routine method to control single molecule conductance.
1. Introduction
Single-molecule electronics is an interdisciplinary field that studies the electronic properties of molecular materials and explores individual molecules as building blocks to construct nanoscale electronic components.1–3 To probe the charge transport principles of single molecules, a method is needed that can incorporate a molecule into a molecular-size gap and then wire this unit into a circuit. The development of the scanning tunnelling microscope-based break junction (STM-BJ) technique has played a critical role in enabling the reliable formation of single-molecule circuits and reproducible measurements of molecular junction properties.4,5 Another commonly used method for measuring single-molecule conductance is the mechanically controlled break junction (MCBJ) technique that provides benefits such as enhanced mechanical stability.6,7 In the past decade, we have seen a growing number of organic,8 organometallic,9,10 inorganic cluster,11 peptide,12,13 and DNA14,15 molecules being investigated for relating their single-molecule electronic properties to their structures.Regulating charge transport across a molecule bridged between two electrodes is essential for developing functional molecular circuits. Towards this goal, stimuli such as light,16 electrochemical gating,17 and mechanical modulation18 have been applied for tuning the transport characteristics of metal–molecule–metal junctions. Among different approaches, pH emerges as a new tool for reversibly modulating molecular conductance, and moreover, it provides a new aspect for our understanding of the single molecule electronic properties. pH, a quantification of the concentration of hydrogen ions (or protons) in a solution, can be controlled during single molecule measurements. By designing target molecules that can be protonated by accepting protons or deprotonated by losing protons under the corresponding conditions, we can probe how such processes affect single molecule transport properties. There are three parts of the target molecule that can respond to pH: linker group, side group, and molecular backbone (Fig. 1 left). For example, carboxyl linker group becomes deprotonated under a basic condition, and amine side group as well as pyridine backbone become protonated under an acidic condition (Fig. 1 right). These types of chemical structure changes may alter the molecular orbitals, the orbital energy positions relative to the Fermi level of the electrodes, and the binding strength and geometry of the molecular junction, which ultimately impact the conductance. Revealing the connections between pH and the chemical and electrical properties of molecules has become an important subject in molecular electronics.
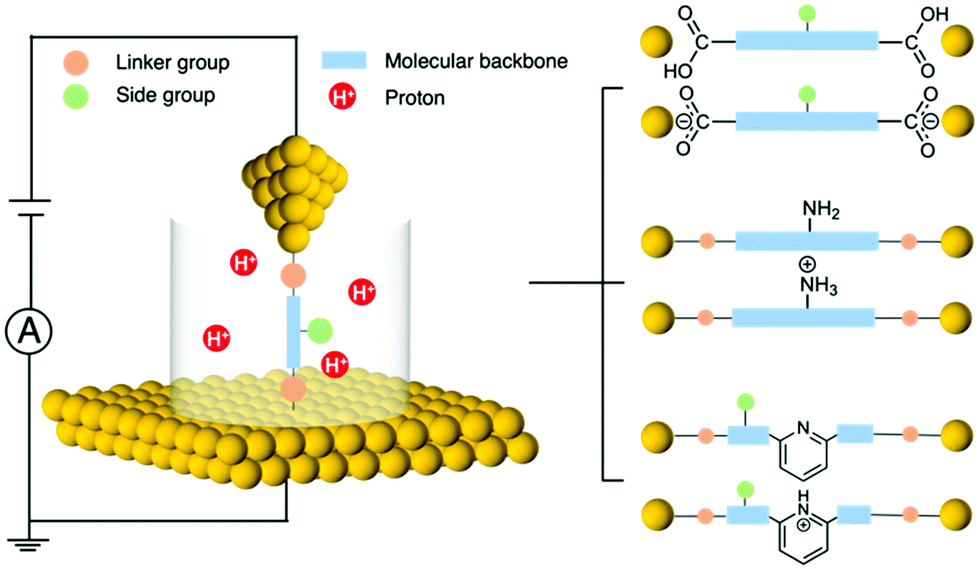 | ||
| Fig. 1 Graphical illustration of an STM-BJ Setup with pH control and example deprotonation reaction occurring at the linker group (top), and protonation reaction occurring at the side group (middle) and the molecular backbone (bottom). | ||
This perspective provides a summary of our current understanding of how protonation and deprotonation profoundly impact the charge transport characteristics of single-molecule junctions, emphasizing the versatile regulatory role of pH in molecular devices. Section 2 provides a brief description of the experimental methods for applying a pH control in molecular conductance measurements. Section 3 is focused on molecular backbones, specifically the organic π-conjugated and heteroatomic backbones that can respond to pH. Discussions about amine and carboxyl groups in response to pH regulation are provided in Section 4 (as linker group) and 5 (in peptides). Section 6 is focused on pH control of the conductance of imidazole when used as a linker group and as a backbone component. Section 7 is focused on the pyrazole unit as an anchor group that can modulate molecular conductance under a pH control. Section 8 is devoted to supramolecular complexes and how pH can affect their conductance. Section 9 is devoted to conductance control by Lewis-acid base interactions. Section 10 is focused on light, and Section 11 is focused on electrochemical gating; both are external stimuli combined with pH for tuning the conductance of molecular junctions. A brief conclusion and outlook appear in Section 12.
2. Implementation of pH control in STM-BJ method
STM-BJ measurements are typically performed with a tip and a substrate in a two-electrode configuration; a three-electrode system which includes a reference electrode,19 and a four-electrode system that includes a reference and a counter electrode,20 have also been established, where the use of a fluidic cell is often required for applying an electrolytic or electrochemical gating voltage on the single molecule junctions. Au is the most commonly used metal electrode, and Ag, Pt, Ni, Cu, and other metals have also been demonstrated as electrical contacts for forming molecular junctions.20–22 A clean Au substrate is either immersed in a high boiling solvent containing a dilute (nM to mM) concentration of the target molecules, or is exposed to a thermal vapor of the molecules, for a solution or dry measurement, respectively. In cases when a polar solvent is used, the STM tip needs to be coated with Apiezon wax in order to reduce ionic conduction and polarization current.23STM-BJ measurements are carried out as follows. First, the STM tip is gradually brought into the substrate until it makes a hard contact, which is usually above a few conductance quanta, where conductance quantum (G0 = 2e2/h = 77.5 μS) is a frequently used conductance unit. Then the tip is withdrawn from the substrate while the conductance is simultaneously recorded as a function of the tip-substrate displacement. When the separation between the tip and the substrate becomes large enough, a molecule in proximity can bridge this gap to form an Au–molecule–Au junction. In the conductance trace, plateaus below 1 G0 signify the formation of such molecular junctions. Finally, as the tip continues to be withdrawn, molecular junction ruptures and conductance drops to the noise floor. The binding geometry of the molecule, as well as the configuration of the atomic surface of the electrode, can all vary from junction to junction, resulting in a different conductance trajectory in each measurement. Thus, thousands of measured trajectories are compiled into a one- or two-dimension (1D or 2D) conductance histogram to determine the most probable conductance of a single molecule junction.
Several studies have reported the use of pH as an external control for tuning the electronic properties of molecular junctions, and their methods are summarized in Table 1. Non-reducing strong inorganic acids such as HClO4, HCl, and H2SO4, as well as strong base NaOH are stable in most systems and do not disrupt the structure of target molecules, thus were commonly used in aqueous solutions as the acid and base reagent for conductance measurement of target molecules such as alkanes, imidazoles, and pyridines (# 1–6 in Table 1). Weak base buffering agent 2-(cyclohexylamino) ethanesulfonic acid (CHES), K2HPO4/KH2PO4, Na2HPO4/NaH2PO4, NaHCO3/Na2CO3, and NaOAc were used in aqueous solutions for increasing pH during the conductance measurement (# 7–10 in Table 1). Besides, organic acids such as trifluoroacetic acid (TFA) and camphorsulfonic acid (CSA), and base triethylamine were used for protonating and deprotonating target molecules, respectively, in measurements performed with organic solvent (# 11–15 in Table 1). In addition, supplying acidic or basic solution vapor to the molecular junctions can also tune the pH during the measurement. This method allows a different solvent to be used in preparing the molecular solution than that used for the acid/base solution (# 16 in Table 1). Conductance measurements have also been performed using monolayer Langmuir–Blodgett (LB) films and mixed self-assembled monolayers (SAM) where pH was controlled (# 16–19 in Table 1). In addition to the general pH regulation methods, Lewis acid–base interactions can be seen as a special case, in which Lewis acid is an electron acceptor and Lewis base is an electron donor. For example, fluoride (Lewis base) and organoborane (Lewis acid) were shown to undergo Lewis acid–base interaction, leading to a change in the molecular conductance of organoborane wire.24
| No. | Target molecule of the measurement | pH | Solvent | Acid and/or base | Measurement methoda | Ref. |
|---|---|---|---|---|---|---|
a Au electrodes were used unless specified.
b Na2CO3 aqueous solution was mixed with TCB solution for increasing the pH, and the organic phase was separated in order to be used in the conductance measurements.
c Spiropyran is a photoacid that undergoes photoinduced proton transfer with the target molecule during the measurement. Spiropyran and an equal amount of TFA were supplemented in the molecular solution for the measurement.
d No solvent was present when conductance measurements were performed on these SAMs. Dimethyl sulfoxide (DMSO) and tetrahydrofuran/chloroform (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, v/v) were used as the solvent for preparing the SAMs in # 16 and # 17, respectively.
e Monolayer Langmuir–Blodgett (LB) film was assembled by using a molecular solution prepared in organic solvent mixture, and subphases were pure water (pH = 5.6) or aqueous NaOH solution (pH = 11.0). The monolayer LB film was transferred onto Au-on-glass substrates for the conductance measurements. :4, v/v) were used as the solvent for preparing the SAMs in # 16 and # 17, respectively.
e Monolayer Langmuir–Blodgett (LB) film was assembled by using a molecular solution prepared in organic solvent mixture, and subphases were pure water (pH = 5.6) or aqueous NaOH solution (pH = 11.0). The monolayer LB film was transferred onto Au-on-glass substrates for the conductance measurements.
|
||||||
| 1 | Malachite green and pararosaniline | 5.5, 13.6 | H2O | HClO4 and NaOH | STM-BJ | 25 |
| 2 | Diamine butane | 1, 10, 13 | H2O | HClO4 and NaOH | STM-BJ | 43 |
| 3 | Dicarboxylic-acid butane | 1, 5, 13 | H2O | HClO4 and NaOH | STM-BJ | 43 |
| 4 | 4,4′-Bipyridine | 1.0, 4.1, 10.0 | H2O | HClO4, KClO4, and NaOH | STM-BJ with electrochemical gate | 80 |
| 5 | Imidazole | 3, 7, 9, 12 | H2O | HCl and NaOH | STM-BJ | 54 |
| 6 | 4,4′-Vinylenedipyridine | 2.35, 2.57, 2.85, 3.01, 3.26, 3.53 | H2O | H2SO4 and Na2SO4 | STM-BJ with Ni electrodes and electrochemical gate | 81 |
| 7 | Peptides | 1.5, 7.4, 13 | H2O | HClO4, NaClO4, NaOAc, NaHCO3, Na2CO3, and NaOH | STM-BJ | 12 |
| 8 | Cucurbit[7]uril (CB[7]) and melphalan@CB[7] complex | 1, 4, 7, 9 | H2O | HCl, Na2HPO4, NaH2PO4, and NaOH | STM fixed junction technique | 64 |
| 9 | Peptides | 2, 6.9 | H2O | HClO4, K2HPO4, and KH2PO4 | STM I(s) technique | 47 |
| 10 | Peptides | 9 | H2O | 2-(Cyclohexylamino) ethanesulfonic acid (CHES) | STM-BJ | 46 |
| 11 | Azulene derivatives | No details | Tetrahydrofuran/mesitylene (1:4, v/v) |
Trifluoroacetic acid (TFA) | MCBJ | 27 |
| 12 | Diketopyrrolopyrrole derivatives | No details | Chloroform/mesitylene (1:4, v/v) |
Camphorsulfonic acid (CSA) and triethylamine | MCBJ | 35 |
| 13 | Pyridine derivatives | No details | 1,2,4-Trichlorobenzene (TCB) | TFA and Na2CO3 aqueous solutionb | STM-BJ | 40 |
| 14 | Spiropyran derivatives | No details | Mesitylene/dicholoromethane (10:1, v/v) |
TFA and triethylamine | STM-BJ | 70 |
| 15 | Azulene derivatives | No details | Tetrahydrofuran/mesitylene (1:4, v/v) |
TFA and spiropyranc | MCBJ with light control | 71 |
| 16 | Benzo-bis(imidazole) derivatives | No details | —d | HCl solution vapor and triethylamine vapor | SAMs with PtIr coated tip, c-AFM method | 53 |
| 17 | Pyrazole derivatives | 5.6, 11.0 | — | NaOH aqueous solutione | LB thin film, touch to contact (TTC) STM method | 57 |
| 18 | Pyrimidine derivatives | No details | — | HClO4 and NaOEt | Mixed SAMs with AuNPs immobilized on top, PtIr STM tip, scanning tunnelling spectroscopy (STS) method | 30 |
| 19 | Spiropyran derivatives | No details | — | Trifluoromethanesulfonic acid and triethylamine | Mixed SAMs, EGaIn junction method, light control | 72 |
3. Control the conductance of π-conjugated molecules by pH
π-Conjugation, given its electronic delocalization, often appears in molecular junctions, and as studies of π-conjugated systems have expanded, we realized that their conductance can be regulated by pH. Here we focus on how the π-conjugation of a backbone can be altered by a basic or acidic environment, leading to rearrangements of the backbone and changes in the junction conductance.Carbon backbone
Li et al. reported that pH could tune the single molecule conductance of dye molecules malachite green (MG) (structure in Fig. 2a) and pararosaniline (PA) via break junction experiments.25MG solution showed blue color under pH = 5.5 and white color under pH = 13.6 because the central carbon was sp2 or sp3 hybridized under acidic or basic condition, respectively (Fig. 2a). The authors performed sequential conductance measurements of MG under five cycles of alternating acidic and basic conditions and found that at pH = 5.5, the average single molecule conductance of MG was about 100 times of the conductance measured at pH = 13.6 (Fig. 2b). This large conductance increase under acidic condition confirmed that protonation of MG effectively promoted charge delocalization. The calculated molecular orbitals of MG showed that the highest occupied molecular orbital (HOMO) did not change significantly, while the lowest unoccupied molecular orbital (LUMO) was ∼0.2 eV (relative to the Fermi level (EF)) at low pH but shifted to ∼2 eV at high pH (Fig. 2c). As a result of this increase of the LUMO energy, transmission probability decreased dramatically near the EF, agreeing with the experimentally observed suppressed conductance in basic solution. | ||
| Fig. 2 (a) Chemical structures and photographs of a solution of malachite green (MG) at pH = 5.5 and pH = 13.6. (b) pH-induced conductance switching of MG between “ON” and “OFF” states. (c) Calculated transmission spectra for Au–MG–Au junctions at pH = 5.5 and pH = 13.6 (EF = −3.35 eV). (d) Left: Protonation of azulene in the presence of TFA. Right: Structure and conductance values determined from MCBJ measurements for 1,3Az, 4,7Az, and 5,7Az. (e and f) Calculated conductance for 1,3AZ, 4,7Az, and 5,7Az junctions at the (e) initial and (f) protonated state. (a–c) are adapted with permission from ref. 25, copyright (2014) Wiley. (d–f) are adapted with permission from ref. 27, copyright (2017) Royal Society of Chemistry. | ||
Azulene (C10H8) is an all-carbon π-conjugated dye molecule, exhibiting an electron-rich five-membered ring and an electron-poor seven-membered ring. This ring system of azulene becomes protonated in the presence of acid and forms a stable azulenium cation (Fig. 2d, left).26 Yang et al. synthesized three azulene derivatives (1,3Az, 4,7Az, and 5,7Az, structures see Fig. 2d) where the seven-membered or five-membered ring was directly incorporated into the backbone and performed conductance measurements using the MCBJ technique.27 All derivatives showed an increase in conductance upon protonation, among which 5,7Az displayed the largest increase (Fig. 2d). The authors applied a parameter-free theory28 to evaluate the correlation between conductance enhancement and the connecting positions of azulene with the rest of the molecule. The calculated conductance suggested that protonation led to separation in the spin-up and spin-down energy levels of azulene and resulted in a smaller HOMO–LUMO gap compared to the initial one for the neutral state. Notably, the initial 5,7Az showed destructive quantum interference near EF (0 eV) (green curve in Fig. 2e), and when 5,7Az became protonated, this anti-resonance valley disappeared (green curve in Fig. 2f); 1,3Az and 4,7Az did not show quantum interference features in either neutral or protonated state (red and blue curves, Fig. 2e and f). Therefore, while all derivatives showed a conductance enhancement upon protonation, 5,7Az demonstrated the highest. This result suggested that protonation can be used as a novel strategy to modulate quantum interference effect in charge transport through single molecule junctions.
Heteroatomic backbone
Heteroatomic backbones that contain nitrogen atom(s) can become (de)protonated under acidic(basic) conditions; if the backbone is π-conjugated, this protonation or deprotonation process can further rearrange the π system and alter the entire electronic structure of the molecule.29 In 2005, Morales et al. synthesized a diblock molecule 1 that had a dipyrimidinyl moiety connected with a diphenyl group (Fig. 3a). Given the asymmetric backbone, 1 had a distinct rectification behavior.30 These authors found that protonating the nitrogen atoms on 1 in perchloric acid solution reversed the rectification direction. The authors proposed that the positive charge on the protonated 1 modified the direction of the dipole moment of the molecule and switched the HOMO- to LUMO-dominated transport, thus leading to the opposite rectification direction. In a separate theoretical study, calculations by Li et al. did not show rectification direction inversion phenomena of 1 when it was protonated. The authors suggested that other effects that were not included in the theory such as solvent and/or electron correlation might be needed in order to reproduce the experimental observation of the inversed rectification upon protonation.31 Zhang et al. further investigated the protonation effects on electron transport of 1 theoretically.32 By analyzing the transmission spectra of the molecular junction with different number and locations of protons residing on the molecule, the authors concluded that protonation in the inner pyrimidinyl group reversed the direction of the rectification and protonation in the outer pyrimidinyl group enhanced the rectification ratio.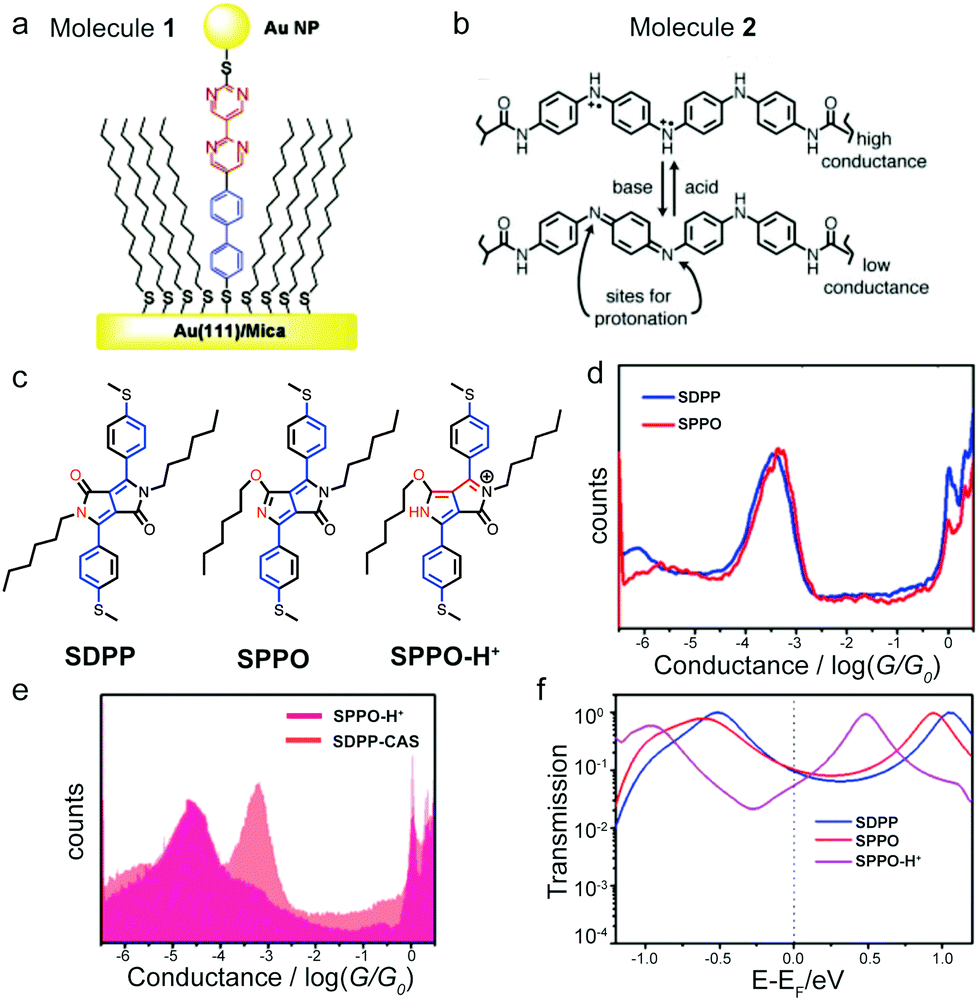 | ||
| Fig. 3 (a) Schematic of molecular junction of 1. (b) Protonated and deprotonated structure of 2 and their corresponding conductance states. (c) Chemical structure of SDPP and SPPO. Resonance structure for SPPO-H+. (d and e) Conductance histograms of (d) SDPP and SPPO, and (e) SPPO-H+ and SDPP-CAS. (f) Transmission spectra for molecular junctions formed with SDPP, SPPO and SPPO-H+. (a) is reprinted with permission from ref. 30, copyright (2005) American Chemical Society. (b) is reprinted with permission from ref. 33, copyright (2006) AAAS. (d–f) are adapted with permission from ref. 35, copyright (2018) American Chemical Society. | ||
In addition to the rectification inversion phenomenon, conductance switching with pH has also been observed for nitrogen-containing heteroatomic backbone-based molecules. In 2006, Guo et al. reported a single-molecule switch controlled by pH using an oligoaniline derivative 2 (structure see Fig. 3b) and single-walled carbon nanotube electrodes.33 The conductance of the oxidized emeraldine form of 2 at pH = 11 was an order of magnitude lower than that at pH = 3 due to the deprotonation of nitrogen atoms (Fig. 3b). Later, Cao et al. carried out experiments on azobenzene-based molecules attached to graphene electrodes and obtained similar results due to the presence of sulfonic acid substitution groups that respond to pH.34 In this study, the conductance of the azobene-based molecule increased by two orders of magnitude at low pH = 1 (protonated state) compared to that measured at high pH = 12 (deprotonated state).
Compounds containing a diketopyrrolopyrrole unit have also been shown to undergo conductance switching events with pH, as described in the study of two diketopyrrolopyrroke isomers: SDPP and SPPO (structures see Fig. 3c) in Zhang et al.35SDPP had alkyl substitutions on two nitrogen atoms, whereas one of the nitrogen atoms was free in SPPO and could be protonated in an acidic environment. The conductance measurements showed that SDPP in the presence of camphorsulfonic acid (CAS), labelled as SDPP-CAS, had a slightly higher conductance than that of SDPP (Fig. 3d and e). In contrast, when CAS was added to SPPO to form the protonated SPPO-H+, its conductance dropped dramatically (Fig. 3d and e). We note that these trends were not the same as the results discussed above of protonation-induced increase in conductance for dye molecules (MG and PA), azulene derivatives (1,3Az, 4,7Az, and 5,7Az), and oligoaniline derivative (2). The authors analyzed all possible structures of these isomers and revealed that the dominant structure for SPPO-H+ was the cross-conjugated structure, as illustrated in the right panel of Fig. 3c. The transmission calculations showed a destructive quantum interference effect in the SPPO-H+ junction, but not in SDPP or SPPO junctions (Fig. 3f), agreeing with previous works that had shown that cross-conjugated structures exhibit destructive quantum interference effect.36–38 Taken together, the destructive interference feature combined with the lower HOMO and LUMO energy levels of SPPO-H+ explained the suppressed conductance observed for SPPO-H+ under protonation. These findings for the first time demonstrated experimentally that protonation could induce destructive quantum interference, suggesting pH as an easy and efficient approach for manipulating frontier molecular orbitals and controlling molecular conductance.
Another nitrogen-containing functional group is pyridine, which is often used for constructing single molecule wires. 4,4′-bipyridine has been shown to exhibit two conductance states and was realized into a mechanically controlled single molecule switch.18,39 In addition, pyridines can undergo protonation process under acidic conditions and became of interest for conductance studies under pH regulation. Specifically, pyridines selectively interact with protons to form pyridiniums, and a series of pyridine-based molecular backbones were studied by STM-BJ technique in the work of Tang et al.40 One of the compounds studied was M3, structure of which is shown in Fig. 4a. M3 could undergo a protonation reaction in the presence of TFA to form M3-H, and the initial M3 state could be re-formed when Na2CO3 aqueous solution treatment was applied. M3-H showed an enhanced conductance of 10−3.90G0 in comparison to the 10−5.10G0 for M3. The authors applied the flicker noise analysis to these two compounds and showed that M3 junctions conducted electrons primarily through-space while M3-H conducted electrons predominately through-bond (Fig. 4b). This result highlighted that pH could tune the transport mechanism between through-space and through-bond, presenting a new property that could be regulated by pH.
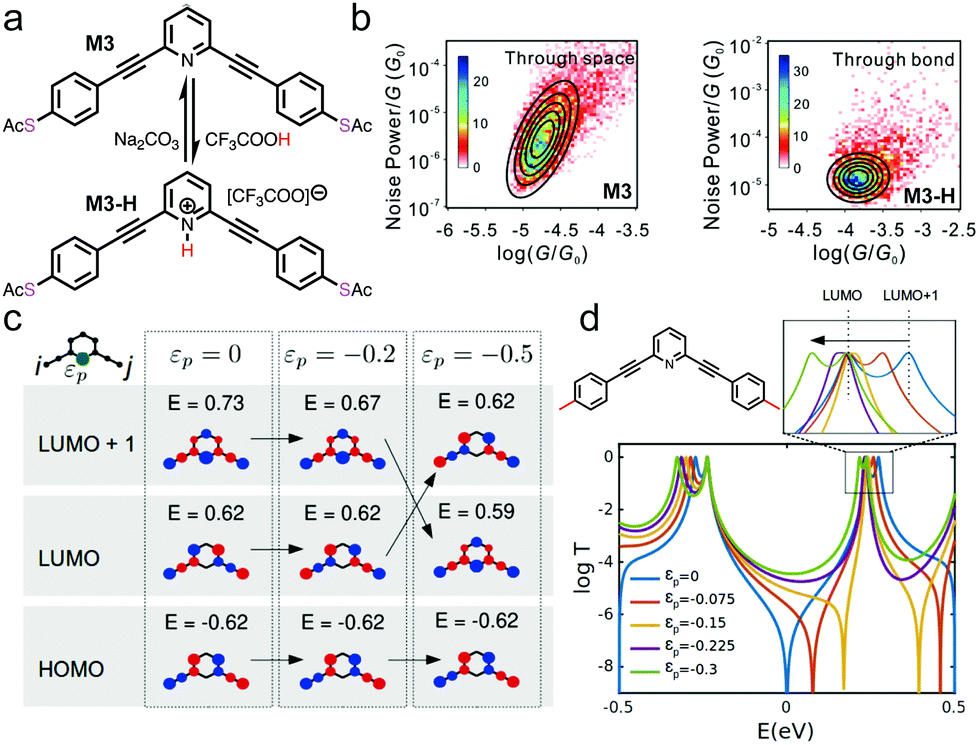 | ||
| Fig. 4 (a) Chemical structure of M3 and its protonated form M3-H. (b) 2D histograms of the flicker noise power versus average conductance for M3 and M3-H. (c) Tight-binding model calculations of frontier molecular orbitals of meta-connected molecular core with (εp ≠ 0) and without (εp = 0) a heteroatom. (d) Calculations of the transmission probability of M3 with different site energies of nitrogen atom (εp). A molecular structure for the calculation is shown on the top left with red lines at both ends representing 1D leads. A zoom-in of the LUMO orbital positions is provided on the top right. (a–d) are reprinted with permission from ref. 40, copyright (2021) American Chemical Society. | ||
To further rationalize this phenomenon, authors used tight-binding model with a parameter εp, which represented the perturbation energy on the nitrogen (indicated in green in Fig. 4c), to computationally evaluate how the molecular orbitals of a model structure of M3 were affected by the presence (εp ≠ 0) or absence (εp = 0) of a heteroatom. When εp was varied from 0 to −0.5, LUMO+1 moved down below LUMO, forming an inversion of the LUMO and LUMO+1 level, as illustrated in Fig. 4c. This led to a switch of the quantum interference pattern from destructive to constructive interference. This type of phenomenon has been observed theoretically in other systems. For example, calculations have shown that HOMO and HOMO−1 inversions were associated with the appearance of destructive quantum interference in permethylated oligosilanes, which occurred when the dihedral angles of the internal Si–Si–Si–Si was varied from 90° to 0°.11 Next, transmission calculations of M3 connected to two 1D leads were carried out where site energy for nitrogen was decreased from 0 to −0.3, and the calculations revealed that the anti-resonance character of the destructive quantum interference disappeared at εp = −0.225 or lower values (Fig. 4d). The authors concluded that the protonation effect in the measurement could be modelled as a negative site energy on the nitrogen atom, therefore the calculated result of the disappearance of the quantum interference explained the observed increase in conductance.
4. Control the conductance of amine- and carboxyl-terminated molecules by pH
Now we turn to chemical linker groups that can respond to pH. Amine group has been shown to form dative bond with bulk Au for attaching molecules to electrodes,41 and has also been shown to form weak covalent bond with Au nanocrystals due to a finite size effect.42 The binding strength of amine–Au bond is likely affected by the protonation state of the nitrogen on amine. Chen et al. performed STM-BJ experiments of diamine butane and dicarboxylic-acid butane in aqueous solutions of different pH values to determine how pH affected their single molecule conductance.43 1D histograms for diamine butane junctions are shown in Fig. 5a. At pH = 13 (purple area), one peak located at 1.45 × 10−3G0 and a second peak located at about two times the conductance of the first peak were observed. The authors assigned the 1.45 × 10−3G0 peak to be the conductance of a single molecule covalently bound to the electrodes through the deprotonated amine group. When pH was reduced to 10, the intensity of the conductance peaks (orange line) reduced, which was ascribed to a decrease in the binding probability. When the pH was adjusted to 1, no peak was observed anymore and the conductance histogram became essentially featureless (green area), which indicated that likely no molecular junction was formed. This work demonstrated that in an aqueous solution of pH ∼ 1, the protonated amine groups donated their lone pairs of electrons in the nitrogen atoms to protons, which weakened NH3+–Au bonds, thus preventing the formation of the molecular junctions.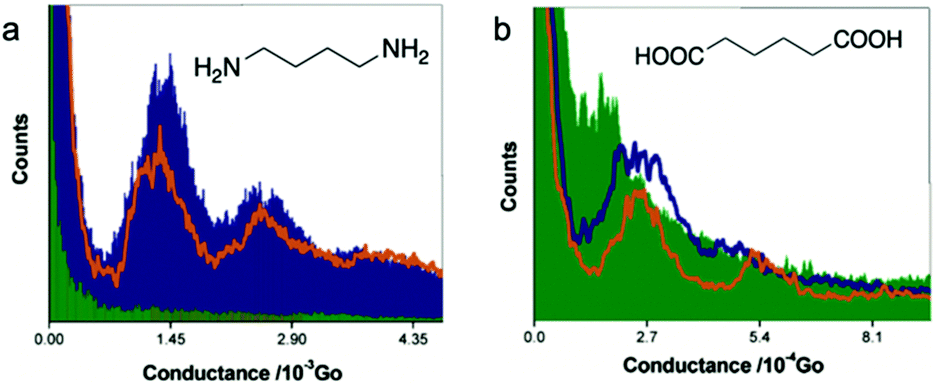 | ||
| Fig. 5 Conductance histograms of (a) diamine butane and (b) dicarboxylic-acid butane measured at pH = 1 (green shaded area), 10 for diamine butane and 5 for dicarboxylic-acid butane (orange line), and 13 (purple shaded area or line). (a and b) are adapted with permission from ref. 43, copyright (2006) American Chemical Society. | ||
Fig. 5b shows the conductance histograms of dicarboxylic-acid butane measured at pH = 1, 5, and 13. Like amine-terminated butane, dicarboxylic-acid butane also showed a pH-dependent conductance. When pH = 1, a weak peak located at 1.5 × 10−4G0 (green area) indicated that protonated –COOH groups could interact with Au electrodes to some degree, which was attributed to the lone pair of electrons on the oxygen atoms.44 We note that in a previous work by Ahn et al. no identifiable conductance signature was observed for 4-(methylthio)benzoic acid in an acidic solution (pH of 1–3), which was explained by the reduced solubility of 4-(methylthio)benzoic acid in the solution of such low pH or by the fact that 4-(methylthio)benzoic acid was protonated in strongly acidic solution.45 The different behaviors between dicarboxylic-acid butane and 4-(methylthio)benzoic acid suggest that the solubility of the molecule, molecular length, and/or backbone structure might influence the binding of carboxyl-terminated molecules to Au under strongly acidic conditions. The pK1 and pK2 of dicarboxylic-acid butane were measured to be 4.42 and 5.41, respectively.43 Thus, when pH was increased to 5, both –COOH and –COO− existed in the solution as one of the anchoring groups –COOH was possibly deprotonated to –COO− with a negative charge conjugated between the two equal O atoms. A negative charge could enhance the electronegativity of the O atom, thereby increasing the strength of the O–Au bond. Indeed, at pH = 5, the authors observed a conductance peak located at 2.7 × 10−4G0 (orange line), nearly twice of the conductance measured at pH = 1, confirming their hypothesis. Finally, at pH = 13, both anchoring groups likely became deprotonated, and a more intense conductance peak located at 2.7 × 10−4G0 (purple line) was observed, suggesting a more frequent binding between the molecule and the Au electrodes. This pH-dependent conductance study of amine- and carboxyl-terminated alkanes highlighted the important role that pH plays in regulating both the formation of the molecular junction and the junction conductance through a protonation and deprotonation process of the anchoring groups.
5. Control the conductance of peptides by pH
Amine and carboxyl anchor groups were shown to undergo a deprotonation reaction in response to an increase in pH, which could promote the molecular binding to the Au electrodes. Specifically, at low pH ∼ 1, –NH3+ and –COOH dominated in the solution, while at pH close to 7 or above, these two groups were deprotonated to form –NH2 and –COO−. Now we discuss how this property of amine and carboxyl group can also play a role in tuning the molecular conductance when they serve as side groups of a backbone chain of peptides.12,46,47 Xiao et al. studied peptides containing amine and carboxyl side groups (structures see Fig. 6a) and probed how pH modulated their single molecule conductance.12Peptide 1, 2, and 3 each contained one, two, and one peptide bond(s) in the backbone, where peptide 3 had an additional carboxyl side group; all were terminated with thiol linker groups. The authors carried out STM-BJ measurements for peptide 1–3 and found that they all showed a decrease in conductance with increasing pH from 2 to 12.5 (Fig. 6a). The authors suggested that this was an outcome of the deprotonation process at the amine position, given that amine inside a cysteine in solution phase has a pKa of 8. The protonated –NH3+ is more electron-withdrawing than –NH2, and how this electron-withdrawing property of a side group impacts the conductance of molecular junctions has been investigated in both experimental48 and theoretical studies.49–51 In Venkataraman et al, the authors showed that electron-donating substituents increase the conductance, and electron-withdrawing substituents decrease the conductance of a 1,4-benzenediamine.48 Same trend was observed in DFT calculations of 1,4-benzenedithiol and 1,4-benzenediamine junctions in Smeu et al.49 and Jin et al.50 Well, in a theoretical study of a series of functionalized bicyclo[2.2.2]octanes, -octasilanes, and -octagermanes, no clear correlation for molecules carrying non-carbon-based substituents was observed, but an opposite trend where a more electron-donating substituent decreases the conductance was observed for molecules with carbon-based substituents.51 In this study of peptides, the more electron-donating substitution group –NH2 in comparison to –NH3+ was shown to decrease the conductance of peptide 1–3. Considering the conjugated π orbitals in benzenediamine and benzenedithiol, and the saturated carbon chains in bicyclics and peptides, different electronic structures of the backbones may dictate how the electron-withdrawing property of the substitution group impacts the molecular conductance. This consideration of the electron-withdrawing property of the amine group under different pH values provides a perspective in understanding the underlying mechanism of this pH-induced conductance change, and we believe further studies are needed to fully understand the pH regulation of the conductance of –NH2 substituted backbones.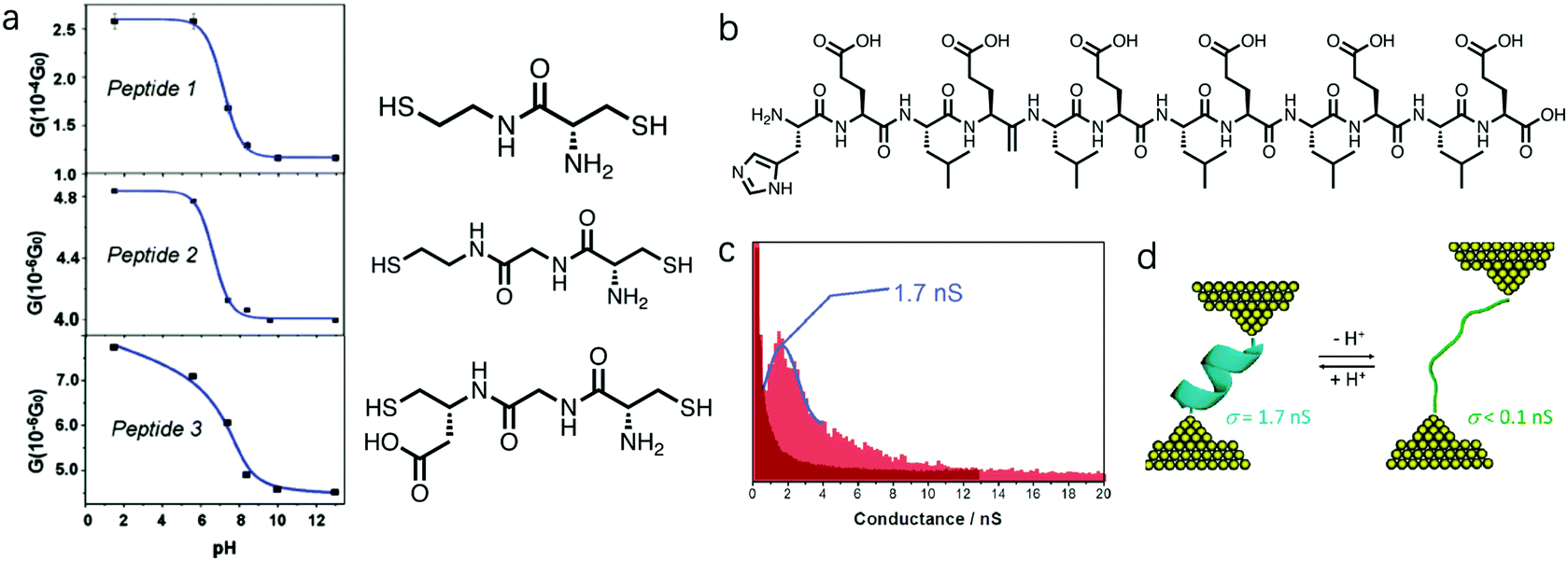 | ||
| Fig. 6 (a) Conductance versus pH plots and chemical structures for peptides 1, 2 and 3. The blue solid lines are guide for the eye. (b) Chemical structure for H(EL)5C. (c) Conductance histograms for H(EL)5C at pH = 6.9 (dark red) and pH =2 (light red) using I(s) technique in an STM method. (d) A schematic diagram of molecular configuration change for H(EL)5C during the pH regulation. (a) is adapted with permission from ref. 12, copyright (2004) American Chemical Society. (c and d) are reprinted with permission from ref. 47, copyright (2011) American Chemical Society. | ||
The authors observed a greater conductance change with pH for peptide 1 than those observed for peptide 2 and 3, likely due to the fact that among the three, amine group occupied the largest fraction of the molecule in peptide 1. In addition, peptide 1 and 2 showed a steeper conductance drop from pH = 5 to pH = 8 in comparison to that of peptide 3; the authors reasoned that this difference possibly came from the pH response of the carboxyl group on peptide 3. Carboxyl group COOH on peptide 3 likely became deprotonated to form COO− when pH was increased, thus affecting the overall conductance of peptide 3. Since the pH at which the deprotonation process occurs is different for –NH3+ and –COOH, the conductance change of peptide 3 was occurring over a wider pH range than that observed for peptide 1 and 2.
In a later study, Scullion et al. designed a peptide H(EL)5C, where H stands for histidine, E for glutamic acid, L for leucine, and C for cysteine; its chemical structure is shown in Fig. 6b.47 STM-based method with I(s) technique was used to measure the single molecule conductance of Au–H(EL)5C–Au junctions. Different from the study described above, unmodified histidine and cysteine were used as linkers to attach the molecule to the Au surface. The authors observed a well-defined conductance peak at 1.7 nS for H(EL)5C at pH = 2, and a significantly lower conductance below 0.10 nS when the pH was increased to 6.9 (Fig. 6c). The peptides within these monolayers existed as α-helices at pH ∼ 2 while deprotonation of the carboxyl groups in the glutamic acid residues led to a more extended conformation of the peptide at pH ∼ 7 (Fig. 6d) and substantially suppressed electron transport across the molecular film. When multiple groups were negatively charged, the charges on each group repelled each other and disrupted the original folding of the peptide. Such unfolding of the peptide increased its junction length and decreased its single-molecule conductance.
6. Control the conductance of imidazole derivatives by pH
The imidazole moieties were studied recently, both as anchor groups and as backbones, in single molecule conductance measurements by several research groups.52–55 Imidazoles were identified as attractive anchor groups for providing a pH-activated linkage between Au electrodes and molecules, and were shown to provide robust conductance pathways as molecular backbones. In 2020, Pan et al. reported the binding mechanism of imidazole with Au, revealing that the molecule attached to Au electrodes when it was deprotonated in position 1 (Fig. 7a, top) under basic conditions.54 The authors measured the conductance of imidazole deposited from a solution with pH of 3, 7, 9, or 12, respectively, and found that the conductance peaks between 10−2G0 and 10−1G0 occurred under basic conditions but disappeared when pH was below 7 (Fig. 7a, bottom). The authors attributed the high conductance peak at 1.9 × 10−2G0 to the binding of the lone pair on the 3N and the deprotonated 1N positions with the Au electrodes and attributed the additional high conductance peaks occurring at multiples of 1.9 × 10−2G0 to be parallel binding of multiple molecules in the junctions. For the low conductance peak observed near 10−4G0, the authors proposed that they corresponded to molecules of imidazole bound in series through an Au atom, supported by the plateau lengths measured for this conductance feature as well as DFT calculations. This work demonstrated that imidazole provided a persistent binding to Au electrodes under basic conditions, and such binding did not occur under acidic conditions. In a separate study, Wu et al. reported an almost identical 1D conductance histogram of imidazole measured in Milli-Q water (Fig. 7b, bottom) compared to the histogram in Pan et al. (Fig. 7a, black curve).55 Authors in this study, however, emphasized a different aspect of imidazole conductance regulation: the impact of water rather than pH. The authors concluded that the multiple peaks (Fig. 7b, bottom) corresponded to water-containing monomer (labelled as MH2O and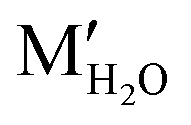 ) and dimer (labelled as DH2O and
) and dimer (labelled as DH2O and  ) species, and the multiple peaks for each monomer and dimer configuration resulted from different hydrogen bonding conformations. One such optimized imidazole–water–imidazole dimer chain structure is shown at top of Fig. 7b. When authors repeated the conductance measurement of imidazole under an anhydrous condition, they observed a drastically different conductance histogram (Fig. 7c, bottom). The authors assigned the three major conductance peaks to monomer (M), dimer (D) and trimer (T) species, as the formation of the oligomeric chain through hydrogen bonding was supported by the DFT calculations; a DFT optimized structure is shown on the top of Fig. 7c. Thus, the authors emphasized that not only protonation state but also hydrogen bonding contributed to the conductance features of imidazole. We note that apart from the major conductance peak at 1.9 × 10−2G0, the detailed interpretations in these two studies about the second peak between 10−2G0 and 10−1G0 and the peak near 10−4G0 were different, highlighting the complex nature of the imidazole conductance profile.
) species, and the multiple peaks for each monomer and dimer configuration resulted from different hydrogen bonding conformations. One such optimized imidazole–water–imidazole dimer chain structure is shown at top of Fig. 7b. When authors repeated the conductance measurement of imidazole under an anhydrous condition, they observed a drastically different conductance histogram (Fig. 7c, bottom). The authors assigned the three major conductance peaks to monomer (M), dimer (D) and trimer (T) species, as the formation of the oligomeric chain through hydrogen bonding was supported by the DFT calculations; a DFT optimized structure is shown on the top of Fig. 7c. Thus, the authors emphasized that not only protonation state but also hydrogen bonding contributed to the conductance features of imidazole. We note that apart from the major conductance peak at 1.9 × 10−2G0, the detailed interpretations in these two studies about the second peak between 10−2G0 and 10−1G0 and the peak near 10−4G0 were different, highlighting the complex nature of the imidazole conductance profile.
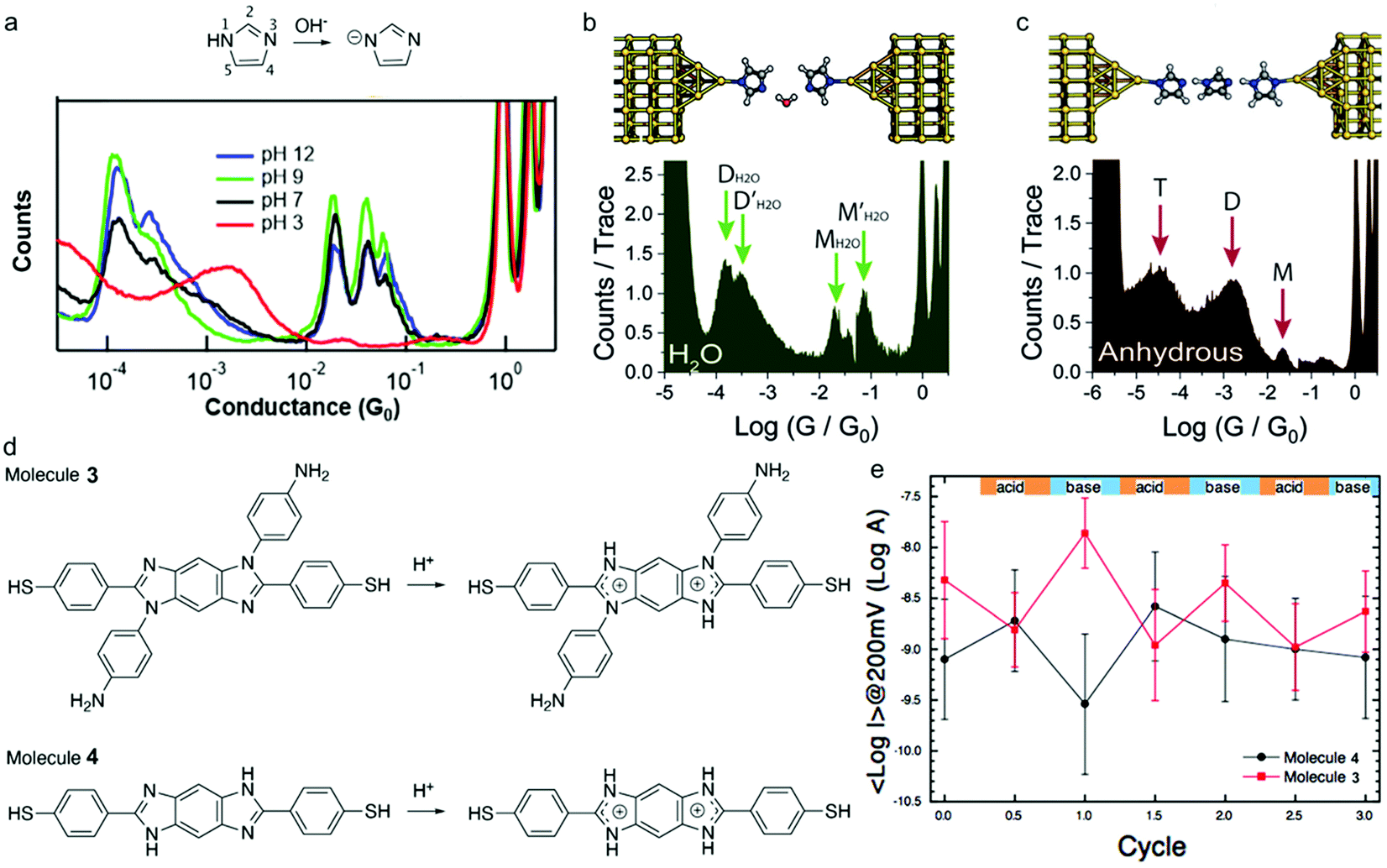 | ||
| Fig. 7 (a) Upper: Chemical structure of imidazole under neutral and basic conditions. Lower: 1D conductance histograms for imidazole junctions measured under pH of 3, 7, 9, or 12, respectively. (b and c) Upper: DFT-optimized structure of a chain of imidazoles formed through hydrogen bonding in (b) Milli-Q water and (c) an anhydrous environment. Lower: 1D conductance histogram of imidazole measured in (b) Milli-Q water and (c) an anhydrous environment. (d) Protonation process for 3 and 4 under acidic condition. (e) Plot of the log mean current for 3 and 4 during three successive acidic and basic cycles. Current was determined from a fitted log-normal distribution to the current histograms measured by C-AFM at 200 mV. (a) is reprinted with permission from ref. 54, copyright (2020) American Chemical Society. (b and c) are reprinted with permission from ref. 55, copyright (2020) Royal Society of Chemistry. (e) is reprinted with permission from ref. 53, copyright (2020) Royal Society of Chemistry. | ||
In the same year, Audi et al. reported a conductance study of benzo-bis(imidazole) derivatives 3 and 4 (structures see Fig. 7d) where imidazole was part of the molecular backbone, and demonstrated how pH regulation on molecular conductance was affected by side chain chemistry.53 Both aniline-substituted 3 and H-substituted 4 could undergo protonation processes in the presence of acid, as shown in Fig. 7d. The authors used conductive atomic force microscopy (C-AFM) to measure the current–voltage characteristics of 3 and 4. Interestingly, the opposite conductance trend was observed in these two molecules under acidic and basic conditions: the conductance of 3 increased when 3 switched from the protonated state to the neutral state, whereas the conductance of 4 decreased when the same deprotonation process occurred (Fig. 7e). DFT calculations showed that protonation decreased the HOMO–LUMO gap in both molecules, and the observed opposite change in conductance upon protonation was rationalized to be a result of the different position of the HOMO level in these two molecules that led to the different transmission probabilities at the Fermi energy level. We note that imidazole is among the few that has been extensively investigated both as an anchor group and as the backbone for its electronic properties under different pH values.
7. Control the conductance of pyrazole derivatives by pH
In this section, we will discuss a similar approach for tuning the molecular conductance by use of another anchor group, pyrazole. Reliable binding between molecules and Au is important for forming robust and persistent molecular junctions, and pyrazole has recently emerged as a new anchor group for its multidentate binding and strong electronic coupling to the Au. Herrer et al. reported an unconventional electrical peak behavior in single molecule conductance traces of 1,4-bis(1H-pyrazol-4-ylethynyl)benzene (labelled as 5) (Fig. 8a and b).56 The authors assigned this phenomenon to a deprotonation process of pyrazole group in contact with the Au tip during junction evolution. 1H NMR spectrum of 3,5-dimethylpyrazole showed a peak at 12 ppm whereas this peak disappeared in the spectrum of a mixture of 3,5-dimethylpyrazole and Au nanoparticles, indicating that the pyrazolyl moiety became deprotonated in the presence of Au (Fig. 8c). Combined with XPS results and DFT calculations, authors concluded that the conductance increase during the elongation of the junction in the single molecule conductance measurement resulted from a change in the protonation state and the binding geometry. This work revealed that the deprotonation process has a significant impact on the single molecule conductance as well as the dynamical change of the conductance during a junction formation and elongation. In a follow-up work, the same authors formed Langmuir–Blodgett (LB) film of 5 to study the pH effect on its conductance.57 The authors used STM Touch-To-Contact (TTC) method58 to record I–V curves under pH = 5.6 and 11.0, and observed a higher conductance at pH = 11.0 (Fig. 8d). This result suggested that the deprotonation process of the binding group pyrazole under basic conditions could induce efficient electronic coupling and enhance the electron transport through the molecular junctions.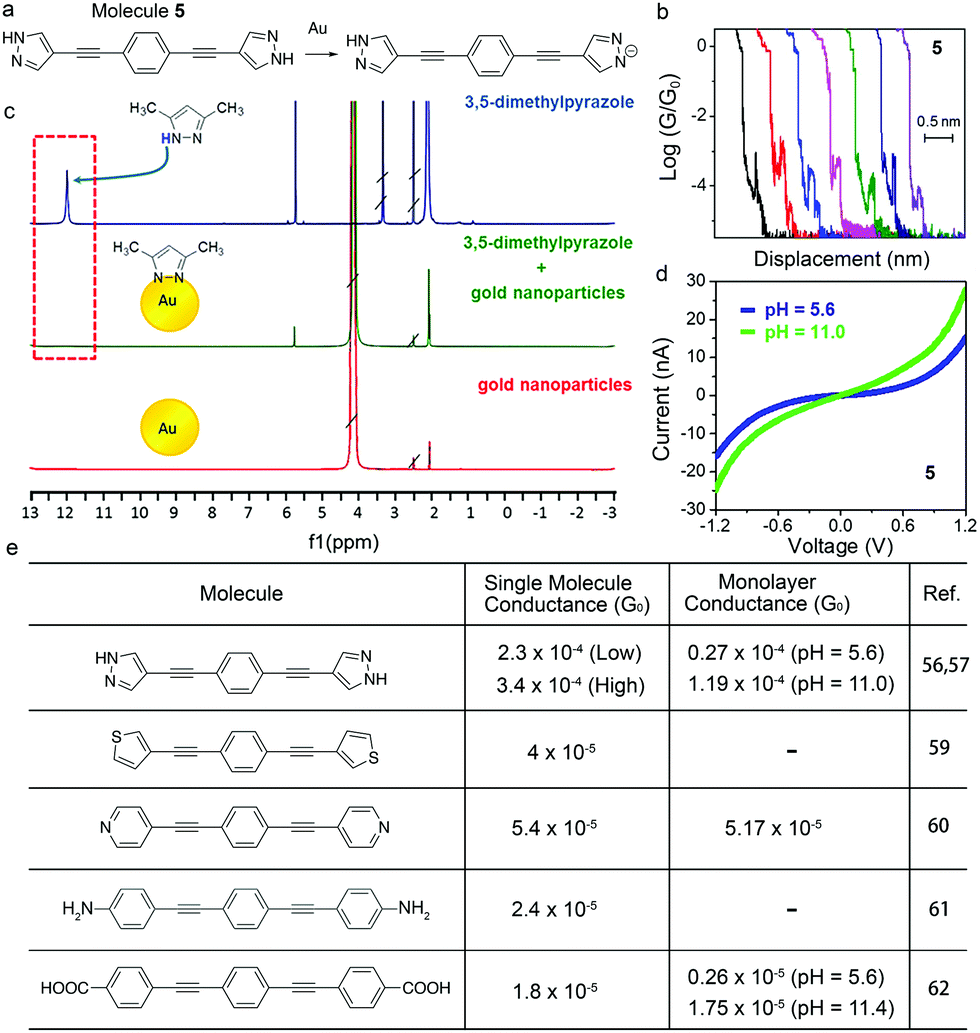 | ||
| Fig. 8 (a) Deprotonation process of 5 in the presence of Au. (b) Single molecule break junction traces of 5 showing peak-like shape events. (c) 1H NMR spectra of 3,5-dimethylpyrazole (blue line), uncapped Au nanoparticles (red line), and a mixture of 3,5-dimethylpyrazole and uncapped Au nanoparticles (green line). (d) Averaged I–V curves for LB film of 5 measured using a TTC method under pH = 5.6 and 11.0. (e) Table of single molecule and monolayer conductance experimentally determined for 5, and its thiophene-, pyridine-, aniline-, and benzoic acid-terminated analogs. (b and c) are adapted with permission from ref. 56, copyright (2018) American Chemical Society. (d) is reprinted with permission from ref. 57, copyright (2021) Royal Society of Chemistry. | ||
A comparison of single molecule conductance and monolayer conductance of 5 to those of its analogs where the binding group pyrazole is replaced by thiophene,59 pyridine,60 aniline,61 or benzoic acid group62 is shown in Fig. 8e for further understanding the impact of the deprotonation process of pyrazole on the molecular conductance. For both single molecule and monolayer measurements, an order of magnitude higher conductance was observed for pyrazole-terminated 5 in comparison to those of other molecules in Fig. 8e with similar molecular lengths. Specifically, DFT calculations of thiophene-,59 aniline-,61 and pyrazole-terminated compounds had corroborated this experimental observation in single molecule conductance measurements.63 DFT calculations indicated that the higher conductance of pyrazole-linked junctions likely resulted from a closer alignment between frontier orbitals of the molecule and the Fermi level of Au for deprotonated pyrazole-terminated compounds compared to the aniline- and thiophene-terminated ones. These features made pyrazole a promising molecular anchor group to be incorporated into molecular materials for transport mechanism studies.
This comparison in Fig. 8e also shows that the monolayer conductance of benzoic acid-terminated compounds is pH dependent.62 The authors suggested that the deprotonation of –COOH into –COO− can lead to two effects: an increase of occupied area for each molecule in LB films, and a more tilted arrangement of the molecules on the gold substrate. These effects, accompanied by the reduced hydrogen-bonding between the molecules due to the –COO–Au linkage, resulted in an increased monolayer conductance in deprotonated LB sample. Taken together with our discussions in Section 4, although the measurement details of the LB film and single molecule junctions were different, both followed the same trend that protonation increases the molecular conductance for carboxylic acid-terminated compounds.
8. Control the conductance of supramolecular complexes by pH
Supramolecular systems, composed of a discrete number of molecules, often result from a few noncovalent interactions including van der Waals interaction, hydrogen bonding, electrostatic interaction, etc. Such supramolecular interactions can also be affected by the pH of the solution and single-molecule conductance of supramolecular systems can therefore be modulated by pH. When two chemical moieties form a supramolecular system through non-covalent interactions, they are referred to as host–guest complexes. Ai et al. investigated the pH dependence of molecular conductance of a host–guest complex in which a host of cucurbit[7]uril (CB[7]) had a guest of melphalan (Mel) encapsulated inside its cavity (structures see Fig. 9a).64Mel had been reported to form 1:1 host–guest complexes with CB[7] at a high equilibrium binding affinity K = 106 at pH = 1 through cation–dipole interactions between NH3+ and CB[7] when the amine group on Mel was protonated under acidic condition.65 The authors carried out STM fixed junction measurements in phosphate buffer (PB) as well as in organic solvent 1,2,4-trichlorobenzene (TCB) to determine the conductance of CB[7] and Mel@CB[7] complex attached to Au electrodes under different pH values. As summarized in Fig. 9b, the authors found that Mel@CB[7] complex had a slightly lower conductance than that of CB[7] under each measured pH, which indicated that encapsulation of Mel inside the cavity of CB[7] potentially weakened the CB[7] binding through the multiple carbonyl groups to the electrodes and suppressed charge transport, consistent with an earlier work by Xiao et al.66 Second, the authors showed that both CB[7] and Mel@CB[7] complex exhibited a very modest decrease in conductance as the pH was increased, and more experiments are likely required to further determine this conductance change with pH. This study provided a starting point for exploring pH control of the conductance of supramolecular systems, and more investigations are likely to be focused on transport mechanisms of similar complex systems.
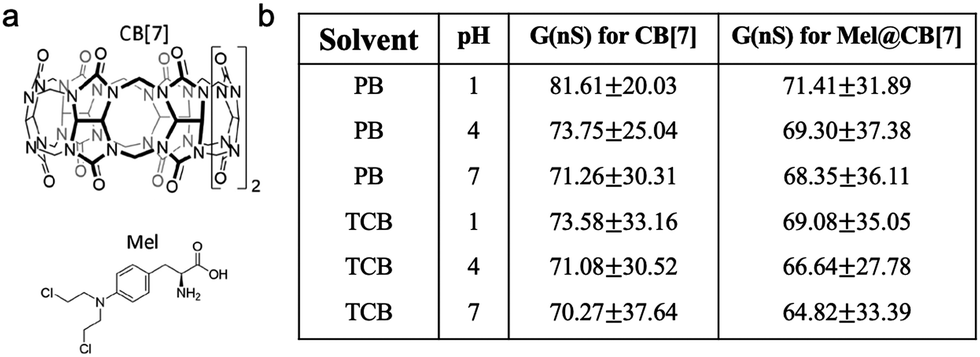 | ||
| Fig. 9 (a) Chemical structures for CB[7] and Mel. (b) Single molecule conductance for CB[7] and Mel@CB[7] measured in phosphate buffer (PB) or organic TCB solvent under different pH values. (a and b) are reprinted with permission from ref. 64, copyright (2020) Frontiers Media S.A. | ||
9. Control the conductance through Lewis acid–base interactions
Liu et al.24 applied a Lewis acid–base interaction strategy to modulate the electronic structure of an organoborane wire for tuning its electronic properties. The authors added fluoride as the Lewis base to a linear phenylene ethynylene wire system, in which the central phenyl moiety is 2,5-di-substituted by dimesitylboryl groups (BMes2) (Fig. 10a).67 Specifically, the authors used tetra-n-butylammonium fluoride (TBAF), a common quaternary ammonium salt that can ionize free fluoride ions in organic solvents, as the Lewis base. It had been reported previously that fluoride ion and organoboron compounds had a strong Lewis acid–base interaction, and the resulting B–F covalent bond could affect the extended π-conjugation and thus the electronic structure of the entire organoboron molecule.68 The authors performed MCBJ measurements (1D histograms shown in Fig. 10b), and found that organofluoroborate 6·2F had a conductance of 10−4.6G0, which was about 75% lower than that of the untreated molecule 6 (10−4.0G0). This apparent decrease in conductance when TBAF was added resulted from the weakening of the extended π-conjugation when electrons in the B–F bonds occupied the boron pπ orbital. DFT calculations (Fig. 10c) further suggested that the extended π-conjugation accounted for the low-lying LUMO of the borane, and the Lewis acid–base interaction between the boron atom and fluoride substantially increased the LUMO energy level, leading to a larger HOMO–LUMO gap and an increased separation between the LUMO and the Fermi level of the Au surface.69 We envision that the pH control of the molecular conductance through Lewis acid–base interactions described in this work might be extended to other molecular materials.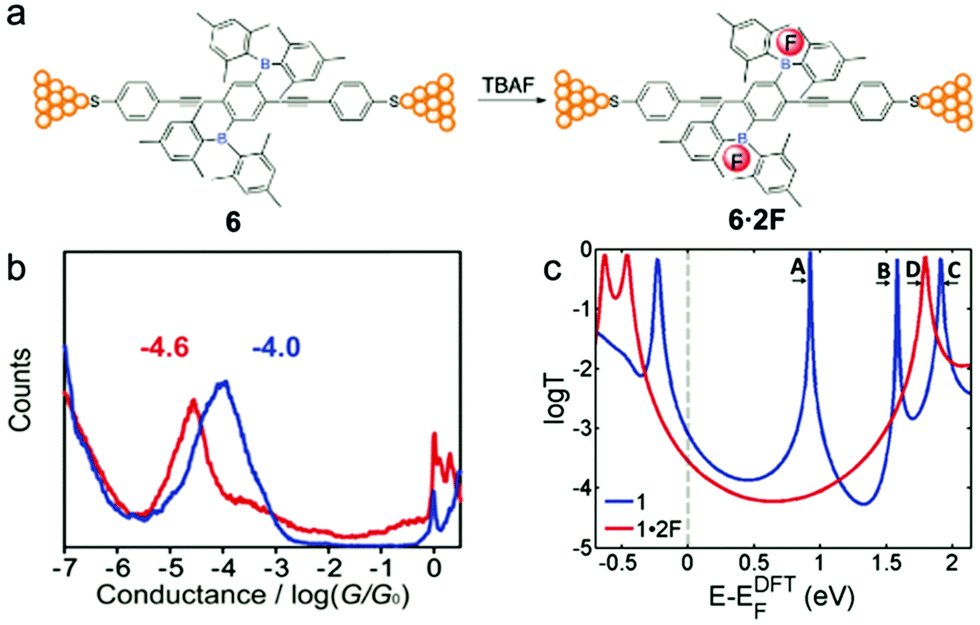 | ||
| Fig. 10 (a) A schematic diagram of single molecule junctions formed with 6 and 6·2F. (b) 1D conductance histograms and (c) DFT transmission coefficients of 6 (blue) and 6·2F (red). (a–c) are reprinted with permission from ref. 24, copyright (2018) Royal Society of Chemistry. | ||
10. pH combined with light for tuning the molecular conductance
pH-sensitive molecules respond not only to pH, but also to other stimuli such as light and force, therefore provide exciting opportunities for integrating functionalities into molecular devices. Light is an attractive means to regulate molecules due to its non-invasiveness, remote manner, and high temporal resolution.69 In this section, we describe several fundamental achievements on a combined use of light and pH for a dual control of molecular conductance. Darwish et al. realized a dual light- and pH-responsive electronic switch in a bifunctional spiropyran derivative through the STM-BJ technique.70 From UV-Vis absorption studies, the authors found that spiropyran SP-1 turned to merocyanine MC-1 upon UV irradiation and switched to protonated merocyanine (MC-H-1) upon addition of TFA (structures see Fig. 11a top); these processes were reversible when visible light was applied to MC-1 or base was added to MC-H-1. As the structures of MC-1 and MC-H-1 manifested increased conjugation in comparison to that of SP-1, an increased conductance for MC-1 and MC-H-1 was expected. STM-BJ measurements showed that SP-1 had two conductance peaks, and the authors explained that these events likely resulted from multiple contact geometries (Fig. 11a). An additional high conductance peak (0.15 G0) appeared when UV light was turned on; this peak was attributed to the extended conjugation and the delocalized frontier orbitals of MC-1. When an acid was applied, conductance for MC-H-1 was determined to be 0.11 G0, also higher than that of SP-1. Furthermore, all conductance plateaus appeared at the high conductance value under the acidic condition, whereas only 15% to 35% of the conductance plateaus were at the high conductance state under UV irradiation, indicating that the pH-induced isomerization was possibly more complete than that induced by UV light. This demonstration of the multi-responsive single molecule switch based on spiropyran derivatives has offered a new scheme in creating nanoscale logic gates in molecular circuitry. | ||
| Fig. 11 (a) Chemical structure of MC-H-1, SP-1, and MC-1 and their respective 1D conductance histograms determined by STM-BJ. (b) Intermolecular proton transfer occurs from MCH to 1,3Az under blue light, and from 1,3Az-H to SP-2 in dark. The target molecule in the measurement is shown in between Au electrodes, and the molecule supplemented in the measurement solution is shown without Au electrodes attached. (c) A diagram of the molecular AND logic gate and the corresponding measured conductance histograms. (d) Mixed monolayer formed with SP-3 and hexanethiolate and the conversion scheme from SP-3 to MC and from MC to MCH+. (e) Current is plotted as a function of voltage potential measured for mixed SAMs of SP-3 and hexanethiolate with conical EGaln top contacts. Green: exposed to 365 nm light for 20 min, then to white light for 12 h. Red: exposed to acid, followed by 365 nm light for 20 min. Black: exposed to acid, then to 365 nm light for 12 h, and finally to white light for 12 h. Blue: exposed to acid, followed by 365 nm light for 20 min, followed by base, and then white light for 12 h. (f) Left: Writing letters “rcclab” in an Au array where each letter is indicated by an 8-bit array. The bottom binary code corresponds to letter b. Right: Writing of “1” at positions where H+ can flow inside through the holes. Writing of “0” where no holes are punched. (a) is reproduced with permission from ref. 70, copyright (2014) American Chemical Society. (b and c) are reprinted with permission from ref. 71, copyright (2019) Wiley. (d–f) are reprinted with permission from ref. 72, copyright (2019) Wiley. | ||
In 2019, Cai et al. further expanded this idea of using pH and light dual-control for creating logic gates through a unique photo-induced proton transfer (PIPT) strategy using a non-photo-responsive azulene derivative and a photoacid spiropyran.71 The pH-sensitive azulene derivative 1,3Az (structure see bottom of Fig. 11b) was the target molecule bound between two Au electrodes, and the photoswitchable spiropyran (SP-2) and an equal amount of TFA were supplemented in the molecular solution for MCBJ measurements. As illustrated in Fig. 11b, merocyanine MCH released a proton to form SP-2 under blue light. Meanwhile, 1,3Az in proximity accepted this proton released by MCH to form 1,3Az-H. This process was referred to as PIPT. In MCBJ measurements, the protonated 1,3Az-H showed a conductance twelve times that of 1,3Az, agreeing with the previous work by Yang et al.27 This work demonstrated that the PIPT strategy enabled a light-induced conductance modulation for non-photoresponsive molecules. Furthermore, the authors used high (protonated) and low (deprotonated) conductance states of 1,3Az junctions as electrical output signals “1” and “0” to build single-molecule Boolean logic gates, and specifically, the AND and OR gate. Fig. 11c shows the construction of an AND gate where acid and blue light were defined as the inputs and the measured conductance state was defined as the output. Among the four combinations of the inputs, only “1, 1” combination gave rise to the “1” output; “0, 0”, “0, 1”, and “1, 0” all gave rise to the “0” output.
In the same year, Kumar et al. evaluated another spiropyran derivative, denoted as SP-3 (structure see Fig. 11d), as building blocks to construct well-ordered SAMs for the encoding of non-volatile information.72 Owing to the fact that when isomerization of SP-3 to MC occurred upon UV light exposure, MC could spontaneously thermalize back to SP-3,73 the authors exposed MC to acid for generating the protonated form labelled as MCH+ to create a locked state74,75 (Fig. 11d). Once MCH+ was formed, it could not convert back to SP-3 unless a basic solution was added. The authors recorded current density versus voltage (J/V) in tunnelling junctions formed with eutectic Gallium–Indium top-contacts76 to characterize the conductance of SP-3, MC, and MCH+. As plotted in Fig. 11e, the authors showed that a mixed monolayer of SP-3 and hexanethiolate under an exposure to UV light followed by white light (green curve) had a low conductance, while under an additional exposure to acid, the conductance of the mixed monolayer increased by a factor of 103, as shown in red (acid, followed by UV light) and black curves (acid, then UV light, then followed by white light). This result indicated that protonation effectively locked the molecular junction in the high conductance state and a white light treatment no longer converted it back to the original low conductance state. Finally, the experiment of the mixed monolayer exposed to acid, followed by UV, followed by base, and then by white light, showed the return of the conductance to the initial state (blue curve), indicating that the conductance locking process was reversed by a chemical treatment of base. Based on these results, the authors then developed a memory device, where SP-3 state was encoded as “0” bit, and MCH+ form was encoded as “1” bit. Bits were defined by their respective conductance values, as a ratio of 103 between the two conductance values was considered large enough for encoding the information. The authors demonstrated a non-volatile memory device where the rcclab six-character string encoded by 7-bit ASCII was written, erased, and rewritten (Fig. 11f) with 100% bits erased and one erroneous bit rewritten. Protonation as a chemical locking mechanism makes spiropyran-based molecular materials particularly encouraging for molecular memory devices.
11. pH combined with electrochemical gating for tuning the molecular conductance
In addition to light, electrochemical gating has also been demonstrated as a powerful tool in probing and regulating charge transport through molecular junctions.77–79 Here we focus on tuning both the electrochemical gate voltage and pH in controlling the single molecule conductance. In an earlier work, Baghernejad et al. varied electrochemical gate voltage and pH in conductance measurements of junctions of 4,4’-bipyridine (44BP) attached to Au electrodes (Fig. 12a).80 The authors found that elongation of the molecular junctions accompanying three conductance plateaus (denoted as H, M, and L), and rationalized the three conductance states to the three species: two molecule bridging the electrode gap, one molecule with overlap between Au and π orbitals, and one molecule with Au-N interactions (Fig. 12b). Here we will focus on the regulation of single molecule junction conductance, in other words, the M and L states. The authors showed that both M and L conductance of 44BP decreased with increasing electrochemical potential at each measured pH (Fig. 12c). If we compare the conductance under different pH, the M and L conductance of 44BP at near zero-potential showed a negligible change in measurements at pH = 10.0, 4.1, and 1. As 44BP was considered protonated under acidic condition, the authors suggested that protonation of one or both pyridyl moieties had no effect on the junction conductance and a deprotonation process occurred upon the formation of the Au–molecule contacts. Similar results were reported by Brooke et al. where the authors measured the conductance of 4,4′-vinylenedipyridine (44VDP) attached to Au electrodes (structure see Fig. 12d).81 The authors observed similar conductance histograms for 44VDP measured in pH = 2.5 solution and in neutral organic solvent 1,3,5-trimethylbenzene, suggesting that deprotonated pyridines bound to Au in both cases, even the pK1 and pK2 of 44VDP were 4.411 and 5.646,82 respectively.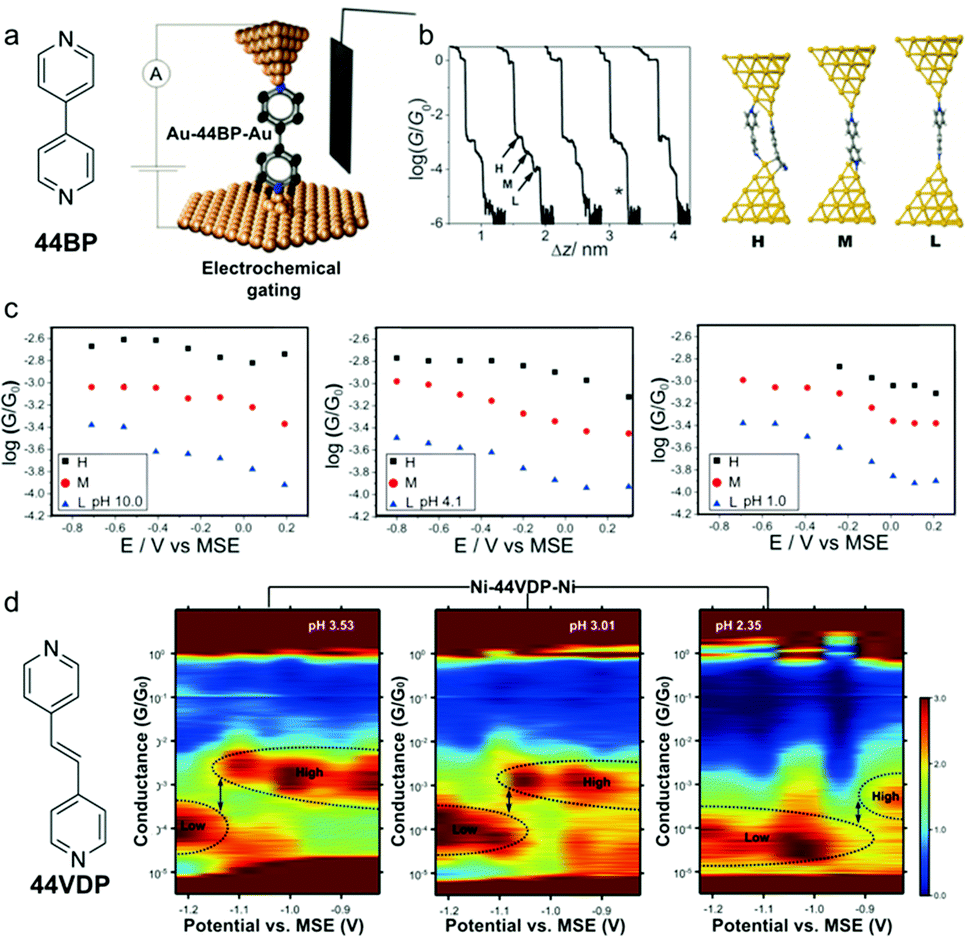 | ||
| Fig. 12 (a) Chemical structure of 44BP and STB-BJ technique employing an electrochemical gate. (b) Conductance vs. displacement traces for 44BP and optimized binding geometry of the H, M, and L conductance states by DFT. (c) The single molecule conductance vs. applied electrode potential for H, M, and L states at pH = 10.0, 4.1, and 1.0. (d) Chemical structure of 44VDP and 2D conductance vs. potential histograms measured for Ni–44VDP–Ni junctions in solutions of pH = 3.53, 3.01, and 2.35. Dotted lines indicate the locations of the conductance peaks. The applied potential in (c) and (d) is the substrate potential relative to a mercury sulfate reference electrode (MSE). (a–c) are adapted with permission from ref. 80, copyright (2014) Royal Society of Chemistry. (d) is adapted with permission from ref. 81, copyright (2018) American Chemical Society. | ||
In a later work, Brooke et al. applied Ni as source and drain electrodes to probe the conductance of 44VDP under a dual control of pH and electrochemical gate voltage,81 as Ni–44VDP–Ni junctions had been shown to display an enhanced response to electrochemical gating than Au–44VDP–Au junctions.83 Ni–44VDP–Ni junctions showed two conductance peaks, labelled as high and low conductance states. Based on the plateau lengths in 2D histograms, pKa values of 44VDP, and previous measurements of 44BP,83 the authors concluded that the high conductance state corresponded to deprotonated 44VDP and the low conductance state corresponded to protonated 44VDP at one of the pyridyl groups. In 2D conductance vs. gate potential histograms (Fig. 12d), a transition from high to low conductance for Ni–44VDP–Ni junctions happened on a more positive gate voltage when pH was decreased. The authors suggested that this result, combined with previous works, indicated that both deprotonated-pyridyl and protonated-pyridyl–H+ could bind to Ni electrodes, and the negatively charged electrodes, as was the case when gate potential was lower, favored the binding to the positively charged protonated molecules. The authors further showed that the STM-BJ traces exhibited individual proton transfer reactions in real-time and showed that such events were stochastic. These demonstrations showed that pH manipulation combined with external stimuli has emerged as a promising strategy to construct single-molecule devices for functions such as chemical sensors and logic gates, and furthermore, STM-BJ can allow us to study chemical processes at the single molecule level and observe dynamics and transient events that are not captured by ensemble approaches.
12. Conclusions and outlook
This Review sets out to showcase the power of pH in regulating molecular electronics by highlighting a wide range of pH-controlled molecular properties. For instance, pH can increase as well as decrease the single molecule conductance, switch the quantum interference pattern, alter the through-bond versus through-space transport mechanism, affect the molecular junction length, and strengthen or weaken the binding between the molecule and the electrodes. pH, through protonation or deprotonation, manipulates the electronic structure of an(many) atom(s), thereby modifying the molecular orbitals and their alignment with the Fermi energy of the metal electrodes. It is precisely the changes in the molecular orbitals and their coupling with the electrodes that lead to significant changes in single-molecule conductance measured by break junction techniques, enabling the fabrication of single-molecule devices such as switches and rectifiers.Although many molecules remain to be explored, a few of them showed a reduced HOMO–LUMO gap once the molecular backbones became protonated, primarily resulting from a lower LUMO orbital. Remarkably, experimental results and calculations indicated that (de)protonation processes sometimes are accompanied by quantum interference pattern switching. For example, protonation of the diketopyrrolopyrrole derivative SPPO occurred with appearance of destructive quantum interference and conductance decrease, while protonation of the pyridine derivative M3 resulted in destructive quantum interference disappearing and conductance increase. The impact of pH in charge transport is significant, and the underlying mechanism of pH-tuning will continue to be a crucial area for investigation in molecular electronics. We are optimistic that the strategy of regulating conductance by pH will enable new understanding of the structure–function relationships of molecular materials.
One of the important benefits from pH-regulated conductance studies is the realization of multi-functional molecular devices. So far, the combined use of pH and light has enabled the construction of single-molecule logic gates and non-volatile memory devices, and the pH and electrochemical gate dual control has been demonstrated as single-molecule switches. We note that theoretical calculations have proposed that protonation is capable of modulating the spin transport properties of specific molecules and the combined implementation of pH and magnetic field could modulate the direction and intensity of spin–polarized current.27,84,85 These works hold promise for new observations to be made in protonation-controlled single molecule spin transport under magnetic fields.
Furthermore, pH can serve as a unique probe for studying intramolecular and supramolecular interactions of complex systems. The examples detailed in this Review involve chemical groups such as amines, carboxylic acids, and amino acids that respond to pH, and these chemical groups are also critical elements in biological molecules such as nucleic acids and proteins. As we know, living organisms carefully regulate their pH values to maintain optimal enzyme activities because pH impacts the structures, structural dynamics, activities, and functions of biological macromolecules. On the other hand, electron-transfer reactions occur in a variety of biological processes such as photosynthesis and nitrogen fixation. Considering the importance of both pH and charge transfer in biology, these studies that reveal the electron transport properties of supramolecular systems at the single molecule level, as well as their regulations by pH, will deepen our understanding of biological processes and biomolecular assemblies. We anticipate that future studies will continue to elucidate mechanisms of pH-regulated charge transport through biomolecules and extend the insights that we gain from these studies to biology. As we approach conductance control with the use of pH, the rules of how pH governs the chemistry and electronics in junctions will be unveiled.
Author contributions
Following CRediT definitions: conceptualization: H. L.; funding acquisition: H. L.; investigation: B. W., W. G., J. M. A., and H. L.; project administration: H. L.; supervision: H. L.; visualization: B. W., W. G., J. M. A., and H. L.; writing – original draft: B. W., W. G., and J. M. A.; writing – review and editing: B. W., W. G., J. M. A., and H. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. L. acknowledges start-up funding from City University of Hong Kong, no. 9610521.Notes and references
- A. Aviram and M. A. Ratner, Chem. Phys. Lett., 1974, 29, 277–283 CrossRef CAS.
- S. V. Aradhya and L. Venkataraman, Nat. Nanotechnol., 2013, 8, 399–410 CrossRef CAS PubMed.
- T. A. Su, M. Neupane, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Nat. Rev. Mater., 2016, 1, 16002 CrossRef CAS.
- B. Q. Xu and N. J. J. Tao, Science, 2003, 301, 1221–1223 CrossRef CAS PubMed.
- L. Venkataraman, J. E. Klare, I. W. Tam, C. Nuckolls, M. S. Hybertsen and M. L. Steigerwald, Nano Lett., 2006, 6, 458–462 CrossRef CAS PubMed.
- J. M. vanRuitenbeek, A. Alvarez, I. Pineyro, C. Grahmann, P. Joyez, M. H. Devoret, D. Esteve and C. Urbina, Rev. Sci. Instrum., 1996, 67, 108–111 CrossRef CAS.
- J. Reichert, R. Ochs, D. Beckmann, H. B. Weber, M. Mayor and H. von Lohneysen, Phys. Rev. Lett., 2002, 88, 176804 CrossRef CAS PubMed.
- G. Gryn'ova and C. Corminboeuf, Chimia, 2019, 73, 245–251 CrossRef PubMed.
- D. C. Milan, A. Vezzoli, I. J. Planje and P. J. Low, Dalton Trans., 2018, 47, 14125–14138 RSC.
- S. J. Higgins and R. J. Nichols, Polyhedron, 2018, 140, 25–34 CrossRef CAS.
- T. C. Siu, J. Y. Wong, M. O. Hight and T. A. Su, Phys. Chem. Chem. Phys., 2021, 23, 9643–9659 RSC.
- X. Y. Xiao, B. Q. Xu and N. J. Tao, J. Am. Chem. Soc., 2004, 126, 5370–5371 CrossRef CAS PubMed.
- J. Juhaniewicz and S. Sek, Bioelectrochemistry, 2012, 87, 21–27 CrossRef CAS PubMed.
- J. M. Artes, Y. H. Li, J. Q. Qi, M. P. Anantram and J. Hihath, Nat. Commun., 2015, 6, 8870 CrossRef CAS PubMed.
- C. L. Guo, K. Wang, E. Zerah-Harush, J. Hamill, B. Wang, Y. Dubi and B. Q. Xu, Nat. Chem., 2016, 8, 484–490 CrossRef CAS PubMed.
- V. Ferri, M. Elbing, G. Pace, M. D. Dickey, M. Zharnikov, P. Samorì, M. Mayor and M. A. Rampi, Angew. Chem., Int. Ed., 2008, 47, 3407–3409 CrossRef CAS PubMed.
- W. Haiss, H. van Zalinge, S. J. Higgins, D. Bethell, H. Höbenreich, D. J. Schiffrin and R. J. Nichols, J. Am. Chem. Soc., 2003, 125, 15294–15295 CrossRef CAS PubMed.
- S. Y. Quek, M. Kamenetska, M. L. Steigerwald, H. J. Choi, S. G. Louie, M. S. Hybertsen, J. B. Neaton and L. Venkataraman, Nat. Nanotechnol., 2009, 4, 230–234 CrossRef CAS PubMed.
- B. Capozzi, Q. S. Chen, P. Darancet, M. Kotiuga, M. Buzzeo, J. B. Neaton, C. Nuckolls and L. Venkataraman, Nano Lett., 2014, 14, 1400–1404 CrossRef CAS PubMed.
- R. J. Brooke, C. J. Jin, D. S. Szumski, R. J. Nichols, B. W. Mao, K. S. Thygesen and W. Schwarzacher, Nano Lett., 2015, 15, 275–280 CrossRef CAS PubMed.
- H. X. Li, T. A. Su, M. Camarasa-Gomez, D. Hernangomez-Perez, S. E. Henn, V. Pokorny, C. D. Caniglia, M. S. Inkpen, R. Korytar, M. L. Steigerwald, C. Nuckolls, F. Evers and L. Venkataraman, Angew. Chem., Int. Ed., 2017, 56, 14145–14148 CrossRef CAS PubMed.
- X.-S. Zhou, J.-H. Liang, Z.-B. Chen and B.-W. Mao, Electrochem. Commun., 2011, 13, 407–410 CrossRef CAS.
- L. A. Nagahara, T. Thundat and S. M. Lindsay, Rev. Sci. Instrum., 1989, 60, 3128–3130 CrossRef CAS.
- X. S. Liu, X. H. Li, S. Sangtarash, H. Sadeghi, S. Decurtins, R. Haner, W. J. Hong, C. J. Lambert and S. X. Liu, Nanoscale, 2018, 10, 18131–18134 RSC.
- Z. Li, M. Smeu, S. Afsari, Y. Xing, M. A. Ratner and E. Borguet, Angew. Chem., Int. Ed., 2014, 53, 1098–1102 CrossRef CAS PubMed.
- E. Amir, R. J. Amir, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2011, 133, 10046–10049 CrossRef CAS PubMed.
- G. Yang, S. Sangtarash, Z. Liu, X. Li, H. Sadeghi, Z. Tan, R. Li, J. Zheng, X. Dong, J. Liu, Y. Yang, J. Shi, Z. Xiao, G. Zhang, C. Lambert, W. Hong and D. Zhang, Chem. Sci., 2017, 8, 7505–7509 RSC.
- S. Sangtarash, H. Sadeghi and C. J. Lambert, Nanoscale, 2016, 8, 13199–13205 RSC.
- H. Schlicke and C. Herrmann, ChemPhysChem, 2014, 15, 4011–4018 CrossRef CAS PubMed.
- G. M. Morales, P. Jiang, S. W. Yuan, Y. G. Lee, A. Sanchez, W. You and L. P. Yu, J. Am. Chem. Soc., 2005, 127, 10456–10457 CrossRef CAS PubMed.
- Z. Y. Li, J. Huang, Q. X. Li and J. L. Yang, Sci. China, Ser. B: Chem., 2008, 51, 1159–1165 CrossRef CAS.
- G. P. Zhang, G. C. Hu, Z. L. Li and C. K. Wang, J. Phys. Chem. C, 2012, 116, 3773–3778 CrossRef CAS.
- X. Guo, J. P. Small, J. E. Klare, Y. Wang, M. S. Purewal, I. W. Tam, B. H. Hong, R. Caldwell, L. Huang, S. O'Brien, J. Yan, R. Breslow, S. J. Wind, J. Hone, P. Kim and C. Nuckolls, Science, 2006, 311, 356–359 CrossRef CAS PubMed.
- Y. Cao, S. H. Dong, S. Liu, Z. F. Liu and X. F. Guo, Angew. Chem., Int. Ed., 2013, 52, 3906–3910 CrossRef CAS PubMed.
- Y. P. Zhang, L. C. Chen, Z. Q. Zhang, J. J. Cao, C. Tang, J. Liu, L. L. Duan, Y. Huo, X. Shao, W. Hong and H. L. Zhang, J. Am. Chem. Soc., 2018, 140, 6531–6535 CrossRef CAS PubMed.
- G. C. Solomon, D. Q. Andrews, T. Hansen, R. H. Goldsmith, M. R. Wasielewski, R. P. Van Duyne and M. A. Ratner, J. Chem. Phys., 2008, 129, 054701 CrossRef PubMed.
- C. M. Guedon, H. Valkenier, T. Markussen, K. S. Thygesen, J. C. Hummelen and S. J. van der Molen, Nat. Nanotechnol., 2012, 7, 304–308 CrossRef PubMed.
- V. Rabache, J. Chaste, P. Petit, M. L. Della Rocca, P. Martin, J. C. Lacroix, R. L. McCreery and P. Lafarge, J. Am. Chem. Soc., 2013, 135, 10218–10221 CrossRef CAS PubMed.
- M. Kamenetska, S. Y. Quek, A. C. Whalley, M. L. Steigerwald, H. J. Choi, S. G. Louie, C. Nuckolls, M. S. Hybertsen, J. B. Neaton and L. Venkataraman, J. Am. Chem. Soc., 2010, 132, 6817–6821 CrossRef CAS PubMed.
- C. Tang, L. Huang, S. Sangtarash, M. Noori, H. Sadeghi, H. Xia and W. Hong, J. Am. Chem. Soc., 2021, 143, 9385–9392 CrossRef CAS PubMed.
- Y. S. Park, A. C. Whalley, M. Kamenetska, M. L. Steigerwald, M. S. Hybertsen, C. Nuckolls and L. Venkataraman, J. Am. Chem. Soc., 2007, 129, 15768–15769 CrossRef CAS PubMed.
- D. V. Leff, L. Brandt and J. R. Heath, Langmuir, 1996, 12, 4723–4730 CrossRef CAS.
- F. Chen, X. L. Li, J. Hihath, Z. F. Huang and N. J. Tao, J. Am. Chem. Soc., 2006, 128, 15874–15881 CrossRef CAS PubMed.
- Z. J. Zhang and T. Imae, Nano Lett., 2001, 1, 241–243 CrossRef CAS.
- S. Ahn, S. V. Aradhya, R. S. Klausen, B. Capozzi, X. Roy, M. L. Steigerwald, C. Nuckolls and L. Venkataraman, Phys. Chem. Chem. Phys., 2012, 14, 13841–13845 RSC.
- J. M. Brisendine, S. Refaely-Abramson, Z.-F. Liu, J. Cui, F. Ng, J. B. Neaton, R. L. Koder and L. Venkataraman, J. Phys. Chem. Lett., 2018, 9, 763–767 CrossRef CAS PubMed.
- L. Scullion, T. Doneux, L. Bouffier, D. G. Fernig, S. J. Higgins, D. Bethell and R. J. Nichols, J. Phys. Chem. C, 2011, 115, 8361–8368 CrossRef CAS.
- L. Venkataraman, Y. S. Park, A. C. Whalley, C. Nuckolls, M. S. Hybertsen and M. L. Steigerwald, Nano Lett., 2007, 7, 502–506 CrossRef CAS PubMed.
- M. Smeu, R. A. Wolkow and G. A. DiLabio, J. Chem. Phys., 2008, 129, 034707 CrossRef PubMed.
- C. Jin, M. Strange, T. Markussen, G. C. Solomon and K. S. Thygesen, J. Chem. Phys., 2013, 139, 184307 CrossRef PubMed.
- M. H. Garner, M. Koerstz, J. H. Jensen and G. C. Solomon, ACS Phys. Chem. Au., 2022 DOI:10.1021/acsphyschemau.2c00016.
- T. R. Fu, S. Smith, M. Camarasa-Gomez, X. F. Yu, J. Y. Xue, C. Nuckolls, F. Evers, L. Venkataraman and S. J. Wei, Chem. Sci., 2019, 10, 9998–10002 RSC.
- H. Audi, Y. Viero, N. Alwhaibi, Z. Chen, M. Iazykov, A. Heynderickx, F. Xiao, D. Guerin, C. Krzeminski, I. M. Grace, C. J. Lambert, O. Siri, D. Vuillaume, S. Lenfant and H. Klein, Nanoscale, 2020, 12, 10127–10139 RSC.
- X. Y. Pan, B. Lawson, A. M. Rustad and M. Kamenetska, Nano Lett., 2020, 20, 4687–4692 CrossRef CAS PubMed.
- C. L. Wu, A. Alqahtani, S. Sangtarash, A. Vezzoli, H. Sadeghi, C. M. Robertson, C. X. Cai, C. J. Lambert, S. J. Higgins and R. J. Nichols, Nanoscale, 2020, 12, 7914–7920 RSC.
- I. L. Herrer, A. K. Ismael, D. C. Milan, A. Vezzoli, S. Martin, A. Gonzalez-Orive, I. Grace, C. Lambert, J. L. Serrano, R. J. Nichols and P. Cea, J. Phys. Chem. Lett., 2018, 9, 5364–5372 CrossRef CAS PubMed.
- L. Herrer, S. Martin, A. Gonzalez-Orive, D. C. Milan, A. Vezzoli, R. J. Nichols, J. L. Serrano and P. Cea, J. Mater. Chem. C, 2021, 9, 2882–2889 RSC.
- L. M. Balesteros, S. Martin, S. Marques-Gonzalez, M. C. Lopez, S. J. Higgins, R. J. Nichols, P. J. Low and P. Cea, J. Phys. Chem. C, 2015, 119, 784–793 CrossRef.
- S. Bock, O. A. Al-Owaedi, S. G. Eaves, D. C. Milan, M. Lemmer, B. W. Skelton, H. M. Osorio, R. J. Nichols, S. J. Higgins, P. Cea, N. J. Long, T. Albrecht, S. Martín, C. J. Lambert and P. J. Low, Chem. – Eur. J., 2017, 23, 2133–2143 CrossRef CAS PubMed.
- H. M. Osorio, S. Martin, M. C. Lopez, S. Marques-Gonzalez, S. J. Higgins, R. J. Nichols, P. J. Low and P. Cea, Beilstein J. Nanotechnol., 2015, 6, 1145–1157 CrossRef CAS PubMed.
- R. R. Ferradás, S. Marqués-González, H. M. Osorio, J. Ferrer, P. Cea, D. C. Milan, A. Vezzoli, S. J. Higgins, R. J. Nichols, P. J. Low, V. M. García-Suárez and S. Martín, RSC Adv., 2016, 6, 75111–75121 RSC.
- L. M. Ballesteros, S. Martín, J. Cortés, S. Marqués-González, S. J. Higgins, R. J. Nichols, P. J. Low and P. Cea, Chem. – Eur. J., 2013, 19, 5352–5363 CrossRef CAS PubMed.
- I. L. Herrer, A. K. Ismael, D. C. Milán, A. Vezzoli, S. Martín, A. González-Orive, I. Grace, C. Lambert, J. L. Serrano, R. J. Nichols and P. Cea, J. Phys. Chem. Lett., 2018, 9, 5364–5372 CrossRef CAS PubMed.
- Q. S. Ai, Q. Fu and F. Liang, Front. Chem., 2020, 8, 736 CrossRef CAS PubMed.
- G. Villarroel-Lecourt, J. Carrasco-Carvajal, F. Andrade-Villalobos, F. Solis-Egana, I. Merino-San Martin, J. Robinson-Duggon and D. Fuentealba, ACS Omega, 2018, 3, 8337–8343 CrossRef CAS PubMed.
- B. H. Xiao, F. Liang, S. M. Liu, J. Im, Y. C. Li, J. Liu, B. T. Zhang, J. H. Zhou, J. He and S. Chang, Nanotechnology, 2018, 29, 365501 CrossRef PubMed.
- T. W. Hudnall, C. W. Chiu and F. P. Gabbai, Acc. Chem. Res., 2009, 42, 388–397 CrossRef CAS PubMed.
- C. H. Zhao, A. Wakamiya, Y. Inukai and S. Yamaguchi, J. Am. Chem. Soc., 2006, 128, 15934–15935 CrossRef CAS PubMed.
- C. Jia, A. Migliore, N. Xin, S. Huang, J. Wang, Q. Yang, S. Wang, H. Chen, D. Wang, B. Feng, Z. Liu, G. Zhang, D.-H. Qu, H. Tian, M. A. Ratner, H. Q. Xu, A. Nitzan and X. Guo, Science, 2016, 352, 1443–1445 CrossRef CAS PubMed.
- N. Darwish, A. C. Aragones, T. Darwish, S. Ciampi and I. Diez-Perez, Nano Lett., 2014, 14, 7064–7070 CrossRef CAS PubMed.
- S. Cai, W. Deng, F. Huang, L. Chen, C. Tang, W. He, S. Long, R. Li, Z. Tan, J. Liu, J. Shi, Z. Liu, Z. Xiao, D. Zhang and W. Hong, Angew. Chem., Int. Ed., 2019, 58, 3829–3833 CrossRef CAS PubMed.
- S. Kumar, M. Merelli, W. Danowski, P. Rudolf, B. L. Feringa and R. C. Chiechi, Adv. Mater., 2019, 31, e1807831 CrossRef PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- L. Kortekaas, O. Ivashenko, J. T. van Herpt and W. R. Browne, J. Am. Chem. Soc., 2016, 138, 1301–1312 CrossRef CAS PubMed.
- L. Kortekaas, J. Chen, D. Jacquemin and W. R. Browne, J. Phys. Chem. B, 2018, 122, 6423–6430 CrossRef CAS PubMed.
- R. C. Chiechi, E. A. Weiss, M. D. Dickey and G. M. Whitesides, Angew. Chem., Int. Ed., 2008, 47, 142–144 CrossRef CAS PubMed.
- J. Bai, A. Daaoub, S. Sangtarash, X. H. Li, Y. X. Tang, Q. Zou, H. Sadeghi, S. Liu, X. J. Huang, Z. B. Tan, J. Y. Liu, Y. Yang, J. Shi, G. Meszaros, W. B. Chen, C. Lambert and W. J. Hong, Nat. Mater., 2019, 18, 364–369 CrossRef CAS PubMed.
- Y. Q. Li, M. Buerkle, G. F. Li, A. Rostamian, H. Wang, Z. X. Wang, D. R. Bowler, T. Miyazaki, L. M. Xiang, Y. Asai, G. Zhou and N. J. Tao, Nat. Mater., 2019, 18, 357–363 CrossRef CAS PubMed.
- H. Y. Fu, X. Zhu, P. H. Li, M. M. Li, L. Yang, C. C. Jia and X. F. Guo, J. Mater. Chem. C, 2022, 10, 2375–2389 RSC.
- M. Baghernejad, D. Z. Manrique, C. Li, T. Pope, U. Zhumaev, I. Pobelov, P. Moreno-Garcia, V. Kaliginedi, C. Huang, W. Hong, C. Lambert and T. Wandlowski, Chem. Commun., 2014, 50, 15975–15978 RSC.
- R. J. Brooke, D. S. Szumski, A. Vezzoli, S. J. Higgins, R. J. Nichols and W. Schwarzacher, Nano Lett., 2018, 18, 1317–1322 CrossRef CAS PubMed.
- M. Israeli, D. K. Laing and L. D. Pettit, Dalton Trans., 1974, 2194–2197, 10.1039/dt9740002194.
- R. J. Brooke, C. J. Jin, D. S. Szumski, R. J. Nichols, B. W. Mao, K. S. Thygesen and W. Schwarzacher, Nano Lett., 2015, 15, 275–280 CrossRef CAS PubMed.
- W. K. Zhao, D. Q. Zou, Z. P. Sun, Y. Q. Xu, G. M. Ji, Y. J. Yu and C. L. Yang, Adv. Theory Simul., 2019, 2, 1900057 CrossRef.
- S. Qiu, Y. Y. Miao, G. P. Zhang, J. F. Ren, C. K. Wang and G. C. Hu, J. Mater. Sci., 2020, 55, 16311–16322 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |