
Bioinspired NO release coating enhances endothelial cells and inhibits smooth muscle cells
Sheng-yu
Chen
 a,
Jing
Wang
b,
Fan
Jia
b,
Zhi-da
Shen
a,
Wen-bin
Zhang
a,
You-xiang
Wang
b,
Ke-feng
Ren
*ab,
Guo-sheng
Fu
*a and
Jian
Ji
ab
a,
Jing
Wang
b,
Fan
Jia
b,
Zhi-da
Shen
a,
Wen-bin
Zhang
a,
You-xiang
Wang
b,
Ke-feng
Ren
*ab,
Guo-sheng
Fu
*a and
Jian
Ji
ab
aKey Laboratory of Cardiovascular Intervention and Regenerative Medicine of Zhejiang Province, Department of Cardiology, Sir Run Run Shaw Hospital, Zhejiang University, Hangzhou 310016, China. E-mail: renkf@zju.edu.cn; fugs@zju.edu.cn
bMOE Key Laboratory of Macromolecule Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
First published on 6th October 2021
Abstract
Thrombus and restenosis after stent implantation are the major complications because traditional drugs such as rapamycin delay the process of endothelialization. Nitric oxide (NO) is mainly produced by endothelial nitric oxide synthase (eNOS) on the membrane of endothelial cells (ECs) in the cardiovascular system and plays an important role in vasomotor function. It strongly inhibits the proliferation of smooth muscle cells (SMCs) and ameliorates endothelial function when ECs get hurt. Inspired by this, introducing NO to traditional stent coating may alleviate endothelial insufficiency caused by rapamycin. Here, we introduced SNAP as the NO donor, mimicking how NO affects in vivo, into rapamycin coating to alleviate endothelial damage while inhibiting SMC proliferation. Through wicking effects, SNAP was absorbed into a hierarchical coating that had an upper porous layer and a dense polymer layer with rapamycin at the bottom. Cells were cultured on the coatings, and it was observed that the injured ECs were restored while the growth of SMCs further diminished. Genome analysis was conducted to further clarify possible signaling pathways: the effect of cell growth attenuated by NO may cause by affecting cell cycle and enhancing inflammation. These findings supported the idea that introducing NO to traditional drug-eluting stents alleviates incomplete endothelialization and further inhibits the stenosis caused by the proliferation of SMCs.
Introduction
Cardiovascular diseases (CVDs) are one of the leading causes of the high mortality worldwide.1 For coronary arteries with severe stenosis, implanting drug-eluting stents has become one of the most commonly used treatments.2 Drug-eluting stents (DESs) used clinically are mostly loaded with rapamycin or its analogues, which are released at the implantation site and have an anti-proliferative effect on cells.3,4 However, this effect is non-selective. While inhibiting the proliferation of SMCs affects ECs as well.5 The loss of endothelialization leads to the incomplete coverage on stent; therefore, thrombus and restenosis occur and gradually progress, which would cause late in-stent restenosis and disease recurrence.6,7Nitric oxide (NO) is a natural small molecule present in human body; it is mainly produced by endothelial nitric oxide synthase (eNOS) on the membrane of ECs in the cardiovascular system.8 NO plays an important role in vascular physiological processes and endothelial function.9,10 NO secreted by ECs relax SMCs, thereby regulating vasomotion under physiological conditions. When ECs are injured, the addition of exogenous NO improves the endothelial function8,11 and strongly inhibits the proliferation of SMCs.12–14 Because of the possibility NO offers to regulate cell behavior differently, we introduced NO to the rapamycin coating15,16 to alleviate the problem of incomplete endothelialization and further suppress the proliferation of SMCs.
NO has a high activity and short half-life that would be quickly oxidized to NO2− and NO3− and inactivated under oxygenated conditions.17 In human body, NO modifies the free sulfhydryl groups of protein cysteine to generate S–nitrosothiols (SNOs), and the weak S–N bond could be broken to release NO again.18,19 Therefore, GSNO (S–nitrosoglutathione) containing the SNO group is considered as the endogenous NO donor.14 We chose SNAP, which has the same SNO group,20 as the NO donor to simulate the transportation and storage of NO in human body.21,22 Although numerous studies have proved that NO has an inhibitory effect on SMCs,12,13 whether the combination of NO further inhibits the proliferation on the basis of rapamycin and how the signaling pathways work on cells have not been studied.
In this study, we fabricated a double-layer structure that had a dense polymer base layer loaded with rapamycin and an upper porous layer,23 which could load SNAP through the wicking process rapidly and conveniently before use to ensure the activity (Scheme 1). Genome analysis was performed to further study the effects of drugs on the signaling pathway and molecular function.
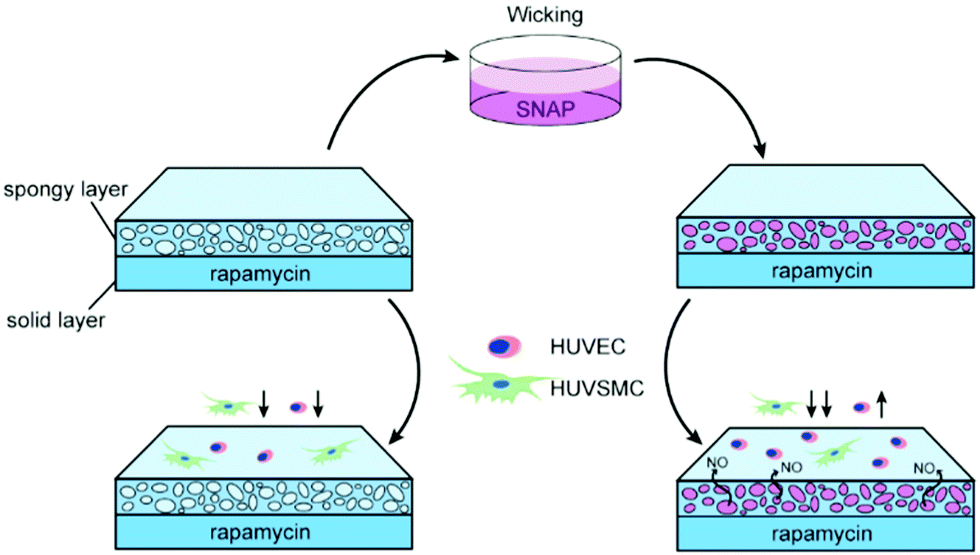 | ||
| Scheme 1 NO load and release into rapamycin coating and regulates cell behavior. | ||
Experimental
Materials
Rapamycin was purchased from MedChenExpress (USA). S–Nitroso-N-acetyl-DL-penicil-lamine (SNAP) and polyvinylpyrrolidone (PVP) were purchased from Sigma-Aldrich (USA). Poly (D,L-lactide-co-glycolide) (PLGA, LA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GA = 75:25, Mn = ∼65 kDa) was purchased from Jinan Daigang Biomaterial Co., Ltd. (Jinan, China). Rabbit anti-human von Willebrand factor (anti-vWF) monoclonal antibody (1:500) and anti-calponin (1:500) were purchased from Invitrogen (CA, USA). A nitric oxide detection kit was purchased from Beyotime Biotechnology (China). A Cell Counting Kit-8 (CCK-8) was purchased from Dojindo (Tokyo, Japan). An RNA-Quick Purification Kit was purchased from Yishan Biotechnology (Shanghai, China). Deionized (DI) water (>18 MΩ cm) used in experiments was provided by a Milli-pore Milli-Q water purification system. All materials were used as received without further purification.
:GA = 75:25, Mn = ∼65 kDa) was purchased from Jinan Daigang Biomaterial Co., Ltd. (Jinan, China). Rabbit anti-human von Willebrand factor (anti-vWF) monoclonal antibody (1:500) and anti-calponin (1:500) were purchased from Invitrogen (CA, USA). A nitric oxide detection kit was purchased from Beyotime Biotechnology (China). A Cell Counting Kit-8 (CCK-8) was purchased from Dojindo (Tokyo, Japan). An RNA-Quick Purification Kit was purchased from Yishan Biotechnology (Shanghai, China). Deionized (DI) water (>18 MΩ cm) used in experiments was provided by a Milli-pore Milli-Q water purification system. All materials were used as received without further purification.
Construction of hierarchical coatings
A PLGA solution was sprayed onto the substrate as the base layer, followed by the addition of a PVP/PLGA mixed solution (w/w 8/7) as the top layer by ultrasonic spraying. PVP was leached by water to obtain a microporous sponge-like structure. Rapamycin was dissolved into PLGA to prepare a drug-containing base layer with a concentration of 10 µg cm−2. The coating for cell experiments was immersed in the dopamine solution for 1 h to obtain a hydrophilic surface, and then rinsed with double-distilled water (ddH2O) several times, blow dried, and stored for use.Measurement of drug loading and release
Rapamycin release: the coatings loaded with rapamycin were placed in the culture medium, and the samples were taken out at different time periods, rinsed with ddH2O, vacuumdried, dissolved with ethyl acetate and measured by a UV-Vis spectrophotometer (UV-2550, Shimadzu, Japan). The released amount of rapamycin is the total amount minus the remaining content obtained from the test. SNAP loading: the samples were placed in a 24-well plate, 20 µl 500 µM SNAP was added to each sample wicking for 5 min, and the residual SNAP present on the surface with ddH2O was washed away. The samples were dissolved in ethyl acetate and measured by a UV-Vis spectrophotometer. NO Release: the release of NO was measured by the Nitric Oxide detection kit (Beyotime Biotechnology, China) through Griess reagent according to its instructions.Cell viability and cell growth
Cells were seeded in a 24-well plate with the hierarchical coatings at a density of 4 × 104 per well and incubated for 24 h. The Cell Counting Kit-8 (CCK-8) (Dojindo, Tokyo, Japan) was used to determine cell viability: 20 µL of the CCK-8 solution was added to each well with another 180 µL of the medium and the cells were incubated for 1–4 h at 37 °C, 5% CO2 environment. The absorbances at 450 nm were measured on a spectrophotometer. Cell viability was calculated using following the equation:| Cell viability (%) = absorbance (treated cells)/absorbance (control cells) × 100%. |
Cells were stained for 2 h with a rabbit anti-human von Willebrand factor (anti-vWF) monoclonal antibody (1:500) or anti-calponin monoclonal antibody (1:500) in order to obtain the fluorescence stained cells. The cells were isolated and seeded into 24-well plates with coatings. Cells were observed by a fluorescence microscope (Nikon, Japan) after 24 h.
RNA extraction and genome analysis
Total RNA of SMCs was extracted using a RNA-Quick Purification Kit (Yishan Biotechnology, Shanghai, China), following the manufacturer's instructions. All the RNA samples were sent to LC-Biotechnologies (Hangzhou, China) CO., Ltd for RNA-seq detection and analysis via Illumina Novaseq™ sequencer. Using DAVID bioinformatics resources (https://david.ncifcrf.gov/), we obtained the functional annotation clustering of genomic data through the KEGG database and GO database. Protein–protein interaction networks were analyzed by STRING (https://string-db.org/).cGMP analysis
cGMP secretion of SMCs was measured on a cGMP ELISA kit (Enzo Life Sciences, New York, USA) according to its manufacturer's instruction. Samples from “RAPA” and “RAPA + SNAP” were added into the plate provided by the kit. The conjugate and antibody were added into the appropriate wells. After incubation for 2 h at room temperature, the contents were removed and washed wells for 3 times. A pNpp substrate solution was added to each well and incubated for another 1 h at room temperature. The stop solution was added and the absorbances were read at 405 nm immediately. A Logit-Log paper was used to approximate the fitting curve. The concentrations of cGMP were determined by interpolation.qPCR detection
A PrimeScript RT- PCR kit (Takara, Dalian, China) was used to synthesize cDNA. qPCR detections were run with UltraSYBR Mixture (CWBIO, Beijing, China) and specific primers. The average threshold cycle values (Ct) were normalized to 18 s (human) to achieve a ΔCt value. The ΔCt values were normalized to “RAPA” as the control set to achieve a ΔΔCt value. The 2−ΔΔCt value was used for analysis.Data analysis
All experiments were performed in triplicate at a minimum. Data obtained were statistically analyzed via GraphPad with ANOVA or Student's t test. P value < 0.05 was considered significant.Results and discussion
Bilayer coating structure
In order to load SNAP on the coating surface, we used the PLGA/PVP mixed solution spraying on a PMMA substrate. The substrate was then leached by water to obtain a porous coating. PVP/PLGA mixed solutions with different ratios (3:7, 4:6, 8:7, and 6:4) were used to find a suitable content of PVP for fabricating the porous structure (Fig. 1(a)). Unevenly distributed pores and dense polymer were observed from the cross-section when the content of PVP is lower (30%, 40%). When the content of PVP is higher (60%), collapsed pores were observed. The contents with these ratios of PVP were not conducive to the uniform load of drugs. When the PVP content is 53%, the pore structure is favorable with a large size and the directly connected to each other, which were able to effectively load drugs and enable drugs to evenly distribute in the coating. This ratio of PVP/PLGA was used to further fabricate the double-layer coating. A hierarchical structure consisted of an upper layer with a porous structure and a lower layer with a dense polymer was observed, an obvious delamination was noticed between the upper and lower layers (Fig. 1(b)).
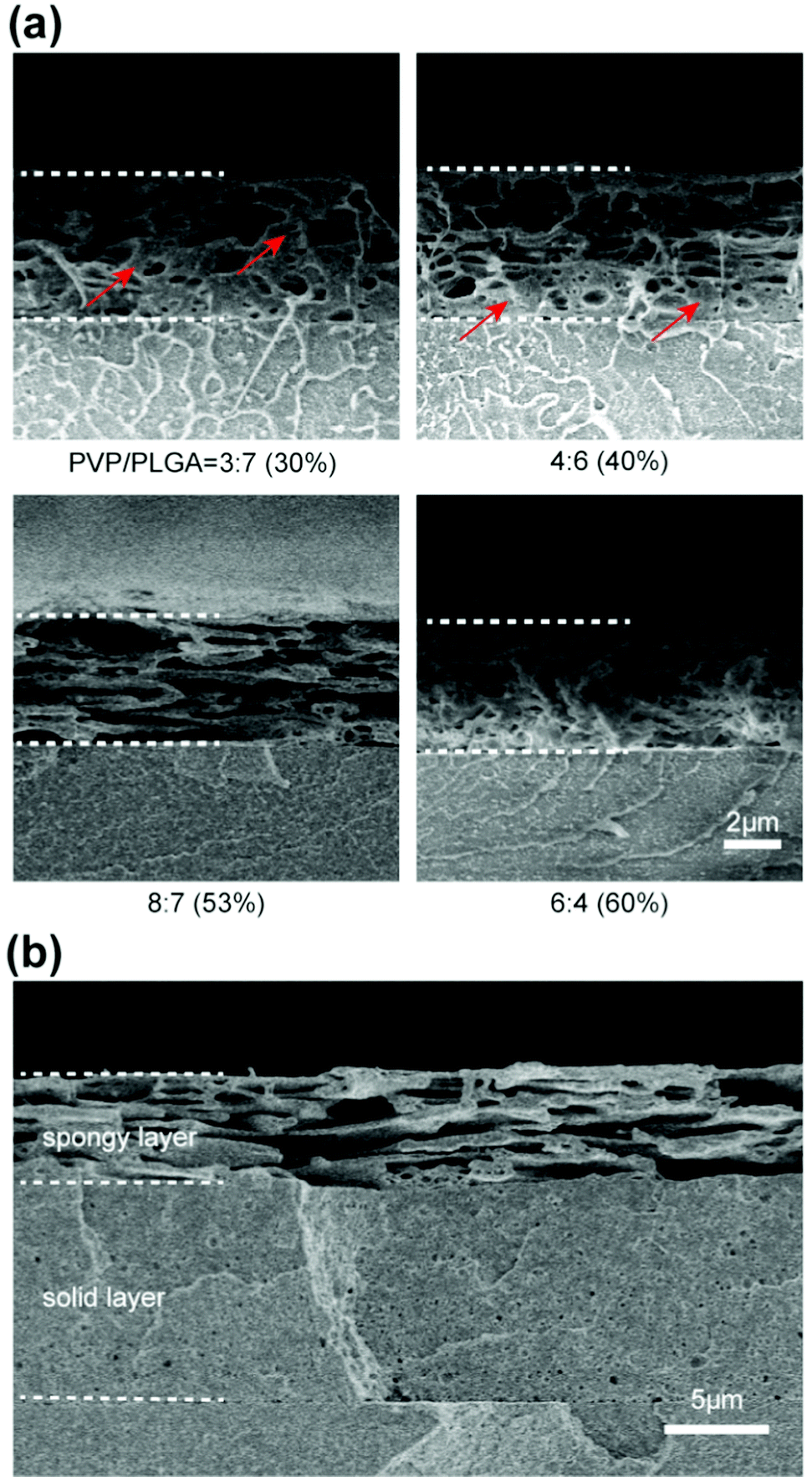 | ||
| Fig. 1 Preparation of the hierarchical structure. (a) Cross-section of the upper porous structure prepared with different ratios of PVP/PLGA, red arrows indicate dense polymer. (b) Cross-section of the hierarchical structure. | ||
In order to improve the effects of traditional stents on insufficient endothelialization and further inhibit SMCs, we decided to add SNAP on the basis of the traditional rapamycin coating. The initial burst of rapamycin release made the naked rapamycin coating not support the adhesion of ECs and SMCs.23 Therefore, we used the upper porous layer to separate the base rapamycin layer and cells.
Drug adsorption and release
Rapamycin was dissolved in the PLGA solution and sprayed directly on the substrate with a concentration of 10 µg cm−2. It was released rapidly in the first 48 h and slowly increased to 58.91 ± 0.25% of the total loading content in the following two weeks (Fig. 2(a)). In order to evaluate the wicking ability of the sponge layer, we dripped Dulbecco's Modified Eagle Medium (DMEM) with phenol red as an indicator on coatings. The liquid was quickly absorbed and gradually diffused to the surroundings (Fig. 2(b)). Considering the loading process relies on wicking, the concentration of the solution may have a great impact on the loading content. As shown in Fig. 2c, we studied the relationship between the SNAP concentration and loading content in the porous layer and found a linear relationship between them. It indicated that the solution concentration of water-soluble biomolecules was able to be adjusted to achieve an accurate control of coating load. We used the SNAP solution with a concentration of 500 µM to load into coatings, with a loading content of 131.217 ± 18.557 ng cm−2 to determine the NO release. Fig. 2(d) shows that NO was released rapidly in the first 40 h and gradually increased to 3.977 ± 0.144 µM in the next week. Once released into the solution, NO quickly generated NO2− and NO3− and got inactivated due to its unstable property.17 NO works only when it entered into the cells; fluorescent probes were used to detect NO in the cells seeded on the coatings. Fig. 2(e) shows that NO quickly entered into cells within 1 h and reached a large amount in 2 h.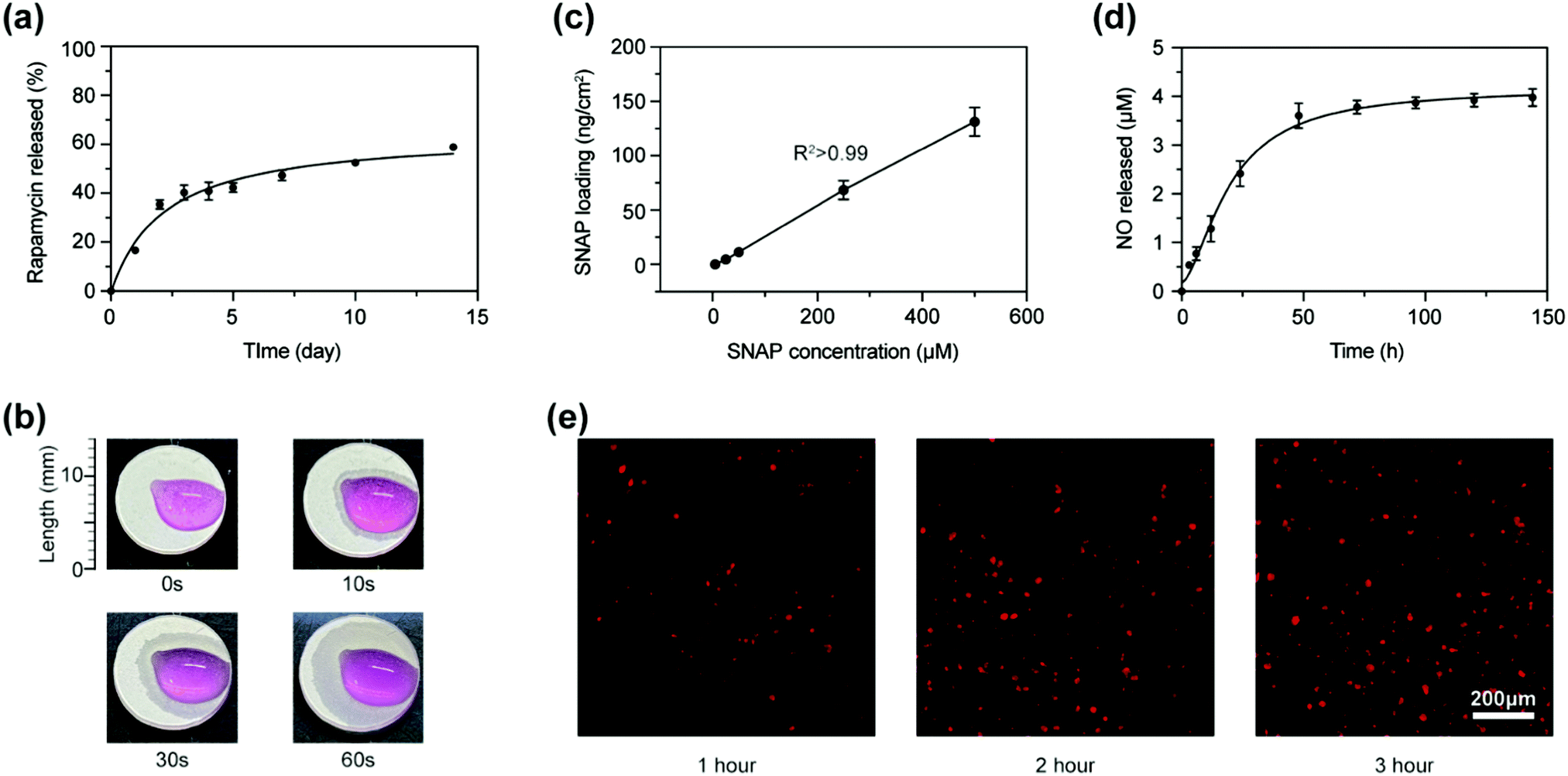 | ||
| Fig. 2 The loading and release profile of the hierarchical coating. (a) Rapamycin release profile as a function of time. (b) The wicking action of DMEM. (c) Loading of SNAP as a function of SNAP solution concentration. (d) SNAP release profile as a function of time. (e) Fluorescent probes of NO in cells at different times, scale bar is 200 µm. | ||
So far, there are many studies on promoting NO release to improve endothelial regeneration,24,25 such as immersing stents into an aqueous solution with dopamine and copper ions (CuII),26 incorporating glutathione peroxidase at the solid interface through the co-immobilization of selenocystamine in the framework of dopamine27 or to form an amine-bearing hexamethylenediamine, glutathione peroxidase-like CuII and adhesive catechol dopamine coating through a one-step molecule/ion coassembling procedure.28 In this way, coatings continuously released NO through catalytically decomposing the endogenous S–nitrosothiols (RSNO) from blood. A long-term continuous NO supplement may cause the inactivation of sGC protein in vivo.29–32 The NO-sGC-cGMP pathway is the key way for vascular SMCs to maintain normal contraction and relaxation functions. NO should keep releasing from coatings until a layer of functional ECs formed, which spontaneously produce NO. Such requirements just fit the releasing profiles of our coatings. Compared with catalytic NO generating coatings, the NO releasing rate of our coating is independent of native donors in blood, which may vary greatly from patient to patient. Therefore, our coating may have more stable performance.
Effect on cell proliferation
In order to verify the effect of the coatings on cell proliferation, we evaluated the qualitative and quantitative analysis through fluorescence staining and cell viability, respectively. The proliferations of ECs and SMCs were both significantly inhibited on RAPA coating. On RAPA-SNAP coating, the proliferation of SMCs is further inhibited, while ECs were relieved to a certain extent (Fig. 3(a)–(f)). When ECs and SMCs were co-cultured on the coatings, it is observed that RAPA coating has an inhibitory effect on both ECs and SMCs (EC/SMC = 1.0). The ratio of EC/SMC increased (EC/SMC = 1.7) when cells was cultured on RAPA-SNAP coatings (Fig. 3(g) and (h)). This part of cell experiment indicated that RAPA-SNAP coating reduced the inhibitory effect of ECs on the premise of ensuring SMCs suppression.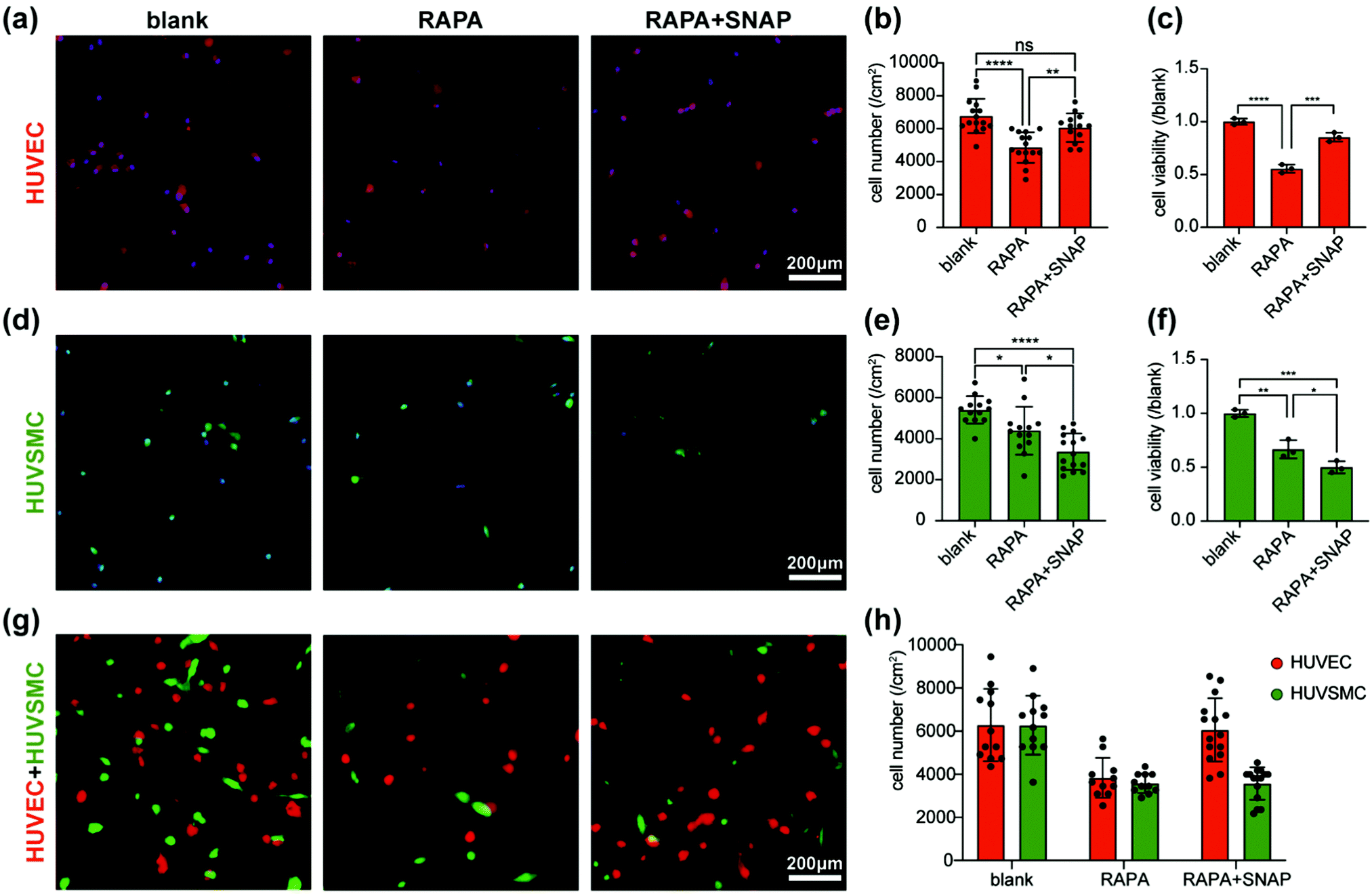 | ||
| Fig. 3 Effect of the hierarchical coatings on the proliferation of HUVECs and HUVSMCs. (a) Immunofluorescent images, (b) immunofluorescent statistical diagrams and (c) cell viability of HUVECs on the coatings. (d) immunofluorescent images, (e) immunofluorescent statistical diagrams and (f) cell viability of HUVSMCs on the coatings. (g) immunofluorescent images and (h) its statistical diagrams of co-culture of HUVECs and HUVSMCs. Scale bar is 200 µm. | ||
Previous studies have shown that rapamycin and its analogues reduce the expression of endothelial nitric oxide synthase (eNOS)33,34 and decrease the content of NO34 produced by EC through inhibiting the Akt-PI3K-mTORC pathway. Other studies had shown that the activity of mTORC1 is necessary and sufficient for the phosphorylation of eNOSS1177, so that rapamycin may inhibit the function of eNOS by directly suppressing the activity of mTORC1.35,36 The exogenous addition of NO may alleviate or reverse the vascular endothelial dysfunction by increasing the bioavailability of NO and reducing the damage of ROS to cells.37–39
Genome analysis
Previous studies proved that rapamycin inhibits the activity of the cyclin-dependent kinase (CDK)/cyclin complex and increases the expression of specific CDK inhibitor proteins (CDKI), such as p21 and p27, to prevent the entry of cells from the G1 Phase into S phase40,41 (DNA synthesis phase), thereby reducing cell proliferation. Moreover, past studies have discovered that the expression of p21 and p27 would be reduced after the treatment of VSMCs with NO donors.3 According to our experimental results above, NO further inhibited the proliferation of SMCs on the basis of the effect of rapamycin. We extracted RNA separately from cells of “RAPA” and “RAPA + SNAP” and used genome analysis to further explore how NO enhanced the effect of rapamycin.Based on the use of rapamycin, among the 60613 genes analyzed between two cell groups treated with SNAP or not, 807 genes had significant differences, of which 602 genes were up-regulated and 204 genes were down-regulated (Fig. 4). The heat map and volcano plot formed after clustering these differential genes are shown in the Fig. 4(a) and (c).
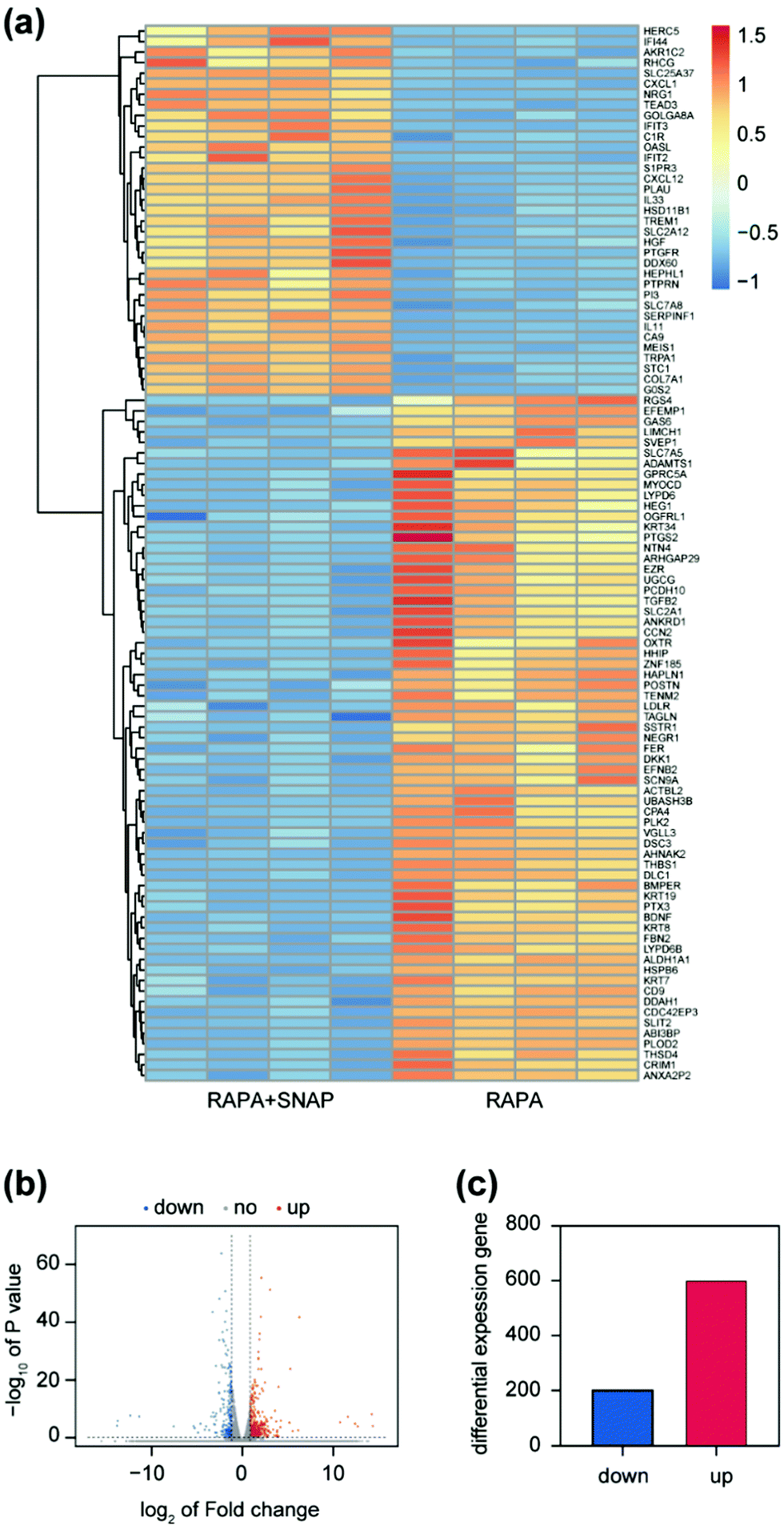 | ||
| Fig. 4 (a) The overall change in cell gene expression when cells cultured on RAPA and RAPA + SNAP coatings. The threshold is set to ≥1.5 times up (red) or ≤−1 times down (blue). (b) Total numbers of genes up-regulated or down-regulated in the two sets are denoted. (c) The volcano map depicts genes that are significantly up-regulated (red) and down-regulated (blue) between RAPA and RAPA + SNAP coatings. | ||
We used DAVID Bioinformatics Resources42 to analyze the differential genes.43 The enrichment analysis of GO and KEGG were performed through DAVID. GO database44 indicates gene ontology and divided the function of genes into three parts: cellular component (CC), molecular function (molecular function, MF), biological process (biological process, BP) (Fig. 5(a)), which showed adding NO may regulate cell proliferation, inflammation and cell adhesion. The most significant influence on gene expression is “regulation of cell proliferation”. In addition to the annotation of the function of the gene itself, the KEGG database45,46 showed us the various signaling pathways genes participated in. Analyzing signaling pathways helps to further explain biological function. The KEGG database is a main public pathway related database and more well-known to the public. The enrichment of differential genes was concentrated in TGF-β, NF-κB and TNF signaling pathways (Fig. 5(b)), which provided ideas for further verification in the follow-up.
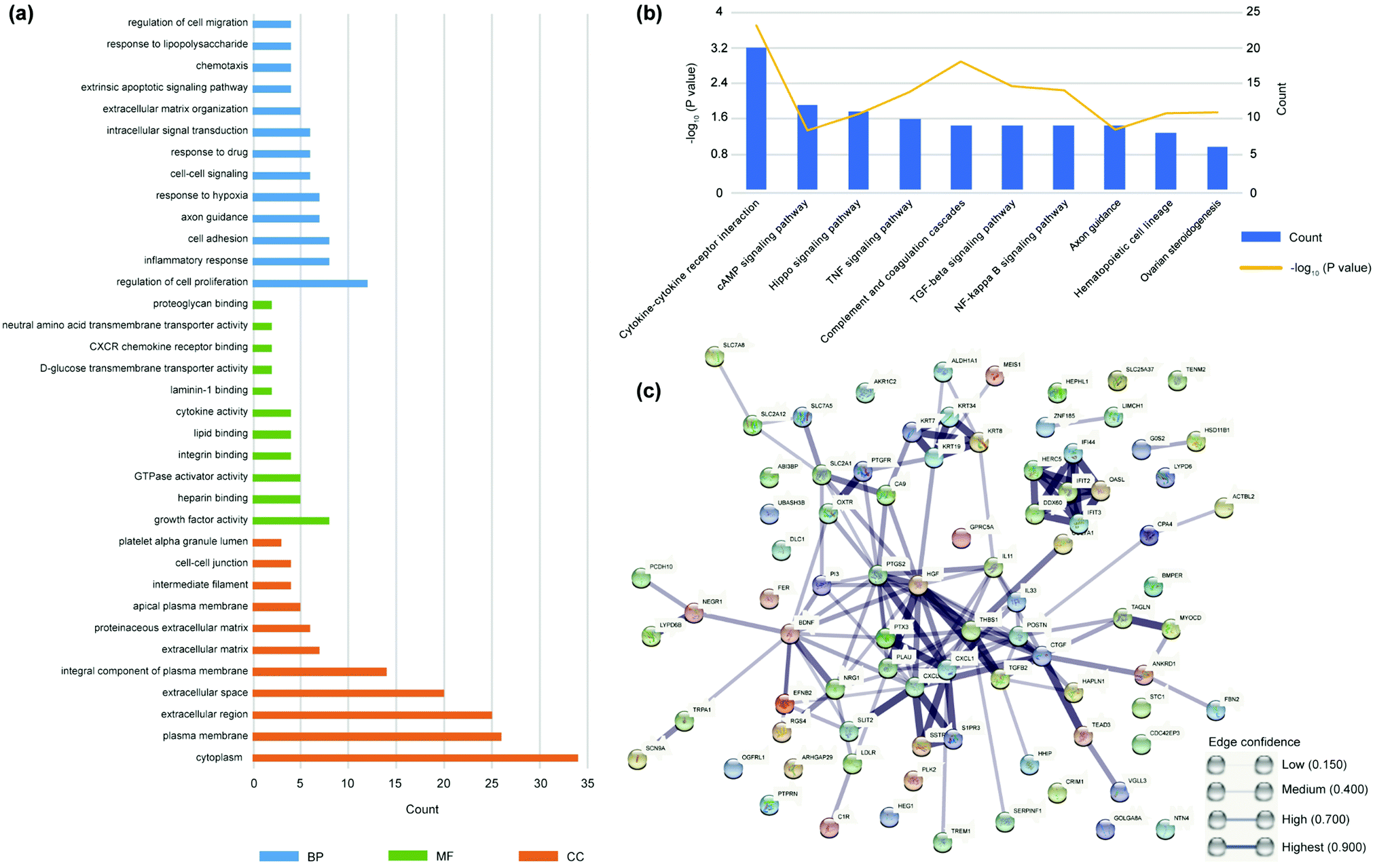 | ||
| Fig. 5 (a) All genes from RAPA vs. RAPA + SNAP coatings were classified according to three aspects: biological process (BP), molecular function (MF), and cellular component (CC). (b) The top 10 significant activated pathways with the most reliable significance from the differential genes enrichment of RAPA vs. RAPA + SNAP coatings, where the left y-coordinate is −log10 (Q-value), the Q-value is the P-value adjusted by multiple hypothesis testing. (c) STRING network analysis showed significant differences between RAPA and RAPA + SNAP coatings mainly in inflammatory and TGF-β2 pathways. | ||
The construction of a protein–protein interaction network (PPI network) helped us understand the role of these genes with expression differences in biological processes. We implemented a PPI network through the STRING database,47,48 which predicted protein–protein interactions. According to the STRING database, highly concentrated hub node genes were identified, including CXCL1, CXCL12, and TGF-β2 (Fig. 5(c)). Chemokines (CXCL1, CXCL12) are a kind of cytokines or signal proteins secreted by cells to induce the targeted chemotaxis of nearby responding cells. The highly concentrated hub nodes indicate that the proteins synthesized by differential genes play an important role in the biological changes of cell inflammation and apoptosis.
The genome analysis showed that the addition of NO may further reduce cell proliferation through inflammatory response. The mechanism of rapamycin on cell proliferation is by blocking the progression of the cell cycle. Therefore, we focused on inflammation and cell cycle in subsequent RNA verification and mechanism exploration.
Exploration of mechanism
We selected the key genes of the enrichement signaling pathways and found the ones with significant difference by qPCR; the downstream genes of their possible pathways were subjected to further qPCR detection. SMAD2, SMAD4 and their downstream p21 were found significantly different (Fig. 6(a)), which were all involved in cell cycle signaling pathways. Combined with the results from qPCR test, supposing NO up-regulates the activity of SMAD4 and SMAD2/3 through the TGF-β signaling pathway, thereby up-regulating the activity of p21, inhibiting the synthesis of cell cycle-related proteins arresting the progress of the G1 phase to S phase and thus inhibiting cell proliferation. Previous studies have proved that NO up-regulated the activity of p21 in SMCs49 but have not clarified its pathway. Our results were consistent with previous research and inferred on its possible pathways. Rapamycin achieved the effect of G1 arrest by inhibiting the synthesis of cell cycle-related proteins as well by inhibiting its target protein RPS6KB.50,51 Thus, these two drugs jointly inhibited the progress of the G1 phase through different pathways, forming a synergistic effect (Fig. 6(d)). We noticed that the cGMP signaling pathway is not listed in the enrichment pathways of differential genes. NO-sGC-cGMP is the classic signaling pathway for NO to act on SMCs.16,52 Therefore, cGMP Elisa kit was used for further verification and found that after adding SNAP, the content of cGMP in SMCs significantly increased (Fig. 6(b)). It is deduced that NO quickly entered into cells and had completed transcription and translation when cells’ RNA was to be extracted.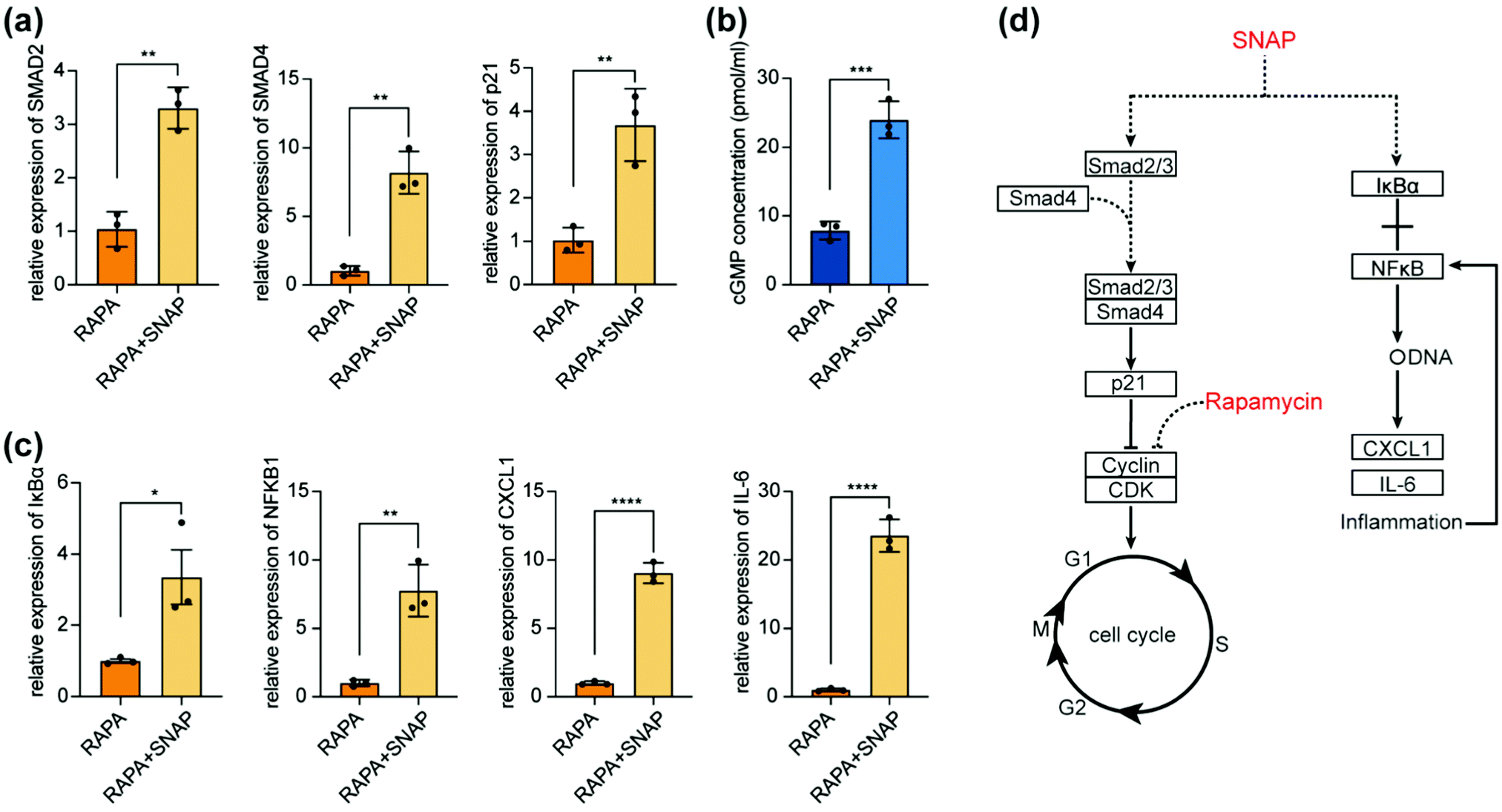 | ||
| Fig. 6 (a)–(c) Genes with obvious differences in enrichment signaling pathways. (b) cGMP content detected by an Elisa kit. (d) Possible synergistic effect between SNAP and RAPA. | ||
qPCR results showed that NO may activate the NF-κB signaling pathway: activating IκBα kinase and forming p50/RelA dimers, which increases the important nuclear transcription factor NF-kB in cells, thereby participating in cellular inflammatory response. We screened chemokines and inflammatory factors and found that CXCL1 and IL-6 significantly increased. The inflammatory process further aggravated the NF-κB signaling pathway, forming a positive feedback. This participation in the inflammatory response by activating the NF-κB pathway makes NO a further inhibitory response to cells (Fig. 6(c)).
Conclusions
Through the coating with hierarchical structure, we have successfully introduced NO to the traditional inhibitory drug rapamycin. Cell experiments confirmed that the coating further suppressed the growth of SMCs while it improved the endothelial repair. Genomic analysis and further verification showed that the addition of NO participated in the cell cycle by regulating the TGF-β signaling pathway, which has a synergistic effect with rapamycin, and strengthen the cellular inflammatory response through the NF-κB signaling pathway to further enhance the inhibition of cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key Research and Development Program of China (2020YFE0204400), the National Natural Science Foundation of China (21875210, U20A20262, 82070408 and 82070359), the Fundamental Research Funds for the Central Universities (2021FZZX003-01-03), Zhejiang Provincial Ten Thousand Talents Program (2018R52001), the Natural Key Research and Development Project of Zhejiang Province (2018C03015).Notes and references
- G. A. Roth, G. A. Mensah, C. O. Johnson, G. Addolorato, E. Ammirati, L. M. Baddour, N. C. Barengo, A. Z. Beaton, E. J. Benjamin, C. P. Benziger, A. Bonny, M. Brauer, M. Brodmann, T. J. Cahill, J. Carapetis, A. L. Catapano, S. S. Chugh, L. T. Cooper, J. Coresh, M. Criqui, N. DeCleene, K. A. Eagle, S. Emmons-Bell, V. L. Feigin, J. Fernandez-Sola, G. Fowkes, E. Gakidou, S. M. Grundy, F. J. He, G. Howard, F. Hu, L. Inker, G. Karthikeyan, N. Kassebaum, W. Koroshetz, C. Lavie, D. Lloyd-Jones, H. S. Lu, A. Mirijello, A. M. Temesgen, A. Mokdad, A. E. Moran, P. Muntner, J. Narula, B. Neal, M. Ntsekhe, G. Moraes de Oliveira, C. Otto, M. Owolabi, M. Pratt, S. Rajagopalan, M. Reitsma, A. L. P. Ribeiro, N. Rigotti, A. Rodgers, C. Sable, S. Shakil, K. Sliwa-Hahnle, B. Stark, J. Sundstrom, P. Timpel, I. M. Tleyjeh, M. Valgimigli, T. Vos, P. K. Whelton, M. Yacoub, L. Zuhlke, C. Murray, V. Fuster and G.-N.-J. G. B. o. C. D. W. Group, J. Am. Coll. Cardiol., 2020, 76, 2982–3021 CrossRef PubMed.
- J. W. Moses, N. Kipshidze and M. B. Leon, Am. J. Cardiovasc. Drugs, 2002, 2, 163–172 CrossRef CAS PubMed.
- F. C. Tanner, P. Meier, H. Greutert, C. Champion, E. G. Nabel and T. F. Luscher, Circulation, 2000, 101, 1982–1989 CrossRef CAS PubMed.
- B. J. Rensing, J. Vos, P. C. Smits, D. P. Foley, M. J. van den Brand, W. J. van der Giessen, P. J. de Feijter and P. W. Serruys, Eur. Heart J., 2001, 22, 2125–2130 CrossRef CAS PubMed.
- R. L. Wilensky, K. L. March, I. Gradus-Pizlo, G. Sandusky, N. Fineberg and D. R. Hathaway, Circulation, 1995, 92, 2995–3005 CrossRef CAS PubMed.
- J. E. Sousa, M. A. Costa, A. Abizaid, A. S. Abizaid, F. Feres, I. M. Pinto, A. C. Seixas, R. Staico, L. A. Mattos, A. G. Sousa, R. Falotico, J. Jaeger, J. J. Popma and P. W. Serruys, Circulation, 2001, 103, 192–195 CrossRef CAS PubMed.
- S. Glagov, Circulation, 1994, 89, 2888–2891 CrossRef CAS PubMed.
- E. W. Albrecht, C. A. Stegeman, P. Heeringa, R. H. Henning and H. van Goor, J. Pathol., 2003, 199, 8–17 CrossRef PubMed.
- S. Kawashima and M. Yokoyama, Arterioscler., Thromb., Vasc. Biol., 2004, 24, 998–1005 CrossRef CAS PubMed.
- T. Chataigneau, M. Feletou, P. L. Huang, M. C. Fishman, J. Duhault and P. M. Vanhoutte, Br. J. Pharmacol., 1999, 126, 219–226 CrossRef CAS PubMed.
- R. D. Rudic, E. G. Shesely, N. Maeda, O. Smithies, S. S. Segal and W. C. Sessa, J. Clin. Invest., 1998, 101, 731–736 CrossRef CAS.
- J. A. Bauer and H. L. Fung, J. Pharmacol. Exp. Ther., 1991, 256, 249–254 CAS.
- U. C. Garg and A. Hassid, J. Clin. Invest., 1989, 83, 1774–1777 CrossRef CAS PubMed.
- E. A. Kowaluk and H. L. Fung, J. Pharmacol. Exp. Ther., 1990, 255, 1256–1264 CAS.
- J. L. Ivy, Methodist Debakey Cardiovasc J., 2019, 15, 200–206 CrossRef PubMed.
- J. O. Lundberg, M. T. Gladwin and E. Weitzberg, Nat. Rev. Drug Discovery, 2015, 14, 623–641 CrossRef CAS PubMed.
- W. K. Kim, Y. B. Choi, P. V. Rayudu, P. Das, W. Asaad, D. R. Arnelle, J. S. Stamler and S. A. Lipton, Neuron, 1999, 24, 461–469 CrossRef CAS.
- J. S. Scharfstein, J. F. Keaney, Jr., A. Slivka, G. N. Welch, J. A. Vita, J. S. Stamler and J. Loscalzo, J. Clin. Invest., 1994, 94, 1432–1439 CrossRef CAS PubMed.
- N. Hogg, Annu. Rev. Pharmacol. Toxicol., 2002, 42, 585–600 CrossRef CAS PubMed.
- H. H. Al-Sa'doni and A. Ferro, Curr. Med. Chem., 2004, 11, 2679–2690 CrossRef.
- E. A. Kowaluk, R. Poliszczuk and H. L. Fung, Eur. J. Pharmacol., 1987, 144, 379–383 CrossRef CAS PubMed.
- J. S. Stamler, O. Jaraki, J. Osborne, D. I. Simon, J. Keaney, J. Vita, D. Singel, C. R. Valeri and J. Loscalzo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7674–7677 CrossRef CAS PubMed.
- J. Wang, Y. Xue, J. Liu, M. Hu, H. Zhang, K. Ren, Y. Wang and J. Ji, Research, 2020, 1458090 CAS.
- J. Mendhi, M. Asgari, G. Ratheesh, I. Prasadam, Y. Yang and Y. Xiao, Appl. Mater. Today, 2020, 19, 100562 CrossRef.
- T. Yang, Z. Du, H. Qiu, P. Gao, X. Zhao, H. Wang, Q. Tu, K. Xiong, N. Huang and Z. Yang, Bioact. Mater., 2020, 5, 17–25 CrossRef PubMed.
- F. Zhang, Q. Zhang, X. Li, N. Huang, X. Zhao and Z. Yang, Biomaterials, 2019, 194, 117–129 CrossRef CAS PubMed.
- Z. Yang, Y. Yang, L. Zhang, K. Xiong, X. Li, F. Zhang, J. Wang, X. Zhao and N. Huang, Biomaterials, 2018, 178, 1–10 CrossRef CAS.
- Y. Yang, P. Gao, J. Wang, Q. Tu, L. Bai, K. Xiong, H. Qiu, X. Zhao, M. F. Maitz, H. Wang, X. Li, Q. Zhao, Y. Xiao, N. Huang and Z. Yang, Research, 2020, 9203906 CAS.
- V. T. Dao, M. H. Elbatreek, M. Deile, P. I. Nedvetsky, A. Guldner, C. Ibarra-Alvarado, A. Godecke and H. Schmidt, Sci. Rep., 2020, 10, 10012 CrossRef CAS PubMed.
- N. Sayed, P. Baskaran, X. Ma, F. van den Akker and A. Beuve, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12312–12317 CrossRef CAS.
- P. Schmidt, M. Schramm, H. Schroder and J. P. Stasch, Eur. J. Pharmacol., 2003, 468, 167–174 CrossRef CAS.
- O. V. Evgenov, P. Pacher, P. M. Schmidt, G. Hasko, H. H. Schmidt and J. P. Stasch, Nat. Rev. Drug Discovery, 2006, 5, 755–768 CrossRef CAS.
- H. Ota, M. Eto, J. Ako, S. Ogawa, K. Iijima, M. Akishita and Y. Ouchi, J. Am. Coll. Cardiol., 2009, 53, 2298–2305 CrossRef CAS PubMed.
- A. Barilli, R. Visigalli, R. Sala, G. C. Gazzola, A. Parolari, E. Tremoli, S. Bonomini, A. Simon, E. I. Closs, V. Dall'Asta and O. Bussolati, Cardiovasc. Res., 2008, 78, 563–571 CrossRef CAS PubMed.
- B. Decker and K. Pumiglia, Phys. Rep., 2018, 6, e13733 CrossRef PubMed.
- C. Li, M. M. Reif, S. M. Craige, S. Kant and J. F. Keaney, Jr., Nitric oxide, 2016, 55-56, 45–53 CrossRef CAS.
- A. L. Sindler, B. S. Fleenor, J. W. Calvert, K. D. Marshall, M. L. Zigler, D. J. Lefer and D. R. Seals, Aging Cell, 2011, 10, 429–437 CrossRef CAS PubMed.
- M. El Assar, J. Angulo and L. Rodriguez-Manas, Free Radical Biol. Med., 2013, 65, 380–401 CrossRef CAS PubMed.
- L. Rochette, J. Lorin, M. Zeller, J. C. Guilland, L. Lorgis, Y. Cottin and C. Vergely, Pharmacol. Ther., 2013, 140, 239–257 CrossRef CAS PubMed.
- X. Wang and C. G. Proud, Trends Cell Biol., 2009, 19, 260–267 CrossRef CAS PubMed.
- J. L. Jewell and K. L. Guan, Trends Biochem. Sci., 2013, 38, 233–242 CrossRef CAS.
- D. W. Huang, B. T. Sherman, Q. Tan, J. Kir, D. Liu, D. Bryant, Y. Guo, R. Stephens, M. W. Baseler, H. C. Lane and R. A. Lempicki, Nucleic Acids Res., 2007, 35, W169–175 CrossRef PubMed.
- M. A. Mooney, J. T. Nigg, S. K. McWeeney and B. Wilmot, Trends Genet., 2014, 30, 390–400 CrossRef CAS PubMed.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin and G. Sherlock, Nat. Genet., 2000, 25, 25–29 CrossRef CAS PubMed.
- M. Kanehisa and S. Goto, Nucleic Acids Res., 2000, 28, 27–30 CrossRef CAS PubMed.
- M. Kanehisa, S. Goto, Y. Sato, M. Kawashima, M. Furumichi and M. Tanabe, Nucleic Acids Res., 2014, 42, D199–205 CrossRef CAS.
- T. Li, R. Wernersson, R. B. Hansen, H. Horn, J. Mercer, G. Slodkowicz, C. T. Workman, O. Rigina, K. Rapacki, H. H. Staerfeldt, S. Brunak, T. S. Jensen and K. Lage, Nat. Methods, 2017, 14, 61–64 CrossRef CAS PubMed.
- G. D. Bader and C. W. Hogue, BMC Bioinf., 2003, 4, 2 CrossRef PubMed.
- M. Ingham and G. K. Schwartz, J. Clin. Oncol., 2017, 35, 2949–2959 CrossRef CAS PubMed.
- J. Kim and K. L. Guan, Nat. Cell Biol., 2019, 21, 63–71 CrossRef CAS PubMed.
- K. Inoki, H. Ouyang, Y. Li and K. L. Guan, Microbiol. Mol. Biol. Rev., 2005, 69, 79–100 CrossRef CAS PubMed.
- C. Farah, L. Y. M. Michel and J. L. Balligand, Nat. Rev. Cardiol., 2018, 15, 292–316 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |